Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Nanotechnology for delivery...
3. RNA nanotherapeutics for...
4. Conclusions and future...
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(1):281-299. doi:10.7150/thno.35568 This issue Cite
Review
RNA Nanotechnology-Mediated Cancer Immunotherapy
Yao-Xin Lin1*, Yi Wang1,2,4*, Sara Blake1,5, Mian Yu3, Lin Mei3 ![]() , Hao Wang2,4
, Hao Wang2,4 ![]() , Jinjun Shi1
, Jinjun Shi1 ![]()
1. Center for Nanomedicine and Department of Anesthesiology, Brigham and Women's Hospital, Harvard Medical School, Boston, MA 02115, USA
2. CAS Center for Excellence in Nanoscience, CAS Key Laboratory for Biomedical Effects of Nanomaterials and Nanosafety, National Center for Nanoscience and Technology, Beijing 100190, China
3. School of Pharmaceutical Sciences (Shenzhen), Sun Yat-sen University, Guangzhou, Guangdong 510006, China
4. Center of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
5. Tufts University, Medford, MA 02155, USA
*These authors contributed equally to this work.
Received 2019-4-8; Accepted 2019-8-6; Published 2020-1-1
Abstract

RNA molecules (e.g., siRNA, microRNA, and mRNA) have shown tremendous potential for immunomodulation and cancer immunotherapy. They can activate both innate and adaptive immune system responses by silencing or upregulating immune-relevant genes. In addition, mRNA-based vaccines have recently been actively pursued and tested in cancer patients, as a form of treatment. Meanwhile, various nanomaterials have been developed to enhance RNA delivery to the tumor and immune cells. In this review article, we summarize recent advances in the development of RNA-based therapeutics and their applications in cancer immunotherapy. We also highlight the variety of nanoparticle platforms that have been used for RNA delivery to elicit anti-tumor immune responses. Finally, we provide our perspectives of potential challenges and opportunities of RNA-based nanotherapeutics in clinical translation towards cancer immunotherapy.
Keywords: RNA, nanoparticle, immunotherapy, cancer, RNAi, CRISPR
1. Introduction
Over the past two decades, RNA-based therapeutics, such as messenger RNA (mRNA), microRNA, and small interfering RNA (siRNA), have emerged as highly attractive classes of drugs for the treatment and prevention of numerous diseases (e.g., cancers, genetic disorders, diabetes, inflammatory diseases, and neurodegenerative diseases)[1-6]. In comparison to the conventional small molecular drugs or proteins (specifically antibodies), RNA therapeutics can play remarkable regulatory roles in the treatment of targeted cells by either increasing the expression of a given protein or knocking out targeted genes to some varying degree. In addition to these regulatory roles, the fact that they are more convenient and easier to design than protein-based drugs, is what makes RNA therapeutics such an appealing form of treatment to researchers. However, the instability of RNAs themselves and the presence of various physiological barriers that inhibit the delivery and transfection of RNAs are what hinder their clinical application in cancer therapy [7, 8]. Moreover, the exogenous RNA is more likely to be cleared by the human body's intrinsic defense systems, e.g., various exonucleases and RNases responsible for RNA degradation, the major organs or tissues (e.g., kidneys and liver), and the innate immune system for RNA clearance [3, 9]. To overcome such immunogenic hurdles and make sure safe delivery of these RNA therapeutics to their target sites occurs, nanoparticle-based delivery systems have been explored as potential RNA delivery tools for in vitro and in vivo applications [10, 11].
Since the clinical success of immune checkpoint blockade (ICB)[12] and chimeric antigen receptor (CAR) T-cell therapies[13, 14], cancer immunotherapy treatments have drawn increasing interests. In contrast to chemotherapeutic drugs with dose-limited toxicities and potential development of drug-resistance by tumor cells, immunotherapeutics can inhibit the ability of tumor cells to evade termination by the immune system or re-program cancer-associated immune systems, and are thus more specific and able to trigger long-lasting memory anti-tumor responses. Despite these desirable features and research breakthroughs, currently used ICB antibodies and cell-based therapeutics (e.g., CAR-T) in tumor immunotherapy are far from perfect, and it is imperative to pursue new strategies for improving their safety and efficacy [15-17]. RNA-based therapeutics have many potential uses in immunomodulation and cancer immunotherapy, such as silencing immune checkpoint genes, activating the innate or adaptive immune system by regulating cytokines expressions, and acting as tumor antigen vaccines[18, 19]. The use of RNA-based therapeutics has recently expanded dramatically, and some have been moved to clinical trial studies during the past decade, revealing these genetic materials as excellent candidates for cancer treatment. Meanwhile, the development of various nanoparticle-based platforms, such as liposomes [20], polymeric nanoparticles (NPs)[21-26], and inorganic NPs[27, 28] for efficient delivery of RNAs provides a bright future for RNA-based therapeutics and their applications in cancer immunotherapy.
In this review article, an overview of RNA-based nanotherapeutics and recent advances, including their delivery nanoplatforms and applications in tumor immunotherapy, will be presented. Also, the various nanomaterials that have been used to deliver RNAs to tumor cells or immune cells for the induction of anti-tumor immune responses, will be highlighted. Finally, the current challenges of RNA-based nanotherapeutics will be discussed and the potential clinical value of RNA-based nanotherapeutics in tumor immunotherapy will be highlighted.
