Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2019; 9(15):4558-4566. doi:10.7150/thno.31052 This issue Cite
Research Paper
LncRNA DCRF regulates cardiomyocyte autophagy by targeting miR-551b-5p in diabetic cardiomyopathy
Yu Feng1,2,#, Weiting Xu3,#, Wei Zhang3,#, Wenjing Wang3, Tong Liu2, Xiang Zhou3, ![]()
1. Department of Endocrinology, The Second Affiliated Hospital of Soochow University, Suzhou, China
2. Jiangsu Key Laboratory of Neuropsychiatric Diseases and Institute of Neuroscience, Soochow University, Suzhou, China
3. Department of Cardiology, The Second Affiliated Hospital of Soochow University, Suzhou, China
# These authors contributed equally to this work.
Received 2018-10-29; Accepted 2019-5-6; Published 2019-6-10
Abstract

Background: We generated a rat model of diabetic cardiomyopathy (DCM) and reported significant upregulation of the long non-coding RNA DCRF. This study was designed to determine the molecular mechanisms of DCRF in the development of DCM.
Methods: Real-time PCR and RNA fluorescent in situ hybridization were conducted to detect the expression pattern of DCRF in cardiomyocytes. Histological and echocardiographic analyses were used to assess the effect of DCRF knockdown on cardiac structure and function in diabetic rats. mRFP-GFP-LC3 fluorescence microscopy, transmission electron microscopy, and Western blotting were carried out to determine cardiomyocyte autophagy. RNA immunoprecipitation and luciferase reporter assays were performed to elucidate the regulatory role of DCRF/miR-551b-5p/PCDH17 pathway in cardiomyocyte autophagy.
Results: Our findings showed that DCRF knockdown reduced cardiomyocyte autophagy, attenuated myocardial fibrosis, and improved cardiac function in diabetic rats. High glucose increased DCRF expression and induced autophagy in cardiomyocytes. RNA immunoprecipitation and luciferase reporter assays indicated that DCRF was targeted by miR-551b-5p in an AGO2-dependent manner and PCDH17 was the direct target of miR-551b-5p. Forced expression of DCRF was found to attenuate the inhibitory effect of miR-551b-5p on PCDH17. Furthermore, DCRF knockdown decreased PCDH17 expression and suppressed autophagy in cardiomyocytes treated with high glucose.
Conclusion: Our study suggests that DCRF can act as a competing endogenous RNA to increase PCDH17 expression by sponging miR-551b-5p, thus contributing to increased cardiomyocyte autophagy in DCM.
Keywords: autophagy, DCRF, diabetic cardiomyopathy, miR-551b-5p
Introduction
Diabetic cardiomyopathy (DCM), a major cardiovascular complication of diabetes, is characterized by myocardial fibrosis, ventricular remodeling, and cardiac dysfunction. In recent years, increasing evidence has demonstrated that mitochondrial dysfunction, oxidative stress, inflammation, autophagy, renin-angiotensin system activation, impaired calcium handling, diabetic microangiopathy, and myocardial metabolic abnormalities are implicated in the pathogenesis of DCM [1,2].
Long non-coding RNAs (lncRNAs) are defined as transcripts longer than 200 nucleotides without protein-coding potential. They can modulate gene expression by functioning as nuclear transcriptional regulators, RNA-binding protein sequestering agents, and microRNA (miRNA) sponges [3]. It has been documented that lncRNAs are critically involved in the pathogenesis of various cardiovascular diseases [4,5]. We generated a rat model of DCM and identified differentially expressed lncRNAs in the myocardium by sequencing analysis. The results indicated that TCONS_00069849 Chr2: 228036171-228036880 was significantly upregulated in DCM rats. This lncRNA was discovered to be associated with DCM and was therefore named DCM-related factor (DCRF). The present study was conducted to investigate the molecular mechanisms of DCRF in the development of DCM. Our findings suggested that DCRF could regulate cardiomyocyte autophagy by acting as a miRNA sponge.
Methods
Animal model and treatment
All experiments in this study were approved by the Animal Ethics Committee of Soochow University. Male Sprague-Dawley rats weighing 200-250 g were received a single intraperitoneal injection of streptozotocin (65 mg/kg). The fasting blood glucose was measured one week after injection. Only rats with glucose levels higher than 16.7 mmol/L were defined as diabetic. The diabetic rats were intramyocardially injected with 100 μL adeno-associated virus-9 (AAV-9, 1×1010 GC/mL) containing DCRF-shRNA (DM + DCRF-shRNA) or scrambled-shRNA (DM + Scr-shRNA). Two other groups consisted of diabetic rats without AAV-9 injection (DM) and normal rats (Control). All animals were kept for 12 weeks under the same laboratory conditions.
