Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2019; 9(16):4580-4594. doi:10.7150/thno.34337 This issue Cite
Review
Circulating Tumor Cells as a Tool for Assessing Tumor Heterogeneity
Marta Tellez-Gabriel1 ![]() , Marie-Françoise Heymann2,3,4, Dominique Heymann2,3,4
, Marie-Françoise Heymann2,3,4, Dominique Heymann2,3,4 ![]()
1. RNA and molecular pathology (RAMP) research group. Department of Medical Biology. The Artic University of Norway (Tromso), Norway.
2. INSERM, European Associated Laboratory “Sarcoma Research Unit”, Department of Oncology and Metabolism, Medical School, University of Sheffield, United Kingdom.
3. INSERM, Institut de Cancérologie de l'Ouest, LabCT, U1232, CRCINA, Université de Nantes, Université d'Angers, 44805 Saint Herblain cedex, France.
4. Institut de Cancérologie de l'Ouest, “Tumor Heterogeneity and Precision Medicine”, 44805 Saint Herblain cedex, France.
Received 2019-2-22; Accepted 2019-4-23; Published 2019-6-19
Abstract

Tumor heterogeneity is the major cause of failure in cancer prognosis and prediction. Accurately detecting heterogeneity for the development of biomarkers and the detection of the clones resistant to therapy is one of the main goals of contemporary medicine. Metastases belong to the natural history of cancer. The present review gives an overview on the origin of tumor heterogeneity. Recent progress has made it possible to isolate and characterize circulating tumor cells (CTCs), which are the drivers of the disease between the primary sites and metastatic foci. The most recent methods for characterizing CTCs are summarized and we discuss the power of CTC profiling for analyzing tumor heterogeneity in early and advanced diseases.
Keywords: single cell, circulating tumor cell, precision medicine, tumor heterogeneity, omic technologies
Introduction
Anyone who has observed a tumor section under a microscope is familiar with the significant heterogeneity of the tumor mass. Within a single tumor mass, several foci with various, specific histological features can cohabit (Figure 1). In addition to their diverse morphology, cancer cells exhibit considerable heterogeneity in terms of genetic profile, gene expression, metabolic property, motility, proliferation and metastatic potential [1]. As proposed by S. Paget with the “seed and soil” theory, a symbiosis is established between cancer cells and their local micro-environment, defining the notion of tumor niche favorable for tumor growth [1]. Cancer cells regulate their environment by direct cellular contacts [1] or by secreting soluble extracellular vesicles [1]. In return, the micro-environment impacts the differentiation, proliferation and death of cancer cells, as well as contributing to a selection process leading to drug resistance, cell dormancy [1] and to an immune-tolerant environment [1]. Cancer cells are then functionally entangled with numerous other cell types (e.g. stromal cells, endothelial cells, immune infiltrate) that enrich the heterogeneous character of tumor tissues. Consequently, tumor heterogeneity is not a fixed state but should be considered as a dynamic ecosystem that evolves as the tumor progresses and is strongly modulated under therapeutic pressure. Tumor heterogeneity is subject to major spatial and temporal modulations that can impair sustained therapeutic response and drug sensitivity.
Typical microscopic observation of tumor heterogeneity. Osteosarcoma is a rare form of bone cancer mainly affecting adolescents and young adults. Osteosarcoma is a perfect illustration of highly heterogeneous tumors with multiple, diverse histological areas in a same tumor mass including osteoid, hypervascularized, proliferative and necrotic foci. In addition, associated lung metastases exhibit a histological morphology different from the primary tumor highlighting the contribution and effect played by the pressure of the local micro-environment on tumor heterogeneity.
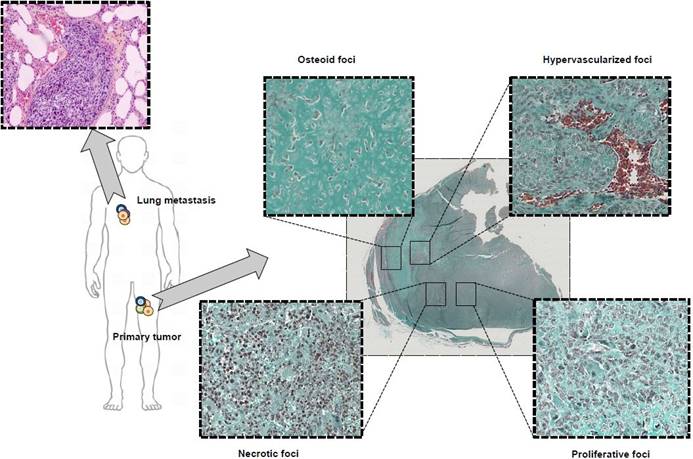
The evolution in good clinical practices (e.g. needle biopsies) has recently led to a significant decrease in the amount of biological material available for molecular investigations. A biopsy cannot reflect and illustrate spatial tumor heterogeneity and should be considered as a partial photograph of the tumor mass at a given time. The follow-up of the dynamic process requires repeated sampling carried out using low invasive methods and should be as representative as possible of the tumor mass. Repeated biopsies are ethically non-acceptable and sometimes unrealistic for high-risk localizations of metastatic foci (e.g. the spine). The establishment of metastatic disease is directly related to the migration of cancer cells called circulating tumor cells (CTCs) into the blood and/or lymphatic circulation, plus their ability to reach distant organs [1]. However, although CTCs are theoretically easily accessible, their low number makes it challenging to isolate and characterize them. The last decade has seen a surge in the new devices available for isolating CTCs, and specific downstream analysis workflows have been proposed in order to evaluate their biological value (e.g. therapeutic response, drug resistance, reflection of the tumor heterogeneity). The present review will give a brief overview of tumor heterogeneity: its origin, biological and clinical impacts and the methods for assessing it. We will put specific focus on the recent methods developed for isolating single cells and how these single cells can help to decipher this heterogeneity. We will also discuss how better characterization of single cells may orient future clinical decisions.
Tumor heterogeneity: origins and enrichment
Origin of cancer cell heterogeneity: from monoclonal to polyclonal disease
Cancer is characterized by the development and growth of abnormal cell populations (e.g. mutations, altered proliferation and/or differentiation). As DNA is the only cell component that can accumulate and transmit changes throughout life, it has been accepted that the carcinogenesis process requires the progressive accumulation of multiple DNA modifications [2]. The current, generally-accepted model for carcinogenesis is the somatic mutation or clonal evolution theory (Table A) [3, 4]. Although it is the prevailing model for carcinogenesis, it has been challenged by several lines of evidence [1, 2, 5, 6]. The main alternative model focuses on the concept of asymmetric division initially observed in healthy tissue renewal [1, 2, 5, 6]. This process is defined as a biological process in which a single cell generates two daughter cells with two distinct destinies: one undifferentiated cell - a stem cell - expressing stemness markers (e.g. Oct4, nano, sox2) and in charge of the self-tissue renewal, and one committed to a specific form of differentiation [14]. A similar process has been proposed in cancer leading to the maintenance of cancer-like “stem-cells” harboring the mutated driven gene and participating in the dynamic enrichment of the spatial and temporal heterogeneity of cancer cells [1,5,15,16]. Regardless of the carcinogenesis model, mutations in the tumor driven gene expressed by cells from pre-neoplastic lesions (e.g. TP53, Rb) lead to the formation of neoplastic foci characterized at the early stage by monoclonal expansions of mutated cells. This type of mutation results in genomic instabilities characterized by a high sensitivity to chromosome breakages and consequently a new series of mutations, deletions, and amplifications [17] (Figure 2A). Random chromosome breakages and secondary genetic events clearly contribute to the development of cancer cells with a new genotype and phenotype, and then to the polyclonal expansion stage of the disease. Epigenetic modifications complete the framework of heterogeneity mapping. As revealed by genetically homogeneous cell lines, intercellular epigenetic alterations (e.g. DNA methylation, miRNA expression, etc) strengthen tumor heterogeneity and the drug response [5,18,19]. Genetically- and epigenetically-modified cells are prone to migrate from their primary site. In the natural history of cancer, the diffusion of cancer cells from a primary tumor to a secondary distant organ is frequently observed. Interestingly, several authors have demonstrated the migration of prostate cancer cells from metastastic foci to seed new, distant locations or pre-existing lesions, and consequently permanently enrich the heterogeneity of the tumor [20, 21]. Thus, the bidirectional seeding between different tumor sites plays a part in enriching tumor heterogeneity.
Models of carcinogenesis.
| • The somatic mutation or clonal evolution theory is based on DNA changes in oncogenes and tumor-suppressor genes that lead to alterations in cell proliferation and/or cell-cycle arrest and/or cell differentiation and/or inhibition cell death. • The stem cell division theory for cancer suggests that Tumor-Initiating Cells (TICs) are the origin of cancer development. TICs are characterized by their capacity for self-renewal and play a part in the development of the heterogeneous lineages of cancer cells by accumulating successive asymmetric cell divisions. Similar to stem cells, TICs carry individual DNA from zygote to death and therefore hold the DNA long enough to accumulate the alterations required for carcinogenesis. |
Tumor models and tissue heterogeneity. A. From a pre-neoplastic lesion to the development of metastases, the tumor tissue will undergo a marked cellular evolution leading to polyclonal disease. Tumor driven genes appearing in determined normal cells will be responsible for chromosomal instability with numerous chromosome breakages (fusions, deletions, etc) concomitant to secondary genetic and epigenetic events. From the detection of the first oncogenic event, new clones will be formed and will enrich the heterogeneity of the tumor. The pressure of the local micro-environment and/or the therapeutic pressure will enrich the tumor mass in dominant/resistant clones, which will leave the primary tumors to spread to distant organs. Tumor heterogeneity is a property of cancers sustained and amplified by the reseeding of cancer cells from one site to distant foci. B. Several models of tumor development have been proposed and may coexist simultaneously in a single tumor mass. Three main models can be described: i) clonal evolution of an initial cancer cell in which subsequent genomic abnormalities occur will lead progressively (in a linear manner) to the emergence of new clones; ii) the various oncogenic events can also lead to the establishment of multiple subclones with common ancestors; this type of model is called the “branched model”; iii) more recently, both models have been completed by the “plasticity model” directly related to the plasticity property of cancer cells. One cancer cell can evolve between two phenotypic states, A/B, linked to various functional states explaining the co-existence and equilibrium of a mixed population expressing a large panel of fusion genes or/and cluster of differentiation and contributing to the polyclonal expansion and heterogeneity of the tumors. This type of mechanism increases the chance of survival for a cancer cell by upmodulating its adaptability to the micro-environment in a permanent manner.
