Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(12):3659-3673. doi:10.7150/thno.32126 This issue Cite
Research Paper
RIP3 promotes colitis-associated colorectal cancer by controlling tumor cell proliferation and CXCL1-induced immune suppression
Zhen-Yu Liu1,2*, Ming Zheng1,2*, Yi-Ming Li1,2*, Xin-Yu Fan1,2, Jian-Chao Wang1,2, Zhu-Chun Li1,2, Hai-Jiao Yang1,2, Jing-Min Yu1,2, Jian Cui1,2, Jian-Li Jiang1,2, Juan Tang1,2, Zhi-Nan Chen1,2 ![]()
1. National Translational Science Center for Molecular Medicine, 710032 Xi'an, China.
2. Department of Cell Biology, School of Basic Medicine, The Fourth Military Medical University, 710032 Xi'an, China.
* These authors contributed equally to this work.
Received 2018-12-9; Accepted 2019-5-12; Published 2019-6-2
Abstract

Rationale: Necroptosis is a programmed form of non-apoptotic cell death that requires receptor-interacting protein 3 (RIP3). RIP3 has been shown to be relevant in multiple tumor types and has differential impact on tumor progression. We investigated whether RIP3 is involved in the progression of colitis-associated cancer (CAC) in mice.
Methods: Tissues from colorectal cancer patients were examined for RIP3 expression. CAC was induced using azoxymethane (AOM) injection followed by dextran sodium sulfate (DSS) treatment in RIP3-deficient or wild-type mice. Colon tissues were collected and analyzed by Western blotting and gene expression profile analyses. Immune cell infiltration and CXCL1 expression were examined by flow cytometry and Real-time PCR, respectively.
Results: RIP3 expression was upregulated in mouse CAC and human colon cancer. RIP3-deficient mice showed significantly attenuated colitis-associated tumorigenesis. Bone marrow transplantation experiments suggested that RIP3's function in hematopoietic cells primarily contributes to the phenotype. RIP3 supported epithelial proliferation and tumor growth via JNK signaling but had no effect on apoptosis. RIP3 deletion increased T cell accumulation and reduced infiltration by immunosuppressive subsets of myeloid cells during acute colitis and CAC. The immune-suppressive tumor microenvironment was dependent on RIP3-induced expression of the chemokine attractant CXCL1, and administration of recombinant CXCL1 during CAC restored tumorigenesis in Rip3-/- mice.
Conclusion: Our results reveal an unexpected function of RIP3 in enhancing the proliferation of premalignant intestinal epithelial cells (IECs) and promoting myeloid cell-induced adaptive immune suppression. These two distinct mechanisms of RIP3-induced JNK and CXCL1 signalling contribute to CAC progression.
Keywords: colorectal cancer, IBD, ulcerative colitis, necroptosis, RIP3.
Introduction
Chronic inflammation is now known to have decisive roles in the pathogenesis of cancer [1]. Inflammatory bowel diseases (IBDs) are a salient example of the link between chronic inflammation and cancer, and one consequence of persistent inflammation of the colon or ulcerative colitis (UC) is an increased risk for developing colorectal cancer [2]. Animal models that reproduce many aspects of this human disease have provided significant clues regarding the crucial roles of inflammatory mediators and related molecular events leading to the development of colon cancer [3].
Programmed necrosis or “necroptosis” is a recently described caspase 8-independent mode of cell death that requires the kinase activity of central adaptor receptor interacting protein kinase 3 (RIP3) and has similar features of necrosis [4, 5]. Necroptosis has emerged as an important regulator of host immunity to pathogens and inflammation. Various stimuli can trigger necroptosis through engagement of the tumor necrosis factor (TNF) receptor-like death receptors, Toll-like receptors (TLRs) 3 and 4, and interferon (IFN) receptors [6-8]. These pathways promote the interaction of RIP3 and the upstream kinase RIP1 via their respective RIP-homotypic interaction motifs (RHIMs) and the formation of an amyloid-like RIP1/RIP3 necrosome complex [9]. Activation of RIP3 leads to the recruitment and phosphorylation of mixed-lineage kinase domain-like (MLKL), which mediates sodium influx through Ca2+ and Na+ ion channels. The increased osmotic pressure of necroptotic dying cells leads to rapid plasma membrane rupture and promotes inflammation by the release of substantial amounts of damage-associated molecular patterns (DAMPs), interleukin-1β (IL-1β) and other cytokines [10].
Recent studies have shown that necroptosis plays an important role in several inflammatory diseases, including IBD, acute pancreatitis, inflammatory skin diseases, and liver injury [4, 8, 11-13]. The significance of the necroptotic pathway in cancer biology has been investigated in preclinical studies of several hematological malignancies and solid tumors. A preclinical study showed that RIP3 inhibits malignant myeloproliferation in acute myeloid leukemia (AML) by promoting cell death of transformed progenitor cells and that the release of IL-1β by dying cells and subsequent activation of the inflammasome promote differentiation of leukemia-initiating cells [14]. Moreover, in non-Hodgkin lymphoma, single nucleotide polymorphisms (SNPs) in the RIP3 gene were identified in a cohort of 458 patients and correlated with an increased risk of lymphomagenesis, suggesting that genetic variations in the RIP3 gene may contribute to the etiology of this cancer [15]. Based on these studies, triggering necroptosis seems to be effective in tumor cells in hematopoietic malignancies, but a number of counter-observations have also been made, suggesting that these factors may sometimes promote various functions of tumor cells. In pancreatic cancer cells, necroptotic signaling promotes myeloid cell-induced adaptive immune suppression and thereby enables pancreatic ductal adenocarcinoma progression [16]. Another study showed that melanoma cells induce necroptosis of endothelial cells, which promoted tumor cell extravasation and metastasis via death receptor 6 (DR6). Targeting DR6-mediated necroptosis inhibited tumor cell-induced necroptosis and the development of metastasis in melanoma [17]. Altogether, these data indicate that depending on the cancer cell type and the tumor microenvironment (TME), necroptosis has differential impact on tumor progression.
RIP3 was shown to have a necroptosis-independent function in intestinal inflammation [18]. The expression of RIP3 was increased in the intestinal epithelium of both adult and pediatric Crohn's disease patients [19, 20]. RIP3 was reported to be profoundly important for innate inflammatory cytokine expression and injury-induced tissue repair during acute colitis [18]. Furthermore, repair-associated proliferation may be linked to tumor growth. However, the role of RIP3 in the pathogenesis of colorectal cancer remains unclear, and whether RIP3 has protumorigenic functions needs to be uncovered.
Here, we investigated the functional importance of RIP3 in promoting tumorigenesis during the progression of colitis-associated cancer (CAC). We demonstrated that RIP3-induced JNK signaling promotes the proliferation of intestinal epithelial cells (IECs) and that RIP3-induced CXCL1 signaling promotes myeloid cell-mediated immune suppression during acute colitis and CAC. These data indicate that RIP3 is a critical checkpoint during inflammation and tumorigenesis.
Methods
Mice and CAC protocol
Wild-type (WT) C57BL/6 mice were purchased from the Air Force Medical University. The generation and validation of RIP3 knockout (Rip3-/-) mice on C57BL/6 background is described in Figure S1. Mice were used at 8-10 weeks of age. Experiments were conducted using age- and gender-matched groups. Animals were housed under specific pathogen free (SPF) conditions. All in vivo procedures were performed in accordance with protocols approved by the Animal Experiment Administration Committee of the University. CAC was induced as described in a previous study [21]. Briefly, mice were intraperitoneally injected with 12.5 mg/kg AOM (Sigma-Aldrich) and after 5 days, received drinking water containing 2.5% DSS (MP Biomedicals, molecular weight 35-50 kDa) for 5 days. Mice were then provided regular drinking water for 16 days, followed by two additional DSS treatment cycles (Figure 1A). Colons were removed on day 100, flushed with PBS, and tumors were counted. Macroscopic tumors were measured with calipers, and software was used to measure microscopic tumors. Portions of the distal colon tissues were either frozen in liquid nitrogen or fixed with formaldehyde (4%) and embedded in paraffin for histological analyses.
