Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusions
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(15):4287-4307. doi:10.7150/thno.36077 This issue Cite
Research Paper
The role of Nav1.7 and methylglyoxal-mediated activation of TRPA1 in itch and hypoalgesia in a murine model of type 1 diabetes
Ruo-Xiao Cheng1,2,7#, Yu Feng1,2#, Di Liu3,4#, Zhi-Hong Wang2#, Jiang-Tao Zhang2, Li-Hua Chen5, Cun-Jin Su1,2, Bing Wang2, Ya Huang2, Ru-Rong Ji3,4, Ji Hu1, ![]() , Tong Liu1,2,6,
, Tong Liu1,2,6, ![]()
1. Jiangsu Key Laboratory of Neuropsychiatric Diseases and the Second Affiliated Hospital of Soochow University, Suzhou 215004, P.R. China.
2. Institute of Neuroscience, Soochow University, Suzhou, 215021, P.R. China.
3. Departments of Anesthesiology, Duke University Medical Center, Durham, North Carolina, 27710, USA.
4. Department of Neurobiology, Duke University Medical Center, Durham, North Carolina, 27710, USA.
5. Jiangsu Key Laboratory of Preventive and Translational Medicine for Geriatric Diseases, Department of Nutrition and Food Hygiene, School of Public Health, Soochow University, Suzhou, 215021, P.R. China.
6. College of Life Sciences, Yanan University, Yanan, 716000, China
7. Present address: State Key Laboratory of Bio-organic and Natural Products Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai, 200032, China
#These authors contributed equally to this study.
Received 2019-4-26; Accepted 2019-4-26; Published 2019-5-31
Abstract

Methylglyoxal (MGO), an endogenous reactive carbonyl compound, plays a key role in the pathogenesis of diabetic neuropathy. The aim of this study is to investigate the role of MGO in diabetic itch and hypoalgesia, two common symptoms associated with diabetic neuropathy.
Methods: Scratching behavior, mechanical itch (alloknesis), and thermal hypoalgesia were quantified after intradermal (i.d.) injection of MGO in naïve mice or in diabetic mice induced by intraperitoneal (i.p.) injection of streptozotocin (STZ). Behavioral testing, patch-clamp recording, transgenic mice, and gene expression analysis were used to investigate the mechanisms underlying diabetic itch and hypoalgesia in mice.
Results: I.d. injection of MGO evoked dose-dependent scratching in normal mice. Addition of MGO directly activated transient receptor potential ankyrin 1 (TRPA1) to induce inward currents and calcium influx in dorsal root ganglia (DRG) neurons or in TRPA1-expressing HEK293 cells. Mechanical itch, but not spontaneous itch was developed in STZ-induced diabetic mice. Genetic ablation of Trpa1 (Trpa1-/-), pharmacological blockade of TRPA1 and Nav1.7, antioxidants, and mitogen-activated protein kinase kinase enzyme (MEK) inhibitor U0126 abrogated itch induced by MGO or in STZ-induced diabetic mice. Thermal hypoalgesia was induced by intrathecal (i.t.) injection of MGO or in STZ-induced diabetic mice, which was abolished by MGO scavengers, intrathecal injection of TRPA1 blockers, and in Trpa1-/- mice.
Conclusion: This study revealed that Nav1.7 and MGO-mediated activation of TRPA1 play key roles in itch and hypoalgesia in a murine model of type 1 diabetes. Thereby, we provide a novel potential therapeutic strategy for the treatment of itch and hypoalgesia induced by diabetic neuropathy.
Keywords: Itch, Hypoalgesia, Diabetes, TRPA1, Methylglyoxal
Introduction
Itch (pruritus) is a unpleasant cutaneous sensation that causes the desire or reflex to scratch [1]. Acute itch protects us against harmful stimuli by rapid remove them from the skin. Primary sensory neurons are responsible for detecting pruritogens by peripheral free nerve terminals, and they relay sensory information via their central branches to spinal cord dorsal horn, then project to higher brain centers to produce the perception of itch [2; 3]. Chronic itch is an important symptom of systemic diseases, including skin, liver, and kidney diseases, and it is debilitating and dramatically reduced quality of life [4]. Unfortunately, there are few therapeutic strategies for management of chronic itch. Lack of information about the mechanisms underlying chronic itch caused by systemic diseases is a major hurdle to develop effective anti-itch treatment.
Diabetic neuropathy is a debilitating complication in both type 1 and type 2 diabetes [5; 6]. Up to 50% of diabetes patients develop peripheral neuropathy during their lifetime [7; 8]. Many patients with diabetic neuropathy suffer from chronic neuropathic pain, such as tactile allodynia (touch-evoked pain) [9]. Intriguingly, clinicians have a strong impression that itch is more frequent or severe in diabetic patients compared with nondiabetic patients [10]. Recent epidemiological study showed that itch was observed in about 12% of general diabetic outpatients in a frequency similar to that of feet pain [10]. Thus, itch is now considered as a newly recognized symptom of diabetic neuropathy, and it may reflect the dysfunction of skin or peripheral nervous system [10]. Alarmingly, itch is an often overlooked symptom for diabetic patients, although itch substantially reduces the quality of life [4]. Additionally, pain hypersensitivity is progressively replaced by loss of pain sensation, termed hypoalgesia, possibly due to loss of peripheral free nerve terminals [11]. Unfortunately, there are current lack of medications for the treatment of itch and hypoalgesia associated with diabetic neuropathy [11; 12]. Although the molecular mechanisms underlying diabetic neuropathic pain are increasingly being uncovered [9; 13-15], the etiology of itch and hypoalgesia associated with diabetic neuropathy remains poorly understood.
Methylglyoxal (MGO) is a reactive α-dicarbonyl metabolite that is synthesized as a byproduct of flux through multiple pathways, including glycolysis and lipid peroxidation [16; 17]. Elevated levels of plasma MGO (over 800 nmol/L) was demonstrated in diabetic patients [6; 18]. MGO mediates rapid non-enzymatic glycation of proteins, lipids, and DNA to promote formation of advanced glycation end products (AGEs), which eventually renders irreversible damage to these macromolecules, including their integrity of structure and function [18-20]. Accelerated AGEs formation contributes to the pathogenesis of certain types of diabetic complications, including peripheral neuropathy, cardiomyopathy, retinopathy, and vascular damage [19-22]. MGO is primarily metabolized to the end product D-lactate by glyoxalase 1 (GLO1) and GLO2, which belong to the evolutionarily conserved glutathione-dependent glyoxalase system [23]. Peripheral nerves are considered to be particularly susceptible to toxicity of MGO, since the activity of GLO1 is low in these tissues [13]. Systemic increase in MG levels by pharmacological inhibition or genetic deletion of GLO1 has been shown to produce painful diabetic neuropathy in the absence of hyperglycemia in rodents [24; 25]. Thus, elevated levels of MGO and formation of AGEs contribute to the pathogenesis of peripheral diabetic neuropathy [26]. However, a recent clinical study failed to confirm an association between the serum MGO level and diabetic peripheral neuropathy in a cohort of well-treated patients with short-term Type 2 diabetes [27]. Notably, previous studies demonstrated that MGO-mediated modification of voltage gated sodium channel Nav1.8 and MGO-mediated activation of transient receptor potential ankyrin 1 (TRPA1) contribute to diabetic neuropathic pain [13; 28; 29]. To date, it remains elusive about the roles of MGO in itch and loss of pain sensation (hypoalgesia) associated with diabetic neuropathy.
In this study, to better understand the mechanisms underlying diabetic itch and hypoalgesia, we sought to identify the role of MGO and its downstream signaling in itch and hypoalgesia in a mouse model of type 1 diabetes by using patch clamp recording, calcium imaging, pharmacological or genetic manipulations, and behavioral testing. Our results reveal that MGO-mediated activation of TRPA1 and Nav1.7 play crucial roles in diabetic itch and hypoalgeisa.
Materials and Methods
Ethics
This study was approved by the Ethics Review Board of all authors' institutions.
Animals
Male ICR mice (6-8 weeks old) were obtained from the Shanghai SLAC Laboratory Animal CO., LTD. (Shanghai, China). Male C57BL/6J, Trpv1-/-, and Trpa1-/- mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA). Both Trpv1-/- and Trpa1-/- mice were continuously backcrossed to C57BL/6J. All animals were maintained under a 12-h light/dark cycle with food and water available ad libitum, and the room was kept at 22 ± 2°C and 40%-60% humidity. All animal experiments were performed according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the guidelines of the International Association for the Study of Pain. All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committee at Soochow University and Duke University.
Generation of STZ-induced diabetic mice
Diabetes model was induced by a single intraperitoneal injection of streptozotocin (STZ; 100 mg/kg, Sigma-Aldrich, St. Louis, MO). STZ was freshly dissolved in citrate buffer (0.1 mol/L, citrate:Na citrate = 1:1, pH 4.2-4.4). The control group received citrate buffer only in an equivalent volume. One week later, diabetes was confirmed by measurements of blood glucose concentrations in samples obtained from the tail vein. Only mice with random blood glucose concentration >16.7 mmol/L (300 mg/dL) were further used in the study.
Behavioral testing
Mice were intradermally injected of 50 μl of agents into the nape of the neck or cheek, and scratching behavior were recorded and quantified as described previously [30; 31]. The alloknesis score was determined by calculating the total number of scratching reaction elicited by ten mechanical stimuli, providing by a von Frey filament (0.7 mN) as previous reports [32; 33]. For testing alloknesis in diabetic model, von Frey stimuli were applied at the nape of the neck area to elicit scratching response at 1, 3, and 5 weeks after injection of streptozotocin (STZ). The terminal 3 cm of a mouse's tail was immersed in hot water bath at 52 °C and the latency of tail flick was recorded. The results were conveyed as a percent of the maximum possible effect (% MPE) according to the equation: % MPE = 100 × (Experimental latency - Baseline latency)/(10 sec - Baseline latency). Behavioral tests were videotaped from a side angle and behavioral tests were done by observers blind to the treatments or animal genotypes. The detailed methods see Supplementary material.
Pharmacological treatments
(1) Morphine (1 mg/kg), naloxone (1 mg/kg), or chlorpheniramine (1 and 10 mg/kg), N-Acetyl-L-cysteine (NAC; 200 mg/kg), N-tert-butyl-a-phenylnitrone (PBN; 100 mg/kg) or alpha-lipoic acid (ALA; 100 mg/kg) was intraperitoneal (i.p.) injected into mice 20 min before intradermally (i.d.) injection of 3 μmol MGO in mice. (2) We depleted mast cells by daily treatment with compound 48/80 (1, 3, 10 and 10 μg per site on the 1st, 2nd, 3rd and 4th days, respectively) before injection of MGO. (3) We destroyed the C-fibers by daily treatment with the resiniferatoxin (RTX, 30, 70 and 100 μg/kg; subcutaneously for 3 consecutive days), one week before injection of MGO. (4) Capsazepine (50 μg), HC-030031 (50 μg), A967079 (50 μg), A803467 (3 μg), TTX (100 ng), PF-05089771 (10 μg), ALA (100, 200, and 300 μg), or U0126 (1 and 10 nmol) were co-administrated with 3 μmol MGO in the nape of neck in mice. (5) Aminoguanidine (180 mM) and D-arginine (180 mM), two MGO scavengers, were incubated with MGO (60 mM) at 37 °C for 3 hours and then was i.d. injected (total volume 50 μl) in mice. (6) Intrathecal (I.t.) injection of mitogen-activated protein kinase kinase (MEK) inhibitor U0126 (1 nmol) was performed 20 min before i.d. injection of MGO.
Dissociation of mouse DRG neurons and patch clamp recording
DRG in cervical segments (C4-C8) were isolated and cultured using a previous described protocol [34]. Briefly, after laminectomies on control or diabetic mice, bilateral DRGs were dissected and incubated in dissecting solution with collagenase (1.25mg/ml, Roche, Switzerland)/dispase-II (2.4 units/ml, Roche, Switzerland) for 90 min at 37 °C, then digested with 0.25% trypsin for 8 min at 37 °C, followed by 0.25% trypsin inhibitor. DRGs were mechanically dissociated with a flame polished Pasteur pipette in the presence of 0.05% DNAse I (Cat#ampd1, Sigma-Aldrich, St. Louis, MO, USA). DRG neurons were plated on glass cover slips and grown in a medium (with 2% B27 supplement, Thermo Fisher Scientific) with 5 μM arabinosylcytosine (AraC) and 5% carbondioxide at 36.5 °C. Whole-cell patch clamp recordings were performed at room temperature (22-24 °C) using an Axon 700B amplifier (Molecular Devices, Sunnyvale, CA, USA). The patch pipettes were pulled from borosilicate capillaries (Chase Scientific Glass Inc.). Pipette resistance was 4-6 MΩ. To prevent large current-induced desensitization or tachyphylaxis in the recordings after repeated application of the agonist (AITC, 50 μM) and MGO (1 mM), current amplitudes that were no larger than 1500 pA were used for analysis. The bath solution (in mM) contained: 140 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 5 EGTA, 10 glucose, and 10 HEPES (pH7.4, adjusted with NaOH and osmolarity 300-310 mOsm). The pipette solution (in mM) contained 126 K-gluconate, 10 NaCl, 1 MgCl2, 10 EGTA, 2 NaATP, and 0.1 MgGTP (pH 7.3, adjusted with KOH and osmolarity 295-300 mOsm). The pClamp10 (Axon Instruments) software was used during experiments and data analysis.