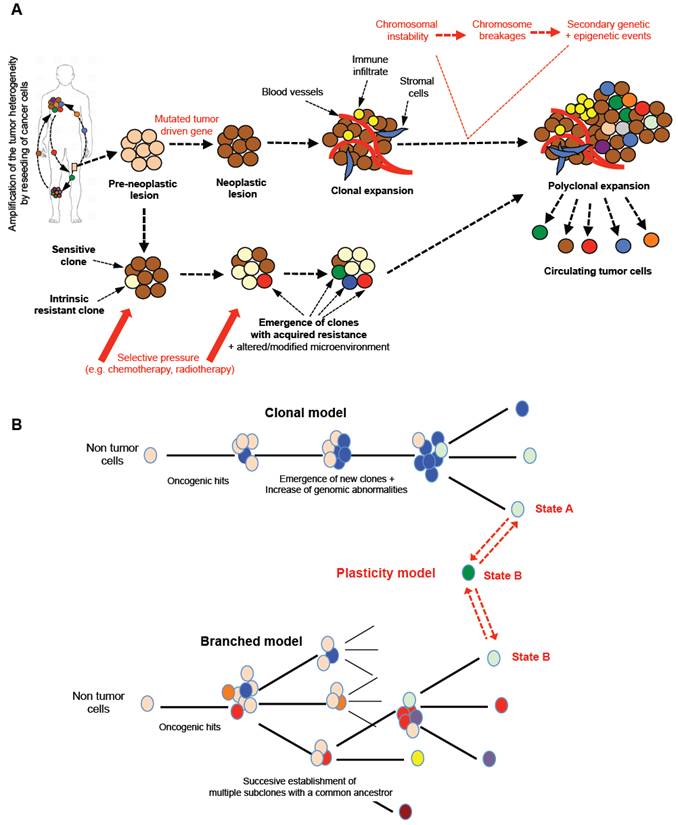
Similarly to embryonic cells, cancer cells are not blocked in a defined state and adapt permanently their phenotype under the microenvironment and therapeutic pressure. Indeed, phenotypic and functional plasticity is a common mechanism observed during embryonic development [22]. Blastocyst cells are composed of various cell types including stem cells, lineage-committed progenitors and differentiated cells linked by hierarchical relationships that make possible the formation of all the organs of the future embryo/fetus and also the extra-embryonic annex from the pre-implantation step to the final gestation. All of these cells can switch between different cell states thanks to a high degree of plasticity. Similarly, cancer cells exhibit marked plasticity as illustrated by the epithelial-mesenchymal transition (EMT) in which cancer cells progressively acquire a mesenchymal phenotype and lose their epithelial properties, thus leading to the metastatic process. In the opposite direction, at the metastatic location, cancer cells undergo mesenchymal-epithelial transition (MET) and re-acquire their epithelial characteristics [23]. Inoculation into immunocompetent mice of pure EpCAM+ or EpCAM- breast cancer cells, sorted by flow cytometry, led to the detection of mixed EpCAM+/EpCAM- cells in the blood stream after a couple of days. This simple experiment perfectly illustrates the extreme plasticity of cancer cells [24] (Figure 2B). The notion of plasticity should be extended to any subtle equilibrium making possible the functional orchestration of various cell populations. Recently, Franzetti et al. studied the impact of EWSR1/FLi1 expression on the functional behavior of Ewing sarcoma cells [25]. They observed that cancer cells fluctuated over time from low to high EWSR1/FLi1 expression in a reversible process, and the low expression phenotype was correlated with a metastatic profile (e.g. high propensity to migrate and invade). Both cell populations can co-exist in patient samples and EWSR1/FLi1Low contribute to the maintenance of tumor growth based on ESWR1/FL1 re-expression. Their manuscript illustrates a new model of phenotypic plasticity and gives evidence of the functional impact of this dynamic phenotypic fluctuation associated with a dominant oncogene.
However, the therapeutic pressure plays a significant role in the selective amplification of tumor heterogeneity and contributes to emergence of specific dominant clones driving the tumor heterogeneity [26]. A tumor mass is composed of a panel of cancer cells with sensitivity or innate resistance to a specific drug or specific therapeutic intervention [29] (Figure 2). Drug resistant clones are then preferentially chosen and in turn selectively modify the tissue heterogeneity. Therapeutic selective pressure is also responsible for acquired resistance mechanisms resulting in the dynamic emergence of new cancer cell clones leading to dynamic heterogeneity. The notion of drug resistance is also related to persister cells observed in cancer and in micro-organisms [5]. Persisters are low proliferating cells with a stem-like profile and immune tolerant activities. Overall, the literature demonstrates that tumor heterogeneity becomes an obstacle to determining the appropriate therapeutics in oncology because of the temporal instability of tumor tissue organization. The dynamic evolution of dominant clones and persister cells fuel the tumor heterogeneity which is enriched by a heterogeneous local micro-environment.
Heterogeneity of the tumor micro-environment: the functional relationship of tumor heterogeneity
As described above, from a clonal disease, the successive mutations in tumor cells play a part in temporal heterogeneity and the establishment of a very complex polyclonal oncogenic disease. In addition to the heterogeneous populations of neoplastic cells, tumor bulk is composed of non-neoplastic resident cells, the extracellular matrix [7-10], fibroblasts (called cancer-associated fibroblast) [7-10], blood vessels [7-10] and immune cells [7-10] that together form the tumor micro-environment (TME) (Figure 3). MALDI imaging mass spectrometry makes it possible to visualize tumor heterogeneity at the protein level [7-10]. Extracellular matrix is a key factor related to metastasis efficiency, controlling collective cell invasiveness [7-10]. This observation is related to the diversity of cancer-associated fibroblasts (CAF) [7-10]. Indeed, Costa et al. identified four subsets of CAF in breast cancer with specific distinct functional properties. In triple negative breast cancers, one of them, called CAF-S1, promotes an immune tolerant environment and stimulates T lymphocytes toward an immunosuppressive phenotype (CD25high FOXP3high). The second, called CAF-S4, increases the T cells' regulatory property to inhibit T effector proliferation. Consequently, the local accumulation of CAF-S1 then contributes to tumor heterogeneity and to local immunosuppression observed in triple negative breast cancers. Such immunoregulation is tightly controlled by the production of local immunocytokinic signals leading to a balance between inflammatory and immunosuppressive effectors [7-10]. The functional impact of CAF on local tumor immunity is directly linked to the spatial and temporal heterogeneity of T lymphocytes and macrophages observed in numerous types of cancer [31-33[7-10]. Interestingly, resident lymphocytes seem pre-adapted to specific tissues and can adapt to wherever they migrate [34[7-10]. As a consequence of local immune regulation, endothelial cells exhibit several phenotypic features and lead to the formation of specific tumor vasculature [7-10]. Interestingly, Hamilton et al. revealed that CTCs are competent to modulate tumor associated macrophages in order to increase invasiveness of cancer cells, angiogenesis and immunosuppression [7-10]. The quality (e.g. topographic localisation) and quantity of the immune infltrates into tumor tissues have strong impacts on patients' clinical outcomes. New technologies such as multispectral imaging will allow to obtain a precise analysis of these infiltrates and may lead to a better patient stratification [7-10]. All components of the tumor microenvironment then play a part in generating more tumor variability, as well as being highly heterogeneous and crucial for determining the development of cancer [7-10]. After the tumor excision and the initiation of the therapy, the key challenging question remains the follow up of the tumor heterogeneity in absence of tumor tissue access? Do CTCs reflect the tumor heterogeneity?
Spatial immunological heterogeneity of tumor tissue. Illustration of the heterogeneity of immune infiltrates associated with human osteosarcoma (cohort previously published in [137]). Numerous immune cell subtypes invade osteosarcoma tissues during tumor development. Interestingly, their spatial distribution shows a high heterogeneity across the tumor tissue, with CD3+ T lymphocytes organized in a diffuse infiltrate as well as small clusters. The localization of CD8+ T cells is diffuse with one area without any infiltrated cells. Macrophages exhibit similar distribution to CD3 and the number of CD20+ B lymphocytes is relatively low but B cells are sometimes organized in pseudo-nodules. CD117+ mastocytes are also observed as diffuse infiltrate in a specific area.