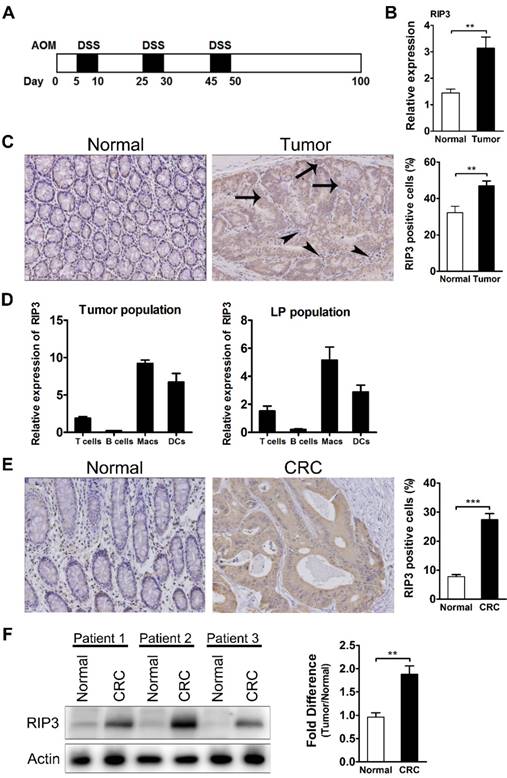
RIP3 expression is upregulated in AOM/DSS tumors and human colorectal carcinoma (CRC). (A) Schematic overview of the CAC regimen. Rip3-/- mice and WT littermates were injected with AOM followed by three cycles of 2.5% DSS in drinking water. Intestinal tumors were analyzed on day 100. (B) The expression of RIP3 in tumor and adjacent normal tissues was determined using qRT-PCR (n = 10 per group). (C) Immunohistochemical staining for RIP3 in the mouse CAC model. Representative images and summary data are shown (n = 10). Arrows and arrowheads indicate RIP3+ colon epithelial cells and mononuclear cells in the lamina propria, respectively. Original magnification, ×200. (D) T cells, B cells, macrophages, and dendritic cells isolated by fluorescence-activated cell sorting were analyzed for RIP3 mRNA by qRT-PCR. Tumor population = tumor-infiltrating cells from pooled CAC tumors from WT mice; LP population = lamina propria-derived cells in colons from which the tumors were excised. (E) Immunohistochemical staining for RIP3 using a human colon cancer tissue microarray. Representative images and summary data are shown. Original magnification, ×200. (F) Western blots showing RIP3 levels in human CRC specimens and adjacent normal human colon tissues. Representative data from three patients and density analysis from five patients are shown. Data are presented as means ± SEM. **p < 0.01, ***p<0.001.
Histological analysis
Colon tissues were sliced into 6 μm thick, 200 μm step serial sections and stained with hematoxylin and eosin (H&E). The extent of inflammation was measured and scored using a previously described method [21]. Paraffin sections were stained using a BrdU In Situ Detection Kit (BD Pharmingen) according to the manufacturer's recommendations to examine BrdU incorporation.
Apoptosis determination
For the TUNEL assay, an In Situ Cell Death Kit (Roche) was used according to the manufacturer's recommendations. For Annexin V and PI staining, cells were stained with 50 µg/ml PI and Annexin V (BD Bioscience) in Annexin V buffer. The cells were analyzed by Fortessa flow cytometer (BD Biosciences).
Real-time PCR
Total RNA was extracted with Trizol reagent (Invitrogen) and reverse-transcribed into cDNAs using a PrimeScript RT reagent Kit (TaKaRa Biotechnology). Real-time PCR was performed using the SYBR Premix Ex Taq II Kit (TaKaRa Biotechnology). The GAPDH mRNA served as an internal control. Primer sequences used in this study are summarized in Table S1.
Immunohistochemistry
Formaldehyde-fixed, paraffin-embedded sections of colon tissues were deparaffinized using xylene and alcohol and then subjected to antigen retrieval in citrate buffer (pH 6.0). Sections were subsequently incubated with 0.3% H2O2 and normal goat serum for blocking. After washes with PBS, the sections were incubated with primary antibodies at 4°C overnight in a moist chamber. Following the incubation, immunoperoxidase staining was completed using a Streptavidin-Peroxidase Kit (ZhongshanJinqiao Co., Beijing, China), and 3, 3'-diaminobenzidine (ZhongshanJinqiao Co., Beijing, China) was employed to detect the target proteins. The primary antibodies used in these experiments were anti-mouse RIP3 (1:100; Enzo Life Sciences), anti-BrdU (1:200; Abcam), anti-Ki-67 (1:100; Abcam), anti-PCNA (1:200; BD biosciences), anti-Cyclin D1 (1:100; Antibody Revolution), anti-p-JNK (1:50; Abcam), anti-p-c-Jun (1:50; Abcam), anti-F4/80 (1:100; Abcam), and anti-CXCL1 (1:50; Abcam). For the analysis of human tissues, human colon cancer tissue microarray slides were purchased from Shanghai Superchip Biotech, paraffin-embedded colon cancer slides were probed with an antibody directed against RIP3 (1:100; Abgent). All specimens were collected after informed consent was obtained.
Western blot analysis
Protein was extracted from tissue samples with RIPA lysis buffer (Beyotime, Shanghai, China) containing 1 mM PMSF and a cocktail of protease and phosphatase inhibitors using standard methods. Solubilized proteins (30 µg) were separated by standard SDS-PAGE on a 10% polyacrylamide separating gel and 5% stacking gel and then transferred to a PVDF western blot membrane (Roche) using standard methods. The following primary antibodies were used: anti-RIP3 (dilution of 1:500; Abgent), anti-JNK, anti-c-Jun, anti-p-JNK, anti-p-c-Jun, anti-AKT, anti-p-AKT, anti-ERK, anti-p-ERK, anti-p38, anti-p-p38, p-MLKL and cleaved caspase-3 (dilution of 1:1000; Cell Signaling Technology). After washing, the membranes were incubated with horseradish-peroxidase conjugated secondary antibodies (Pierce). Protein bands were visualized using an enhanced chemiluminescence (ECL) Plus Western blotting detection kit (Amersham Biosciences) according to the manufacturer's instructions. A PageRuler Prestained Protein Ladder Plus (Fermentas Life Sciences) was used for sizing the proteins.
Bone marrow transplant
A single cell suspension of bone marrow cells was obtained from the tibia and femur of four Rip3-/- and five WT donor mice at an age of 6 weeks, and the red blood cells were lysed with erythrocyte lysis buffer (Buffer EL, Qiagen). Approximately 0.85 × 106 bone marrow cells were injected into 22 Rip3-/- and 21 WT lethally irradiated (900 rads) 8-12-week-old recipient mice via the tail vein. The mice were then fed acidified water (0.015% HCl in autoclaved water) supplemented with 1.1 g/L neomycin sulfate and 125 mg/L polymyxin B sulfate for 2 weeks after transplantation (modified from the procedure reported by Cotta and colleagues [22]). At eight weeks post-transplantation, the mice were subjected to the CAC protocol.
Administration of SP600125, anti-CXCL1, anti-CD90, or recombinant CXCL1 to mice with CAC
For SP600125 treatment, WT and Rip3-/- mice were administered the JNK inhibitor SP600125 (40 mg/kg) (Sigma-Aldrich) or vehicle (DMSO) by oral gavage once daily during CAC induction. For anti-CXCL1 treatment, WT and Rip3-/- mice receiving AOM/DSS were treated either with a mouse anti-CXCL1 neutralizing antibody (120 µg/mouse once a week, i.p.) or mouse IgG1 isotype control (both from R&D Systems) until sacrifice (day 100). For anti-CD90 treatment, T cells were depleted with neutralizing anti-CD90 monoclonal antibody as previously described [23]. Rip3-/- mice receiving AOM/DSS were treated either with a mouse anti-CD90 neutralizing antibody (100 µg/mouse once a week, i.p.) or isotype control (both from BioXcell) until sacrifice (day 100). For recombinant CXCL1 treatment, Rip3-/- mice receiving AOM/DSS were treated either with mouse recombinant CXCL1 (300 ng/mouse twice a week, i.p.) or control PBS (both from R&D Systems) until sacrifice (day 100).