2. Nanotechnology for delivery of therapeutic RNAs
2.1 RNA therapeutics
RNA-based therapeutics have demonstrated a wide array of promising applications in the field of cancer treatment. They function as either inhibitors (e.g., siRNA and microRNA) or upregulators (e.g., mRNA) of target protein expression (Figure 1). siRNA is double-stranded in nature and approximately 22 nucleotides in length. Its precursor is initially recognized by Dicer RNase and is then incorporated into the RNA-induced silencing complex (RISC). The siRNA-RISC complex can bind the targeting site of mRNA, and lead to a sequence-specific cleavage by endonuclease Argonaute-2 (AGO2), thus decreasing expressions of a targeted protein [29]. MicroRNA is another common short regulatory noncoding RNA, used for blocking target gene expression via binding to target sites in the 3'-untranslated regions (UTR) of protein-coding transcripts [30]. Firstly, primary microRNA (pri-microRNA) with a characteristic hairpin structure is recognized and processed by enzymes of Drosha and DGCR8 into ∼70 nt precursor microRNA (pre-microRNA). The resultant pre-microRNA is further cleaved by Dicer RNase, thus resulting in the formation of a mature dsRNA (microRNA). The mature microRNA is finally incorporated into RISC to induce cleavage of targeted mRNA, such as siRNAs, or translational repression, which induces a decrease of targeted proteins. Generally, the target sequences of the microRNA are frequently found in the 3' UTR of mRNA and can often be found within non-coding or intronic regions. Therefore, each microRNA can be capable of targeting hundreds of unique mRNAs and inducing regulation of the transcriptome. However, in comparison to microRNA's multi-mRNA targeting abilities, siRNA has specific binding activity; therefore, each siRNA can only bind one mRNA target.
The biological mechanism of siRNA, microRNA, and mRNA for inhibition of target protein expressions or up-regulation of a given protein.
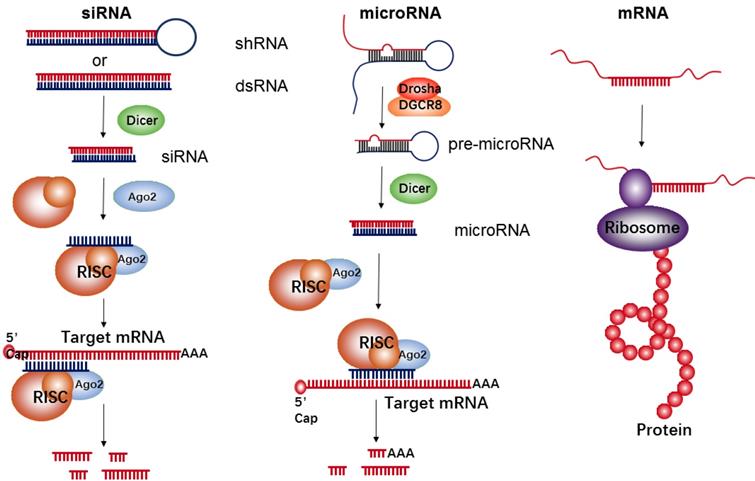
The goal of mRNA delivery is to upregulate targeted protein expressions like DNA delivery, but in contrast to DNA, mRNA therapeutics have several unique features, such as the absent risk of insertional mutagenesis, more consistent and predictable kinetics of protein expression, and relatively convenient in vitro synthesis[31]. Meanwhile, the transfection efficiency with mRNA is higher than that of DNA, especially in immune cells[32-34]. Each mRNA has an open reading frame (ORF) that includes two untranslated regions (UTRs) located at the 5' and 3' ends of mRNA, with the purpose of being recognized by the translational machinery (Ribosome). In addition to those UTRs, the mRNA's 5' methyl cap and 3' poly(adenosine) tail are also crucial for efficient translation[35].
2.2 Nanocarriers for RNA delivery
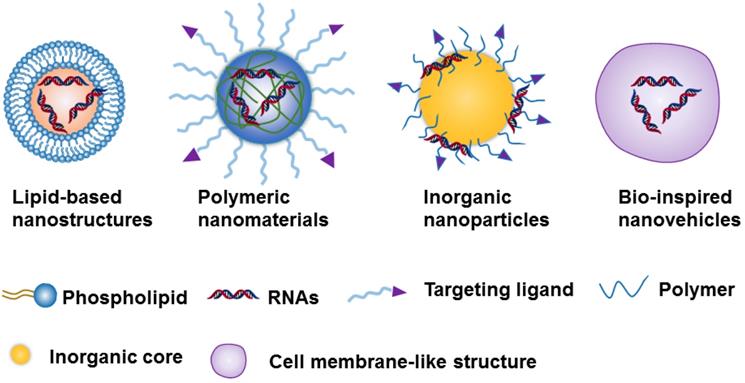
Due to the challenges of naked RNA molecules for in vivo applications, i.e., extremely short half-lives (e.g., minutes), poor chemical stability, and easy degradation by nucleases[18, 36], nanotechnology provides a versatile and targeted system for the safe delivery of them [37]. The nanoparticle-based delivery systems not only protect RNA molecules from enzymatic degradation and immune system threats, but they also enable RNA accumulation to occur in the tumor site [35, 36]. This RNA accumulation is able to occur due to the enhanced permeability and retention effect (EPR), which is a byproduct of the diameter of the NPs, which can range from 10 to 200 nm[35, 36]. Currently, the constituents of nanocarriers applied to RNAs delivery can be classified as lipid-based nanosystems [38-42], polymeric nanomaterials [43-45], inorganic nanoparticles [28, 46, 47] ,or Bio-inspired nanovehicles [48, 49] (Figure 2). We will here summarize these nanoparticle-based platforms and further discuss their strengths and potential drawbacks for RNA delivery (Table 1).