Live Cell Ca2+ imaging
We used Live Cell Imaging System to measure the level of intracellular free calcium using the fluorescent marker Fluo 3-AM (Cat#39294, Sigma-Aldrich, St. Louis, MO, USA) dissolved in dimethyl sulfoxide (DMSO) as a stock solution, according to the manufacturer's instructions. HEK-293 cells were purchased from ATCC (ATCC CRL-1573). Cells were grown as a monolayer maintained in Dulbecco's modified Eagles medium (DMEM) (Life Technologies, Carlsbad, CA, USA), supplemented with 10% fetal bovine serum (FBS) (Life Technologies), 100 units/ml penicillin, and 100 μg/ml streptomycin in a humidified incubator at 37 °C with 5% CO2. Cells were cultured in confocal microscope at least 12 h before treatment. cDNAs for mouse TRPV1 (mTRPV1) or mouse TRPA1 (mTRPA1) were transiently transfected into HEK293 cells using the TurboFect Transfection Reagent (Cat#R0531, ThermoFisher Scientific, Lithuania) for at least 36 h. HEK293 were then washed with PBS for 3 times, and was replaced with 1 mL Fluo 3-AM (10 μmol/L), the cells were incubated at 37 °C for 60 min, and washed three times with cold PBS. The change of intracellular calcium of mTRPA1-expressing or mTRPV1-expressing HEK293 cells after treating with MGO (1 mM), capsaicin (1μM), or ATIC (200μM) were measured by Live Cell Imaging System (Cell'R; Olympus, Japan).
Measurement of intracellular ROS in ND7-23 cells
The intracellular ROS levels were measured using the fluorescent marker 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA, Molecular Probes, USA) dissolved in DMSO according to the manufacturer's instructions [31]. Briefly, a cell line derived from DRG cells ND7-23 cells (mouse neuroblastoma X rat neuronal hybrid) were seeded in 6-well plates at least 12 h before treatment. Before MGO (1 mM) was added, cells were treated with PBS, PBN (200 μM) for 15 min. After 30 min incubation, the medium was replaced with 1 mL DCFH-DA (25 μM), the cells were incubated at 30 min, then washed 3 times with PBS and suspended in 500 μL PBS for flow cytometry (FC500; Beckman Coulter, Brea, CA). Fluorescence intensity was measured and analyzed by Cxp (FC500; Beckman Coulter).
Western blotting analysis
ND7-23 cells (mouse neuroblastoma X rat neuronal hybrid) were plated at a density of 1.5 x 105 cells per 6 cm dish. After at least 12 h, the cells were incubated with or without MGO (750 μM) for 15, 30 and 60 min at 37 °C. Then, cells were lysed with Radio-Immunoprecipitation Assay (RIPA) buffer containing a cocktail of phosphatase inhibitors and protease inhibitors after washed with Phosphate Buffered Saline (PBS). The lumbar spinal dorsal horns and dorsal root ganglia were rapidly removed from mice, which were terminally anesthetized with isoflurane 5 min after injection of MGO only or co-administration of MGO with HC030031 or capsazepine, or 30 minutes after intraperitoneal (i.p.) injection of antioxidants NAC or PBN and transcardially perfused with sterile saline. At the 5th week after STZ injection, the mice were terminally anesthetized with 4% chloral hydrate (i.p. 10 mL/kg) and transcardially perfused with saline. The lumbar spinal dorsal horns and dorsal root ganglia were also rapidly collected and homogenized in lysis buffer containing a cocktail of phosphatase inhibitors and protease inhibitors for total protein extraction assays as previously reported [24,25]. The protein concentrations were measured by Pierce bicinchoninic acid (BCA) protein assay (ThermoFisher Scientific, Cat#23250; Waltham, MA, USA), and equal amounts of protein (25 μg) were loaded onto each lane and separated on 10% sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE). After transfer, the blots were blocked with 5% nonfat milk diluted in Tris-HCl Buffer Saline (TBS) at room temperature for 1 h and the PVDF membranes were incubated overnight at 4 °C with primary monoclonal anti-p-ERK (mouse, 1:1000; Cat#sc-7383, Santa Cruz Biotechnology, USA) or anti-TRPA1 antibody (rabbit, 1:500; Cat#ACC-037, Alomone labs, Jerusalem, Israel). For loading control, the blots were probed with α-tubulin antibody (1:2000, Cat#4413s, Vazyme; Nanjing, China). The blots were washed and incubated with horseradish peroxidase-conjugated goat anti-mouse or goat anti-rabbit IgG secondary antibody (1:2000, Vazyme). Protein bands were visualized using an enhanced chemiluminescence detection kit (Pierce) and the band densities were assessed and analyzed using the Molecular Imager ChemiDoc XRS+ System (Bio-Rad, Hercules, CA). Data from five mice were used for statistical analysis.
Real-time quantitative PCR
Total RNA was extracted from cervical DRG tissue using RNeasyPlus Mini kit (Cat#74134, Qiagen, Valencia) according to manufacturer's instruction. RNA was treated with DNase I (Invitrogen) and the cDNA was synthesized using ThermoScript RT-PCR System kit (Invitrogen). Reactions were carried out in a volume of 20 μl per reaction containing 10 μl SYBR Green master mix (2×) (Cat#mf015, mei5bio, Beijing, China), 0.5 μl cDNA, 1.2 μl 5 μM primer mix, and 8.3 μl water using Opticon real-time PCR Detection System (ABI Life7500, USA). Relative mRNA expression levels of different target gene compared to GAPDH were calculated using 2-ΔΔCt methods. Triplicate RT-PCR analyses were performed. Q-RT-PCR primer sequences (5' to 3') used were:
TRPA1: GGAAATACCCCACTGCATTGT (forward) and CAGCTATGTGAAGGGGTGACA (reverse);
TRPV1: ACCACGGCTGCTTACTATCG (forward) and GCTGGAATCCTCGGGTGTAG (reverse);
Cav3.2: TCTCGCTACCCAATGACAGC (forward) and CTCCGTGTAGTCTGGGATGC (reverse);
Nav1.8: ATGACGGGGAGGTGAACAAC (forward) and AACTGGTAATGGCCCGACTG (reverse);
Nav1.7: TGGATCAGAATCCGCAGGTG (forward) and TTTCCCCTTCGGATCCTTACC (reverse).
Reagents
Streptozocin (STZ, Cat#S0130), methylglyoxal solution (MGO, Cat#M0252), glyoxal solution (GO, Cat#50649), U0126 (Cat#U120), aminoguanidine hydrochloride (Cat#396494), D-Arginine monohydrochloride (Cat#A6757), serotonin hydrochloride (5-HT, Cat#H9523), compound 48/80 (Cat#C2313), chloroquine (CQ, Cat#C6628), resiniferatoxin (RTX, Cat#R8756), capsaicin (Cat#12084), chlorpheniramine maleate (Cat#C3025), N-acetyl-L-cysteine (NAC, Cat#A7250), allylisothiocyanate (AITC,Cat#377430), N-tert-butyl-a-phenylnitrone (PBN, Cat#B7263) was obtained from Sigma-Aldrich (St. Louis, MO, USA). A967079 (Cat#4716/10), PF-05089771 (Cat#5931), endothelin 1 (ET-1, Cat#1160), capsazepine (CPZ, Cat#0464) and HC-030031 (Cat#2896) was obtained from Tocris (Bristol, UK). Alpha-lipoic acid (ALA) was obtained from Hameln Pharmaceuticals GmbH (LangesFeld, Hameln, Germany). Morphine hydrochloride was obtained from China Northeast Pharmaceutical Group Shenyang No.1 Pharmaceutical CO., Ltd (Shenyang City, Liaoning Province, China). Naloxone hydrochloride was obtained from China Sinopharm Group Guorui Pharmaceutical CO., Ltd (Huainan City, Anhui Province, China). Resiniferatoxin (RTX), PBN, capsazepine, HC-030031, and A967079 were freshly dissolved in 10% dimethyl sulfoxide (DMSO). Allylisothiocyanate (AITC), capsaicin, MGO were dissolved in Artificial cerebrospinal fluid (ACSF) when use in Live Cell Imaging System. Other reagents were dissolved in sterile saline if not specified.
Statistical analysis
Data were analyzed using Graphpad Prism 6 (GraphPad, La Jollar, CA). All data were expressed as the mean ± standard error of the mean (S.E.M.). Statistical analysis were performed by Student's t test, one-way analysis of variance (ANOVA) followed by Dunnett multiple comparison tests, or two-way repeated-measured ANOVA followed by post hoc Bonferroni's multiple comparison tests as appropriately indicated. Difference with P < 0.05 was considered to be statistically significant.
Results
Intradermal (i.d.) injection of MGO induces itch behavior in mice
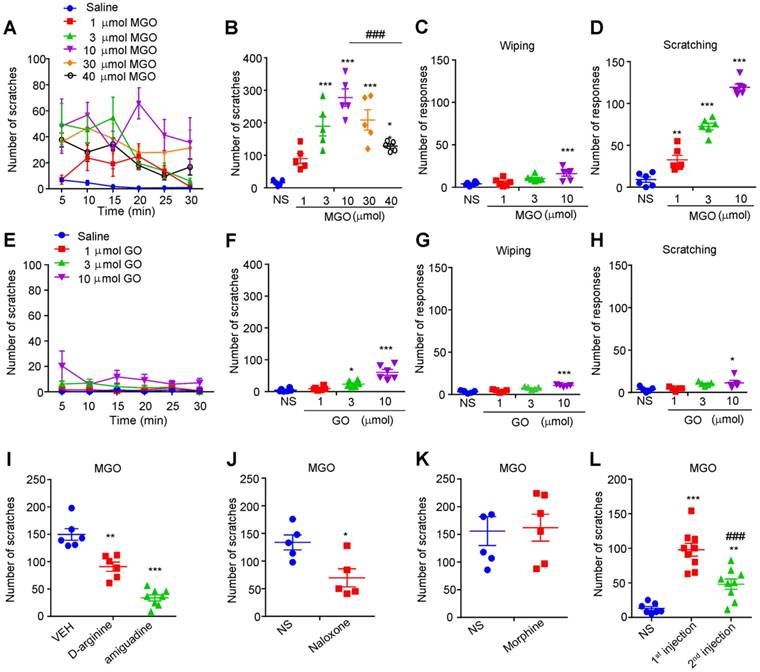
To identify the potential pruritogens related to diabetic neuropathy, we first examined the possible prurigenic activities of MGO, glyoxal (GO), and glucose by using neck model by i.d. injection of chemicals into nape of the neck in mice. Cheek model was further employed to behaviorally distinguish itch and pain responses in mice by i.d. injection of chemicals into cheek of mice [35]. I.d. injection of painful reagents into the cheek evokes wiping behavior, while i.d. injection of pruritogens evokes scratching behavior in mice [35]. We found that i.d. injection MGO (1 μmol-40 μmol) into the nape of the neck dose-dependently evoked robust scratching behavior in mice (Figure 1, A and B). The dose-response curve showed an obvious invert “U” shape (Figure 1B). I.d. injection of MGO into the cheek dose-dependently induced scratching behavior, but only elicited wiping at the highest dose (10 μmol) in mice (Figure 1, C and D). In sharp contrast, i.d. injection GO (1 μmol-10 μmol) into the neck slightly induced scratching behavior at the highest dose (10 μmol) in mice (Figure 1, E and F), and i.d. injection of GO into the cheek slightly produced mixed wiping and scratching behavior in mice at the highest dosage (10 μmol) (Figure 1, G and H). Additionally, i.d. injection of glucose into the nape of neck was not able to induce scratching behavior in mice at all tested dosage (Supplementary Figure 1, A and B). We next demonstrated that pre-incubation of MGO scavengers D-arginine (0.18 M) or aminoguanidine (0.18 M) with MGO (0.06 M) significantly reduced MGO-induced scratching behavior in mice (Saline: 149.8 ± 10.58 vs. D-arginine: 90.83 ± 8.499; t10 = 4.348; P = 0.0014; Saline: 149.8 ± 10.58 vs. aminoguanidine: 33.75 ± 5.519; t12 = 10.47; P < 0.0001; Figure 1I). Intraperitoneal (i.p.) injection of naloxone (1 mg/kg; Figure 1J), an opioid receptor antagonist, but not morphine (1mg/kg; Figure 1K), attenuated MGO-induced scratching in mice (Saline: 133.8 ± 13.52 vs. naloxone: 69.60 ± 16.47; t8 = 3.013; P = 0.0167). It was noteworthy that repeated injection of MGO (3 μmol) induced behavioral desensitization of scratching response in mice (First injection: 97.89 ± 9.387 vs. second injection: 48.11 ± 7.473; t16 = 4.149; P = 0.0008; Figure 1L).
Intradermal (i.d.) injection of MGO induces itch behaviors in mice. (A-B) Time course (A) and total number (B) of scratching bouts within 30 min induced by i.d. injection of MGO (1-30 μmol) into the nape of neck of mice (n = 5-6 for each group, *P < 0.05, ***P < 0.001 vs. control group, ### P < 0.001 vs. MGO (10 μmol) group, one-way AVOVA followed by post-hoc Dunnett's test). (C-D) Total number of forelimb wiping (C) and hind paw scratching behavior (D) induced by i.d. injection of MGO (1-10 μmol) into the cheek of mice (n = 5-6 for each group, **P < 0.01, ***P < 0.001 vs. control group, one-way AVOVA followed by post-hoc Dunnett's test). Saline served as negative control. (E-F) Time course (E) and total number (F) of scratching bouts within 30 min induced by i.d. injection of GO (1-10 μmol) into the nape of neck of mice (n = 5-6 for each group, *P < 0.05, ***P < 0.001 vs. control group, one-way AVOVA followed by post-hoc Dunnett's test). (G-H) Total number of forelimb wiping (E) and hindpaw scratching behavior (F) induced by i.d. injection of GO (1-10 μmol) into the cheek of mice (n = 5-6 for each group, *P < 0.05, ***P < 0.001 vs. control group, one-way AVOVA followed by post-hoc Dunnett's test). (I) Pre-incubation of MGO scavengers (aminoguanidine and D-arginine) decreased MGO (3 μmol)-induced scratching behavior in 30 min (n = 5-7 for each group, **P < 0.01, ***P < 0.001 vs. VEH group, Student's t test). (J-K) Naloxone (i.p., 1 mg/kg; J) reduced MGO-induced scratching behavior in mice (n = 5-6 for each group, *P < 0.05 compared with NS group, Student's t test), but not for morphine (i.p., 1 mg/kg; K). (L) Second injection of MGO (3 μmol) induced less scratching behavior than that of first injection (n = 7-9 for each group, **P < 0.01, ***P < 0.001 compared with NS group, ###P < 0.001 compared with 1st injection group, Student's t test). All data are expressed by means ± SEM. GO, glyoxal; MGO, methylglyoxal; NS, normal saline; VEH, Vehicle.