Cell harvest and flow cytometry
Colonic immunocytes were isolated as described in a previous report [24]. Briefly, tumor and inflamed colonic tissues were carefully excised from WT and Rip3-/- mice. After an incubation with complete medium containing antibiotics and antimycotics for 15 min at 37°C, tissues were washed, minced, and digested with collagenase I (1000 U/ml) (Gibco) and DNase I (25 U/ml) for 30 min at 37°C. The supernatant was removed and the cell pellets were suspended with complete medium. The cell suspensions were filtered through 100 μm cell strainers. After blocking FcγRIII/II with an anti-CD16/CD32 monoclonal antibody (mAb) (BD Biosciences), cell labelling was performed by incubating 106 cells with 1 μg fluorescently conjugated mAbs directed against mouse CD3, CD8, CD19, CD11b, CD11c, MHC II, Gr1, IL-10, IFNγ, F4/80, CD206, CD44, CD107a, PD-L1, PD-1 (all Biolegend). Intracellular cytokine staining was performed using the Fixation/Permeabilization Solution Kit (BD Biosciences). Flow cytometry was performed using the Fortessa flow cytometer (BD Biosciences).
Isolation of tumor epithelial cells
For colonic immune cells, tumors were separated from colon tissues of AOM/DSS-induced WT mice under a dissecting microscope and cut into small pieces (approximately 1 mm). Tumor pieces were digested with 12 mg/ml collagenase I (Gibco) and passed through 100 and 70 µm cell strainers. Isolated cells were cultured on plates coated with 5 mg/ml rat tail collagen type I (Gibco) in F12 medium supplemented with 10% horse serum, 100 ng/ml bovine pituitary extract, 1 mg/ml BSA, 1x penicillin/streptomycin (Gibco), 5 μg/ml ITS (Cellgro), and 10 ng/ml EGF (Peprotech).
CTL assay
CD8+ T cells were isolated from colonic mucosa of AOM/DSS-treated mice as effector cells by CD8+ T Cell Isolation Kit II (R&D Systems) according to the manufacturer's instructions. Isolated CD8+ T cells were co-cultured with 1 x 104 tumor cells in 96-well plates at various ratios (CD8+ T cells:tumor cells = 1:1, 10:1, or 50:1) for 8 hrs. The cytotoxicity of CD8+ T cells against AOM/DSS-induced tumor cells (CD8+ T cells:tumor cells = 50:1) was determined using a CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega).
ELISA
IFNγ protein level in the cell medium was measured by a mouse IFNγ Quantikine ELISA kit (R&D Systems) according to the manufacturer's protocol.
Microarray and statistical analyses
Total RNA was extracted from cohorts of 4 WT or Rip3-/- mice per group treated with AOM/DSS and from cohorts of 4 untreated WT or Rip3-/- mice/group using Trizol reagent (Invitrogen), according to the manufacturer's instructions. Microarray experiments were performed by the Beijing CapitalBio Corporation. Samples were processed for the Agilent Mouse (V2) Gene Expression Microarray (Agilent Technologies). Using a hierarchical clustering analysis, we selected interesting subsets of genes that clustered together and extracted the biological meaning from the given gene list using web-based bioinformatics resources. The enriched biological processes terms and the relationships among the annotated terms were identified using the FatiGO functional enrichment tool and the DAVID functional annotation clustering. P values were calculated using the two-tailed Fisher's exact test and gene functional classifications were performed under high stringency to control the behavior of DAVID fuzzy clustering.
Statistical analysis
The data are reported as mean values ± SEM. P values were calculated with a Mann-Whitney test or a two-way repeated-measure ANOVA. P values lower than 0.05 were considered statistically significant. All statistical analyses were performed using GraphPad Prism v 5.0 software.
Results
RIP3 is overexpressed in mouse CAC and human colon cancer
Proteins that promote intestinal epithelial regeneration and tissue repair often contribute to oncogenesis. Given the recently discovered role of RIP3 in regulating these processes coupled with the observation that loss of RIP3 results in sensitization to gut injury [18], we hypothesized that RIP3 expression may be altered in AOM/DSS tumors. For these experiments, 8-10-week-old mice were injected with AOM and treated with three cycles of DSS as described in the Methods section and shown in Figure 1A. Tumors and adjacent tissues were harvested, and the expression of RIP3 in tumors was determined by qRT-PCR. RIP3 levels were significantly higher in tumor tissue than in adjacent mucosa (Figure 1B). Immunohistochemical detection of RIP3 also showed an increase in RIP3 expression in tumors, and this expression was localized in the epithelial cells within the tumor (arrows) as well as in the infiltrating inflammatory cells (arrowheads) (Figure 1C). To specify the immune cell type that is increasing its RIP3 expression, we isolated lamina propria cells from colons of CAC-bearing mice and from CAC adenomas using a protocol that increases the yield of myeloid cells [25].
Analysis of RIP3 mRNA in cells sorted by fluorescence activated cell sorting (FACS) from lamina propria and CAC adenomas revealed that macrophages and dendritic cells were the major RIP3 producers during CAC growth, followed by T cells (Figure 1D). To investigate whether RIP3 levels varied in human colorectal cancer, we analyzed a colon cancer tissue microarray that included 168 tumors and 103 nontumor controls stained for RIP3 expression, and found that RIP3 was highly expressed in colorectal cancer (Figure 1E). Western blotting confirmed that RIP3 expression was higher in colorectal cancer than in the surrounding normal mucosa (Figure 1F). These results suggest that RIP3 may contribute to colitis-associated tumorigenesis.
RIP3 deficiency reduces colitis-associated tumorigenesis
To investigate whether the absence of RIP3 altered the susceptibility to developing CAC, we employed CRISPR/Cas9 technology to delete the mouse RIP3 gene (Figure S1). Eight- to ten-week-old WT or Rip3-/- mice underwent the AOM/DSS protocol to induce colorectal tumors (Figure 1A). Compared with WT mice, RIP3-deficient mice showed a markedly decreased tumor number and tumor size, with a concomitant decrease in tumor load (Figure 2A-D). However, there was no significant difference in the grade of colon tumors between WT and RIP3-/- mice, and no invasive carcinomas were observed in WT or RIP3-/- mice (Figure 2E). The above data demonstrate that RIP3 is required for CAC tumorigenesis.
RIP3 functions as a tumor promoter in a murine AOM/DSS-induced colorectal carcinogenesis model. (A-E) WT and Rip3-/- mice were subjected to CAC induction using 2.5% DSS. Intestinal tumors were analyzed on day 100. Tumor number (A), tumor size (B), and tumor load (C), the sum of the diameters of all tumors in a given mouse, were determined. The data shown in A-C correspond to one representative experiment out of three performed. Data are means ± SEM, n = 10, *p < 0.05, **p < 0.01. (D) Representative images of colons from WT and Rip3-/- mice. (E) The colons were H&E-stained, and representative histological sections from each group are shown. Original magnification, ×200 (upper rows), ×400 (lower rows). (F-I) Tumor number (F) per mouse, tumor size (G), and tumor load (H) in AOM/DSS-treated WT and Rip3-/- radiation bone marrow chimeras, with representative images of colons (I) (n = 9 for WT > WT and Rip3-/- > Rip3-/-, n = 8 for WT > Rip3-/- and Rip3-/- > WT). The data shown in E-H were obtained from the bone marrow transplantation experiments that were performed once. Data are presented as means ± SEM. *p < 0.05, **p < 0.01.