Nanoparticle-based platforms for RNA delivery
| Nanocarriers | Classifications | Advantages | Disadvantages |
|---|---|---|---|
| Lipid-based nanostructures | Liposomes; solid lipid nanoparticles; lipid emulsions | Easy preparation, good biocompatibility and biodegradability | Limited stability, easy leakage of payloads, and rapid clearance |
| Polymer-based nanomaterials | Natural or naturally derived polymers: chitosan, poly-l-lysine, atelocollagen, etc. Synthetic polymers: PLGA, PEI, PVA, PLA, PEG, etc. | Good biocompatibility and biodegradability for natural or naturally derived polymers, low cost of production, stimulation of drug release, easy modification | Nondegradable for some responsive polymers, dose-dependent toxicity |
| Inorganic NPs | MSNs, CNTs, QDs, and metal nanoparticles (e.g., iron oxide and gold nanoparticles) | Easy surface modification, good reproducibility, and easy cell uptake | Non-biodegradability, potential toxicity |
| Bio-inspired nano-vehicles | DNA-based nanostructures, exosome-mimetic nanovesicles, red blood cell member-based ghosts | Good biodegradability, low toxicity, strong targeting and low immune induction | High cost, stability concern |
Schematic representation of the 4 nanoparticle-based platforms used in the RNA delivery.
2.2.1 Lipid-based nanostructures
Lipid-based nanostructures are one of the most commonly used non-viral delivery systems both in academic studies and clinical trials for chemotherapeutics or genetic drugs. Several classes of lipid-based nanocarriers including liposomes, lipid nanoemulsions, and solid lipid NPs have been used for RNA delivery [50]. Generally, cationic lipids could be used as RNAs delivery carriers owing to their positively charged motif that has a strong interaction with negatively charged nucleic acids. As the lipids and phospholipids are the basic units of the cell membrane, lipid-based nanostructures with the similar units have a natural tendency to interact well with the cells membrane and thereby facilitating cellular uptake of RNAs. Meanwhile, the obvious other advantages of lipid-based nanostructures, such as easy preparation, good biocompatibility and biodegradability, have led them to be a promising tool in the delivery of RNA-based therapeutics. However, there are also several issues, including limited stability, easy leakage of payloads, and rapid clearance by the kidneys or liver should be addressed before these lipid-based delivery systems are put to perform at clinical or in vivo levels.
2.2.2 Polymer-based nanomaterials
Polymer-based nanomaterials are well-studied systems for RNA delivery [51], which are classified into two major categories: natural or naturally derived polymers, and synthetic polymeric conjugates [43, 44]. Natural or naturally derived polymers such as chitosan, which is composed of N-acetyl-d-glucosamine and d-glucosamine, poly-l-lysine, which consists of repeating units of lysine, and atelocollagen, occur in nature and are produced by all living organisms. The advantages of these nanoparticles are good biocompatibility and biodegradability, and low cost of production [52-54]. Among the synthetic polymers, poly(dl-lactide-co-glycolide) (PLGA), polyethyleneimine (PEI), polyvinyl alcohol (PVA), polyethylene glycol (PEG), poly-l-lactic acid (PLA), etc., are the most common polymers used in delivery of RNAs due to their high stability, good biocompatibility and biodegradability [55-59]. Meanwhile, it is important to note that these synthetic polymers are easy to functionalize with ligand bindings for targeting, and with responsive units that respond to chemical, biological, and physical stimuli for the controllable release of cargoes. However, some synthetic polymers (e.g., PLGA) could not be directly applied in RNA delivery due to no cationic units on them, thus leading to low electrostatic interaction between polymers and RNAs. One method to overcome this problem is modification with various cationic motifs (e.g., PEI) or co-assembly with cationic polymers into nanostructures, it is also noted that the most cationic units or polymers are nondegradable, which may be associated with potential toxicity issues [22].
2.2.3 Inorganic nanoparticles
Recently, various inorganic NPs, such as mesoporous silica nanomaterials (MSNs) [60-62], carbon nanotubes (CNTs) [63], quantum dots (QDs) [64], and metal nanostructures[65] (e.g., iron oxide and gold NPs), are reported as carriers for delivery of RNAs. These inorganic NPs are synthesized by biodegradable polymers and inorganic particles. Consequently, the properties of these nanoparticles are easy to control, and their advantages include surface modification, good reproducibility, and easy cell uptake. However, the level of degradability of all of the inorganic materials has yet to be determined, and potential toxicity could be a problem [58]. As a result, more extensive in vivo studies are still needed.
