Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(12):3639-3652. doi:10.7150/thno.31693 This issue Cite
Research Paper
Integration of metabolomics and expression of enolase-phosphatase 1 links to hepatocellular carcinoma progression
Hao Zhuang1,2*, Zhaoyan Qiang3*, Xiaowen Shao2*, Huan Wang4, Yamei Dang2, Zun Wang2, Fei Wu2, Wen Wei4 ![]() , Yongmei Li2
, Yongmei Li2 ![]()
1. Department of Hepatic Biliary Pancreatic Surgery, Cancer Hospital Affiliated to Zhengzhou University, Zhengzhou, Henan Province 450000, China
2. Department of Pathogen Biology, School of Basic Medical Sciences, Key Lab of Immune Microenvironment and Disease (Ministry of Education) Tianjin Medical University, Tianjin 300070, China
3. Department of Pharmacology, School of Basic Medical Sciences, Tianjin Medical University, Tianjin 300070, China
4. School of Life Sciences, Chongqing University, Chongqing, 400044, China
*These authors contribute equally to this manuscript.
Received 2018-11-21; Accepted 2019-4-19; Published 2019-5-26
Abstract

Reprogramming of cellular metabolism is one of the hallmarks for cancer, in which tumor cells rewire their metabolic fluxes to generate sufficient energy and biosynthetic intermediates. Therefore, elucidating the correlation between cellular metabolism and hepatocellular carcinoma (HCC) progression may provide insights into novel approaches to cancer therapy.
Methods: We assembled an integrated pathway-level metabolic profiling by mining metabolomic, transcriptomic and proteomic data of three HCC cell lines with increasing metastatic potentials. Immunohistochemical staining was performed in a tissue microarray from 185 HCC clinical specimens. Kaplan-Meier survival and Cox regression analyses were applied to test the association between gene expression and survival outcome. In vitro assays were conducted to investigate the functional role of enolase-phosphatase 1 (ENOPH1) in HCC malignant behaviors. Reversed genetics analysis was performed to determine the function of ENOPH1 in HCC metastasis. An intrahepatic mouse model further confirmed the role of ENOPH1 in metastasis.
Results: We have determined that HCC cell metastasis is associated with alterations in metabolite levels and expressions of metabolic enzymes in the cysteine/methionine metabolism pathway, and show that one of metabolic enzymes, enolase-phosphatase 1 (ENOPH1), is persistently upregulated with an increase in metastatic potential. The upregulation of ENOPH1 expression was observed as an independent prognostic factor for HCC patients. ENOPH1 overexpression promoted cell migration and invasion, whereas ENOPH1 downregulation inhibited cell migration and invasion. Furthermore, an enhanced phosphorylation of AKT with ENOPH1 upregulation was observed. ENOPH1-mediated malignant capacity in HCC cells can be rescued by an AKT inhibitor.
Conclusion: Taken together, our findings illustrate that ENOPH1 promotes HCC progression and could serve as a novel biomarker and therapeutic target for HCC.
Keywords: hepatocellular carcinoma, enolase-phosphatase 1, AKT, omics technologies
Introduction
Reprogramming of cellular metabolism is one of the hallmarks of tumor cells [1, 2]. Tumor cells rewire their metabolic fluxes to generate sufficient energy and biosynthetic building blocks for malignant behaviors [3]. A mounting evidence indicates that cancer cells utilize aerobic glycolysis instead of oxidative phosphorylation to generate adenosine triphosphate (ATP) and biosynthetic intermediates for rapid growth [4]. Several metabolic enzymes in the glycolysis pathway, including pyruvate kinase M2 and lactate dehydrogenase, are highly expressed and associated with poor prognosis in hepatocellular carcinoma (HCC) patients [5, 6]. Glutaminolysis is another alterable bioenergetic profile in cancer cells. Glutaminolysis replenishes the tricarboxylic acid cycle to support accumulating biosynthetic precursors and NADPH for proliferating cancer cells [7-10]. Studies showing that cellular metabolism increases cancer cell aggressiveness are limited. Metastatic cancer cells utilize a special way in enhancing mitochondrial respiration, which is necessary to increase ATP production [11]. Moreover, dihydropyrimidine dehydrogenase, a pyrimidine-degrading enzyme, is upregulated during induction of epithelial-mesenchymal transition in high-grade carcinoma cells and in malignant cells to extravasate into the mouse lung [12]. Therefore, the association between cellular metabolism and HCC progression are worthy of further exploration and could provide insight into the development of novel approaches to cancer therapy.
Cancer cells have elevated intracellular polyamine levels, which are induced by the upregulated expression levels of polyamine biosynthetic enzymes contributing to supporting their rapid proliferation and preserving neoplastic phenotype [13, 14]. Moreover, high polyamine levels are positively associated with poorer prognosis for breast cancer patients [15]. Importantly, suppression of polyamine production would be a selective anti-tumor therapeutic strategy [16, 17]. For example, difluoromethyl ornithine, a suicide inhibitor of ornithine decarboxylase (a rate-limiting polyamine biosynthetic enzyme), has been assessed in pre-clinical and clinical trials [14, 16]. Unfortunately, clinical trials have shown that currently available inhibitors of enzymes in the polyamine metabolic pathway have limited effect. Thus, there is a need to identify novel diagnostic biomarkers or drug targets based on studies on polyamine metabolism. Enolase-phosphatase 1 (ENOPH1) is a bifunctional enolase-dephosphorylase enzyme of the methionine (Met) salvage pathway [18] that is required for polyamine biosynthesis via catalysis of S-adenosylmethionine (SAM) [19]. To date, except for the association between ENOPH1 overexpression and ischemic brain injury [20], report about the tumorigenesis relevance of ENOPH1 is limited.
After the Human Genome Project, studies on tumorigenesis and metastasis have been revolutionized by the development of omics technologies [21]. Omics technologies could provide the comprehensive observation of genomics, transcriptomics, proteomics, and metabolomics analyses to expand our understanding of disease processes [22]. Cancer is a complex systemic disease that is characterized by various dysfunctional mutually associated molecular networks that are constructed by alterations in DNA, RNA, protein, to metabolite [23, 24]. For example, the integrated analysis of copy number and gene expression has revealed novel breast cancer subgroups with distinct clinical outcomes [25]. An analysis combining gene expression profiling and DNA methylation revealed that genes belonging to the nucleoporin family play pivotal roles in the liver metastasis of colorectal cancer [26]. Another integrative analysis of transcriptomics and metabolomics identified four urinary biomarkers for breast cancer patients [27]. Therefore, the integration of multi-omics profiling may help to elucidate the molecular mechanisms involving tumorigenesis and cancer progress.
The present study also adopted an integrated analytical strategy of multi-omics data and caged a specific metabolic enzyme, ENOPH1. We found that increased ENOPH1 expression promoted HCC progression. The relationship between ENOPH1 and the clinical characteristics was examined, and the functional relevance of ENOPH1 in HCC metastasis was also assessed based on RNA-seq data from reversed genetic operation.
Methods
Chemicals, reagents, and antibodies
Lipofectamine 3000, BCA reagents, bovine serum albumin (BSA), and N-2-hydroxyethylpiperazine-N-2-ethane sulfonic acid (HEPES) were obtained from Invitrogen (Carlsbad, CA, USA). Rabbit polyclonal antibody against ENOPH1 was from Abcam (Cambridge, UK), rabbit polyclonal antibody against AKT and mouse monoclonal antibodies against p-AKT (Ser473) were from Cell Signaling Technology (Danvers, MA, USA), and mouse monoclonal antibodies against β-actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Enhanced chemiluminescence (ECL) reagents were purchased from Pierce Biotechnology (Rockford, IL, USA). Protease inhibitor cocktail tablets, X-tremeGENE siRNA transfection reagent, Adenosine-13C10,15N5 5'-triphosphate sodium salt solution (13C10, 15N5 5'-ATP), S-adenosyl-L-Methionine (SAM) were purchased from Sigma-Aldrich (Shanghai, China). The ENOPH1-expressing plasmid was purchased from Sino Biological Inc (Beijing, China). The shRNA against ENOPH1 and non-targeting shRNA were designed and synthesized by GenePharma (Shanghai, China). The siRNA against ENOPH1 and non-targeting siRNA were designed and synthesized by RiboBio (Guangzhou, China). Perifosine was purchased from Santa Cruz Biotechnology (CA, USA).