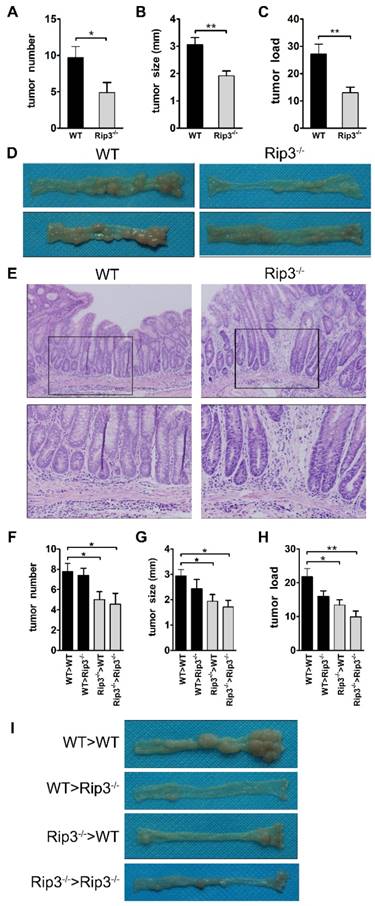
We next determined the cellular source responsible for the reduced tumorigenesis in the absence of RIP3 by generating radiation bone marrow-chimeric mice. WT and Rip3-/- mice were exposed to whole-body irradiation to deplete the hematopoietic-progenitor compartment. Irradiated recipients were then reconstituted with either WT or Rip3-/- bone marrow, and CAC was induced using AOM/DSS. Tumor numbers were markedly reduced when RIP3 was inactivated in hematopoietic cells (Figure 2F and I). RIP3 deficiency in bone marrow-derived cells also decreased tumor size and load (Figure 2G-I). We observed an increase in tumor number in Rip3-/->WT mice in comparison with Rip3-/-> Rip3-/- mice, but the effect was not statistically significant (Figure 2F). These results indicate that the absence of RIP3 in the hematopoietic cellular compartments primarily protects against colitis-associated tumorigenesis.
RIP3 promotes epithelial proliferation and tumor growth without affecting apoptosis
Differences in tumor multiplicity and load may be explained by altered proliferation and/or death of tumor progenitors. The decreased tumor multiplicity in Rip3-/- mice suggested that RIP3 may be involved in early tumor promotion, which in this model is linked to inflammation. To determine the role of RIP3 in inflammation, we treated WT and Rip3-/- mice with DSS to induce acute colitis. Upon DSS treatment, Rip3-/- mice exhibited more severe colitis with greater body weight loss than WT mice, shortening of the colon and loss of crypt structure, ulceration, and infiltration of inflammatory cells (Figure S2A-D), similar to previous reports [18].
To examine whether RIP3 regulates the proliferation of IECs in the inflamed colon, we injected naive and DSS-treated mice with 5-bromo-2-deoxyuridine (BrdU), which incorporates into newly synthesized DNA, and sacrificed the animals 3 h later. Staining with BrdU or Ki-67-specific antibodies did not reveal any significant differences in basal crypt proliferation rates between naive WT and Rip3-/- mice (Figure 3A-B). However, IEC proliferation within crypts of Rip3-/- mice was slightly but significantly decreased after DSS exposure relative to that of WT mice (Figure 3A). These results were confirmed by immunohistochemical determination of Ki-67-expressing cells in the crypts (Figure 3B).
RIP3 promotes epithelial cell proliferation and tumor growth without affecting apoptosis. (A) The extent of intestinal epithelial cell proliferation in the colons of DSS-treated mice was determined by BrdU labeling and immunohistochemistry. The percentage of BrdU-positive cells among all crypt cells in the colons was enumerated. Original magnification, ×100. (B) Proliferation was determined by Ki-67 staining and the percentage of Ki-67-positive cells among all crypt cells in the colons of DSS-treated mice was calculated. Original magnification, ×100. (C-D) Colons of adenoma-bearing mice were stained with antibodies against PCNA and Ki-67. The percentage of PCNA- (C) and Ki-67-positive cells (D) within colonic crypts was determined. Original magnification, ×200. (E) Western blot analysis of RIP3 and p-MLKL expression in the colons of AOM/DSS-treated WT and RIP3-/- mice with representative densitometry. (F) The number of TUNEL+ cells/visual field in colon tissues from control mice and mice treated with AOM/DSS for 100 days was enumerated. The positive control for the TUNEL reaction was performed by incubating sections with DNase I. Representative images of TUNEL staining are shown. Arrows indicate TUNEL-positive cells. Original magnification, ×400. (G) The percentage of Annexin V-positive (PI-negative) cells in colon tissues from control mice and mice treated with AOM/DSS. (H) Western blot analysis of cleaved caspase-3 expression in the colons of AOM/DSS-treated WT and RIP3-/- mice with representative densitometry. Data are presented as means ± SEM. *p < 0.05, **p < 0.01, ***p<0.001.
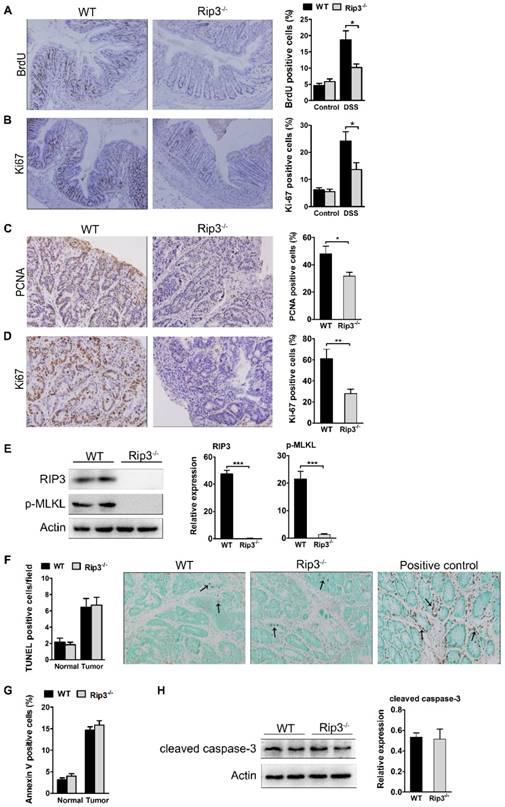
We next analyzed cell proliferation in WT and RIP3-deficient tumors. PCNA nuclear staining was obviously decreased in Rip3-/- adenomas (Figure 3C). The expression of Ki-67 was also lower in Rip3-/- tumors than WT tumors (Figure 3D), indicating a decreased growth capacity for the tumors in the absence of RIP3. To examine whether the deletion of RIP3 could prevent necroptosis activation, we analyzed the expression of phospho-MLKL, known to be a terminal executor of necroptosis. The deletion of RIP3 during CAC significantly reduced phospho-MLKL expression and therefore prevented necroptosis activation (Figure 3E). To analyze intracellular apoptosis rates, we performed TdT-mediated dUTP nick end labeling (TUNEL) staining. The number of TUNEL+ cells was similar in WT and Rip3-/- tumors (Figure 3F). There were also no significant differences in the numbers of annexin V+ cells between WT and Rip3-/- mice during acute colitis (Figure 3G). Our western blot assay also showed that RIP3 deletion did not affect cleaved caspase-3 expression levels in CAC adenomas (Figure 3H). Together, these results suggest that RIP3 promotes the proliferation of IECs and tumor growth independent of its effect on apoptosis.