2.2.4 Bio-inspired nanovehicles
In recent studies, some bio-inspired nanovehicles, such as DNA-based nanostructures [66, 67], exosome-mimetic nanovesicles [68, 69], and red cell member-based ghosts [70] have been explored as gene and drug carriers to enhance delivery to targeted cells. The exosome is a bilayer membrane-coated vesicle secreted by cells, whose function is triggering intercellular communication by transferring payload mRNA, miRNA, or proteins from one cell to another. Studies have shown that these exosome nanovesicles exhibit good biodegradability, which can be attributed to their morphology and surface properties which are similar to those of natural cells. Originated from cell membrane proteins, cell membrane-based ghosts are another cell-derived nanovesicles, which possess identical physiochemical properties, such as topological/physiological activities. These cell membrane-based ghosts have a promising future in nanotechnology for a variety of biomedical applications, such as targeted delivery of RNA therapeutics, and tissue regeneration [71]. In comparison to other drug delivery systems, the advantages of these bio-inspired nanovehicles are low toxicity, strong targeting abilities, and low immune response induction. However, the high cost of production and vesicle stability should be considered in further clinical applications.
3. RNA nanotherapeutics for tumor immunotherapy
Tumor immunotherapy can induce a synergistic killing effect on tumor cells through primarily targeting the immune system rather than the tumor cells themselves. Current immunotherapeutics, including antibodies, proteins, and engineered immune cells (e.g., CAR-T) are promising strategies for cancer treatment and some of them have been successful in clinical applications. However, these therapies still face some critical issues, such as insufficient efficacy (e.g., ICB has shown activity in approximately 15%-25% of patients [72]), high costs and side-effect [15, 17]. RNA-based agents provide some advantages in immunomodulation, such as high selectivity for silence or increased expression of specific targets and low risk of off-target hitting. Using RNA therapeutics in conjunction with nanomaterials is a valuable strategy to improve the efficacy of immunomodulation for cancer treatments. When used as partners in synergistic combinations, more efficient and personalized results will be reached, enabling the combination to compete with current immunotherapies already on the market.
3.1 Nanostructure-mediated siRNA delivery for immunotherapy
Recently, many approaches have demonstrated that silencing the crucial factors of tumor progression in cancer cells or knocking out/down the immunosuppressive genes in cancer [73] and cancer-associated immune cells can effectively induce an anti-tumor immune response [74, 75]. Nanomaterials can act as Trojan horses by delivering siRNAs, for regulating the immune response, to cancer or immune cells [76, 77]. Here, we summarize the potential siRNA targets in tumor or immune cells, as well as nanostructure delivery systems for siRNA-mediated immunotherapy (Table 2).
Summary of siRNA-based nanotherapeutics for tumor immunotherapy
| Cells | Nanocarriers | Targeted gene | Immunological effects | Ref. |
|---|---|---|---|---|
| Tumor cells | PCL-PEG/PCL-PEI | PD-L1 | Checkpoint blockade | [78] |
| FA-PEI polymers | PD-L1 | Checkpoint blockade | [79] | |
| Acidity-Responsive Micelleplex | PD-L1 | Checkpoint blockade | [80] | |
| Acid-activatable micelleplex (PDPA based) | PD-L1 | Checkpoint blockade | [81] | |
| Au-CGKRK nanoconjugates | PD-L1, STAT3 | Anti-proliferation and checkpoint blockade | [82] | |
| Liposome-protamine-hyaluronic acid NPs | TGF-β | Decrease TGF-β and enhance the antigen-specific immune response | [83] | |
| ROS-responsive NPs | TGF-β | Modify the immunosuppress microenvironment | [84] | |
| HA-coated liposome | CD47 | Decrease immune escape of tumor cells | [85] | |
| Glutamine-functionalized branched polyethyleneimine | CD47 | Induce evasion of phagocytic clearance | [86] | |
| Chitosan lactate | CD-73 | Attenuate the immunosuppressive microenvironment of the tumor | [87] | |
| The extracellular vesicles (EVs) | β-catenin | Combo therapy with ICB | [88] | |