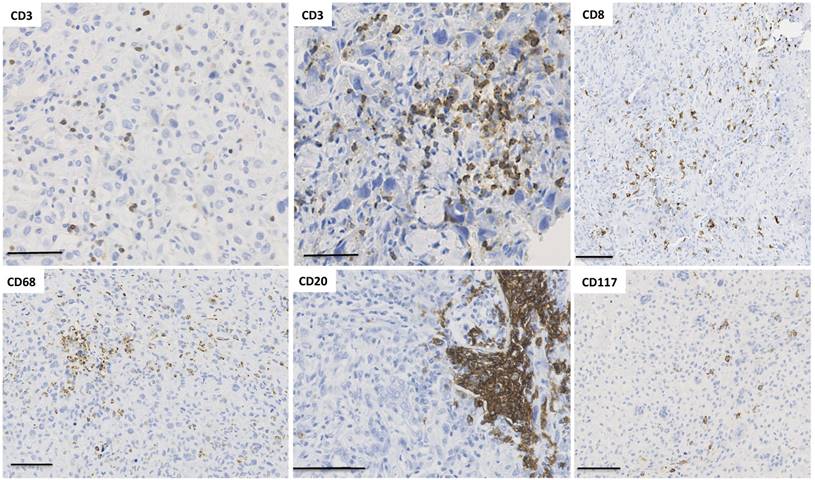
The characterization of circulating tumor cells for predicting tumor heterogeneity
Tumor heterogeneity has challenged the potential benefits of precision medicine. Current methods used to analyze tumor masses and further therapeutic design are based on a global overview of the characteristics of cancer tissues, corresponding to an average picture of all the tumor clones and their micro-environment without considering their diversity [11]. This variability limits the prognosis and predictive power of a biomarker. Moreover, differences in the evolution of tumor cells and micro-environments at the metastatic sites question the utility of the biomarkers previously identified in the primary tumor for treating metastatic disease [12, 13]]. Monitoring changes in cell populations during disease progression and treatment will improve both cancer diagnosis and therapeutic design. Current protocols to check the consistency of the biomarkers, establish diagnoses and define treatment are based on very small biopsies (e.g. needle biopsies) of the primary tumor and metastatic sites. The main limitation is that most metastases are difficult to access, and biopsies are invasive, inconvenient, costly and do not make possible longitudinal follow-up of tumor heterogeneity. To overcome these problems, detecting and characterizing heterogeneity in CTCs could be a good alternative and opportunity.
CTC characteristics as a snapshot of tumor heterogeneity
In the last decade, numerous clinical studies revealed the link between CTC numbers and metastatic prognosis [49, 50]. Similarly, the phenotypic properties of CTCs can be related to overall patient survival [51]. In the most recent meta-analyses published, the authors found a correlation between CTC count and clinical progression. Recently, by combining more than 20 published studies, CTC count was shown to be an independent and quantitative prognostic factor in patients suffering from early breast cancer [52]. At the protein level, there are some discrepancies depending on the series analyzed. In breast cancer for instance, HER2 expression between initial tumor tissues and corresponding CTCs are contradictory, with a concordance of around 90% for some of them [52,53], inconsistency for others [54] or fluctuating variations in the course of the disease [55]. Similar to EpCAM, the high plasticity of cancer cells results in variations in HER2 expression and reflects a snapshot of tumor heterogeneity at a specific time point. However, several studies support the idea that cellular heterogeneity in CTCs reflects the spectrum of mutations in the primary tumor and metastatic lesions. Mohamed Suhaimi et al. detected a high concordant mutation list in CTCs and tissues (e.g. KRAS and BRAF markers), predicting the outcome of anti-EGFR therapy in colorectal cancer patients [14]. Bingham et al. showed that CTCs were representative of the entire spectrum of mutations present in the primary tumor and distal metastases for patients with breast cancer. Their findings suggested the utility of CTCs for identifying targetable mutations and for being used as a biomarker to reveal cell populations sensitive to current or previous therapies [15].
Despite these promising results, the existence of heterogeneity in the CTC compartment has been demonstrated, as shown by Pestrin et al., who observed variability in the mutational status of the PIK3CA gene in breast cancer patients. The PIK3CA mutation profile has prognostic significance and is potentially predictive for the response to agents targeting the PI3K pathway 8 Similarly, Pailler et al. observed considerable heterogeneity in ROS1-gene abnormalities in CTCs from non-small cell lung cancer (NSCLC), which could explain the mechanism by which tumor cells can escape sensitivity to ROS1-inhibitor therapy [16]. Furthermore, it has also been demonstrated that CTC profiles evolve as the disease progresses, illustrating the temporal heterogeneity of cancer diseases. Tsao et al. then mapped the phenotypic evolution of melanoma CTCs and detected the presence of drug-resistant clones harboring different molecular signatures of potential clinical value [17]. The discordance observed between CTCs and tumor heterogeneity may be explained by the dynamic heterogeneity of the CTCs [61, 62]. Primary tumors as well as metastatic foci are permanently reorganized, resulting dynamically in the differentiation and release of new cancer cell clones into the bloodstream, explaining the partial genomic overlap of CTCs and tumor foci at a specific time point. Consequently, even CTCs cannot fully reflect tumor heterogeneity: they are the mirror and snapshot of the disease's progression as well as the clonal evolution of the tumor foci. Overall, the data currently available has pointed out the major advantages of CTC investigation at the single cell level, possibly representing the most accurate strategy for determining the temporal heterogeneity of the disease.
Single CTC analysis could effectively reveal the high heterogeneity, stochastic changes, and driver mutations in cancer cell populations in order to detect drug resistance, and develop new personalized therapeutic strategies as well as their use as prediction markers. De Luca et al. validated a protocol to assess the clonal evolution of metastatic breast cancer, based on single CTC analysis by next generation sequencing (NGS), suitable for the development of new therapeutic strategies in precision medicine [18]. Miyamoto et al. analyzed single-cell RNA-sequencing (RNA-Seq) profiles in prostate cancer and concluded that there was CTC heterogeneity in signaling pathways that could contribute to treatment failure [19]. Pailler et al. investigated ALK-copy number gains (CNG) in individual CTCs by filter-adapted fluorescent in situ hybridization (FA-FISH) and reported a significant association between the dynamic evolution of the numbers of ALK-CNG and progression-free survival (PFS) in NSCLC patients treated with crizotinib, an ALK/ROS1 inhibitor [20]. Paolillo et al. found ESR1 mutations in single CTCs from metastatic breast cancer patients associated with endocrine therapy resistance [21]. As mentioned above, the tumor cell component is not the only source of heterogeneity. TME elements present high variability that can be assessed by single-cell analysis. Tirosh et al. presented an extensive study of the heterogeneity associated with the components that shape the melanoma micro-environment, assessed by single-cell RNA-seq. The authors discovered different micro- environments associated with distinct malignant cell profiles that could be used as prognostic markers [22].
CTC cluster enrich CTC heterogeneity and have increased metastatic potential
In addition to single CTCs detectable into the bloodstream, CTCs can be observed in clusters composed by cancer cells and/or in association with non-malignant cells. Recent findings suggest that CTC clusters may have a greater contribution to the metastatic process for mechanical and immune features. Indeed, Au et al. demonstrated that cluster CTCs are able to reorganize into single-file chain-like geometries in a rapid and reversible manner with reduced hydrodynamic resistance. Consequently, the progression of cluster CTCs through capillaries is slowed down and cancer cell clusters can traverse easily thin constrictions for extravasation and migration into distant organs [23-25]. CTC clusters are rare compared to single CTCs, do not come from intravascular aggregations and arise from oligoclonal cancer cell groupings (called homotypic CTC clusters) reinforcing the CTC heterogeneity [23-25]. Heterotypic CTC clusters have been also observed in patient samples. In that cases, CTC clusters are not only composed by tumor cell clones but are associated with tissue-derived macrophages [23-25], fibroblasts [23-25] or neutrophils [23-25]. Non-malignant cells escorting CTCs may strengthen the biomechanical properties of CTC clusters and may improve their survival. Very recently, Szczerba et al. demonstrated that the association between neutrophils and CTCs drove cell cycle progression within blood and increased the metastatic potential of CTCs in breast cancer patients [23-25].
However, heterotypic CTC clusters have never been associated with circulating lymphocytes. What could be the relationship between T lymphocytes and CTCs? A close relationship between immune cells and cancer cells has been described through the immune checkpoints [23-25]. Cancer cells can express programmed death-ligand (PDL-1) which binds to PD-1 and/or B7/1 expressed at the cell surface of T lymphocytes. This binding results in the inhibition of downstream signaling and in the decrease of T cell proliferation and an increase of their apoptosis. Recently, Mishra et al. compared the dialog between immune cells and CTCs isolated from non-metastatic and metastatic cell lines inoculated in syngeneic immune-competent mice [23-25]. In this model, the metastatic cell line exhibited a significant highrer expression of PDL-1 compared to the non-metastatic cell line and inoculation of activated immune cells had no impact on CTCs established from metastatic cell line in contrast to CTCs produced from non-metastatic cell line. PDL-1 was found to correlate with histological tumor grading and is frequently expressed by CTCs [23-25]. Since PDL-1 is a molecular regulator of regulatory T lymphocytes, a CTC immune escape mediated by PDL-1 can be hypothesized [23-25]. In such context, T lymphocytes may indirectly contribute to the tumor CTC heterogeneity by selecting specific dominant cancer cell clones that escape to the immune surveillance and lead to the release of specific CTCs into the bloodstream. To illustrate this purpose, Sun et al. studied a series of non-small cell lung cancer patients [23-25]. They observed that the number of CTCs were positively associated with the metastatic process and negatively associated with the level of circulating T lymphocytes. CTC cluster thanks the interactions between cancer cells and non-malignant cells contribute to the CTC survival, proliferation, immune escape and drug-resistance [23-25].
Brief overview of the most significant breakthrough technologies for deciphering single-cell characteristics
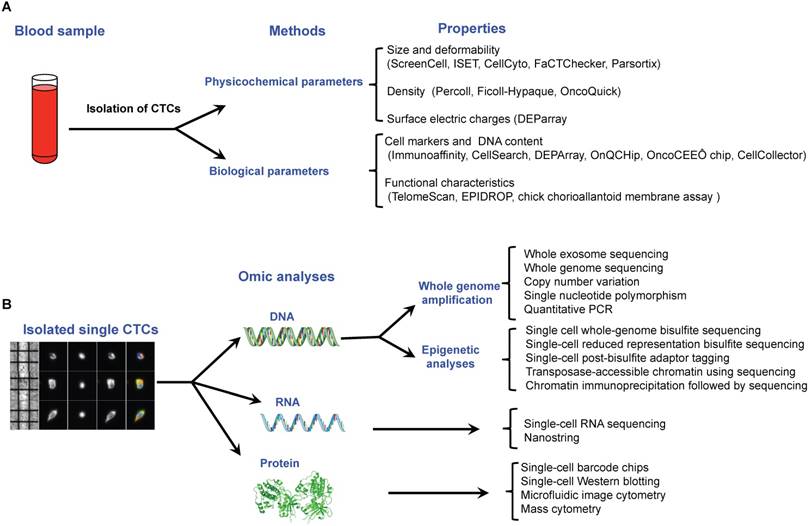
CTCs are rare cell events in blood with a frequency of around one tumor cell for 106-108 normal blood cells. Consequently, before single CTC analysis can take place, the first step is to enrich and isolate CTCs. Multiple technologies have been described to do this in individual CTCs, based on the different properties that distinguish them from surrounding normal hematopoietic cells, including biological properties (cell surface protein expression, viability, invasive capacity) and physical properties (size, density, electric charges, deformability) reviewed previously [23-25] (Figure 4A). Once single cells have been isolated, downstream analyses can be performed depending on the molecular components assessed: DNA, RNA or protein (Figure 4B).