Multi-omics data of three cell lines
Metabolomic data were obtained from our previous study [28]. Transcriptomics data were downloaded from the Gene Expression Omnibus (Bioproject: PRJNA399198), and proteomics data were from PRIDE (PXD005647). To investigate the correlation between metastatic capabilities and the levels of mRNA, protein, and metabolite, the metastatic capability of Huh7, MHCC97L, and HCCLM3 was quantitated as 1, 2, 3, respectively. Absolute values of correlation coefficient greater than 0.8 (P < 0.05) were used as a cutoff to indicate that the levels of mRNA, protein, and metabolite were significantly correlated with metastatic capability. Mbrole 2.0 (http://csbg.cnb.csic.es/mbrole2/) was used in KEGG pathway analysis of the metabolites that were potentially related to metastasis. R package “KEGG profile” was used for enriching differentially expressed genes (DEGs) in the KEGG pathway.
Tissue samples and analysis
Twenty-eight paired HCC and para-tumor tissues were collected between January and March 2016 from the Affiliated Cancer Hospital of Zhengzhou University (cohort #1, Table S1, Henan Cancer Hospital, Henan, China). Another 185 paraffin-embedded HCC tissues (cohort #2, Table S2) were collected between 2011 and 2012 from the same hospital. HCC diagnosis was confirmed by pathology. Informed consent was provided by each patient, and ethics approval was approved by the Ethics Committee (No. 2016CT054) of Henan Cancer Hospital.
Immunohistochemical analysis
Five-micrometer sections of the paraffin-embedded samples were placed on microscopic slides, followed by 15 min of dewaxing, rehydration in an alcohol gradient, antigen retrieval in 1% sodium citrate buffer (pH 6.0), and incubation in 3% hydrogen peroxide. The sections were then incubated with anti-ENOPH1 overnight at 4°C. Protein was visualized using the Polink-1 HRP DAB detection Kit (ZSGB-BIO, Beijing, China).
RNA extraction and quantitative reverse transcription PCR (qPCR)
RNA extraction and qPCR were performed as previously described [29]. The primer sequences used for qPCR are listed in Table S3.
Western blot
Western blot analysis was performed as described in our previous study [30].
Cell culture and transfection
HCC cell lines, including Huh7, PLC, HepG2, MHCC97L, MHCC97H, HCCLM3, and HCCLM6 cells were cultured in DMEM supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin at 37°C in a humidified 5% CO2 atmosphere. The ENOPH1 construct was transfected into cells using Lipofectamine 3000. The ENOPH1 shRNA or siRNAs and its scrambled control siRNAs (Table S4) were transfected into cells using X-tremeGENE siRNA Transfection Reagent. The overexpression or knockdown efficiency of ENOPH1 was confirmed by qPCR and Western blot analysis.
Cell proliferation assays
A growth curve assay was conducted according to the Kratzat's method [31]. 1×105 cells were seeded evenly in six-well plates. Cells were counted and replaced at the same quantity at that at the start point every two days.
For the colony formation assay, 103 cells were plated into six-well plates, and the culture medium was replaced every three days. After two weeks, cell colonies were stained with 0.01% crystal violet and counted manually.
Wound healing, migration, and invasion assays
For the wound healing assay, cells were seeded evenly into six-well plates. Upon reaching 80% - 90% confluency, the cells were scratched with a pipette tip in the middle of the well and gently washed with PBS, then incubated in culture medium containing 1% BSA. The wound closure was monitored microscopically at different time-points and photographed at 0 h and 48 h.
Invasion and migration assays were performed using Transwell chambers containing polycarbonate membranes with eight-micrometer pores (Millipore, Billerica, MA, USA) coated with or without Matrigel matrix (Corning, Lowell, MA, USA ). 2×105 of pre-starved cells in the DMEM were added to the upper chamber, and 600 μL of the complete culture media were added to the lower chamber. After 24 h of incubation at 37°C, the cells on the upper surface were wiped off. The invading or migrating cells on the under surface were fixed, stained, and photographed at six random regions at 100×.
Liquid chromatography-mass spectrometry (LC/MS) analysis
A relative quantitative polar matabolomic data was acquired with a 5500 qTRAP hybrid repine quadrupole mass spectrometer (AB/SCIEX). In a brief, metabolites were extracted from 1×107 cell with 500 μL of pre-cooled methanol containing 2 μL of 13C10, 15N5 5'-ATP. Then, after separated with Acquity UPLC HSS T3 (1.7 μm; 2.1 mm i.d. × 100 mm length; Waters), the metabolites were detected with positive/negative ion switching from a single 15 min LC/MS acquisition. The MRM pairs of standard metabolites were methionine (ESI+, 148/100), SAM (ESI+, 397/134), S-Adenosyl-L-homocysteine (ESI+, 383/134).The detected peaks of metabolites and 13C10, 15N5 5'-ATP were integrated to generate chromatographic peak areas to quantify using MultiQuant 2.0 (AB/SCIEX, framingham, MA, USA).
RNA-seq of reversed genetics operated cells
RNA-seq of ENOPH1-regulated HCC cells and their corresponding control cells were performed at Novogene (Beijing, China) using Illumina 2500. We sequenced 12 samples, including three pairs of ENOPH1-knockdown cells and their controls, and three pairs of ENOPH1-overexpressing cells and their controls. All sequencing data that support this operation have been deposited to the National Center for Biotechnology Information (Bioproject: PRJNA502993). Adapters and low-quality reads were cleaned by using Trimmomatic [32]. Cleaned reads were mapped to the reference genome (GRCh38) with HISAT2 [33] and then remaining reads were sorted by samtools [34]. All mRNA reads were quantitated by Stringtie [35], and mRNAs with read depth < 10 were removed. Then, R package DEGseq was used for search DEGs (logFC > 1, q < 0.05).
In vivo metastasis assays
An orthotopic xenograft model was constructed using four-week-old male BALB/c-nude mice [30, 36]. Briefly, the mice were anesthetized using an intraperitoneal injection of 1.2% 2,2,2-tribromoethanol (240mg/kg; Sigma-Aldrich). A 5- to 8-mm transverse incision was made below the xiphoid. The left hepatic lobe of mice was pulled out of the abdominal cavity and inoculated with 2 × 106 cells, which was mixed previously with Matrigel matrix (Corning) at a 1:1 ratio in a total volume of 40 μL. The mice were sacrificed at the sixth week after surgery and the number of intrahepatic metastases was counted manually.
Statistics
Statistical analyses were performed using R and the data were presented as the means ± SEM (Standard Error of Mean). Cell proliferation and wound-healing assays were analyzed by two-way ANOVA. Student's t-test was used to compare the difference of the mean values between groups. Clinicopathological correlations were calculated using a Pearson's chi-square test. A Kaplan-Meier survival analysis and log-rank tests were used to calculate overall survival (OS) and disease-free survival (DFS). Statistical significance was considered as P < 0.05.
Results
Multi-omics reveals ENOPH1 expression correlating with HCC metastasis
Here, we identified metabolites associated with HCC progress by computing and comparing metabolomic data from three cell lines with increasing metastatic potentials, Huh7, MHCC97L, and HCCLM3 cells [37]. We found that the levels of 42 metabolites were significantly altered with metastatic potential (Figure 1A). Among the identified 42 metabolites, the levels of 27 metabolites showed negative correlations to metastatic potential, which were reduced in MHCC97L cells compared to Huh7 cells, and further reduced in HCCLM3 cells compared to MHCC97L cells (Figure 1A, top panel). In contrast, the levels of 15 metabolites showed positive correlations and were elevated in MHCC97L cells compared to Huh7 cells, and further elevated in HCCLM3 cells compared to MHCC97L cells (Figure 1A, bottom panel). We then enriched those 42 differential metabolites into the KEGG pathways, and they concentrated in seven metabolic pathways, including cysteine/methionine metabolism, purine metabolism, and nicotinate/nicotinamide metabolism (Figure 1B).
