Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(15):4324-4341. doi:10.7150/thno.32734 This issue Cite
Research Paper
Regulatory T-cells regulate neonatal heart regeneration by potentiating cardiomyocyte proliferation in a paracrine manner
Jiatao Li1,2, Kevin Y. Yang1, Rachel Chun Yee Tam1, Vicken W. Chan1, Hui Yao Lan2,3, Shohei Hori4, Bin Zhou5, Kathy O. Lui1,2 ![]()
1. Department of Chemical Pathology, Prince of Wales Hospital, The Chinese University of Hong Kong, Hong Kong, China
2. Li Ka Shing Institute of Health Sciences, Prince of Wales Hospital, The Chinese University of Hong Kong, Hong Kong, China
3. Department of Medicine and Therapeutics, Prince of Wales Hospital, The Chinese University of Hong Kong, Hong Kong, China
4. Laboratory of Immunology and Microbiology, Graduate School of Pharmaceutical Sciences, The University of Tokyo, Japan
5. The State Key Laboratory of Cell Biology, CAS Center for Excellence in Molecular Cell Science, Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, University of Chinese Academy of Sciences, Shanghai, China
Received 2019-1-2; Accepted 2019-5-9; Published 2019-6-9
Abstract

The neonatal mouse heart is capable of transiently regenerating after injury from postnatal day (P) 0-7 and macrophages are found important in this process. However, whether macrophages alone are sufficient to orchestrate this regeneration; what regulates cardiomyocyte proliferation; why cardiomyocytes do not proliferate after P7; and whether adaptive immune cells such as regulatory T-cells (Treg) influence neonatal heart regeneration have less studied.
Methods: We employed both loss- and gain-of-function transgenic mouse models to study the role of Treg in neonatal heart regeneration. In loss-of-function studies, we treated mice with the lytic anti-CD25 antibody that specifically depletes Treg; or we treated FOXP3DTR with diphtheria toxin that specifically ablates Treg. In gain-of-function studies, we adoptively transferred hCD2+ Treg from NOD.Foxp3hCD2 to NOD/SCID that contain Treg as the only T-cell population. Furthermore, we performed single-cell RNA-sequencing of Treg to uncover paracrine factors essential for cardiomyocyte proliferation.
Results: Unlike their wild type counterparts, NOD/SCID mice that are deficient in T-cells but harbor macrophages fail to regenerate their injured myocardium at as early as P3. During the first week of injury, Treg are recruited to the injured cardiac muscle but their depletion contributes to more severe cardiac fibrosis. On the other hand, adoptive transfer of Treg results in mitigated fibrosis and enhanced proliferation and function of the injured cardiac muscle. Mechanistically, single-cell transcriptomic profiling reveals that Treg could be a source of regenerative factors. Treg directly promote proliferation of both mouse and human cardiomyocytes in a paracrine manner; and their secreted factors such as CCL24, GAS6 or AREG potentiate neonatal cardiomyocyte proliferation. By comparing the regenerating P3 and non-regenerating P8 heart, there is a significant increase in the absolute number of intracardiac Treg but the whole transcriptomes of these Treg do not differ regardless of whether the neonatal heart regenerates. Furthermore, even adult Treg, given sufficient quantity, possess the same regenerative capability.
Conclusion: Our results demonstrate a regenerative role of Treg in neonatal heart regeneration. Treg can directly facilitate cardiomyocyte proliferation in a paracrine manner.
Keywords: CD4+ regulatory T-cells, heart regeneration, cardiomyocyte proliferation, cardiac fibrosis, macrophages, single-cell RNA-seq
Introduction
Heart disease remains the leading cause of deaths worldwide. Yet the mammalian heart is notorious for its inability to repair and regenerate after injury; and the loss of cardiac muscle is replaced by scar tissues that further compromise heart function. Intriguingly, recent studies demonstrate that the mouse heart can transiently regenerate after birth till postnatal day 7 (P7) in a range of injury models including amputation of the ventricular apex via apical resection (AR) [1], myocardial infarction (MI) [2], cryoinfarction (CI) [3] and cardiomyocyte-specific cell death [4]. Importantly, neonatal heart regeneration is also observed in human [5]. In contrast to adults, neonatal cardiomyocytes can proliferate so the heart muscle is regenerated after injury with robust angiogenesis and minimal fibrosis [1, 6, 7]. Nevertheless, mechanisms driving neonatal heart regeneration are largely elusive. Since decades ago, the immune system has been known to orchestrate tissue repair as immune cells regulate both angiogenesis and fibrosis. Understanding how immune cells participate in neonatal heart regeneration would shed light on the development of potential therapeutics to promote heart repair and regeneration [8].
During the last decade, innate immunity, particularly macrophages and their various polarization states, have been considered as a central regulator of the tissue healing processes. Indeed, previous studies demonstrate that macrophages are required for neonatal heart regeneration [4, 9]. Nonetheless, the role of adaptive immunity in neonatal heart regeneration has not been investigated. Long recognized as potent suppressors of the immune system, CD4+ regulatory T-cells (Treg) are recently rediscovered as direct or indirect regulators of organ regeneration (for review, see [10, 11]). We and others have shown that Treg promote repair of skeletal muscle [12, 13], skin [14], lung [15], bone [16], central nervous system [17] and peripheral vascular system [18] after injury. Treg are recruited to the damaged tissues in response to neoantigens for resolution of inflammation and regulation of innate immune response [19]. Mechanistically, Treg can directly activate skeletal muscle progenitors/satellite cells via amphiregulin (AREG) [12] or stimulate hair follicle stem cells via Notch signaling [14]. Moreover, we have also reported that Treg directly promote endothelial cell proliferation and blood vessel regeneration via apelin [18].
In this study, we further investigate the unappreciated role of Treg in neonatal heart regeneration. We utilized CI as a major injury model because the fibrotic responses during neonatal heart repair resemble the healing processes of the adult heart. Unlike their wild type counterparts, T-cell deficient NOD/SCID mice failed to regenerate their injured myocardium at P3. Similarly, treatment with the anti-CD25 antibody (clone PC61) or diphtheria toxin that specifically depletes Treg in wild type or FOXP3DTR mice, respectively, led to increased cardiac fibrosis and reduced neonatal heart regeneration. On the other hand, adoptive transfer of Treg promoted neonatal heart regeneration in NOD/SCID mice after injury. Single cell transcriptomic profiling reveals that Treg could be a source of regenerative factors. Treg directly promoted human and mouse cardiomyocyte proliferation in a paracrine manner. By comparing the regenerating P3 and non-regenerating P8 heart, we uncover that the quantity but not the maturity of Treg determines the outcome of heart regeneration.
Results
Innate immunity alone is insufficient for neonatal heart regeneration of NOD/SCID mice
Previous study shows that depletion of macrophages leads to excessive fibrosis, lack of neoangiogenesis and ultimately compromised regeneration of the neonatal heart after injury [9]. Nevertheless, whether innate immune cells including macrophages are sufficient for neonatal heart regeneration is unanswered. To address this question, we induced CI as previously described [3, 20] to the regenerating P3 or non-regenerating P8 neonatal heart of NOD/SCID mice that harbor innate immune cells such as macrophages without functional adaptive immune cells such as T-cells. We then examined heart regeneration at 4 weeks after injury (Figure S1A). We used NOD/ShiLtJ (NOD), an ascendant of NOD/SCID, as a control and autoimmune diabetes does not occur during the study period (i.e. before 4-8 weeks of age). We also included ICR, an ascendant of NOD, as an additional control because ICR does not develop autoimmunity. Our results showed that the P3 hearts of both NOD and ICR regenerated better than their P8 counterparts (Figure S1B), consistent with previous reports. However, both the P3 and P8 hearts of NOD/SCID did not regenerate (Figure S1B). We then quantified the degree of injury by Masson's trichrome staining that identifies collagen fibers. The P3 hearts of NOD (Figure S1C-D) and ICR (Figure S1E-F) had significantly less fibrotic tissues than their P8 hearts, respectively. Compared to that of NOD, however, the P3 heart of NOD/SCID showed significantly more excessive scar tissue formation (Figure S1G-H), suggestive of impaired neonatal heart repair.
Furthermore, we examined heart function via fractional shorting (%FS) or ejection fraction (%EF) and morphological change via left ventricular end-diastolic or -systolic diameter (LVID d/s) by echocardiography (Figure S2). We first ensured a similar degree of injury was induced to all groups of mice by examining heart function and morphological change at day 4 after injury (Figure S2A-B). Our results demonstrated that there was no significant difference in all parameters measured between the sham and CI groups of NOD and NOD/SCID, respectively (Figure S2C-F). At 2 months after injury (Figure S2G-H), our echocardiographic data revealed that the P3 heart of NOD fully regenerated as there was no significant difference in %FS (Figure S2I), %EF (Figure S2J), %LVID d (Figure S2K) and %LVID s (Figure S2L) between the CI and sham groups, respectively. However, the P3 heart of NOD/SCID showed significantly less functional and morphological regeneration by comparing with its sham control or the CI group of NOD (Figure S2I-L). Taken together, our findings showed that innate immune cells alone were insufficient for driving functional neonatal heart regeneration in NOD/SCID mice.
The regenerating heart attracts more FOXP3+ Treg than the non-regenerating one
We asked whether Treg were responsible for neonatal heart regeneration as the absence of which in NOD/SCID mice might contribute to impaired regeneration. We backcrossed the Foxp3hCD2 reporter “knockin” allele onto NOD background as previously described [21], allowing us to trace and purify FOXP3+ Treg via their surface expression of hCD2. We quantified CD3+CD4+hCD2+ Treg of the damaged myocardium after CI to a P3 heart by flow cytometry (Figure S3A). There was a significant increase in %CD4+hCD2+ Treg of total CD4+ T-cells (Figure S3B) or in the absolute number of CD4+hCD2+ Treg per mg heart tissue (Figure S3C) at day 7 after CI compared to the sham control.