| T cells | Lipid-coated calcium phosphate (LCP) | PD-1 | Checkpoint blockade | [89] |
| PEG-PLA | CTLA-4 | Checkpoint blockade | [90] | |
| TAMs | Gold NPs | TNF-α | Silence pro-inflammatory cytokines | [91] |
| Gold NPs | VEGF | Reduce the recruitment of inflammatory TAMs | [92] | |
| Peptides NPs | CSF-1R | Elimination of M2-like TAMs | [93] | |
| DCs | Gold nanorods or GNRs-PEI | IDO | Promote DCs maturation, and increase secretion of pro-inflammatory cytokines | [94, 95] |
| Cationic lipid NPs | PD-L1, PD-L2 | Checkpoint blockade | [96] | |
| PEI based NPs | PD-L1 | Induce immunosuppressive DCs to antigen-presenting cells | [97] | |
| PEI based NPs | IDO | Increase secretion of proinflammatory cytokines | [98] | |
| PEG-PLL-PLLeu polypeptide micelles | STAT3 | Induce DCs maturation and activation, elevate expressions of CD86 and CD40 and IL-12 production | [99] | |
| PLGA NPs | STAT3 | Induce DCs maturation and promote antigen cross-presentation | [100] | |
| Cationic lipid NPs | SOCS1 | Promote production and release of pro-inflammatory cytokines | [101] | |
| PLGA NPs | SOCS1 | Enhance the production and release of pro-inflammatory cytokines | [102] | |
| lipid envelope-type NPs | A20 | Enhance production of pro-inflammatory molecules after lipopolysaccharide stimulation | [103] | |
| Others | Lipid/PEG NPs | CCR2 | Prevent monocytes accumulation | [104] |
| PEG/MT/PC NPs | VEGF, PIGF | Anti-proliferation and reverse immune environment | [105] | |
| Chitosan NPs | Galectin-1 | Reduce polarization to M2 TAMs | [106] |
3.1.1 Tumor cells-targeted siRNA immunenanotherapy
Tumor cells can develop numerous strategies to promote immune system escape and drive the generation of an immunosuppressive microenvironment. For example, tumor cells can effectively induce and recruit distinct immunosuppressive cells, by promoting the secretion of some pro-tumor cytokines and chemokines[107], such as interleukin-6 (IL-6), IL-10, transforming growth factor-β (TGF-β), vascular endothelial growth factor (VEGF), and C-X-C motif chemokine 5 (CXCL5). Meanwhile, the tumor-infiltrating suppressive cells evade immune surveillance by secreting or expressing a few immunosuppressive molecules that disrupt antigen presentation of dendritic cells (DCs) and suppress proliferation and activation of T cells [108, 109]. Additionally, tumor cells could evade immunological eradication by downregulating antigen expression, and upregulating the expression of immune checkpoint proteins (e.g., PD-L1) or “don't eat me” signals (e.g., CD47) [110, 111]. CD47 is over-expressed on the cancer cell surface, which enables escape from immune system recognition by labeling the cells with the “self” marker.
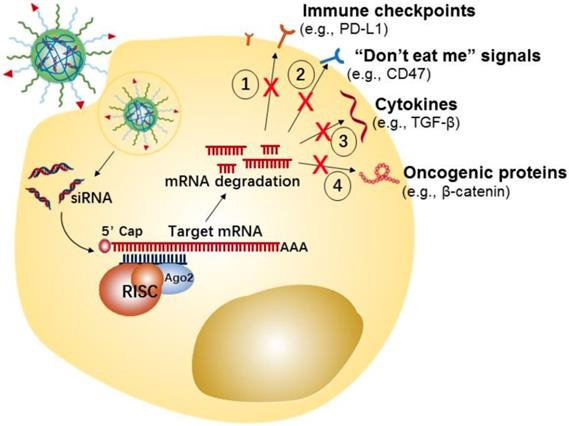
In the last few years, tumor cell-targeted siRNA nanotherapeutics have been centered on the downregulation of immune checkpoint proteins, “don't eat me” signals, anti-inflammatory cytokines, etc., for inducing anti-tumor immune-responses has been gaining increasing attention (Figure 3). For example, Yang et al. developed a systemic delivery strategy based on HA-coated Lipid NPs for delivery of CD47 siRNA to melanoma cancer cells, which induced an efficient knockdown of CD47 in cancer cells [85]. CD47 silence effectively suppressed the tumor growth in a melanoma mice model. Similarly, using siRNA-based nanotherapy for the direct knockdown of immune checkpoints or anti-inflammatory cytokines on tumor cells has also enhanced anti-tumor immune responses and showed significant inhibition of tumor growth in vivo. For instance, Xu et al. developed a mannose-modified liposome-protamine-hyaluronic acid NPs (LPH) for encapsulating with TGF-β siRNA to B16F10 melanoma tumor cells [83]. Meanwhile, the authors developed another lipid-calcium-phosphate NP (LCP) to deliver tumor antigens (i.e., Trp 2 peptide and CpG oligonucleotide) to the dendritic cells, with the purpose of eliciting a systemic immune response. The in vivo results displayed that silencing of TGF-β by LPH boosted the vaccination efficacy of LCP, and significantly inhibited tumor growth.
Potential strategies of tumor cells-targeted siRNA nanotherapeutics for cancer immunotherapy.