Involvement of capsaicin-sensitive C-fibers, mast cells and sodium channel Nav1.7 in MGO induces itch behavior in mice
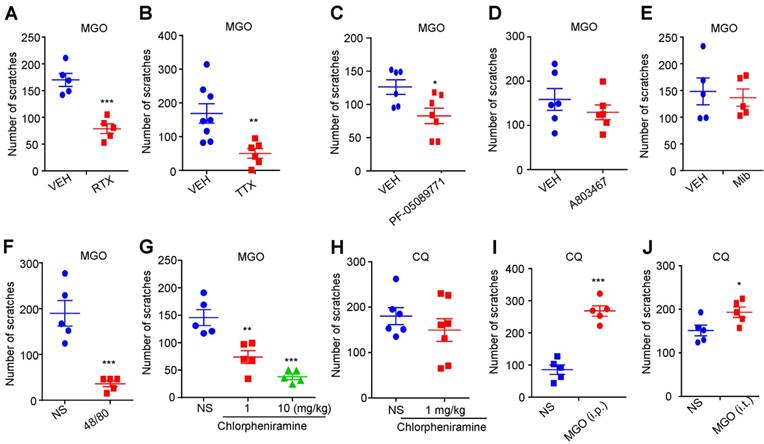
We next examined the roles of capsaicin-sensitive C-fibers, voltage-gated sodium channels (VGSCs), mast cells, and histamine in MGO-induced scratching behavior in mice. MGO-induced scratching was significantly decreased in resiniferatoxin (RTX)-treated mice compared with control mice (Vehicle: 78.80 ± 9.013 vs. RTX: 170.0 ± 12.22; t8 = 6.005; P = 0.0003; Figure 2A), suggesting capsaicin-sensitive C-fibers were involved in this process. Local co-administration of pan sodium channels blocker tetrodotoxin (TTX; 100 ng) abolished MGO-induced scratching in mice (Vehicle: 168.8 ± 28.90 vs. TTX: 50.33 ± 14.08; t12 = 3.308; P = 0.0063; Figure 2B). Local co-administration of Nav1.7 selective blocker PF-05089771 (10 μg; Figure 2C), but not a Nav1.8 blocker A803467 (3 μg; Figure 2D) and a pan T-type calcium channels blocker mibefradil (10 nmol; Figure 2E), significantly reduced MGO-induced scratching in mice (Vehicle: 126.2 ± 10.94 vs. PF-05089771: 82.86 ± 11.62; t11 = 2.683; P = 0.0213). MGO-induced scratching was significantly decreased in compound 48/80-pretreatment mice compared with control mice (Vehicle group: 35.80 ± 6.484 vs. compound 48/80-treated group: 189.8 ± 27.77; t8 = 5.401; P = 0.0006; Figure 2F). MGO-induced scratching (Figure 2G), but not chloroquine (CQ)-induced scratching (Figure 2H), was dose-dependently decreased by i.p. injection of a histamine H1 receptor antagonist chlorpheniramine (1 mg/kg) in mice (F(2, 12) = 24.01; P < 0.0001). CQ-induced scratching in mice was significantly enhanced by both i.p. injection of MGO (50 mg/kg) (Saline: 85.60 ± 14.80 vs. MGO: 268.0 ± 16.27; t8 = 8.292; P < 0.0001; Figure 2I) or i.t. injection of MGO (1 nmol) (Saline: 151.4 ± 12.21 vs. MGO: 193.2 ± 12.09; t8 = 2.432; P = 0.0410; Figure 2J).
The roles of Nav1.7, mast cells, and histamine in MGO-induced itch in mice. (A) MGO (3 μmol)-induced scratching was significantly decreased in RTX-treated mice (n = 5 for each group, ***P < 0.001 compared with VEH group, Student's t test). (B) Co-administration of pan sodium channels blocker TTX (100 ng) significantly reduced MGO (3 μmol)-induced scratching in mice. (n = 6-8 for each group, **P < 0.01 compared with VEH group, Student's t test). (C) Nav1.7 selective inhibitor PF-05089771 (10 μg) significantly reduced MGO (3 μmol)-induced scratching in mice (n = 6-7 for each group, *P < 0.05 compared with VEH group, Student's t test). (D) Nav1.8 selective inhibitor A803467 (3 μg) did not affect MGO (3 μmol)-induced scratching in mice (n = 6 for each group, P > 0.05 compared with VEH group, Student's t test). (E) Pan T-type calcium channel inhibitor Mib (10 nmol) did not affect MGO (3 μmol)-induced scratching in mice (n = 5 for each group, P > 0.05 compared with VEH group, Student's t test). (F) MGO (3 μmol)-induced scratching behavior was significantly decreased in compound 48/80-pretreated mice (n = 5 for each group, ***P < 0.001 compared with NS group, Student's t test). (G-H) I.p. injection of a histamine H1 antagonist chlorpheniramine (1 and 10 mg/kg) dose-dependently suppressed MGO (3 μmol)-induced scratching behavior (G; n = 5 for each group, **P < 0.01; ***P < 0.001 compared with NS group, one-way AVOVA followed by post-hoc Dunnett's test), but did not affect CQ (200 μg)-induced scratching behavior in mice (H; n = 6-7 for each group, P > 0.05 compared with NS group, Student's t test). (I-J) I.p. injection of MGO (I; 50 mg/kg) and i.t. injection of MGO (J; 1 nmol) significantly increased MGO (3 μmol)-induced scratching in mice (n = 5 for each group, ***P < 0.001, *P < 0.05 compared with NS group, Student's t test). All data are expressed by means ± SEM. 48/80, compound 48/80; CQ, chloroquine; Mib, mibefradil; MGO, methylglyoxal; NS, normal saline; RTX, resiniferatoxin; TTX, tetrodotoxin; VEH, Vehicle.
MGO-mediated activation of TRPA1 in dorsal root ganglia (DRG) neurons mediates MGO-induced itch behavior in mice
To identify the potential molecular targets of MGO, we performed whole-cell patch clamp recording on dissociated C4-C8 DRG neurons to identify the direct effects of MGO on TRPV1 or TRPA1, two important ion channels in itch [36]. Under the voltage clamp mode, bath application of 1 mM MGO evoked obvious inward currents in dissociated DRG neurons from mice, which responded to a TRPA1 selective agonist allyl-isothiocyanate (AITC) (Figure 3A). Incubation of a TRPA1 blocker HC030031 (100 μM) abolished MGO-evoked inward currents in dissociated DRG neurons (Figure 3B). To test the possibility that MGO directly activates either TRPV1 or TRPA1, we transfected HEK-293 cell with either mouse TRPA1 or mouse TRPV1 construct and measured TRP channels activities using live-cell Ca2+ imaging. MGO did not affect intracellular Ca2+ in mTRPV1-transfected HEK-293 cells (Figure 3C). In contrast, MGO evoked robust intracellular Ca2+ increase in mTRPA1-transfected HEK-293 cells (Figure 3D).
Direct activation of TRPA1 channel is required for MGO-induced itch behaviors in mice. (A) Inward currents evoked by MGO (1 mM) and AITC (50 μM) in dissociated C4-C5 DRG neurons. Notably, TRPA1 blocker HC030031 (100 μM) abolished MGO-induced inward currents in C4-C5 DRG neurons. (B) Amplitude of inward currents induced by MGO (1 mM) was significantly decreased by TRPA1 blocker HC030031 (100 μM). (n = 5 neurons from 3 mice, ***P < 0.001 compared with corresponding group, Student's t test). (C) Representative traces showing HEK-293 cells transfected with mTRPV1 did not show detectable intracellular Ca2+ responses to MGO (n = 3 cells). (D) Representative traces showing perfusion of MGO (1 mM) evoked large intracellular Ca2+ responses in mTRPA1-transfected HEK-293 cells (n = 3 cells). CAP (1 μM) and AITC (200 μM) were used. 3 independent experiments were performed. (E) MGO (3 μmol)-induced scratching in 30 min was decreased by co-administration of TRPA1 blocker HC030031 (50 μg), and A967079 (50 μg), but not for TRPV1 blocker CPZ (50 μg) in mice (n = 5 for each group, ***P < 0.001 vs. VEH group, Student's t test). (F) MGO (3 μmol) induced significantly less scratching behavior in Trpa1-/- mice than that of WT mice (n = 5 for each group, ***P < 0.001 compared with WT mice, Student's t test). (G) MGO (3 μmol) induced comparable scratching behavior between WT and Trpv1-/- mice. All data are expressed by means ± SEM. AITC, allyl-isothiocyanate; CAP, capsaicin; CPZ, capsazepine; MGO, methylglyoxal; TRPA1, transient receptor potential ankyrin 1; TRPV1, transient receptor potential vanilloid 1; VEH, Vehicle; WT, wild type.
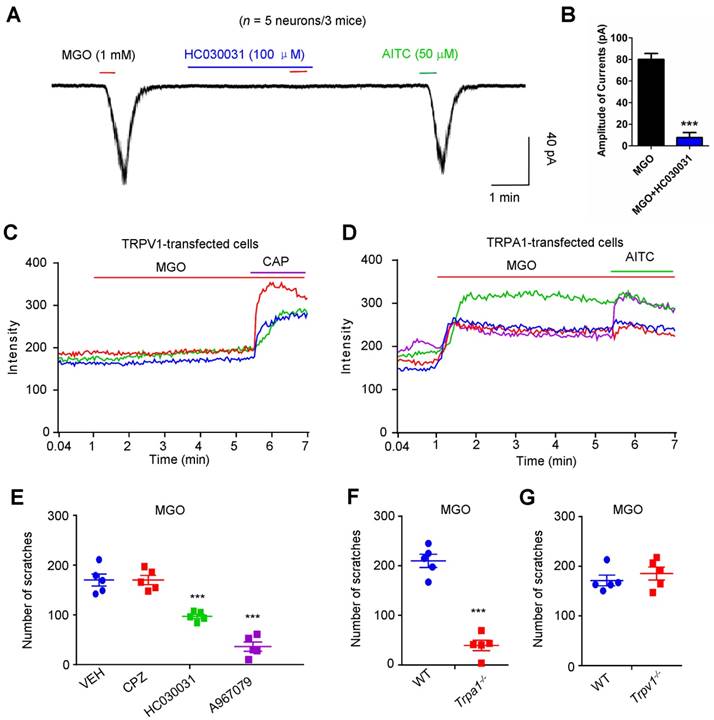
We subsequently examined the role of TRP channels in MGO-induced itch in mice by using genetic and pharmacological approaches. Co-administration of selective TRPA1 blocker HC-030031 (50 μg; Vehicle: 170.0 ± 12.22 vs. HC030031: 97.00 ± 4.183; t8 = 5.651; P = 0.0005) or A967079 (50 μg; Vehicle: 157.8 ± 13.52 vs. A967079: 36.40 ± 9.190; t9 = 7.103; P < 0.0001), but not a TRPV1 selective blocker capsazepine (CPZ; 50 μg), significantly decreased MGO-induced scratching in mice (Figure 3E). Furthermore, MGO-induced scratching was abolished in Trpa1-/- mice (WT mice: 419.8 ± 26.38 vs. Trpa1-/- mice: 78.80 ± 20.90; t8 = 10.13; P < 0.0001; Figure 3F), but not in Trpv1-/- mice (Figure 3G).
