Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Engineering cells via genetic...
Engineering cells via...
Engineering cells via chemical...
Engineering cells via physical...
Conclusion and Outlook
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(25):7889-7905. doi:10.7150/thno.38583 This issue Cite
Review
Advances in Engineering Cells for Cancer Immunotherapy
Xiao Xu1#, Teng Li1#, Shiyang Shen1, Jinqiang Wang2,3, Peter Abdou2,3, Zhen Gu2,3 ![]() , Ran Mo1
, Ran Mo1 ![]()
1. State Key Laboratory of Natural Medicines, Jiangsu Key Laboratory of Drug Discovery for Metabolic Diseases, Center of Advanced Pharmaceuticals and Biomaterials, China Pharmaceutical University, Nanjing 210009, China.
2. Department of Bioengineering, University of California, Los Angeles, CA 90095, USA.
3. Jonsson Comprehensive Cancer Center, California NanoSystems Institute, and Center for Minimally Invasive Therapeutics, University of California, Los Angeles, CA 90095, USA.
#Equal contribution to this work.
Received 2019-8-30; Accepted 2019-9-17; Published 2019-10-16
Abstract

Cancer immunotherapy aims to utilize the host immune system to kill cancer cells. Recent representative immunotherapies include T-cell transfer therapies, such as chimeric antigen receptor T cell therapy, antibody-based immunomodulator therapies, such as immune checkpoint blockade therapy, and cytokine therapies. Recently developed therapies leveraging engineered cells for immunotherapy against cancers have been reported to enhance antitumor efficacy while reducing side effects. Such therapies range from biologically, chemically and physically -engineered cells to bioinspired and biomimetic nanomedicines. In this review, advances of engineering cells for cancer immunotherapy are summarized, and prospects of this field are discussed.
Keywords: Cell therapy, Drug delivery, Cancer immunotherapy, Cell engineering, Nanomedicine
Introduction
Immunotherapies that either induce or inhibit the host immune response have been used in the clinical treatment of many diseases including infections [1-3], cancers [4, 5] and autoimmune diseases [6, 7]. As opposed to cytotoxic drugs that directly kill pathogens or mutant cells, immunotherapeutics function by activating the patient's own immune system to eradicate the pathogens or mutant cells [8-10]. The specific antigens produced by the pathogenic cells can be recognized and internalized by antigen-presenting cells (APCs), such as dendritic cells (DCs), which are subsequently presented on major histocompatibility complexes (MHCs) on the APC surfaces [11]. When APCs with the MHC-bound antigens interact with T lymphocytes, the T lymphocytes become primed to recognize the antigens and attack the pathogenic cells [12-14]. However, cancer cells often suppress host immune cells using various mechanisms in order to evade destruction and continue to proliferate [15-17].
Cancer immunotherapy, also referred to as immuno-oncology, aims to induce the immune system of host to identify and eliminate cancers [18-20]. The primary cancer immunotherapeutic strategies currently being used in the clinic include cancer vaccines, immune checkpoint blockade (ICB) and adoptive cell transfer (ACT) therapy [21-23]. Cancer vaccines are intended to enhance the autoimmune response against cancer cells, and are typically categorized into nucleic acid, viral or cellular vaccines [24-26]. Nucleic acid vaccines contain DNA or RNA sequences that express specific proteins to activate APCs, which further activate T lymphocytes to promote anticancer activity [27]. Virus vaccines act as viruses specifically proliferating in and killing cancer cells without harming normal cells. Oncolytic virus is clinically applied as an active drug for cancer therapy [28]. Cell vaccines are engineered antigen-presenting cells that activate T cells to produce an immune response after entering into the body [29]. Sipuleucel-T (Provenge®) is the first cell-based therapeutic vaccine approved by the U.S. Food and Drug Administration (FDA) to treat prostate cancer [30]. The ICB therapy uses antibodies to block the immune-inhibitory interaction between cancer cells and immune cells in an effort to unlock the host antitumor response, and has demonstrated notable clinical outcomes [31-33]. FDA-approved immune checkpoint inhibitors include ipilimumab [34], pembrolizumab [35], nivolumab [36], atezolizumab [37], avelumab [38], durvalumab [39] and cemiplimab [40]. The ACT therapy utilizes the cytotoxic capabilities of T lymphocytes to kill cancer cells, including tumor-infiltrating lymphocyte (TIL) therapy [41, 42], T cell receptor (TCR)-engineered T (TCR-T) cell therapy [43, 44] and chimeric antigen receptor T (CAR-T) cell therapy [45, 46]. The first success of ACT for cancer treatment witnessed the regression of melanoma treated with the ex vivo expanded TILs [47, 48]. Kymriah®, the first CAR-T cell therapy approved by the FDA, has demonstrated effective clinical therapeutic outcomes for the treatment of refractory or recrudescent B-cell precursor acute lymphoblastic leukemia [49, 50]. Despite the tremendous clinical achievements of cancer immunotherapy, several significant concerns still remain, which are associated with adverse effects, off-target effects and limited efficacy [51-53].
To this end, many novel strategies from the perspectives of drug discovery and drug delivery have been developed. Among them, cell-based drug delivery systems provide a promising platform to enhance delivery efficiency, increase therapeutic efficacy, and reduce off-target and side effects of cancer immunotherapy. By utilizing recent advances in immunotechnology, micro/nanotechnology and molecular pharmaceutics, such cellular systems range from biologically, chemically, and physically-engineered cells to bioinspired and biomimetic nanomedicines (Figure 1) [54-56]. In this review article, we will focus on recent progress in the field of cell engineering for cancer immunotherapy, and discuss potential future directions of cell engineering approaches for delivery of cancer immunotherapies.
Schematic of the representative strategies of engineering cells for cancer immunotherapy. The representative cells used for drug delivery and cancer immunotherapy involve erythrocytes, platelets, leukocytes, cancer cells and stem cells.
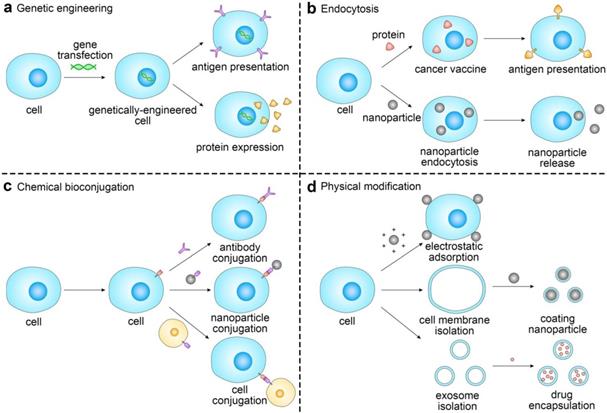
Engineering cells via genetic modification
Genetic engineering aims to change cell phenotypes by altering genetic information [57]. A variety of immune cells can be genetically engineered for cancer immunotherapy, including macrophages, natural killer (NK) cells and T cells [58-60]. Among them, genetically-engineered T cells have been extensively studied. T cells can be isolated from the peripheral blood or tumor tissue of patients [61]. After screening and gene transfection, functionalized T cells are re-administered into the patients to eradicate cancer cells. TCR-T and CAR-T cell therapies are two emerging ACT therapies in which the genetically-engineered cells have preferable targeting capabilities and clinical therapeutic response [5, 62, 63].
TCRs are a characteristic biomolecule of T cells, and consist of α- and β-chains associated with the CD3 complex composed of γ-, δ-, ε- and ζ-chains [61]. TCRs are membrane proteins responsible for recognizing specific antigens and mediating intracellular signaling pathways for activation of T cells. This process is mediated by MHCs, kind of polymorphic molecules that are expressed on the APC surface associated with antigens. Interactions between antigens and TCRs result in phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs), and therefore activate intracellular signaling in the T cells and release of cytokines, such as interferon-γ (IFN-γ)/interleukin-2 (IL-2) and cytotoxic proteins, such as perforin/granzyme [61, 64, 65]. There are many cancer-associated antigens, which include but are not limited to carcinoembryonic antigen (CEA), B-lymphocyte antigen, glycoprotein 100 (gp 100) and human epidermal growth factor receptor-2 (HER-2) [66-68]. However, evidence has shown that cancer cells share similar surface antigens with normal cells, which limits the ability of autologous T cells to distinguish between cancer cells that escape immune eradication and normal cells [69]. The TCR-T-based technique is considered to be a promising strategy to decrease cancer cell immune escape by genetically modifying T cells to express receptors with high affinity to the antigens [70]. In this strategy, TCR genes derived from tumor-specific T cells or screened by bacteriophage libraries of antibodies are further optimized by substitution of nucleotides to elevate the TCR affinity to the tumor-associated antigens. This affinity-enhanced TCR approach reinforces intracellular signal transduction and therefore enables T cells with augmented activity to kill the cancer cells [61].
TCR-T therapy is often utilized as a therapy for hematological malignancies [71, 72]. For example, Tawara et al. developed TCR-T cells capable of specifically binding to Wilms tumor 1 (WT1) peptide, a specific epitope on leukemic cells of acute leukemia and myelodysplastic syndrome [73]. The engineered TCR-T cells were able to maintain ex vivo peptide-specific immune reactivity in the peripheral blood of patients. Hematopoietic function recovery was observed in 40% of patients after treatment. Additionally, TCR-T therapy can also be used for treatment of solid tumors such as melanoma [74], multiple myeloma [75], colorectal [76] and synovial sarcoma [77]. Orlando et al. identified that the tumor-associated antigen, preferentially expressed antigen in melanoma (PRAME) was a specific epitope on medulloblastoma cells correlated with poor overall survival [78]. Enhanced in vitro and in vivo anticancer activities were observed after treatment with the PRAME-specific TCR-T cells. Meanwhile, lower toxicity of these TCR-T cells introduced with an inducible caspase 9 gene was observed compared with the untransduced control T cells [79].
