Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Results and discussions
3. Conclusions
4. Experimental Section
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(15):4354-4374. doi:10.7150/thno.35125 This issue Cite
Research Paper
Dual-responsive, Methotrexate-loaded, Ascorbic acid-derived Micelles Exert Anti-tumor and Anti-metastatic Effects by Inhibiting NF-κB Signaling in an Orthotopic Mouse Model of Human Choriocarcinoma
Lili Wei#2, Chenyuan Wang#4,9, Xianjue Chen5, Bing Yang6, Kun Shi7, Leah R. Benington8, Lee Yong Lim8, Sanjun Shi3 ![]() , Jingxin Mo1,2
, Jingxin Mo1,2 ![]()
1. Clinical Research Center for Neurological Diseases of Guangxi Province, The Affiliated Hospital of Guilin Medical University, Guilin 541001, China
2. Department of Pharmacy, The Affiliated Hospital of Guilin Medical University, Guilin 541001, China
3. Department of Pharmacy, Institute of Surgery Research, Daping Hospital, Third Military Medical University, Chongqing 400042, China
4. Department of Reproductive Medicine, The Second Affiliated Hospital of Guangzhou University of Chinese Medicine, Guangzhou 510623, China
5. School of Chemistry, University of New South Wales, Sydney, NSW 2052, Australia
6. Department of Gynecology, The Affiliated Hospital of Guilin Medical University, Guilin 541001, China
7. Department of Gynecology, Guangzhou Women and Children's Medical Center, Guangzhou 510623, China
8. Division of Pharmacy, School of Allied Health, University of Western Australia, Perth, WA 6009, Australia.
9. Sun Yat-sen University Cancer Center, State Key Laboratory of Oncology in South China, Collaborative Innovation Center of Cancer Medicine, Guangzhou 510623, China
# These authors contributed equally to this work.
Received 2019-3-22; Accepted 2019-5-22; Published 2019-6-9
Abstract

Gestational trophoblastic neoplasia (GTN), the most aggressive form of which is choriocarcinoma, can result from over-proliferation of trophoblasts. Treating choriocarcinoma requires high doses of systemic chemotherapeutic agents, which result in nonspecific drug distribution and severe toxicity. To overcome these disadvantages and enhance chemotherapeutic efficacy, we synthesized redox- and pH-sensitive, self-assembling, ascorbic acid-derived (PEG-ss-aAPP) micelles to deliver the drug methotrexate (MTX).
Methods: We developed and tested self-assembling PEG-ss-aAPP micelles, which release their drug cargo in response to an intracellular reducing environment and the acidity of the early lysosome or tumoral microenvironment. Uptake into JEG3 choriocarcinoma cancer cells was examined using confocal microscopy and transmission electron microscopy. We examined the ability of MTX-loaded PEG-ss-aAPP micelles to inhibit metastasis in an orthotopic mouse model of human choriocarcinoma.
Results: Drug-loaded micelles had encapsulation efficiency above 95%. Particles were spherical based on transmission electron microscopy, with diameters of approximately 229.0 nm based on dynamic light scattering. The drug carrier responded sensitively to redox and pH changes, releasing its cargo in specific environments. PEG-ss-aAPP/MTX micelles efficiently escaped from lysosome/endosomes, and they were effective at producing reactive oxygen species, strongly inducing apoptosis and inhibiting invasion and migration. These effects correlated with the ability of PEG-ss-aAPP/MTX micelles to protect IκBα from degradation, which in turn inhibited translocation of NF-κB p65 to the nucleus. In an orthotopic mouse model of human choriocarcinoma, PEG-ss-aAPP/MTX micelles strongly inhibited primary tumor growth and significantly suppressed metastasis without obvious side effects.
Conclusions: Our results highlight the potential of PEG-ss-aAPP micelles for targeted delivery of chemotherapeutic agents against choriocarcinoma.
Keywords: Orthotopic choriocarcinoma, Ascorbyl phosphate palmitate derivative, Methotrexate, Dual-responsive micelles, NF-κB signaling pathway
1. Introduction
Gestational trophoblastic neoplasia (GTN) is a spectrum of pregnancy-related malignant disorders, including invasive moles, gestational choriocarcinoma, placental-site trophoblastic tumors and epithelioid trophoblastic tumors [1-3]. The most aggressive form of GTN is choriocarcinoma, which grows rapidly and tends to metastasize widely at an early stage [4-5]. First-line treatment is either single-agent systemic chemotherapy or a polychemotherapy regimen of methotrexate, etoposide, actinomycin D, cyclophosphamide, and vincristine [6-7]. Although choriocarcinoma is sensitive to chemotherapy, its metastasis to distant organs such as brain, lung, liver, and kidney often leads to extremely poor prognosis [2,8], which highlights the need for novel strategies to simultaneously suppress choriocarcinoma growth and reduce its metastases.
One promising anti-cancer strategy is to induce the production of reactive oxygen species (ROS), which include the superoxide anion (O2 •_), hydrogen peroxide (H2O2), hydroxyl radical (OH·), singlet oxygen (O·) and ozone (O3) [9-10]. High ROS levels suppress cell proliferation and migration, and they activate stress-induced signaling pathways that inhibit tumor development [11-12]. At physiological concentrations, ascorbic acid (AA), more commonly known as vitamin C, acts as an anti-oxidant, but at pharmacological concentrations in tumors, it induces ROS production [13-14]. High-dose intravenous AA has shown potential as an anti-cancer treatment [15-16]. However, AA is unstable in aqueous solution, and it decomposes readily in response to heat or oxidation [17-18]. This has led researchers to develop AA derivatives that can generate more ROS. For example, 6-O-palmitoyl L-ascorbic acid generates hydrogen peroxide (H2O2) and superoxide (O2•-), inhibiting growth of mouse Ehrlich carcinoma to a greater extent than AA [19-20]. A potentially even more promising AA derivative is trisodium L-ascorbyl 2-phosphate 6-palmitate (APP), which is prepared by incorporating palmitic acid into an ascorbyl phosphate derivative [21-23]. APP has lipophilic and hydrophilic groups, allowing it to form micelles in aqueous solution, and APP can be more effective than AA at slowing tumor growth. This superiority may reflect the 2-O-phosphatidyl moiety, which makes the derivative more hydrophilic [24]. Our preliminary experiments showed that APP induce higher levels of oxygen radicals than AA, perhaps because the phosphate groups in APP carry greater negative charge than hydroxyl groups (Figure S8).
Anti-cancer agents such as APP and the anti-folate drug 5-fluorouracil, also called methotrexate (MTX), induce ROS by inhibiting signaling pathways involving NF-κB [25-27]. NF-κB, which lies inactive in the cytoplasm in complex with the inhibitor IκB, translocates upon activation to the nucleus, where it triggers synthesis of genes that can drive inflammation, proliferation, invasion and metastasis [28]. For example, NF-κB activation leads to up-regulation of matrix metalloproteinases-2 and -9, which in turn drive osteosarcoma metastasis [29]. These same two enzymes are up-regulated in many human cancers, including choriocarcinoma [30]. APP and MTX can lead to ROS accumulation and suppress IkBa phosphorylation, preventing the translocation of NF-κB to the nucleus, ultimately preventing choriocarcinoma cells from proliferating or metastasizing [25-27].
Targeting delivery of agents such as APP and MTX to tumor areas may reduce side effects and increase agent concentration at sites of disease, thereby increasing efficacy. One strategy to achieving targeted delivery is delivering agents within self-assembling micelles [31]. In such micelles, hydrophilic segments serve as a protective shell to minimize removal by the reticuloendothelial system, while hydrophobic segments act as a reservoir to contain water-insoluble drugs such as MTX. In one approach, MTX was conjugated via an ester bond to another anticancer drug, camptothecin [32]. The resulting conjugates of MTX-camptothecin self-assembled into nanoparticles that co-delivered the drugs to different sites of action, where they participated in multiple pathways to kill folate receptor-overexpressing tumor cells in a synergistic way. These micelles can accumulate passively in tumors via the enhanced permeability and retention (EPR) effect, leading to concentrations at tumor sites that are 7- to 10-fold higher than in the blood [32-33]. Micelles can be further modified to trigger drug release in response to specific stimuli, such as a change in local pH or redox potential [34]. For example, engineering a cis-aconityl linkage into the micelle can result in a 100-fold increase in drug release when the local pH changes from 7.5 to 5.5, allowing rapid release in acidic environments such as the endosome or lysosome. Engineering a disulfide linkage within micelles can keep the drug encapsulated by the carrier as long as glutathione concentration remains 2-20 µM, similar to its levels in the extracellular environment [35]. In contrast, the glutathione concentration of 20 mM typical of the cytoplasm of tumor cells triggers cleavage of the disulfide bond, releasing the drug [36-38].
Here we developed and tested self-assembling micelles, termed PEG-ss-aAPP micelles, that are based on an APP derivative and that release their drug cargo in response to redox or pH changes (Scheme 1). In this multifunctional drug delivery system, the combination of an intracellular reducing environment and the acidity of the early lysosome or tumoral microenvironment lead synergistically to the release of drug cargo. We demonstrated the ability of MTX-loaded PEG-ss-aAPP micelles to inhibit metastasis in an orthotopic mouse model of human choriocarcinoma.
Schematic illustration of the formation of pH- and redox-responsive, methotrexate-loaded, APP-derived micelles via self-assembly in aqueous solution. Also depicted is the ability of these micelles to induce ROS production and act via the NF-κB signaling pathway to inhibit tumor growth and metastasis. aAPP, cis-aconityl APP; APP, trisodium L-ascorbyl 2-phosphate 6-palmitate; MMP, matrix metalloproteinase; MTX, methotrexate; ROS, reactive oxygen species; ss, disulfide bond.