Mechanical itch is enhanced in streptozocin (STZ)-induced diabetic mice
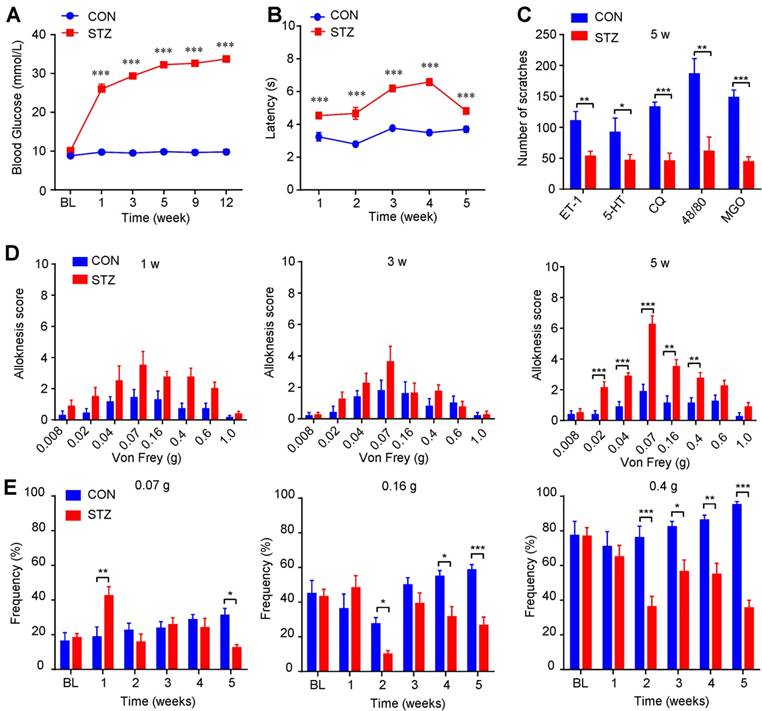
To further investigate the itch-related behavioral phenotypes under diabetes condition, we established streptozocin (STZ)-induced diabetic mouse model [37]. Our results showed that blood glucose level of STZ-induced diabetes mice was increased compared with control mice (Figure 4A), while body weight increased much slower than that of control mice (Supplementary Figure 2A). Rota-rod test showed that the motor function was comparable between control and diabetic mice (Supplementary Figure 2B). The latency of tail flick response to hot water (52 °C) was significantly prolonged in diabetic mice compared with control mice (Figure 4B). There was no spontaneous scratching behavior observed in diabetic mice (Supplementary Figure 2C). Furthermore, several pruritogens, including ET-1 (100 pmol), 5-HT (30 μg), chloroquine (CQ; 200 μg), compound 48/80 (48/80; 100 μg), and MGO (3 μmol), induced less scratching behavior in diabetic mice at the 5th week after STZ injection than that of control mice (Figure 4C), suggesting impaired function of C-fibers may be responsible for decreased chemical pruritogens-induced itch. Mechanical itch (alloknesis; namely touch-evoked itch) is a remarkable feature of chronic itch, which is caused by skin diseases, systemic diseases, aging, or neurological disorders [33; 38]. Intriguingly, diabetic mice displayed significant more mechanical itch behaviors compared with that of control mice at the 5th week after STZ injection (Figure 4D). In contrast, we found that diabetic mice showed early mechanical pain hypersensitivity only within one week after STZ treatment (Figure 4E), suggesting different time course of mechanical allodynia and mechanical itch under diabetic condition. Thus, our data suggested that mechanical itch, as an itch-related modality, is enhanced in STZ-induced diabetic mice.
Mechanical itch is enhanced in STZ-induced diabetic mice. (A) Increased level of blood glucose (mmol/L) was confirmed in STZ-induced diabetic mice (n = 8-10, *** P < 0.001 compared with control mice, two-way repeated-measured ANOVA). (B) The latency of tail flick responding to 52 °C water bath was significantly prolonged in diabetic mice compared with control mice (n = 8-10, ***P < 0.001 compared with control group, two-way repeated-measured ANOVA). (C) Pruritogens ET-1 (100 pmol), 5-HT (30 μg), chloroquine (200 μg), compound 48/80 (100 μg), and MGO (3 μmol) induced less scratching in diabetic mice than control mice at the 5th week after STZ injection (n = 6-8 per group, *P < 0.05, **P < 0.01, ***P < 0.001 compared with control group, Student's t test). (D) Diabetic mice displayed enhanced mechanical itch behavior compared with control mice at the 5th week after STZ injection (n = 6-8, **P < 0.01, ***P < 0.001 compared with control group, two-way repeated-measured ANOVA). (E) Diabetic mice displayed early enhanced mechanical allodynia behavior and late hypoalgesia after STZ injection compared with that of control mice (n = 6-8, *P < 0.05, **P < 0.01, ***P < 0.001 compared with control group, two-way repeated-measured ANOVA). All data are expressed by means ± SEM. 48/80, compound 48/80; BL, baseline; CON, control; CQ, chloroquine; ET-1, endothelin-1; MGO, methylglyoxal; STZ, streptozotocin.
The expression and function of TRPA1 is enhanced in STZ-induced diabetic mice
To examine the expression and function of TRPA1 in the DRGs under diabetes condition, we utilized q-PCR analysis, Western blotting, and electrophysiological methods. We demonstrated that mRNA expression of TRPA1 was significantly increased in the C4-C8 DRGs from diabetic mice (P = 0.0003; Figure 5A). In contrast, the mRNA expression of TRPV1 was significantly decreased in the DRGs from diabetic mice (P < 0.001; Figure 5A). Western blotting analysis also confirmed the up-regulation of TRPA1 protein in the DRGs from diabetic mice (P < 0.0001; Figure 5, B and C). Whole-cell patch clamp recording analysis confirmed that MGO-induced inward currents in the DRG neurons dissociated from STZ-induced diabetic mice were significantly amplified (P < 0.01; Figure 5, D-F). Incubation with TRPA1 selective blockers HC030031 (P < 0.001; Figure 5, G and H) or A-967079 (P < 0.001; Figure 5, I and J) significantly inhibited MGO-induced inward currents in the DRG neurons dissociated from STZ-induced diabetic mice.
Expression and function of TRPA1 in DRG neurons are enhanced in STZ-induced diabetic mice. (A) Q-PCR analysis showed that significant reduction of mRNA expression of TRPA1 in the C4-C5 DRGs was found in STZ-induced diabetic mice compared with that of control mice (n = 3 per group, **P < 0.01, ***P < 0.001 compared with CON group, Student's t test). (B-C) Western blotting (B) and quantification (C) confirmed that protein expression TRPA1 in C4-C5 DRG was significantly increased in diabetic mice compared with that of control mice (n = 3 per group, ***P <0.001 vs. vehicle, Student's t test). (D) Patch-clamp recordings were performed in the cultured dissociated C4-C5 DRG neurons from control mice and STZ-induced diabetic mice at 5 weeks after STZ injection. A representative example of MGO (1 mM)-induced inward currents in the cultured dissociated C4-C5 DRG neurons from the control and diabetic mice. (E) Bar graph showing an average peak current values obtained from the control and STZ-induced diabetic mice (n = 3, **P < 0.01 compared with CON group, Student's t test). (F) Bar graph showing an average density of currents (pA/pF) obtained from the control and STZ-induced diabetic mice (n = 3, ***P < 0.001 compared with CON group, Student's t test). (G) A representative example of MGO (1 mM)-induced inward currents in the cultured dissociated C4-C5 DRG neurons from STZ-induced diabetic mice were abolished by TRPA1 blocker HC030031 (100 μM). (H) Bar graph showed average peak current density values induced by MGO (1 mM) with or without HC030031 (100 μM) in the cultured dissociated C4-C5 DRG neurons from diabetic mice (n = 3, ***P < 0.001 compared with MGO group, Student's t test). (I) An representative example of MGO (1 mM)-induced inward currents in the cultured dissociated C4-C5 DRG neurons from diabetic mice were abolished by TRPA1 blocker A-967079 (10 μM). (J) Bar graph shows average peak current density values induced by MGO with or without A-967079 (10 μM) in the cultured dissociated C4-C5 DRG neurons from STZ-induced diabetic mice (n = 3, ***P < 0.001 compared with MGO group, Student's t test). All data are expressed by means ± SEM. CON, control; MGO, methylglyoxal; STZ, streptozotocin; TRPA1, transient receptor potential ankyrin 1; TRPV1, transient receptor potential vanilloid 1; VEH, Vehicle.
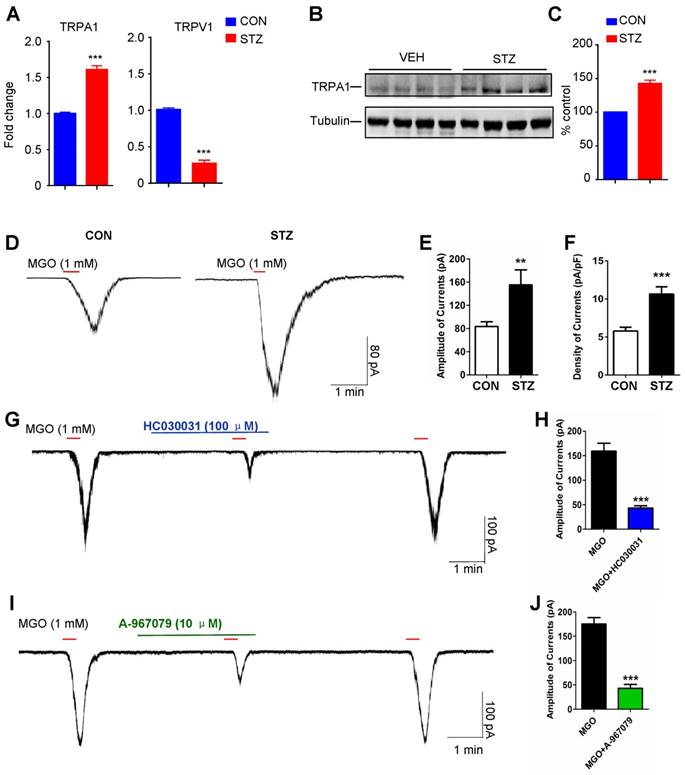
Activation of TRPA1 and sodium channel Nav1.7 mediates mechanical itch induced by i.d. injection of MGO in mice
Subsequently, we asked whether activation of TRPA1 was required for mechanical itch induced by i.d. injection of MGO or in STZ-induced diabetic mice. As expected, we found that i.d. MGO induced obvious mechanical itch in mice (Saline: 3.167 ± 0.6009 vs. MGO: 7.143 ± 0.5948; t11 = 4.677; P = 0.0007; Figure 6A). Pre-incubation of MGO scavengers (D-arginine or aminoguanidine) with MGO significantly reduced MGO-induced mechanical itch in mice (Saline: 6.714 ± 0.2857 vs. D-arginine: 2.333 ± 0.6667; t11 = 6.379; P < 0.0001; Saline: 6.714 ± 0.2857 vs. aminoguanidine: 3.286 ± 0.5216; t12 = 5.765; P < 0.0001; Figure 6B). Co-administration of TRPA1 blockers HC030031 (50 μg; Vehicle: 7.143 ± 0.5948 vs. HC030031: 2.200 ± 0.5831; t11 = 5.736; P = 0.0002), A967079 (50 μg; Vehicle: 5.400 ± 0.5099 vs. A967079: 1.500 ± 0.5976; t12 = 4.520; P = 0.0009), but not a TRPV1 blocker CPZ (50 μg), significantly reduced MGO-induced mechanical itch in mice (Figure 6C). MGO-induced mechanical itch in mice was abolished in Trpa1-/- mice compared with WT control mice (WT: 6.500 ± 0.6708 vs. Trpa1-/-: 3.429 ± 0.3689; t11 = 4.177; P = 0.0015; Figure 6D). Co-administration of TTX (100 ng; Vehicle: 5.667 ± 0.4216 vs. TTX: 1.833 ± 0.4014; t10 = 6.585; P < 0.0001; Figure 6E), and PF-05089771 (10 μg; Vehicle: 5.000 ± 0.6172 vs. PF-05089771: 2.143 ± 0.3401; t12 = 4.054; P = 0.0016; Figure 6F), but not A803467 (3 μg; Figure 6G) and mibefradil (10 nmol; Figure 6H), significantly reduced MGO-induced mechanical itch in mice.
Nav1.7 and activation of TRPA1 are required for mechanical itch induced by intradermal injection of MGO in mice. (A) I.d. injection of MGO (3 μmol) induced mechanical itch in mice 30 minutes after injection of MGO (n = 6-7 for each group, ***P < 0.001 compared with NS group, Student's t-test). (B) Pre-incubation with aminoguanidine (180 mM) and D-arginine (180 mM) with MGO (60 mM) suppressed MGO-induced mechanical itch in mice (n = 6-7 for each group, ***P < 0.001 compared with NS group, Student's t test). (C) Co-administration of HC030031 (50 μg) or A967079 (50 μg) decreased MGO (3 μmol)-induced mechanical itch in mice (n = 5-7 for each group, ***P < 0.001 vs. VEH group, Student's t test), but not for CPZ (50 μg). (D) MGO (3 μmol)-induced mechanical itch was significantly reduced in Trpa1-/- mice compared with that of WT mice (n = 6-7 for each group, **P < 0.01 compared with WT group, Student's t test). (E) Local co-administration of TTX (100 ng) significantly reduced MGO (3 μmol)-induced mechanical itch in mice (n = 6 for each group, **P < 0.01 compared with control group, Student's t test). (F) Local co-administration of Nav1.7 selective blocker PF-05089771 (10 μg) significantly reduced MGO (3 μmol)-induced mechanical itch in mice (E; n = 6 for each group, ** P < 0.01 compared with control group, Student's t-test). (G) Nav1.8 selective blocker A803467 (3 μg) did not affect MGO (3 μmol)-induced mechanical itch in mice. (H) Pan T-type calcium channel blocker mibefradil (Mib; 10 nmol) did not affect MGO (3 μmol)-induced mechanical itch in mice. All data are expressed by means ± SEM. CPZ, capsazepine; MGO, methylglyoxal; Mib, mibefradil; NS, normal saline; TTX, tetrodotoxin; VEH, Vehicle.