Recent technological approaches used for isolating and characterising circulating tumor cells. A. Isolation of single CTCs is based on a two steps method including a pre-enrichment step followed by an isolation approach. All of these methods are related to the physicochemical or biological properties of CTCs. B. Single CTCs can be characterized by omic methods at the DNA, RNA and protein levels.
Single-cell DNA analysis
Studying genetic variability in single cells has stimulated the development of several high throughput sensitive methods for detecting large patterns of mutations including target-specific amplification using PCR, as a means of querying specific loci of interest [26], and the next generation of sequencing approaches: whole exome sequencing (WES), [27] and whole genome sequencing (WGS) [28] to obtain information regarding the complete exome and genome respectively. As the amount of DNA that can be extracted from a single CTC is limited, an initial step involving whole genome amplification (WGA) is required. Depending on the downstream application, the WGA method selected can differ. Interestingly, recent studies compared different WGA methods and revealed that that multiple displacement amplification (MDA) methods are better suited for single nucleotide polymorphism (SNP) detection, while PCR-based methods are the better option for copy number variant (CNV) detection [29]. The authors suggested that AMPLI1 or MALBAC should be used in favor of the REPLi-G or PicoPlex kits when high target coverage is required [30]. They also found higher sensitivity of DOPlify and Picoseq for detecting 100% of CNVs than Ampli-1 and REPLI-g [31]. After genome amplification, the type of genomic investigation must be defined according to the objective of the study. For instance, Polzer et al. used a qPCR assay to analyze the variability of HER2 and PIK3CA in breast cancer single CTCs. Their findings demonstrated that assessing the heterogeneity for these two markers may uncover tumor evolution mechanisms useful for personalized therapy decisions [32]. In another study, Janiszewska et al. assessed single-nucleotide PIK3CA mutations and HER2 copy number alterations in single cells in formalin-fixed paraffin-embedded breast tumor samples using the STAR-FISH (specific-to-allele PCR-FISH) methodology. This analysis proved to be useful for predicting clinical outcomes in breast cancer patients subjected to neo-adjuvant chemotherapy followed by adjuvant therapy with trastuzumab [33].
In the last few years, there has been a significant increase in the number of studies that use next generation sequence approaches for unraveling single cell heterogeneity. The study performed by Li et al. identified 4 new driver mutations in renal cell carcinoma stem cells using WES that constitute important prognostic factors and therapeutic targets [34]. Liu et al. combined multi-region WES and single-cell WGS to examine the intra-tumor heterogeneity of rectal tumors. Their results suggest a specific architecture for each tumor related to different diagnoses, prognoses and drug responses [35]. Sequencing single CTC genomes and exomes provides considerable amounts of information, but also faces several technical challenges, in addition to difficulties with CTC capture [23]. The key limitations of single-cell sequencing using WGA are low coverage of the human genome and the consistency of the coverage between single cells [36]. This issue could be solved by third-generation sequencing technologies, such as the Pacific Bioscience system [37] and Nanopore sequencing [38]. The second challenge is data analysis, as currently the bioinformatics tools used for single-cell sequencing were initially developed and adapted for bulk cell sequencing. New computational and statistical methods have been developed to meet the requirements of single-cell analysis to reduce these biases and address biological and clinical questions more accurately [39-41].
Single-cell epigenetic analysis
Robust technologies have been developed for mapping epigenetic marks in single cells. Bisulfite conversion followed by sequencing (BS-seq) is considered the gold-standard method for single base resolution and absolute quantification of DNA methylation levels [39-41]. Farlik et al. described a whole-genome bisulfite sequencing (WGBS) assay that makes it possible to analyze heterogeneous DNA methylation patterns in single cells (scWGBS) and compared it with the other two methods: single-cell reduced representation bisulfite sequencing (scRRBS) and single-cell post-bisulfite adaptor tagging (scPBAT) [42]. The authors concluded that scWGBS is the method of choice for analyzing large numbers of single cells at low sequencing coverage, scRRBS is useful for comparing CpG islands across single cells, and scPBAT is best suited for deep sequencing of single cells with maximum coverage [43]. For mapping histone marks at the single cell level, chromatin immunoprecipitation followed by sequencing (ChIP-seq) has recently been implemented. Rotem et al. used this methodology to investigate the cell-to-cell variability of different types of regulatory elements and they confirmed its suitability for revealing aspects of epigenetic heterogeneity not captured by transcriptional analysis alone [44]. To assess the spatial organization of chromosomes, Kind et al. successfully modified the DamID method [45]. The heterogeneity of chromatin structure in single cells can be monitored by assay for transposase- accessible chromatin using sequencing (ATAC-seq) [46] and DNase I hypersensitive site sequencing (DNase-seq) [47]. Nowadays, it is also possible to assess variability in the 3D structure of chromosomes at the single cell level using a HiC-based method [48]. Other methodologies that have been developed to analyze epigenetic changes in bulk samples can potentially be adapted to single cell analysis [49]. Despite the fact that analyzing epigenetic state in CTCs is still in its infancy, various studies have already been published in the field. Pixberg et al. analyzed the epigenetic status of the genes associated with epithelial mesenchymal transition (EMT) in individual breast cancer CTCs, and explored potential intra- and inter-patient heterogeneity using the agarose-embedded bisulfite sequencing (AEBS) protocol. They found heterogeneous methylation patterns in CTCs with clear infrequent hypermethylation at key promoters of the inhibitor genes of the EMT, suggesting that both epithelial and mesenchymal CTCs can contribute equally to the metastatic process [50].
Single-cell RNA analysis
Single-cell RNA sequencing (scRNAs) is the method selected if the aim of the study is to explore transcriptome heterogeneity [51]. Interestingly, Ziegenhain et al. performed a comparative analysis of the most prominent scRNA-seq methods and identified Drop-seq as the best method for analyzing the transcriptome of large numbers of cells with low sequencing depth, SCRB-seq and MARS-seq are preferable for the transcriptome of fewer cells, and Smart-seq2 would be the appropriate method of choice when annotating the transcriptome of very small quantities of cells [52]. All the data generated after scRNAs should be processed to avoid false-positives due to nonlinear amplification, false-negative allelic drop-out due to amplification bias, non-uniform coverage, and noise that arises during single-cell transcript amplification. Specific computational models have been specifically developed to address these issues [53].
Numerous studies show extensive use of scRNAs for unraveling CTC heterogeneity. Patel et al. reported the heterogeneity of single glioblastoma CTCs and the existence of high variability in signaling molecules relevant to the targeted therapy, a wide spectrum of stemness and differentiation states, variable proliferative capacity and expression of quiescence markers, all of which were related to the success or failure of therapeutic strategies [54]. Chung et al. used scRNA to characterize heterogeneity in tumor cells and TME components (mainly immune cells) in breast cancer. The authors identified various signatures in both compartments related to tumor development and the response to cancer therapy [55]. Despite the multiple advantages conferred by single- cell RNAseq, it also presents certain limitations that need to be resolved. The main ones are that RNA losses have to be kept to a minimum during cDNA conversion, and that the amplification should provide enough DNA for sequencing without too much quantitative bias or altering of the original picture of the cells' transcriptomic profile. Moreover, scRNA-seq methods use an oligo-dT primer that specifically captures only polyadenylated RNA, avoiding the unwanted amplification of tRNA and rRNA. However, it represents a problem for the non-polyadenylated RNAs such as long non-coding RNA and microRNAs that have been shown to play important roles in cancer [56, 57]. Some commercial kits have been developed to overcome the poli(A) tail restriction [58]. Another challenge is the low signal-to-noise ratio of single-cell RNA-seq technologies. It is thus important that cell isolation, library preparation, and other automated workflows be as standardized as possible to minimize any bias introduced by human error [59]. In this regard, Suzuki et al. proposed the use of standard cell lines in future quality controls [60]. Finally, many scRNA-seq analyses are still performed using methods originally developed for bulk RNA-seq even if their adaptability to single-cell transcriptomics is unclear [61]. As the reliability of the bioinformatic method directly determines the accuracy of the experimental results, it is important to develop bioinformatics tools specific to the analysis of single-cell RNA-seq data, such as the two very recent methods developed by Wu et al. [62] and Miao et al. [63].
Single-cell proteomic analysis
Recent progress in microfluidic technologies and mass spectrometric approaches have led to new single-cell proteomics studies that could be performed with greater sensitivity and specificity. The micro- engraving technique, single-cell barcode chips (SCBCs) and single-cell Western blotting (scWB) are the microfluidic platforms providing the most advanced capabilities [64]. The latest version of microfluidic image cytometry (MIC) makes it possible to analyze the heterogeneous expression of up to 90 proteins in each single cell [65]. Another technology is CyTOF, a mass cytometry platform that has been developed to assess phenotypic heterogeneity at the single cell level. It can simultaneously image the localization and modifications of 34 proteins (and potentially up to 100) in each cell at subcellular resolution [66]. Sinkala et al. validated the clinical utility of proteomics in single cells by using scWB, and analyzed variability in metastatic breast cancer cells. They observed high heterogeneity in the expression of 8 key proteins related to breast cancer progression [67].