Loss-of-function of FOXP3+ Treg contributes to increased cardiac fibrosis of the neonatal heart
To functionally demonstrate the role of Treg during neonatal heart regeneration, we first depleted CD4+CD25hiFOXP3+ Treg in NOD.Foxp3hCD2 mice via the lytic anti-CD25 antibodies (clone PC61, Figure S4A) as previously described [18, 22]. Treatment with PC61 significantly reduced %CD4+hCD2+ Treg in the spleen and circulation (Figure S4B-C). At 4 weeks after CI to a P3 heart, Treg depletion contributed to a significantly increase in cardiac fibrosis (Figure S5A-B). Immunostaining of markers specific for fibroblasts and cardiomyocytes, COLA1 and cTnT, showed significantly increased fibroblast deposition (Figure S5C) yet significantly reduced myocardium in the infarct zone (Figure S5C-D). Moreover, at day 7 after CI, costaining of cTnT and the proliferation marker Ki67 or phospho histone 3 (pH3) demonstrated a significantly reduced number of Ki67+cTnT+ (Figure S5E-F) or pH3+cTnT+ (Figure S5E, G) proliferating cardiomyocytes in the border zone.
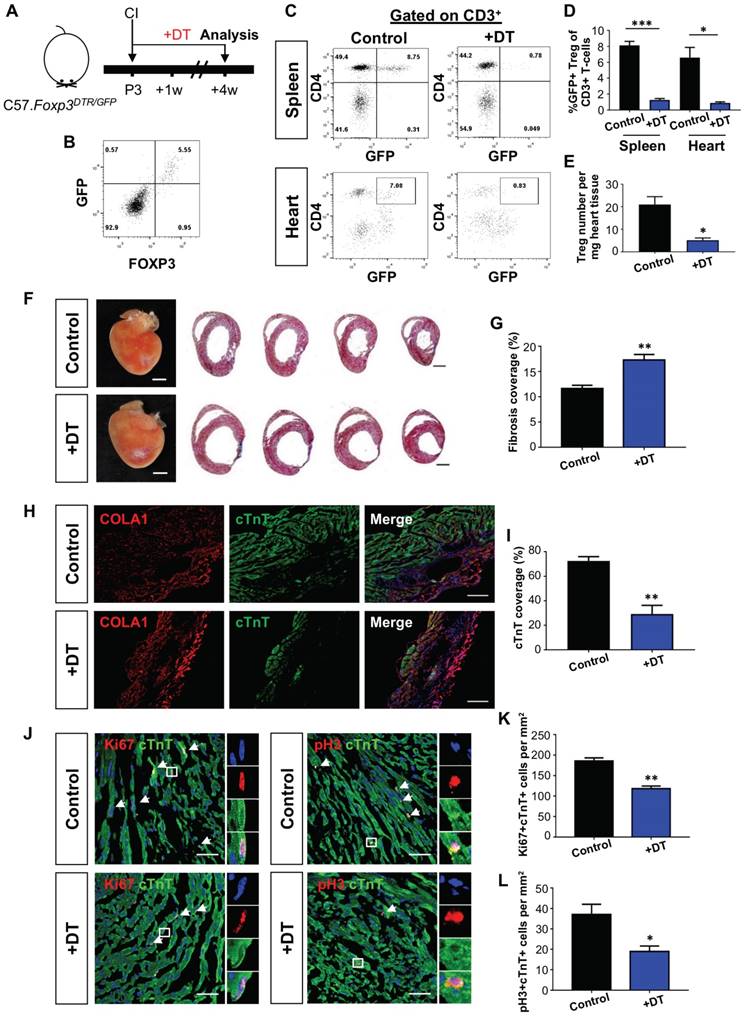
Alternatively, we depleted Treg in C57.FOXP3DTR/GFP mice (Figure 1A). In this model, GFP can be used to trace Treg as we confirmed co-expression of GFP and FOXP3 in Treg by flow cytometry (Figure 1B). Seven days after treatment with diphtheria toxin, we found significantly reduced %GFP+ Treg among total CD3+ T-cells of both the spleen and heart when compared to the control (Figure 1C-D). Moreover, there was a significantly reduced number of GFP+ Treg per mg heart tissue (Figure 1E). We then performed CI to a P3 heart and examined heart regeneration at 4 weeks after injury. In line with the PC61 experiments, Treg ablation via diphtheria toxin significantly increased cardiac fibrosis (Figure 1F-G) and COLA1 deposition (Figure 1H); while significantly reduced cTnT+ myocardium of the infarct zone (Figure 1I). At day 7 after injury, there was a significantly reduced number of Ki67+cTnT+ (Figure 1J-K) or pH3+cTnT+ (Figure 1J, L) proliferating cardiomyocytes in the border zone. Taken collectively, our loss-of-function experiments showed that depletion of Treg impaired the endogenous regenerative capability of a P3 heart after CI.
Loss-of-function of CD4+ Treg leads to increased fibrosis and reduced cardiomyocyte proliferation after cryoinfarction. (A) Schematic diagram showing the experimental design. Flow cytometric analysis showing (B) co-expression of GFP and FOXP3 in Foxp3DTR/GFP mice and (C) depletion efficiency of CD3+CD4+FOXP3+ Treg in the spleen and heart after treatment with diphtheria toxin. Quantification of (C) showing (D) %Treg among total CD3+ T-cells, n=4 or (E) absolute number of Treg per mg tissue at day 7 after CI, n=4. (F) Images of scar tissues, scale bars: 2000 um; and Masson's trichrome staining showing representative serial cross sections of fibrotic tissues in blue, scale bars: 1000 um. (G) Quantification of fibrotic tissue coverage based on (F), n=9. Immunostaining on frozen sections for (H) COLA1+ (red) and cTnT+ (green) cells within the infarct zone, scale bars: 100 um; or (J) Ki67+ (red) or pH3 (red) and cTnT+ (green) cells within the border zone at day 7 post CI, scale bars: 50 um. (J) Arrows indicate cardiomyocytes positive for Ki67 or pH3 and square denotes magnified images on the right. Quantification of absolute number of (I) %cTnT+ coverage, n=5; and (K) Ki67+cTnT+ or (L) pH3+cTnT+ cardiomyocytes per mm2 area, n=7. (D, E, G, I, K, L) Data are presented as mean±S.E.M., *P<0.05, **P<0.01, ***P<0.001.
Adoptive transfer of FOXP3+ Treg promotes neonatal heart regeneration of NOD/SCID mice
To further illustrate the potential role of Treg in neonatal heart regeneration, we adoptively transferred hCD2+ Treg purified from the spleen of NOD.Foxp3hCD2 mice to NOD/SCID mice via intraperitoneal injection as previously described [23] so that the recipients only harbored Treg without conventional T-cells. We were only able to harvest several thousand Treg from neonates, so we chose to use 6-8 week old NOD.Foxp3hCD2 as donors and purified 1 million Treg from them for adoptive transfer (Figure 2A). By flow cytometric analysis on day 7 following adoptive transfer, we found that about 88% of total CD3+CD4+ T-cells were hCD2+ Treg in the spleen and heart, respectively (Figure 2B). Moreover, we also quantified the engraftment of CD3+CD4+hCD2+ Treg weekly during the first 4 weeks following adoptive transfer and found that the %hCD2+ Treg of total splenocytes increased gradually and reached about 5% by day 28 after adoptive transfer (Figure 2C), similar to a wild type control.
Adoptive transfer of CD4+ Treg potentiates neonatal heart regeneration after cryoinfarction. (A) Schematic diagram showing the experimental design. (B) Flow cytometric analysis showing engraftment and infiltration of CD3+CD4+hCD2+ Treg in the spleen and myocardium, respectively, at day 7 following CI to a P3 heart of NOD/SCID mice. Quantification of (C) %Treg among total splenocytes or (D) absolute number of hCD2+ Treg per mg heart tissue at the indicated time points after adoptive transfer by flow cytometry, n=3 per time point. (E) Images of scar tissues, scale bars: 2000 um; and Masson's trichrome staining showing representative serial cross sections of fibrotic tissues in blue at 4 weeks post CI, scale bars: 1000 um. (F) Quantification of fibrotic tissue coverage based on (E), n=7. (G) Immunostaining on frozen sections for Ki67+ (red) or pH3 (red) and cTnT+ (green) cells within the border zone at day 7 post CI, scale bars: 50 um. Arrows indicate cardiomyocytes positive for Ki67 or pH3 and square denotes magnified images on the right. Quantification of absolute number of (H) Ki67+cTnT+ or (I) pH3+cTnT+ cardiomyocytes per mm2 area, n=7. (J) Echocardiographic analysis and quantification showing (K) %fractional shortening or (L) %ejection fraction at 8 weeks post CI, n=7. (C, D, F, H, I, K, L) Data are presented as mean±S.E.M., *P<0.05, **P<0.01.
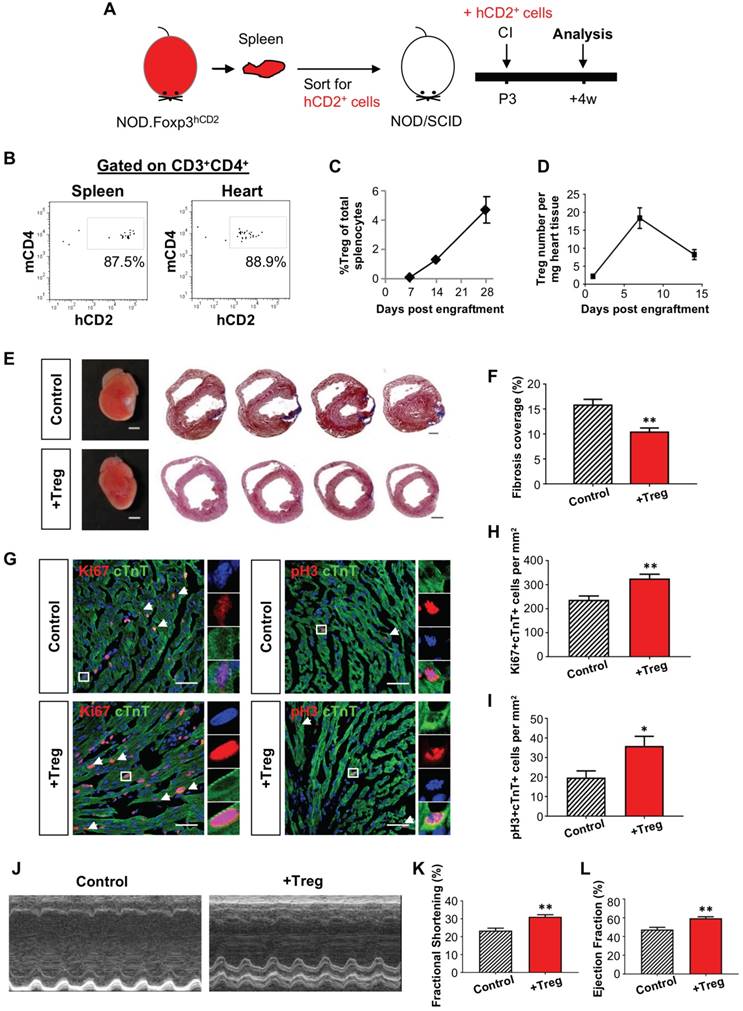
We then performed CI to a P3 heart of NOD/SCID mice with or without (control) adoptive transfer of Treg; and examined the kinetics of Treg infiltration into the injured myocardium. Similar to NOD (Figure S3C), there was an increased number of intracardiac Treg after adoptive transfer and a peak was found at day 7 after CI (Figure 2D). To examine the effect of adoptive transfer of Treg, we performed Masson's trichrome staining and found significantly reduced cardiac fibrosis at 4 weeks after CI when compared to the control (Figure 2E-F). To investigate if heart regeneration was mediated via proliferation of cardiomyocytes, we performed immunostaining for Ki67, pH3 and cTnT at day 7 after CI (Figure 2G) and found that adoptive transfer of Treg significantly increased the number of Ki67+cTnT+ (Figure 2H) or pH+cTnT+ (Figure 2I) cardiomyocytes in the border zone. To evaluate heart regeneration functionally, we performed echocardiography at 8 weeks after CI (Figure 2J). Our data revealed that adoptive transfer of Treg did not contribute to morphological change of the left ventricle; but significantly increased %FS (Figure 2K) and %EF (Figure 2L).