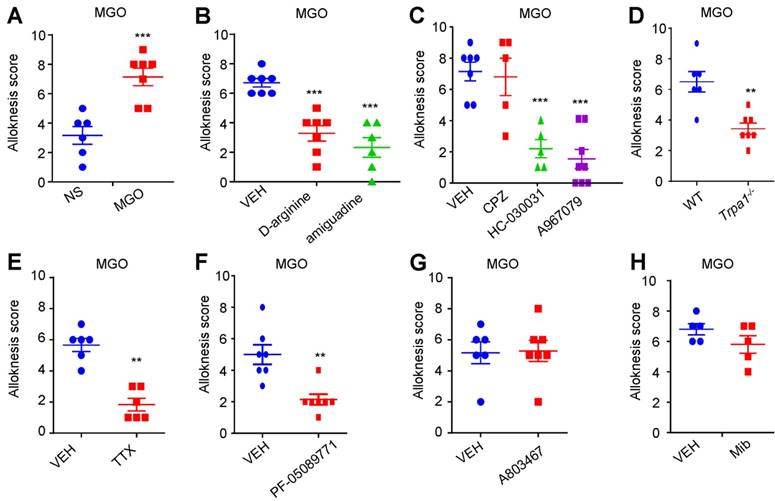
Activation of TRPA1 and sodium channel Nav1.7 mediates mechanical itch in STZ-induced diabetic mice
We further explored the contribution of MGO-mediated activation of TRPA1 to mechanical itch in STZ-induced diabetic mice. We found that i.p. injection of MGO scavengers, including aminoguanidine (50-200 mg/kg) and D-Arginine (300-600 mg.kg), significantly inhibited mechanical itch in STZ-induced diabetic mice (Figure 7A). I.d. injection of a TRPA1 blocker HC-030031 (100 μg), but not a TRPV1 blocker CPZ (100 μg), significantly inhibited mechanical itch in STZ-induced diabetic mice (P < 0.001; Figure 7B). In addition, diabetes-induced mechanical itch was also abolished in Trpa1-/- mice (P < 0.0001; Figure 7C). I.d. administration of TTX (100 ng; P < 0.001) or PF-05089771 (10 μg; P < 0.01), but not for A803467 (3 μg), significantly inhibited mechanical itch in STZ-induced diabetic mice (Figure 7D). Additionally, q-PCR analysis confirmed that mRNA expression of Nav1.7 was significantly increased (t10 = 9.930; P = 0.0001), but the mRNA expression of Cav3.2 and Nav1.8, was significantly decreased in the DRGs of STZ-induced diabetic mice compared with that of control mice (Figure 7E).
Nav1.7 and activation of TRPA1 are required for mechanical itch in STZ-induced diabetic mice. (A) I.p. injection of MGO scavengers aminoguanidine (50-200 mg/kg) and D-arginine (300-600 mg/kg) significantly suppressed mechanical itch in STZ-induce diabetic mice (n = 5-7 for each group, **P < 0.01, ***P < 0.001 compared with VEH group, one-way AVOVA followed by post-hoc Dunnett's test). (B) i.d. injection of TRPA1 blocker HC030031 (100 μg), but not TRPV1 blocker CPZ (100 μg), suppressed mechanical itch in STZ-induce diabetic mice (n = 6-7 for each group, ***P < 0.001 compared with VEH group, Student's t test). (C) Mechanical itch in diabetic mice was significantly reduced in Trpa-/- mice compared with that of WT mice (n = 7 for each group, ***P < 0.001 compared with WT group, Student's t test). (D) I.d. injection of sodium channel blocker TTX (100 ng) and Nav1.7 blocker PF-05089771 (10 μg), but not Nav1.8 blocker A803467 (3 μg), suppressed mechanical itch in STZ-induced diabetic mice (n = 5-8 for each group, **P < 0.01, ***P < 0.001 compared with VEH group, Student's t test). (E) Q-PCR analysis showed that mRNA expression of Nav1.7 was significantly increased, but the mRNA expression of Cav3.2 and Nav1.8, was significantly decreased in the DRGs of STZ-induced diabetic mice compared with that of control mice. All data are expressed by means ± SEM. CPZ, capsazepine; MGO, methylglyoxal; Mib, mibefradil; NS, normal saline; STZ, streptozotocin; TTX, tetrodotoxin; VEH, Vehicle.
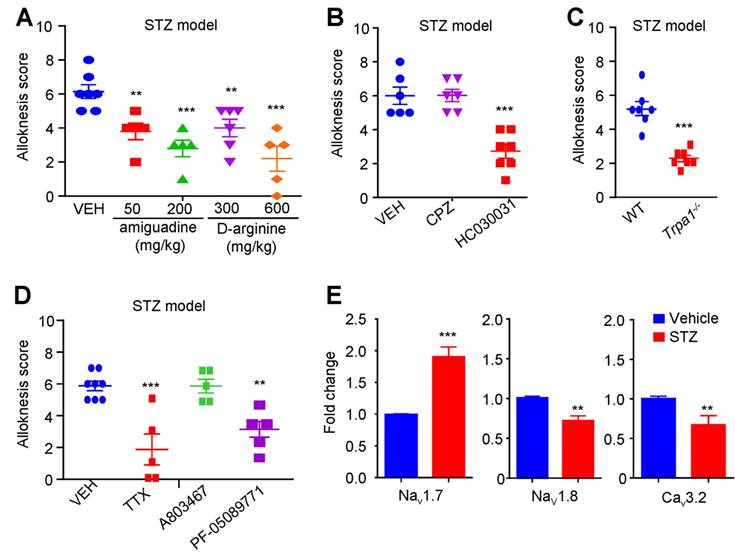
Oxidative stress contributes to itch induced by i.d. injection of MGO or in STZ-induced diabetic mice
Our previous work showed that oxidative stress played a key role in acute and chronic itch [31; 39]. We next found that incubation of a dorsal root ganglia-derived cell line ND7-23 cells (mouse neuroblastoma X rat neuronal hybrid) with MGO (1 mM) significantly increased the levels of intracellular reactive oxygen species (ROS), as reflected by enhanced dichloro-dihydro-fluorescein diacetate (DCFH-DA) fluorescence intensity compared with control, while antioxidants phenyl-tert-butynitrone (PBN) remarkable decreased it (Figure 8, A and B). MGO-induced scratching behavior was significantly reduced by pretreatment with N-Acetyl-L-cysteine (NAC; i.p. 200 mg/kg; Saline: 141.0 ± 19.69 vs. NAC: 33.71 ± 6.948; t10 = 5.858, P = 0.0002; Figure 8C), PBN (i.p. 100 mg/kg; Saline: 141.0 ± 19.69 vs. PBN: 31.80 ± 13.36; t8 = 4.589; P = 0.0018; Figure 8C), α-lipoic acid (ALA; i.p. 100 mg/kg; Saline: 203.8 ± 26.00 vs. ALA: 60.80 ± 21.34; t11 = 3.838; P = 0.0028; Figure 8D). I.d. co-administration of ALA also reduced MGO-induced scratching behavior in mice (Saline: 141.71 ± 60.68 vs. ALA 200 μg: 53.63 ± 29.34 vs. ALA 300 μg: 47.88 ±31.47; F = 6.818, P = 0.0014; Figure 8E). MGO-induced mechanical itch was significantly reduced by systemic injection of ALA (i.p. 100 mg/kg; Saline: 6 ± 1.07 vs. ALA: 2.38 ± 0.26; t10 = 3.506; P = 0.0039; Figure 8F) and local co-administration of ALA in mice (Saline: 11.0 ± 5 vs. ALA 100 μg: 4.8 ± 0.98 vs. ALA 200 μg: 2.85 ± 1.25 vs. ALA 300 μg: 4.17 ± 1.34; F = 8.166, P < 0.001; Figure 8G). In STZ-induced diabetic mice, i.p. injection of antioxidant ALA (100 mg/kg) also inhibited mechanical itch (t11 = 6.599; P < 0.0001; Figure 8H). Thus, these data suggested that oxidative stress contributes to itch (including mechanical itch) induced by i.d. injection of MGO or in STZ-induced diabetic mice.
The roles of oxidative stress in itch induced by intradermal injection of MGO and in STZ-induced diabetic mice. (A-B) Flow cytometry (A) and quantification (B) revealed that incubation of MGO (1 mM) increased intracellular ROS in ND7-23 cells, which was inhibited by pre-incubation of antioxidant PBN (200 μM) (n = 4-5 for each group, **P < 0.01, ***P < 0.001 compared with corresponding groups, Student's t test). (C) I.p. injection of antioxidants NAC (200 mg/kg) and PBN (100 mg/kg) significantly decreased MGO (3 μmol)-induced scratching in 30 min (n = 5-7 for each group, **P < 0.01, ***P < 0.001 compared with VEH group, Student's t test). (D) I.p. injection of another antioxidant ALA (100 mg/kg) also decreased MGO (3 μmol)-induced scratching within 30 min (n = 5-8 for each group, ***P < 0.001 vs. VEH group, Student's t test). (E) Local co-administration of ALA (100-300 μg) dose-dependently decreased MGO (3 μmol)-induced scratching in 30 min (n = 7-8 for each group, ** P < 0.01 compared with VEH group, one-way AVOVA followed by post-hoc Dunnett's test). (F) I.p. injection of antioxidant ALA (100 mg/kg) also decreased MGO (3 μmol)-induced mechanical itch in mice (n = 5-7 for each group, ***P < 0.001 compared with VEH group, Student's t test). (G) Co-administration of ALA (100-300 μg) dose-dependently decreased MGO (3 μmol)-induced mechanical itch in mice (n = 5-7 for each group, **P < 0.05, **P < 0.01 compared with VEH group, one-way AVOVA followed by post-hoc Dunnett's test). (H) Mechanical itch was suppressed by i.p. injection of ALA (100 mg/kg) in STZ-induced diabetic mice (n = 7-10 for each group, ***P < 0.001 compared with NS group, Student's t test). All data are expressed by means ± SEM. ALA, α-lipoic acid; MGO, methylglyoxal; NS, normal saline; PBN, N-tert-butyl-a-phenylnitrone; PBS, phosphate buffer saline; STZ, streptozotocin; VEH, Vehicle.
Intracellular ERK signaling contributes to itch induced by i.d. injection of MGO or in STZ-induced diabetic mice
To further investigate the downstream signaling of MGO-mediated activation of TRPA1 under diabetes, we subsequently examined the role of ERK signaling activation in itch induced by i.d. injection of MGO or in STZ-induced diabetic mice. We used Western blotting to determine p-ERK expression in vivo and in vitro in order to test whether MGO is able to induce p-ERK activation. Our results demonstrated that i.d. injection of MGO induced the expression of p-ERK in the DRGs (P < 0.0001; Figure 9A) and spinal cord (P = 0.0004; Figure 9B). We also found that incubation ND7-23 cells with MGO (1 mM) from 10 minutes to 60 minutes significantly induced the expression of p-ERK (F(3, 8) = 526.0; P < 0.0001; Figure 9C). These results suggest that MGO is able to induce p-ERK activation in the DRGs and spinal cord in mice. Western blotting analysis showed that MGO-induced p-ERK expression in the spinal cord was significantly suppressed by i.d. injection of HC030031 (P < 0.01), but not CPZ (P = 0.2745) (Figure 9D). MGO-induced p-ERK expression in the spinal cord was also significantly suppressed by antioxidants NAC (P < 0.05) and PBN (P < 0.01) (Figure 9E). Thus, our data indicated oxidative stress and TRPA1 activation contribute to MGO-induced ERK activation in the spinal cord. We next found that i.t. injection of U0126 (1 nmol) significantly inhibited MGO-induced scratching (Vehicle: 170.0 ± 12.22 vs. U0126: 49.60 ± 13.01; t8 = 6.745; P = 0.0001; Figure 9F) and mechanical itch in mice (P < 0.001; Figure 9G). Additionally, i.d. co-injection of U0126 (10 nmol) also significantly inhibited MGO-induced scratching in mice (P < 0.001; Figure 9H). In STZ-induced diabetic mice, i.d. injection of U0126 (1 nmol) was also able to reduce mechanical itch behavior (P < 0.001; Figure 9I). Thus, these data suggested that ERK signaling activation in the DRGs and spinal cord contributes to itch induced by i.d. injection of MGO or in STZ-induced diabetic mice.
The role of ERK activation in the DRG and spinal cord in itch induced by intradermal injection of MGO and in STZ-induced diabetic mice. (A-B) After i.d. injection of MGO (3 μmol) in the nape of the neck, the expression of p-ERK in C4-C5 DRG (A) and spinal cord (B) was examined by Western blotting analysis (upper panel). Quantification (lower panel) showed that the expression of p-ERK in the DRGs and spinal cord were significantly increased by i.d. injection of MGO (3 μmol). (C) Incubation of ND7-23 cells with MGO (1 mM) from 10 min to 60 min significantly increased p-ERK expression (n = 3-4 per group, ***P < 0.001 compared with NS group, one-way AVOVA followed by post-hoc Dunnett's test). (D) Western blotting analysis (upper panel) and quantification (lower panel) showed that MGO-induced the expression of p-ERK in spinal cord was significantly suppressed by i.p. injection of HC030031, but not for CPZ in mice (n = 3-4 per group, *P < 0.05 compared with NS group, Student's t test; ##P < 0.01 compared with VEH group, Student's t test). (E) Western blotting analysis (upper panel) and quantification (lower panel) showed that MGO-induced the expression of p-ERK in spinal cord was significantly suppressed by i.p. injection of antioxidants NAC and PBN in mice (n = 3-4 per group, **P < 0.01 compared with NS group, Student's t test; #P < 0.05, ##P < 0.01 compared with VEH group, Student's t test). (F) I.t. injection of MEK inhibitor U0126 significantly inhibited MGO (3 μmol)-induced scratching in mice (n = 5 per group, ***P < 0.001 compared with VEH group, Student's t test). (G) I.t. injection of MEK inhibitor U0126 (1 nmol) inhibited MGO (3 μmol)-induced mechanical itch in mice (n = 6-8 per group, ***P < 0.001 compared with VEH group, Student's t-test). (H) I.d. injection of U0126 (10 nmol) inhibited MGO (3 μmol)-induced scratching in mice (n = 5-6 per group, ***P < 0.001 compared with VEH group, Student's t test). (I) I.t. injection of U0126 (1 nmol) inhibited mechanical itch in STZ-induced diabetic mice (n = 5-6 per group, ***P < 0.001 compared with VEH group, Student's t test).All data are expressed by means ± SEM. CPZ, capsazepine; ERK, extracellular signal-regulated kinase; HC, HC030031; MGO, methylglyoxal; NAC, N-Acetyl-L-cysteine; NS, normal saline; PBN, N-tert-butyl-a-phenylnitrone; STZ, streptozotocin; VEH, Vehicle.