Conclusions and perspectives
Repeatable, minimally-invasive and cost-effective approaches for real time assessment of relevant biomarkers and monitoring cancer therapies in the bloodstream have been developed to overcome the intrinsic limitations of primary tumor and metastasis biopsies. This field of investigation has been termed “liquid biopsy” and includes circulating tumor cells (CTCs), exosomes, circulating cell-free DNA (cfDNA), miRNAs and proteins. CTC characteristics can be considered to be a snapshot of overall tumor bulk (primary tumor and metastases). Compared to other liquid biopsies, CTCs are a little bit more laborious to obtain but can be analyzed at the DNA, RNA, and protein level, as well as with regard to their functional cellular characteristics as a means of providing information that relates to the whole cell [67, 68].
Pooling the cells might provide different results and could mask clinically relevant rare mutations [18]. A major question emerges regarding the number of CTCs that need to be analyzed in order to capture the overall profile of the dominant disease driving the (sub)clones in a patient suffering from widespread metastatic disease. Gao et al. conducted a study on this subject and concluded that around 20-40 single cells are required to detect the main subclones with 95% power [69]. Despite promising results, showing a high concordance between paired CTCs and primary tumors or metastatic sites [15, 70], many other studies found discordant results between the mutational status of CTCs and those of the corresponding primary tissue or metastasis [71-73]. Like tumor tissues, CTCs are in fact heterogeneous in all the cancer types analyzed [57,60, 131-143] (Table 1). Very recently, Sun et al. demonstrated the existence of specific CTC territories, marking the spatial heterogeneity of CTCs. These authors compared the EMT status of CTCs isolated from various vascular territories and observed surprisingly high heterogeneity depending on their location [138] and hypothesized that spatial CTC heterogeneity could impact both the recurrence of the disease and the metastatic process. This could be explained by the fact that CTCs reflect the dynamic evolution of the advanced stages of cancer more closely than the primary tumor (temporal heterogeneity) [18].
Recent studies analyzing the heterogeneity of circulating tumor cells (CTCs) at the single cell level.
| Analytical methods | Isolation methods | Number of patients | Clinical relevance | Reference |
|---|---|---|---|---|
| Allele-specific PCR | Size-based microsieve technology | 44 | Analysis of KRAS and BRAF heterogeneity analyzed in CTCs can predict outcomes of anti‐EGFR therapy in colorectal cancer patients. | [54] |
| Foundation One™ | CellSearch followed by single-cell isolation by DEPArray Markers used : EpCAM+/-, CD45-, DAPI | 32 | CTC analysis can be used to identify targetable mutations, and as a biomarker to reveal the sensitivity to therapy of different breast cancer cell populations | [55] |
| PI3KCA Sanger sequencing | CellSearch followed by single-cell isolation by DEPArray Markers used : EpCAM+/-, CD45-, DAPI | 39 | Detection of variability in PIK3CA gene mutational status in single CTCs isolated from breast cancer patients. PIK3CA mutations have prognostic significance and are potentially predictive for response to agents targeting the PI3K pathway | [56] |
| Filter-adapted-fluorescence in situ hybridization (FA-FISH) | Filtration, Isolation by size of epithelial tumor cells (ISET) | 8 | Heterogeneity of ROS1-gene abnormalities in CTCs from non-small cell lung carcinoma could explain the tumor cells' resistance to ROS1-inhibitor therapy | [57] |
| Antibody-conjugated and surface-enhanced Raman spectroscopy (SERS) | No CTC isolation, detection of CTCs in blood samples | 10 | Detection of cell heterogeneity in CTC drug-resistant clones with potential clinical value for treatment decisions (melanoma) | [58] |
| Next Generation Sequencing (NGS) | CellSearch followed by single-cell isolation by DEPArray Markers used : EpCAM+/-, CD45-, DAPI | 4 | High intra-tumor heterogeneity in breast single CTCs in genes related to therapeutic response. This can be used to assess the clonal evolution of metastatic breast cancer and further therapeutic intervention based on the mutational status. | [61] |
| Single-cell RNA-sequencing (RNA-Seq) | CTC-iChip Markers used : EpCAM+/-, CDH11+/-, CD45- | 22 | Complex inter-tumor and intra-tumor heterogeneity in drug resistance mechanisms of analyzed prostate single CTCs relates to anti-androgen therapy failure. | [62] |
| FA-FISH | Enrichment by ISET, enumeration by CellSearch Marker used: EpCAM+/-, CD45- | 18 | Inter-tumor heterogeneity in CTC numbers with ALK-copy number gains has significant association with crizotinib efficacy and progression-free survival (PFS) in non-small cell lung carcinoma. | [63] |
| Sanger sequencing | CellSearch Marker used: EpCAM+/-, CD45- | 30 | Clonal heterogeneity analysis in single CTCs from metastatic breast cancer patients revealed early ESR1 mutations associated with endocrine therapy resistance. This can be used to predict which patients will benefit from a given therapy. | [64] |
| RNA-Seq | Flow cytometry: CD45- | 19 | Detection of intra- and inter-individual heterogeneity in melanoma cells and tumor micro-environment components linked to resistance to targeted therapies. | [65] |
EMT biomarkers (e.g. TWIST1, SNAI1/2, N-cadherin, vimentin) are differentially expressed in CTCs [144-146]. EMT was shown to enhance metastatic properties of tumor cells. Markiewicz et al. analyzed the link between the detection of breast cancer CTCs with a mesenchymal phenotype and EMT status of primary tumors [147]. In their series, mesenchymal phenotype of CTCs was more frequent in primary tumors with E-cadherin loss compared to those with normal E-cadherin expression. However, EMT status of matched samples at different stages of dissemination was frequently discordant, especially for pairs associating CTCs. In more of 500 breast cancer patients, CTCs were detected in only 19% of blood samples [148]. These authors identified a subset of primary breast cancer patients with EMT (29%) and stem cell (14%) phenotype and they did find any correlation between these markers and other prognostic clinical markers. Similarly, it has been shown that around 30% of metastatic prostate cancer patients had no detectable EpCAM+ CTCs [149]. More recently, Lowes et al. studied the EMT process and CTC release in pre-clinical models of prostate cancer [150]. They confirmed that that the method used for isolating CTCs is crucial and that CellSearch®-based assay used in their study failed to detect around 40-50% of CTCs with mesenchymal phenotype. Overall, these studies confirmed the high plasticity of cancer cells and demonstrated that the current methods used for detecting/isolating all subtypes of CTCs which undergo EMT are not efficient enough. Novel technological approaches are required to better follow the metastatic disease.
Nowadays, most of the studies published focus on revealing cell-to-cell differences at the DNA and RNA level. For overall understanding of single-cell heterogeneity, future studies should focus on the combination of different multi-omic assays on the same cell, such as the study performed by Hou et al. in which the authors used a single-cell triple omics sequencing technique called scTrio-seq which links the complex contribution of genomic and epigenomic heterogeneities to transcriptomic heterogeneity within a population of cells [151]. After conquering the barrier of multi-omics analysis for single cells, the final challenge will be temporal and spatial measurement of the molecular profile in a single cell. New technologies should not only solve the problem of the existing analysis methods which characterize only a snapshot profile of CTCs, but also provide real-time dynamics to measure patient status and then to follow the heterogeneity of the disease. The primary aspect of this new technology is in vivo monitoring and analysis of single CTCs, as has been shown in different studies [152, 153], but the high cost and lack of sensitivity prevent it from serving as a routine clinical test. In addition, future studies should also consider the importance of micro-environment and immunological elements in the heterogeneity state of cancer cells, as numerous emergent studies have demonstrated their consequences in tumor evolution and therapeutic response [7, 9, 10, 154]. In this regard, efforts must be made to clarify the complex interplay among cells within the tumor ecosystem and between functional states in space and time before its translational application [22] (Table B). Understanding tumor heterogeneity is of the utmost importance, as this phenomenon is associated with a decrease in diagnostic precision and is an obstacle for designing appropriate therapeutic strategies. Enumeration and molecular profiling of CTCs may be useful for a better patient stratification. High content analysis of CTCs can give a snapshot of the tumor heterogeneity at a given time and could allow to adapt therapeutic approach all along the treatment. Indeed, CTCs like all cancer cells are highly plastic and can modify their phenotype according the micro-environmental and therapeutic pressure. EGFR-mutated non-small lung cancer is a good illustration of cancer cell plasticity related to drug resistance [22]. EGFR-mutated NSCLC is a genetically heterogeneous disease with more than 200 distinct mutations. The identification of the most common L858R mutations-predict sensitivity to EGFR tyrosine kinase inhibitors. However, some patients become progressively resistant to the first line of tyrosine kinase inhibitor by developing new sets of mutations of EGFR illustrating again the plasticity of cancer cells. CTCs collection is weakly invasive and would allow the follow up of EGFR mutation status in order to adapt the therapy “in real time” [155]. Genetic/ epigenetic/molecular profiling of CTCs open new era of personalized medicine. Unfortunately, the implementation of single CTCs in clinical practice is still limited because technologies are expensive, time-consuming and require standardization processes. Current protocols should be replaced by new ones that make it possible to obtain results in a short time and thus avoid any delay in treating the disease for cancer patients.
Precision of medicine: the future
| • Losing a significant amount of CTCs can be associated with subsequent misinterpretations of heterogeneity, and thus bad clinical decisions. • Future studies should focus on the combination of different multi-omics assays on the same cell to obtain a heterogeneity profile at different molecular levels. • Efforts should be made to implement in clinical practice real-time heterogeneity single CTC analysis to measure patient status at any time in the course of the disease. • Many studies have shown that TME plays an important role in tumor heterogeneity. Upcoming research studies assessing tumor heterogeneity thus need to include the analysis of different components in the TME. • It is mandatory to run clinical trials to clarify the clinical utility of CTC data. • The implementation of single cell heterogeneity analysis in clinical practice is a priority for improving precision medicine. Scientific and clinical communities should concentrate their efforts on solving the problem of high cost and time-consuming technologies. • A social debate is open for the near future regarding the difficulties many patients have with affording the high cost of getting diagnosed by precision medicine technologies. |
Glossary
Circulating tumor cells (CTCs): originate from the primary tumor or metastatic foci and at least some of them are able to invade the surrounding tissue, enter either the lymphatics or the bloodstream, survive in circulation, extravasate into a tissue and finally grow at the new site.