RIP3 deficiency inhibits JNK activation to restrict tumorigenesis
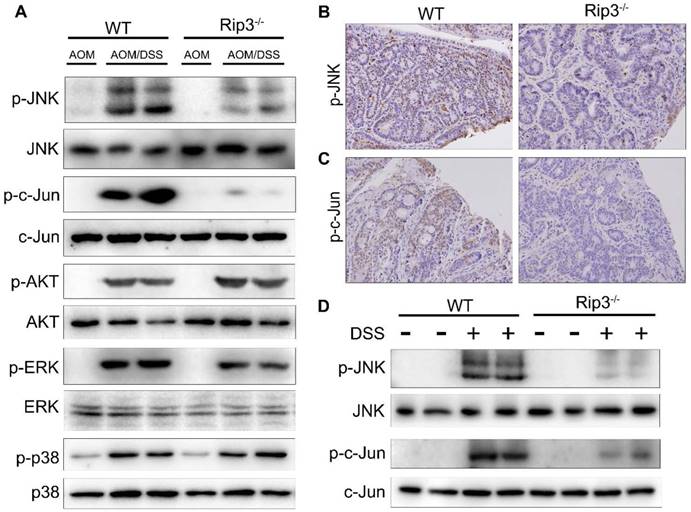
We further explored the molecular mechanisms that mediate the effects of RIP3 on tumor growth and proliferation during CAC tumorigenesis. Mitogen-activated protein kinase (MAPK) pathways are major regulators of cellular proliferation in cancer development [26]. Western blot analysis of the total colon lysates of WT and Rip3-/- CAC-bearing mice revealed marked downregulation of phosphorylated JNK (p-JNK) and its main substrate, phosphorylated c-Jun (p-c-Jun), in Rip3-/- mice, but the extent of activation of AKT, ERK, and p38 was not significantly altered by the absence of RIP3 (Figure 4A and Figure S3A-E). In agreement with these findings, immunohistochemical analysis confirmed that p-JNK and p-c-Jun positive cells were significantly decreased in Rip3-/- mice compared to the WT littermates (Figure 4B-C). However, since WT mice have higher tumor loads than Rip3-/- mice, it is difficult to conclude whether the changes in protein expression are a direct reflection of RIP3 deficiency or are partially due to reduced tumor load. To circumvent this difficulty, we examined the consequences of RIP3 deficiency during acute colitis and confirmed that there was a considerable reduction in p-JNK and p-c-Jun in IECs from Rip3-/- mice (Figure 4D and Figure S3F-G). Therefore, RIP3 is an important activator of the JNK pathway in IECs during acute colitis and tumor growth.
RIP3 deficiency inhibits the activation of JNK signaling during acute colitis and CAC. (A) Colonic lysates from AOM/DSS-treated WT and Rip3-/- mice were analyzed by immunoblotting with the indicated antibodies. (B-C) Images of phospho-JNK (B) and phospho-c-Jun (C) staining in CAC tumors from WT and RIP3-deficient mice. Original magnification, ×200. (D) Acute colitis was induced with 2.5% DSS. Colonic lysates were analyzed by immunoblotting with the indicated antibodies. The results shown are from 1 of 3 experiments performed.
Because RIP3 deficiency suppresses the activation of JNK/c-Jun signaling, leading to decreased intestinal cell proliferation, we sought to examine the effects of inactivation of the JNK pathway during CAC tumorigenesis. AOM/DSS-treated WT and Rip3-/- mice were gavaged with the JNK inhibitor SP600125 (40 mg/kg) for 100 days. Compared with the vehicle-treated group, SP600125 treatment significantly reduced tumor number, size, and load. However, SP600125 administration did not further enhance tumor protection in Rip3-/- mice (Figure S4A-C). Moreover, SP600125 treatment reduced the number of Ki-67-positive cells in WT tumors (Figure S4D). The increased p-JNK and p-c-Jun staining observed in IECs of CAC adenomas was also drastically reduced after treatment with SP600125 (Figure S4E-F). Taken together, these findings reveal a significant function of RIP3 in JNK signaling activation and proliferation of premalignant IECs.
RIP3 deletion enhances the immunogenicity of the TME during colitis and CAC
To examine whether RIP3 is involved in immune cell infiltration, we analyzed immunocyte profiles in the colonic mucosa of mice treated with AOM/DSS. RIP3 deletion diminished infiltration by tumor-associated macrophages (TAMs; Figure 5A). Conversely, the fractions of peri-tumoral CD3+ T cells and CD8+ T cells were increased in Rip3-/- mice (Figure 5B-C). CD8+ T cells expressed less IL-10 and PD-1 and more CD44 and CD107a in Rip3-/- mice than in WT mice (Figure 5D-G). There were no significant differences in the numbers of peri-tumoral B cells between WT and Rip3-/- mice (Figure 5H). Analysis of the myeloid compartment showed a decreased fraction of myeloid-derived suppressor cells (MDSC) and dendritic cells in Rip3-/- mice (Figure 5I-J). Furthermore, consistent with our immune-histochemical data, the number of bulk tumor-infiltrating TAMs and their M2-like CD206+ Arg1+ subset were reduced by RIP3 deletion (Figure 5K-M). Macrophage expression of programmed death ligand 1 (PD-L1) was also reduced by RIP3 deletion (Figure 5N). Altogether, these data suggest that RIP3 deletion increases lymphocyte accumulation and reduces infiltration by immunosuppressive subsets of myeloid cells in CAC.
RIP3 deletion enhances the immunogenicity of the tumor microenvironment during CAC. (A) Colon tissues from adenoma-bearing mice were stained with an anti-F4/80 antibody. Representative images and quantitative data are shown. Original magnification, ×200. (B-H) The fractions of peri-tumoral CD3+ T cells (B) and CD8+ T cells (C), the expression of IL-10 (D), PD-1 (E), CD44 (F) and CD107a (G) on CD8+ T cells, and the fraction of peri-tumoral CD19+ B cells (H) were determined by flow cytometry. SSC, side scatter. (I, J, K, L, and N) The fractions of peri-tumoral Gr1+ CD11b+ MDSC (I), F4/80- CD11c+ MHC II+ dendritic cells (J), and CD11c- Gr1- CD11b+ F4/80+ TAMs (K), and the expression of CD206 (L) and PD-L1 (N) in TAMs were assessed by flow cytometry. (M) Lysates of distal colons from CAC-bearing mice were prepared, and Arg1 mRNA expression was analyzed using qRT-PCR. Data are means ± SEM (n = 5), *p < 0.05, **p < 0.01, ***p < 0.001. Flow cytometry experiments were carried out twice.
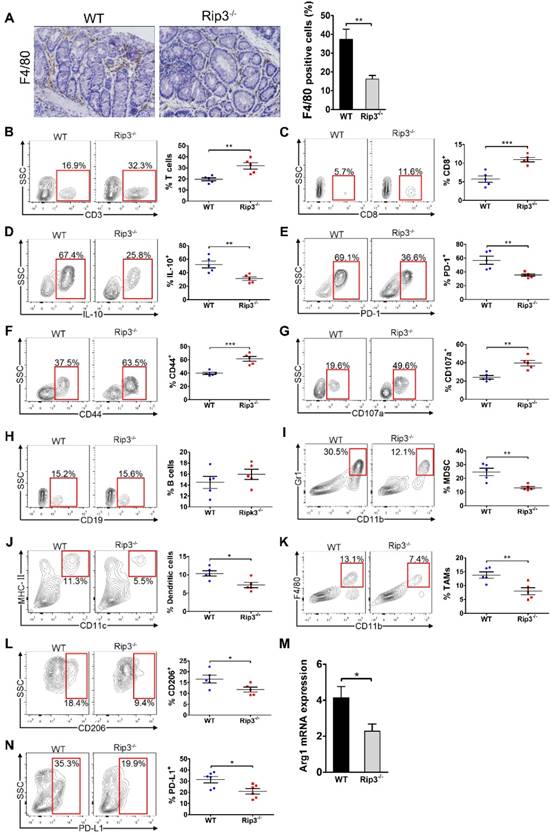
Because Rip3-/- mice showed increased intestinal inflammation in acute DSS-induced colitis, we next examined inflammatory cell infiltrate in the colonic mucosa of mice during acute colitis. RIP3 deletion resulted in elevated T cell infiltration, decreased recruitment of MDSCs and dendritic cells, and a diminished fraction of TAMs with M1-like polarization (Figure S5A-E). Colonic CD8+ T cells also showed higher activation in Rip3-/- mice than in WT mice, as shown by elevated CD107a and CD44 expression (Figure S5F-H). The fraction of B cells was not different between DSS-challenged WT and Rip3-/- mice (Figure S5I). In addition, in naive WT and Rip3-/- mice, deletion of RIP3 did not affect infiltration of CD3+ T cells, CD8+ T cells, B cells, and dendritic cells, but was associated with decreased MDSC infiltration and a trend towards a reduction in the number of macrophages (Figure S5J-O). Taken together, these results indicate that RIP3 deletion enhances the immunogenicity of the inflammatory TME during acute DSS-induced colitis and tumor growth.