Cardiomyocyte culture
The neonatal rats were anesthetized with isoflurane and the hearts were surgically removed and rinsed in cold saline. Cardiomyocytes were digested, centrifuged, suspended, and purified as previously described [6], then diluted to 1×106 cells/ml and cultured at 37°C in a humidified atmosphere of 5% CO2- 95% air. Cardiomyocytes were transfected with adenovirus containing DCRF-siRNA or scrambled-siRNA and exposed to high glucose (30 mmol/L). The sequences of DCRF-siRNA and scrambled-siRNA were 5'-GAGAGAAGUUGGGUGAUUAUC-3' and 5'-UUCUCCGAACGUGUCACGU-3'.
Histological analysis
Myocardial tissue was fixed in 10% buffered formalin, embedded in paraffin and sliced into 5-μm-thick sections. Subsequently, hematoxylin and eosin (HE) and Masson's trichrome were used to stain myocardial sections and the slides were observed under an optical microscope. Myocyte cross-sectional area (CSA) and collagen volume fraction (CVF) were measured with image analysis software (Image-Pro Plus, Media Cybernetics).
Echocardiographic measurement
Cardiac structure and function were determined using echocardiography as previously described [7]. The following parameters were measured: left ventricular end-systolic diameter (LVESD), left ventricular end-diastolic diameter (LVEDD), left ventricular fractional shortening (LVFS), and left ventricular ejection fraction (LVEF). All measurements were averaged for 3 consecutive cardiac cycles.
Transmission electron microscopy
Cardiac tissue was fixed with 2.5% glutaraldehyde, post-fixed with 2% osmium tetroxide, dehydrated in graded ethanol, and embedded in epoxy resin. Subsequently, the ultrathin sections were prepared and stained with uranyl acetate and lead citrate. The samples were observed under a transmission electron microscope (Hitachi HT7700, Japan).
Tandem mRFP-GFP-LC3 fluorescence microscopy
Cardiomyocyte autophagy was analyzed using tandem mRFP-GFP-LC3 fluorescence microscopy. Myocardial cells were transfected with adenovirus expressing mRFP-GFP-LC3 (GenePharma, Suzhou, China) at 10 MOI for 24 h and were observed under a confocal laser scanning microscope (LSM 510; Carl Zeiss, Germany).
RNA fluorescent in situ hybridization (RNA-FISH)
The RNA-FISH assay was conducted using Cy3-labelled RNA probes to the DCRF sequence. After prehybridization, cardiomyocytes were incubated with RNA probes in the hybridization buffer overnight. The signals were detected using a Fluorescent In Situ Hybridization Kit (GenePharma, Suzhou, China). The sequences of RNA probes were as follows: DCRF-Probe1: 5'-CGCTCAACAGATGAACATGT-3'; DCRF-Probe2: 5'-CGGCTCCTGAAATAACACAT-3'; negative control: 5'-GTGTAACACGTCTATACGCCCA -3'; positive control: 5'-CTTCCTTGGATGTGGTAGCCGTTTC-3'.
Luciferase reporter assay
The 3'-untranslated region (UTR) of PCDH17 was cloned downstream of luciferase gene to generate a Luc-PCDH17-Wt vector, while the 3'-UTR without binding site of miR-551b-5p was constructed to generate the Luc-PCDH17-Mut vector. HEK293 cells were seeded into 96-well plates and transfected with Luc-PCDH17 vectors and miRNA mimics. The luciferase activity was then detected using a Luciferase Reporter Assay System (Promega, USA).
RNA-binding protein immunoprecipitation (RIP)
The RIP assay was performed using a Magna RIP™ RNA-Binding Protein Immunoprecipitation Kit (Millipore, USA). Myocardial cells were incubated with RIP buffer containing magnetic beads conjugated with AGO2 antibody or negative control IgG, and then were lysed in RIP lysis buffer. The immunoprecipitated RNAs were purified followed by quantitative PCR to determine the target sequences.