Multi-omics reveals ENOPH1 expression correlating with HCC metastasis. (A) Heatmap showing change of metabolite levels that significantly related with metastasis. (B) seven KEGG pathway enrichment based on the differential metabolites. (C) Number of DEGs obtaining from transcriptomic and proteomic data in the seven pathways. (D) qRT-PCR analysis of ENOPH1. Data represent the means ± SEM of triplicate independent analyses. (E) Western blots showing ENOPH1 expression in seven HCC cell lines. β-actin was used as a loading control.
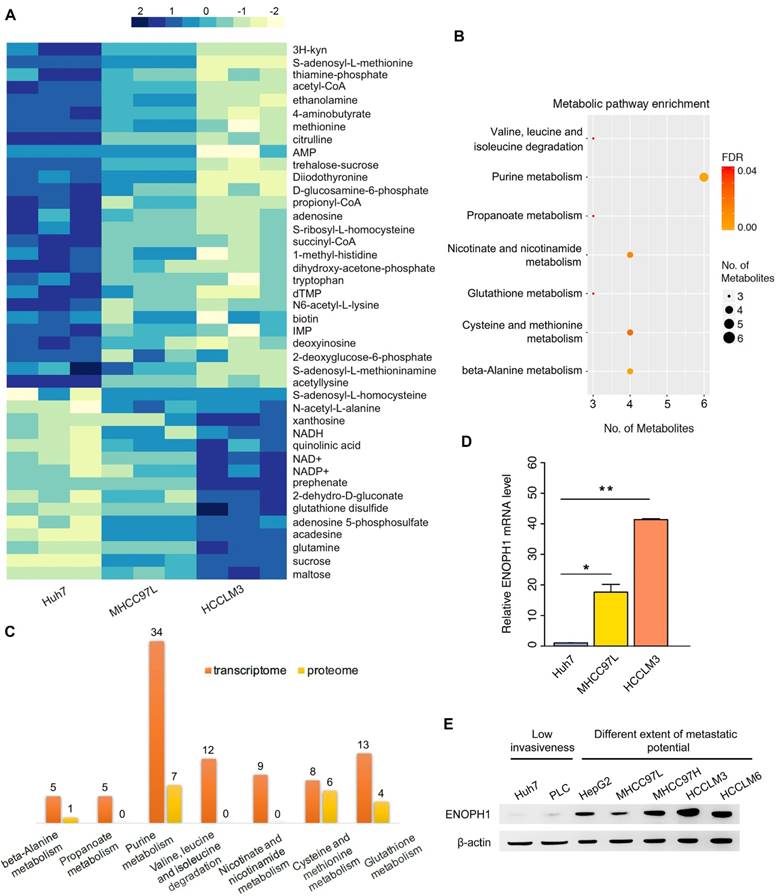
To understand the gene expression profiles involved in these seven enriched metabolic pathways, pathway-related differentially expressed genes (DEGs) were identified using the transcriptomic and proteomic data of Huh7, MHCC97L, and HCCLM3 cells (Figure 1C). The results showed that the pathway of cysteine/methionine metabolism displayed the highest percentage of enriched DEGs both in the transcriptomic data (8/45 = 17.8%) and the proteomic data (6/45 = 13.3%) (Table S5).
Among the DEGs in the cysteine/methionine metabolism pathway, ENOPH1 (Gene ID: 58478) expression was shown to be upregulated with metastatic capability at both the mRNA and protein levels. qPCR analysis confirmed that the expression of ENOPH1 was increased in MHCC97H cells and further upregulated in HCCLM3 and HCCLM6 cells (Figure 1D, Supplemental Fig S1A). We also examined the protein levels of ENOPH1 in a series of HCC cell lines. ENOPH1 could be detected in all seven HCC cell lines and in significantly upregulated metastatic HCC cells, particularly the MHCC97H, HCCLM3, and HCCLM6 cells (Figure 1E). Taken together, we observe that the increased expression of ENOPH1 is positively correlated to metastatic potentials of the HCC cells. Thus, we focused on ENOPH1 for the subsequent studies.
ENOPH1 is overexpressed in HCC patients
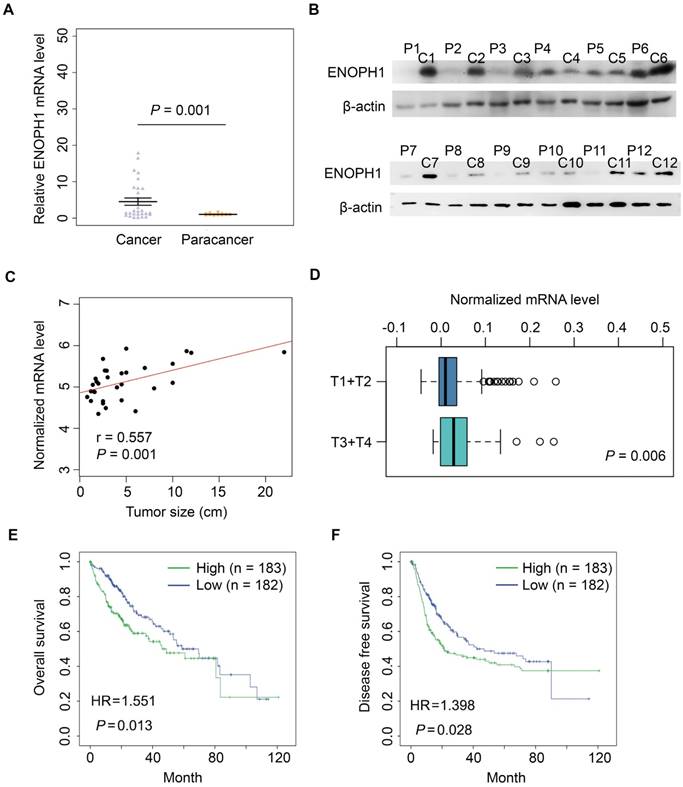
ENOPH1 expressions were measured in 28 paired of HCC samples (cohort #1) by qPCR. The average fold change of ENOPH1 mRNA levels in HCC was significantly higher compared to para-tumor tissues (5.91 vs. 1.00, Figure 2A). Simultaneously, the increased protein levels were also observed in 9 of 12 randomly tested HCC tissues compared to para-tumor tissues (Figure 2B). Moreover, the ENOPH1 expression levels were significantly positively correlated to tumor size in the HCC clinical dataset (www.oncomine.org) (Figure 2C). In addition, the expression level of ENOPH1 was significantly higher in T3/T4 stage tumors (n = 48) than T1/T2 stage tumors (n = 276), as indicated by The Cancer Genome Atlas (TCGA) clinical data (Figure 2D). ENOPH1-overexpression patients (TCGA cohort) showed a shorter OS (Figure 2E) and DFS (Figure 2F) after surgery. Taken together, our results indicate that ENOPH1 is overexpressed in HCC patients.
ENOPH1 is overexpressed in HCC patients. (A) ENOPH1 mRNA expressions in paired HCC samples and para-tumor tissues. Data represent the means ± SEM (P = 0.001, Student's t-test). (B) ENOPH1 protein expression in paired HCC samples and para-tumor samples. (C) ENOPH1 expression is positively correlated to tumor size from an HCC clinical dataset of oncomine (n = 31, r = 0.557, P = 0.001). (D) Expression levels of ENOPH1 in T3+T4 HCC samples are higher than those in T1+T2 HCC samples from the TCGA clinical data (P = 0.006, Student's t-test). (D) High ENOPH1 expression affects the OS of the patients from the TCGA clinical data (P = 0.013, Kaplan-Meier survival analysis). (F) high ENOPH1 expression affects the DFS of the patients from the TCGA clinical data (P = 0.028, Kaplan-Meier survival analysis).