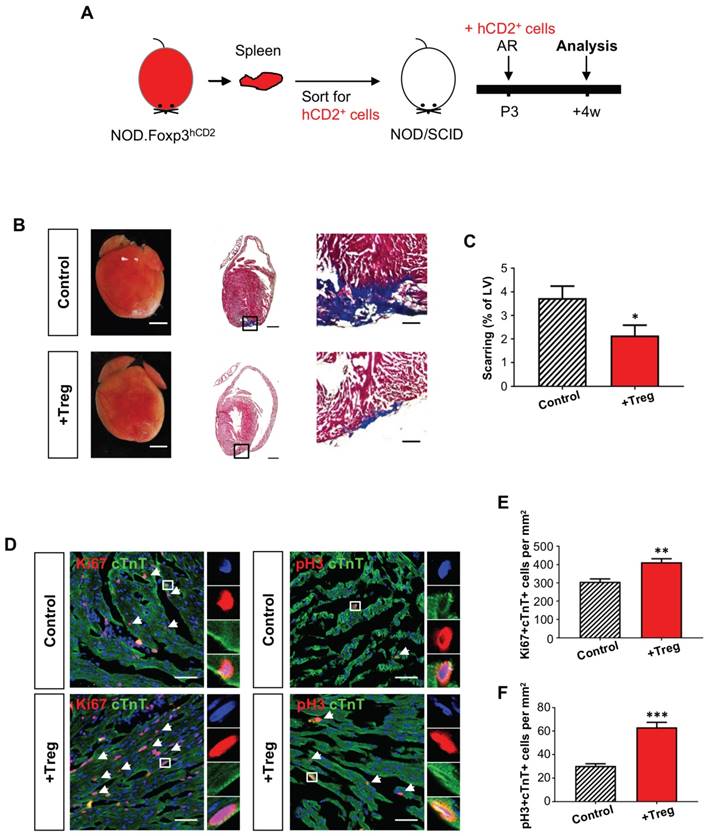
We also included an additional injury model to recapitulate the role of Treg in neonatal heart regeneration. AR was performed on a P3 heart of Treg-infused or control NOD/SCID mice; and heart regeneration was examined at 4 weeks after injury (Figure 3A). Similar to the CI model, we demonstrated that adoptive transfer of Treg significantly reduced cardiac fibrosis (Figure 3B-C) and significantly enhanced the number of Ki67+cTnT+ (Figure 3D, E) or pH+cTnT+ (Figure 3D, F) proliferating cardiomyocytes in the border zone at day 7 after injury. Altogether, our gain-of-function experiments in both the CI and AR models uncovered an unappreciated role of Treg in potentiating neonatal heart regeneration.
Adoptive transfer of FOXP3+ Treg leads to reduced fibrosis of the neonatal heart after apical resection. (A) Schematic diagram showing the experimental design. (B) Images of scar tissues, scale bars: 2000 um; Masson's trichrome staining showing a representative section of fibrotic tissues in blue, scale bars: 1000 um; and a magnified image, scale bars: 200 um. (C) Quantification of fibrotic tissue coverage based on (B), n=6. (D) Immunostaining on frozen sections for Ki67+ (red) or pH3 (red) and cTnT+ (green) cells within the border zone at day 7 post CI, scale bars: 50 um. Arrows indicate cardiomyocytes positive for Ki67 or pH3 and square denotes magnified images on the right. Quantification showing the absolute number of (D) Ki67+cTnT+ or (E) pH3+cTnT+ cardiomyocytes per mm2 area, n=7. (C, E, F) Data are presented as mean±S.E.M., *P<0.05, **P<0.01, ***P<0.001.
Single cell transcriptomic profiling reveals Foxp3+ Treg as a source of paracrine factors
In order to understand how Treg regulate neonatal heart regeneration, we performed genome-wide single-cell transcriptomic profiling (scRNA-seq) as recently described [21, 24, 25]. In this study, the spleen served as a source of naïve T-cells (control) while the heart served as a source of neoantigen-activated T-cells. At day 7 after CI, we respectively purified about ~1850 and ~581 CD3+ T-cells from the spleen and heart of ICR mice that underwent CI at P3. We then focused our analysis on Foxp3+ Treg and performed unsupervised analysis that did not rely on any known marker. t-distributed stochastic neighbor embedding (t-SNE) plots showed that Treg of the spleen and heart formed distinct populations (Figure 4A): splenic Treg mainly constituted cluster C1; while heart Treg primarily formed cluster C2. We identified the most significantly upregulated genes in C1 and C2 (Table 1); and performed gene ontology (GO) functional annotations as demonstrated by pathway analyses (Figure 4B, Table S1, S2) and heatmap (Figure 4C).
The most significantly upregulated genes expressed by heart Treg during neonatal heart regeneration. Foxp3+ Treg are purified from the spleen or heart at day 7 post CI to P3 ICR mice. C1: upregulated genes in splenic naïve Treg; C2: upregulated genes in Treg following activation by neoantigens released during cryoinfarction in the heart.
| Cluster | Significantly upregulated genes |
|---|---|
| C1 | Elane, Mpo, Prtn3, Fcnb, Ngp, Kpna2, Lcn2, S100a8, S100a9, Camp, BC100530, Stfa3, Ccnb1, Wfdc21, Ltf, Ube2c, Cenpf, Gm5483, Cdc20, Lockd, Ccnb2, Cdc25b, Tubb4b, Cdk1, Cd6, Retnlg, Def6, Birc5, Cdca8, Nusap1, Hdac1, Tnfrsf4, Ms4a4b, Glipr2, Cxcr3, 2810417H13Rik, Gm10093, Smc2, Gimap7, Cd28, Ass1, Hmgb2, Pttg1, Sell, Pglyrp1, Tuba1c, Ezr, Hmgn2, Tap2, Ddx39 |
| C2 | C3ar1, Cd14, Fcrls, Spp1, Ccl7, Cxcl2, Sdc4, Ier3, Cx3cr1, Cxcl16, Atf3, Ccl12, Ccl3, Pid1, Trem2, Ccl2, Ms4a7, Klf4, Mafb, Apoe, Csf1r, Kctd12, Rhob, P2ry6, C1qa, Fcgr3, Cd86, Mef2c, Tmem176b, AF251705, Ckb, Clec4n, Cdkn1a, Ltc4s, C5ar1, Ms4a6d, C1qb, Ccl4, Ccl24, Adgre1, Mrc1, Wfdc17, Zeb2, Fcer1g, C1qc, Dab2, Dhrs3, Lgmn, Lyve1, Grn, Pla2g7, Gas6, Gpr34, Pmp22, Tmem86a, Syk, Camk1, Ctsl, F13a1, Cbr2, Ccr1, Tspan4, Rin2, Tbxas1, Mt1 |
Single cell transcriptomic profiling reveals that Treg are a source of paracrine factors. (A) Biaxial scatter plots by t-SNE analysis showing single-cell transcriptomic clustering of Foxp3+ Treg purified from the spleen or heart at day 7 post CI to P3 ICR mice. Cells were subgrouped into specific clusters (C1-4). (B) Analysis showing selected most significantly upregulated pathways determined by GO functional annotations in terms of biological processes of C1 splenic and C2 heart Treg (Tables S1). (C) Upregulated genes in (B) were displayed by a heatmap. (D) Violin plots showing selected most significantly upregulated genes that regulate macrophage activity or regeneration processes. (E) qRT-PCR validation of selected genes identified in (C, D) based on expression level, novelty and relative function in regeneration. Data are presented as mean±S.E.M., n=4, *P<0.05, **P<0.01.