The exciting anti-tumor effect produced by the combination of two NPs suggested the combo therapy by two or more therapeutics will induce a more powerful anti-tumor immune response and offer a powerful platform for cancer treatment. Based on this, some strategies that combine RNA-based nanotherapeutics with photodynamic or chemical agents are being reported in recent studies [80, 81]. For instance, Dai et al. reported a pH-responsive nanosystem that when co-loaded with PD-L1 siRNA and a mitochondrion-targeting photosensitizer and given to tumor cells, induced the synergistic anti-tumor effect by combing photodynamic and immunotherapy [80]. The in vitro and in vivo results reveal that the nanosystem not only efficiently induced an immune response by photodynamic therapy, but also subsequently induced a siRNA-mediated immune checkpoint blockade that further activated systemic anti-tumor immune responses, and thereby led to significant growth inhibition for melanoma. A similar result has also been reported by Qiao et al. They designed a ROS-responsive nanotheranostic system which combined temozolomide (TMZ)-mediated chemotherapy and immunomodulation by siTGF-β-based therapy on glioblastoma [84]. In this study, cationic poly[(2-acryloyl)ethyl(p-boronic acid benzyl)diethyl ammonium bromide] (BA-PDEAEA, BAP) was used to condense with TGF-β siRNA, the zwitterionic lipid-based envelopes (ZLEs) were then coated on polymer-siRNA nanocomplex, and TMZ was loaded into the core of the nanotheranostic NPs (LiB(T+AN@siTGF-β), LBTA). The NPs were finally modified with an angiopep-2 peptide that enabled the NPs to cross the blood-brain barrier (BBB) and accumulate into the glioblastoma tumor region. Both in vitro and in vivo results demonstrated that this nanotheranostic NPs effectively down-regulated TGF-β expression of tumor cells and dramatically enhanced the efficacy of TMZ mediated chemotherapy. Meanwhile, it showed that the survival time of glioblastoma tumor-bearing mice was significantly prolonged after the synergistic combination treatment.
In addition to directly silencing the immunosuppressive genes, combination therapy with knock out/down oncogenic genes, through siRNA and immune checkpoint blockade therapy, may provide another promising method for cancer treatment. For example, Matsuda et al. used the extracellular vesicles (EVs) to develop a biological nanoparticle-mediated delivery system for intrahepatic delivery of β-catenin siRNA to hepatocellular carcinoma (HCC)[88]. In this study, the β-catenin siRNA EVs and anti-PD-1-based therapy were systemically administrated together. The in vivo results demonstrated the therapeutic EVs improved CD8+ T cells infiltration and priming, and thus enhanced the anti-tumor effect of anti-PD-1.
3.1.2 Immune cells-targeted siRNA immune-nanotherapy
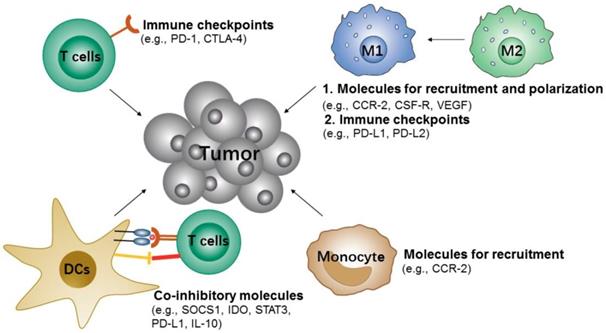
During tumor progression, the immune cells can also recognize and eliminate tumor cells, which are known as tumor immunosurveillance. However, tumor cells can also change the host's immune system and escape immune system control by re-education or re-programing the immune cells, e.g., recruitment of various immunosuppressive cells to the primary microenvironment of the tumor, induction of immune cells into pro-tumorigenic types and inhibition of the anti-tumor activity of immune cells [109-111]. Therefore, suppressing the immunosuppressive cells or modulating immune cells to anti-tumorigenic types would be an attractive approach to inhibit tumor immune escape and slow tumor growth (Figure 4).
Potential siRNA targets of immune cells for cancer immunotherapy.
3.1.2.1 T cells
Like cancer cells' highly/over-expressed inhibitory molecules (e.g., PD-L1), some activated T cells also over-express corresponding inhibitory molecules [112, 113], such as programmed cell death protein 1 (PD-1), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and mucin-containing protein 3 (TIM-3). All these inhibitory signals are known as immune checkpoints, which can downregulate, T cell activities and therefore prevent the elimination of tumor cells by effector T cells [113]. Thus, targeting the immune checkpoints of T cells may also boost the anti-tumor effects. Given this, Li et al. constructed cationic lipid-assisted PEG-PLA-based NPs for efficiently delivering CTLA-4 siRNA to T cells [90]. The NPs consisted of poly(ethylene glycol)-block-poly(d,l-lactide) (PEG5k-PLA11k) and the cationic lipid N,N-bis(2-hydroxyethyl)-N-methyl-N-(2-cholesteryoxycarbonyl-aminoethyl) ammonium bromide (BHEM-Chol) polymers which could encapsulate with CTLA-4 siRNA by electrostatic interaction. Although the NPs were only internalized by 4-6% of T cells in vivo, it promoted an ~ 2-fold increase in effector CD8+ T cells (approximately 40.3% vs. 18.9% of PBS), and the ratio of CD4+ FOXP3+ Tregs was decreased by ~ 2.5 fold compared to PBS. Accordingly, these NPs could effectively inhibit tumor growth and prolong survival time in mice with melanoma.
3.1.2.2 TAMs
Macrophages are professional phagocytes for eliminating pathogens and cellular debris. In the tumor microenvironment, tumor-associated macrophages (TAMs) usually are either pro-tumor M2 type or anti-tumor M1 type. The macrophages or monocytes are usually recruited to the tumor region and polarized to M1-type at the initial stages of tumor formation, and in advanced tumor progression stage, the macrophages will convert from M1 to M2 type, which thus exerts pro-tumor effects by helping block CD8 T cells [114, 115]. Conclusively, reducing the survival and recruitment of TAMs in tumor site or performing targeted delivery of therapeutics to the M2-like TAMs, for depleting them from tumors or converting them to M1 type, would be promising strategies for cancer immunotherapy [116].