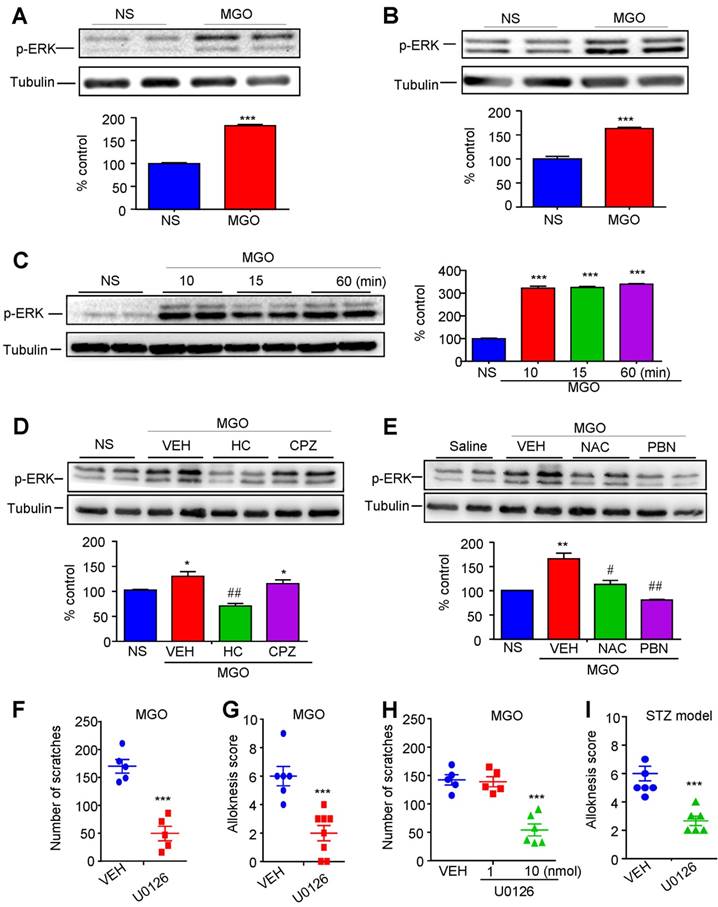
Activation of TRPA1 in spinal cord mediates thermal hypoalgesia induced by injection of MGO or in STZ-induced diabetic mice
Finally, we aimed to study the role of MGO-mediated activation of TRPA1 in hypoalgesia under diabetes condition. We found that the latency of tail-flick in response to 52 °C hot water was increased by i.p. or i.t. injection of MGO increased in normal mice (Figure 10, A and B). Interestingly, the higher dose of MGO (i.p. 100 mg/kg or i.t. 3 nmol) produced less anti-nociceptive effects than that of lower dose of MGO (i.p. 50 mg/kg or i.t. 1 nmol), suggesting a non-linear dose-response curve for MGO-induced anti-nociception in normal mice. As expected, pre-incubation MGO scavenges aminoguanidine and D-arginine with MGO abolished MGO-induced anti-nociception in control mice (Figure 10C). I.t. co-administration of TRPA1 blocker HC-030031 (10 μg; Figure 9D), but not capsazepine (10 μg; Figure 10E), abolished MGO-induced anti-nociception in normal mice (P < 0.001). MGO-induced anti-nociception was impaired in Trpa1-/- mice compared with WT mice (Figure 10F). Thus, these data provided strong evidence to suggest that activation of TRPA1 in the spinal cord contributed to MGO-mediated thermal hypoalgesia in mice.
MGO-mediated activation of TRPA1 in the spinal cord contributes to thermal hypoalgesia in STZ-induced diabetic mice. (A) I.p. injection of MGO (10-100 mg/kg) significantly increased the latency of tail-flick in response to 52 °C hot water in control mice. (B) I.t. injection of MGO (0.3-3 nmol) significantly increased the latency of tail-flick in response to 52 °C hot water in control mice. (C) Co-administration of MGO scavengers Aminoguanidine (10 μg) and D-arginine (10 μg) abolished MGO (i.t., 1 nmol)-induced increased latency of tail-flick in response to 52 °C hot water in control mice (n= 5-8 per group, ***P < 0.001 compared with saline group; ##P < 0.01, ###P < 0.001 compared with MGO group; &&&p < 0.001 compared with MGO group; two-way repeated-measured ANOVA). (D) I.t. injection of TRPA1 blocker HC030031 (10 μg) suppressed MGO (i.t., 1 nmol)-induced increased latency of tail-flick response to 52 °C hot water in the control mice (n = 6-8, *P < 0.05, ***P < 0.001 compared with NS group; #P < 0.01, ###P < 0.001 compared with MGO group; two-way repeated-measured ANOVA). (E) I.t. injection of TRPV1 blocker capsazepine (10 μg) did not affect MGO (i.t., 1 nmol)-induced increased latency of tail-flick response to 52 °C hot water in the control mice (n = 5-8, ***P < 0.001 compared with saline group; two-way repeated-measured ANOVA). (F) MGO (i.p., 50 mg/kg)-induced the increase of latency of tail-flick response to 52 °C hot water was abolished in Trpa1-/- mice (n = 6-8, *P < 0.05, **P < 0.01 compared with WT mice; two-way repeated-measured ANOVA). (G) I.t. injection of MGO scavengers aminoguanidine (10 μg) and D-arginine (10 μg) significantly reduced the latency of tail-flick in response to 52 °C hot water in STZ-induced diabetic mice (n = 6-8, ***P < 0.001 compared with CON group; ###P < 0.001 and &&&P < 0.001 compared with STZ group; two-way repeated-measured ANOVA). (H) I.t. injection of TRPV1 blocker capsazepine (10 μg) failed to change the latency of tail-flick in response to 52 °C hot water in diabetic mice (n = 6-8, ***P < 0.001 compared with CON group; two-way repeated-measured ANOVA). (I) I.t. injection of two selective TRPA1 blockers, HC030031 (10 μg) and A967079 (10 μg), significantly reduced the latency of tail-flick response to 52 °C hot water in STZ-induced diabetic mice (n = 6-8, ***P < 0.001 compared with control group; ###P < 0.001, &&P < 0.05 and &&&P < 0.001 compared with STZ group; two-way repeated-measured ANOVA). All data are expressed by means ± SEM. BL, baseline; CON, control; CPZ, capsazepine; MGO, methylglyoxal; STZ, streptozotocin; WT, wild type.
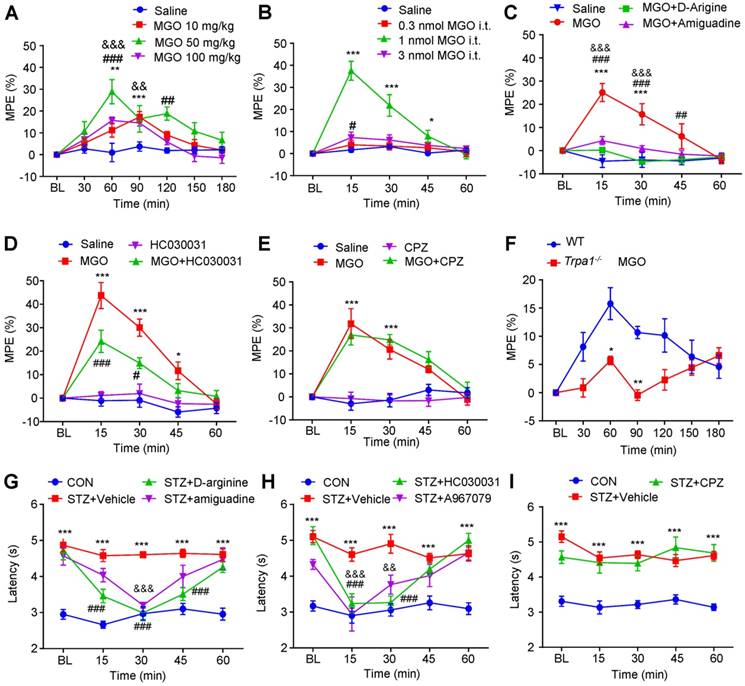
For STZ-induced diabetic mice, i.p. injection of MGO scavenges aminoguanidine (200 mg/kg) and D-arginine (300 mg/kg) significantly reversed diabetes-induced thermal hypoalgesia in mice (Figure 10G). I.t. injection of HC-030031 (10 μg) and A967079 (10 μg), two selective TRPA1 blockers, significantly reversed diabetes-induced thermal hypoalgesia in STZ-induced diabetic mice (Figure 10H). In sharp contrast, i.t. injection of TRPV1 blocker CPZ failed to change the latency of tail-flick in response to 52 °C hot water in STZ-induced diabetic mice (Figure 10I). Thus, these data suggested that MGO-mediated activation of TRPA1 in the spinal cord contributes to thermal hypoalgesia in STZ-induced diabetic mice.
Discussion
Itch and hypoalgesia are two common symptoms associated with diabetic neuropathy [5; 8]. However, the underlying molecular mechanisms are poorly understood. Here, using patch clamp recording, calcium imaging, pharmacological or genetic manipulations, and behavioral testing, we defined a novel signaling pathway by which itch and hypoalgesia is induced by diabetic neuropathy in mice. First, we found that i.d. injection of MGO (an endogenous reactive carbonyl compound) evoked itch behaviors in a dose-dependent manner, while i.t. injection of MGO induced thermal hypoalgesia in normal mice. Second, MGO directly activated TRPA1 channel to induce inward currents and calcium influx in primary cultured DRG neurons. Third, MGO scavengers, genetic ablation of Trpa1 (Trpa1-/-), and pharmacological blockade of TRPA1 abrogated MGO-induced itch and thermal hypoalgesia in mice. Fourth, STZ-induced diabetic mice developed obvious mechanical itch and thermal hypoalgesia. Fifth, antioxidants NAC, PBN or ALA significantly reduced itch induced by MGO or in STZ-induced diabetic mice. Lastly, MGO is sufficient to induce ERK activation in the DRGs and spinal cord, and administration of MEK inhibitor U0126 significantly reduced itch induced by MGO or in STZ-induced diabetic mice. Together, our studies revealed MGO-mediated activation of TRPA1 in periphery and spinal cord contributes to diabetic itch and hypoalgesia, which may provide a novel therapeutic strategy for clinical management of diabetic itch and hypoalgesia.
MGO, a potential itch mediator related to diabetes or other pathological conditions
There has been much interest in identifying potential itch mediators and characterizing their receptors under chronic itch conditions, including skin, liver, kidney diseases, and metabolic diseases [12; 40; 41]. In the current study, we identified and functionally characterized MGO as a potential itch mediator under diabetes condition. We found that i.d. injection of glucose was not sufficient to induce itch behavior in normal mice, suggesting hyperglycemia may be not a root cause of diabetic itch. In stark contrast, i.d. injection of MGO induced robust scratching behavior and mechanical itch in normal mice. We provided several evidences to support MGO as a potential pruritogen under diabetes condition. First, the dose-response curve for MGO-induced itch shows a typical “invert-U” shape, which is similar with H2O2 [39], imiquimod [30] or chloroquine-induced itch [42] in mice. Thus, we postulated that higher doses of MGO may produce pain sensation to suppress itch responses, which is consistent with many previous studies supporting MGO as critical contributor to diabetic neuropathic pain [13; 28; 29; 43]. Second, in cheek model, i.d. injection of low-dose MGO induced only scratching behavior, whereas MGO at the highest dose induced both wiping and scratching behavior in mice. It suggests MGO induces either pain or itch, which is dependent on its local concentrations. MGO at the lower concentrations mainly induces itch, while MGO at the higher concentrations induces pain. Third, we found that MGO-induced itch was suppressed by naloxone, but not morphine, which served as pharmacological evidence to support MGO at lower doses are one of pruritogens. Fourth, we confirmed that neutralizing MGO by D-arginine or aminoguanidine was able to abolish MGO-induced itch in normal mice and mechanical itch in STZ-induced diabetic mice. Since the levels of MGO are also elevated under other diseases, including chronic kidney disease [44], MGO may be considered as a pruritogen not limited to diabetes. Together, we identified MGO as a new potential itch mediator under diabetes condition, possible other pathological conditions associated with elevated level of MGO.
Mechanical itch is a feature for diabetic neuropathy
In our present study, we tried to establish an animal model to investigate itch and hypoalgesia induced by diabetes. Unfortunately, we did not observe spontaneous scratching behavior in STZ-induced diabetic mice. Additionally, several pruritogens-induced acute scratching behaviors were reduced in diabetic mice. An explanation for this phenotype may be due to the impairment of nociceptive C-fibers caused by diabetic neuropathy, which are also involved in mediating itch transmission. Mechanical itch, also called touch-evoked itch, is considered as a remarkable feature of chronic itch, which is caused by skin diseases, systemic diseases, aging, or neurological disorders [33; 38]. In sharp contrast, we observed obvious mechanical itch in STZ-induced diabetic mice. Previous reports demonstrated mechanical itch may be gated by neuropeptide Y (NPY)-expressing inhibitory interneurons in the spinal cord [33]. Aging-induced loss of Merkel cells in the skin or mechanosensitive ion channel Piezo2 deficiency in Merkel cells led to chronic mechanical itch in mice [45]. It warrants further investigation whether dysfunction of NPY-expressing inhibitory interneurons in the spinal cord, possible modulation piezo2 by MGO, and Merkel cells in the skin, are involved in diabetic mechanical itch.