Recently, two FDA-approved CAR-T cell-based therapies, Kymriah and Yescarta are being utilized for the treatment of patients with acute lymphoblastic leukemia and non-Hodgkin lymphoma, respectively [80, 81]. The basic structure of CAR includes antigen-binding, transmembrane and intracellular signaling domains. The antigen-binding domain is a single-chain variable fragment (scFv) derived from the B cell. Since recognition by CAR is MHC-independent, scFv has been widely used regardless of the type of human leukocyte antigen (HLA). CARs recognize antigens on cancer cell membranes, such as CEA, CD19 and vascular endothelial growth factor receptor 2 (VEGFR2), leading to recruitment of signal-initiating molecules, phosphorylation of signaling domains and activation of kinase cascades [82, 83]. In design of CAR, the signal-initiating molecules contain the ζ-chain of the CD3 complex and the γ-chain of the high-affinity receptor for immunoglobulin E (FcεRI) [61]. Identification of antigen epitopes on cancer cells is important for CAR design. CD19 on B cell malignancies is an ideal target for CAR. It has been reported that 50-90% of patients respond to anti‑CD19 CAR-T cell therapy [84]. However, serious side effects including cytokine-release syndrome and neurotoxicity, which are potentially life-threatening in severe cases are frequently concomitant, which greatly hinders its widespread application in clinic [85]. The second and third generations of CARs have been developed for enhanced in vivo persistence and function of CAR-T cells and reduced side effects. The costimulatory molecule genes are transduced into the T cells simultaneously. The expressed CARs include costimulatory signaling domains as a part of the intracellular domain [86]. Ying et al. constructed an anti-CD19 CAR molecule (CD19-BBz(86)) with intracellular 4-1BB co-stimulatory and CD3ζ signaling domains [87]. The CD19-BBz(86) CAR-T cells were safer and more effective than the counterparts without the costimulatory signaling domain owing to release of fewer cytokines and more anti-apoptotic molecules. Six of eleven patients with B cell lymphoma receiving the treatment of CD19-BBz(86) CAR-T cells presented complete remission but no significant increase of cytokine serum level or neurotoxicity.
Efficient activation and expansion of T cells is of the essence in enhancing immunotherapy. The use of commercial expansion beads (Dynabeads) for ex vivo expansion of T cells is limited by low efficiency and limited functionality of the T cell products. Cheung et al. developed APC-mimic scaffolds (APC-ms) composed of lipid membrane-coated mesoporous silica micro-rods [88]. By encapsulation of IL-2 and bioconjugation of anti-CD3 and anti-CD28 antibodies, APC-ms presented superior effects on polyclonal expansion of primary mouse and human T cells than Dynabeads. Elevation of antigen-specific expansion of cytotoxic T cells was achieved by a single simulation using APC-ms compared with the monocyte-derived DCs. Moreover, APC-ms exhibited favorable expansion ability on the restimulated CAR-T cells than Dynabeads, and comparable antitumor efficacy in vivo. Due to costly and time-consuming processes of ex vivo preparation of CAR-T cells, in situ programming of T cells with nanoparticles was proposed by Smith et al. [89]. The CAR gene-encoded plasmid DNA was mixed with a cationic polymer to form nanosized complexes, followed by modification with the T-cell-targeting anti-CD3e f(ab')2 fragments that mediate endocytosis by lymphocytes. When administered to the mice bearing B-cell acute lymphoblastic leukemia, the nanoparticles programmed the circulating T cells and induced tumor regression equivalent to the traditional CAR-T cell therapy.
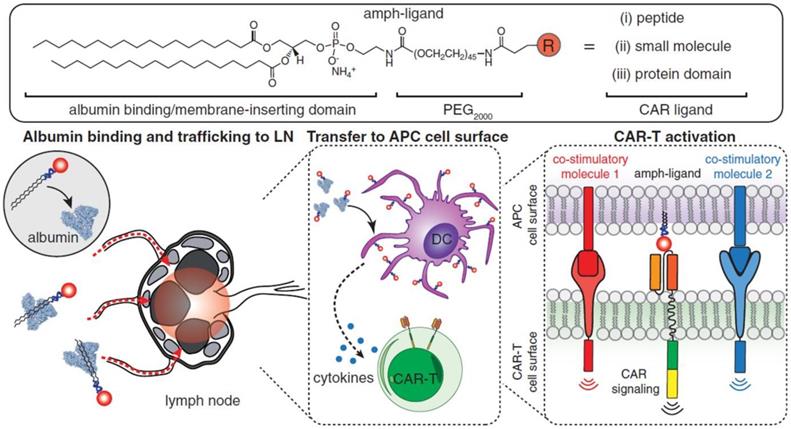
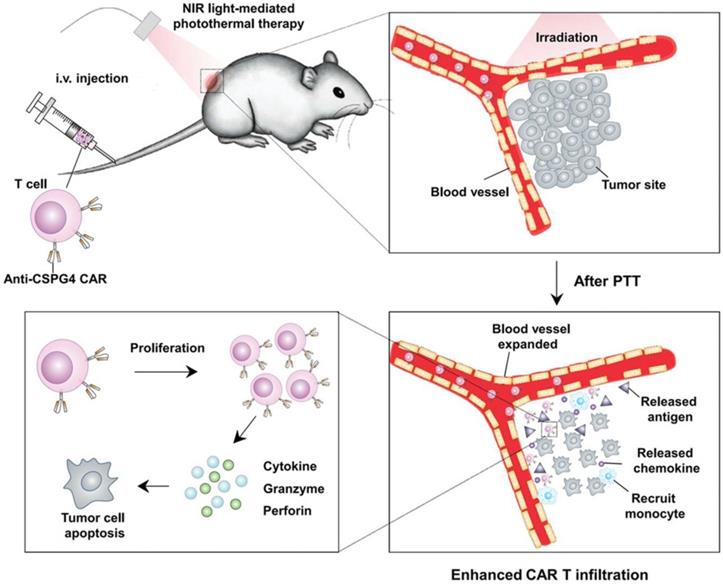
Although effective in treating hematological malignancies, the utilization of CAR-T cells for the treatment of solid tumors is more challenging, which is due in part to limited expansion, poor penetration, and decreased viability of administered CAR-T cells. Recently, Ma et al. developed lymph node-targeted amphiphile CAR-T cell ligands (amph-ligands) to directly promote donor cells via their chimeric receptor in vivo for enhanced efficacy of the CAR-T cell therapy against solid tumors (Figure 2) [90]. Amph-ligands were composed of phospholipid, polyethylene glycol (PEG) and CAR ligand moieties. After injection, the long-chain alkane of the phospholipid moiety readily bound to the albumin in the blood, which mediated the transport of amph-ligands to the lymph nodes. The CAR ligands were further decorated on the APC surfaces, which primed the circulating CAR-T cells in the lymph nodes. This approach showed its potential to increase the CAR-T cell expansion and augment the antitumor immunity in multiple mouse solid tumor models. On the other hand, IL-7 and CCL19 are regarded to be crucial for the maintenance of the T cell zone in lymphoid organs where DCs and T cells are recruited from the periphery [91, 92]. IL-7 enhances proliferation and survival of T cells, while CCL19 is a chemotactic factor for DCs and T cells [93, 94]. Adachi et al. developed CAR-T cells expressing both IL-7 and CCL19, which could significantly augment the DC and T cell infiltration into the solid tumor compared with the traditional counterpart without cytokine expression [95]. To enhance penetration of CAR-T cells into solid tumors, Chen et al. applied photothermal pre-treatment to disrupt extracellular matrix (ECM) for enhanced tumor penetration of CAR-T cells (Figure 3) [96]. Indocyanine green (ICG), a photothermal agent, was loaded into poly(lactic-co-glycolic) acid (PLGA) nanoparticles, which were intratumorally injected into the tumor tissue. Upon light irradiation, mild heating generated by the ICG-loaded nanoparticles resulted in the disruption of the ECM followed by decreased interstitial fluid pressure and increased blood perfusion. This photothermal pre-treatment significantly improved the tumor penetration of subsequent intravenously-injected CAR-T cells, leading to enhanced antitumor efficacy in the solid tumors. Cho et al. reported that addition of a pair of leucine zippers between the scFv and the intracellular domain controlled the recognition of different antigens by T cells by altering the structure of the leucine zipper-scFv, thereby increasing the functionality of T cells [97]. Specifically controlling the type and level of immune response could also be achieved by customized sculpt immune cell response to overcome tumor immunosuppression [98].
Schematic of the structure of amph-ligands and the process of amph-ligand vaccine-boosting approach. The amph-ligand consists of lipid, PEG and CAR ligand. The long-chain lipid moiety could bind to serum albumin that facilitates the accumulation of the amph-ligand into the lymph nodes. The CAR ligand was subsequently decorated on the surface of DCs, which activated the CAR signaling to increase the expansion of the CAR-T cells. Reprinted with permission from ref [90]. Copyright 2019 The American Association for the Advancement of Science.
Schematic of enhanced infiltration and activation of the CAR-T cells by mild heating of the tumor. ICG-PLGA nanoparticles were intratumorally injected followed by light irradiation to generate mild heat to disrupt the ECM, which led to reduced interstitial fluid pressure and increased blood perfusion, and therefore enhanced infiltration of the circulating CAT-T cells in the tumor tissue. Reprinted with permission from ref [96]. Copyright 2019 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
In addition to T and B cells, NK cells are another type of lymphocyte, which are critical to the innate immune system and defend the human body against cancer. By secreting cytokines, NK cells regulate immune response and promote maturation of APCs [99, 100]. NK cells can also induce the polarization of macrophages to M1 type [101, 102] and target tumor tissue via membrane protein, such as natural killer group 2 member D (NKG2D) receptor or DNAX accessory molecule-1 (DNAM-1) [103, 104]. Furthermore, NK cells have been reprogrammed with CAR to strengthen recognition specificity and reactivity to cancer cells [105]. Jiang et al. genetically modified NK-92MI cells with a CAR containing anti-CD138 fragment, which showed significantly enhanced cytotoxicity toward the CD138-positive multiple myeloma cells compared with the CD138-negative counterparts [106]. Chu et al. developed CS1-specific CAR-expressing NK cells for immunotherapy of multiple myeloma [107]. The engineered NK cells displayed specific recognition of multiple myeloma cells overexpressing the CS1 surface protein, which efficiently slowed growth of human multiple myeloma and prolonged mouse survival in the tumor mouse model.