ENOPH1 overexpression is associated with clinicopathological characteristics and prognosis of HCC patients
We performed immunohistochemical using a tissue microarray from another 185 HCC patients (cohort #2) independent of cohort #1. The intensity of ENOPH1 immunostaining was examined under double-blinded conditions. The staining intensity was categorized as 0 (negative staining), 1 (weak staining), 2 (medium staining), or 3 (strong staining) (Figure 3A). The percentage of immunopositive cells in each field under a microscope was scored as 0 (< 25%), 1 (> 25% and < 50%), 2 (> 50% and < 75%), 3 (> 75%). Considering both the staining intensity and percentage of immunopositive cells, ENOPH1 expressions were divided into low (score 0 - 3) and high (score 4 - 9) expression groups (Figure S1B). Kaplan-Meier analysis showed that ENOPH1 overexpression was significantly correlated to poorer OS (Figure 3B). Several factors, including TNM stage, BCLC stage, the presence of satellite metastasis, microvascular invasion, macrovascular invasion, tumor size, tumor amount, intraoperative blood transfusion, surgery time, intraoperative hemorrhage, the alpha-fetoprotein (AFP), and ENOPH1 expression, were significantly correlated with OS (Figure 3C, left panel) in a univariate model. However, only four factors, namely, ENOPH1 expression level, intraoperative blood transfusion, tumor size, and the presence of satellite metastasis, had independent impact on prognostic factors for the OS of HCC patients (Figure 3C, right panel) according to a multivariate Cox regression model.
ENOPH1 overexpression is association with clinicopathological characteristics and prognosis of HCC patients. (A) Representative IHC staining intensity of ENOPH1 expression in HCC samples (magnifications: 100×). (B) Kaplan-Meier analysis indicating a correlation of ENOPH1 overexpression with poorer OS. (C)Hazard ratios (HRs) for overall survival by univariate analysis (left) or multivariate analysis (right). (D) Kaplan-Meier analysis indicating a correlation of ENOPH1 overexpression with poorer DFS. (E) HRs for disease free survival by univariate analysis (left) or multivariate analysis (right).
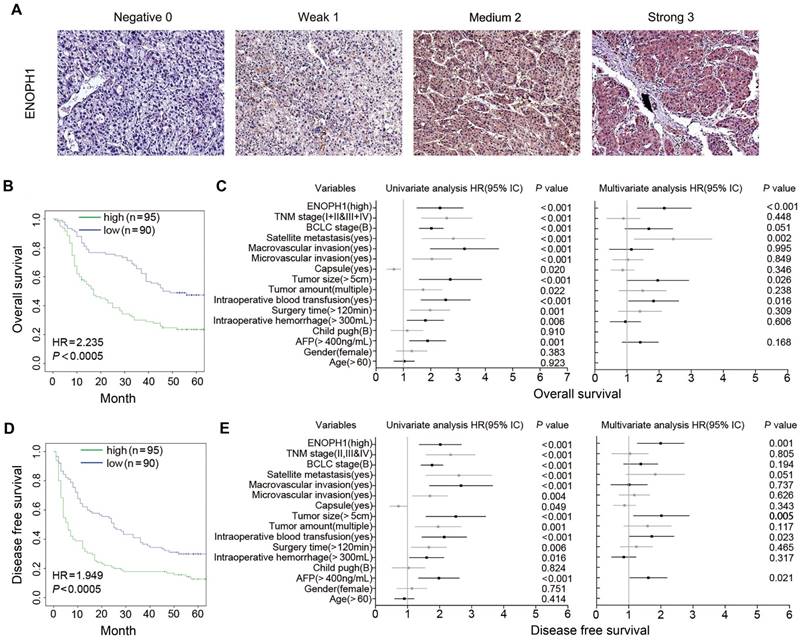
ENOPH1 overexpression was also significantly correlated with poorer DFS (Figure 3D). Univariate Cox regression analyses determined that 13 clinicopathologic characteristics were significantly correlated to DFS, including ENOPH1 expression (Figure 3E, left panel). ENOPH1 expression, tumor size, intraoperative blood transfusion, and the AFP levels were significant and independent prognostic factors for the DFS of HCC patients based on multivariate Cox regression model (Figure 3E, right panel). By combining the both results of OS and DFS analyses, we conclude that high ENOPH1 expression might be an important and independent risk factor for HCC patients.
ENOPH1 promotes cell proliferation, migration, and invasiveness in HCC cells
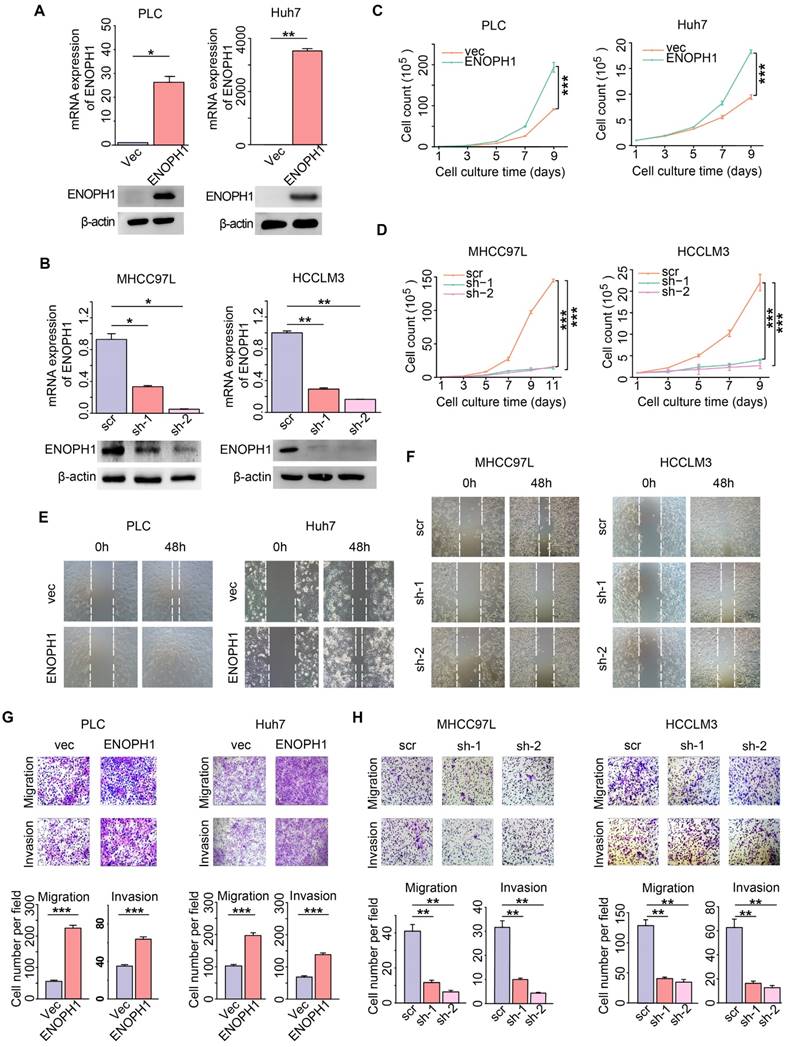
To explore the function of ENOPH1 in HCC, we repressed ENOPH1 expression with two independent shRNAs in HCCLM3 and MHCC97L cells and overexpressed ENOPH1 in Huh7 cells and PLC cells. Synthesized specific siRNA also used to decrease ENOPH1 expression in HCCLM3 cells. Efficiency of ENOPH1 knockdown or overexpression was confirmed by qRT-PCR and Western blotting (Figure 4A-B, Figure S2A). The increase in ENOPH1 expression significantly increased cell growth, whereas ENOPH1 downregulation severely impaired cell proliferation (Figure 4C-D, Figure S2B). The enhancement of cell proliferation by ENOPH1 overexpression and the reduction in cell proliferation by ENOPH1 knockdown were confirmed by colony formation assays (Figure S2C-G). The wound-healing assay showed that ENOPH1 overexpression induced cell migration, whereas knockdown of ENOPH1 significantly reduced cell migration (Figure 4E-F, Figure S3A-C). In the results of Transwell-chamber and Matrigel invasion assays, migration and invasiveness abilities in ENOPH1-overexpressing HCC cells were increased compared to their corresponding control cells (Figure 4G). However, overexpression of ENOPH1 enhanced these abilities (Figure 4H, Figure S3D). Thus, these results indicate that ENOPH1 overexpression promotes proliferation, migration, and invasion in HCC cells.