Recent studies indicated that the CCL2-CCR2 [117] and CCL3-CCR1/CCR5 signaling [118] were required for the recruitment, retention, and the phenotypic recruitment of TAMs. They also indicated that the cytokines [119], including colony stimulating factor 1 (CSF1) and vascular endothelial growth factor (VEGF), are known to recruit monocytes as well. Given this, Qian et al. reported a molecularly-targeted strategy based on dual-targeting nanoparticles (M2NPs) that deliver colony stimulating factor-1 receptor (CSF-1R) siRNA to M2-like TAMs for the specific blockade of the survival of M2-like TAMs, which results in the depletion of them from melanoma tumors [93]. The siRNA-carrying M2NPs can inhibit the production of immunosuppressive factors (e.g., IL-10 and TGF-β), but also increase the expression of immunostimulatory cytokines (IFN-γ and IL-12) and CD8+ T cells infiltration (2.9-fold). Moreover, it effectively induced the anti-tumor activity of T cells by down-regulating expressions of the exhaustion markers (PD-1 and Tim-3) and stimulating secretion of IFN-γ (6.2-fold). In vivo results confirmed that M2NPs led to a dramatic elimination of M2-like TAMs (52%), inhibition of tumor growth (87%), and prolonged survival.
Conde et al. presented peptide-functionalized gold nanoparticles (AuNPs) for delivery of VEGF siRNA to M2-like TAMs where a decrease in the accumulation in lung tumor tissue was observed, alongside enhanced tumor growth inhibition in a lung cancer orthotopic murine model [92]. VEGF is a key angiogenic factor, which is highly expressed on M2-like TAMs and well known to promote cancer progression and metastasis. In this study, the authors proved that siRNA mediated gene silencing for inhibiting TAMs accumulation could be achieved by targeting the VEGF pathway, which also indicated that modulation for TAMs would induce an anti-tumor immune response and could be a potential target for cancer treatment.
3.1.2.3 DCs
DCs are the most important antigen-presenting cells (APCs), which could capture, process, and present tumor antigens to either naive CD8+ or CD4+ T cells. The functions of DCs are either mediating immune tolerance or inducing an anti-tumor immune response. Generally, DCs express a variety of co-inhibitory molecules [120], such as suppressors of cytokine signaling (SOCS) 1, signal transducers and activators of transcription-3 (STAT3), and indoleamine 2,3-dioxygenase (IDO). All of these molecules may suppress the antigen presentation process of DCs. Studies have shown that knockdown of these molecules by RNAi would be an effective strategy for DCs-based immunotherapy [121-124]. Based on this, Heo et al. reported a PLGA polymeric NPs that combined the delivery of tumor antigens and SOCS1 siRNA to DCs that induced an enhanced anti-tumor immune response [102]. SOCS1 functions as a broadly immuno-suppressive protein which can directly inhibit antigen presentation of DCs to T cells, and it also acts as a negative regulator of Janus kinases (JAKs), which induce immune tolerance. In this study, PLGA polymeric NPs with the loading of SOCS1 siRNA and OVA peptide were efficiently taken up by bone-marrow-derived dendritic cells (BMDCs) and this showed proof of the significant knockdown effect on the expression of SOCS1. The downregulation of SOCS1 in BMDC further led to a drastic enhancement in cytokine production, and finally resulted in the essential induction of anti-tumor immune response.
3.1.2.4 Others
Neutrophils has been described as part of the innate immune response, but recent studies have shown that neutrophils can be a key negative regulator for adaptive immune responses by suppressing T cell proliferation, and therefore they also be so-called myeloid-derived suppressor cells (MDSCs)[125]. MDSCs have been identified to facilitate the development of an immunosuppressive tumor microenvironment [126]. Many studies have also demonstrated MDSCs are critically involved in immunosuppressive effects through the high expression of metabolic enzymes [127, 128], such as IDO, and arginase-1 (ARG1), cytokines[129, 130], and chemokines, such as TGF-β and IL-10. Meanwhile, the recruitment of MDSCs relies on the chemokines [131] (e.g., CCL2). Due to the important role in tumor-associated immune suppression of MDSC, many studies were focused on exploring therapeutic strategies to eliminate these cells or to modulate their functions [132-135]. For example, Leuschner et al. introduced monocyte-targeting lipid NPs for the delivery of CCR2 siRNA to the inflammatory monocyte [104]. The results showed efficient inhibition of CCR2 expression by siRNA-mediated CCR2 gene silencing in monocytes that prohibited their accumulation in inflammatory sites and reduced the number of TAMs.
3.2 Nanostructure-mediated microRNA delivery for immunotherapy
Non-coding RNAs are usually classified into small non-coding RNA (e.g., microRNA) and long non-coding RNA (lncRNA, > 200 nt), depending on their size. Recent studies of lncRNAs have indicated that lncRNAs function as crucial regulators in various immunity processes, including immune cells differentiation and function, regulation for tumor microenvironment, but the detail regulatory mechanisms remain unclear [136]. While current studies are mainly focused on the development of the lncRNAs for immune regulation and the functional relationship between lncRNAs and immunity, and there were limited reports on lncRNAs-based nanomaterials for immunotherapy, we might believe that lncRNAs could become a novel therapeutic agent in cancer immunotherapy.