2. Results and discussions
2.1. 1H NMR of PEG-ss-aAPP building blocks
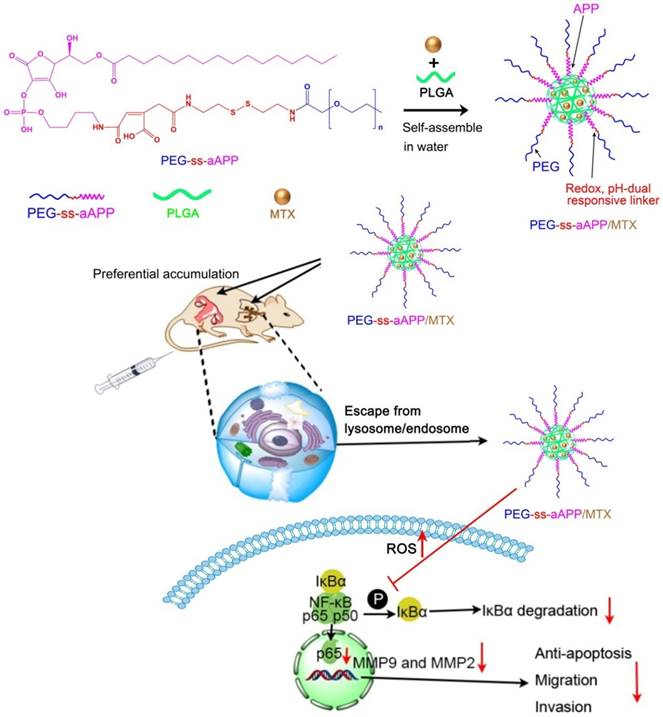
6-Pal-AA-2P, protected PEG-ss-cis-aconityl 6-Pal-AA-2P and PEG-ss-cis-aconityl 6-Pal-AA-2P palmitate ester were analyzed by 1H NMR to confirm their chemical structure (Figure 1). The following characteristic peaks were identified. (A) 6-Pal-AA-2P (CDCl3, 500 MHz): peak a, δ = 4.88 (d, 2(5H)-furanon, 1H), peak b, δ = 4.55 (m, methine, 1H), peak c, δ = 4.56, 4.21 (2t, methylene, 2H), peak d, δ = 1.26 (m, methylene, 14 × 2H), peak e, δ = 0.86 (s, methyl, 1H). (B) Protected PEG-ss-cis-aconityl 6-Pal-AA-2P (CDCl3, 500 MHz): peak a, δ = 6.32 (s, l-ethylene, 1H), peak b, δ = 5.15 (m, 1, 3 (5H)-dioxolan, 1H), peak b`, b, δ = 5.06 (d, 2(5H)-furanon, 1H), peak c, δ = 4.20, 4.00 (2t, 1, 3 (4H)-dioxolan, 2H), peak d, δ = 3.70 (m, methylene, 27 × 2H), peak e, δ = 1.44, 1.37 (2s, methyl, 2H). (C) PEG-ss-cis-aconityl 6-Pal-AA-2P palmitate ester (CDCl3, 500 MHz): peak a, δ = 6.32 (s, l-ethylene, 1H), peak b, δ = 4.84 (d, 2(5H)-furanon, 1H), peak c, δ = 4.57 (2d, methine, 1H), peak d, δ = 4.55, 4.23 (2t, methylene, 2H), peak e, δ = 3.70 (m, methylene, 27 × 2H), peak f, δ = 1.26 (m, methylene, 14 × 2H).
1H NMR spectrum of (A) 6-Pal-AA-2P (APP), (B) protected PEG-ss-cis-aconityl 6-Pal-AA-2P and (C) PEG-ss-cis-aconityl 6-Pal-AA-2P palmitate ester (PEG-ss-aAPP).
The characteristic methylene peak on palmitic acid was observed at about δ = 1.26 ppm (m, methylene, 14 × 2H; peak d; Figure 1A). The spectrum of protected PEG-ss-cis-aconityl 6-Pal-AA-2P confirmed the introduction of the cis-aconityl group because of the presence of peaks at about δ = 6.32 ppm due to the l-ethylene group in the cis-aconityl linkage (peak c, [1H]; Figure 1B). In the spectrum of PEG-ss-cis-aconityl 6-Pal-AA-2P palmitate ester, the characteristic methylene peak on palmitic acid re-occurred with the same chemical shift at δ = 1.26 ppm (m, methylene, 14 × 2H) (peak f; Figure 1C). These 1H NMR results indicate that the desired compounds were successfully synthesized.
L-ascorbic acid-2-phosphate is one of the most promising L-ascorbic acid derivatives, with perhaps the strongest ability to inhibit cancerous cell transformation [21]. This derivative can be acylated with nontoxic long-chain fatty acids to improve its lipophilicity. In our case, we prepared PEG-ss-cis-aconityl 6-Pal-AA-2P palmitate ester and examined its release under different conditions as well as its ability to stimulate ROS production.
2.2. PEG-ss-aAPP micelle characterization and MTX loading
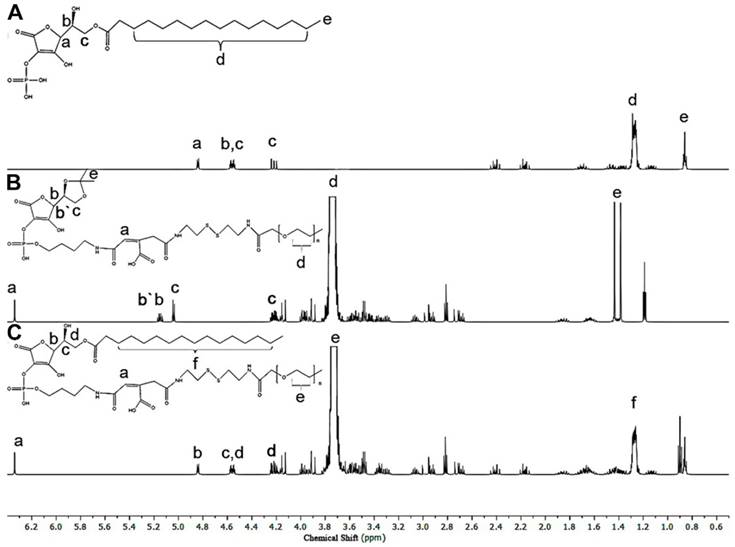
Micelle size and size distribution were examined using dynamic laser scattering. Average size was 186.6 ± 9.6 nm (PDI = 0.281) for APP/MTX micelles and 229.1 ± 5.6 nm (PDI = 0.221) for PEG-ss-aAPP/MTX micelles, suggesting a narrow size distribution (Figure 2A and B). Transmission electron micrographs revealed that APP/MTX and PEG-ss-aAPP/MTX micelles were spherical and had a size consistent with the results of dynamic light scattering (Figure 2C and D). MTX loading content and loading efficiency were, respectively, 8.57 ± 0.8 mg/g and 95.6 ± 7.1% for APP/MTX micelles, and 11.2 ± 1.8 mg/g and 98.3 ± 12.4% for PEG-ss-aAPP/MTX micelles. In scanning electron micrographs, APP/MTX micelles (Figure 2E) and PEG-ss-aAPP/MTX micelles (Figure 2F) appeared as smooth and round particles, with diameters that were consistent with the data from dynamic light scattering and transmission electron microscopy.
Thin-film method to prepare methotrexate-loaded APP (APP/MTX) micelles and methotrexate-loaded PEG-ss-aAPP (PEG-ss-aAPP/MTX) micelles. Size distribution of (A) APP/MTX micelles and (B) PEG-ss-aAPP/MTX micelles, based on dynamic light scattering. Transmission electron micrographs of (C) APP/MTX micelles and (D) PEG-ss-aAPP/MTX micelles. Scale bar, 500 nm; Scanning electron micrographs of (E) APP/MTX micelles and (F) PEG-ss-aAPP/MTX micelles. Scale bar, 200 nm.
2.3. In vitro stability and release profiles of PEG-ss-aAPP micelles in different media
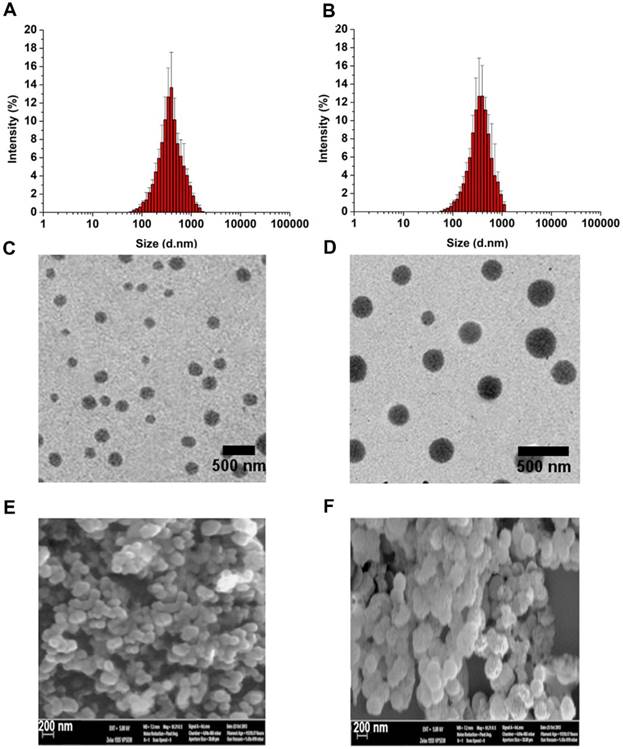
The effects of the cis-aconityl linkage and disulfide bond on drug release behavior of the PEG-ss-aAPP micelles were evaluated at two pH values in the presence or absence of dithiothreitol (DTT) (Figure 3A). At neutral pH without DTT, micelles showed biphasic release of MTX with an initial burst of about 20% of MTX within 2 h, followed by stable release that lasted 2 h. Adding 20 mM DTT increased the cumulative MTX release to 40%. Reducing the pH and adding DTT significantly increased MTX release to 80% within 12 h, which was double the amount of MTX released after only a change in pH or addition of DTT on its own. These results indicate that pH-dependent breaking of the cis-aconityl linkage and reduction of the disulfide bond synergistically promoted MTX release.
Stability of APP/MTX and PEG-ss-aAPP/MTX micelles and MTX release profiles under different pH and redox conditions. (A) Percentages of MTX released from APP/MTX and PEG-ss-aAPP/MTX micelles were compared at the same time points. At 37 °C with stirring (100 rpm), micelles were exposed to PBS (pH 7.4) for the first 4 h, then 20 mM dithiothreitol for the next 32 h, after which pH was adjusted to 5.5 by addition of acetate buffer. The DTT concentration-dependent responsiveness of PEG-ss-aAPP/MTX micelles. Changes in the following parameters of PEG-ss-aAPP/MTX micelles were measured during incubation in 0.9 % saline for up to 96 h: (B) size distribution in the presence of DTT (48-h incubation), (C) mean diameter in the presence of DTT (48-h incubation), (D) size distribution, (E) mean particle size, (F) polydispersity index (PDI) and (G) zeta potential. Results are mean ± SD (n = 3).
MTX was released much more slowly from APP/MTX micelles than from PEG-ss-aAPP micelles: only 30% of MTX was released from APP micelles by 72 h, regardless of pH or presence of DTT. The MTX release curve was slightly steeper at pH 5.5 than at pH 7.4, but it was unaffected by addition of DTT.
We created a derivative that responds to pH and redox changes, allowing potent MTX release from micelles in certain microenvironments. Our use of a redox-sensitive linkage in PEG-ss-aAPP/MTX micelles promotes drug release specifically in tumor microenvironments, where glutathione is much more concentrated (~20 mM) than in the circulation (2-20 µM). At the same time, our micelles contain an acid-responsive linkage that leads to much greater drug release in the acidic interior of lysosomes (pH 5.5) than in the circulation (pH 7.4).
Dynamic light scattering showed the PEG-ss-aAPP/MTX micelles to have a mean diameter of 235.3± 14.0 nm and a narrow size distribution after 48 h incubation in PBS (Figure 3B). Adding DTT at 10 or 15 mM decreased mean micelle size (Figure 3C), reflecting the production of ROS that hydrolyzed the disulfide bond in the micelles. DTT at 20 mM caused the breakup of PEG-ss-aAPP/MTX micelles into smaller particles, leading to even more drug release.