ENOPH1 promotes cell proliferation, migration and invasiveness in HCC cells. (A) The efficiency of ENOPH1 overexpression by qPCR and Western blot analysis. (B) The efficiency of ENOPH1 knockdown by qPCR and Western blot analysis. (C-D) A growth curve of: ENOPH1-overexpressing cells or ENOPH1-knockdown cells and their corresponding control cells. (E-F) Wound-healing assays using ENOPH1-overexpressing cells or ENOPH1-knockdown cells and their corresponding control cells. Representative images were taken at 0 and 48 h after the scratches were created. (G-H) Transwell-chamber and Matrigel invasion assays using ENOPH1-overexpressing cells or ENOPH1-knockdown cells and their corresponding control cells. Representative images show the results of the assays. Magnification: 100× (upper panel). Histogram shows the the numbers of migration or invading cells (below panel). Data represent the means ± SEM (*:P < 0.05, **:P < 0.01, ***:P < 0.001, Student's t-test or two-way ANOVA).
ENOPH1 promotes metastasis through the regulation of cysteine/methionine metabolism
ENOPH1 is an enzyme of the methionine salvage pathway and catalyzes the reconstitution of methionine with methylthioadenosine, the methylthio group of the metabolite of S-adenosylmethionine [19]. Our metabolomics data suggested that the cysteine/methionine metabolism pathway was correlated with metastatic potential in the HCC cells (Figure 1B). To confirm the association of the cysteine/methionine metabolism with the metastatic potential, we examined the levels of methionine, S-adenosyl-L-methionine (SAM), and S-adenosyl-L-homocysteine (SAH) using four HCC cells (MHCC97L, MHCC97H, HCCLM3, and HCCLM6 cells) with increasing metastasis potential [37, 38]. The yields of methionine and SAM were decreased in MHCC97H, HCCLM3, and HCCLM6 cells compared to MHCC97L cells. The levels of SAH were increased in MHCC97H, HCCLM3, and HCCLM6 cells compared to MHCC97L cells (Figure 5A), suggesting that the cysteine/methionine metabolism is associated with metastatic potential in HCC cells.
ENOPH1 promotes metastasis through the regulation of cysteine/methionine metabolism in HCC cells. (A) The relative levels of Met, SAM, and SAH detected with LC-MS/MS analysis using MHCC97L, MHCC97H, HCCLM3, and HCCLM6 cells. The relative levels of SAM detected with LC-MS/MS analysis using (B) ENOPH1-overexpressing PLC cells and its corresponding control cells; (C) ENOPH1-overexpressing Huh7 cells and its corresponding control cells; (D) ENOPH1-knockdown MHCC97L cells and its corresponding control cells; (E) ENOPH1-knockdown HCCLM3 cells and its corresponding control cells. (F) Transwell-chamber and Matrigel invasion assays were performed using ENOPH1-overexpressing PLC cells with or without 240 μM of SAM treatment for 48 h. Representative images are showing the results of the assays. Magnification: 100×. (G) Histogram showing the the numbers of migration or invading cells. Data represent the means ± SEM (*:P < 0.05, **:P < 0.01, ***:P < 0.001, Student's t-test).
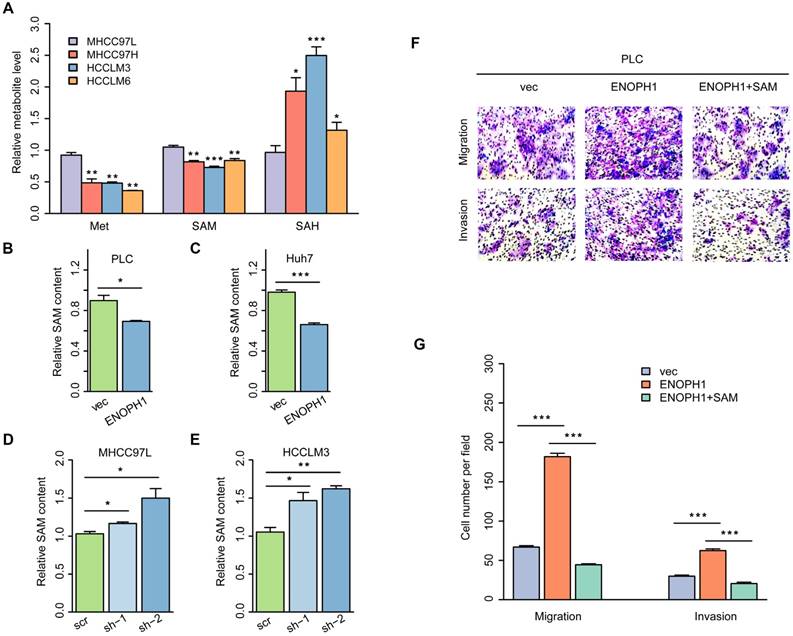
Studies have suggested that a reduction of hepatic SAM level is associated with malignant degeneration in HCC rodent models [39, 40]. SAM could significantly inhibit HCC establishment through its pro-apoptotic effect and its ability of inhibiting angiogenesis in a rat model [41]. We detected the levels of SAM in ENOPH1-overexpressing cells or ENOPH1-knockdown cells and their corresponding control cells. The levels of SAM were reduced by the unregulated expression of ENOPH1 and induced by knocking down ENOPH1 expression (Figure 5B-E). To confirm that ENOPH1 regulates HCC progression by modulating the cysteine/methionine metabolism pathway, ENOPH1-overexpressing PLC cells were incubated with SAM for 48 h, and then the abilities of migration and invasiveness were determined. SAM impaired the cell migratory and invasive potential that was promoted by ENOPH1 overexpression (Figure 5F-G). Altogether, these data indicate that ENOPH1 promotes cell migration and invasion by regulating the cysteine/methionine metabolism pathway.
ENOPH1 enhances phosphorylation of AKT in HCC cells
We performed reversed genetics analysis of ENOPH1-knockdown HCCLM3 cells, ENOPH1-overexpressing Huh7 cells and their corresponding control cells. Overall, 29 pathways were enriched based on DEGs in ENOPH1-overexpressing Huh7 cells, and 39 pathways were enriched for ENOPH1-knockdown HCCLM3 cells (Figure 6A). Among the overlapped 11 relevant pathways (Figure 6B), the AKT pathway included the maximum number of DEGs both in ENOPH1-knockdown and -overexpressing cells compared to their corresponding control cells. Therefore, the AKT pathway could be considered as a significantly enriched pathway that was influenced by ENOPH1 expression.
ENOPH1 promotes cell metastasis via increased phosphorylation of AKT in HCC cells. (A) Venn diagrams showing the overlap of the numbers of DEG-enriched pathways based on analysis of transcriptomic data between the ENOPH1-knockdown HCCLM3 cells or ENOPH1-overexpressing Huh7 cells and their corresponding control cells. (B) The details of 11 overlapped pathways. (C) Western blots of p-AKT and AKT expression in the ENOPH1-overexpressing cells or ENOPH1-knockdown cells and their corresponding control cells (left panel). Histograms show the relative intensities of phophorylation levels of AKT versus total AKT expression levels (right panel). (D) Western blots of p-AKT and AKT expression in ENOPH1-overexpressing Huh7 cells treated with 30 μM of perifosine (left panel). Histograms shows the relative intensities of phosphorylation levels of AKT versus total AKT expression levels (right panel). (E) Transwell-chamber and Matrigel invasion assays of ENOPH1-overexpressing Huh7 cells treated with 30 μM of perifosine. Representative images are showing the results of the assays. Magnification: 100× (upper panel). Histograms show the the numbers of migration or invading cells (below panel). (F) Western blots of p-AKT and AKT expression in ENOPH1-overexpressing PLC cells treated with 240 μM of SAM (left panel). Histograms show the relative intensities of phosphorylation levels of AKT versus total AKT expression levels (right panel). Data represent the means ± SEM (*:P < 0.05, **:P < 0.01, ***:P < 0.001, Student's t-test).