Real-time PCR
Total RNAs were extracted using TRIzol reagent (Invitrogen, USA). The cytoplasmic and nuclear fractions were prepared using the NE-PER extraction kit (Pierce, USA). To quantify the amount of lncRNA, miRNA, and mRNA, real-time PCR was performed with the SYBR Premix Ex Taq™ kit (TaKaRa, Japan). GAPDH and U6 were used as internal controls in this study. The relative expression of genes was calculated using the 2-ΔΔCT method. The sequences of primers used in this study were as follows:
DCRF, Forward primer: 5'-TGTTCATCTGTTGAGCGTCTAA-3', Reverse primer: 5'-ACACTAGATAATCACCCAACTT-3'; PCDH17, Forward primer: 5'-CTTGCCGATGTTGCCTAT-3', Reverse primer: 5'-CCATCTGTTGCTGCTTTC-3'; GAPDH, Forward primer: 5'-GCGAGATCCCGCTAACATCA-3', Reverse primer: 5'-CTCGTGGTTCACACCCATCA-3'; miR-551b-5p, RT-primer: 5'-GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTGCACTGGATACGACAGGTCT-3', Forward primer: 5'-GGGGAAATCAAGCTTGGGTG-3', Reverse primer: 5'-CAGTGCGTGTCGTGGAGT-3'.
Western blotting
Cardiomyocytes were lysed with RIPA buffer (Thermo Scientific, USA). Cell lysates containing 20 μg of protein were separated by SDS-PAGE, transferred to PVDF membranes, and blocked with 5% non-fat milk. The membranes were incubated with primary antibodies at 4 °C overnight, followed by secondary antibodies conjugated with horseradish peroxidase for 1 h at room temperature. All of the antibodies were obtained from Cell Signaling Technology and used at the recommended dilutions following the manufacturer's protocols. The immunoblots were visualized using the enhanced chemiluminescence reagent (Thermo Scientific, USA).
Statistical analysis
All statistical analyses were conducted using SPSS 20.0 (IBM, USA). The experimental data were expressed as mean ± SD. The differences between groups were compared using one-way ANOVA. The Scheffé post hoc test was used to perform multiple comparisons. A value of p < 0.05 was considered statistically significant.
Results
DCRF expression pattern in cardiomyocytes
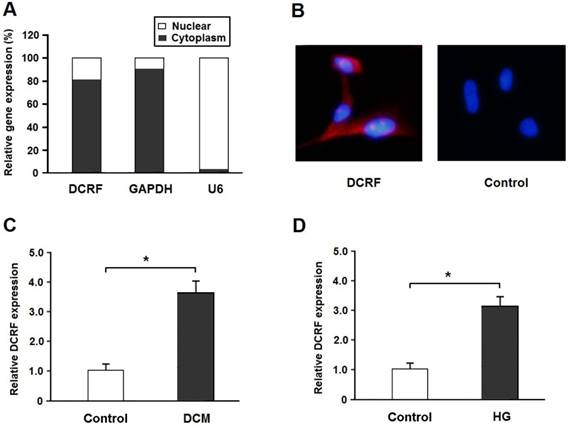
DCRF is located on Chr2: 228036171-228036880 in the rat genome assembly Rnor_6.0 and its sequence is shown in Table 1. Real-time PCR and the FISH assay indicated that DCRF was primarily expressed in the cytoplasm of cardiomyocytes (Figure 1A and B). Also, DCRF expression was upregulated in the myocardium of diabetic rats and in cardiomyocytes treated with high glucose (Figure 1C and D).
DCRF sequence
| TCONS_00069849 Chr2: 228036171-228036880 |
|---|
| GTAAGGAAGGATTTTCACAATCTTTTCCAAAAAGTTTCTATGTGTTATTTCAGGAGCCGGTTTGCAGAAAGATCTGGGTTTGATTTCCAGCACAAAATTATGGTGGAAGAAAGAAAAAAGAAAGAGGAAACGATGAGGAAGGGTGTGGCCAGATGGAGTGAGTCAAGCCTTTCTATTACTGAAGAAGGAGCTCTTCAGCCACCATGGCAGTCCAGGCCTGTCAGCAGCCGTTCAAGGTCATGGGCACTTCCTCGAGAGCAAGGATTTAAGGACATGTTCATCTGTTGAGCGTCTAAAGGAACAAATTTTGTACTGGGTGCTTCTTCCCCTCGATATTGAGTTTTTATCTCTTACATGTCCCTTTTGAGAGAAGTTGGGTGATTATCTAGTGTTTTTATTTTTCTGTTTACCATATATTTAGTTGGGATCAGCTGTCTTTTCTTGGCTTTTTATTTATTTCAAACTCTAATTAGGGCA |
DCRF expression pattern in cardiomyocytes. (A) The expression of DCRF, GAPDH (cytoplasmic control), and U6 (nuclear control) was detected by real-time PCR in the nuclear and cytoplasmic fractions of cardiomyocytes. (B) RNA-FISH assay was performed to determine DCRF expression in cardiomyocytes using Cy3-labeled sense (Control) and antisense probes (DCRF). (C, D) DCRF expression was detected by real-time PCR in myocardial tissue of DCM rats and in cardiomyocytes treated with high glucose (HG, 30 mmol/L). (n = 5, *P < 0.05).