There were no significant changes in mean particle size upon incubation of the micelles in 0.9% saline for up to 96 h (p > 0.05), but the size distribution broadened (Figure 3D and E). Moreover, the polydispersity index and zeta potential increased with extended incubation times (Figure 3F, G), indicating that the PEG-ss-aAPP/MTX micelles were not stable beyond 48 h and therefore had to be used soon after preparation or lyophilized for storage.
PEG-ss-aAPP/MTX micelles were less stable to hydrolysis than APP/MTX micelles (Figure S9). After 48-h incubation with 20 mM DTT, PEG-ss-aAPP/MTX micelles disintegrated into smaller particles, whereas the size distribution of APP/MTX micelles remained narrow. We hypothesize that the ROS-mediated hydrolysis of disulfide bonds disrupted the PEG-ss-aAPP/MTX micelle assembly, leading to higher release of its MTX payload.
2.4. Micelle uptake in vitro
JEG3 cells were treated with micelles loaded with the hydrophobic near-infrared fluorescent dye Cy 5.5 and then stained with Lysotracker Green, a specific marker for acidic organelles. The two fluorescence signals were used to examine uptake of the micelles and their intracellular localization under the fluorescence microscope (Figure 4A). After 24-h incubation, most red fluorescence due to free Cy 5.5 overlapped with green fluorescence. Intracellular fluorescence of Cy 5.5 was much greater when the dye was conjugated and encapsulated in micelles, especially PEG-ss-aAPP/Cy 5.5 micelles (Figure 4A). In cells treated with PEG-ss-aAPP/Cy 5.5 micelles, endosome/lysosomes were fragmented and Cy 5.5 had leaked into the cytoplasm.
Effects of treating JEG3 cells for 24 h with different micelle formulations. (A) JEG3 cells were observed by confocal microscopy. Lysosomes were detected using Lyso-Tracker Green. Scale bar, 100 µm. (B) JEG3 cells were observed by transmission electron microscopy. Red arrows indicate efflux of free MTX and MTX-loaded micelles. Scale bar, 2 µm.
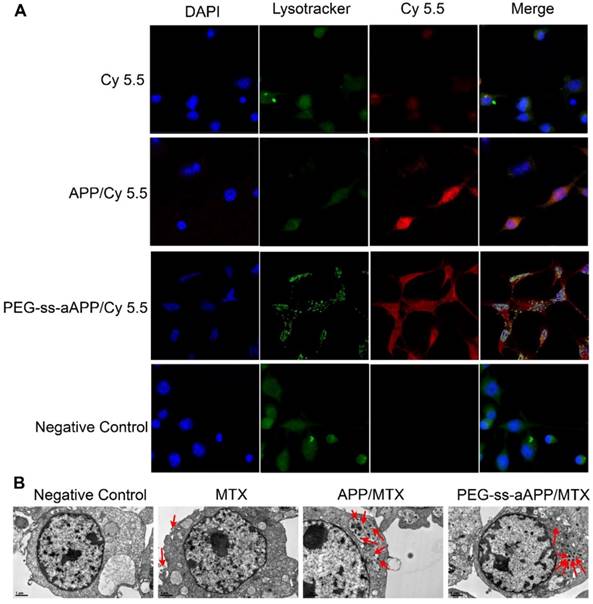
For more detailed understanding of the intracellular distribution of micelles, JEG3 cells were treated with free MTX, APP/MTX micelles or PEG-ss-aAPP/MTX micelles, then examined using transmission electron microscopy (Figure 4B). Treating cells with free MTX led to blebbing, which may contain MTX debris. Most APP/MTX signal was contained in the single-layer membrane of endosome/lysosomes, while most PEG-ss-aAPP/MTX signal was detected in the cytoplasm, which implies that acid-sensitive PEG-ss-aAPP micelles may help MTX escape from endosome/lysosomes.
Drug-delivery micelles should ideally escape endosomes efficiently after they have been internalized by target cells, since such escape allows them to avoid degradation in the lysosome or recycling out of the cell. We found that co-localization with lysosomes was less extensive for PEG-ss-aAPP/Cy 5.5 and PEG-ss-aAPP/MTX micelles than for APP/Cy 5.5 micelles (Fig. 4). These results suggest that our micelle system can facilitate escape from endosomes. This may be because dissociation of the disulphide bond and cis-aconityl linker in the dual-responsive PEG-ss-aAPP/MTX micelles disturbs the homeostasis of endosome/lysosomes.
2.5. Intracellular ROS or glutathione
We measured dichlorofluorescein fluorescence in order to quantify cellular ROS generation in response to free MTX, APP/MTX micelles or PEG-ss-aAPP/MTX micelles (Figure 5A and B). ROS levels were 3.2-fold higher after treatment with PEG-ss-aAPP/MTX micelles than in control cells. This increase in ROS was probably due to the relatively rapid release of APP triggered by endogenous glutathione or low pH. ROS levels were lower after treatment with APP/MTX micelles than after treatment with PEG-ss-aAPP/MTX micelles, probably because APP/MTX micelles were trapped to a greater extent in lysosomes.
Confocal microscopy of intracellular (A) ROS (green) and (C) glutathione (GSH, green) in JEG3 cells treated for 12 h with different formulations. Levels of (B) intracellular ROS and (D) glutatione were quantified.
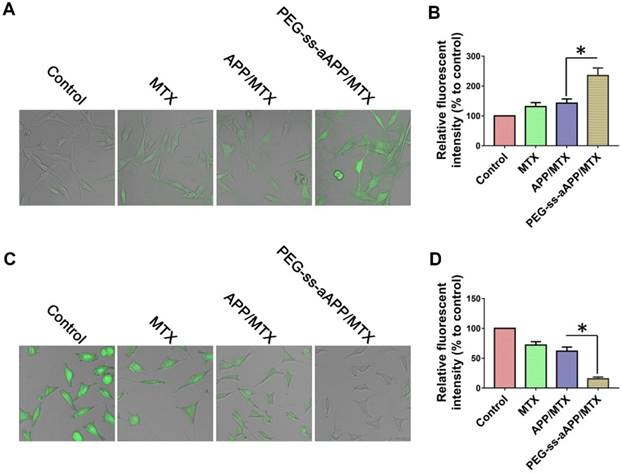
Levels of glutathione inside JEG3 cells after treatment with PEG-ss-aAPP/MTX micelles were approximately 5-fold lower than levels in untreated control cells and 3-fold lower than levels in cells treated with free MTX (Figure 5C and D). The high glutathione levels in tumors, which are thought to help mediate multidrug resistance, may help drive selective drug release from PEG-ss-aAPP micelles.
We found that PEG-ss-aAPP/MTX micelles depleted JEG3 cells of glutathione and induced ROS production, leading to cell death. This is consistent with the ability of MTX to competitively inhibit the generation of NADPH via the folate pathway. NADPH allows tumor cells, including choriocarcinoma cells, to cope with oxidative stress by scavenging radicals with glutathione. This can lead to resistance against MTX-based drugs.
2.6. In vitro cytotoxicity, anti-cancer and pro-apoptotic effects
The cytotoxicity of free MTX, APP/MTX micelles and PEG-ss-aAPP/MTX micelles was investigated in JAR and JEG3 cell lines using the MTS assay. Free MTX reduced the viability of both cell types after 48 h in a concentration-dependent manner (Figure 6A and C). While free MTX showed some anti-proliferative effects against both cell types, MTX encapsulated in micelles showed much greater therapeutic efficacy: proliferation after 48-h treatment with APP/MTX micelles was 44% of the control level, while proliferation after treatment with PEG-ss-aAPP/MTX micelles was 31% of the control level at an equivalent concentration of 1 μM (Figure 6A and C). In JAR cells, IC50 was 4.40 ± 0.34 for MTX, 0.61 ± 0.06 for APP/MTX micelles and 0.19 ± 0.01 μM for PEG-ss-aAPP/MTX micelles (Figure 6B). Similar trends were observed in JEG3 cells (Figure 6D), where IC50 was much higher for MTX (3.80 μM) than for PEG-ss-aAPP/MTX micelles (0.13 μM). These results suggest that PEG-ss-aAPP can synergize with MTX to slow cancer cell proliferation.
Effects of MTX, APP/MTX micelles and PEG-ss-aAPP/MTX micelles on viability, apoptosis and cell cycle distribution of JAR and JEG3 cells. (A-D) Dose-response curves and IC50 values. (E, G) Effects on apoptosis. (F, H) Effects on cell cycle distribution.
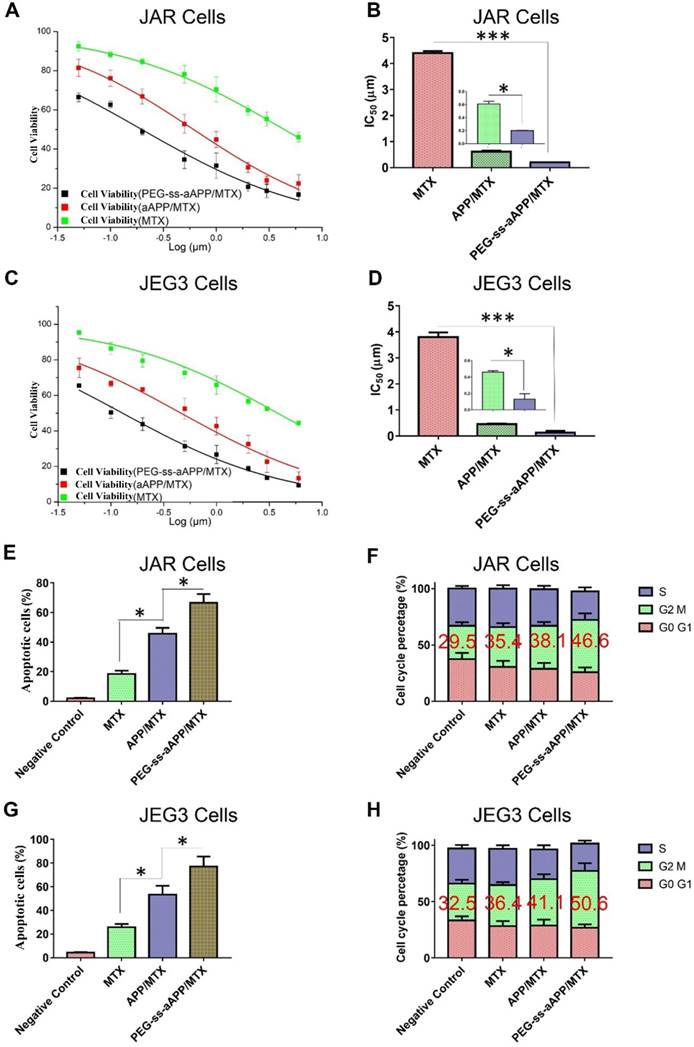
Treating JAR cells with PEG-ss-aAPP/MTX micelles for 48 h led to arrest of 46.6 ± 5.71% of cells in the G2/M-phase, compared to 35.4 ± 3.56% of cells after treatment with free MTX (p = 0.0314; Figure 6F). Similarly, treating JEG3 cells with PEG-ss-aAPP/MTX micelles led to arrest of 50.6 ± 6.64% of cells in G2/M-phase, compared to 36.4±2.75% with free MTX (p = 0.0298). The proportions of arrested cells after treatment with APP/MTX micelles were similar to the proportions after treatment with free MTX. These results suggest that mitotic arrest helps to explain the observed ability of micelle-encapsulated MTX to inhibit tumor cell proliferation.