Tumor micro-environment (TME): is the cellular environment in which the tumor exists, including surrounding blood vessels, immune cells, fibroblasts, bone marrow-derived inflammatory cells, lymphocytes, signaling molecules and the extracellular matrix. TME and tumors are closely-related and interact constantly. Cells in the micro-environment can affect the growth and evolution of tumor cells, while cancer cells can induce changes in the micro-environment by releasing extracellular signals, promoting tumor angiogenesis and inducing peripheral immune tolerance.
Liquid biopsy: non-invasive test performed on blood samples for the capture and analysis of molecules originating from tumors such as CTCs, exosomes, miRNAs, proteins and circulating cell-free DNA. The main potential applications of this technique are: the screening and early detection of cancer; relapse-risk estimation; identification of therapeutic targets for precision medicine; and real-time monitoring of response to therapy and anticipation of emergent therapy resistance.
Precision medicine: refers to the adjustment of medical treatment to the individual characteristics of each patient. It does not literally mean the creation of drugs or medical devices that are unique to a patient, but the ability to classify individuals into subpopulations that differ in their susceptibility to a particular disease, in the biology or prognosis of those diseases they may develop, or in their response to a specific treatment. Preventive or therapeutic interventions can then be concentrated on those who will benefit, sparing expense and side effects for those who will not.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lopez-Lazaro M. The stem cell division theory of cancer. Crit Rev Oncol Hematol. 2018;123:95-113
2. Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1998;8:98-101
3. Tellez-Gabriel M, Charrier C, Brounais-Le Roeyr B. et al. Analysis of gap junctional intercellular communications using a dielectrophoresis-based microchip. Eur J Cell Biol. 2017;96:110-18
4. Wee I, Syn N, Sethi G. et al. Role of tumor-derived exosomes in cancer metastasis. Biochim Biophys Acta Rev Cancer. 2018;1871:12-9
5. Vallette FM, Olivier C, Lézot F. et al. Dormant, quiescent, tolerant and persister cells: four synonyms for the same target in cancer. Biochem Pharmacol. 2019;162:169-176
6. Courau T, Nehar-Belaid D, Florez L. et al. TGFb and VEGF cooperatively control the immunotolerant environment and the efficacy of cancer immunotherapies. JCI Insight. 2016;1:e85974
7. Mackall CL, Meltzer PS, Helman LJ. Focus on sarcomas. Cancer Cell. 2002;2:175-8
8. Lopez-Lazaro M. Cancer etiology: Variation in cancer risk among tissues is poorly explained by the number of gene mutations. Genes Chromosomes Cancer. 2018;57:281-93
9. Tomasetti C, Macrhionni L, Nowak MA. et al. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc Natl Acad Sci U S A. 2015;112:118-23
10. Vogelstein B, Kinzler KW. The path to cancer -three strikes and You're out. N Engl J Med. 2015;373:1895-8
11. Chatterjee A, Rodger EJ, Eccles MR. Epigenetic drivers of tumourigenesis and cancer metastasis. Semin Cancer Biol. 2018;51:149-59
12. Noone AM, Cronin KA, Altekruse SF. et al. Cancer incidence and survival trends by subtype using data from the surveillance epidemiology and end results program, 1992-2013. Cancer Epidemiol Biomarkers Prev. 2017;26:632-41
13. Vertii A, Kaufman PD, Hehnly H. et al. New dimensions of asymmetric division in vertebrates. Cytoskeleton. 2018;75:87-102
14. Santoro A, Vlachou T, Carminati M. et al. Molecular mechanisms of asymmetric divisions in mammary stem cells. EMBO Rep. 2016;17:1700-720
15. Tomasetti C, Li L, Vogelstein B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science. 2017;355:1330-334
16. Tomasetti C, Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015;347:78-81
17. Mitelman F, Johansson B, Mertens F. et al. Cancer chromosome breakpoints cluster in gene-rich genomic regions. Genes Chromosomes Cancer. 2019;58:149-54
18. Grzywa TM, Praskal W, Wlodarski PK. Intratumor and Intertumor Heterogeneity in Melanoma. Transl Oncol. 2017;10:956-75
19. Brown HK, Tellez-Gabriel M, Cartron PF. et al. Characterization of circulating tumor cells as a reflection of the tumor heterogeneity: myth or reality? Drug Discov Today. 2019;24:763-772
20. Gundem G, Van Loo P, Kremeyer B. et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520:353-357
21. Hong MK, Macintyre G, Wedge DC. et al. Tracking the origins and drivers of subclonal metastatic expansion in prostate cancer. Nat Commun. 2015;6:6605
22. Bedzhov I, Graham SJ, Leung CY. et al. Developmental plasticity, cell fate specification and morphogenesis in the early mouse embryo. Philos Trans R Soc Lond B Biol Sci. 2014;369:1657
23. Nieto MA, Huang RY, Jackson RA. et al. EMT:2016. Cell. 2016;166:21-45
24. Gabriel MT, Calleja LR, Chalopin A. et al. Circulating tumor cells: a review of non-EpCAM-based approaches for cell enrichment and isolation. Clin Chem. 2016;62:571-81
25. Franzetti GA, Laud-Duval K, van der Ent W. et al. Cell-to cell heterogeneity of EWSR1-FLI1 activity determines proliferation/migration choices in Ewing sarcoma cells. Oncogene. 2017;36:3505-514
26. Gambera S, Abarrategi A, Gonzalez-Camacho F. et al. Clonal dynamics in osteosarcoma defined by RGB marking. Nat Commun. 2018;9:3994
27. Zahreddine H, Borden KL. Mechanisms and insights into drug resistance in cancer Front Pharmacol. 2013; 4:28.
28. Zhu J, Liang L, Jiao Y. et al. Enhanced invasion of metastatic cancer cells via extracellular matrix interface. PLoS One. 2015;10:e0118058
29. Costa A, Kieffer Y, Scholer-Dahirel A. et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell. 2018;33:463-79
30. Biffi G, Oni TE, Speilman B. et al. IL-1 induced JAK/SATAT signaling is antagonized by TGF-beta to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 2019;9:182-301
31. Mani NL. Quantitative assessment of the spatial heterogeneity of tumor-infiltrating lymphocytes in breast cancer. Breast Cancer Res. 2016;18:78
32. Berthel A, Zoernig I, Valous NA. et al. Detailed resolution analysis reveals spatial T cell heterogeneity in the invasive margin of colorectal cancer liver metastases associated with improved survival. Oncoimmunol. 2017;6:e1286436
33. Tashireva LA, Denisov EV, Gerahchenko TS. et al. Intratumoral heterogeneity of macrophages and fibroblasts in breast cancer is associated with the morphological diversity of tumor cells and contributes to lymph node metastasis. Immunobiol. 2017;222:631-40
34. Germain RN, Huang Y. ILC2s-resident lymphocytes pre-adapted to a specific tissue or migratory effectors that adapt to where they move? Curr Opin Immunol. 2018;56:76-81
35. Wagenblast E, Soto M, Gutierrez-Angel S. et al. A model of breast cancer heterogeneity reveals vascular mimicry as a driver of metastasis. Nature. 2015;520:358-62
36. Hamilton G, Rath B, Klameth L. et al. Small cell lung cancer: Recruitment of macrophages by circulating tumor cells. Oncoimmunol. 2015;5:e1093277
37. Vasaturo A, Di Blasio S, Verweij D. et al. Multispectral imaging for highly accurate analysis of tumour-infiltrating lymphocytes in primary melanoma. Histopathology. 2017;70:643-649
38. Le Faouder J, Laouirem S, Alexandrov T. et al. Tumoral heterogeneity of hepatic cholangiocarcinomas revealed by MALDI imaging mass spectrometry. Proteomics. 2014;14:965-72
39. Lawson DA, Kessenbrock K, Davis RT. et al. Tumour heterogeneity and metastasis at single-cell resolution. Nat Cell Biol. 2018;20:1349-360
40. Liu J, Dang H, Wang XW. The significance of intertumor and intratumor heterogeneity in liver cancer. Exp Mol Med. 2018;50:e416
41. Martelotto LG, Ng CK, Piscuoglio S. et al. Breast cancer intra-tumor heterogeneity. Breast Cancer Res. 2014;16:210
42. Jimenez-Sanchez A, Memon D, Pourpe S. et al. Heterogeneous tumor-immune microenvironments among differentially growing metastases in an ovarian cancer patient. Cell. 2017;170:927-938 e20
43. Malandrino G, Finocchiaro ST, Rossi P. et al. Multifunctional cadmium single source precursor for the selective deposition of CdO or CdS by a solution route. Chem Commun. (Camb). 2005;45:5681-683
44. Josson S, Gururajan M, Sung SY. et al. Stromal fibroblast-derived miR-409 promotes epithelial-to-mesenchymal transition and prostate tumorigenesis. Oncogene. 2015;34:2690-699
45. Chittezhath M, Dhillon MK, Lim JY. et al. Molecular profiling reveals a tumor-promoting phenotype of monocytes and macrophages in human cancer progression. Immunity. 2014;41:815-29
46. Cyll K, Ersvaer E, Vlatkovic L. et al. Tumour heterogeneity poses a significant challenge to cancer biomarker research. Br J Cancer. 2017;117:367-75
47. Kümler I, Balslev E, Knop AS. et al. Expression patterns of biomarkers in primary tumors and corresponding metastases in breast cancer. Appl Immunohistochem Mol Morphol. 2018;26:13-19