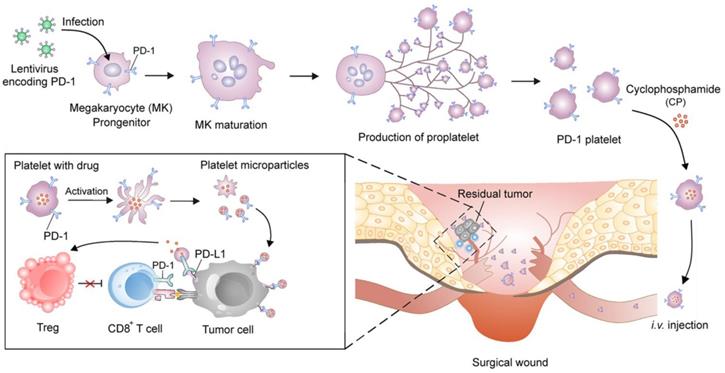
Zhang et al. engineered platelets decorated with PD1 for preventing post-surgical cancer recurrence (Figure 4) [108]. Megakaryocytes (MKs) that are responsible for in vitro large-scale production of platelets were genetically modified using lentivirus encoding PD1. The obtained PD1-presenting MKs was able to generate mature platelets with PD1, which increased the accumulation of PD1 to surgical wounds by relying on the physiological function of platelets as monitors of vascular injury. PD1 was released via platelet-derived microparticles from cell membranes upon platelet activation and blocked PDL1 on tumor cells to reinvigorate the exhausted CD8+ T lymphocytes. Cyclophosphamide (CP), an immunosuppressant, was simultaneously delivered by the PD1-expressing platelets to exhaust the regulatory T cells (Tregs) and enhance the cytotoxic effects of the CD8+ T cells. Xue et al. conjugated granulocyte macrophage-colony stimulating factor (GM-CSF) mRNAs to polypeptide linker covalently to construct fusion gene GC2A, which was further inserted into the adenovirus vector to transfect human embryonic kidney 293T (HEK293T) cells [109]. The recombinant adenovirus proliferated in HEK293T cells and released GM-SCF that promoted the DC proliferation and differentiation in the tumor-bearing mice.
Schematic of the development of the platelets with PD1 presentation and reactivation of the CD8+ T cells. The PD1-expressing platelets were obtained from the MKs that were genetically transfected with the lentivirus encoding PD1, and further loaded with CP, an immunosuppressant. The PD1-expressing platelets with CP could efficiently accumulate to the surgical wounds and release both PD1 and CP. The released PD1 blocked the PDL1 on the tumor cells to reactivate the CD8+ T cells, while the released CP depleted the Tregs to enhance the activity of the CD8+ T cells. Reprinted with permission from ref [108]. Copyright 2018 American Chemical Society.
Engineering cells via endocytosis-mediated functionalization
Endocytosis is an approach for cell engineering in which autologous cells are treated with proteins, drugs or nanoparticles in vitro, followed by reinfusion to the body. Depending on the phagocytosed substances, the representative approaches include vaccine endocytosis for antigen presentation and nanoparticle endocytosis for drug delivery [110-112].
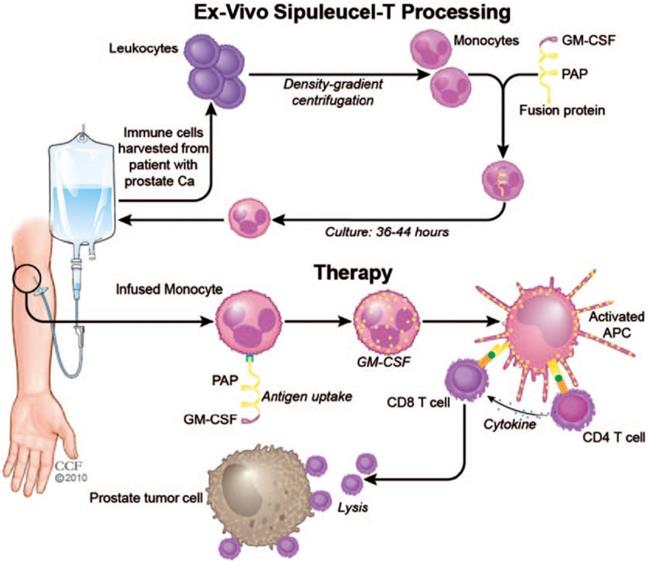
Research of cancer vaccines reached a milestone with the development of Sipuleucel-T for treating patients with metastatic castration-resistant prostate cancer in 2010 (Figure 5) [113]. Leukocytes are harvested from the patients' peripheral blood, and monocytes are isolated by density gradient centrifugation. The harvested monocytes are cultured with fusion protein for 36-44 hours to allow for endocytosis of the fusion protein. The recombinant fusion protein used is PA2024, which is composed of a prostatic acid phosphatase (PAP) domain and a GM-CSF domain. PAP is highly expressed on prostate cancer cell membranes and serves as a target antigen for T cells. GM-CSF enhances proliferation of monocytes and promotes their differentiation to APCs. After administration, engineered monocytes first differentiate into APCs and in succession, activate the PAP-specific CD4+ and CD8+ T cells. The CD8+ T cells induce cell lysis of the prostate tumor cells with the help of cytokines secreted by CD4+ T cells. Sipuleucel-T showed prolonged median survival of 4.1 months with mild side effects [30, 113]. With the exception of Sipuleucel-T, no other cell-based vaccines have yet passed clinical trials due to limited therapeutic efficacy [114]. Accordingly, it remains a challenge to develop cancer vaccines with clinically significant anticancer activity [115-117].
Schematic of the process of the Sipuleucel-T therapy against prostate cancer. The monocytes were harvested from the patients and treated with the fusion protein, and re-infused into the patients. The infused monocytes could differentiate to the APCs, which activated the PAP-specific CD4+ and CD8+ T cells to kill the prostate cancer cells. Reprinted with permission from ref [113]. Copyright 2011 SAGE Publications.
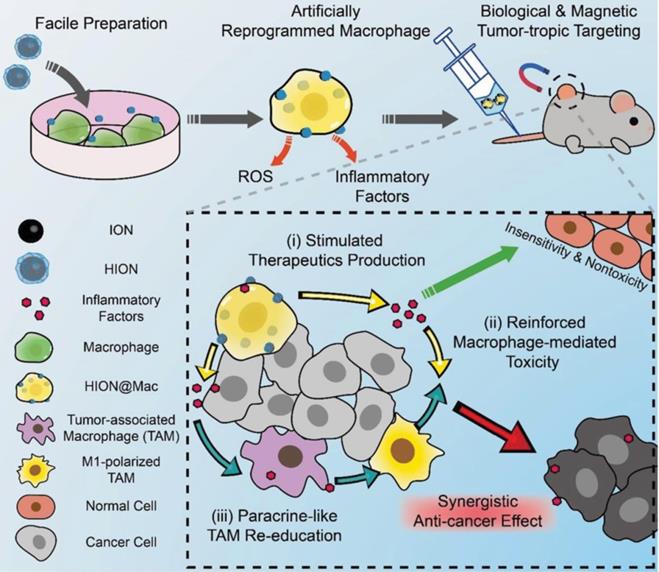
Like fusion proteins, nanoparticles can also be internalized by cells to construct a cell-based drug delivery system to combine the advantages of nanoparticles and cells for augmented immunotherapeutic efficacy [118]. Nanoparticles enhance the accumulation of anticancer drugs in solid tumors through the enhanced penetration and retention (EPR) effect, which is the phenomenon whereby nanoparticles tend to preferentially accumulate in the tumor microenvironment due to its leaky vasculature [119]. However, recent studies suggest that the EPR-based passive-targeting effect of nanoparticles is inadequate [120-122]. Combination of the distinct advantages of nanoparticles and cells is a promising strategy to achieve enhanced tumor accumulation [123-125]. The immunosuppressive microenvironment is one of the primary obstacles to the effectiveness of immunotherapeutics [126,127]. A number of immunosuppressive factors have been identified using the molecular imaging techniques [128, 129], and a variety of strategies have been proposed to overcome the immunosuppressive tumor microenvironment in order to activate immune response and improve cancer immunotherapy [130-132]. Li et al. prepared a dendritic cell-based nanodiamond delivery system [133]. The nanodiamond (denoted as Nano-DOX) was covalently conjugated with doxorubicin (DOX), a clinically-used anticancer drug and cyclictripeptides (RGD), a tumor-targeting ligand. Nano-DOX was efficiently internalized by DCs that were isolated from mouse bone marrow to form Nano-DOX-DC. After intravenous injection into athymic mice bearing orthotopic human glioma xenografts, Nano-DOX-DC crossed the blood-brain barrier and entered into the glioma tissue, which was followed by the release of Nano-DOX into the tumor microenvironment. Nano-DOX induced emission of damage associated molecular patterns (DAMPs), including calreticulin, high-mobility group box 1 protein (HMGB1) and adenosine triphosphate, which increased the immunogenicity and antigenicity of the glioma cells and subverted the tumor-associated immunosuppression [134]. The enhanced immunogenicity stimulated maturation of DCs, which in turn promoted maturation of the innate and supplementary lymphocytes. Nano-DOX-DC exhibited greater antitumor efficacy compared with the blank DCs without Nano-DOX. The monocytes and macrophages carrying Nano-DOX also showed their preferable antitumor efficacy for glioma treatment [135, 136]. Jin et al. prepared magnetic nanoparticles to enhance the enrichment of DCs in the lymph nodes. The DCs containing the magnetic nanoparticles could be directed to the lymph node upon the external magnetic field, therefore increasing the lymphatic targeting of DCs and enhancing the anticancer effects [137]. Recently, Li et al. developed macrophage-based drug delivery systems to reverse the immunosuppressive microenvironment in the tumor tissue (Figure 6) [138]. Superparamagnetic iron oxide nanoparticles were prepared and further modified with hyaluronic acid, followed by internalization by naive macrophage (designated as HION@Mac). Inflammatory signals drove HION@Mac to accumulate in tumor tissue where HION@Mac secreted reactive oxygen species (ROS) and inflammatory factors to induce apoptosis of tumor cells. HION@Mac also demonstrated its potential to polarize intratumoral tumor-associated microphages (TAMs) to M1-type macrophages. These two pathways jointly contributed to immune activation and tumor cell apoptosis, resulting in a synergistic anticancer effect.