The high efficacy of PEG-ss-aAPP/MTX micelles was confirmed in flow cytometric analysis: 3 μM MTX led to proportions of apoptotic JAR and JEG3 cells of 18.4 ± 2.4% and 25.7 ± 2.8%, respectively, compared to 45.7 ± 3.9% and 53.4 ± 7.5% with 3 μM APP/MTX micelles (Figure 6E and G). In other words, loading MTX into redox-responsive micelles increased efficacy more than 2-fold. Adding a pH-responsive release element to the micelles further increased the apoptotic efficacy to 66.7 ± 5.8% in JAR cells and 77.2 ± 8.4% in JEG3 cells, which was at least 3-fold higher than with free MTX.
2.7. Effects of PEG-ss-aAPP/MTX micelles on tumor cell migration and invasion
A wound healing assay was carried out to examine whether different formulations hindered JEG3 cells from migrating into the scratched area (Figure 7A and B). While the wound was healed nearly completely in the negative control group after 24 h, the wound area was still 47.7 ± 4.2% of the baseline area in cultures treated with free MTX, 54.0 ± 6.6% in cultures treated with APP/MTX micelles, and 82.8 ± 8.3% in cultures treated with PEG-ss-aAPP/MTX micelles. This indicates that the MTX-loaded micelles inhibited tumor cell migration.
Effects of PEG-ss-aAPP/MTX micelles on invasion and migration of JEG3 cells. (A) Cells were scratched with a pipette tip, photographed (0 h), then treated for 24 h with 3 μM free MTX, APP/MTX micelles or PEG-ss-aAPP/MTX micelles. Control cells were left untreated. Cell migration was assessed using phase-contrast microscopy. Scale bar, 100 µm. (B) Quantitation of gap area. Ratios of the gap area relative to the gap at 0 h were plotted. (C-D) Typical images and quantitation of inhibition of JEG3 cell invasion after the treatments in panel (A). Magnification, 40×.
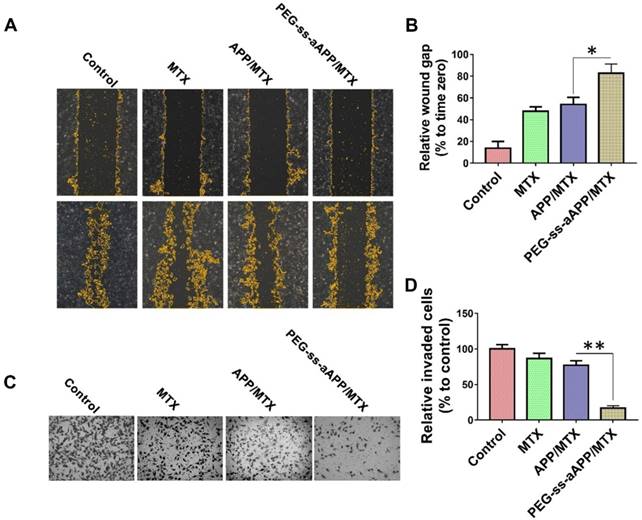
To examine the effect of different formulations on invasiveness of JEG3 cells in a three-dimensional setting, a Boyden transwell chamber assay was performed. The numbers of invasive cells in cultures treated with free MTX, APP/MTX micelles, or PEG-ss-aAPP/MTX micelles were, respectively, 86.7 ± 7.2%, 77.2 ± 6.4% and 16.8 ± 3.3% of the number in the control group (Figure 7C and D).
2.8. Effects of PEG-ss-aAPP/MTX micelles on NF-κB activation in JAR and JEG3 cells
The degradation of IκBα is a crucial step in the translocation of NF-κB into the nucleus, where it acts as a transcription factor to up-regulate genes related to cancer progression and metastasis. To investigate whether PEG-ss-aAPP/MTX micelles affect this translocation, we measured levels of total IκBα and phospho-IκBα in cells treated for 24 h with 3 μM free MTX, APP/MTX micelles or PEG-ss-aAPP/MTX micelles. Western blotting of total cell lysates showed that levels of phospho-IκBα were significantly lower in cells treated with PEG-ss-aAPP/MTX micelles than in other cultures (Figure 8A and B). Consistent with an inhibitory effect on NF-κB translocation, PEG-ss-aAPP/MTX micelles were associated with significantly lower levels of nuclear p65 than other treatments (Figure 8A and B). Immunofluorescence assays confirmed that 6-h treatment with PEG-ss-aAPP/MTX micelles (3 μM MTX) led to significantly lower amounts in the nucleus, such that most p65 was in the cytoplasm (Figure 8C).
Effects of PEG-ss-aAPP/MTX micelles on NF-κB activation in JAR and JEG3 cells. (A) Levels of IκBα and p-IκBα in total cell lysate of JAR and JEG3 cells, determined by Western blotting. (B) Levels of p65 in cytoplasmic and nuclear fractions, determined by Western blotting. Levels were quantitated relative to the levels of histone H3 and β-actin; values are mean ± SD of three independent experiments. *p < 0.05 vs. control group. (C) Localization of NF-κB immunofluorescence. JAR and JEG3 cells were treated with 3 μM PEG-ss-aAPP/MTX micelles for 6 h, and p65 was detected based on fluorescence. The experiment was performed three times with similar results, and typical results are shown. (D) Inhibition of NF-κB activity by PEG-ss-aAPP/MTX micelles based on a luciferase reporter assay. Values are mean ± SD of three independent experiments. *p < 0.05 vs TNF-α group.
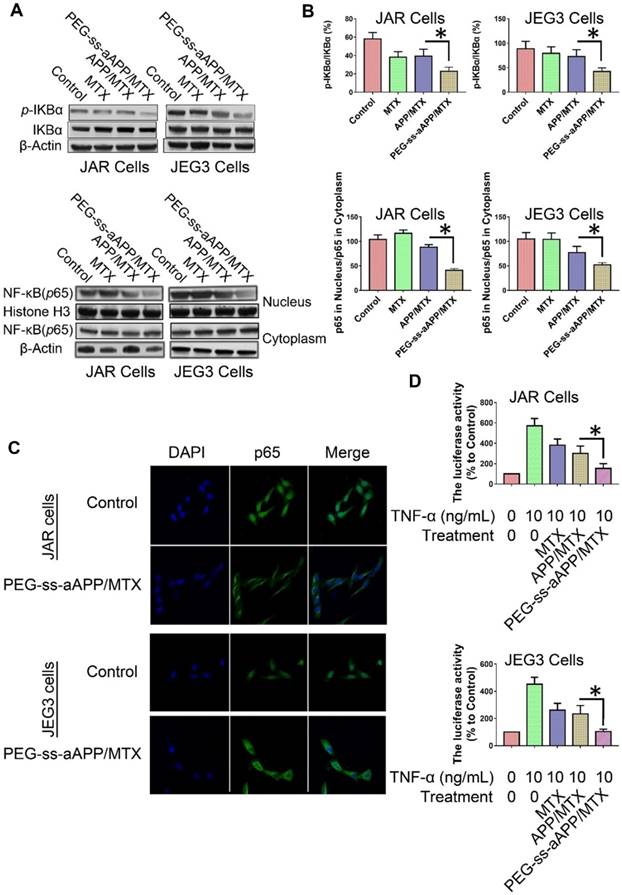
These experiments examined only the intracellular localization of NF-κB. Next, we examined the functional consequences of the micelle-mediated inhibition of NF-κB translocation. JAR and JEG3 cells were first transfected with a luciferase reporter under control of NF-κB, then treated with free MTX, APP/MTX micelles or PEG-ss-aAPP/MTX micelles. At 2 h later, cells were treated with TNF-α to activate NF-κB. Luciferase activity was significantly lower in cells treated with PEG-ss-aAPP/MTX micelles than in other treatment groups (Figure 8D), reflecting sequential inhibition of IκBα phosphorylation and p65 translocation to the nucleus.
2.9. Effects of PEG-ss-aAPP/MTX micelles on human choriocarcinoma cells involve inhibition of NF-κB signaling
To examine directly whether the inhibition of NF-κB signaling helps mediate the observed ability of PEG-ss-aAPP/MTX micelles to induce apoptosis and inhibit migration and invasion by human choriocarcinoma cells, p65 expression was transiently silenced in JEG3 cells using siRNA. After confirming that the siRNA reduced p65 expression below the expression in cells transfected with scrambled siRNA (Figure 9A), we found that such knockdown was associated with greater apoptosis (Figure 9B) and with lower invasion (Figure 9C) and migration (Figure 9D). In addition, p65 knockdown was associated with lower expression of MMP-2 and MMP-9 (Figure 9E and F).
NF-κB inhibition helps mediate the ability of PEG-ss-aAPP/MTX micelles to inhibit migration/invasion and promote apoptosis in human choriocarcinoma cells. (A) Knockdown of p65 expression by siRNA in JAR and JEG3 cells was confirmed by Western blotting. (B) Effects of p65 knockdown on the ability of PEG-ss-aAPP/MTX micelles to inhibit proliferation of JAR and JEG3 cells. Cells were transiently transfected with anti-p65 siRNA for 24 h, then treated with vehicle (0.1% DMSO) or 3 μM PEG-ss-aAPP/MTX micelles. Proliferation was determined using the MTT assay; values are mean ± SD of three independent experiments (n = 6). (C-D) Effects of p65 knockdown on the ability of PEG-ss-aAPP/MTX micelles to inhibit cell migration and invasion in cells treated as described in panel (B). Invasion was assessed using a wound healing assay, while migration was assessed using a transwell assay with or without Matrigel. (E-F) Effects of p65 knockdown on expression of MMP-2 and -9 in cells treated as described in panel (B). Transcript levels were assayed using RT-PCR; values are mean ± SD of three independent experiments. *p < 0.05, ***p < 0.001 vs the corresponding group.