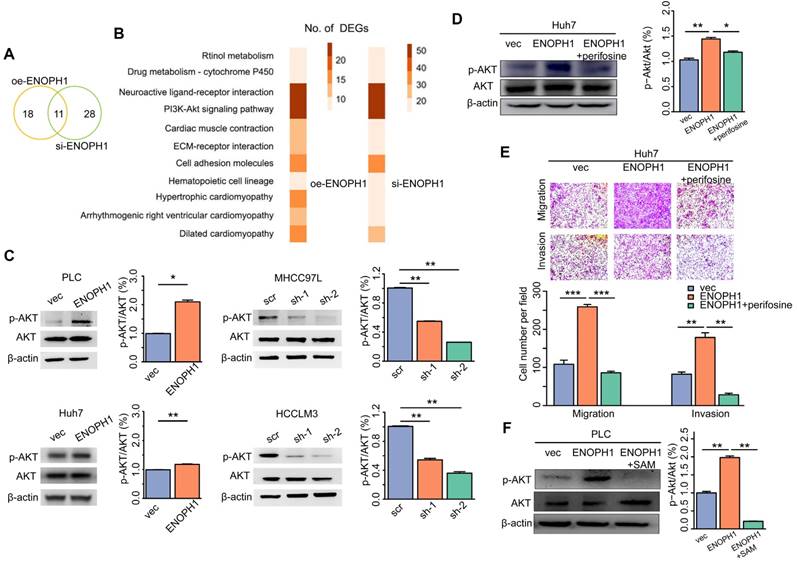
The activation of AKT pathway is an important contributor to HCC progression [42]. The relationship between the expression levels of ENOPH1 and the AKT activation was confirmed by Western blotting, which suggested that the up regulation of ENOPH1 enhanced AKT phosphorylation, whereas ENOPH1 knockdown inhibited it (Figure 6C). Next, an AKT pathway blocker, perifosine [43], was used to inhibit the phenotypes of ENOPH1-overexpressing Huh7 cells. Perifosine restrained AKT activation in ENOPH1-overexpressing Huh7 cells (Figure 6D). The Transwell-chamber and Matrigel invasion assays confirmed that perifosine treatments reversed the increase of cell migration and invasion in response to ENOPH1 overexpression (Figure 6E). In addition, the phosphorylation levels of AKT were determined in ENOPH1-overexpressing PLC cells after the treatment of SAM for 48 h. The results showed that treatment with SAM significantly impaired the abilities of migration and invasiveness promoted by ENOPH1 upregulation (Figure 6F). Thus, our results suggest that ENOPH1 overexpression enhances the AKT activation, consequently promoting cell metastasis in HCC cells.
ENOPH1 promotes intrahepatic metastasis in vivo
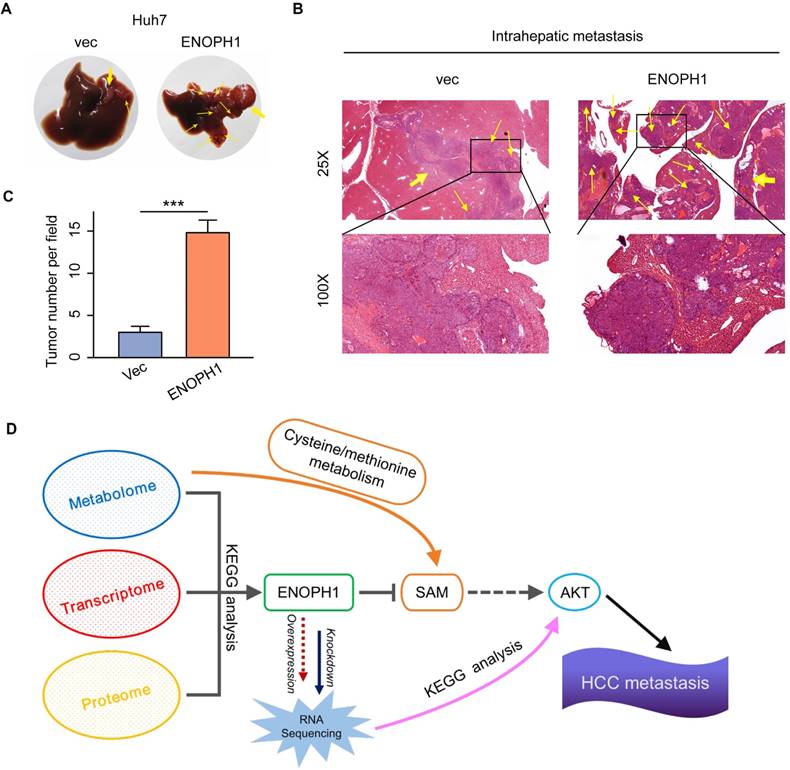
To determine the metastatic relevance of ENOPH1 in vivo, we examined the effects of ENOPH1 in an intrahepatic tumor xenograft mouse model. The ENOPH1-overexpression Huh7 cells and the corresponding control cells were injected orthotopically in the mouse left hepatic lobe, respectively. As shown in Fig 7A, ENOPH1 upregulation induced the intrahepatic micro-metastasis of Huh7 cells. The intrahepatic metastases were further observed using hematoxylin-eosin staining (Figure 7B). More extensive tumor foci were shown in mice injected with the ENOPH-overexpression Huh7 cells compared with mice injected with the corresponding control cells (Figure 7C).
ENOPH1 promotes intrahepatic metastasis. (A) The orthotropic mouse models were transplanted with ENOPH1-overexpressing Huh7 cells and their corresponding control cells. Representative images show the orthotropic transplanted tumor (marked with yellow arrows in bold) and the intrahepatic metastases (marked with thin yellow arrows). (B) Representative hematoxylin and eosin-staining images of the orthotropic transplanted tumor (bold yellow arrows) and the intrahepatic metastases (thin yellow arrows) in the orthotropic mouse models injected with ENOPH1-overexpressing Huh7 cells and their corresponding control cells. Magnification: 25 × and 100 ×. (C) Histogram shows the intrahepatic metastases counted under the microscope. Data represent the means ± SD (***: P < 0.001, Student's t-test). (D) Schematic representation of the proposed working model, in which ENOPH1 contributes to HCC progress.
Discussion
Cancer metabolism is in tandem with the proliferation of genomic, transcriptomic, proteomic, and epigenomic measurements of tumor. A pathway-based pipeline for studying cancer metabolism from an integrative multi-omics perspective could generate a complete atlas of cancer metabolism [44-46]. In our pervious study, we integrated the multi-omics data of HCC cells and identified a metabolic enzyme, uridine diphosphate (UDP)-glucose pyrophosphorylase 2, which is associated with HCC metastasis [28]. Here, we adopted another integrated analysis strategy of the multi-omics data. Here, seven metabolic pathways are associated with HCC progression, which were identified in the first step by the metabolomic analysis of three HCC cell lines with increasing metastatic potentials. These seven pathways included purine metabolism, nicotinate/nicotinamide metabolism, cysteine/methionine metabolism, beta-alanine metabolism, glutathione metabolism, valine, leucine/isoleucine degradation, and propanoate metabolism. Previous studies have shown that the cysteine/methionine and glutathione metabolism pathways were associated with tumor progression and metastasis in clear cell renal cell carcinoma [44], and purine synthesis is required by cancer cells [47]. Then, we integrated the result of metabolomic analysis into transcriptomic and proteomic analysis and focused on ENOPH1. Analysis of TCGA HCC data and tissue assay of clinical samples both suggest that ENOPH1 over-expression is associated with poor prognosis. Functional analysis revealed that ENOPH1 promoted HCC cells proliferation and metastasis. By using mass spectroscopy, the effect of ENOPH1 upregulation on cellular metabolism were measured, and found that the SAM content is decreased with ENOPH1 upregulation. As shown in previous studies, AKT phosphorylation could be enhanced by the decrease of SAM level [48-50], thus we observed a positive link between ENOPH1 expression and AKT phosphorylation in this study. Taken together, ENOPH1 appears exert its function through repressing SAM level, resulting in inducted activation of AKT (Figure 7D).