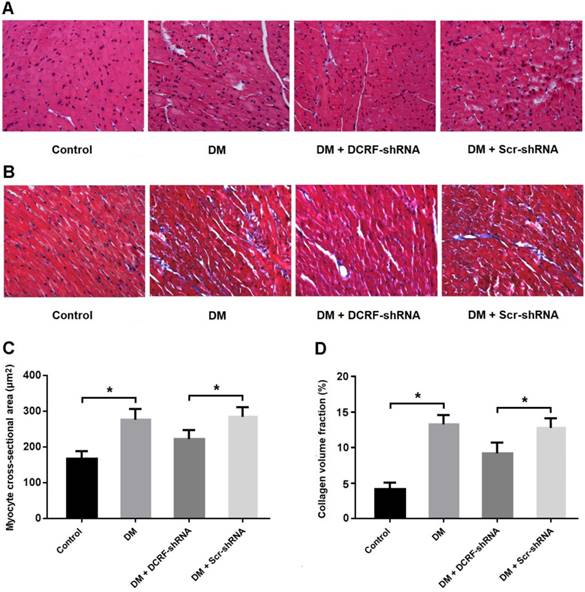
DCRF knockdown improves histological abnormalities in diabetic rats
Myocardial tissue was stained with HE and Masson's trichrome, and CSA and CVF were measured. HE staining showed the typical histological changes in diabetic myocardium, including cardiomyocyte hypertrophy, myocardial fiber rupture, and increased cell gap, whereas DCRF knockdown was associated with fewer histological abnormalities (Figure 2A). Also, Masson staining indicated that reduced expression of DCRF could attenuate myocardial fibrosis in diabetic rats (Figure 2B). Furthermore, CSA and CVF were markedly increased in the DM group and decreased in the DM + DCRF-shRNA group (Figure 2C and D).
DCRF knockdown improves histological abnormalities in diabetic rats. (A, C) Myocardial tissue was stained with hematoxylin-eosin and myocyte cross-sectional area was measured. (B, D) Myocardial tissue was stained with Masson's trichrome and collagen volume fraction was calculated. (n = 5, *P < 0.05).
DCRF knockdown improves cardiac function in diabetic rats
Transthoracic echocardiography was used to evaluate the effect of DCRF knockdown on cardiac structure and function. As shown in Figure 3A, DCRF expression was upregulated in the diabetic myocardium and downregulated after DCRF-shRNA transfection. Echocardiographic measurements indicated that LVESD and LVEDD were increased in the DM group and reduced in the DM + DCRF-shRNA group (Figure 3B and C).
DCRF knockdown improves cardiac function in diabetic rats. (A) The expression of DCRF in myocardial tissue was determined by real-time PCR (n = 5, *P < 0.05). (B-D) Cardiac structure and function were evaluated by echocardiography (n = 5, *P < 0.05). LVESD = left ventricular end-systolic diameter; LVEDD = left ventricular end-diastolic diameter; LVFS = left ventricular fractional shortening, LVEF = left ventricular ejection fraction.
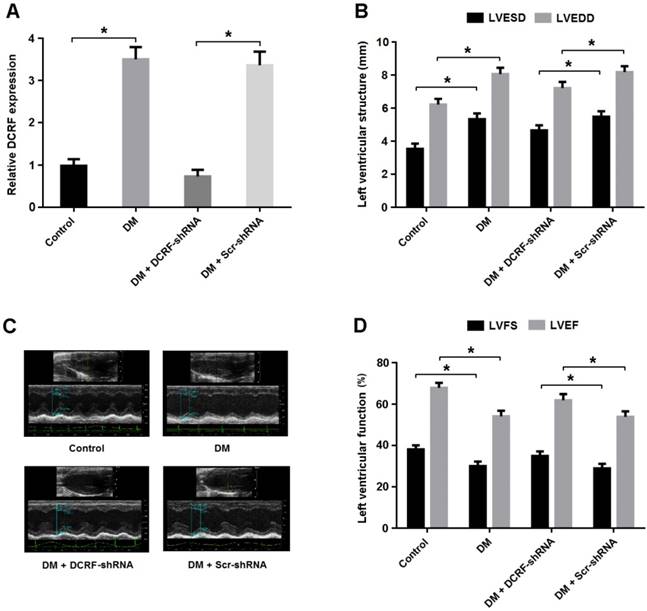
Moreover, LVFS and LVEF were decreased in diabetic rats, while DCRF knockdown was found to improve left ventricular function (Figure 3D).