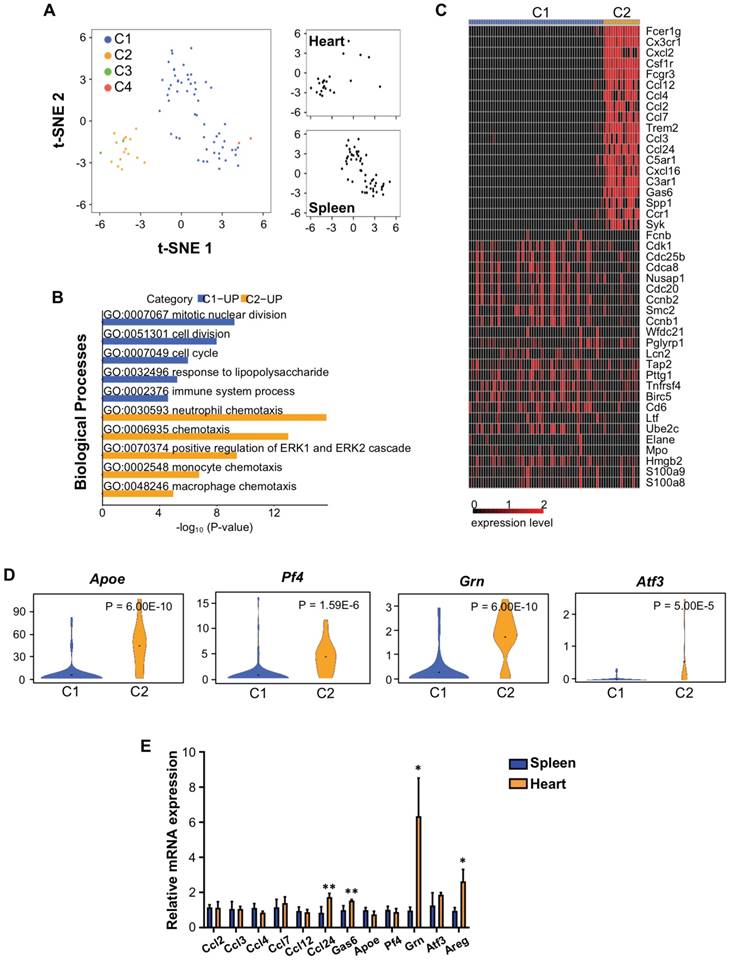
Our results revealed that C1 was distinguished by upregulated cell cycle/cell division genes, indicating that Treg of a regenerating neonatal heart were less proliferative than naive T-cells. Moreover, C2 was marked by upregulated chemotactic factors including chemokines and cytokines that attract neutrophils, monocytes and macrophages, suggesting that Treg of the regenerating neonatal heart could recruit innate immune cells including macrophages, previously reported to drive neonatal heart regeneration [4, 9]. We also highlighted several significantly upregulated genes in C2 compared to C1 that have been reported to regulate macrophage activity such as apolipoprotein E (Apoe) [26] and CXCL4 (Pf4) [27]; and to facilitate tissue regeneration such as granulin (Grn) [28-30] and activating transcription factor 3 (Atf3) [31, 32] (Figure 4D).
We validated our scRNAseq data. At day 7 after CI, we purified hCD2+ Treg from the spleen and heart of NOD.Foxp3hCD2 mice that underwent CI at P3. We then examined gene expression by qRT-PCR with a particular focus on chemotactic factors and secreted factors that were associated with tissue regeneration. Our results confirmed that there were significantly increased gene expression levels of chemokine ligand 24 (Ccl24), growth arrest specific 6 (Gas6), Grn and Areg in Treg of the regenerating heart when compared to that of the spleen (Figure 4E).
FOXP3+ Treg facilitate proliferation of mouse and human cardiomyocytes in a paracrine manner
Next, we examined if Treg can directly regulate neonatal heart regeneration in a paracrine manner. To test this, we cocultured mouse neonatal cardiomyocytes with purified 4-6 week old hCD2+ Treg or supernatant (SN) of Treg cultures for 1-3 days. We then performed immunostaining for proliferation markers Ki67, pH3 or Aurora B with cTnT (Figure 5A). Our results showed that Treg or Treg SN significantly increased the total number of cardiomyocytes after cocultured for 3 days when compared to the control (Figure 5B). Moreover, Treg or Treg SN significantly increased %Ki67+cTnT+ (Figure 5C), pH3+cTnT+ (Figure 5D) or Aurora B+cTnT+ (Figure 5E) cells among total cTnT+ cardiomycytes after cocultured for 1 day. To investigate if Treg promote cardiomyocyte proliferation by regulating cell cycle progression, we performed qRT-PCR to examine gene expression of cyclin-dependent kinase inhibitors such as p18, p21, p27 and p57. Our results showed that gene expression levels of p27 and p57 were significantly reduced in cardiomyocytes after cultured in Treg SN for 1 day (Figure S6A).
Treg directly promote proliferation of mouse neonatal cardiomyocytes in a paracrine manner. Immunocytochemistry for cTnT+ (red) and Ki67+ (green), pH3+ (green) or Aurora B+ (green) cells at day 1 after coculture of (A) CD3+CD4+hCD2+ Treg, Treg supernatant (SN), or (G) the combination of CCL24, GAS6 and AREG (Pool 3) with mouse neonatal cardiomyocytes of P1 ICR hearts, scale bars: 50 um. Quantification of (B) the absolute number of total cTnT+ cardiomyocytes after cocultured for 3 days; or (C) %Ki67+cTnT+, (D) %pH3+cTnT+ or (E) %Aurora B+cTnT+ proliferating cardiomyocytes among total cTnT+ cardiomyocytes based on (A). Quantification of proliferating cardiomyocytes after cultured with (F) the respective paracrine factors or (H-J) Pool 3 for 1 day. Data are presented as mean±S.D., n = 3 independent experiments, *P<0.05, **P<0.01.
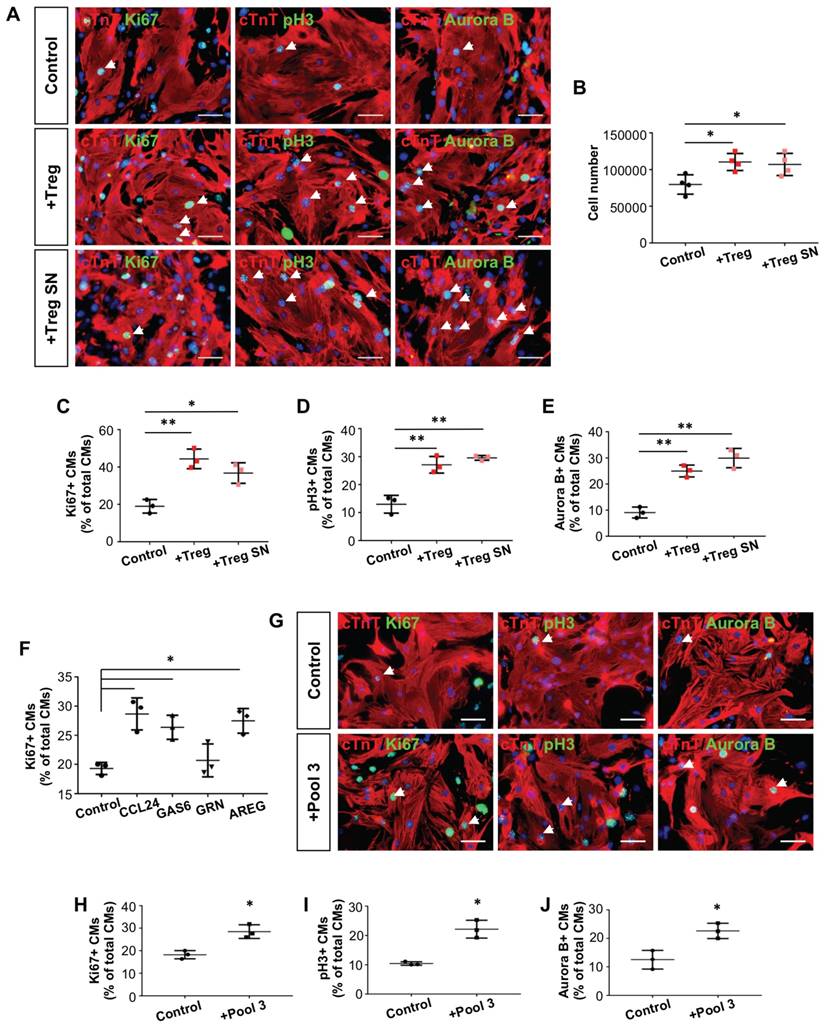
Since our scRNA-seq and qRT-PCR data showed that Ccl24, Gas6, Grn and Areg were significantly upregulated in Treg during neonatal heart regeneration, we examined if these paracrine factors alone or in combination facilitate neonatal cardiomyocyte proliferation. We and others found that CCR3 (receptor of Ccl24, Figure S6B-C), AXL receptor tyrosine kinase (receptor of Gas6) [33], epidermal growth factor receptor (receptor of Areg) [34] and EphA2 (receptor of Grn) [35] are expressed by cardiomyocytes. Moreover, CCL24, GAS6 or AREG alone significantly increased %Ki67+cTnT+ cells among total cTnT+ cardiomycytes after cultured for 1 day (Figure 5F). To illustrate if these factors have any synergistic effect, we pooled them together (Pool 3) and performed the same proliferation assays (Figure 5G). Our results demonstrated that Pool 3 significantly increased %Ki67+cTnT+ (Figure 5H), pH3+cTnT+ (Figure 5I) or Aurora B+cTnT+ (Figure 5J) cells among total cTnT+ cardiomycytes after cultured for 1 day. However, the order of magnitude was similar to those contributed by each of the factors alone.
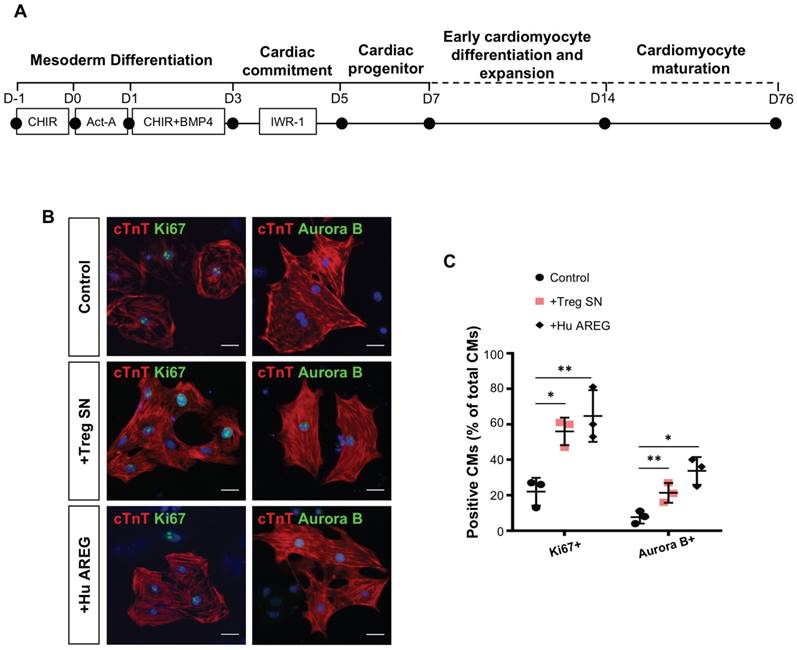
Furthermore, we asked if we can recapitulate the same regenerative effect of Treg in potentiating proliferation of human cardiomyocytes. We differentiated beating cardiomyocytes from human embryonic stem cells (hESC-CM, Figure 6A) as previously described [36]; and cocultured relatively mature, less proliferative hESC-CM (day 76) with Treg SN or recombinant human AREG protein for 3 days. Immunostaining for Ki67 or Aurora B with cTnT (Figure 6B) revealed that Treg SN or AREG significantly increased %Ki67+cTnT+ or %Aurora B+cTnT+ cells among total cTnT+ cardiomyocytes (Figure 6C). Taken together, our results uncovered that Treg directly promoted proliferation of mouse neonatal and human fetal-like cardiomyocytes in a paracrine manner.