ENOPH1 was first reported to be included in a 4q21 microdeletion syndrome with a distinctive phenotype comprised of severe mental retardation, hypotonia, pronounced developmental delay, absent of speech, and facial dysmorphism [51]. Recent studies have shown that ENOPH1 is widely expressed in brain tissues of C57BL/6J mice and ENOPH1 upregulation was associated with ischemic brain injury [20]. However, studies suggesting the tumorigenesis relevance of ENOPH1 are limited. ENOPH1 has been implicated in stress responses [20]. Upregulation of ENOPH1 could increase intracellular ROS generation in a model of cerebral ischemia [52]. Elevated production of free radical is common pathophysiological event occurring in the metastatic tumor cells [28, 53]. Intracellular ROS levels are gradually increased with metastatic potential in HCC cells [28]. We have shown that ENOPH1 is upregulated in malignant HCC cells and in HCC tissues. Multivariate analysis revealed that ENOPH1 expression was an independent risk factor for OS and DFS in HCC patients. The overexpression of ENOPH1 significantly promoted cell migration and invasiveness, whereas the downregulation of ENOPH1 remarkably impaired cell migration and invasiveness. Therefore, ENOPH1 might be a novel prognostic biomarker and therapeutic target for HCC.
Our mechanistic investigation was based on reverse genetic analysis of ENOPH1-regulated HCC cells and their corresponding control cells. ROS/mTORC2/p-AKT signaling is involved in cell migration and invasion in HCC [54]. Our observation also showed the enhanced phosphorylation of AKT with ENOPH1 overexpression in the HCC cells. In addition, AKT activation has been suggested to be a potential therapeutic target. Blockage of AKT activation could be a promising approach for HCC treatment [42]. Perifosine is a synthetic alkylphosphocholine anti-tumor agent that targets the AKT pathway [55]. Perifosine inhibited growth of HCC cells through inhibition of AKT phosphorylation [56]. Moreover, perifosine has been used in breast and prostate cancer patients in a phase I/II clinical trial [57, 58]. Here, we showed that perifosine restrained ENOPH1-mediated migration and invasiveness. Therefore, an analysis of the ENOPH1 expression levels could possibly predict increased sensitivity to AKT-inhibitors, which may be selectively added to the chemotherapy strategy in HCC patients.
In summary, this integrative multi-omics study expands our knowledge of the relationship between the expression of metabolic enzymes and metastasis. According to the multi-omics analysis, our study defines a novel role of ENOPH1 in HCC, in which ENOPH1 upregulation increases phosphorylation of AKT, leading to HCC progression. Thus, ENOPH1 could be utilized as a prognostic and therapeutic target of HCC. AKT inhibitors could also be employed as a potential drug for the treatment of ENOPH1-driven HCC patients.
Abbreviations
AFP: alpha-fetoprotein; BCLC: Barcelona clinic liver cancer; BSA: bovine serum albumin; DEGs: differentially expressed genes; DFS: disease-free survival; ECL: Enhanced chemiluminescence; ENOPH1: Enolase-phosphatase 1; HCC: hepatocellular carcinoma; HEPES: N-2-hydroxyethylpiperazine-N-2-ethane sulfonic acid; HR: Hazard ratio; IHC: immunohistochemistry; Met: methionine; OE: overexpression; OS: overall survival; SAH: S-Adenosyl-L-homocysteine; SAM: S-adenosyl-L-Methionine; SEM: Standard Error of Mean; SD: Standard Deviation; TCGA: The Cancer Genome Atlas; TNM: tumour node metastasis.
Acknowledgements
This work was supported by grants from National Natural Science Foundation of China [81872377]; Tianjin Natural Science Foundation of China [16JCQNJC09600, 18JCYBJC25600]; Science and Technology Development Foundation of Henan Province [172102310103]; Henan Provincial Medical Science and Technology Project [2018020480].
Author Contributions
Y Li and W Wei designed and supervised the project. H Zhuang, Z Qiang, X Shao, and Y Dang performed the experiments. W Wei, H Wang, H Zhuang, and F Wu performed bioinformatical analysis. Y Li, W Wei, H Zhuang, Z Qiang, X Shao, and Z Wang analyzed the clinic and cellular experimental data. Y Li, W Wei, and H Zhuang wrote and revised the paper.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
2. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11-20
3. Payen VL, Porporato PE, Baselet B, Sonveaux P. Metabolic changes associated with tumor metastasis, part 1: tumor pH, glycolysis and the pentose phosphate pathway. Cell Mol Life Sci. 2016;73:1333-48
4. Warburg O. On the origin of cancer cells. Science. 1956;123:309-14
5. Chen Z, Lu X, Wang Z, Jin G, Wang Q, Chen D. et al. Co-expression of PKM2 and TRIM35 predicts survival and recurrence in hepatocellular carcinoma. Oncotarget. 2015;6:2538-48
6. Sheng SL, Liu JJ, Dai YH, Sun XG, Xiong XP, Huang G. Knockdown of lactate dehydrogenase A suppresses tumor growth and metastasis of human hepatocellular carcinoma. FEBS J. 2012;279:3898-910
7. Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK. et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008;105:18782-7
8. Jin L, Alesi GN, Kang S. Glutaminolysis as a target for cancer therapy. Oncogene. 2016;35:3619-25
9. Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. 2013;123:3678-84
10. Wong CC, Qian Y, Li X, Xu J, Kang W, Tong JH. et al. SLC25A22 Promotes Proliferation and Survival of Colorectal Cancer Cells With KRAS Mutations and Xenograft Tumor Progression in Mice via Intracellular Synthesis of Aspartate. Gastroenterology. 2016;151:945-60 e6
11. LeBleu VS, O'Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC. et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol. 2014;16:992-1003 1-15
12. Shaul YD, Freinkman E, Comb WC, Cantor JR, Tam WL, Thiru P. et al. Dihydropyrimidine accumulation is required for the epithelial-mesenchymal transition. Cell. 2014;158:1094-109
13. Cervelli M, Pietropaoli S, Signore F, Amendola R, Mariottini P. Polyamines metabolism and breast cancer: state of the art and perspectives. Breast Cancer Res Treat. 2014;148:233-48
14. Nowotarski SL, Woster PM, Casero RA Jr. Polyamines and cancer: implications for chemotherapy and chemoprevention. Expert Rev Mol Med. 2013;15:e3
15. Moinard C, Cynober L, de Bandt JP. Polyamines: metabolism and implications in human diseases. Clin Nutr. 2005;24:184-97
16. Gamble LD, Hogarty MD, Liu X, Ziegler DS, Marshall G, Norris MD. et al. Polyamine pathway inhibition as a novel therapeutic approach to treating neuroblastoma. Front Oncol. 2012;2:162
17. Song X, Han X, Yu F, Zhang X, Chen L, Lv C. Polyamine-Targeting Gefitinib Prodrug and its Near-Infrared Fluorescent Theranostic Derivative for Monitoring Drug Delivery and Lung Cancer Therapy. Theranostics. 2018;8:2217-28
18. Wang H, Pang H, Bartlam M, Rao Z. Crystal structure of human E1 enzyme and its complex with a substrate analog reveals the mechanism of its phosphatase/enolase activity. J Mol Biol. 2005;348:917-26
19. Sauter M, Moffatt B, Saechao MC, Hell R, Wirtz M. Methionine salvage and S-adenosylmethionine: essential links between sulfur, ethylene and polyamine biosynthesis. Biochem J. 2013;451:145-54
20. Barth A, Bilkei-Gorzo A, Drews E, Otte DM, Diaz-Lacava A, Varadarajulu J. et al. Analysis of quantitative trait loci in mice suggests a role of Enoph1 in stress reactivity. J Neurochem. 2014;128:807-17