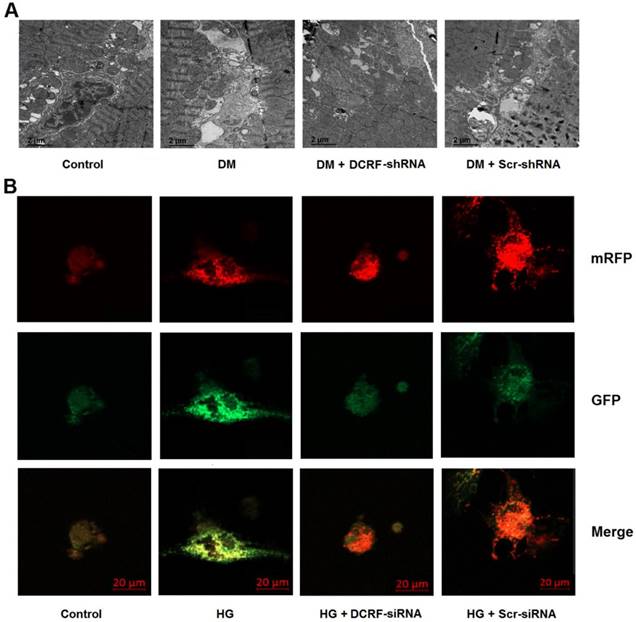
DCRF is involved in the modulation of cardiomyocyte autophagy
Cardiomyocyte autophagy was detected by transmission electron microscopy and mRFP-GFP-LC3 fluorescence microscopy. As shown in Figure 4A, autophagy was elevated in the diabetic myocardium, while DCRF knockdown significantly reduced the number of autophagosomes. Moreover, high glucose treatment was associated with increased autophagy in cardiomyocytes, whereas DCRF-siRNA transfection could attenuate high glucose-induced autophagy (Figure 4B).
DCRF is involved in the modulation of cardiomyocyte autophagy. (A) Diabetic rats were intramyocardially injected with adeno-associated virus containing DCRF-shRNA or scrambled-shRNA. After 12 weeks, transmission electron microscopy was used to detect autophagosomes in the myocardium. (B) Myocardial cells were transfected with adenovirus containing DCRF-siRNA or scrambled-siRNA and then exposed to high glucose (HG, 30 mmol/L). After 48 hours, tandem mRFP-GFP-LC3 fluorescence microscopy was performed to evaluate cardiomyocyte autophagy.
DCRF is targeted by miR-551b-5p in an AGO2-dependent manner
Bioinformatics analysis showed that DCRF sequence contained the putative binding site for miR-551b-5p (Figure 5A). The expression of miR-551b-5p was elevated in HEK293 cells transfected with miR-551b-5p mimics (Figure 5B). The DCRF cDNA was cloned downstream of the luciferase gene (Luc-DCRF-Wt) and transfected into HEK293 cells with miR-551b-5p mimics. We then mutated the miR-551b-5p binding site in DCRF to generate Luc-DCRF-Mut. The luciferase assay indicated that miR-551b-5p transfection could inhibit the activity of Luc-DCRF-Wt, but not Luc-DCRF-Mut (Figure 5C).
DCRF is targeted by miR-551b-5p in an AGO2-dependent manner. (A) Bioinformatic prediction showed that DCRF sequence contained the putative binding site of miR-551b-5p. (B) The expression of miR-551b-5p was increased in HEK293 cells transfected with miR-551b-5p mimics (n = 3, *P < 0.05). (C) The DCRF cDNA was cloned downstream of the luciferase gene (Luc-DCRF-Wt) and transfected into HEK293 cells with miR-551b-5p mimics or control oligonucleotides. The miR-551b-5p binding site in DCRF was mutated to generate Luc-DCRF-Mut. Luciferase activity was detected 48 hours after transfection (n = 3, *P < 0.05). (D) The RIP assay was conducted to verify whether DCRF and miR-551b-5p directly bind to AGO2 in myocardial cells (n = 3, *P < 0.05).