48. Erdem GU, Altundag K, Ozdemir NY. et al. Comparative study of receptor discordance between primary and corresponding metastatic lesions in breast cancer. J BUON. 2017;22:365-76
49. Alix-Panabières C, Pantel K. Challenges in circulating tumour cell research. Nat Rev Cancer. 2014;14:623-31
50. Bidard FC, Michiels S, Riethdorf S. et al. Circulating tumor cells in breast cancer patients treated by neoadjuvant chemotherapy: a meta-analysis. J Natl Cancer Inst. 2018;110:560-67
51. de Wit S, Manicone M, Rossi E. et al. EpCAMhigh and EpCAMlow circulating tumor cells in metastatic prostate and breast cancer patients. Oncotarget. 2018;9:35705-716
52. Munzone E, Nolé F, Goldhirsch A. et al. Changes of HER2 status in circulating tumor cells compared with the primary tumor during treatment for advanced breast cancer. Clin Breast Cancer. 2010;10:392-97
53. Punnoose EA, Atwal SK, Spoerke JM. et al. Molecular biomarker analyses using circulating tumor cells. PLoS One. 2010;5:e12517
54. Beije N, Onstenk W, Krann J. et al. Prognostic impact of HER2 and ER status of circulating tumor cells in metastatic breast cancer patients with a HER2-negative primary tumor. Neoplasia. 2016;18:647-53
55. Wallwiener M, Hartkopf AD, Riethdorf S. et al. The impact of HER2 phenotype of circulating tumor cells in metastatic breast cancer: a retrospective study in 107 patients. BMC Cancer. 2015;15:403
56. Mohamed Suhaimi NA, Foong YM, Lee DY. et al. Non-invasive sensitive detection of KRAS and BRAF mutation in circulating tumor cells of colorectal cancer patients. Mol Oncol. 2015;9:850-60
57. Bingham C, Fernandez SV, Fittipaldi P. et al. Mutational studies on single circulating tumor cells isolated from the blood of inflammatory breast cancer patients. Breast Cancer Res Treat. 2017;163:219-30
58. Pestrin M, Salvianti F, Galardi F. et al. Heterogeneity of PIK3CA mutational status at the single cell level in circulating tumor cells from metastatic breast cancer patients. Mol Oncol. 2015;9:749-57
59. Pailler E, Auger N, Lindsay CR. et al. High level of chromosomal instability in circulating tumor cells of ROS1-rearranged non-small-cell lung cancer. Ann Oncol. 2015;26:1408-15
60. Tsao SC, Wang J, Wang Y. et al. Characterising the phenotypic evolution of circulating tumour cells during treatment. Nat Commun. 2018;9:1482
61. Kermanshah L, Poudineh M, Ahmed S. et al. Dynamic CTC phenotypes in metastatic prostate cancer models visualized using magnetic ranking cytometry. Lab Chip. 2018;18:2055-64
62. Lack J, Gillard M, Cam M. et al. Circulating tumor cells capture disease evolution in advanced prostate cancer. J Transl Med. 2017;15:44
63. De Luca F, Rotunno G, Savianti F. et al. Mutational analysis of single circulating tumor cells by next generation sequencing in metastatic breast cancer. Oncotarget. 2016;7:26107-19
64. Miyamoto DT, Zheng Y, Wittner BS. et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science. 2015;349:1351-6
65. Pailler E, Oulhen M, Borget I. et al. Circulating tumor cells with aberrant ALK copy number predict progression-free survival during crizotinib treatment in ALK-rearranged non-small cell lung cancer patients. Cancer Res. 2017;77:2222-230
66. Paolillo C. et al. Detection of activating estrogen receptor gene (ESR1) mutations in single circulating tumor cells. Clin Cancer Res. 2017;23:6086-93
67. Tirosh I, Izar B, Prakadan SM. et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189-96
68. Au SH, Storey BD, Moore JC. et al. Clusters of circulating tumor cells traverse capillary-sized vessels. Proc Natl Acad Sci U S A. 2016;113:4947-4952
69. Aceto N, Bardia A, Miyamoto DT. et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014;158:1110-1122
70. Sarioglu AF, Aceto N, Kojic N. et al. A microfluidic device for label-free, physical capture of circulating tumor cell clusters. Nat Methods. 2015;12:685-691
71. Satake T, Suetsugu A, Nakamura M. et al. Color-coded imaging of the circulating tumor cell microenvironment. Anticancer Res. 2018;38:5635-5638
72. Szczerba BM, Castro-Giner F, Vetter M. et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature. 2019;566:553-557
73. Dyck L, Mills KHG. Immune checkpoints and their inhibition in cancer and infectious diseases. Eur J Immunol. 2017;47:765-779
74. Heymann MF, Lézot F, Heymann D. The contribution of immune infiltrates and the local microenvironment in the pathogenesis of osteosarcoma. Cell Immunol. in press
75. Mishra DK, Rocha HJ, Miller R. et al. Immune cells inhibit the tumor metastasis in the 4D cellular lung model by reducing the number of live circulating tumor cells. Sci Rep. 2018;8:16569
76. Mazel M, Jacot W, Pantel K. et al. Frequent expression of PD-L1 on circulating breast cancer cells. Mol Oncol. 2015;9:1773-82
77. Sun WW, Xu ZH, Lian P. Characteristics of circulating tumor cells in organ metastases, prognosis, and T lymphocyte mediated immune response. Onco Targets Ther. 2017;10:2413-2424
78. Arnoletti JP, Fanaian N, Reza J. et al. Pancreatic and bile duct cancer circulating tumor cells (CTC) form immune-resistant multi-cell type clusters in the portal venous circulation. Cancer Biol Ther. 2018;19:887-897
79. Wang X, Sun Q, Liu Q. et al. CTC immune escape mediated by Med Hypotheses. 2016;93:138-9.
80. Shen Z, Wu A, Chen X. Current detection technologies for circulating tumor cells. Chem Soc Rev. 2017;46:2038-56
81. Tellez-Gabriel M, Ory B, Lamoureux F, Heymann MF, Heymann D. Tumour heterogeneity: the key advantages of single-cell analysis. Int J Mol Sci. 2016;17:2142
82. Gawad C, Koh W, Quake SR. Dissecting the clonal origins of childhood acute lymphoblastic leukemia by single-cell genomics. Proc Natl Acad Sci U S A. 2014;111:17947-52
83. Lohr JG, Adalsteinsson VA, Cibulskis K. et al. Whole-exome sequencing of circulating tumor cells provides a window into metastatic prostate cancer. Nat Biotechnol. 2014;32:479-84
84. Jiang R, Lu YT, Ho H. et al. A comparison of isolated circulating tumor cells and tissue biopsies using whole-genome sequencing in prostate cancer. Oncotarget. 2015;6:44781-93
85. de Bourcy CF, De Vlaminck, Kanbar JN. et al. A quantitative comparison of single-cell whole genome amplification methods. PLoS One. 2014;9:e105585
86. Borgström E, Paterlini M, Mold JE. et al. Comparison of whole genome amplification techniques for human single cell exome sequencing. PLoS One. 2017;12:e0171566
87. Deleye L, Tilleman, L, Vander Plaetsen AS. et al. Performance of four modern whole genome amplification methods for copy number variant detection in single cells. Sci Rep. 2017;7:3422
88. Polzer B, Medoro G, Pasch S. et al. Molecular profiling of single circulating tumor cells with diagnostic intention. EMBO Mol Med. 2014;6:1371-86
89. Janiszewska M, Liu L, Almendro V. et al. In situ single-cell analysis identifies heterogeneity for PIK3CA mutation and HER2 amplification in HER2-positive breast cancer. Nat Genet. 2015;47:1212-9
90. Li C, Wu S, Yang Z. et al. Single-cell exome sequencing identifies mutations in KCP, LOC440040, and LOC440563 as drivers in renal cell carcinoma stem cells. Cell Res. 2017;27:590-93
91. Liu M, Liu Y, Di J. et al. Multi-region and single-cell sequencing reveal variable genomic heterogeneity in rectal cancer. BMC Cancer. 2017;17:787
92. Zhang X, Marjani SL, Hu Z. et al. Single-cell sequencing for precise cancer research: progress and prospects. Cancer Res. 2016;76:1305-12
93. Russo G, Patrignani A, Poveda L. et al. Highly sensitive, non-invasive detection of colorectal cancer mutations using single molecule, third generation sequencing. Appl Transl Genom. 2015;7:32-9
94. Kilianski A, Roth PA, Liem AT. et al. Use of unamplified RNA/cDNA-hybrid nanopore sequencing for rapid detection and characterization of RNA viruses. Emerg Infect Dis. 2016;22:1448-51
95. Yalcin D, Hakguder ZM, Out HH. Bioinformatics approaches to single-cell analysis in developmental biology. Mol Hum Reprod. 2016;22:182-92
96. Chen C, Xing D, Tan L. et al. Single-cell whole-genome analyses by linear amplification via Transposon Insertion (LIANTI). Science. 2017;356:189-94
97. Vu TN, Wills QF, Kalari KR. et al. Beta-Poisson model for single-cell RNA-seq data analyses. Bioinformatics. 2016;32:2128-35
98. Guo H, Zhu P, Wu X. et al. Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. Genome Res. 2013;23:2126-35
99. Smallwood SA, Lee HJ, Angermueller C. et al. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat Methods. 2014;11:817-20
100. Farlik M, Sheffield NC, Nuzzo A. et al. Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics. Cell Rep. 2015;10:1386-97
101. Rotem A, Ram O, Shoresh N. et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat Biotechnol. 2015;33:1165-72
102. Kind J, Pagie L, de Vries SS. et al. Genome-wide maps of nuclear lamina interactions in single human cells. Cell. 2015;63:134-47