Treg directly promote proliferation of human fetal-like cardiomyocytes in a paracrine manner. (A) Schematic diagram showing the differentiation protocol for generating human fetal-like cardiomyocytes from embryonic stem cells (hESC-CM). (B) Immunocytochemistry and (C) quantification of cTnT+ (red) and Ki67+ (green) or Aurora B+ (green) cells at day 3 after coculture of Treg supernatant or human amphiregulin (AREG) with hESC-CM, scale bars: 20 um. Data are presented as mean±S.D., n = 3 independent experiments, *P<0.05, **P<0.01.
FOXP3+ Treg promotes neonatal heart regeneration regardless of the age
In fact, whether the quantity and/or the age of Treg determine their efficacy in heart regeneration remains unclear. To address this question, we compared the quantity of intracardiac CD4+hCD2+ Treg of the regenerating P3 and non-regenerating P8 hearts. At day 7 after injury, there was a significant reduction in the absolute number of CD4+hCD2+ Treg of the P8 than P3 heart (Figure 7A). To ask whether Treg differ in the identity during neonatal heart regeneration, we performed genome-wide scRNAseq. At day 7 after CI, we purified about ~1263 and ~2292 CD3+ T-cells from the hearts of ICR mice that underwent CI at P3 and P8, respectively. We then performed unsupervised analysis; and t-SNE plots demonstrated that Foxp3+ Treg of the P3 and P8 hearts did not form distinct clusters (Figure 7B). We also did not find significantly differential expression (adjusted p-value <0.05 and common mean expression >=0.01) by analyzing 15864 genes, indicating that the whole transcriptome of Treg did not change at a single-cell level regardless of whether regeneration was resulted in the neonatal heart.
The quantity but not the age of Treg determines the outcome of neonatal heart regeneration. (A) Flow cytometric analysis showing infiltration of CD3+CD4+hCD2+ Treg into the injured myocardium of ICR mice at day 7 post CI to the P3 or P8 heart compared to the sham control, n=4. (B) Biaxial scatter plots by t-SNE analysis showing single-cell transcriptomic clustering of Foxp3+ Treg purified from the injured myocardium at day 7 post CI to P3 or P8 ICR mice. (C) Images of scar tissues, scale bars: 2000 um; Masson's trichrome staining showing a representative section of fibrotic tissues in blue, scale bars: 1000 um. (D) Quantification of fibrotic tissue coverage based on (C). (E) Quantification of the absolute number of total cTnT+ cardiomyocytes after cultured in Treg SN for 3 days. (F) Immunocytochemistry for cTnT+ (red) and Ki67+ (green), pH3+ (green) or Aurora B+ (green) cells at day 1 after culture of Treg supernatant (SN) with mouse neonatal cardiomyocytes of P1 ICR hearts, scale bars: 50 um. Quantification of (G) %Ki67+cTnT+, (H) %pH3+cTnT+ or (I) %Aurora B+cTnT+ proliferating cardiomyocytes among total cTnT+ cardiomyocytes based on (F). Data are presented as (A) mean±S.E.M., n=4; (D) mean±S.E.M., n=6; or (E, G-I) mean±S.D., n = 3 independent experiments; *P<0.05, **P<0.01.
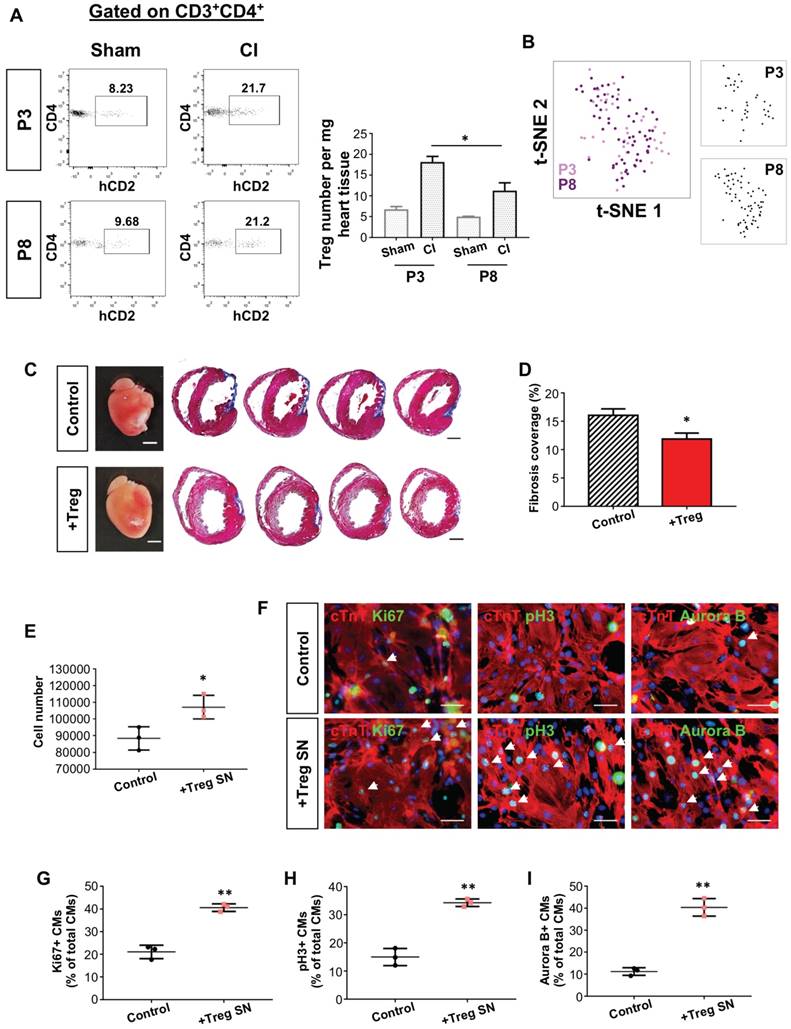
We then examined if mature Treg contributed to neonatal heart regeneration by adoptively transferring 1 million hCD2+ Treg from 12-week old NOD.Foxp3hCD2 mice to P3 NOD/SCID mice after CI. Masson's trichrome staining showed significantly reduced cardiac fibrosis at 4 weeks after injury when compared to the control (Figure 7C-D). Moreover, we cocultured mouse neonatal cardiomyocytes with supernatant of 12 week old hCD2+ Treg. Treg SN significantly increased the total number of cardiomyocytes after cultured for 3 days (Figure 7E). Furthermore, immunostaining for proliferation markers and cTnT (Figure 7F) showed that Treg SN significantly increased %Ki67+cTnT+ (Figure 7G), pH3+cTnT+ (Figure 7H) or Aurora B+cTnT+ (Figure 7I) cells among total cTnT+ cardiomycytes after cultured for 1 day. Altogether, our findings may suggest that the quantity but not the age of Treg determined the outcome of heart regeneration and cardiomyocyte proliferation.
FOXP3+ Treg promotes neonatal heart regeneration by regulating M2-like macrophages
Lastly, we examined if Treg regulate recruitment of macrophages during neonatal neonatal heart regeneration. We performed CI to a P3 heart of Treg-depleted C57.FOXP3DTR/GFP mice (Figure S7A). One week after CI, we found no significant difference in %F4/80+Ly6Chigh M-1 like macrophages but significantly increased %F4/80+CD206+Ly6Clow M-2 like macrophages in the myocardium (Figure S7B-C). On the other hand, we performed CI to a P3 heart after adoptive transfer of Treg to NOD/SCID mice (Figure S7D). We did not observe differential macrophage recruitment at one week after CI. At two weeks after CI, adoptive transfer of Treg contributed to significantly reduced %F4/80+CD206+Ly6Clow M2-like macrophages albeit with no difference in M1-like macrophages (Figure S7E-F).
Discussion
It has long been believed that mammals are unable to regenerate their hearts after injury. It is, therefore, important to learn that the postnatal mouse heart is capable of regenerating shortly after birth till P7 [1]. Macrophages are essential in this regeneration as their depletion leads to excessive cardiac fibrosis, impeded neoangiogenesis and impaired cardiomyocyte survival [4, 9]. However, we showed that macrophages alone were insufficient to drive functional heart regeneration at as early as P3 in the T-cell-deficient NOD/SCID mice. Previously, it has been demonstrated that proliferation of pre-existing cardiomyocytes is the major mechanism by which the neonatal heart regenerates [6, 7]. What regulates cardiomyocyte proliferation during mammalian heart regeneration; why the proliferative capacity of cardiomyocytes halts after P7; and whether adaptive immune cells influence neonatal heart regeneration have not been investigated.
Unlike mammals, the adult zebrafish heart can regenerate with robust cardiomyocyte proliferation after injury that overcomes scar formation [37]. Recently, Treg are found essential for this regeneration as their depletion contributes to impaired regeneration of the zebrafish heart [38]. In addition, Treg are considered beneficial for cardiac repair in mammals. Expansion of Treg via adoptive transfer or administration of the CD28 superagonistic antibody ameliorates adverse cardiac remodeling after injury [39, 40]. Moreover, Treg promote the accumulation of regenerative macrophages that are associated with reduced rate of left ventricular rupture and increased survival after MI [41]. More recently, Treg in the maternal circulation that expanded to maintain maternal-fetal tolerance have been shown to promote maternal and prenatal cardiomyocyte proliferation during pregnancy [42]. Therefore, we hypothesized that Treg might play a pivotal role in neonatal heart regeneration.