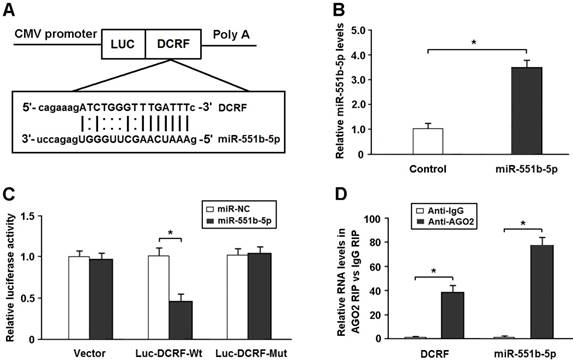
DCRF was primarily expressed in the cytoplasm of cardiomyocytes. Thus, we speculated that DCRF might regulate gene expression by functioning as a miRNA sponge. The AGO2 protein is a core component of the RNA-induced silencing complex. The RIP assay showed that DCRF and miR-551b-5p were especially abundant in the immuno-precipitate pulled down by anti-AGO2 but not anti-IgG (Figure 5D).
PCDH17 is a direct target of miR-551b-5p in cardiomyocytes
Among the potential targets of miR-551b-5p, we were interested in PCDH17 involved in the modulation of autophagy (Figure 6A). The 3'-UTR of PCDH17 was fused to the coding region of luciferase and transfected into HEK293 cells with miR-551b-5p mimics. The results indicated that PCDH17 was a direct target of miR-551b-5p (Figure 6B). In addition, the expression of PCDH17 was increased after transfection with miR-551b-5p antagomir, thus leading to enhanced autophagy, whereas PCDH17 knockdown could attenuate autophagy following miR-551b-5p antagomir transfection (Figure 6C and D).
PCDH17 is a direct target of miR-551b-5p in cardiomyocytes. (A) PCDH17 was predicted as a target gene of miR-551b-5p using miRBase. (B) HEK293 cells were transfected with miR-551b-5p mimics and luciferase constructs of PCDH17 3'UTR (Luc-PCDH17-Wt) or mutant (Luc-PCDH17-Mut). Luciferase activity was detected 48 hours after transfection (n = 3, *P < 0.05). (C) Myocardial cells were transfected with miR-551b-5p antagomir and/or PCDH17 siRNA. The PCDH17 expression was detected by Western blotting (n = 3, *P < 0.05). (D) Cardiomyocyte autophagy was evaluated by detection of LC3 expression (n = 3, *P < 0.05). (E) HEK293 cells were transfected with luciferase constructs of PCDH17 3'UTR, miR-551b-5p mimic, and pcDNA-DCRF. The luciferase assay was conducted to determine whether DCRF competitively inhibits the binding of miR-551b-5p to PCDH17 (n = 3, *P < 0.05).
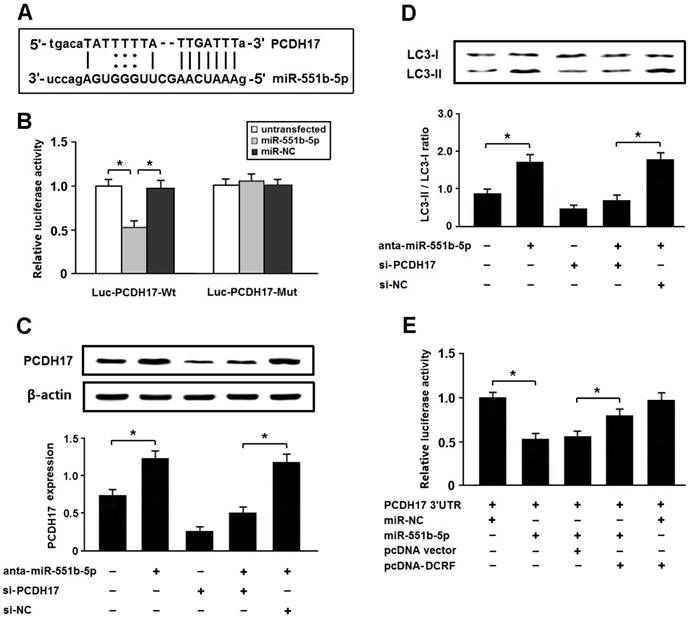
To determine whether DCRF competitively inhibits the binding of miR-551b-5p to PCDH17, we carried out luciferase reporter assay in HEK293 cells. The results revealed that DCRF could counteract the inhibitory effect of miR-551b-5p on PCDH17 (Figure 6E), which suggested that DCRF might function as a competing endogenous RNA (ceRNA) to control PCDH17 expression by sponging miR-551b-5p.