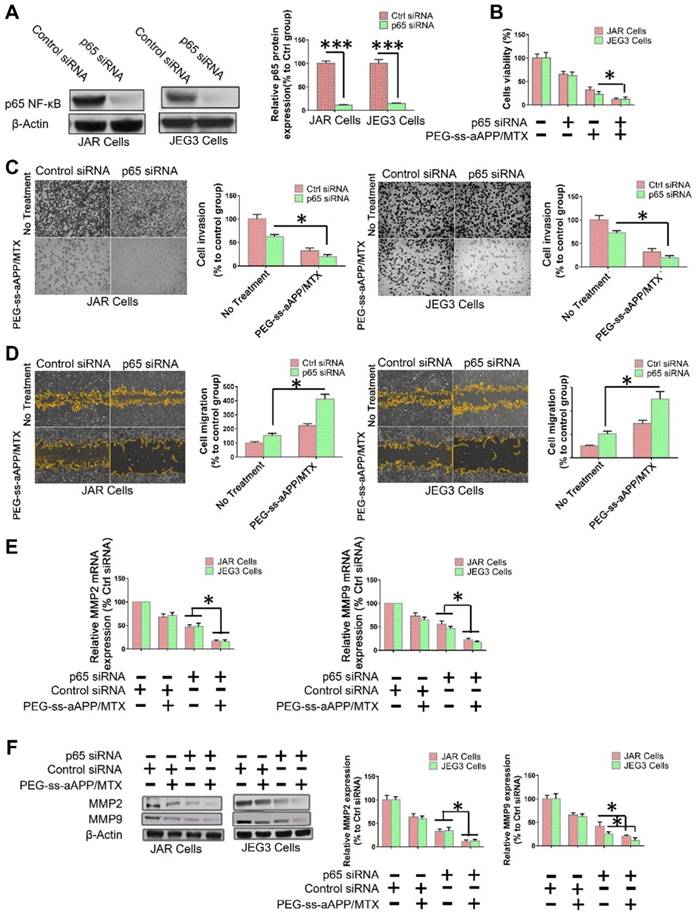
PEG-ss-aAPP/MTX micelles show strong potential for inhibiting metastasis in human choriocarcinoma cells by inhibiting NF-κB signaling. NF-κB controls the transcription of several genes involved in inflammatory responses, cell survival, apoptosis and metastasis. It up-regulates MMP-2 and -9 during choriocarcinoma progression and metastasis, which facilitates migration and invasion. We showed by Western blotting and immunofluorescence that PEG-ss-aAPP/MTX micelles led to lower phosphorylation of IκBa and thereby lower nuclear accumulation of NF-κB p65. As a result, MMP-2 and MMP-9 were down-regulated, making cells less invasive. In addition, cells became more apoptotic, consistent with the oncogenic ability of NF-kB to inhibit apoptosis. Indeed, we found that anti-p65 siRNA led to greater apoptosis following treatment with PEG-ss-aAPP/MTX micelles.
2.10. Biodistribution of PEG-ss-aAPP/Cy 5.5 ex vivo
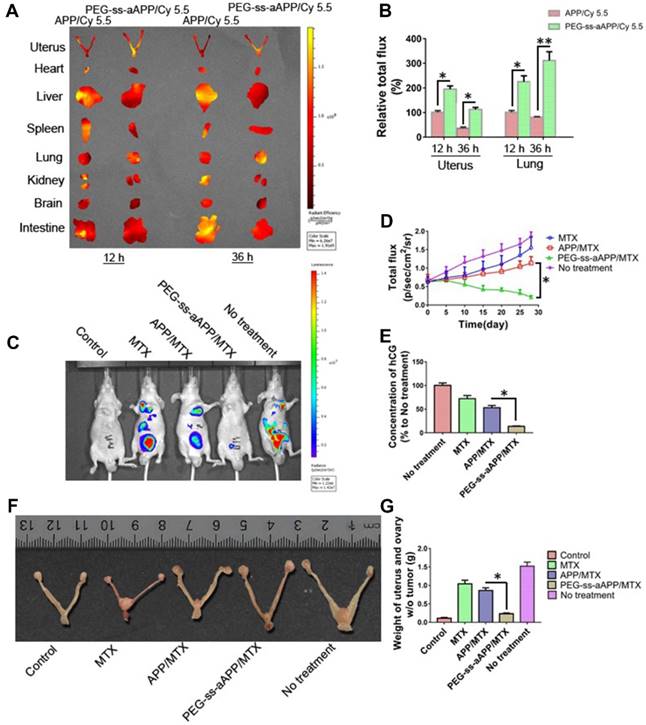
The anti-tumor efficacy of PEG-ss-aAPP/MTX micelles was investigated in vivo using an orthotopic uterus tumor xenograft model in nude mice. As a first analysis after setting up this model, luciferase-expressing JEG3 cancer cells (JEG3/luc) were injected into animals and allowed to grow into orthotopic tumors for approximately one week. First, we confirmed the success of xenograft growth by imaging luciferase fluorescence in vivo. Second, we treated tumor-bearing animals with APP and PEG-ss-aAPP micelles loaded with Cy 5.5 instead of MTX, sacrificed the animals, and analyzed tumors and normal tissues ex vivo. At 12 and 36 h post-injection, PEG-ss-aAPP/Cy 5.5 micelles were associated with markedly higher fluorescence intensity in tumor regions of interest (ROIs) than APP/Cy 5.5 micelles (Figure 10A). In contrast, at the same time points, PEG-ss-aAPP/Cy 5.5 micelles were associated with negligible fluorescence intensity in the kidney and with much lower intensity in the liver than APP/Cy 5.5 micelles. PEG-ss-aAPP/Cy 5.5 micelles sustainably accumulated in lung tissue. The accumulation of these micelles in tumor and lung likely reflects their large size and persistence in systemic circulation, the EPR effect, and the responsiveness of micelles to pH and redox potential.
(A) Fluorescence imaging of uterus with tumors and major organs from JEG3 tumor-bearing nude mice at 12 and 36 h after they were injected with APP/Cy 5.5 or PEG-ss-aAPP/Cy 5.5 micelles. (B) Semi-quantitative analysis of APP/Cy 5.5 and PEG-ss-aAPP/Cy 5.5 accumulation in tumors and organs at 12 and 36 h after injection (n = 3). (C) Growth of tumors in animals treated with free MTX, APP/MTX micelles, or PEG-ss-aAPP/MTX micelles based on bioluminescence imaging. (D) Quantification of total bioluminescence in the animals in panel C. (E) Serum concentrations of human chorionic gonadotropin (hCG). (F) Typical image of uterus from healthy nude mice and JEG3 tumor-bearing nude mice after the last treatment. (G) Weight of excised uterus after the last treatment.
2.11. Anti-tumor efficacy of PEG-ss-aAPP/MTX in vivo
Next, we measured tumor growth during 28 days after tail vein injection with PBS, free MTX, APP/MTX micelles or PEG-ss-aAPP/MTX micelles (equivalent dose of 5 mg/kg MTX). MTX inhibited tumor growth to a greater extent when delivered in micelles than when delivered free, and its efficacy was greater with PEG-ss-aAPP micelles than with APP micelles (Figure 10C and D). By the end of the experiment, the volume of tumor ROIs was only approximately 30% of the pretreatment volume in animals treated with PEG-ss-aAPP/MTX micelles. The final ROI volume was larger than pretreatment volume by 5.2-fold in animals treated with PBS, 4.3-fold in animals treated with free MTX and 2.2-fold in animals treated with APP/MTX micelles.
To confirm successful establishment of the orthotopic model of human choriocarcinoma, we examined levels of human chorionic gonadotropin (hCG), which serves as a tumor marker of choriocarcinoma. As expected, hCG level was high in the untreated group (Figure 10E), suggesting that our tumor model was successful. The concentration of hCG in animals treated with PEG-ss-aAPP/MTX micelles was only 10% of the level in the untreated group and 25% of the level in the APP/MTX treatment group, suggesting that PEG-ss-aAPP/MTX micelles showed the greatest treatment efficacy.
These findings based on measurement of tumor ROIs and hCG levels were confirmed by measurement of tumor weight, which was significantly lower in animals treated with PEG-ss-aAPP/MTX micelles than in other animal groups (p < 0.05; Figure 10G). Figure 10F shows typical images of excised uterus and ovary on day 28.
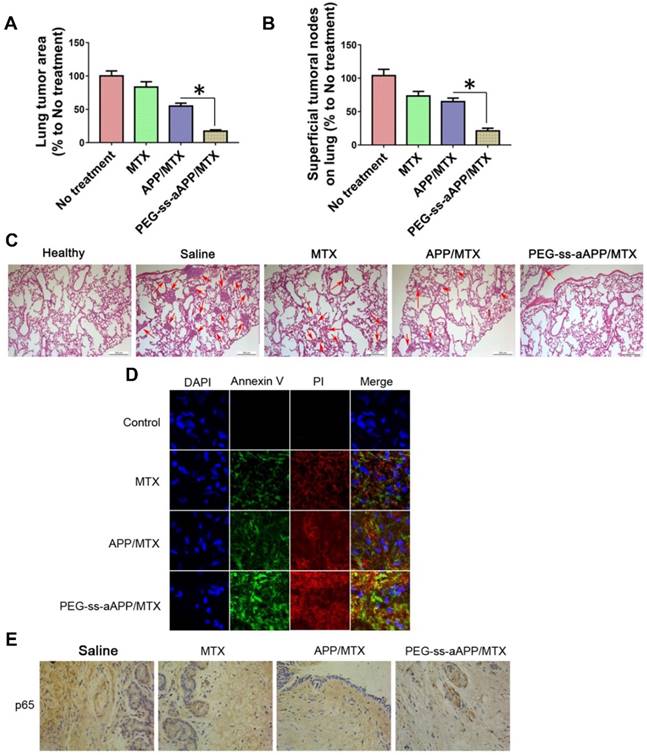
The number and area of metastatic superficial lung tumors were significantly lower in animals treated with PEG-ss-aAPP/MTX micelles than in other animal groups (p < 0.05; Figure 11A and B). In addition, metastatic lung tumors from animals treated with PEG-ss-aAPP/MTX micelles contained greater proportions of cells in early or late apoptosis than tumors from other animal groups (Figure 11C and D). Immunohistochemistry showed that PEG-ss-aAPP/MTX micelles were associated with significantly lower p65 expression in metastatic lung tumors (Figure 11E).
Anti-metastatic and pro-apoptotic effects of PEG-ss-aAPP/MTX micelles in vivo. (A) Area of superficial lung tumors, quantified with respect to untreated tumor-bearing control animals (defined as 100%). (B) Number of superficial lung tumors, quantified as in panel (A) (n = 5). (C) Representative sections of lung tissues stained with hematoxylin-eosin. Red arrows indicate tumors. Scale bar, 500 μm. (D) Staining of tumors collected on day 36 and stained to detect cells in early apoptosis (annexin V) or late apoptosis (propidium iodide, PI). (Magnification, 200) (E) Immunohistochemistry of tumor sections against p65.