The role of MGO-mediated activation of TRPA1 in diabetic itch
Mounting evidence indicates that TRP channels expressed by primary sensory neurons and non-neuronal cells (e.g., immune cells) play a key role in the regulation of pain and itch [36; 46; 47]. There is a simplified concept that TRPV1 and TRPA1 channel mediate histaminergic and nonhistaminergic itch, respectively [1; 48]. Nevertheless, it remains unclear whether and which TRP channels play a role in the regulation of itch associated with diabetic neuropathy. Previous reports demonstrated that MGO directly activated TRPA1 through an intracellular binding site of TRPA1, which may contribute to the pathogenesis of diabetic neuropathy [49] and pain [13; 28; 29; 50]. Recent studies also described that direct activation of TRPA1 by miRNA-711 (a kind of microRNA) by binding extracellularly to it can drive acute and chronic itch in mice [51]. Imiquimod was also shown to directly activate TRPA1 to evoke itch in mice and zebrafish [52]. Our previous work also demonstrated that H2O2 induced itch via activation of TRPA1 in mice [39]. Previous report demonstrated that streptozotocin (STZ), the toxin used to generate type 1 diabetic animal model in the current study, directly activates the TRPA1 channel in sensory neurons, TRPA1 cell lines, and membrane patches [53]. Intraplantar injections of low dose STZ evoked acute pain within hours in mice systemic STZ treatment (180 mg/kg) evoked a loss of cold and mechanical sensitivity within an hour of injection, which lasted for at least 10 days [53]. In contrast, we tested mechanical itch after STZ-injection 5 weeks, when the levels of blood glucose remained high and STZ may be excreted form the body. Thus, both indirect activation of TRPA1 by signaling from G-protein coupled receptors and direct activation of TRPA1 are sufficient to transduce itch signaling in primary sensory neurons.
In the present study, we provided strong evidence supporting that MGO-mediated direct activation of TRPA1 contributed to diabetic itch in mice. First, pharmacological approach revealed that MGO scavengers (e.g., D-arginine and aminoguanidine) and selective TRPA1 blockers (e.g., HC030031 and A-967079) effective attenuated itch induced by MGO or STZ-induced diabetic mice. Second, using TRPA1 knockout mice, we demonstrated that itch induced by MGO or in STZ-induced diabetic mice was attenuated in Trpa1-/- mice compared with that of WT mice. Third, we demonstrated that the expression and function of TRPA1 in DRG neurons are significantly up-regulated in the setting of diabetes in mice. Our studies are also consistent with previous studies, which showed that the hyperexcitability of primary sensory fibers in the skin, especially Mas-related G protein-coupled receptor (Mrgpr) A3 and MrgprD-positive primary afferents, contributes to peripheral sensitization and is required for the development of chronic itch [54; 55]. Together, it suggests that direct activation and up-regulation of expression and function of TRPA1 in primary sensory neurons contributes to the pathogenesis of diabetic itch.
The role of Nav1.7 in diabetic itch
It is well demonstrated that VGSCs play a key role in the genesis and propagation of action potentials (APs) and regulation of membrane excitability in neurons of peripheral and central nervous system [56; 57]. According to the pharmacological sensitivity of VGSCs to TTX, VGSCs can be divided into TTX-sensitive VGSCs (e.g., Nav1.1, Nav1.2, Nav1.3, Nav1.4, Nav1.6, and Nav1.7) and TTX-resistant VGSCs (e.g., Nav1.5, Nav1.8, and Nav1.9) [56; 57]. Multiple VGSC subtypes are demonstrated to be involved in the pain signaling pathways [57]. Recently, it was demonstrated that certain subtypes of VGSCs, such as Nav1.7 and Nav1.9, were involved in itch signaling transduction [58; 59]. However, the role of VGSCs in diabetic itch remains unclear.
Previous study identified that modification of Nav1.8 by MGO contributed to neuronal hyperexcitability of DRG neurons and pain hypersensitivity in mice [13; 28]. Surprisingly, our present data indicates that Nav1.7, but not Nav1.8, play a key role in diabetic itch in mice. First, pharmacological method showed that local co-administration of Nav1.7 selective blocker PF-05089771 abolished itch induced by MGO or in STZ-induced diabetic in mice, but not for Nav1.8 selective blocker A803467. Consistently, local application of low-dose TTX dramatically inhibited itch induced by MGO and in STZ-induced diabetic mice. Second, q-PCR analysis revealed that mRNA expression of Nav1.7 was significantly increased, but the mRNA expression of Nav1.8 was significantly decreased in the DRGs of STZ-induced diabetic mice compared with that of control mice. Although Nav1.8 is involved in multiple forms of chronic pain, including inflammatory pain, bone cancer pain and diabetic neuropathic pain [57], our data did not support an important role of Nav1.8 in diabetic itch in mice. Although our previous data showed T-type calcium channels were involved in acute itch responses in mice [60], our pharmacological data found that pan T-type calcium channels blocker mibefradil did not affect MGO-induced itch in mice. Additionally, q-PCR analysis found that the expression of Cav3.2 was down-regulated in the DRGs from STZ-induced diabetic mice. Thus, these data suggested TTX-sensitive VGSC subtype Nav1.7 was required for the pathogenesis of diabetic itch, but not TTX-resistant VGSC subtype Nav1.8 and T-type calcium channels.
The role of oxidative stress and ERK signaling in diabetic itch
Our and others' previous reports demonstrated that oxidative stress play a key role in the development of acute and chronic itch [31; 39; 61]. In the present study, we provided evidence supporting oxidative stress may be also involved in diabetic itch. First, application of MGO in cultured ND7-23 cells increased the levels of intracellular ROS and was attenuated by antioxidants. Second, antioxidants NAC and PBN suppressed MGO-induced itch behavior and ERK activation in the spinal cord in mice. Third, administration of clinical used antioxidant ALA was also able to attenuate itch induced by MGO or mechanical itch in STZ-induced diabetic mice. Thus, oxidative stress may be another important contributor to diabetic itch.
Extracellular signal-regulated kinases ERK1/2 transduce extracellular stimuli into intracellular signaling through transcriptional and post-translational mechanisms [62]. ERK1/2 activation in the DRGs and spinal cord contributes to the development of many types of chronic pain, including inflammatory pain [63-66], neuropathic pain [67], and cancer pain [66]. It was found that ERK1/2 activation in the DRGs, skin, and spinal cord was required for acute and chronic itch [31; 68-70]. In the present study, MGO was shown to be able to induce phosphorylation of ERK1/2 (p-ERK) in the DRGs and spinal cord in mice. Suppression of ERK activation by MEK inhibitor U0126 attenuated itch induced by MGO or STZ-induced diabetic mice. These data are consistent with our previous results that inhibition of pruritogens-induced ERK activation was involved in anti-itch effects of antioxidants in mice [31]. Furthermore, blockade of TRPA1 or antioxidants treatment significantly decreased MGO-induced ERK activation in spinal cord in mice. Thus, these data suggests that TRPA1 activation and oxidative stress are required for MGO-induced ERK activation and itch behavior.
Does MGO play a role in diabetic hypoalgesia?
To date, molecular mechanisms underlying loss of pain perception (hypoalgesia) in diabetic neuropathy remain poorly understood. Notably, animal models for diabetic neuropathy showed either thermal hyperalgesia or hypoalgesia, dependent on species or time course of disease [11; 71; 72]. It was reported that thermal hyperalgesia and mechanical allodynia were developed in STZ-induced diabetic rats [15; 49; 73], while thermal hypoalgesia and tactile allodynia were developed in STZ-induced diabetic mice [74]. It was showed thermal hyperalgesia was developed in the early phase and thermal hypoalgeisa occurred in the late phase in diabetic mice [72]. In our study, thermal hypoalgesia developed in STZ-induced diabetic mice after STZ injection 5 weeks, while tactile allodynia developed in diabetic mice one week after STZ injection. MGO is widely appreciated to be a key precursor of advanced glycation end products (AGEs) [19]. Interestingly, a previous study found that the receptor for advanced glycation end products (RAGE), a receptor associated with sustained NF-κB activation contributed to hypoalgesia in diabetic mice [11].
Our present results showed that systemic or intrathecal injection of MGO induced thermal hypoalgesia in normal mice, indicating that elevated MGO level may contributes to loss of pain perception under diabetic neuropathy. We further showed that blockade of TRPA1 channel in spinal cord abolished MGO-induced thermal hypoalgesia, suggesting activation of spinal TRPA1 channel contributes to MGO-mediated thermal hypoalgesia in normal mice. In STZ-induced diabetic mice, administration of MGO scavengers or TRPA1 blockers reversed thermal hypoalgesia, suggesting MGO-mediated activation of TRPA1 in the spinal cord mediates thermal hypoalgesia under diabetes condition. It is noteworthy that a recent study revealed that spinal TRPA1 activation produces hypoalgesia (or anti-nociception) in mice, possibly via central modulation of synaptic transmission in spinal cord [75]. Thus, our data reveals an alternative mechanism underlying thermal hypoalgesia induced by diabetic neuropathy, which involved in MGO-mediated direct activation of TRPA1 in the spinal cord. Thus, neutralizing MGO or blockade TRPA1 in the spinal cord could be a potential therapeutic strategy for thermal hypoalgesia in diabetic neuropathy.
Conclusions
In summary, the results we present in this study focus on the role of Nav1.7 and MGO-mediated activation of TRPA1 in diabetic itch and hypoalgesia in mice. Although clinical evidence supporting MGO as a pruritogen is still lacking, we have provided strong preclinical evidences that Nav1.7 and activation of TRPA1 in the DRG neurons are required for itch induced by MGO or in STZ-induced diabetic mice. Additionally, MGO-mediated activation of TRPA1 in the spinal cord contributes to thermal hypoalgesia in STZ-induced diabetic mice. Thus, these findings will help to develop novel effective treatments of itch and hypoalgesia related to diabetic neuropathy by neutralizing excessive MGO, blocking TRPA1 channel, and/or inhibiting ERK signaling pathways.
Supplementary Material
Supplementary methods and figures.
Acknowledgements
This work was supported by grants from National Natural Science Foundation of China (81870874, 81803307 and 81471041) and Nature Science Foundation of Jiangsu province (BK20170004, BK20140372, and 2015-JY-029). This work was also supported by Jiangsu Key Laboratory of Neuropsychiatric Diseases (BM2013003). Y.F. was supported by Suzhou Science and Technology For People's Livelihood (SYSD2018097). This project is subject to the Second Affiliated Hospital of Soochow University Preponderant Clinic Discipline Group Project Funding (XKQ2015007).