21. Tran B, Dancey JE, Kamel-Reid S, McPherson JD, Bedard PL, Brown AM. et al. Cancer genomics: technology, discovery, and translation. J Clin Oncol. 2012;30:647-60
22. Horgan RP, Kenny LC. 'Omic' technologies: genomics, transcriptomics, proteomics and metabolomics. The Obstetrician & Gynaecologist. 2011;13:189-95
23. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P. et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807-12
24. Schuster SC. Next-generation sequencing transforms today's biology. Nat Methods. 2008;5:16-8
25. Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ. et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346-52
26. Tsuji S, Midorikawa Y, Seki M, Takayama T, Sugiyama Y, Aburatani H. Network-based analysis for identification of candidate genes for colorectal cancer progression. Biochem Biophys Res Commun. 2016;476:534-40
27. Nam H, Chung BC, Kim Y, Lee K, Lee D. Combining tissue transcriptomics and urine metabolomics for breast cancer biomarker identification. Bioinformatics. 2009;25:3151-7
28. Li Y, Zhuang H, Zhang X, Li Y, Liu Y, Yi X. et al. Multiomics Integration Reveals the Landscape of Prometastasis Metabolism in Hepatocellular Carcinoma. Mol Cell Proteomics. 2018;17:607-18
29. Zhuang H, Li Q, Zhang X, Ma X, Wang Z, Liu Y. et al. Downregulation of glycine decarboxylase enhanced cofilin-mediated migration in hepatocellular carcinoma cells. Free Radic Biol Med. 2018;120:1-12
30. Liu Y, Zhang X, Yang B, Zhuang H, Guo H, Wei W. et al. Demethylation-Induced Overexpression of Shc3 Drives c-Raf-Independent Activation of MEK/ERK in HCC. Cancer Res. 2018;78:2219-32
31. Kratzat S, Nikolova V, Miething C, Hoellein A, Schoeffmann S, Gorka O. et al. Cks1 is required for tumor cell proliferation but not sufficient to induce hematopoietic malignancies. PLoS One. 2012;7:e37433
32. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114-20
33. Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357-60
34. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078-9
35. Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 2016;11:1650-67
36. Zhuang H, Wu F, Wei W, Dang Y, Yang B, Ma X. et al. Glycine decarboxylase induces autophagy and is downregulated by miRNA-30d-5p in hepatocellular carcinoma. Cell Death Dis. 2019;10:192
37. Li Y, Tian B, Yang J, Zhao L, Wu X, Ye SL. et al. Stepwise metastatic human hepatocellular carcinoma cell model system with multiple metastatic potentials established through consecutive in vivo selection and studies on metastatic characteristics. J Cancer Res Clin Oncol. 2004;130:460-8
38. Li Y, Tang ZY, Ye SL, Liu YK, Chen J, Xue Q. et al. Establishment of cell clones with different metastatic potential from the metastatic hepatocellular carcinoma cell line MHCC97. World J Gastroenterol. 2001;7:630-6
39. Pascale RM, Simile MM, Seddaiu MA, Daino L, Vinci MA, Pinna G. et al. Chemoprevention of rat liver carcinogenesis by S-adenosyl-L-methionine: is DNA methylation involved? Basic Life Sci. 1993;61:219-37
40. Pascale RM, Simile MM, De Miglio MR, Nufris A, Daino L, Seddaiu MA. et al. Chemoprevention by S-adenosyl-L-methionine of rat liver carcinogenesis initiated by 1,2-dimethylhydrazine and promoted by orotic acid. Carcinogenesis. 1995;16:427-30
41. Lu SC, Ramani K, Ou X, Lin M, Yu V, Ko K. et al. S-adenosylmethionine in the chemoprevention and treatment of hepatocellular carcinoma in a rat model. Hepatology. 2009;50:462-71
42. Ko E, Seo HW, Jung G. Telomere length and reactive oxygen species levels are positively associated with a high risk of mortality and recurrence in hepatocellular carcinoma. Hepatology. 2018;67:1378-91
43. Lin X, Yao Y, Wang B, Emlen DJ, Lavine LC. Ecological Trade-offs between Migration and Reproduction Are Mediated by the Nutrition-Sensitive Insulin-Signaling Pathway. Int J Biol Sci. 2016;12:607-16
44. Hakimi AA, Reznik E, Lee CH, Creighton CJ, Brannon AR, Luna A. et al. An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell. 2016;29:104-16
45. Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S. et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013;497:633-7
46. Dang CV. MYC on the path to cancer. Cell. 2012;149:22-35
47. Tedeschi PM, Markert EK, Gounder M, Lin H, Dvorzhinski D, Dolfi SC. et al. Contribution of serine, folate and glycine metabolism to the ATP, NADPH and purine requirements of cancer cells. Cell Death Dis. 2013;4:e877
48. Pascale RM, Feo CF, Calvisi DF, Feo F. Deregulation of methionine metabolism as determinant of progression and prognosis of hepatocellular carcinoma. Transl Gastroenterol Hepatol. 2018;3:36
49. Sadek KM, Lebda MA, Nasr NE, Nasr SM, El-Sayed Y. Role of lncRNAs as prognostic markers of hepatic cancer and potential therapeutic targeting by S-adenosylmethionine via inhibiting PI3K/Akt signaling pathways. Environ Sci Pollut Res Int. 2018;25:20057-70
50. Frau M, Feo F, Pascale RM. Pleiotropic effects of methionine adenosyltransferases deregulation as determinants of liver cancer progression and prognosis. J Hepatol. 2013;59:830-41
51. Bonnet C, Andrieux J, Beri-Dexheimer M, Leheup B, Boute O, Manouvrier S. et al. Microdeletion at chromosome 4q21 defines a new emerging syndrome with marked growth restriction, mental retardation and absent or severely delayed speech. J Med Genet. 2010;47:377-84
52. Zhang Y, Wang T, Yang K, Xu J, Ren L, Li W. et al. Cerebral Microvascular Endothelial Cell Apoptosis after Ischemia: Role of Enolase-Phosphatase 1 Activation and Aci-Reductone Dioxygenase 1 Translocation. Front Mol Neurosci. 2016;9:79
53. Chew SH, Okazaki Y, Akatsuka S, Wang S, Jiang L, Ohara Y. et al. Rheostatic CD44 isoform expression and its association with oxidative stress in human malignant mesothelioma. Free Radic Biol Med. 2017;106:91-9
54. Jiang F, Chen L, Yang YC, Wang XM, Wang RY, Li L. et al. CYP3A5 Functions as a Tumor Suppressor in Hepatocellular Carcinoma by Regulating mTORC2/Akt Signaling. Cancer Res. 2015;75:1470-81
55. Vinall RL, Hwa K, Ghosh P, Pan CX, Lara PN Jr, de Vere White RW. Combination treatment of prostate cancer cell lines with bioactive soy isoflavones and perifosine causes increased growth arrest and/or apoptosis. Clin Cancer Res. 2007;13:6204-16
56. Fei HR, Chen G, Wang JM, Wang FZ. Perifosine induces cell cycle arrest and apoptosis in human hepatocellular carcinoma cell lines by blockade of Akt phosphorylation. Cytotechnology. 2010;62:449-60
57. Chiarini F, Del Sole M, Mongiorgi S, Gaboardi GC, Cappellini A, Mantovani I. et al. The novel Akt inhibitor, perifosine, induces caspase-dependent apoptosis and downregulates P-glycoprotein expression in multidrug-resistant human T-acute leukemia cells by a JNK-dependent mechanism. Leukemia. 2008;22:1106-16
58. Kondapaka SB, Singh SS, Dasmahapatra GP, Sausville EA, Roy KK. Perifosine, a novel alkylphospholipid, inhibits protein kinase B activation. Mol Cancer Ther. 2003;2:1093-103
Author contact
![]() Corresponding author: Dr. Wen Wei, Tel: 86-023-65678491; Email: weiwenedu.cn Dr. Yongmei Li, Tel: 86-022-83336836; Email: liymedu.cn
Corresponding author: Dr. Wen Wei, Tel: 86-023-65678491; Email: weiwenedu.cn Dr. Yongmei Li, Tel: 86-022-83336836; Email: liymedu.cn