Schematic of the artificially reprogrammed HION@Mac for cancer therapy. Reprinted with permission from ref [138]. HION@Mac was obtained by the macrophage internalizing HION, the hyaluronic acid-coated iron oxide nanoparticles. HION@Mac after injection could migrate to the tumor with expression of inflammatory factors via chemotaxis of the macrophage. In the tumor tissue, HION@Mac not only induced tumor cell apoptosis by releasing ROS and inflammatory factors, but also facilitated the polarization from the TAMs to the M1-type macrophages for enhanced antitumor efficacy. Copyright 2019 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Engineering cells via chemical bioconjugation
Endocytosis may involve concerns about the instability of nanoparticles and unexpected release of drugs by endocytic vesicle-mediated degradation [139]. Bioconjugation of cargoes on cell membranes is an alternative approach [140]. To improve the tumor-targeting efficiency of anti-PDL1 and reduce its off-target effects, Wang et al. developed platelets conjugated with anti-PDL1 (P-aPDL1) that could travel to the surgical site in order to inhibit post-surgical tumor recurrence [141]. P-aPDL1 was obtained by conjugating aPDL1 to the surface of platelets using a SMCC crosslinker containing amine- and sulfhydryl-reactive groups. aPDL1 was stably bound to the non-activated platelets, which facilitates delivery of aPDL1 to the residual tumors at the surgical site. Upon activation of platelets, expression of PDL1 in the tumor tissue was upregulated, and aPDL1 was released due to generation of platelet-derived microparticles from cell membranes of platelets. The released aPDL1 blocked PDL1 on the tumor and antigen-presenting cells. P-aPDL1 was demonstrated to enable controlled delivery of PDL1 and potent recurrence inhibition on the post-surgical mouse models with melanoma and triple-negative breast carcinoma (TNBC). Han et al. applied P-aPDL1 to inhibit tumor relapse and metastasis after thermal ablation [142]. Photothermal therapy is limited because remnants of microtumors are often responsible for local recurrence and distant metastasis. P-aPDL1 therapy exhibited efficient targeting capacity to an incompletely ablated tumor based on damaged vascular microenvironment after photothermal treatment and therefore produced significantly augmented therapeutic efficacy on the xenograft breast tumor mouse model. Such platelet-mediated drug delivery system could also effectively target the residual microtumors after treatment with high-intensity-focused-ultrasound ablation.
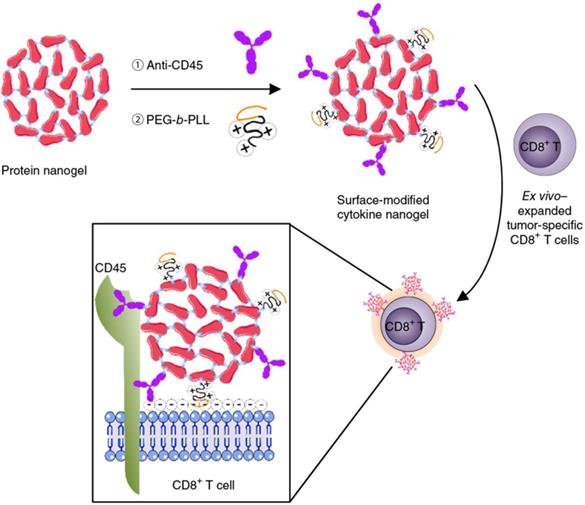
Cellular backpacks using nanoparticles without being internalized also hold promise of targeted drug delivery and enhanced cancer immunotherapy, which integrates the respective merits of nanoparticles as drug depots and cells with natural directional migration potency as active carriage [143, 144]. Huang et al. reported nanoparticle-conjugated T cells for enhanced targeting of chemotherapeutics to disseminated tumor cells in lymph nodes [145]. Topoisomerase I inhibitor, SN-38 was encapsulated in lipid nanoparticles decorated with maleimide headgroups, which were linked on the T cell surface with a high level of reduced thiol groups by maleimide-thiol coupling. The engineered T cells showed superior lymph-targeting capacity, which rendered the quantity of SN-38 in lymph nodes 90-fold higher than that of free drug that was intravenously injected at 10-fold higher dosage. Tumor burden was significantly reduced and survival period was markedly prolonged by the SN-38-loaded nanoparticle-functionalized T cells in comparison with either free SN-38 or SN-38-loaded nanoparticles. Tang et al. developed a cytokine nanogel-decorated T cell-mediated delivery system for enhanced T cell function and cancer immunotherapy (Figure 7) [146]. Repetitive units of interleukin-15 (IL-15) were crosslinked with themselves by a synthetic disulfide-containing bis-N-hydroxy succinimide crosslinker to form a “carrier-free” protein nanogel, which was later decorated with poly(ethylene glycol)-co-poly(lysine) (PEG-b-PLL) and anti-CD45 antibodies. The cationic PEG-b-PLL polymer facilitated electrostatic absorption of nanogels onto T cells, while anti-CD45 antibody increased cell surface retention of nanogels by preventing internalization. Elevation of surface reduction potential of T cells following antigen recognition in lymph node and tumor tissues led to nanogel collapse and IL-15 release. This controlled manner brought about an 8-fold higher maximum tolerated dosage of IL-15 and a 16-fold amplification of T cells. The mouse T cells and human CAR-T cells backpacking the nanogels with large quantities of IL-15 presented superior antitumor effects in mouse melanoma and glioblastoma models. Apart from IL-15, similar strategy was adopted for delivery of IL-2 to overcome the IL-2-induced vascular leak syndrome. A sustained and slow release of IL-2 was achieved by the IL-2 nanogel backpacks, leading to more CD8+ memory precursor differentiation and less T-cell exhaustion compared with free IL-2 [147].
Schematic of the modification of cytokine nanogel on the T cell surface for cancer immunotherapy. IL-15 was self-crosslinked to prepare the IL-15 nanogel, which was subsequently modified with the anti-CD45 antibodies and the cationic PEG-b-PLL polymer. The surface-modified IL-15 nanogels could be stably attached on the surface of T cells via a combination of electronic and antibody-receptor interactions. The obtained T cell backpack showed superior effects in augmenting immunotherapeutic efficacy. Reprinted with permission from ref [146]. Copyright 2018 Springer Nature.
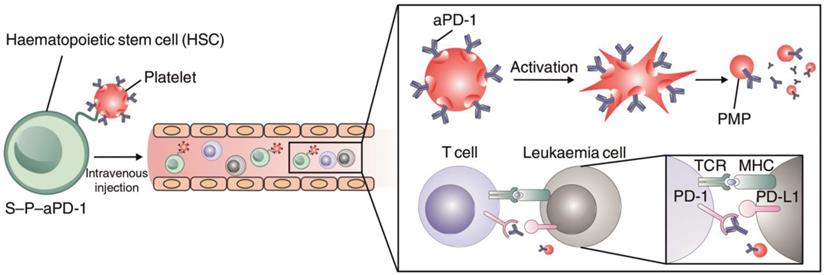
In addition to modification with antibodies or nanoparticles for cell engineering, Hu et al. conjugated anti-PD1 antibody (aPD1)-decorated platelets to hematopoietic stem cells (HSCs) to enhance the delivery of aPD1 to the leukemia site in order to inhibit leukemia growth and relapse (Figure 8) [148]. The HSC-platelet-aPD1 conjugate (S-P-PD1) could efficiently migrate to bone marrow after intravenous administration into leukemia-bearing mice and release aPD1 locally at the leukemia site upon platelet activation. Treatment with S-P-PD1 resulted in evidently enhanced anti-leukemia effects and prolonged survival of the mice through elevation of the ratio of active T cells and generation of cytokines. This “cell combination” delivery strategy displayed potent effects in improving the anticancer activity of checkpoint blockade therapy by combining the leukemia-targeting capabilities of HSCs and the controlled drug delivery property of platelets.
Schematic of the HSC-platelet assembly-assisted delivery of aPD1. The S-P-PD1 elevated the distribution of aPD1 in the leukemia site by the targeting ability of HSCs, thereby yielding enhanced anticancer effects in suppressing the leukemia proliferation and recurrence. Reprinted with permission from ref [148]. Copyright 2018 Springer Nature.
Engineering cells via physical modification
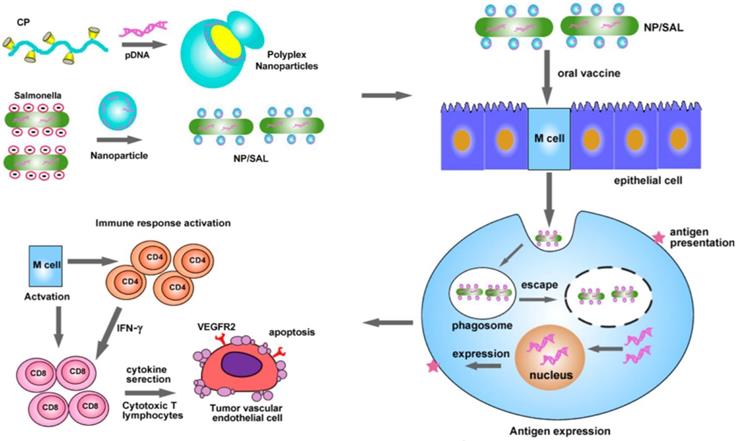
Non-covalent physical modification is a convenient strategy for engineering cells, often enabling high activity of cargoes linked to the cellular carriers compared to covalent bioconjugation. Hu et al. used cationic polymers to condense the DNA plasmid encoding VEGFR2 and Salmonella, a low-cost live attenuated bacteria as a carrier for oral delivery of DNA-based vaccines (Figure 9) [149]. Electrostatic interaction between cationic polymer and anionic DNA promoted formation of nanoparticles whose surface potential could be adjusted by the ratio of polymer and DNA. The bacteria-shuttled vaccine had excellent stability and could survive under gastric acidity after oral administration. With the aid of cationic nanoparticles, the vaccine can be efficiently taken up by the M cells that serve as protectors for antigen internalization and transportation in the intestine. VEGFR2, which was efficiently expressed by the M cells, acted as an antigen to activate the CD4+ and CD8+ T cells to eradicate the VEGFR2-expressing tumor vascular endothelial cells. This bacteria-mediated DNA-based vaccine revealed higher effect in inhibiting tumor growth due to increased angiogenesis suppression and tumor necrosis.
Schematic of oral delivery of the cationic nanoparticle-coated attenuated Salmonellae for cancer immunotherapy. The cationic nanoparticles loaded with the DNA plasmid encoding VEGFR2 were coated on the surface of the attenuated Salmonella. After oral administration, the bacteria delivered the VEGFR2 gene into the M cells, which expressed the VEGFR2 antigen to activate the CD4+ and CD8+ T cells. The activated CD8+ T cells eliminated the VEGFR2-expressing tumor vascular endothelial cells to inhibit the tumor angiogenesis and growth. Reprinted with permission from ref [149]. Copyright 2015 American Chemical Society.