Within the first two weeks after injury to a regenerating P3 heart, there was a significant increase in the absolute number of Treg with a peak at day 7 after injury. To ask if these Treg facilitated functional neonatal heart regeneration, we performed both loss- and gain-of-function studies. Depletion of CD4+CD25hiFOXP3+ Treg via the lytic anti-CD25 antibody in Foxp3hCD2 mice or ablation of Treg via diphtheria toxin in Foxp3DTR mice after CI to a P3 heart led to impaired heart regeneration manifested by significantly increased cardiac fibrosis and reduced number of proliferating cardiomyocytes when compared to the control. Although the P3 heart of NOD/SCID mice failed to regenerate after injury, we demonstrated that adoptive transfer of FOXP3+ Treg resulted in significantly mitigated cardiac fibrosis, increased number of proliferating cardiomyocytes and enhanced cardiac function 8 weeks after CI when compared to that of the Treg-deficient control. Similarly, adoptive transfer of Treg also promoted regeneration of the NOD/SCID heart in an additional injury model though AR.
Mechanistically, single cell transcriptomic profiling revealed that Treg of the regenerating P3 heart could be a source of regenerative factors. We uncovered some of these factors and validated their role in cardiomyocyte proliferation based on the expression level, novelty and function in regeneration of other organ systems such as Gas6 [43], Grn [28-30] and Areg [12]. We showed that Treg or the culture supernatant of Treg promoted proliferation of the murine neonatal and human fetal-like cardiomyocytes. Moreover, we further illustrated that CCL24, GAS6 and AREC directly potentiated cardiomyocyte proliferation. Although the combination of these paracrine factors (Pool 3) also significantly increased neonatal cardiomycyte proliferation when compared to the control, the order of magnitude was similar to that contributed by each of the factors alone. Therefore, there might be no synergy in using these paracrine factors for stimulating cardiomyocyte proliferation. Furthermore, we demonstrated that Treg supernatant contributed to reduced expression of cell cycle inhibitors such as p27 and p57. Future studies are needed to delineate the exact mechanisms by which Treg regulate cardiomyocyte proliferation.
In this study, we observed significantly more Treg in the regenerating P3 than the non-regenerating P8 heart at day 7 after injury. Intriguingly, the whole transcriptomes of these intracardiac Treg showed no statistical difference regardless of whether the neonatal heart regenerates. More importantly, adoptive transfer of adult Treg promoted regeneration of the neonatal NOD/SCID heart; and adult Treg or their supernatant potentiated proliferation of neonatal cardiomyocytes. Our findings may suggest that the quantity of Treg determined the outcome of heart regeneration; and the mammalian heart loses its regenerative capacity by losing the accumulation of intracardiac Treg during development. We could speculate that these intracardiac Treg might come from the circulation as demonstrated by the adoptive transfer experiments in NOD/SCID mice. Nevertheless, why there are more Treg in the injured myocardium of P3 than P8 heart awaits future investigations.
Previous study showed that Treg promote macrophage polarization towards a M2-like phenotype which is associated with increased collagen expression in scar formation during adult cardiac repair [41]. Nevertheless, it is well acknowledged that the neonatal heart displays less fibrotic responses when compared to the adult heart. In the neonatal myocardium after injury, we did not observe significant difference in accumulation of M1-like macrophages. However, there were significantly more M2-like macrophages during scar formation after Treg ablation; and significantly less M2-like macrophages during heart regeneration following adoptive transfer of Treg. In fact, M2 macrophages have been reported to be correlated with cardiac fibrosis via secretion of TGFβ, osteopontin and galatin-3 [44, 45]. Our findings demonstrated that Treg contributed to reduced accumulation of the pro-fibrotic M2-like macrophages that appeared to be less beneficial for neonatal heart regeneration.
Taken together, we have highlighted an unappreciated role of Treg in potentiating neonatal heart regeneration possibly by facilitating cardiomyocyte proliferation and reducing the accumulation of pro-fibrotic macrophages after injury. In fact, targeting Treg for tissue regeneration offers unappreciated advantages such as specificity towards autoantigens released during injury. Indeed, we have recently demonstrated that the therapeutic effect of Treg in driving vascular regeneration is localized in the injured tissues only [18]. Our findings might also resolve some of the long-standing questions in the cardiac regeneration field such as how the neonatal heart regenerates and what regulates the proliferation of neonatal cardiomyocytes during heart regeneration.
Materials and Methods
Mice
NOD.Foxp3hCD2 reporter mice were described previously [21]. ICR, NOD/SCID and C57.Foxp3DTR/GFP mice were purchased from the Jackson Laboratory. Treg depletion via the lytic anti-CD25 antibodies (clone PC61, BioxCell) was carried out by intraperitoneal (i.p.) injection at 0.5 mg/pup on days 0, 2, 4, 6 and 10 after CI. Treg ablation in C57.Foxp3DTR/GFP mice was induced by i.p. injection of diphtheria toxin (diluted in PBS, 30 ng/g body weight) every other day for 4 weeks after injury. Treg for adoptive transfer were purified from the spleen of NOD.Foxp3hCD2 mice using anti-hCD2 magnetic beads following the manufacturer's instructions (Miltenyi Biotech). Each neonatal NOD/SCID mouse was adoptively transferred with 1 million Treg through i.p. injection immediately after CI, AR or sham surgery. All animal procedures were approved by the CUHK Animal Experimentation Ethics Committee and performed in compliance with the Guide for the Care and Use of Laboratory Animals (NIH publication, eighth edition, updated 2011).
Neonatal mouse heart cryoinfarction (CI)
CI was performed as previously described [20]. Briefly, neonatal mice at P3 or P8 were subjected to anesthesia by freezing for ∼3-5 minutes, and were then placed on the frozen operation table once breathing was steady. Mouse limbs were fixed in location with forceps and 70% ethanol was applied to disinfect the surgical area. An incision (∼1 cm) was made along the sternum and vertical of the chest muscles under a stereomicroscope. Two or three intercostal incisions were made in the left sternum chest to separate the pericardium and expose the left ventricle. Blunt port copper wire (1 mm thickness) was frozen in liquid nitrogen and then put on the left ventricle in order to induce frostbite; this was maintained for ∼7-8 seconds until the left ventricle appeared white. After injury, bubbles and blood in the chest were squeezed out. The chest was closed and the skin was sewn up with 8-0 sutures. After surgery, neonatal mice were placed under a 37°C heating pad to keep warm. They were then placed back with their mothers as soon as they woke up and the skin color returned to normal. In the sham-operated control group, we performed the same experimental procedures as above except that we replaced liquid nitrogen with PBS at room temperature.
Neonatal mouse heart apical resection (AR)
AR was performed as previously described [20]. Briefly, neonatal mice at P3 were subjected to anesthesia by freezing for 3-5 minutes, and were then placed on the frozen operation table once breathing was steady. Mouse limbs were fixed in location with forceps and 70% ethanol was applied to disinfect the surgical area. An incision (∼1 cm) was made along the sternum and vertical of the chest muscles under a stereomicroscope. Two or three intercostal incisions were made in the left sternum chest to separate the pericardium and expose the apex. Curved forceps were extended into the intrathoracic to pull out the heart and the apex was then truncated with microsurgical scissors. After injury, bubbles and blood in the chest were squeezed out. The chest was closed and the skin was sewn up with 8-0 sutures. After surgery, neonatal mice were placed under a 37°C heating pad to keep warm. They were then placed back with their mothers as soon as they woke up and the skin color returned to normal. In the sham-operated control group, we performed the same experimental procedures as above except that we did not truncate the heart apex.
Echocardiography
The left ventricular function and morphology were determined at the indicated time points after CI or sham operation by echocardiography via a digital ultrasound system (Vevo770 High-Resolution Ultrasound Imaging System, VisualSonics). We have controlled the heart rate at 400 beats per minute. Our measurements of the left ventricle included: fractional shortening (FS), ejection fraction (EF), end-diastolic diameter (LVID d) and end-systolic diameter (LVID s). Analysis was performed using the VisualSonics Vevo770 software.
Immunostaining
Heart tissues were dissected and fixed in 4% paraformaldehyde at 4°C overnight. The fixed tissues were washed three times with PBS and equilibrated in 30% sucrose at 4°C for 2 days before freezing and cryosectioning. Eight micrometer frozen sections were blocked at 2% goat serum and then stained with the respective primary antibodies at 10 ug/ml at 4˚C overnight. Anti-mouse primary antibodies used: COLA1 (abcam, ab34710), cTnT (abcam, ab8295), Ki67 (eBiosciences 14-5698-82), pH3 (Millipore, 06-570) and Aurora B (abcam, ab2254). Anti-human primary antibodies used: cTnT (RnD systems, MAB1874), Ki67 (abcam, ab15580) and Aurora B (abcam, ab2254). Alexa-Fluor-488- or Alexa-Fluor-546-conjugated secondary antibodies (Invitrogen) were used at room temperature for 30 minutes in the dark. Slides were mounted with DAPI-containing fluorescence mounting medium (Dako) and fluorescence was detected with an upright fluorescence microscope, inverted fluorescence microscope or confocal microscope (all Leica). Images were processed with the ImageJ software and cTNT coverage was analyzed based on this formula: cTnT+ area/total area. For blind cell count of proliferating cardiomyocytes, contaminating cell types without cTnT expression and background staining without DAPI+ nuclei were excluded.
Masson's trichrome staining
Masson's trichrome staining was performed to determine collagen deposition per manusfacturer's instruction (Polysciences 25088-1). Briefly, frozen sections were washed in PBS and fixed in 4% paraformaldehyde for 8 min. Slides were then incubated in Bouins' solution (5% acetic acid, 9% formaldehyde and 0.9% picric acid) at room temperature overnight. The next day after washed with distilled water, slides were incubated in Weigert's iron hematoxylin solution for 10 minutes, washed and then stained with Biebrich scarlet-acid fuchsin for 5 minutes. After three washes with distilled water, slides were incubated in phosphotungstic/phosphomolybdic acid for 10 minutes followed by staining with aniline blue solution for 5 minutes. After that, slides were washed with distilled water for three times and dehydrated with ethanol and xylene based on standard procedures. Images were acquired on Nikon eclipse TE2000-S microscope. To quantify fibrosis after CI, images were analyzed by the ImageJ software and fibrosis coverage was calculated as fibrotic tissue covered length out of the total circumference of heart sections in sequentially stained sections based on this formula: scar perimeter/total perimeter [20]. For AR sections, neonatal scar size was calculated relative to the size of left ventricle as previously described [46].