We further examined whether the upregulation of cytotoxic CD8+ T cells in the colon of Rip3-/- mice resulted in a reduction in epithelial tumor cells in our AOM/DSS model. As shown in Figure S6A, colonic CD8+ T cells isolated from Rip3-/- mice had higher cytotoxicity against epithelial tumor cells isolated from AOM/DSS-treated WT mice than colonic CD8+ T cells isolated from WT mice, and the cytotoxicity of colonic CD8+ T cells against tumor cells was ratio-dependent. Furthermore, tumor-associated CD8+ T cells isolated from Rip3-/- mice produced higher levels of INFγ than CD8+ T cells from WT mice (Figure S6B). These results suggest that RIP3 deletion enhanced colonic CD8+ T cell cytotoxicity against epithelial tumor cells in CAC. To investigate whether protection against oncogenesis in the absence of RIP3 is T cell dependent, we depleted T cells and induced CAC tumors in Rip3-/- mice. Protection against tumor growth was abrogated by T cell depletion in Rip3-/- mice (Figure S6C-E), indicating that T cells are tumor-protective during CAC in Rip3-/- mice.
RIP3 promotes colorectal tumorigenesis via CXCL1-induced immune suppression
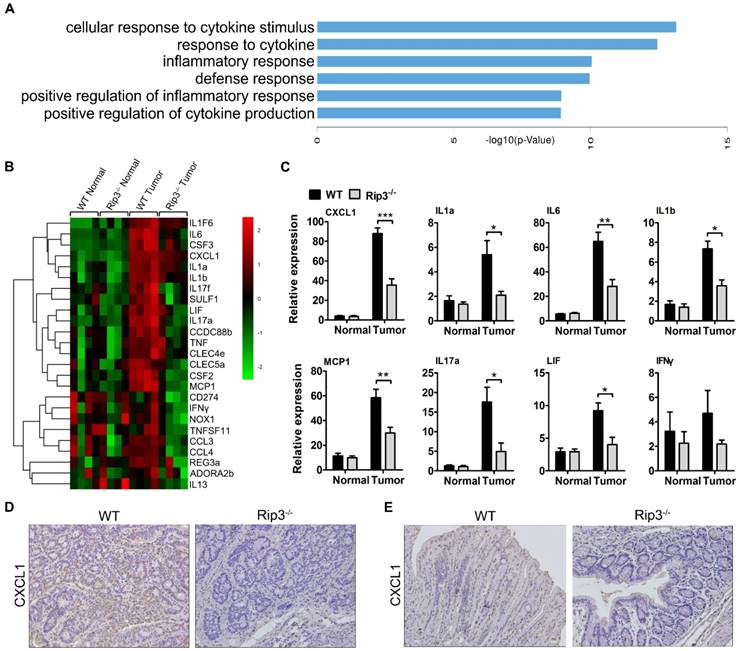
To explore the mechanism by which RIP3 impacts immune suppression in CAC, we performed a DNA microarray experiment with normal and tumor RNA samples from WT and Rip3-/- mice. The results showed a significant reduction in genes mainly related to inflammation and cytokine secretion in the Rip3-/- mouse tumors, these genes included Il1a, CXCL1, Tnf, and Il17a (Figure 6A-B). Real-time PCR analysis suggested that CXCL1 is one of the most significantly decreased chemokines in Rip3-/- tumors (Figure 6C and Figure S7). Immunohistochemical staining also revealed a considerably reduced level of CXCL1 in Rip3-/- adenomas (Figure 6D). Furthermore, in acute DSS-induced colitis, CXCL1 expression was significantly reduced by the absence of RIP3 (Figure 6E). Therefore, CXCL1 is expressed in a RIP3-dependent manner during colitis and CAC.
Deletion of RIP3 reduces CXCL1 expression. (A-B) Differences in gene expression between tumors and normal colon tissues derived from WT and Rip3-/- animals are presented. (A) Comparison between differentially regulated genes enriched in biological processes terms in tumor tissues from Rip3-/- mice compared to WT mice. The top 6 significantly enriched Gene Ontology (GO) terms in the biological process category are shown here. (B) A significant decrease in the expression of genes involved in inflammation-induced CAC was observed in Rip3-/- mice after the AOM/DSS treatment compared with similarly treated WT mice. For each experimental condition, four mice were used. The data shown in A and B were obtained from a microarray experiment that was performed once. (C) The gene expression profiles of colon tissues from mice treated with AOM/DSS were analyzed using real-time PCR. Gene expression was normalized to GAPDH levels. One out of two representative experiments is shown. Data are presented as the means ± SEM (n = 5 mice per group). *p < 0.05, **p < 0.01, ***p < 0.001. (D) Immunohistochemical staining for CXCL1 in CAC tumors from WT and Rip3-/- mice. Magnification, ×200. (E) Immunohistochemical staining for CXCL1 in colons from WT and Rip3-/- mice treated with DSS for 7 days. Magnification, ×200.
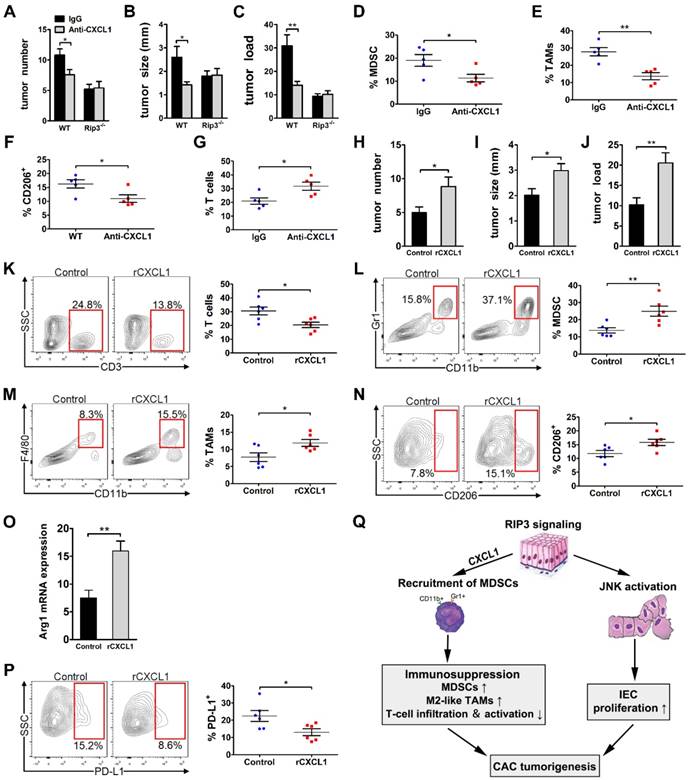
CXCL1 has been reported to be linked to immune suppression and tumorigenesis in various tumor types. To determine whether CXCL1 mediates the protumorigenic immune suppression associated with RIP3 signaling by mobilizing myeloid cells, we induced colorectal tumors in WT and RIP3-/- mice while blocking CXCL1. CXCL1 blockade during CAC induction drastically reduced tumor size, multiplicity, and load (Figure 7A-C). Moreover, similar to RIP3 deletion, CXCL1 blockade reduced MDSC and M2-like TAMs accumulation and increased T cell recruitment (Figure 7D-G). However, anti-CXCL1 treatment did not further enhance tumor protection in Rip3-/- mice (Figure 7A-C). We next administer recombinant CXCL1 (rCXCL1) to RIP3-/- mice to determine whether there was a recovery of phenotype in CAC. Indeed, exogenous CXCL1 treatment during CAC increased tumor size and tumor load in Rip3-/- mice (Figure 7H-J). Moreover, the inflammatory TME in rCXCL1-treated Rip3-/- colon recapitulated the immune-suppressive milieu associated with intact necroptosis signaling. Specifically, rCXCL1-treated colons tended to contain a lower fraction of tumor-infiltrating T cells (Figure 7K) and exhibited increased recruitment of both MDSCs (Figure 7L) and M2-like TAMs, which expressed high PD-L1 compared with control colons (Figure 7M-P). These data suggest that RIP3-induced CXCL1 signaling promotes myeloid cell-induced adaptive immune suppression in CAC.