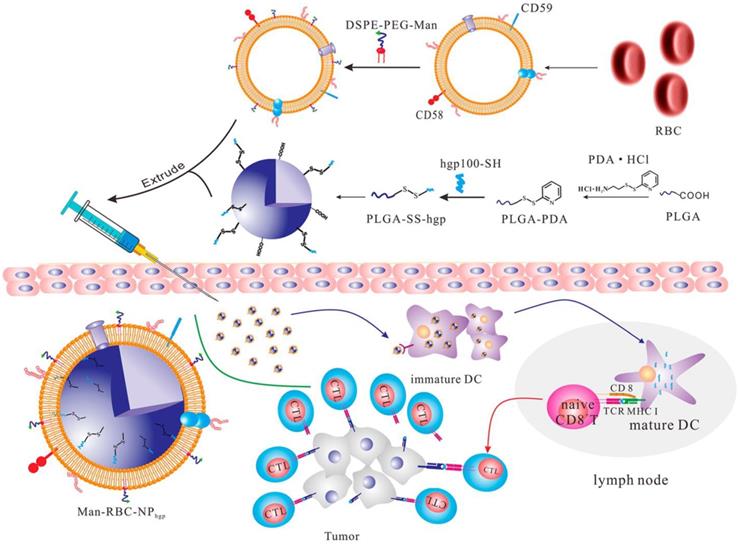
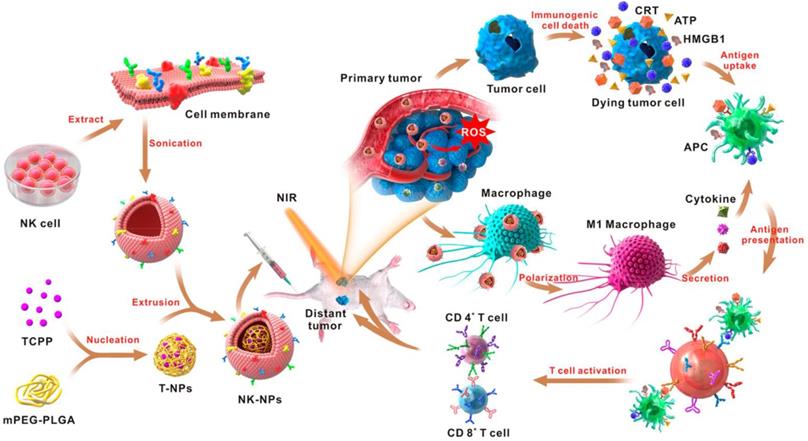
In addition to living cells, cell membranes have been widely used to develop biomimetic drug delivery systems. Preserving cell membrane integrity and a significant portion of membrane proteins facilitates protecting drugs from degradation or activating an immune response [55, 150]. Cell membranes from different types of cells have been reported to camouflage nanoparticles, including blood cells [151-153], immune cells [154, 155] and even cancer cells [150, 156]. The abundance of red blood cells (RBCs) and their lack of organelles render the RBC membranes favorable for drug delivery applications. The RBC membrane-cloaked nanoparticles revealed longer circulation time and less immunological rejection compared with the free nanoparticles [157]. Hu et al. prepared nanovaccines with notable antivirulence efficacy by absorbing intact pore-forming toxins on RBC membrane-coated nanoparticles, which laid a foundation for new vaccine design [152]. Guo et al. reported a RBC membrane-based core-shell drug delivery system for melanoma immunotherapy (Figure 10) [158]. The nanoparticles prepared by melanoma-associated antigenic peptide hgp100 conjugated PLGA polymers were cloaked by RBC membrane to form a core-shell structure. The RBC membrane was modified with DSPE-PEG-mannose, a kind of polysaccharide that can bind to the mannose receptor on the immune cells such as macrophages and DCs for antigen recognition [159, 160]. After administration, the RBC membrane-camouflaged PLGA nanoparticles were easily phagocytosed by immature DCs via mannose receptor-mediated interaction, in which hgp100 was released due to the high intracellular level of glutathione [161]. Activation of DCs by hgp100 in lymph nodes promoted production of cytotoxic T lymphocytes (CTLs) and led to massive tumor regression. Deng et al. prepared NK cell membrane-camouflaged nanoparticles (NK-NPs) by disguising photosensitizer-loaded nanoparticles with the NK cell membranes (Figure 11) [154]. Once entering circulation system, NK-NPs preferably accumulated in tumor tissue and were engulfed by cancer cells via the receptor-mediated interaction. Upon near-infrared (NIR) irradiation, a build-up of ROS generated by tetra(4-carboxyphenyl)porphine (TCPP) as a photosensitizer induced apoptosis of tumor cells. Antigens on dead tumor cells were presented to the T cells by APCs, which activated the T cells to kill remnant tumor cells. On the other hand, proteins on the NK cell membranes stimulated M1-type polarization of macrophages, which further secreted pro-inflammation cytokines to activate APCs for a durable immune response. Xie et al. reported cancer cell membrane-coated glucose oxidase-loaded mesoporous silica nanoparticles (MSNs) for cancer immunotherapy [156]. Specific antigens expressed by tumor cells activated the immune system, which indicates that coating nanoparticles with cancer cell membranes is a superb approach to elicit an immune response for cancer immunotherapy. Moreover, the homing effect of tumor cells is inherited by the cell membrane, which supports elevated tumor-targeting capabilities. Murine melanoma cells were lysed with good preservation of membrane proteins, including homologous target proteins and immune escape proteins, such as CD47. The cancer membrane-coated MSNs were translocated to the tumor tissue by membrane proteins after intravenous injection. Nutritional supply of tumor cells was cut off by glucose oxidase, which results in cell apoptosis by converting glucose to gluconic acid and hydrogen peroxide. Treatment with the obtained MSNs can efficiently enhance the PD1-based immune checkpoint blockade effect by boosting the production of effector cells via DCs, which were activated by the proteins presented on the cancer cell membrane. This starvation strategy combined with immunotherapy resulted in a synergistic anti-tumor effect. Apart from coating nanoparticles with one kind of cell membrane, mixed cell membrane decorated nanoparticles were also investigated to enhance immune response. Liu et al. prepared a cytomembrane consisting of fused cells from DCs and cancer cells to coat nanoparticles (NP@FM). NP@FM presented whole tumor antigens from DC membrane and endogenous tumor antigens from cancer cell membrane, and enhanced the activation of T cells [162].
Schematic of the RBC membrane-coated PLGA nanoparticles for production of antitumor immunity. The hgp100-conjugated PLGA nanoparticles were coated with the RBC membranes that were modified with the DSPE-PEG-mannose conjugate. The subcutaneously-injected nanoparticles were taken up by the immature DCs with expression of mannose receptors, and released hgp100 to activate the DCs and subsequently the CTLs to kill the cancer cells. Reprinted with permission from ref [158]. Copyright 2015 American Chemical Society.
Schematic of the NK cell-membrane-cloaked nanoparticles for augmented immunotherapy based on photodynamic therapy. The TCPP-loaded NK-cell-membrane-coated nanoparticles increased the intratumoral accumulation of TCPP, which produced ROS upon light irradiation to induce tumor cell apoptosis. The proteins expressed on the NK cell membranes could also promote the M1-type macrophage polarization to augment the anticancer immune response. Reprinted with permission from ref [154]. Copyright 2018 American Chemical Society.
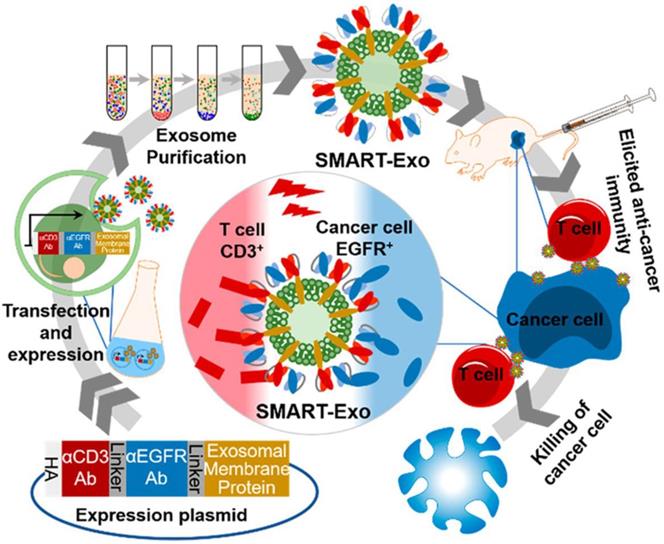
Exosomes, which are membrane vesicles with a size of 30-100 nm derived from various cell types and resemble cell membranes, are vital for cell communication and take along a collection of biological information from the donor cells [163-165]. Thus, exosomes have been increasingly investigated and utilized as drug delivery carriers for cancer immunotherapy [166, 167]. Cheng et al. transfected HEK293 cells with the DNA plasmid encoding three different proteins: CD3 antibody for T cell recognition, endothelial growth factor receptor (EGFR) antibody for breast cancer cell targeting and membrane protein for surface anchoring (Figure 12) [168]. The successfully-transfected cells could express all three proteins, and the harvested exosomes (denoted as SMART-Exo) also carried these proteins. SMART-Exo could induce crosslink between the T cells and breast cancer cells, causing the cancer cell apoptosis. In vitro cell experiments verified the T cell activation ability of SMART-Exo and in vivo studies revealed the engineered exosomes significantly suppressed tumor growth in the xenograft TNBC mouse models. Morishita et al. isolated murine melanoma cell-derived exosomes that contain cancer cell antigens and can activate APCs to provoke antitumor immunity [169]. The melanoma cells were transfected with plasmid DNA encoding streptavidin-lactadherin protein, a fusion protein anchored on membrane that would further be transferred to exosomes. The biotinylated CpG DNA that encodes proteins facilitates presenting the tumor antigens to DCs was conjugated to the exosome membrane via the biotin-streptavidin interaction. The functionalized exosomes exhibited superior antitumor efficacy in tumor-bearing mice in comparison to co-administration of exosomes and CpG DNA.