Author contributions
RXC, YF, ZHW, JTZ, LHC, CJS, BW, and YH performed and collected data from experiments and analyzed the data. DL performed electrophysiological recording and analyzed the data. RRJ, JH and TL designed the study, supervised the project. TL wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Dong X, Dong X. Peripheral and Central Mechanisms of Itch. Neuron. 2018;98:482-94
2. Liu X, Miao XH, Liu T. More than Scratching the Surface: Recent Progress in Brain Mechanisms Underlying Itch and Scratch. Neurosci Bull. 2019 https://doi.org/10.1007/s12264-019-00352-1
3. LaMotte RH, Dong X, Ringkamp M. Sensory neurons and circuits mediating itch. Nat Rev Neurosci. 2014;15:19-31
4. Yosipovitch G, Bernhard JD. Clinical practice. Chronic pruritus. N Engl J Med. 2013;368:1625-34
5. Peltier A, Goutman SA, Callaghan BC. Painful diabetic neuropathy. BMJ. 2014;348:g1799
6. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813-20
7. Feldman EL, Nave KA, Jensen TS, Bennett DL. New Horizons in Diabetic Neuropathy: Mechanisms, Bioenergetics, and Pain. Neuron. 2017;93:1296-313
8. Abbott CA, Malik RA, van Ross ER, Kulkarni J, Boulton AJ. Prevalence and characteristics of painful diabetic neuropathy in a large community-based diabetic population in the U.K. Diabetes Care. 2011;34:2220-4
9. Waldfogel JM, Nesbit SA, Dy SM, Sharma R, Zhang A, Wilson LM. et al. Pharmacotherapy for diabetic peripheral neuropathy pain and quality of life: A systematic review. Neurology. 2017;88:1958-67
10. Yamaoka H, Sasaki H, Yamasaki H, Ogawa K, Ohta T, Furuta H. et al. Truncal pruritus of unknown origin may be a symptom of diabetic polyneuropathy. Diabetes Care. 2010;33:150-5
11. Bierhaus A, Haslbeck KM, Humpert PM, Liliensiek B, Dehmer T, Morcos M. et al. Loss of pain perception in diabetes is dependent on a receptor of the immunoglobulin superfamily. J Clin Invest. 2004;114:1741-51
12. Neilly JB, Martin A, Simpson N, MacCuish AC. Pruritus in diabetes mellitus: investigation of prevalence and correlation with diabetes control. Diabetes Care. 1986;9:273-5
13. Bierhaus A, Fleming T, Stoyanov S, Leffler A, Babes A, Neacsu C. et al. Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat Med. 2012;18:926-33
14. Wang S, Kobayashi K, Kogure Y, Yamanaka H, Yamamoto S, Yagi H. et al. Negative Regulation of TRPA1 by AMPK in Primary Sensory Neurons as a Potential Mechanism of Painful Diabetic Neuropathy. Diabetes. 2018;67:98-109
15. Zhang HH, Hu J, Zhou YL, Qin X, Song ZY, Yang PP. et al. Promoted Interaction of Nuclear Factor-kappaB With Demethylated Purinergic P2X3 Receptor Gene Contributes to Neuropathic Pain in Rats With Diabetes. Diabetes. 2015;64:4272-84
16. Allaman I, Belanger M, Magistretti PJ. Methylglyoxal, the dark side of glycolysis. Front Neurosci. 2015;9:23
17. Chakraborty S, Karmakar K, Chakravortty D. Cells producing their own nemesis: understanding methylglyoxal metabolism. IUBMB Life. 2014;66:667-78
18. Ramasamy R, Yan SF, Schmidt AM. Methylglyoxal comes of AGE. Cell. 2006;124:258-60
19. Vander Jagt DL. Methylglyoxal, diabetes mellitus and diabetic complications. Drug Metabol Drug Interact. 2008;23:93-124
20. Kalapos MP. Methylglyoxal in living organisms: chemistry, biochemistry, toxicology and biological implications. Toxicol Lett. 1999;110:145-75
21. Shamsaldeen YA, Mackenzie LS, Lione LA, Benham CD. Methylglyoxal, A Metabolite Increased in Diabetes is Associated with Insulin Resistance, Vascular Dysfunction and Neuropathies. Curr Drug Metab. 2016;17:359-67
22. Brings S, Fleming T, Freichel M, Muckenthaler MU, Herzig S, Nawroth PP. Dicarbonyls and Advanced Glycation End-Products in the Development of Diabetic Complications and Targets for Intervention. Int J Mol Sci. 2017;18:984
23. Nigro C, Leone A, Raciti GA, Longo M, Mirra P, Formisano P. et al. Methylglyoxal-Glyoxalase 1 Balance: The Root of Vascular Damage. Int J Mol Sci. 2017;18:188
24. Dhar A, Dhar I, Jiang B, Desai KM, Wu L. Chronic methylglyoxal infusion by minipump causes pancreatic beta-cell dysfunction and induces type 2 diabetes in Sprague-Dawley rats. Diabetes. 2011;60:899-908
25. Moraru A, Wiederstein J, Pfaff D, Fleming T, Miller AK, Nawroth P. et al. Elevated Levels of the Reactive Metabolite Methylglyoxal Recapitulate Progression of Type 2 Diabetes. Cell Metab. 2018;27:926-34 e8
26. Jack M, Wright D. Role of advanced glycation endproducts and glyoxalase I in diabetic peripheral sensory neuropathy. Transl Res. 2012;159:355-65
27. Hansen CS, Jensen TM, Jensen JS, Nawroth P, Fleming T, Witte DR. et al. The role of serum methylglyoxal on diabetic peripheral and cardiovascular autonomic neuropathy: the ADDITION Denmark study. Diabet Med. 2015;32:778-85
28. Huang Q, Chen Y, Gong N, Wang YX. Methylglyoxal mediates streptozotocin-induced diabetic neuropathic pain via activation of the peripheral TRPA1 and Nav1.8 channels. Metabolism. 2016;65:463-74
29. Eberhardt MJ, Filipovic MR, Leffler A, de la Roche J, Kistner K, Fischer MJ. et al. Methylglyoxal activates nociceptors through transient receptor potential channel A1 (TRPA1): a possible mechanism of metabolic neuropathies. J Biol Chem. 2012;287:28291-306
30. Liu T, Xu ZZ, Park CK, Berta T, Ji RR. Toll-like receptor 7 mediates pruritus. Nat Neurosci. 2010;13:1460-2
31. Zhou FM, Cheng RX, Wang S, Huang Y, Gao YJ, Zhou Y. et al. Antioxidants Attenuate Acute and Chronic Itch: Peripheral and Central Mechanisms of Oxidative Stress in Pruritus. Neurosci Bull. 2017;33:423-35
32. Akiyama T, Carstens MI, Ikoma A, Cevikbas F, Steinhoff M, Carstens E. Mouse model of touch-evoked itch (alloknesis). JInvest Dermatol. 2012;132:1886-91
33. Bourane S, Duan B, Koch SC, Dalet A, Britz O, Garcia-Campmany L. et al. Gate control of mechanical itch by a subpopulation of spinal cord interneurons. Science. 2015;350:550-4
34. Park CK, Xu ZZ, Liu T, Lu N, Serhan CN, Ji RR. Resolvin d2 is a potent endogenous inhibitor for transient receptor potential subtype v1/a1, inflammatory pain, and spinal cord synaptic plasticity in mice: distinct roles of resolvin d1, d2, and e1. J Neurosci. 2011;31:18433-8
35. Shimada SG, LaMotte RH. Behavioral differentiation between itch and pain in mouse. Pain. 2008;139:681-7
36. Moore C, Gupta R, Jordt SE, Chen Y, Liedtke WB. Regulation of Pain and Itch by TRP Channels. Neurosci Bull. 2018;34:120-42
37. Bishnoi M, Bosgraaf CA, Abooj M, Zhong L, Premkumar LS. Streptozotocin-induced early thermal hyperalgesia is independent of glycemic state of rats: role of transient receptor potential vanilloid 1(TRPV1) and inflammatory mediators. Mol Pain. 2011;7:52
38. Lewis AH, Grandl J. A cellular mechanism for age-induced itch. Science. 2018;360:492-3
39. Liu T, Ji RR. Oxidative stress induces itch via activation of transient receptor potential subtype ankyrin 1 in mice. Neurosci Bull. 2012;28:145-54
40. Kremer AE, Martens JJ, Kulik W, Rueff F, Kuiper EM. et al. Lysophosphatidic acid is a potential mediator of cholestatic pruritus. Gastroenterology. 2010;139:1008-18
41. Alemi F, Kwon E, Poole DP, Lieu T, Lyo V, Cattaruzza F. et al. The TGR5 receptor mediates bile acid-induced itch and analgesia. J Clin Invest. 2013;123:1513-30
42. Green AD, Young KK, Lehto SG, Smith SB, Mogil JS. Influence of genotype, dose and sex on pruritogen-induced scratching behavior in the mouse. Pain. 2006;124:50-8
43. Ohkawara S, Tanaka-Kagawa T, Furukawa Y, Jinno H. Methylglyoxal activates the human transient receptor potential ankyrin 1 channel. J Toxicol Sci. 2012;37:831-5
44. Tezuka Y, Nakaya I, Nakayama K, Nakayama M, Yahata M, Soma J. Methylglyoxal as a Prognostic Factor in Patients with Chronic Kidney Disease. Nephrology (Carlton). 2018 https://doi.org/10.1111/nep.13526
45. Feng J, Luo J, Yang P, Du J, Kim BS, Hu H. Piezo2 channel-Merkel cell signaling modulates the conversion of touch to itch. Science. 2018;360:530-3
46. Julius D. TRP channels and pain. Annu Rev Cell Dev Biol. 2013;29:355-84
47. Dai Y. TRPs and pain. Semin Immunopathol. 2016;38:277-91
48. Han L, Dong X. Itch mechanisms and circuits. Annu Rev Biophys. 2014;43:331-55
49. Koivisto A, Hukkanen M, Saarnilehto M, Chapman H, Kuokkanen K, Wei H. et al. Inhibiting TRPA1 ion channel reduces loss of cutaneous nerve fiber function in diabetic animals: sustained activation of the TRPA1 channel contributes to the pathogenesis of peripheral diabetic neuropathy. Pharmacol Res. 2012;65:149-58
50. Griggs RB, Laird DE, Donahue RR, Fu W, Taylor BK. Methylglyoxal Requires AC1 and TRPA1 to Produce Pain and Spinal Neuron Activation. Front Neurosci. 2017;11:679
51. Han Q, Liu D, Convertino M, Wang Z, Jiang C, Kim YH. et al. miRNA-711 Binds and Activates TRPA1 Extracellularly to Evoke Acute and Chronic Pruritus. Neuron. 2018;99:449-63 e6
52. Esancy K, Condon L, Feng J, Kimball C, Curtright A, Dhaka A. A zebrafish and mouse model for selective pruritus via direct activation of TRPA1. Elife. 2018;7:e32036
53. Andersson DA, Filipovic MR, Gentry C, Eberhardt M, Vastani N, Leffler A. et al. Streptozotocin Stimulates the Ion Channel TRPA1 Directly: INVOLVEMENT OF PEROXYNITRITE. J Biol Chem. 2015;290:15185-96
54. Qu L, Fan N, Ma C, Wang T, Han L, Fu K. et al. Enhanced excitability of MRGPRA3- and MRGPRD-positive nociceptors in a model of inflammatory itch and pain. Brain. 2014;137:1039-50
55. Shi H, Yu G, Geng X, Gu L, Yang N, Wang C. et al. MrgprA3 shows sensitization to chloroquine in an acetone-ether-water mice model. Neuroreport. 2017;28:1127-33
56. Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. Sodium channels in normal and pathological pain. Annu Rev Neurosci. 2010;33:325-47
57. Bennett DL, Clark AJ, Huang J, Waxman SG, Dib-Hajj SD. The Role of Voltage-Gated Sodium Channels in Pain Signaling. Physiol Rev. 2019;99:1079-151
58. Lee JH, Park CK, Chen G, Han Q, Xie RG, Liu T. et al. A monoclonal antibody that targets a NaV1.7 channel voltage sensor for pain and itch relief. Cell. 2014;157:1393-404
59. Salvatierra J, Diaz-Bustamante M, Meixiong J, Tierney E, Dong X, Bosmans F. A disease mutation reveals a role for NaV1.9 in acute itch. J Clin Invest. 2018;128:5434-47
60. Wang XL, Tian B, Huang Y, Peng XY, Chen LH, Li JC. et al. Hydrogen sulfide-induced itch requires activation of Cav3.2 T-type calcium channel in mice. Sci Rep. 2015;5:16768
61. Emrick JJ, Mathur A, Wei J, Gracheva EO, Gronert K, Rosenblum MD. et al. Tissue-specific contributions of Tmem79 to atopic dermatitis and mast cell-mediated histaminergic itch. Proc Natl Acad Sci U S A. 2018;115:E12091-E100
62. Ji RR, Gereau RW, Malcangio M, Strichartz GR. MAP kinase and pain. Brain ResRev. 2009;60:135-48
63. Ji RR, Baba H, Brenner GJ, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci. 1999;2:1114-9
64. Pang XY, Liu T, Jiang F, Ji YH. Activation of spinal ERK signaling pathway contributes to pain-related responses induced by scorpion Buthus martensi Karch venom. Toxicon. 2008;51:994-1007
65. Zhuang ZY, Xu H, Clapham DE, Ji RR. Phosphatidylinositol 3-kinase activates ERK in primary sensory neurons and mediates inflammatory heat hyperalgesia through TRPV1 sensitization. J Neurosci. 2004;24:8300-9
66. Lozano-Ondoua AN, Symons-Liguori AM, Vanderah TW. Cancer-induced bone pain: Mechanisms and models. Neurosci Lett. 2013:557 Pt A:52-9
67. Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114:149-59
68. Zhang L, Jiang GY, Song NJ, Huang Y, Chen JY, Wang QX, Ding YQ. Extracellular signal-regulated kinase (ERK) activation is required for itch sensation in the spinal cord. Mol Brain. 2014;7:25
69. Chen Y, Fang Q, Wang Z, Zhang JY, MacLeod AS, Hall RP. et al. Transient Receptor Potential Vanilloid 4 Ion Channel Functions as a Pruriceptor in Epidermal Keratinocytes to Evoke Histaminergic Itch. J Biol Chem. 2016;291:10252-62
70. Huang K, Hu DD, Bai D, Wu ZY, Chen YY, Zhang YJ. et al. Persistent Extracellular Signal-Regulated Kinase Activation by the Histamine H4 Receptor in Spinal Neurons Underlies Chronic Itch. J Invest Dermatol. 2018;138:1843-50
71. Gao F, Zheng ZM. Animal models of diabetic neuropathic pain. Exp Clin Endocrinol Diabetes. 2014;122:100-6
72. Pabbidi RM, Yu SQ, Peng S, Khardori R, Pauza ME, Premkumar LS. Influence of TRPV1 on diabetes-induced alterations in thermal pain sensitivity. Mol Pain. 2008;4:9
73. Sun W, Miao B, Wang XC, Duan JH, Wang WT, Kuang F. et al. Reduced conduction failure of the main axon of polymodal nociceptive C-fibres contributes to painful diabetic neuropathy in rats. Brain. 2012;135:359-75
74. Tsantoulas C, Lainez S, Wong S, Mehta I, Vilar B, McNaughton PA. Hyperpolarization-activated cyclic nucleotide-gated 2 (HCN2) ion channels drive pain in mouse models of diabetic neuropathy. Sci Transl Med. 2017;9:eaam6072
75. Andersson DA, Gentry C, Alenmyr L, Killander D, Lewis SE, Andersson A. et al. TRPA1 mediates spinal antinociception induced by acetaminophen and the cannabinoid Delta(9)-tetrahydrocannabiorcol. Nat Commun. 2011;2:551
Author contact
![]() Corresponding authors: Tong Liu, Ph.D., Institute of Neuroscience, Soochow University, Suzhou, 215021, Jiangsu Province, China; (Tel) +86-512-65883193; Email: liutong80edu.cn or Ji Hu, M.D., The Second Affiliated Hospital of Soochow University, Suzhou 215021, China; (Tel) +86-512-67784167; Email: hujiedu.cn
Corresponding authors: Tong Liu, Ph.D., Institute of Neuroscience, Soochow University, Suzhou, 215021, Jiangsu Province, China; (Tel) +86-512-65883193; Email: liutong80edu.cn or Ji Hu, M.D., The Second Affiliated Hospital of Soochow University, Suzhou 215021, China; (Tel) +86-512-67784167; Email: hujiedu.cn