CXCL1 promotes the progression of CAC. (A-G) CAC was induced in WT and Rip3-/- mice. A neutralizing CXCL1 antibody (anti-CXCL1) or control antibody was intraperitoneally injected once a week for 14 weeks. Tumor number (A), tumor size (B), and tumor load (C) were analyzed on day 100. (D-G) The fractions of Gr1+ CD11b+ MDSCs (D) and Gr1- CD11b+ F4/80+ TAMs (E), the expression of CD206 in TAMs (F), and the fraction of CD3+ T cells (G) were assessed by flow cytometry. (H-P) CAC was induced in Rip3-/- mice and animals were injected intraperitoneally with 300 ng recombinant CXCL1 (rCXCL1) or control vehicle (PBS) twice a week for 14 weeks. Tumor number (H), tumor size (I), and tumor load (J) were analyzed on day 100. (K, L, M, N, and P) The fractions of peri-tumoral CD3+ T cells (K), Gr1+ CD11b+ MDSCs (L), Gr1- CD11b+ F4/80+ TAMs (M), and the expression of CD206 (N) and PD-L1 (P) in TAMs were assessed by flow cytometry. (O) Lysates of distal colons from CAC-bearing mice were prepared, and Arg1 mRNA expression was analyzed using qRT-PCR. Flow cytometry experiments were carried out twice. Data are presented as means ± SEM (n = 5), *p < 0.05, **p < 0.01. (Q) Working model of the role of RIP3 in regulating tumor cell proliferation via JNK signaling and promoting myeloid cell-induced immune suppression via CXCL1 signaling.
Discussion
Colitis-associated colorectal cancer is the most serious complication of IBD [27]. Initial studies have reported that RIP3-dependent necroptosis contributes to the pathogenesis of IBD in humans [19, 20]. RIP3 has a critical role in promoting innate inflammatory cytokine expression and proliferation of IECs during acute DSS-induced colitis [18]. Furthermore, several studies highlighted an important function of RIP3 in tumor promotion [16, 17]. However, the role of RIP3 in regulating CAC initiation and progression in more physiological settings remains unclear. In this paper, our study reveals an unexpected function of RIP3 in regulating the proliferation of premalignant IECs via JNK signaling and promoting myeloid cell-induced adaptive immune suppression via CXCL1 signaling during inflammation-associated colon carcinogenesis (Figure 7Q). Targeting RIP3 signaling may be a novel therapeutic strategy for the treatment and prevention of CAC.
Previous studies have suggested that RIP3 mRNA expression in the colon was strongly induced by DSS treatment [18]. Additionally, elevated levels of RIP3 have been found in the intestinal epithelium of both adult and pediatric Crohn's disease patients [19, 20]. Our results suggest that RIP3 expression is upregulated in mouse colitis-associated tumors and in human colorectal cancer. RIP3 was expressed in mononuclear cell infiltrates and intestinal epithelium. RIP3-deficient mice developed fewer and smaller adenomas than WT mice. Bone marrow transplantation experiments suggested that hematopoietic-derived RIP3 contributes to colitis-associated tumorigenesis.
RIP3 has been shown to be essential for injury-induced inflammation and tissue repair in the intestine during acute colitis [18]. Our data suggest that in acute DSS-induced colitis, Rip3-/- mice developed more severe colitis than WT mice but exhibited decreased IEC proliferation. Since repair-associated proliferation may be linked to tumor growth, deletion of RIP3 during CAC induction drastically reduced tumor multiplicity and load as well as tumor growth, as shown by decreased Ki-67 and PCNA expression. Therefore, the reduced proliferation of IECs may partially explain why colitis is increased but colorectal adenomas are reduced in Rip3-/- mice.
The attenuated proliferation of premalignant IECs observed in Rip3-/- tumors raises the question of what the contributing mechanisms downstream of RIP3 may be. MAPK pathways are major regulators of cellular growth and proliferation [26]. The JNK pathway has been implicated in oncogenic transformation and cell proliferation during the pathogenesis of cancer in various tissues [28]. The role of the JNK pathway in the promotion of colon carcinogenesis has been described recently. The JNK pathway and its main substrate, the c-Jun transcription factor, have been associated with increases in intestinal cell numbers through regulating the proliferation of intestinal progenitor cells. Activation of JNK/c-Jun signaling accelerated colorectal tumorigenesis, and inactivation of c-Jun led to decreased progenitor cell proliferation and delayed tumorigenesis in mice [29]. Furthermore, a recent study demonstrated that activation of the JNK pathway acts as a downstream mediator of RIP3-driven necroptosis in a mouse model of ethanol-induced liver injury [30]. Another study elegantly demonstrated the presence of a positive feedback loop between RIP3 and JNK signaling. RIP3-dependent JNK activation promotes the release of proinflammatory mediators in non-alcoholic steatohepatitis [31]. Our work showed that the expression levels of both p-JNK and p-c-Jun in mouse models of colitis and CAC were dramatically reduced by deletion of RIP3, but activation of AKT, ERK, and p38 in WT and Rip3-/- colon polyps did not differ. Moreover, we found that the specific JNK inhibitor SP600125 significantly reduced colitis-associated tumorigenesis in mice by inhibiting the proliferation of IECs. These results reveal a critical function of RIP3 in JNK signaling activation and proliferation of premalignant IECs.
In the context of CAC, RIP3 deletion led to immunogenic reprogramming of innate and adaptive inflammatory entities, as evidenced by an increase in tumor infiltrating CD8+ T cells as well as a reduction in MDSCs and TAMs. Furthermore, TAMs exhibited a shift toward an M1-like immunogenic phenotype. Since MDSCs have suppressive effects on the immune response via T cells by direct cell-cell contact [32]. Decreased MDSCs and TAMs lead to the generation of immunogenic T cells, which have powerful antitumor effects. In addition, in acute DSS-induced colitis, Rip3-/- mice exhibited more severe colitis than WT mice and showed increased immunogenic T cell infiltration, decreased recruitment of MDSCs, and a diminished fraction of TAMs with M1-like polarization. These data show for the first time that RIP3 deletion enhances the immunogenicity of the inflammatory TME during CAC induction. RIP3 is linked to immune suppression and tumorigenesis in CAC via expression of CXCL1, a potent chemoattractant for myeloid cells that was highly expressed in a RIP3-dependent manner. Deletion of RIP3 reduced CXCL1 expression in colitis and CAC. CXCL1 has a complex role in extra-colonic malignancies. In breast cancer, metastatic cells overexpressing CXCL1 exhibited chemoresistance via a paracrine loop by attracting Gr1+ CD11b+ myeloid cells, which enhance cancer survival by secreting S100A8/9 [33]. Furthermore, in ovarian cancer, CXCL1 was found to induce epithelial cell proliferation by transactivation of EGFR [34]. Similarly, in melanoma, CXCL1 plays a role in the genesis of primary melanocytic lesions when coupled with the loss of INK-4a/ARF [35]. Moreover, CXCL1-related cytokines recruited immune-suppressive MDSCs to the pre-metastatic niche in the liver of colorectal carcinoma-bearing hosts [36]. The expression of CXCL1 was elevated in AOM/DSS tumors compared to normal mucosa (Figure 6C). Blocking CXCL1 in mice suppressed CAC progression and was associated with myeloid cell-mediated adaptive immune suppression by decreased MDSC and M2-like macrophage infiltration. In addition, rCXCL1 treatment restored tumorigenesis in Rip3-/- mice and promoted macrophage-induced adaptive immune suppression. These data provide a rationale to develop CXCL1-neutralizing antibodies as therapeutic approaches for subverting tumor-induced immunosuppression.
The advent of clinical-grade therapeutics targeting RIP3 signaling may herald an exciting new era in cancer therapy. Furthermore, there is ample rationale for the potential synergistic efficacy of targeting RIP3 signaling in combination with available checkpoint-directed immunotherapies. Ligation of T cell checkpoint receptors, including cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and PD-1, is known to dampen the activation of T cells and deliver inhibitory signals to T cells [37-40]. Monoclonal antibodies directed at T cell checkpoint receptors have shown significant antitumor effects in several solid tumors, including colorectal cancer [41, 42]. Thus, expansion and activation of T cells via blockade of RIP3 signaling is a promising avenue to increase T cell activity and enhance the efficacy of cancer immunotherapy.