Flow cytometry, cell sorting and analysis
To collect splenocytes, the excised spleen was dissociated in PBS with a syringe plunger through a 40um cell strainer to obtain single cell suspension. To study immune cell infiltrates in the neonatal heart, heart tissues were minced into small fragments and dissociated with 1:1 type II collagenase (1000 U/ml in PBS, Worthington) and dispase (11 U/ml in PBS, Gibco) at 37°C for 30 minutes. Enzymatic action was stopped by adding 10% FBS and the dissociated cells were washed twice with PBS. The dissociated single splenocytes or neonatal heart cells were removed from the contaminated erythrocytes by incubating with the red blood cell lysis buffer (eBiosciences) for 5 minutes; and were then blocked with 2% normal rabbit serum. Cells were subsequently stained with fluorochrome-conjugated antibodies against the following antigens: mCD3, mCD4, mCD8, mCD31, mCD45, mF4/80, mLy6C or hCD2 (Biolegend or eBiosciences) at a dilution of 1:100, unless specified by the manufacturer, at 4°C for 30 minutes. Murine Treg were detected with the Treg staining kit according to manusfacturer's instructions (eBioscience). Cells were then washed three times with 2% FBS-containing PBS and analyzed on flow cytometer (BD FACSAriaTM Fusion). Propidium iodide (PI, BD) positive dead cells were excluded for live cell analysis/sorting; and FACS data were then analyzed with the FlowJo software (Tree star).
Cell cultures
For Treg, naïve CD3+CD4+hCD2+ Treg were purified from the spleen of Foxp3hCD2 reporter mice by flow cytometry and were stimulated before cocultured with cardiomyocytes as described previously [47]. Briefly, each well of a 96-well plate was coated with 10 ug/ml anti-CD3 (Biolegend, 100314) and 1 ug/ml anti-CD28 (Biolegend, 102112) at 4°C overnight. The plate was then washed with PBS twice. Treg were cultured in vitro with RPMI1640 supplemented with 10% heat inactivated fetal bovine serum, 1% sodium pyruvate (Life Technologies), 10 mM HEPES (Life Technologies), 50 uM 2-mercaptoethanol (Life Technologies), 40 ng/ml IL-2 (Peprotech, 212-12) and 10 ng/ml TGFb (RnD systems, 7666-MB-005) at 37°C for 4 days before coculture experiments.
For murine neonatal cardiomyocytes, they were isolated with an enzymatic digestion approach as previously described [48]. Briefly, P1 ventricles were minced into small fragments and pre-digested in 0.05% trypsin-EDTA at 4°C overnight. The pre-digested mixture was washed with 10 mM HEPES and 1X penicillin/streptomycin-containing DMEM/F12 medium (light medium) pre-warmed at 37°C, followed by repeated digestions in a stepwise manner: the tissues were digested with 100 U/ml type II collagenase at 37°C for 10 minutes. After that, the supernatant was collected and mixed in a ratio of 1:1 with DMEM/F12 medium supplemented with 10mM HEPES, 1X penicillin/streptomycin, 10% horse serum (Invitrogen) and 5% fetal bovine serum (dark medium). The supernatant mixture was then kept on ice and the tissue pellet was further digested for 2-3 times with the same procedures until the tissues became single cells. All supernatant mixtures were then pooled together and centrifuged at 800 rpm for 5 minutes. Differential plating was performed to remove fibroblasts by resuspending the cell pellet with 10 ml dark medium followed by seeding onto a T25 flask at 37°C for 1 hour. After that, the unattached cells were transferred to a new T25 and replated at 37°C for another hour. The unattached cardiomyocytes were then centrifuged at 400 rpm for 5 minutes, and resuspended with appropriate volume of dark medium for cell counting. Cardiomyocytes were plated on Matrigel (1:100 in DMEM/F12)-coated chamber slide at a density of 10,000 cells per well and cultured in dark medium at 37°C for 24 hours. To synchronize proliferation of cardiomyocytes before experiment, they were starved with serum-free medium overnight. After that, they were cocultured with in vitro stimulated Treg in a ratio of cardiomyocytes: Treg as 3:1, Treg supernatant-containing dark medium (1:1) or 50 ng/ml murine CCL24 (Biolegend, 585102), 100 ng/ml murine GAS6 (RnD systems, 8310-GS-050), 1 ug/ml murine GRN (Lifespan biosciences, LS-G3786-10) or 100 ng/ml murine amphiregulin (RnD systems, 989-AR-100) at 37°C for 1 day before analysis. For total cardiomyocyte count, cardiomyocytes at a single-cell level were harvested by enzymatic digestion with 0.25% trypsin-EDTA for at 37°C for 5 minutes before centrifugation.
For human cardiomyocytes, cardiomyocytes were derived from hESCs as previously described [36]. Cardiomyocytes were generated with a sequential administration of growth factors (Figure 6C). Briefly, hESCs were seeded at a density of 3.75 x 104 cells/cm2 on matrigel (BD, growth factor reduced, 1:60)-coated plate in mTeSRTM (Stemcell Technologes) supplemented with 10 uM Y-27632 (Selleck Chem, S1049) and 1uM CHIR99021 (Selleck Chem,S2924). On the day of mesoderm induction, cells were overlaid with Matrigel (1:60) and 100 ng/ml Activin A (Peprotech, 120-14E) in insulin-free B27-containing RPMI1640 medium (RPMI/B27-) for 24 hours, followed by 1uM CHIR99021 and 5 ng/ml BMP-4 (Peprotech, AF-120-05ET) for 48 hours. Subsequently, the medium was refreshed and supplemented with 5 uM IWR-1 (Calbiochem, 681669) for 48 hours, followed by unsupplemented RPMI/B27- medium for 48 hours. On day 7, cardiac progenitors were expanded and maintained in insulin+ B27-containing RPMI1640 medium (RPMI/B27+). The medium was refreshed every 3 days. Beating cardiomyocytes could often be observed on day 14 of differentiation. To synchronize proliferation of cardiomyocytes, beating cardiomyocytes were starved in RPMI1640 alone for 9 hours followed by replenishment with RPMI/B27+ alone, supernatant of Treg cultures or 50 ng/ml human recombinant amphiregulin (Peprotech, 100-55B) for 3 days before analysis.
Single-cell encapsulation and library preparation
Single cells were purified by FACS sorting before library preparation and single-cell libraries were prepared with the Chromium Single Cell 3' Reagent Kits v2 (10x Genomics) as per manufacturer's instructions. Briefly, sorted cells in suspension were first prepared as gel beads in emulsion (GEMs) on Single Cell 3' Chips v2 (10x Chromium) using the Chromium Controller (10x Genomics). Barcoded RNA transcripts in each single cell were reverse transcribed within GEM droplets. cDNA was purified with DynaBeads MyOne Silane beads (Invitrogen) and then amplified for subsequent library construction. Sequencing libraries were prepared by fragmentation, end-repair, ligation with indexed adapters and PCR amplification using the Chromium Single Cell 3' library kit v2 (10x Genomics). Nucleic acid was cleaned up after each steps using SPRIselect beads (Beckman Coulter). Libraries were then quantified by Qubit and real-time quantitative PCR on a LightCycler 96 System (Roche).
Single-cell RNA-sequencing and Functional Annotations
Pooled libraries were sequenced on the Illumina HiSeq 2500 platform. All single-cell libraries were sequenced with a customized paired-end dual index format (98/26/0/8 basepair) according to manufacturer's instructions. Data were processed, aligned and quantified using the Cell Ranger Single-Cell Software Suite (v 2.1.1) [49]. Briefly, data were demultiplexed based on the 8 base-pair sample index, 16 base-pair Chromium barcodes and 10 base-pair unique molecular identifiers (UMI). After quality control, reads were aligned on Mus Musculus Cell Ranger transcriptome reference (mm10-1.2.0). Data analyses, including tSNE and graph-based clustering, were performed according to Cell Ranger's pipelines with default settings. Differentially expressed genes in each cluster relative to all other clusters were identified by Cell Ranger's pipelines with default settings (minimum mean expression = 1 and adjusted p-value cutoff = 0.05). The top N genes by log2-fold change for each cluster were further analyzed (N=10000/K^2, where K is the number of clusters). Gene ontology (GO) enrichment analyses of the cluster-specific highly expressed genes were performed by DAVID Bioinformatics Resources (v6.8) [50].
Statistical analysis
The data were expressed as arithmetic mean±S.E.M. or mean±S.D. of biological replicates (n = 5, unless otherwise specified) performed under the same conditions for in vivo or in vitro experiments, respectively. Statistical analysis was performed using the unpaired student's t-test with data from two groups; while date from more than two groups was performed using an ANOVA followed by Tukey's method for multiple comparisons. Significance was accepted when P < 0.05.
Abbreviations
Treg: regulatory T-cells; AR: apical resection; MI: myocardial infarction; CI: cryoinfarction; AREG: amphiregulin; scRNA-seq: single-cell RNA sequencing; t-SNE: t-distributed stochastic neighbor embedding; SN: supernatant; FS: fractional shortening; EF: ejection fraction; LVID d: end-diastolic diameter; LVID s: end-systolic diameter.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank Dr. Joaquim S.L. Vong, Xisheng Li, Di Liu and Bao Sheng (CUHK) for their technical help during this study. We also thank Professor Dennis Lo and Professor Rossa Chiu (CUHK) for their machinery support in experiments using single-cell RNAseq. This work was supported by Research Grants Council of Hong Kong (24110515, 14111916, C4024-16W, C4026-17WF); Health and Medical Research Fund (03140346, 04152566); Croucher Foundation (Innovation Award and Start-up Allowance); Direct Grant, Faculty Innovation Award, Seed Fund from Lui Chi Woo Institute of Innovative Medicine, postdoctoral fellowships (K.Y.Y. and R.C.Y.T) and postgraduate studentship (J.L.) from CUHK.