2.12. Preliminary evaluation of toxicity in vivo
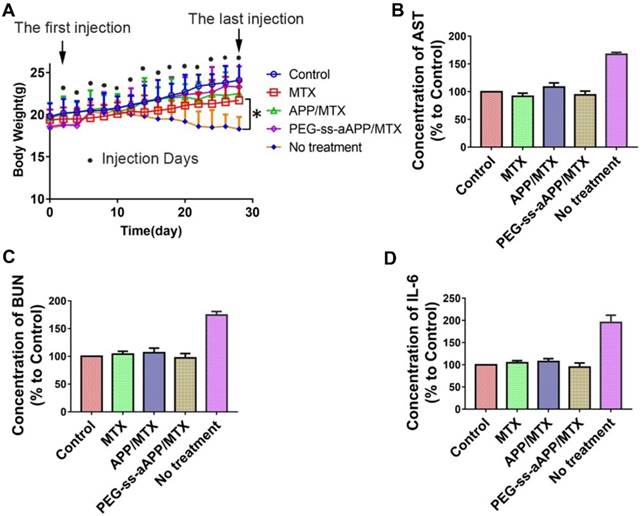
Treating our mouse model with free MTX, APP/MTX micelles or PEG-ss-aAPP/MTX micelles did not induce death and led to similar body weight as in healthy control animals. In contrast, the untreated tumor-bearing control animals lost significant body weight (Figure 12A). All three treatments were associated with levels of liver function (based on levels of aspartate aminotransferase) and kidney function (based on blood urea nitrogen) that were similar to the levels in healthy control animals (Figure 12B and C). These levels were significantly lower than in untreated tumor-bearing control animals (p < 0.05). Serum levels of IL-6 were not significantly different between animals treated with PEG-ss-aAPP/MTX micelles or healthy control mice (p > 0.05), and these levels were significantly lower than those in untreated tumor-bearing mice.
Preliminary safety evaluation of PEG-ss-aAPP/MTX micelles. Animals were treated intravenously as shown and examined 6 h later. (A) Average body weight of mice during the experiment; values are mean ± SD (n=6). (B-C) Levels of aspartate aminotransferase (AST) and blood urea nitrogen (BUN). (D) Serum concentrations of IL-6.
3. Conclusions
We demonstrate that pH and redox-responsive PEG-ss-aAPP/MTX micelles can inhibit the proliferation of human choriocarcinoma by inducing apoptosis, and they can inhibit choriocarcinoma metastasis by suppressing migration and invasion. These effects appear to be mediated, at least in part, through inhibition of NF-κB signaling. Therapeutic efficacy in the absence of major side effects was confirmed in an orthotopic choriocarcinoma mouse model. Our findings show strong potential of PEG-ss-aAPP/MTX micelles as a drug delivery system against choriocarcinoma.
4. Experimental Section
Reagents
Poly (DL-lactic-co-glycolic acid) (50:50) (PLGA), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Dimethyl sulfoxide (DMSO) and formaldehyde were purchased from Merck (MA, USA). Fetal bovine serum (FBS), penicillin, 0.25% trypsin-ethylenediaminetetraacetic acid (EDTA), 4',6-diamidino-2-phenylindole (DAPI), and fluorescein isothiocyanate (FITC) were purchased from Sigma-Aldrich. Unless stated otherwise, all other materials were obtained from Sigma-Aldrich.
Cy 5.5 was purchased from Life Technologies (USA). BCA Protein Quantitation Kit, GSH and GSSG Assay Kit, DCFH-DA Reactive Oxygen Species Assay Kit, and cell lysis buffer were purchased from ThermoFisher Scientific (Pittsburgh, PA). The Aspartate Aminotransferase Assay Kit (AST, ab105135) and Urea Assay Kit (BUN, ab83362) were obtained from abcam (Cambridge, UK).
L-ascorbic acid-2-O-phosphate (AA-2P, ≥ 99%) was purchased from Aladdin Chemical Reagent (Shanghai, China) and used as received. Dichloromethane (DCM) was dried over calcium hydride (CaH2) and distilled before use. LysoTracker Green and propidium iodide (94%) were purchased from ThermoFisher Scientific. All antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibody storage and dilution conditions are listed in Supplementary Table 1.
Cell lines and transfection
JAR and JEG3 cells were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and cultured in DMEM/F12 (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS; Gibco). Cultures were incubated at 37 °C in a humidified atmosphere of 5% CO2. To produce the firefly luciferase (luc)-positive JEG3 cell line (JEG3/luc), lentiviruses expressing the Fluc (LV-luc) were produced as described and then used to infect JEG3 cells [39]. LV-luc-transduced JEG3 cells were sorted based on their production of luc using a BD FACSAria III cell sorter (BD Biosciences, San Jose, CA, USA).
Animals
BALB/c nude mice (female, 14-16 g) were obtained from the Model Animal Research Center of Nanjing University (Nanjing, China). The animals were housed in individually ventilated cage units and were maintained under pathogen-free conditions. Food and water were provided ad libitum. All animal experiments were performed according to the Guide for Care and Use of Laboratory Animals of Guilin Medical University, and treated according to the regulations of Chinese law and the local Ethics Committee.
Preparation of PEG-ss-aAPP micelles and loading with MTX
PEG-ss-aAPP was synthesized as detailed in Supplementary Information. Then PEG-ss-aAPP (150 mg) was mixed with 2 mg of MTX and PLGA (10 mg) in 50 mL of ethyl acetate in a 150-mL round-bottom flask. The flask was left on a rotary evaporator (BUCHI, Switzerland) overnight in a 40 ºC water bath to eliminate ethyl acetate. The resulting thin film was rehydrated for 30 min with 30 mL of deionized water at 40 ºC, after which the suspension was sonicated for 10 min using a probe sonicator (Biologics, Cary, NC, USA) at 80 % strength. The solution was passed through a 0.5-μm filter (Millipore, Billerica, MA, USA) to remove insoluble material, and the resulting PEG-ss-aAPP/MTX micelles were freeze-dried and stored at -20 ºC for further experiments. The same procedure was used to prepare APP/MTX.
Preparation samples for transmission and scanning electron microscopy
The morphology of PEG-ss-aAPP/MTX and APP/MTX micelles was investigated by transmission electron microscopy (JEM-1200EX, JEOL). PEG-ss-aAPP/MTX or APP/MTX micelles were diluted with water and then placed on 400-mesh copper-coated grids, which were allowed to dry at room temperature. The surface morphology of lyophilized PEG-ss-aAPP/MTX and APP/MTX micelles was studied using scanning electron microscopy (Carl Zeiss, Oberkochen, Germany) at an acceleration voltage of 15 kV.
Glutathione- or pH-triggered MTX release in vitro
A dialysis method was used to analyze release of MTX from PEG-ss-aAPP/MTX and APP/MTX micelles. Lyophilized PEG-ss-aAPP/MTX and APP/MTX micelles were dispersed in phosphate-buffered saline (PBS) to a final MTX concentration of 2 mg/mL. Then, 1 mL of micelles was placed into a cellulose membrane tube, which was immersed in a series of media (20 mL): first PBS at pH 7.4 for 4 h, then PBS at pH 7.4 with 20 mM DTT for 32 h, and finally acetate buffer at pH 5.5 with DTT for a further 40 h. Tubes were incubated in these media at 37 ºC under gentle stirring. The amount of released MTX was determined at different times using HPLC as described in Supplementary Information.
ROS-responsiveness and stability of PEG-ss-aAPP/MTX micelles in vitro
To evaluate ROS-responsiveness, 5 mg of freshly prepared PEG-ss-aAPP/MTX micelles was incubated in 10 mL PBS (0.01 M, pH 7.4) containing 0 - 20 mM of dithiothreitol at 37°C. After 48 h incubation, the particle size of the micelles was characterized by dynamic light scattering. For stability evaluation, 5 mg of freshly produced PEG-ss-aAPP/MTX micelles was incubated in 10 mL of 0.9 % saline at 37 °C for 96 h. At specified time points, the particle size distribution profiles and zeta potentials of the samples were characterized by dynamic light scattering.
Micelle uptake into cells
Micelle co-localization with endosome/lysosomes and intracellular delivery of cargo were observed using confocal laser scanning microscopy. JEG3 cells were cultured as described above. Micelles were loaded with Cy 5.5 instead of MTX to give APP/Cy 5.5 and PEG-ss-aAPP/Cy 5.5, which were suspended in DMEM. JEG3 cells were cultured as described above and exposed for 24 h to the two types of micelles (with the same number of Cy 5.5 equivalents). As negative controls, cultures were exposed either to the same number of free Cy 5.5 equivalents in DMEM, or to DMEM alone. Cells were then incubated for 2 h with Lysotracker to stain endosome/lysosomes and DAPI to stain nuclei. Cells were washed with PBS (pH 7.4) three times, fixed and protected with mounting medium, and observed under the confocal microscope (Leica TCS SP5, Germany).
Intracellular distribution of MTX-loaded micelles and intracellular delivery of MTX were observed using transmission electron microscopy (JEOL, Japan). JEG3 cells were cultured until 70-80% confluence, trypsinized and seeded onto 4-well chamber slides (BD Falcon, USA) at a density of 1.0 × 105 cells/well. After 24-h incubation at 37 ºC, cultures were incubated a further 24 h with APP/MTX or PEG-ss-aAPP/MTX micelles or free MTX in DMEM (at the same MTX concentration of 1 mg/mL). Cells were then washed with PBS (pH 7.4) three times, then fixed and dried for transmission electron microscopy, which was performed as described previously [40].
Intracellular detection of ROS or glutathione
ROS generation within JEG3 cells was measured using an ROS Assay Kit (see above). Typically, JEG3 cells were seeded into 35-mm glass-bottom culture dishes (NEST, China) at a density of 1×104 cells/well and cultured overnight in 2 mL DMEM containing 10% FBS. The medium was replaced with fresh DMEM containing 10% FBS, followed by addition of free MTX, APP/MTX micelles or PEG-ss-aAPP/MTX micelles (to the same final MTX concentration of 10 µM). After 24-h incubation, the medium was removed. Then, fresh DMEM medium (2 mL) containing DTT (10 μM) was added, followed by further incubation for 20 min. The cells were washed three times with ice-cold PBS and viewed under the confocal microscope (Leica TCS SP5, Germany) to assess the degradation of dichlorofluorescein into DCFH-DA (excitation 488 nm, emission 500-540 nm). Fluorescence intensity was also analyzed using flow cytometry (BD Biosciences, Franklin Lakes, NJ, USA).
JEG3 cells were cultured as described above, then treated for 12 h with 10 µM free MTX, APP/MTX micelles or PEG-ss-aAPP/MTX micelles. The intracellular glutathione level was determined using the GSH/GSSG Assay Kit (see above) according to the manufacturer's recommendations. Glutathione levels in untreated JEG3 cells were also assayed using the same method. Cell fluorescence was imaged using confocal microscopy (Leica TCS SP5, Germany), then quantified using flow cytometry.
Cytotoxicity, anticancer effect and apoptosis detection in vitro
The anticancer effect of micelles was evaluated using the MTS assay. JEG3 and JAR cells were seeded in 96-well plates at a density of 5 × 103 cells per well and incubated for 24 h. MTX and micelles were dissolved in culture medium to a known number of MTX equivalents, and cells were treated with free MTX, APP/MTX micelles or PEG-ss-aAPP/MTX micelles for 48 h. Then solutions were removed and cells were washed with PBS. The MTS assay was performed using the CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega, USA) [41]. Absorbance at 570 nm was measured using a microplate reader (PerkinElmer, USA). Cell viability was calculated as the ratio of the OD of the experimental wells to OD of the control wells.