Schematic of the design and production of SMART-Exo for cancer immunotherapy. SMART-Exo with surface expression of both CD3+ and EGFR antibodies were obtained from the HEK293 cells transfected with the DNA plasmid encoding the antibodies. SMART-Exo produced the anticancer immunity by inducing the crosslink between the T cells and the EGFR-expressing cancer cells. Reprinted with permission from ref [168]. Copyright 2018 American Chemical Society.
Conclusion and Outlook
In order to enhance the efficacy and reduce the adverse effects of cancer immunotherapy, cell-based immunotherapies have attracted considerable attention today, mainly because of their potential biocompatibility and dynamic physiological functions involving immune interactions. In addition to genetically engineered cell therapies such as CAR-T and TCR-T cell therapies, other kinds of cell-based delivery systems have also been utilized for cancer immunotherapy. Augmenting the antitumor immune response and overcoming immunosuppression are the two main goals for cancer immunotherapy. The combination of immunotherapeutic drugs with cell-based delivery systems can potentially enhance the efficiency or efficacy of cancer immunotherapy, mainly reflects in protecting the drug from unexpected degradation, increasing accumulation in target sites, such as tumor and lymph node and/or boosting the immune response and overcoming the tumor immunosuppression to combat with tumor growth, recurrence or metastasis.
Although remarkable advantages have been validated when combining immunotherapy with cells or cell-derived delivery systems, many issues should be taken into account for accelerating clinical translation. For example, preparation processes of these engineered cells or cell-based delivery systems are often complicated [170, 171], causing difficulty in large-scale manufacturing, particularly for the cells that are in small quantity and difficult to harvest, which must be further exploited and optimized for practical application. Moreover, characterization of engineered cells must be comprehensively performed in order to obtain a detailed understanding of the delivery mechanism, which is essential to boost therapeutic efficacy and address safety risks in vivo. Strict criteria for quality control should be established in order to achieve reproducible engineering approaches with quality assurance.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81673381), the National Ten Thousand Talents Program for Young Top-notch Talents, the Program for Jiangsu Province Innovative Research Talents, the Program for Jiangsu Province Innovative Research Team, the Funding of Double First-Rate Discipline Innovation Team (CPU2018GF05) to R.M. and the UCLA's start-up package to Z.G.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Walker LM, Burton DR. Passive immunotherapy of viral infections:'super-antibodies' enter the fray. Nat Rev Immunol. 2018;18:297-308
2. Parida SK, Poiret T, Liu Z, Meng Q, Heyckendorf J, Lange C. et al. T-cell therapy: options for infectious diseases. Clin Infect Dis. 2015;61:217-24
3. Gehring AJ, Protzer U. Targeting innate and adaptive immune responses to cure chronic HBV infection. Gastroenterology. 2019;156:325-37
4. Maus MV, Fraietta JA, Levine BL, Kalos M, Zhao Y, June CH. Adoptive immunotherapy for cancer or viruses. Annu Rev Immunol. 2014;32:189-225
5. June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359:1361-5
6. Racke MK, Bonomo A, Scott DE, Cannella B, Levine A, Raine CS. et al. Cytokine-induced immune deviation as a therapy for inflammatory autoimmune disease. J Neuroimmunol. 1994;54:1961-6
7. Seo SK, Choi JH, Kim YH, Kang WJ, Park HY, Suh JH. et al. 4-1BB-mediated immunotherapy of rheumatoid arthritis. Nat Med. 2004;10:1088-94
8. Riley RS, June CH, Langer R, Mitchell MJ. Delivery technologies for cancer immunotherapy. Nat Rev Drug Discov. 2019;18:175-96
9. Khansur E, Shah AH, Lacy K, Komotar RJ. Novel immunotherapeutics for treatment of glioblastoma: the last decade of research. Cancer Invest. 2019;37:1-7
10. Kim HY, Park JW. Current immunotherapeutic strategies in hepatocellular carcinoma: recent advances and future directions. Therap Adv Gastroenterol. 2017;10:805-14
11. York IA, Rock KL. Antigen processing and presentation by the class I major histocompatibility complex. Annu Rev Immunol. 1996;14:369-96
12. Cohen M, Reiter Y. T-cell receptor-like antibodies: targeting the intracellular proteome therapeutic potential and clinical applications. Antibodies. 2013;2:517-34
13. Valitutti S, Müller S, Cella M, Padovan E, Lanzavecchia A. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature. 1995;375:148-51
14. Wang C, Sun W, Ye Y, Bomba HN, Gu Z. Bioengineering of artificial antigen presenting cells and lymphoid organs. Theranostics. 2017;7:3504-16
15. Gajewski TF, Schreiber H, Fu Y. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014-22
16. Smyth MJ, Ngiow SF, Ribas A, Teng MW. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat Rev Clin Oncol. 2016;13:143-58
17. Gao S, Yang D, Fang Y, Lin X, Jin X, Wang Q. et al. Engineering nanoparticles for targeted remodeling of the tumor microenvironment to improve cancer immunotherapy. Theranostics. 2019;9:126-51
18. Blattman JN, Greenberg PD. Cancer immunotherapy: a treatment for the masses. Science. 2004;305:200-5
19. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480-9
20. Rosenberg SA. Cancer immunotherapy comes of age. Nat Clin Pract Oncol. 2005;2:115
21. Mitchell M. Cancer vaccines, a critical review-Part I. Curr Opin Investig Drugs. 2002;3:140-9
22. Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019;19:133-50
23. Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299-308
24. Ying H, Zaks TZ, Wang R, Irvine KR, Kammula US, Marincola FM. et al. Cancer therapy using a self-replicating RNA vaccine. Nat Med. 1999;5:823-7
25. Toda M, Rabkin SD, Kojima H, Martuza RL. Herpes simplex virus as an in situ cancer vaccine for the induction of specific anti-tumor immunity. Hum Gene Ther. 1999;10:385-93
26. Palucka K, Banchereau J. Dendritic-cell-based therapeutic cancer vaccines. Immunity. 2013;39:38-48
27. Pardi N, Hogan MJ, Porter FW, Weissman D. mRNA vaccines—a new era in vaccinology. Nat Rev Drug Discov. 2018;17:261-79
28. Bartlett DL, Liu Z, Sathaiah M, Ravindranathan R, Guo Z, He Y. et al. Oncolytic viruses as therapeutic cancer vaccines. Mol Cancer. 2013;12:103
29. Garg AD, Coulie PG, Van den Eynde BJ, Agostinis P. Integrating next-generation dendritic cell vaccines into the current cancer immunotherapy landscape. Trends Immunol. 2017;38:577-93
30. Cheever MA, Higano CS. PROVENGE (Sipuleucel-T) in prostate cancer: the first FDA-approved therapeutic cancer vaccine. Clin Cancer Res. 2011;17:3520-6
31. Hargadon KM, Johnson CE, Williams CJ. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int Immunopharmacol. 2018;62:29-39
32. Gibney GT, Weiner LM, Atkins MB. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016;17:542-51
33. Song H, Yang P, Huang P, Zhang C, Kong D, Wang W. Injectable polypeptide hydrogel-based co-delivery of vaccine and immune checkpoint inhibitors improves tumor immunotherapy. Theranostics. 2019;9:2299-314
34. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-23
35. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L. et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2521-32
36. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE. et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373:1627-39
37. Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, Von Pawel J. et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. The Lancet. 2017;389:255-65
38. Kaufman HL, Russell J, Hamid O, Bhatia S, Terheyden P, D'Angelo SP. et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016;17:1374-85
39. Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R. et al. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N Engl J Med. 2017;377:1919-29
40. Migden MR, Rischin D, Schmults CD, Guminski A, Hauschild A, Lewis KD. et al. PD-1 blockade with cemiplimab in advanced cutaneous squamous-cell carcinoma. N Engl J Med. 2018;379:341-51
41. Stanton SE, Adams S, Disis ML. Variation in the incidence and magnitude of tumor-infiltrating lymphocytes in breast cancer subtypes: a systematic review. JAMA Oncol. 2016;2:1354-60
42. Lee WS, Park S, Lee WY, Yun SH, Chun HK. Clinical impact of tumor-infiltrating lymphocytes for survival in stage II colon cancer. Cancer. 2010;116:5188-99
43. Ping Y, Liu C, Zhang Y. T-cell receptor-engineered T cells for cancer treatment: current status and future directions. Protein Cell. 2018;9:254-66
44. Xue S, Gao L, Hart D, Gillmore R, Qasim W, Thrasher A. et al. Elimination of human leukemia cells in NOD/SCID mice by WT1-TCR gene-transduced human T cells. Blood. 2005;106:3062-7
45. Xia A, Wang X, Lu Y, Lu X, Sun B. Chimeric-antigen receptor T (CAR-T) cell therapy for solid tumors: challenges and opportunities. Oncotarget. 2017;8:90521-31
46. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ. et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507-17
47. Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science. 1986;233:1318-21
48. Clemente CG, Mihm Jr MC, Bufalino R, Zurrida S, Collini P, Cascinelli N. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer. 1996;77:1303-10
49. O'Leary MC, Lu X, Huang Y, Lin X, Mahmood I, Przepiorka D. et al. FDA approval summary: tisagenlecleucel for treatment of patients with relapsed or refractory B-cell precursor acute lymphoblastic leukemia. Clin Cancer Res. 2019;25:1142-6
50. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H. et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378:439-48
51. Puzanov I, Diab A, Abdallah K, Bingham C, Brogdon C, Dadu R. et al. Managing toxicities associated with immune checkpoint inhibitors: consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J Immunother Cancer. 2017;5:95
52. Fedorov VD, Themeli M, Sadelain M. PD-1-and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. 2013;5:215ra172
53. Yang Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest. 2015;125:3335-7
54. Agrahari V, Agrahari V, Mitra AK. Next generation drug delivery: circulatory cells-mediated nanotherapeutic approaches. Expert Opin Drug Deliv. 2017;14:285-9
55. Tan S, Wu T, Zhang D, Zhang Z. Cell or cell membrane-based drug delivery systems. Theranostics. 2015;5:863-81
56. Su Y, Xie Z, Kim GB, Dong C, Yang J. Design strategies and applications of circulating cell-mediated drug delivery systems. ACS Biomater Sci Eng. 2015;1:201-17
57. Knott GJ, Doudna JA. CRISPR-Cas guides the future of genetic engineering. Science. 2018;361:866-9
58. Ray M, Lee YW, Hardie J, Mout R, Yeşilbag Tonga G, Farkas ME. et al. CRISPRed macrophages for cell-based cancer immunotherapy. Bioconjug Chem. 2018;29:445-50
59. Rezvani K, Rouce R, Liu E, Shpall E. Engineering natural killer cells for cancer immunotherapy. Mol Ther. 2017;25:1769-81
60. Zhao B, Wang Y, Tan X, Zheng X, Wang F, Ke K. et al. An Optogenetic Controllable T Cell System for Hepatocellular Carcinoma Immunotherapy. Theranostics. 2019;9:1837-50
61. Kershaw MH, Westwood JA, Darcy PK. Gene-engineered T cells for cancer therapy. Nat Rev Cancer. 2013;13:525-41
62. June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018;379:64-73
63. Schumacher TN. T-cell-receptor gene therapy. Nat Rev Immunol. 2002;2:512-9
64. Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T-bet in TH1 lineage commitment and IFN-γ production in CD4 and CD8 T cells. Science. 2002;295:338-42
65. Seddiki N, Santner-Nanan B, Martinson J, Zaunders J, Sasson S, Landay A. et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J Exp Med. 2006;203:1693-700
66. Benchimol S, Fuks A, Jothy S, Beauchemin N, Shirota K, Stanners CP. Carcinoembryonic antigen, a human tumor marker, functions as an intercellular adhesion molecule. Cell. 1989;57:327-34
67. Morgan RA, Dudley ME, Yik Y, Zheng Z, Robbins PF, Theoret MR. et al. High efficiency TCR gene transfer into primary human lymphocytes affords avid recognition of melanoma tumor antigen glycoprotein 100 and does not alter the recognition of autologous melanoma antigens. J Immunol. 2003;171:3287-95
68. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C. et al. Human epidermal growth factor receptor 2 (HER2) -specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol. 2015;33:1688-96
69. Igney FH, Krammer PH. Immune escape of tumors: apoptosis resistance and tumor counterattack. J Leukoc Biol. 2002;71:907-20
70. Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer. 2016;16:566-81
71. Nelson MH, Paulos CM. Novel immunotherapies for hematologic malignancies. Immunol Rev. 2015;263:90-105
72. Landoni E, Savoldo B. Treating hematological malignancies with cell therapy: where are we now? Expert Opin Biol Ther. 2018;18:65-75
73. Tawara I, Kageyama S, Miyahara Y, Fujiwara H, Nishida T, Akatsuka Y. et al. Safety and persistence of WT1-specific T-cell receptor gene-transduced lymphocytes in patients with AML and MDS. Blood. 2017;130:1985-94
74. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM. et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126-9
75. Barber A, Zhang T, Megli CJ, Wu J, Meehan KR, Sentman CL. Chimeric NKG2D receptor-expressing T cells as an immunotherapy for multiple myeloma. Exp Hematol. 2008;36:1318-28
76. Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA. et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19:620-6
77. Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME. et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29:917-24
78. Orlando D, Miele E, De Angelis B, Guercio M, Boffa I, Sinibaldi M. et al. Adoptive immunotherapy using PRAME-specific T cells in medulloblastoma. Cancer Res. 2018;78:3337-49
79. Straathof KC, Pulè MA, Yotnda P, Dotti G, Vanin EF, Brenner MK. et al. An inducible caspase 9 safety switch for T-cell therapy. Blood. 2005;105:4247-54
80. D'Aloia MM, Zizzari IG, Sacchetti B, Pierelli L, Alimandi M. CAR-T cells: the long and winding road to solid tumors. Cell Death Dis. 2018;9:282
81. Yip A, Webster RM. The market for chimeric antigen receptor T cell therapies. Nat Rev Drug Discov. 2018;17:161-2
82. Burga RA, Thorn M, Point GR, Guha P, Nguyen CT, Licata LA. et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol Immunother. 2015;64:817-29
83. Chinnasamy D, Tran E, Yu Z, Morgan RA, Restifo NP, Rosenberg SA. Simultaneous targeting of tumor antigens and the tumor vasculature using T lymphocyte transfer synergize to induce regression of established tumors in mice. Cancer Res. 2013;73:3371-80
84. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL. et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15:47-62
85. Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M. et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018;24:739-48
86. Guedan S, Posey Jr AD, Shaw C, Wing A, Da T, Patel PR. et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI insight. 2018;3:96976
87. Ying Z, Huang X, Xiang X, Liu Y, Kang X, Song Y. et al. A safe and potent anti-CD19 CAR T cell therapy. Nat Med. 2019;25:947-53
88. Cheung AS, Zhang D, Koshy ST, Mooney DJ. Scaffolds that mimic antigen-presenting cells enable ex vivo expansion of primary T cells. Nat Biotechnol. 2018;36:160-9
89. Smith TT, Stephan SB, Moffett HF, McKnight LE, Ji W, Reiman D. et al. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat Nanotechnol. 2017;12:813-20
90. Ma L, Dichwalkar T, Chang JY, Cossette B, Garafola D, Zhang AQ. et al. Enhanced CAR-T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science. 2019;365:162-8
91. Link A, Vogt TK, Favre S, Britschgi MR, Acha-Orbea H, Hinz B. et al. Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nat Immunol. 2007;8:1255-65
92. Luther SA, Bidgol A, Hargreaves DC, Schmidt A, Xu Y, Paniyadi J. et al. Differing activities of homeostatic chemokines CCL19, CCL21, and CXCL12 in lymphocyte and dendritic cell recruitment and lymphoid neogenesis. J Immunol. 2002;169:424-33
93. Fry TJ, Mackall CL. Interleukin-7: master regulator of peripheral T-cell homeostasis? Trends Immunol. 2001;22:564-71
94. Haessler U, Pisano M, Wu M, Swartz MA. Dendritic cell chemotaxis in 3D under defined chemokine gradients reveals differential response to ligands CCL21 and CCL19. Proc Natl Acad Sci U S A. 2011;108:5614-9
95. Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol. 2018;36:346-51
96. Chen Q, Hu Q, Dukhovlinova E, Chen G, Ahn S, Wang C. et al. Photothermal therapy promotes tumor infiltration and antitumor activity of CAR T cells. Adv Mater. 2019;31:1900192
97. Cho JH, Collins JJ, Wong WW. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell. 2018;173:1426-38
98. Roybal KT, Williams JZ, Morsut L, Rupp LJ, Kolinko I, Choe JH. et al. Engineering T cells with customized therapeutic response programs using synthetic notch receptors. Cell. 2016;167:419-32
99. Shi F, Ljunggren HG, La Cava A, Van Kaer L. Organ-specific features of natural killer cells. Nat Rev Immunol. 2011;11:658-71
100. Cerwenka A, Lanier LL. Natural killer cell memory in infection, inflammation and cancer. Nat Rev Immunol. 2016;16:112-23
101. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677-86
102. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889-96
103. Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL. et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. 1999;285:727-9
104. El-Sherbiny YM, Meade JL, Holmes TD, McGonagle D, Mackie SL, Morgan AW. et al. The requirement for DNAM-1, NKG2D, and NKp46 in the natural killer cell-mediated killing of myeloma cells. Cancer Res. 2007;67:8444-9
105. Hu Y, Tian Z, Zhang C. Chimeric antigen receptor (CAR)-transduced natural killer cells in tumor immunotherapy. Acta Pharmacol Sin. 2018;39:167-76
106. Jiang H, Zhang W, Shang P, Zhang H, Fu W, Ye F. et al. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol Oncol. 2014;8:297-310
107. Chu J, Deng Y, Benson DM, He S, Hughes T, Zhang J. et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28:917-27
108. Zhang X, Wang J, Chen Z, Hu Q, Wang C, Yan J. et al. Engineering PD-1-presenting platelets for cancer immunotherapy. Nano Lett. 2018;18:5716-25
109. Xue Q, Li X, Yang C, Ji B, Li Y, Yan Y. et al. Efficacy of recombinant adenovirus expressing a fusion gene from GM-CSF and Epstein-Barr virus LMP2A in a mouse tumor model. Hum Vaccin Immunother. 2017;13:2260-8
110. Davis K, Wood S, Dill E, Fesko Y, Bitting RL, Harrison MR. et al. Optimizing the efficiency and quality of sipuleucel-T delivery in an academic institution. Clin J Oncol Nurs. 2015;19:297-303
111. Xue J, Zhao Z. et al. Neutrophil-mediated anticancer drug delivery for suppression of postoperative malignant glioma recurrence. Nat Nanotechnol. 2017;12:692-700
112. Zhang L, Zhang Y, Xue Y, Wu Y, Wang Q, Xue L. et al. Transforming weakness into strength: photothermal-therapy-induced inflammation enhanced cytopharmaceutical chemotherapy as a combination anticancer treatment. Adv Mater. 2019;31:1805936
113. Garcia JA. Sipuleucel-T in patients with metastatic castration-resistant prostate cancer: an insight for oncologists. Ther Adv Med Oncol. 2011;3:101-8
114. Golchin A, Farahany TZ. Biological products: cellular therapy and FDA approved products. Stem Cell Rev Rep. 2019;15:166-75