Contributions
J.L., R.C.Y.T. and V.C. performed experiments; H.Y.L., S.H. and B.Z. contributed reagents; J.L., K.Y.Y., R.C.Y.T. and K.O.L. analyzed data; K.O.L. designed research and wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN. et al. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331:1078-1080
2. Porrello ER, Mahmoud AI, Simpson E, Johnson BA, Grinsfelder D, Canseco D. et al. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc Natl Acad Sci U S A. 2013;110:187-192
3. Jesty SA, Steffey MA, Lee FK, Breitbach M, Hesse M, Reining S. et al. c-kit+ precursors support postinfarction myogenesis in the neonatal, but not adult, heart. Proc Natl Acad Sci U S A. 2012;109:13380-13385
4. Lavine KJ, Epelman S, Uchida K, Weber KJ, Nichols CG, Schilling JD. et al. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl Acad Sci U S A. 2014;111:16029-16034
5. Haubner BJ, Schneider J, Schweigmann U, Schuetz T, Dichtl W, Velik-Salchner C. et al. Functional Recovery of a Human Neonatal Heart After Severe Myocardial Infarction. Circ Res. 2016;118:216-221
6. Uygur A, Lee RT. Mechanisms of Cardiac Regeneration. Dev Cell. 2016;36:362-374
7. Ali SR, Hippenmeyer S, Saadat LV, Luo L, Weissman IL, Ardehali R. Existing cardiomyocytes generate cardiomyocytes at a low rate after birth in mice. Proc Natl Acad Sci U S A. 2014;111:8850-8855
8. Sattler S, Rosenthal N. The neonate versus adult mammalian immune system in cardiac repair and regeneration. Biochim Biophys Acta. 2016;1863:1813-1821
9. Aurora AB, Porrello ER, Tan W, Mahmoud AI, Hill JA, Bassel-Duby R. et al. Macrophages are required for neonatal heart regeneration. J Clin Invest. 2014;124:1382-1392
10. Li J, Tan J, Martino MM, Lui KO. Regulatory T-cells: potential regulator of tissue repair and regeneration. Frontiers in Immunology. 2018 (In press)
11. Jahn C, Weidinger G. Regulatory T Cells Know What Is Needed to Regenerate. Dev Cell. 2017;43:651-652
12. Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y. et al. A special population of regulatory T cells potentiates muscle repair. Cell. 2013;155:1282-1295
13. Kuswanto W, Burzyn D, Panduro M, Wang KK, Jang YC, Wagers AJ. et al. Poor Repair of Skeletal Muscle in Aging Mice Reflects a Defect in Local, Interleukin-33-Dependent Accumulation of Regulatory T Cells. Immunity. 2016;44:355-367
14. Ali N, Zirak B, Rodriguez RS, Pauli ML, Truong HA, Lai K. et al. Regulatory T Cells in Skin Facilitate Epithelial Stem Cell Differentiation. Cell. 2017;169:1119-1129 e1111
15. Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S. et al. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell. 2015;162:1078-1089
16. Zaiss MM, Frey B, Hess A, Zwerina J, Luther J, Nimmerjahn F. et al. Regulatory T cells protect from local and systemic bone destruction in arthritis. J Immunol. 2010;184:7238-7246
17. Dombrowski Y, O'Hagan T, Dittmer M, Penalva R, Mayoral SR, Bankhead P. et al. Regulatory T cells promote myelin regeneration in the central nervous system. Nat Neurosci. 2017;20:674-680
18. Leung OM, Li J, Li X, Chan VW, Yang KY, Ku M. et al. Regulatory T Cells Promote Apelin-Mediated Sprouting Angiogenesis in Type 2 Diabetes. Cell Rep. 2018;24:1610-1626
19. Murphy TJ, Ni Choileain N, Zang Y, Mannick JA, Lederer JA. CD4+CD25+ regulatory T cells control innate immune reactivity after injury. J Immunol. 2005;174:2957-2963
20. Yu W, Huang X, Tian X, Zhang H, He L, Wang Y. et al. GATA4 regulates Fgf16 to promote heart repair after injury. Development. 2016;143:936-949
21. Leung CS, Yang KY, Li X, Chan VW, Ku M, Waldmann H. et al. Single-cell transcriptomics reveal that PD-1 mediates immune tolerance by regulating proliferation of regulatory T cells. Genome Med. 2018;10:71
22. Lui KO, Boyd AS, Cobbold SP, Waldmann H, Fairchild PJ. A role for regulatory T cells in acceptance of ESC-derived tissues transplanted across an major histocompatibility complex barrier. Stem Cells. 2010;28:1905-1914
23. Deliyanti D, Talia DM, Zhu T, Maxwell MJ, Agrotis A, Jerome JR. et al. Foxp3(+) Tregs are recruited to the retina to repair pathological angiogenesis. Nat Commun. 2017;8:748
24. Ku M, Ke E, Sabouri-Ghomi M, Abadejos J, Freeman B, Nham A. et al. Deconstructive SCNT reveals novel Treg subsets. J Allergy Clin Immunol. 2018
25. Tsang JCH, Vong JSL, Ji L, Poon LCY, Jiang P, Lui KO. et al. Integrative single-cell and cell-free plasma RNA transcriptomics elucidates placental cellular dynamics. Proc Natl Acad Sci U S A. 2017;114:E7786-E7795
26. Baitsch D, Bock HH, Engel T, Telgmann R, Muller-Tidow C, Varga G. et al. Apolipoprotein E induces antiinflammatory phenotype in macrophages. Arterioscler Thromb Vasc Biol. 2011;31:1160-1168
27. Gleissner CA, Shaked I, Little KM, Ley K. CXC chemokine ligand 4 induces a unique transcriptome in monocyte-derived macrophages. J Immunol. 2010;184:4810-4818
28. Altmann C, Vasic V, Hardt S, Heidler J, Haussler A, Wittig I. et al. Progranulin promotes peripheral nerve regeneration and reinnervation: role of notch signaling. Mol Neurodegener. 2016;11:69
29. Kuse Y, Tsuruma K, Sugitani S, Izawa H, Ohno Y, Shimazawa M. et al. Progranulin promotes the retinal precursor cell proliferation and the photoreceptor differentiation in the mouse retina. Sci Rep. 2016;6:23811
30. Li YH, Chen HY, Li YW, Wu SY, Wangta L, Lin GH. et al. Progranulin regulates zebrafish muscle growth and regeneration through maintaining the pool of myogenic progenitor cells. Sci Rep. 2013;3:1176
31. Hunt D, Raivich G, Anderson PN. Activating transcription factor 3 and the nervous system. Front Mol Neurosci. 2012;5:7
32. Zhou J, Edgar BA, Boutros M. ATF3 acts as a rheostat to control JNK signalling during intestinal regeneration. Nat Commun. 2017;8:14289
33. Batlle M, Recarte-Pelz P, Roig E, Castel MA, Cardona M, Farrero M. et al. AXL receptor tyrosine kinase is increased in patients with heart failure. Int J Cardiol. 2014;173:402-409
34. Schreier B, Rabe S, Schneider B, Bretschneider M, Rupp S, Ruhs S. et al. Loss of epidermal growth factor receptor in vascular smooth muscle cells and cardiomyocytes causes arterial hypotension and cardiac hypertrophy. Hypertension. 2013;61:333-340
35. Jehle J, Staudacher I, Wiedmann F, Schweizer P, Becker R, Katus H. et al. Regulation of apoptosis in HL-1 cardiomyocytes by phosphorylation of the receptor tyrosine kinase EphA2 and protection by lithocholic acid. Br J Pharmacol. 2012;167:1563-1572
36. Palpant NJ, Pabon L, Friedman CE, Roberts M, Hadland B, Zaunbrecher RJ. et al. Generating high-purity cardiac and endothelial derivatives from patterned mesoderm using human pluripotent stem cells. Nat Protoc. 2017;12:15-31
37. Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science. 2002;298:2188-2190
38. Hui SP, Sheng DZ, Sugimoto K, Gonzalez-Rajal A, Nakagawa S, Hesselson D. et al. Zebrafish Regulatory T Cells Mediate Organ-Specific Regenerative Programs. Dev Cell. 2017;43:659-672 e655
39. Matsumoto K, Ogawa M, Suzuki J, Hirata Y, Nagai R, Isobe M. Regulatory T lymphocytes attenuate myocardial infarction-induced ventricular remodeling in mice. Int Heart J. 2011;52:382-387
40. Tang TT, Yuan J, Zhu ZF, Zhang WC, Xiao H, Xia N. et al. Regulatory T cells ameliorate cardiac remodeling after myocardial infarction. Basic Res Cardiol. 2012;107:232
41. Weirather J, Hofmann UD, Beyersdorf N, Ramos GC, Vogel B, Frey A. et al. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res. 2014;115:55-67
42. Zacchigna S, Martinelli V, Moimas S, Colliva A, Anzini M, Nordio A. et al. Paracrine effect of regulatory T cells promotes cardiomyocyte proliferation during pregnancy and after myocardial infarction. Nat Commun. 2018;9:2432
43. Couchie D, Lafdil F, Martin-Garcia N, Laperche Y, Zafrani ES, Mavier P. Expression and role of Gas6 protein and of its receptor Axl in hepatic regeneration from oval cells in the rat. Gastroenterology. 2005;129:1633-1642
44. Lenga Y, Koh A, Perera AS, McCulloch CA, Sodek J, Zohar R. Osteopontin expression is required for myofibroblast differentiation. Circ Res. 2008;102:319-327
45. Braga TT, Agudelo JS, Camara NO. Macrophages During the Fibrotic Process: M2 as Friend and Foe. Front Immunol. 2015;6:602
46. Bassat E, Mutlak YE, Genzelinakh A, Shadrin IY, Baruch Umansky K, Yifa O. et al. The extracellular matrix protein agrin promotes heart regeneration in mice. Nature. 2017;547:179-184
47. Fantini MC, Dominitzki S, Rizzo A, Neurath MF, Becker C. In vitro generation of CD4+ CD25+ regulatory cells from murine naive T cells. Nat Protoc. 2007;2:1789-1794
48. Zangi L, Lui KO, von Gise A, Ma Q, Ebina W, Ptaszek LM. et al. Modified mRNA directs the fate of heart progenitor cells and induces vascular regeneration after myocardial infarction. Nat Biotechnol. 2013;31:898-907
49. Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R. et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049
50. Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1-13
Author contact
![]() Corresponding author: kathyoluiedu.hk
Corresponding author: kathyoluiedu.hk