103. Cusanovich DA, Daza R, Adey A. et al. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science. 2015;348:910-4
104. Jin W, Tang Q, Wan M. et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature 2105;28:142-6.
105. Nagano T, Lubling Y, Stevens TJ. et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 2013;502:59-64
106. Clark SJ, Lee HJ, Smallwood SA. et al. Single-cell epigenomics: powerful new methods for understanding gene regulation and cell identity. Genome Biol. 2016;17:72
107. Pixberg CF, Raba K, Müller F. et al. Analysis of DNA methylation in single circulating tumor cells. Oncogene. 2017;36:3223-231
108. Zhu S, Qing T, Zheng Y. et al. Advances in single-cell RNA sequencing and its applications in cancer research. Oncotarget. 2017;8:53763-779
109. Ziegenhain C, Vieth B, Parkh S. et al. Comparative analysis of single-cell RNA sequencing methods. Mol Cell. 2017;65:631-643
110. Ortega MA, Poirion O, Zhu X. et al. Using single-cell multiple omics approaches to resolve tumor heterogeneity. Clin Transl Med. 2017;6:46
111. Patel AP, Tirsoh I, Trombetta SJ. et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396-401
112. Chung W, Eum HH, Lee HO. et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun. 2017;8:15081
113. Bhan A, Soleimani M, Mandal SS. Long noncoding RNA and cancer: a new paradigm. Cancer Res. 2017;77:3965-981
114. Acunzo M, Romano G, Wermicke D. et al. MicroRNA and cancer-a brief overview. Adv Biol Regul. 2015;57:1-9
115. Abernathy J, Overturf K. Comparison of ribosomal RNA removal methods for transcriptome sequencing workflows in teleost fish. Anim Biotechnol. 2016;27:60-5
116. Wu AR, Neff NF, Kalisky T. et al. Quantitative assessment of single-cell RNA-sequencing methods. Nat Methods. 2014;11:41-6
117. Suzuki A, Matsushima K, Makinoshima H. et al. Single-cell analysis of lung adenocarcinoma cell lines reveals diverse expression patterns of individual cells invoked by a molecular target drug treatment. Genome Biol. 2015;16:66
118. Vallejos CA, Risso D, Sciadone A. et al. Normalizing single-cell RNA sequencing data: challenges and opportunities. Nat Methods. 2017;14:565-571
119. Wu Z, Zhang Y, Stitzel ML. et al. Two-phase differential expression analysis for single cell RNA-seq. Bioinformatics. 2018;34:3340-348
120. Miao Z, Deng K, Wang X. et al. DEsingle for detecting three types of differential expression in single-cell RNA-seq data. Bioinformatics. 2018;34:3223-224
121. Heath JR, Ribas A, Mischel PS. Single-cell analysis tools for drug discovery and development. Nat Rev Drug Discov. 2016;15:204-16
122. Ullal AV, Peterson V, Agasti SS. et al. Cancer cell profiling by barcoding allows multiplexed protein analysis in fine-needle aspirates. Sci Transl Med. 2014;6:219ra9
123. Giesen C, Wang HA, Schapiro D. et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat Methods. 2014;11:417-22
124. Sinkala E, Sollier-Christen E, Renier C. et al. Profiling protein expression in circulating tumour cells using microfluidic western blotting. Nat Commun. 2017:8 14622
125. Gulbahce N, Magbanua MJM, Chin R. et al. Quantitative whole genome sequencing of circulating tumor cells enables personalized combination therapy of metastatic cancer. Cancer Res. 2017;77:4530-541
126. Gao R, Davis A, McDonald TO. et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat Genet. 2016;48:1119-30
127. Paoletti C, Cani AK, Larios JM. et al. Comprehensive mutation and copy number profiling in archived circulating breast cancer tumor cells documents heterogeneous resistance mechanisms. Cancer Res. 2018;78:1110-122
128. Carlsson A. et al. Paired high-content analysis of prostate cancer cells in bone marrow and blood characterizes increased androgen receptor expression in tumor cell clusters. Clin Cancer Res. 2017;23:1722-732
129. Liu Y, Meucci S, Sheng L. et al. Meta-analysis of the mutational status of circulation tumor cells and paired primary tumor tissues from colorectal cancer patients. Oncotarget. 2017;8:77928-941
130. Guibert N, Delaunay M, Lusque A. et al. PD-L1 expression in circulating tumor cells of advanced non-small cell lung cancer patients treated with nivolumab. Lung Cancer. 2018;120:108-12
131. Kaigorodova EV, Savelieva OE, Tashireva LA. et al. Heterogeneity of circulating tumor cells in neoadjuvant chemotherapy of breast cancer. Molecules. 2018:23
132. Jakabova A, Biecikova Z, Pospisilova E. et al. Molecular characterization and heterogeneity of circulating tumor cells in breast cancer. Breast Cancer Res Treat. 2017;166:695-700
133. Aaltonen KE, Novosadova V, Bendahl PO. et al. Molecular characterization of circulating tumor cells from patients with metastatic breast cancer reflects evolutionary changes in gene expression under the pressure of systemic therapy. Oncotarget. 2017;8:45544-565
134. Gasch C, Oldopp T, Mauermann O. et al. Frequent detection of PIK3CA mutations in single circulating tumor cells of patients suffering from HER2-negative metastatic breast cancer. Mol Oncol. 2016;10:1330-43
135. Scher HI, Graf RP, Schreiber NA. et al. Phenotypic heterogeneity of circulating tumor cells informs clinical decisions between AR signaling inhibitors and taxanes in metastatic prostate cancer. Cancer Res. 2017;277:5687-98
136. Kermanshah L, Poudineh M, Ahmed S. et al. Dynamic CTC phenotypes in metastatic prostate cancer models visualized using magnetic ranking cytometry. Lab Chip 2018 (18). 2055 -64
137. Markou A, Lazaridou M, Paraskevopoulos P. et al. Multiplex gene expression profiling of in vivo isolated circulating tumor cells in high-risk prostate cancer patients. Clin Chem. 2018;264:297-306
138. Sun YF, Guo W, Xu Y. et al. Circulating tumor cells from different vascular sites exhibit spatial heterogeneity in epithelial and mesenchymal composition and distinct clinical significance in hepatocellular carcinoma. Clin Cancer Res. 2018;24:547-59
139. Kondo Y, Hayashi K, Kawakami K. et al. KRAS mutation analysis of single circulating tumor cells from patients with metastatic colorectal cancer. BMC Cancer. 2017;17:311
140. Tan K, Leong SM, Kee Z. et al. Longitudinal monitoring reveals dynamic changes in circulating tumor cells (CTCs) and CTC-associated miRNAs in response to chemotherapy in metastatic colorectal cancer patients. Cancer Lett. 2018;423:1-8
141. Joosse SA, Souche FR, Babayan A. et al. Chromosomal aberrations associated with sequential steps of the metastatic Cascade in colorectal cancer patients. Clin Chem. 2018;64:1505-1512
142. Messaritakis I, Stoltidis D, Kotsakis A. et al. TTF-1- and/or CD56-positive circulating tumor cells in patients with small cell lung cancer (SCLC). Sci Rep. 2017;7:45351
143. Chalopin A, Tellez-Gabriel M, Brown HK. et al. Isolation of circulating tumor cells in preclinical model of osteosarcoma: effect of chemotherapy. J Bone Oncol. 2018;12:83-90
144. Hou Y, Huo H, Cao C. et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 2016;26:304-19
145. Polioudaki H, Agelaki S, Chiotaki R. et al. Variable expression levels of keratin and vimentin reveal differential EMT status of circulating tumor cells and correlation with clinical characteristics and outcome of patients with metastatic breast cancer. BMC Cancer. 2015;15:399
146. Li S, Chen Q, Li H. et al. Mesenchymal circulating tumor cells (CTCs) and OCT4 mRNA expression in CTCs for prognosis prediction in patients with non-small-cell lung cancer. Clin Transl Oncol. 2017;19:1147-1153
147. Markiewicz A, Wełnicka-Jaśkiewicz M, Seroczyńska B. et al. Epithelial- mesenchymal transition markers in lymph node metastases and primary breast tumors - relation to dissemination and proliferation. Am J Transl Res. 2014;6:793-808
148. Kasimir-Bauer S, Hoffmann O. et al. Expression of stem cell and epithelial-mesenchymal transition markers in primary breast cancer patients with circulating tumor cells. Breast Cancer Res. 2012;14:R15
149. Allard WJ, Matera J, Miller MC. et al. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin Cancer Res. 2004;10:6897-6904
150. Lowes LE, Goodale D, Xia Y. et al. Epithelial-to-mesenchymal transition leads to disease-stage differences in circulating tumor cell detection and metastasis in pre-clinical models of prostate cancer. Oncotarget. 2016;7:76125-76139
151. Han D, Chen K, Che J. et al. Detection of epithelial-mesenchymal transition status of circulating tumor cells in patients with esophageal squamous carcinoma. Biomed Res Int. 2018;2018:7610154
152. Juratli MA, Sarimollaoglu M, Siegel ER. et al. Real-time monitoring of circulating tumor cell release during tumor manipulation using in vivo photoacoustic and fluorescent flow cytometry. Head Neck. 2014;36:1207-15
153. Sasportas LS, Hori SS, Pratx G. et al. Detection and quantitation of circulating tumor cell dynamics by bioluminescence imaging in an orthotopic mammary carcinoma model. PLoS One. 2014;9:e105079
154. Savas P, Virassamy B, Ye C. et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat Med. 2018;24:986-93
155. Castellanos E, Feld E, Horn L. Driven by Mutations: The predictive value of mutation subtype in EGFR-mutated non-small cell lung cancer. J Thorac Oncol. 2017;12:612-623
Author contact
![]() Corresponding authors: Dr M. Tellez-Gabriel, e-mail: marta.t.gabrielno or Prof. D. Heymann, e-mail: dominique.heymann
Corresponding authors: Dr M. Tellez-Gabriel, e-mail: marta.t.gabrielno or Prof. D. Heymann, e-mail: dominique.heymann