In separate experiments, JAR and JEG3 cells were incubated for 48 h with the different formulations as above, then digested and collected by centrifugation at 157 g for 5 min. Cells were rinsed twice with PBS and re-suspended in 500 μL of binding buffer within the wells, after which 5 μL of propidium iodide with or without 2.5 μL of Annexin V were added for each well. The wells were mixed and incubated in the dark for 15 min. Levels of apoptosis and distribution in the cell cycle were analyzed using flow cytometry.
Wound healing and cell invasion assay
The effects of different formulations on the migratory activity of JEG3 cells were examined using a wound healing assay [42]. Cells were plated into six-well plates and cultured for 24 h until completely confluent. The cell monolayer was scratched with a 200 μL pipette tip and washed twice with PBS to remove floating cells. The medium was then replaced with DMEM containing 0.5% FBS. Cells were subjected to the indicated treatments for 24 h, and cells migrating from the leading edge were photographed using a light microscope. The gap area was evaluated using ImageJ software for quantitation of the scratch wound area. Results are presented as the mean reclosed area ± SD, with 100 % of the area defined as that before scratching (0 h).
The effects of different formulations on the invasion ability of JEG3 cells were examined using an in vitro assay. Cells (4 × 104) in 300 μL serum-free DMEM were plated into the upper chambers of a Falcon® 24 Well Permeable Support Companion Plate and cultured for 24 h, after which 800 μL DMEM with 10% serum was added to the lower chambers. Cells that had not penetrated the filter were wiped away using cotton swabs, and cells that had migrated to the lower surface of the filter were stained with 0.5% crystal violet for 30 min and counted using a light microscope in five randomly selected fields at a magnification of 40×. Each assay was done in triplicate.
Protein extraction
JAR and JEG3 cells were plated into six-well plates for 24 h, then exposed to free MTX, APP/MTX micelles or PEG-ss-aAPP/MTX micelles (at the same MTX concentration of 3 μM). At the end of the treatment, total cell protein was extracted using RIPA buffer (1×PBS, 1% Nonidet P40, 0.5% sodium deoxycholate, 0.1% SDS) in the presence of 0.1 M PMSF plus EDTA-free protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN, USA) and the phosphatase inhibitor sodium orthovanadate (5 mM). To investigate the translocation of p65 to the nucleus, nuclear and cytoplasmic protein fractions were separately extracted using the nuclear and cytoplasmic protein extraction kit (Merck, Darmstadt, Germany).
Luciferase assay
JAR and JEG3 cells were transfected with NF-κB luciferase reporter plasmids (pNFκB-luc, Agilent Technologies, Santa Clara, CA) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. After 24 h, the cell culture medium was replaced with fresh medium with or without 3 μM PEG-ss-aAPP/MTX micelles, and cells were cultured another 6 h. Luciferase activity was determined using the Dual Luciferase Reagent kit (Promega Corporation, Madison, Wisconsin, USA). Each experiment was performed in triplicate.
Small interfering RNAs
NF-κB p65 was silenced in JAR and JEG3 cells using siRNA (sense: 5'-CCAUCAACUAUGAUGAGUUdTdT-3'; antisense: 5'- AACUCAUCAUAGUUGAUGGTdGd -3') (GenePharma, USA). Scrambled siRNA served as a negative control (sense: 5'-UUCUCCGAACGUGUCACGUTT-3'; antisense: 5'-ACGUGACACGUUCGGAGAATT-3'). JAR and JEG3 cells were transiently transfected with anti-p65 siRNA or scrambled siRNA using Lipofectamine 2000 (Invitrogen) following the manufacturer's instructions. At 24 h post-transfection, cells were collected for Western blot analysis or treated with 3 μM free MTX, APP/MTX micelles or PEG-ss-aAPP/MTX micelles, then incubated a further 24 h before being analyzed by Western blot.
Western blotting
Protein concentration was determined using the Bio-Rad BCA reagent (Bio-Rad, USA). Equal amounts of protein (40 μg/lane) were separated by SDS-PAGE and then the gel was transferred to 0.22- or 0.45-μm polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA). The membrane was blocked with 5% nonfat milk for 2 h at room temperature. The membranes were incubated with specific primary antibody (Cell Signaling Technology, USA) overnight at 4 °C against one of the following antigens: histone H3, p65, IκBα, p-IκBα, MMP-2, or MMP-9. Blots were also stained with primary antibody against β-actin as a loading control. Then membranes were incubated for 1.5 h at room temperature with horseradish peroxidase-conjugated anti-rabbit IgG (1:1000 in PBS; Cell Signaling Technology, USA). After extensive washing, the signals were detected using ECL reagent (Millipore) and quantified using ImageJ software (NIH, USA). All samples were analyzed in triplicate.
RNA isolation and quantitative real-time PCR
Total RNA was extracted using Trizol (TaKaRa Biotechnology, Dalian), and reverse transcription was conducted using an RT-PCR synthesis kit (Applied Biological Materials, Canada) according to the manufacturer's instructions. Quantitative real-time PCR was performed in a CFX96 Touch Real-Time PCR detection system (Bio-rad, USA) using the SYBR Green PCR Master Mix kit (Applied Biological Materials, Canada). Quantitative real-time PCR was conducted using the following cycling parameters: 10 min at 95 °C followed by 40 cycles of denaturation at 95 °C for 15 s and annealing at 60 °C for 1 min. Primers (Invitrogen China, Shanghai, China) are listed in Supplementary Table 2. Levels of transcripts were normalized to those of GAPDH. All samples were analyzed in triplicate.
Establishment of orthotopic xenograft mouse model of uterus cancer
All mouse procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Guilin Medical University. A total of 25 female athymic nude mice (6-8 weeks old) were anesthetized using intraperitoneal 1.25% sodium pentobarbital (45 mg/kg), laid down with the dorsal side up, and the lower abdomen and back were swabbed with 70% alcohol. A longitudinal incision about 15-20 mm long was made in the lower abdomen (medial laparotomy). The retroperitoneum was opened and the uterus was identified. Under a dissecting microscope, the left uterus horn was exteriorized and injected with luciferase-transfected JEG3 cancer cells (JEG3/luc, 1 x 106) suspended in 50 μl of Matrigel (BD Matrigel Basement Membrane Matrix, BD Biosciences, San Jose, CA, USA). The cells were injected using a 0.3-mm insulin syringe (Omnican 50, B-Braun, Melsungen, Germany) directly into the bifurcation of the uterus horns at the posterior face of the uterus through the myometrium. The uterus horn was returned to the original position in front of muscles, and the skin was closed with 5-0 absorbable sutures. After surgery, animals were placed in a warm environment and monitored until full recovery. They were injected intraperitoneally with 150 mg/kg of D-luciferin substrate, then tumor growth was analyzed using in vivo bioluminescence imaging (Caliper Life Sciences, USA). Regions of interest (ROIs) were selected in the tumor area and quantified using ImageJ.
Biodistribution in vivo and ex vivo
A hydrophobic near-infrared fluorescence dye, Cy 5.5, was used to monitor the in vivo dynamic distribution and tumor targeting of APP/Cy 5.5 and PEG-ss-aAPP/Cy 5.5. Mice were randomly divided into four groups (n = 5), and after the total luminescent intensity of the ROIs in the tumor area reached 0.6 × 107 p/sec/cm2/sr, APP/Cy 5.5 or PEG-ss-aAPP/Cy 5.5 micelles were injected via the tail vein at a dose equivalent to 10 μg/kg Cy 5.5. Mice were euthanized at 12 and 36 h after injection. Tumors and main organs were harvested and imaged using the Lumina system (Caliper Life Sciences).
To evaluate the anti-metastatic effects of PEG-ss-aAPP/MTX micelles on JEG3 cells, after the total luminescent intensity in tumor ROIs reached 0.6 × 107 p/sec/cm2/sr, JEG3/luc cells pre-treated with different formulations were injected into the mice via the tail vein (5 × 104 cells/100 μL). For this pre-treatment, cells were first seeded into 12-well culture plates, grown to approximately 70% confluence, and treated with 3 μM MTX, APP/MTX or PEG-ss-aAPP/MTX micelles. Control animals were injected with saline. After 4 h of incubation, the medium was changed and cells were cultured a further 48 h. The pre-treated cells were harvested, washed with PBS, diluted in serum-free medium and injected into the mice. The test compounds (MTX, APP/MTX or PEG-ss-aAPP/MTX micelles; 5 mg/kg/mouse) or saline were administered intravenously via tail vein once every other day until day 28. The total luminescence intensity of the nude mice was recorded every five days. At the end of treatment, animals were sacrificed, total luminescent intensity was determined and lungs were harvested and stored at -80 °C or fixed in paraformaldehyde for further analysis. Tumor areas in the lungs were quantitated using ImageJ. The uterus with or without tumors was weighed.
Apoptosis of tumor xenografts based on confocal microscopy
Tumors were cut into slices 5.0 µm thick, rinsed with PBS three times, incubated in binding buffer and stained with annexin V and propidium iodide (Lianke Technology, Hangzhou, China) for 15 min at 25 °C under low-light conditions. Samples were washed again, stained with 5 mM DAPI and analyzed under a confocal microscope (Leica TCS SP5, Germany).
Immunohistochemistry
Tissue sections were deparaffinized with xylene and rehydrated in graded solutions of ethyl alcohol. For antigen retrieval, slides were heated in a microwave oven for 5 min in sodium citrate buffer (pH 6.0), incubated in 3% hydrogen peroxide for 10 min to inhibit endogenous peroxidase activity, blocked in 10% horse serum for 1 h at room temperature, incubated with a primary rabbit polyclonal antibody against p65 overnight at 4 °C, and finally incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG for 1 h at room temperature. Immunostaining was developed by adding diaminobenzidine, and slides were counterstained with hematoxylin. Stained sections were observed under a light microscope (ECLIPSE 80i, Nikon, Tokyo, Japan).
Plasma biochemistry
Effects of micelle treatments on biochemical function of liver and kidneys were assessed using a FUJI DRI-CHEM 7000V (Fuji, Japan). Briefly, blood collected by cardiac puncture was centrifuged at 1500 g for 15 min (4 ºC), and the serum supernatant was analyzed for aspartate aminotransferase and blood urea nitrogen. Serum samples were also assayed for human chorion gonadotropin (hCG) and proinflammatory interleukin-6 (IL-6) using a commercial ELISA kit according to the manufacturer's instructions (BOSTER, Wuhan, China). Absorbance at 450 nm was measured using a Spectra max plus microplate reader (Molecular Devices, USA).
Statistical analysis
Data are expressed as mean ± SD. Differences were assessed for significance using the t-test analysis of variance. Significance was defined at three levels: *p < 0.05, **p < 0.01 and ***p < 0.001.