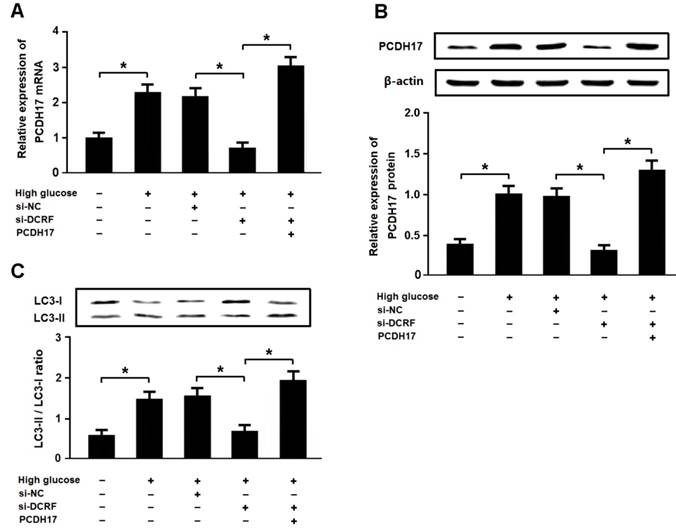
High glucose promotes autophagy by regulating the DCRF/PCDH17 axis
Cardiomyocytes were exposed to high glucose following transfection with DCRF-siRNA and/or pcDNA-PCDH17, and the expression of LC3 was detected by Western blotting. The results revealed that high glucose could increase PCDH17 expression and induce cardiomyocyte autophagy, while DCRF knockdown was associated with reduced PCDH17 expression and cardiomyocyte autophagy. Moreover, DCRF-siRNA and pcDNA-PCDH17 co-transfection promoted autophagy in cardiomyocytes treated with high glucose (Figure 7A-C).
High glucose promotes autophagy by regulating the DCRF/PCDH17 axis. (A, B) Myocardial cells were transfected with adenovirus containing DCRF-siRNA or PCDH17 and then exposed to high glucose (30 mmol/L) for 48 hours. The expression of PCDH17 was analyzed by real-time PCR and Western blotting (n = 3, *P < 0.05). (C) Cardiomyocyte autophagy was assessed by detection of LC3 expression (n = 3, *P < 0.05).
Discussion
Emerging evidence has demonstrated that lncRNAs participate in the molecular mechanisms of multiple cardiovascular diseases [5]. However, it remains unclear what is the function of lncRNAs in the pathogenesis of DCM. The present study was aimed to determine the pathological role of DCRF in the development of DCM. Our findings indicated that the expression of DCRF was increased in diabetic myocardium, while DCRF knockdown could reduce cardiomyocyte autophagy and improve cardiac function in DCM rats. We then further investigated the potential mechanisms of DCRF involved in high glucose-induced autophagy. Our results suggested that DCRF could act as a ceRNA to increase PCDH17 expression by sponging miR-551b-5p, thus contributing to increased cardiomyocyte autophagy and the progression of DCM.
In previous studies, we investigated the pathogenesis of lncRNAs in DCM and discovered that H19, MIAT, and MALAT1 participated in the regulation of cardiomyocyte apoptosis [6-8]. In this study, we performed RNA sequencing to determine differentially expressed lncRNAs in DCM rats. Among them, we focused on DCRF, which was associated with autophagy activation. Our results demonstrated that high glucose could promote cardiomyocyte autophagy by regulating the DCRF/miR-551b-5p/PCDH17 axis, which appears to be involved in the pathogenesis of DCM.
The ceRNA theory has been proposed as a novel regulatory mechanism, which modulates the expression of target genes through miRNA competition [9]. In this study, our results suggested that DCRF could bind to miR-551b-5p and serve as a ceRNA to upregulate PCDH17 expression, thereby leading to increased autophagy in cardiomyocytes. This observation may provide valuable insight into the molecular mechanisms of DCM.