115. Xu Y, Wang Y, Yang Q, Liu Z, Xiao Z, Le Z. et al. A versatile supramolecular nanoadjuvant that activates NF-κB for cancer immunotherapy. Theranostics. 2019;9:3388-97
116. Li H, Li Y, Wang X, Hou Y, Hong X, Gong T. et al. Rational design of polymeric hybrid micelles to overcome lymphatic and intracellular delivery barriers in cancer immunotherapy. Theranostics. 2017;7:4383-98
117. Chavez M, Silvestrini MT, Ingham ES, Fite BZ, Mahakian LM, Tam SM. et al. Distinct immune signatures in directly treated and distant tumors result from TLR adjuvants and focal ablation. Theranostics. 2018;8:3611-28
118. Duan X, Chan C, Lin W. Nanoparticle-mediated Immunogenic Cell Death Enables and Potentiates Cancer Immunotherapy. Angew Chem Int Ed. 2019;58:670-80
119. Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751-60
120. Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF. et al. Analysis of nanoparticle delivery to tumours. Nat Rev Mater. 2016;1:16014
121. Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Del Rev. 2011;63:136-51
122. Danhier F. To exploit the tumor microenvironment: since the EPR effect fails in the clinic, what is the future of nanomedicine? J Control Release. 2016;244:108-21
123. Sang W, Zhang Z, Dai Y, Chen X. Recent advances in nanomaterial-based synergistic combination cancer immunotherapy. Chem Soc Rev. 2019;48:3771-810
124. Chen Z, Hu Q, Gu Z. Leveraging engineering of cells for drug delivery. Acc Chem Res. 2018;51:668-77
125. Li T, Dong H, Zhang C, Mo R. Cell-based drug delivery systems for biomedical applications. Nano Res. 2018;11:5240-57
126. Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267-96
127. Gajewski TF, Meng Y, Blank C, Brown I, Kacha A, Kline J. et al. Immune resistance orchestrated by the tumor microenvironment. Immunol Rev. 2006;213:131-45
128. Giglio BC, Fei H, Wang M, Wang H, He L, Feng H. et al. Synthesis of 5-[18F] Fluoro-α-methyl tryptophan: new Trp based PET agents. Theranostics. 2017;7:1524-30
129. Burvenich IJG, Parakh S, Lee FT, Guo N, Liu Z, Gan HK. et al. Molecular imaging of T cell co-regulator factor B7-H3 with 89Zr-DS-5573a. Theranostics. 2018;8:4199-209
130. Ozpiskin OM, Zhang L, Li J. Immune targets in the tumor microenvironment treated by radiotherapy. Theranostics. 2019;9:1215-31
131. Kong M, Tang J, Qiao Q, Wu T, Qi Y, Tan S. et al. Biodegradable hollow mesoporous silica nanoparticles for regulating tumor microenvironment and enhancing antitumor efficiency. Theranostics. 2017;7:3276-92
132. Liu L, Chen Q, Ruan C, Chen X, Zhang Y, He X. et al. Platinum-Based Nanovectors Engineered with Immuno-Modulating Adjuvant for Inhibiting Tumor growth and Promoting Immunity. Theranostics. 2018;8:2974-87
133. Li T, Li K, Zhang Q, Wang C, Yue Y, Chen Z. et al. Dendritic cell-mediated delivery of doxorubicin-polyglycerol-nanodiamond composites elicits enhanced anti-cancer immune response in glioblastoma. Biomaterials. 2018;181:35-52
134. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12:860-75
135. Wang C, Li K, Li T, Chen Z, Wen Y, Liu X. et al. Monocyte-mediated chemotherapy drug delivery in glioblastoma. Nanomedicine. 2018;13:157-78
136. Li T, Li K, Wang C, Liu X, Wen Y, Xu Y. et al. Harnessing the cross-talk between tumor cells and tumor-associated macrophages with a nano-drug for modulation of glioblastoma immune microenvironment. J Control Release. 2017;268:128-46
137. Jin H, Qian Y, Dai Y, Qiao S, Huang C, Lu L. et al. Magnetic enrichment of dendritic cell vaccine in lymph node with fluorescent-magnetic nanoparticles enhanced cancer immunotherapy. Theranostics. 2016;6:2000-14
138. Li C, Zhang Y, Dong X, Zhang L, Liu M, Li B. et al. Artificially reprogrammed macrophages as tumor-tropic immunosuppression-resistant biologics to realize therapeutics production and immune activation. Adv Mater. 2019;31:1807211
139. Batrakova EV, Gendelman HE, Kabanov AV. Cell-mediated drug delivery. Expert Opin Drug Deliv. 2011;8:415-33
140. Li PY, Fan Z, Cheng H. Cell membrane bioconjugation and membrane-derived nanomaterials for immunotherapy. Bioconjug Chem. 2018;29:624-34
141. Wang C, Sun W, Ye Y, Hu Q, Bomba HN, Gu Z. In situ activation of platelets with checkpoint inhibitors for post-surgical cancer immunotherapy. Nat Biomed Eng. 2017;1:0011
142. Han X, Chen J, Chu J, Liang C, Ma Q, Fan Q. et al. Platelets as platforms for inhibition of tumor recurrence post-physical therapy by delivery of anti-PD-L1 checkpoint antibody. J Control Release. 2019;304:233-41
143. Stephan MT, Moon JJ, Um SH, Bershteyn A, Irvine DJ. Therapeutic cell engineering with surface-conjugated synthetic nanoparticles. Nat Med. 2010;16:1035-41
144. Zheng C, Wang Q, Wang Y, Zhao X, Gao K, Liu Q. et al. In situ modification of the tumor cell surface with immunomodulating nanoparticles for effective suppression of tumor growth in mice. Adv Mater. 2019. 1902 542
145. Huang B, Abraham WD, Zheng Y, López SCB, Luo SS, Irvine DJ. Active targeting of chemotherapy to disseminated tumors using nanoparticle-carrying T cells. Sci Transl Med. 2015;7:291ra94
146. Tang L, Zheng Y, Melo MB, Mabardi L, Castaño AP, Xie Y-Q. et al. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat Biotechnol. 2018;36:707-16
147. Xie Y, Arik H, Wei L, Zheng Y, Suh H, Irvine DJ. et al. Redox-responsive interleukin-2 nanogel specifically and safely promotes the proliferation and memory precursor differentiation of tumor-reactive T-cells. Biomater Sci. 2019;7:1345-57
148. Hu Q, Sun W, Wang J, Ruan H, Zhang X, Ye Y. et al. Conjugation of haematopoietic stem cells and platelets decorated with anti-PD-1 antibodies augments anti-leukaemia efficacy. Nat Biomed Eng. 2018;2:831-40
149. Hu Q, Wu M, Fang C, Cheng C, Zhao M, Fang W. et al. Engineering nanoparticle-coated bacteria as oral DNA vaccines for cancer immunotherapy. Nano Lett. 2015;15:2732-9
150. Fang RH, Hu CM, Luk BT, Gao W, Copp JA, Tai Y. et al. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014;14:2181-8
151. Hu CM, Fang RH, Wang KC, Luk BT, Thamphiwatana S, Dehaini D. et al. Nanoparticle biointerfacing by platelet membrane cloaking. Nature. 2015;526:118-21
152. Hu CM, Fang RH, Luk BT, Zhang L. Nanoparticle-detained toxins for safe and effective vaccination. Nat Nanotechnol. 2013;8:933-8
153. Hu Q, Sun W, Qian C, Wang C, Bomba HN, Gu Z. Anticancer platelet-mimicking nanovehicles. Adv Mater. 2015;27:7043-50
154. Deng G, Sun Z, Li S, Peng X, Li W, Zhou L. et al. Cell-membrane immunotherapy based on natural killer cell membrane coated nanoparticles for the effective inhibition of primary and abscopal tumor growth. ACS Nano. 2018;12:12096-108
155. Pitchaimani A, Nguyen TDT, Aryal S. Natural killer cell membrane infused biomimetic liposomes for targeted tumor therapy. Biomaterials. 2018;160:124-37
156. Xie W, Deng W, Zan M, Rao L, Yu G, Zhu D. et al. Cancer cell membrane camouflaged nanoparticles to realize starvation therapy together with checkpoint blockades for enhancing cancer therapy. ACS Nano. 2019;13:2849-57
157. Han X, Wang C, Liu Z. Red blood cells as smart delivery systems. Bioconjug Chem. 2018;29:852-60
158. Guo Y, Wang D, Song Q, Wu T, Zhuang X, Bao Y. et al. Erythrocyte membrane-enveloped polymeric nanoparticles as nanovaccine for induction of antitumor immunity against melanoma. ACS Nano. 2015;9:6918-33
159. Stahl PD, Ezekowitz RAB. The mannose receptor is a pattern recognition receptor involved in host defense. Curr Opin Immunol. 1998;10:50-5
160. Shi C, Liu T, Guo Z, Zhuang R, Zhang X, Chen X. Reprogramming tumor-associated macrophages by nanoparticle-based reactive oxygen species photogeneration. Nano Lett. 2018;18:7330-42
161. Hirosue S, Kourtis IC, van der Vlies AJ, Hubbell JA, Swartz MA. Antigen delivery to dendritic cells by poly (propylene sulfide) nanoparticles with disulfide conjugated peptides: Cross-presentation and T cell activation. Vaccine. 2010;28:7897-906
162. Liu W, Zou M, Liu T, Zeng J, Li X, Yu W. et al. Cytomembrane nanovaccines show therapeutic effects by mimicking tumor cells and antigen presenting cells. Nat Commun. 2019;10:3199
163. Robbins PD, Morelli AE. Regulation of immune responses by extracellular vesicles. Nat Rev Immunol. 2014;14:195-208
164. Johnstone RM. Exosomes biological significance: a concise review. Blood Cells Mol Dis. 2006;36:315-21
165. Kaur A, Leishangthem GD, Bhat P, Mahajan V, Singh ND, Banga HS. Role of exosomes in pathology-a review. J Pathol. 2014;1:07-11
166. Lindenbergh MF, Stoorvogel W. Antigen presentation by extracellular vesicles from professional antigen-presenting cells. Annu Rev Immunol. 2018;36:435-59
167. Syn NL, Wang L, Chow EK, Lim CT, Goh BC. Exosomes in cancer nanomedicine and immunotherapy: prospects and challenges. Trends Biotechnol. 2017;35:665-76
168. Cheng Q, Shi X, Han M, Smbatyan G, Lenz HJ, Zhang Y. Reprogramming exosomes as nanoscale controllers of cellular immunity. J Am Chem Soc. 2018;140:16413-7
169. Morishita M, Takahashi Y, Matsumoto A, Nishikawa M, Takakura Y. Exosome-based tumor antigens-adjuvant co-delivery utilizing genetically engineered tumor cell-derived exosomes with immunostimulatory CpG DNA. Biomaterials. 2016;111:55-65
170. Chen Z, Wang Z, Gu Z. Bioinspired and biomimetic nanomedicines. Acc Chem Res. 2019;52:1255-64
171. Dwarshuis NJ, Parratt K, Santiago-Miranda A, Roy K. Cells as advanced therapeutics: State-of-the-art, challenges, and opportunities in large scale biomanufacturing of high-quality cells for adoptive immunotherapies. Adv Drug Del Rev. 2017;114:222-39
Author contact
![]() Corresponding author: Zhen Gu (guzhenedu); Ran Mo (rmoedu.cn)
Corresponding author: Zhen Gu (guzhenedu); Ran Mo (rmoedu.cn)