Abbreviations
RIP3: receptor-interacting protein 3; CAC: colitis-associated cancer; AOM: azoxymethane; DSS: dextran sodium sulfate; IECs: intestinal epithelial cells; IBDs: inflammatory bowel diseases; UC: ulcerative colitis; TNF: tumor necrosis factor; MLKL: mixed-lineage kinase domain-like; DAMPs: damage-associated molecular patterns; AML: acute myeloid leukemia; DR6: death receptor 6; TME: tumor microenvironment; WT: wild-type; BrdU: 5-bromo-2-deoxyuridine; TUNEL: TdT-mediated dUTP nick end labeling; MAPK: mitogen-activated protein kinase; TAMs: tumor-associated macrophages; PD-L1: programmed death ligand 1; CTLA-4: T-lymphocyte-associated antigen 4.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank Yu-Kui Shang, Xi-Ying Yao, Bing Xu, Ling Li, and Hui-Jie Bian for thoughtful discussions about this research project.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81872316 and 81472700), the National Science and Technology Major Special Project of China (2018ZX09101001 and 2017ZX10203205-004-002), the National Basic Research Program (2015CB553701), and the Shaanxi Science and Technology New Star Project (2017KJXX-68).
Author contributions
ZYL, MZ, YML, XYF, JLJ, JT, and ZNC developed the hypothesis, designed experiments, analyzed the data, and wrote the manuscript. ZYL, MZ, YML, XYF, JCW, ZCL, HJY, JMY, and JC performed the experiments. ZYL, MZ, YML, XYF, JCW, ZCL, JLJ, JT, and ZNC contributed to the experimental design, generation of the reagents, and manuscript editing. ZYL, MZ, YML, XYF, JLJ, JT, and ZNC conceived and supervised the project.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883-99
2. Ullman TA, Itzkowitz SH. Intestinal inflammation and cancer. Gastroenterology. 2011;140:1807-16
3. Saleh M, Trinchieri G. Innate immune mechanisms of colitis and colitis-associated colorectal cancer. Nat Rev Immunol. 2011;11:9-20
4. He S, Wang L, Miao L, Wang T, Du F, Zhao L. et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100-11
5. Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC. et al. RIP3, an Energy Metabolism Regulator That Switches TNF-Induced Cell Death from Apoptosis to Necrosis. Science. 2009;325:332-6
6. Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S. et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489-95
7. Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M. et al. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell. 2011;43:449-63
8. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311-20
9. Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS. et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150:339-50
10. Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J. et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16:55-65
11. Gunther C, Neumann H, Neurath MF, Becker C. Apoptosis, necrosis and necroptosis: cell death regulation in the intestinal epithelium. Gut. 2013;62:1062-71
12. Deutsch M, Graffeo CS, Rokosh R, Pansari M, Ochi A, Levie EM. et al. Divergent effects of RIP1 or RIP3 blockade in murine models of acute liver injury. Cell Death Dis. 2015;6:e1759
13. Dannappel M, Vlantis K, Kumari S, Polykratis A, Kim C, Wachsmuth L. et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature. 2014;513:90-4
14. Hockendorf U, Yabal M, Herold T, Munkhbaatar E, Rott S, Jilg S. et al. RIPK3 Restricts Myeloid Leukemogenesis by Promoting Cell Death and Differentiation of Leukemia Initiating Cells. Cancer Cell. 2016;30:75-91
15. Cerhan JR, Ansell SM, Fredericksen ZS, Kay NE, Liebow M, Call TG. et al. Genetic variation in 1253 immune and inflammation genes and risk of non-Hodgkin lymphoma. Blood. 2007;110:4455-63
16. Seifert L, Werba G, Tiwari S, Giao Ly NN, Alothman S, Alqunaibit D. et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature. 2016;532:245-9
17. Strilic B, Yang L, Albarran-Juarez J, Wachsmuth L, Han K, Muller UC. et al. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature. 2016;536:215-8
18. Moriwaki K, Balaji S, McQuade T, Malhotra N, Kang J, Chan FK. The necroptosis adaptor RIPK3 promotes injury-induced cytokine expression and tissue repair. Immunity. 2014;41:567-78
19. Gunther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H. et al. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature. 2011;477:335-9
20. Pierdomenico M, Negroni A, Stronati L, Vitali R, Prete E, Bertin J. et al. Necroptosis Is Active in Children With Inflammatory Bowel Disease and Contributes to Heighten Intestinal Inflammation. Am J Gastroenterol. 2014;109:279-87
21. Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ. et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285-96
22. Cotta CV, Zhang Z, Kim HG, Klug CA. Pax5 determines B- versus T-cell fate and does not block early myeloid-lineage development. Blood. 2003;101:4342-6
23. Bedrosian AS, Nguyen AH, Hackman M, Connolly MK, Malhotra A, Ibrahim J. et al. Dendritic cells promote pancreatic viability in mice with acute pancreatitis. Gastroenterology. 2011;141:1915-26 e1-14
24. Vezys V, Olson S, Lefrancois L. Expression of intestine-specific antigen reveals novel pathways of CD8 T cell tolerance induction. Immunity. 2000;12:505-14
25. Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF. et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54:133-46
26. Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279-90
27. Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001;48:526-35
28. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239-52
29. Sancho R, Nateri AS, de Vinuesa AG, Aguilera C, Nye E, Spencer-Dene B. et al. JNK signalling modulates intestinal homeostasis and tumourigenesis in mice. EMBO J. 2009;28:1843-54
30. Christofferson DE, Yuan J. Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol. 2010;22:263-8
31. Gautheron J, Vucur M, Reisinger F, Cardenas DV, Roderburg C, Koppe C. et al. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol Med. 2014;6:1062-74
32. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162-74
33. Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG. et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell. 2012;150:165-78
34. Bolitho C, Hahn MA, Baxter RC, Marsh DJ. The chemokine CXCL1 induces proliferation in epithelial ovarian cancer cells by transactivation of the epidermal growth factor receptor. Endocr Relat Cancer. 2010;17:929-40
35. Yang J, Luan J, Yu Y, Li C, DePinho RA, Chin L. et al. Induction of melanoma in murine macrophage inflammatory protein 2 transgenic mice heterozygous for inhibitor of kinase/alternate reading frame. Cancer Res. 2001;61:8150-7
36. Connolly MK, Mallen-St Clair J, Bedrosian AS, Malhotra A, Vera V, Ibrahim J. et al. Distinct populations of metastases-enabling myeloid cells expand in the liver of mice harboring invasive and preinvasive intra-abdominal tumor. J Leukoc Biol. 2010;87:713-25
37. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734-6
38. Schneider H, Downey J, Smith A, Zinselmeyer BH, Rush C, Brewer JM. et al. Reversal of the TCR stop signal by CTLA-4. Science. 2006;313:1972-5
39. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB. et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793-800
40. Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T. et al. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004;64:1140-5
41. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK. et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409-13
42. Sclafani F. PD-1 inhibition in metastatic dMMR/MSI-H colorectal cancer. Lancet Oncol. 2017;18:1141-2
Author contact
![]() Corresponding author: Jian-Li Jiang, Juan Tang or Zhi-Nan Chen, National Translational Science Center for Molecular Medicine, Xi'an 710032, China; Department of Cell Biology, School of Basic Medicine, Air Force Medical University, 169 West Changle Road Xi'an 710032, China. E-mail: jiangjledu.cn (JLJ), tangjuan1edu.cn (JT) or zhinanchenedu.cn (ZNC).
Corresponding author: Jian-Li Jiang, Juan Tang or Zhi-Nan Chen, National Translational Science Center for Molecular Medicine, Xi'an 710032, China; Department of Cell Biology, School of Basic Medicine, Air Force Medical University, 169 West Changle Road Xi'an 710032, China. E-mail: jiangjledu.cn (JLJ), tangjuan1edu.cn (JT) or zhinanchenedu.cn (ZNC).