It is well recognized that autophagy plays a crucial role in the pathogenesis of DCM [10,11]. Autophagy is an intracellular lysosomal degradation process responsible for the clearance of unnecessary proteins and damaged organelles to maintain cellular homeostasis. However, increased autophagy contributes to cardiac damage and dysfunction due to autophagic cell death [12]. In this study, we found that DCRF could induce cardiomyocyte autophagy by upregulating PCDH17 expression in DCM. PCDH17, which belongs to the protocadherin gene family, has been shown to be associated with the activation of autophagy in cancer cells [13,14]. Our results revealed that high glucose could increase DCRF expression, which promotes the binding of miR-551b-5p to DCRF, thus reducing the repression of PCDH17. The ceRNA regulatory network, DCRF/miR-551b-5p/PCDH17, may provide a potential therapeutic strategy for DCM.
In conclusion, our study has shown increased expression of DCRF in diabetic myocardium. By functioning as a ceRNA, DCRF can upregulate PCDH17 through sponging miR-551b-5p and consequently activate cardiomyocyte autophagy, thus contributing to the progression of DCM.
Acknowledgements
This study was financially supported by National Natural Science Foundation of China (81770370), Scientific Research Program for Young Talents of China National Nuclear Corporation (51001), and Gusu Health Talents Training Project (GSWS2019045).
Author contributions
YF, WX, WZ performed the study and wrote the manuscript. WW and TL analyzed the experimental data. XZ designed the study and revised the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jia G, Hill MA, Sowers JR. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ Res. 2018;122:624-38
2. Marwick TH, Ritchie R, Shaw JE, Kaye D. Implications of Underlying Mechanisms for the Recognition and Management of Diabetic Cardiomyopathy. J Am Coll Cardiol. 2018;71:339-51
3. Moran VA, Perera RJ, Khalil AM. Emerging functional and mechanistic paradigms of mammalian long non-coding RNAs. Nucleic Acids Res. 2012;40:6391-400
4. Ounzain S, Burdet F, Ibberson M, Pedrazzini T. Discovery and functional characterization of cardiovascular long noncoding RNAs. J Mol Cell Cardiol. 2015;89:17-26
5. Sallam T, Sandhu J, Tontonoz P. Long Noncoding RNA Discovery in Cardiovascular Disease: Decoding Form to Function. Circ Res. 2018;122:155-66
6. Li X, Wang H, Yao B, Xu W, Chen J, Zhou X. lncRNA H19/miR-675 axis regulates cardiomyocyte apoptosis by targeting VDAC1 in diabetic cardiomyopathy. Sci Rep. 2016;6:36340
7. Zhou X, Zhang W, Jin M, Chen J, Xu W, Kong X. lncRNA MIAT functions as a competing endogenous RNA to upregulate DAPK2 by sponging miR-22-3p in diabetic cardiomyopathy. Cell Death Dis. 2017;8:e2929
8. Zhang M, Gu H, Xu W, Zhou X. Down-regulation of lncRNA MALAT1 reduces cardiomyocyte apoptosis and improves left ventricular function in diabetic rats. Int J Cardiol. 2016;203:214-6
9. Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505:344-52
10. Kubli DA, Gustafsson ÅB. Unbreak my heart: targeting mitochondrial autophagy in diabetic cardiomyopathy. Antioxid Redox Signal. 2015;22:1527-44
11. Kobayashi S, Liang Q. Autophagy and mitophagy in diabetic cardiomyopathy. Biochim Biophys Acta. 2015;1852:252-61
12. Liu Y, Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. 2015;22:367-76
13. Hu X, Sui X, Li L, Huang X, Rong R, Su X. et al. Protocadherin 17 acts as a tumour suppressor inducing tumour cell apoptosis and autophagy, and is frequently methylated in gastric and colorectal cancers. J Pathol. 2013;229:62-73
14. Wu JC, Wang FZ, Tsai ML, Lo CY, Badmaev V, Ho CT. et al. Se-Allylselenocysteine induces autophagy by modulating the AMPK/mTOR signaling pathway and epigenetic regulation of PCDH17 in human colorectal adenocarcinoma cells. Mol Nutr Food Res. 2015;59:2511-22
Author contact
![]() Corresponding author: Xiang Zhou, Department of Cardiology, The Second Affiliated Hospital of Soochow University, No. 1055 Sanxiang Road, Suzhou, 215004, P.R. China. Tel: +86 512 68282030 Fax: +86 512 68284303 E-mail: zhou-xiangedu.cn
Corresponding author: Xiang Zhou, Department of Cardiology, The Second Affiliated Hospital of Soochow University, No. 1055 Sanxiang Road, Suzhou, 215004, P.R. China. Tel: +86 512 68282030 Fax: +86 512 68284303 E-mail: zhou-xiangedu.cn