Supplementary Material
Supplementary methods, figures and tables.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81860629 and 81402876), the Natural Science Foundation of Guangxi Province (2018JJA140084), and the Open Funds of the Guangxi Key Laboratory of Tumor Immunology and Microenvironmental Regulation (2018KF003).
Author contributions
J. Mo designed all experiments. L. Wei, C. Wang and B. Yang synthesized nanoparticles and performed experiments involving animals. K. Shi performed cell culture experiments. L. Wei, C. Wang and B. Yang performed immunohistochemistry and western blotting. J. Mo and S. Shi interpreted the data and wrote the manuscript, which L. Lim, X. Chen and L. Benington helped edit. All authors critically reviewed the manuscript and approved the final version.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Seckl MJ, Sebire NJ, Berkowitz RS. Gestational trophoblastic disease. Lancet. 2010;376:717-29
2. El-Helw LM, Hancock BW. Treatment of metastatic gestational trophoblastic neoplasia. Lancet Oncol. 2007;8(8):715-24
3. Maesta I, Nitecki R, Horowitz NS, Goldstein DP, de Freitas Segalla Moreira M, Elias KM. et al. Effectiveness and toxicity of first-line methotrexate chemotherapy in low-risk postmolar gestational trophoblastic neoplasia: The New England Trophoblastic Disease Center experience. Gynecol Oncol. 2018;148(1):161-167
4. Ishiguro T, Serikawa T, Yahata T, Enomoto T. Gestational choriocarcinoma: Rare spinal metastasis during a viable pregnancy. J Obstet Gynaecol Res. 2017;43(2):421-424
5. Savage P, Kelpanides I, Tuthill M, Short D, Seckl MJ. Brain metastases in gestational trophoblast neoplasia: an update on incidence, management and outcome. Gynecol Oncol. 2015;137(1):73-6
6. Terakawa T, Miyake H, Muramaki M, Takenaka A, Fujisawa M. Salvage chemotherapy with methotrexate, etoposide and actinomycin D in men with metastatic nonseminomatous germ cell tumors with a choriocarcinoma component: a preliminary report. Int J Urol. 2010;17(10):881-5
7. Chu MM, Ma Y, Tse KY, Chan KK, Ngan HY. Cyclophosphamide, hydroxyurea, actinomycin D, methotrexate, and vincristine in the treatment of gestational trophoblastic neoplasia. Int J Gynecol Cancer. 2015;25(3):498-503
8. Li XM, Liu XY, Liu ZX. Choriocarcinoma with multiple lung, skull and skin metastases in a postmenopausal female: A case report. Oncol Lett. 2015;10(6):3837-3839
9. Moloney JN, Cotter TG. ROS signalling in the biology of cancer. Semin Cell Dev Biol. 2018;80:50-64
10. Xu J, Wu Y, Lu G, Xie S, Ma Z, Chen Z. et al. Importance of ROS-mediated autophagy in determining apoptotic cell death induced by physapubescin B. Redox Biol. 2017;12:198-207
11. Gill JG, Piskounova E, Morrison SJ. Cancer, Oxidative Stress, and Metastasis. Cold Spring Harb Symp Quant Biol. 2016;81:163-175
12. Liu H, Talalay P. Relevance of anti-inflammatory and antioxidant activities of exemestane and synergism with sulforaphane for disease prevention. Proc Natl Acad Sci U S A. 2013:19 110(47):19065-70
13. Levine M, Conry-Cantilena C, Wang Y, Welch RW, Washko PW, Dhariwal KR. et al. Vitamin C pharmacokinetics in healthy volunteers: evidence for a recommended dietary allowance. Proc Natl Acad Sci U S A. 1996:8
14. Sawant RR, Vaze O, D'Souza GG, Rockwell K, Torchilin VP. Palmitoyl ascorbate-loaded polymeric micelles: cancer cell targeting and cytotoxicity. Pharm Res. 2011;28:301-8
15. Zhou M, Li X, Li Y, Yao Qe, Ming Y, Li Z. et al. Ascorbyl palmitate-incorporated paclitaxel-loaded composite nanoparticles for synergistic anti-tumoral therapy. Drug Deliv. 2017;24(1):1230-1242
16. Subramani T, Yeap SK, Ho WY, Ho CL, Omar AR, Aziz SA. et al. Vitamin C suppresses cell death in MCF-7 human breast cancer cells induced by tamoxifen. J Cell Mol Med. 2014;18(2):305-13
17. Kim AN, Lee K-Y, Kim H-J, Chun J, Kerr WL, Choi S-G. The Effects of Added Water and Grinding Temperature on Stability and Degradation Kinetics of Antioxidant Activity, Phenolic Compounds, and Ascorbic Acid in Ground Apples. J Food Sci. 2018;83(12):3019-3026
18. Miura K, Tai A. 2-O-alpha-D-Glucopyranosyl-l-ascorbic acid as an antitumor agent for infusion therapy. Biochem Biophys Rep. 2017:22 10:232-236
19. Miwa N, Yamazaki H, Nagaoka Y, Kageyama K, Onoyama Y, Matsui-Yuasa I. et al. Altered production of the active oxygen species is involved in enhanced cytotoxic action of acylated derivatives of ascorbate to tumor cells. Biochim Biophys Acta. 1988:18 972(2):144-51
20. Kato S, Asada R, Kageyama K, Saitoh Y, Miwa N. Anticancer effects of 6-o-palmitoyl-ascorbate combined with a capacitive-resistive electric transfer hyperthermic apparatus as compared with ascorbate in relation to ascorbyl radical generation. Cytotechnology. 2011;63(4):425-35
21. Shibuya S, Sakaguchi I, Ito S, Kato E, Watanabe K, Izuo N. et al. Topical Application of Trisodium Ascorbyl 6-Palmitate 2-Phosphate Actively Supplies Ascorbate to Skin Cells in an Ascorbate Transporter-Independent Manner. Nutrients. 2017:22 9(7). pii: E645
22. Inoue Y, Hibino M, Murata I, Kanamoto I. A Nanocarrier Skin-Targeted Drug Delivery System using an Ascorbic Acid Derivative. Pharm Res. 2017;35:1
23. Song QX, Wei DZ, Zhou WY, Xu WQ, Yang SL. Enzymatic synthesis and antioxidant properties of L-ascorbyl oleate and L-ascorbyl linoleate. Biotechnol Lett. 2004;26(23):1777-80
24. Asada R, Kageyama K, Tanaka H, Kimura M, Saitoh Y, Miwa N. Carcinostatic effects of diverse ascorbate derivatives in comparison with aliphatic chain moiety structures: Promotion by combined hyperthermia and reduced cytotoxicity to normal cells. Oncol Lett. 2012;3(5):1042-1046
25. Shibuya S, Ozawa Y, Toda T, Watanabe K, Tometsuka C, Ogura T. et al. Collagen peptide and vitamin C additively attenuate age-related skin atrophy in Sod1-deficient mice. Biosci Biotechnol Biochem. 2014;78(7):1212-20
26. Majumdar S, Aggarwal BB. Methotrexate Suppresses NF-κB Activation Through Inhibition of IκBα Phosphorylation and Degradation. J Immunol. 2001:1 167(5):2911-20
27. Yi SJ, Kim KH, Choi HJ, Yoo JO, Jung HI, Han JA. et al. [Ca(2+)]-dependent generation of intracellular reactive oxygen species mediates maitotoxin-induced cellular responses in human umbilical vein endothelial cells. Mol Cells. 2006:28 21(1):121-8
28. Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer. 2013:2 12:86
29. Fang C, Zhang J, Yang H, Peng L, Wang K, Wang Y. et al. Leucine aminopeptidase 3 promotes migration and invasion of breast cancer cells through upregulation of fascin and matrix metalloproteinases-2/9 expression. J Cell Biochem. 2019Mar;120(3):3611-3620
30. Kajihara H, Yamada Y, Kanayama S, Furukawa N, Noguchi T, Haruta S. et al. Clear cell carcinoma of the ovary: potential pathogenic mechanisms (Review). Oncol Rep. 2010;23(5):1193-203
31. Inoue Y, Hibino M, Murata I, Kanamoto I. A Nanocarrier Skin-Targeted Drug Delivery System using an Ascorbic Acid Derivative. Pharm Res. 2017:28 35(1):1
32. Li Y. Methotrexate-Camptothecin Prodrug Nanoassemblies as a Versatile Nanoplatform for Biomodal Imaging-Guided Self-Active Targeted and Synergistic Chemotherapy. ACS Appl Mater Interfaces. 2017:11 9(40):34650-34665
33. Greish K. Enhanced Permeability and Retention (EPR) Effect for Anticancer Nanomedicine Drug Targeting. Methods Mol Biol. 2010;624:25-37
34. Zhang R, Gao S, Wang Z, Han D, Liu L, Ma Q. et al. Multifunctional Molecular Beacon Micelles for Intracellular mRNA Imaging and Synergistic Therapy in Multidrug-Resistant Cancer Cells. Adv Funct Mater. 2017; 18;27(31). pii. 1701 027
35. Ulbrich K, Šubr Vr. Polymeric anticancer drugs with pH-controlled activation. Adv Drug Deliv Rev. 2004Apr23;56(7):1023-50
36. Cheng R, Feng F, Meng F, Deng C, Feijen J, Zhong Z. Glutathione-responsive nano-vehicles as a promising platform for targeted intracellular drug and gene delivery. J Control Release. 2011:30 152(1):2-12
37. Cheng R, Meng F, Deng C, Klok H-A, Zhong Z. Dual and multi-stimuli responsive polymeric nanoparticles for programmed site-specific drug delivery. Biomaterials. 2013;34:3647-57
38. Ganta S, Devalapally H, Shahiwala A, Amiji M. A review of stimuli-responsive nanocarriers for drug and gene delivery. J Control Release. 2008;126:187-204
39. Karki S, Li MMH, Schoggins JW, Tian S, Rice CM, MacDonald MR. Multiple interferon stimulated genes synergize with the zinc finger antiviral protein to mediate anti-alphavirus activity. PloS one. 2012;7:e37398
40. Shen Y, Yang J, Zhao J, Xiao C, Xu C, Xiang Y. The switch from ER stress-induced apoptosis to autophagy via ROS-mediated JNK/p62 signals: A survival mechanism in methotrexate-resistant choriocarcinoma cells. Exp Cell Res. 2015:10 334(2):207-18
41. Ecimovic P, Murray D, Doran P, Buggy DJ. Propofol and bupivacaine in breast cancer cell function in vitro - role of the NET1 gene. Anticancer Res. 2014;34(3):1321-31
42. Chen W, Li L, Zhang X, Liang Y, Pu Z, Wang L. et al. Curcumin: a calixarene derivative micelle potentiates anti-breast cancer stem cells effects in xenografted, triple-negative breast cancer mouse models. Drug Deliv. 2017;24(1):1470-1481
Author contact
![]() Corresponding authors: E-mail address: Jingxin.mocom (J. Mo), shisanjunscom (S. Shi).
Corresponding authors: E-mail address: Jingxin.mocom (J. Mo), shisanjunscom (S. Shi).