In addition, recently studies have shown that microRNAs are associated with the modulation of various pathways of cancer and immune cells. MicroRNAs usually produce competition for oncogenic and tumor suppressive effects by blocking either tumor suppressive mRNA or oncogenic mRNA [137-139]. Generally, oncogenic microRNAs are highly/over-expressed in cancer cells, while tumor-suppressive microRNAs are under-expressed. Restoration of tumor-suppressive microRNAs could both inhibit cancer cell proliferation and induce cell apoptosis. Because of these possibilities, this technique has been viewed as novel therapeutics for cancer treatment [6, 140]. Ultimately, it is necessary to carefully assess the specific roles of microRNAs in cancer or immune cells, and develop an effective and safe microRNA-based therapeutic for cancer treatment.
3.2.1 Tumor cells-targeted microRNA immunonanotherapy
As mentioned above, tumor cells will up-regulate the expression of immune checkpoint proteins, or “don't eat me” signals, to construct an immunosuppressive microenvironment. microRNA acts as a gene regulator, which can either be used to directly inhibit expressions of immune escape factors and pro-inflammatory cytokines or act as oncogenic factors for facilitating immune system evasion by tumor cells[141]. For example, many microRNAs including miR-34a, miR-200, miR-142-5p and miR-424, et al. have been found to be involved in PD-L1 expression levels in several cancer cells [142, 143]. Among them, miR-424 not only activates T cells immune response through direct targeting of PD-L1, but also can restore the chemosensitivity of ovarian cancer cells [144]. However, some microRNAs, such as miR-20b, miR21, and miR-130b, have been found to inhibit PTEN expression in colorectal cancer, but they in turn promote PD-L1 upregulation [145]. MicroRNA antagonists are short, single-stranded oligonucleotide molecules complementary to microRNA sequences that have recently been used for targeting and reducing microRNA activity. Therefore, using antagonists of microRNA will also decrease oncogenic microRNA activity and provide promising results of targeted genes for immunomodulation. To date, there has been a limited report by using nanotechnology for the delivery of microRNA to tumor cells for triggering an anti-tumor immune response, but there is high confidence that it would be a potential strategy for cancer immunotherapy.
3.2.2 Immune cells-targeted microRNA immunonanotherapy
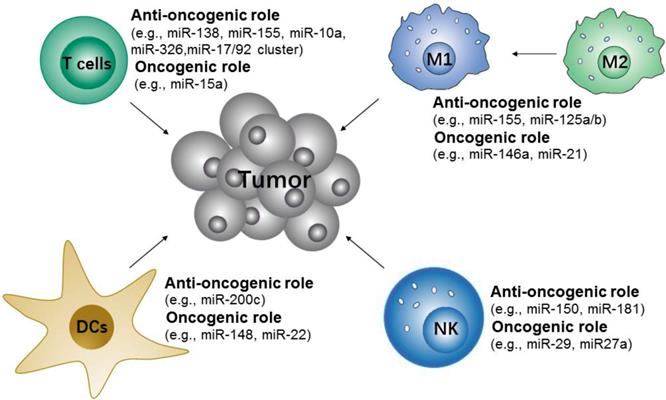
Many studies have shown that microRNAs are important in the modulation of innate and adaptive immune-responses via controlling the differentiation and activation of immune cells and the maintenance of immune proinflammation factors [141]. These microRNAs either contribute to or repress the immune cells to initiate anti-tumor responses (Figure 5). Specifically, the upregulation of miR-146a [146] and miR-21[147] attenuate M1-like TAMs activation by targeting TLR/NF-κB pathway, but miR-155[148, 149] promote M1-like TAMs polarization by targeting SOCS1 or targeting the negative regulator of NF-κB and TNF-α-induced protein 3. Along with TAMs, some microRNAs, such as miR-150 and miR-181a/b, have been found to control the differentiation of NK cells [150]. The use of miR-148 and miR-22 inhibitors may also promote DCs maturation [151-153]. In addition, the differentiation and functions of different T cell subsets are regulated by microRNAs [154, 155]. For example, miR-155 are more in favor of Th1 phenotype [156], miR-326 promote Th17 differentiation [157], and miR-10a and miR-17/92 cluster regulate T follicular helper maturation [158].
Potential microRNA targets of immune cells for cancer immunotherapy. The microRNAs either contribute to or repress the immune cells to initiate anti-tumor responses.
Given the potential applications of microRNA-based therapeutics in cancer immunotherapy, a variety of microRNA-based nanomaterials that can efficiently target immune cells for immunomodulation have been developed (Table 3). For instance, Zhang et al. designed lipid-coated calcium phosphonate nanoparticles (CaP/miR@MNPs) which were further conjugated with mannose for specific delivery of miR155 to TAMs [159]. The results demonstrated that CaP/miR@MNPs could successfully transfer pro-tumor M2-like TAMs to antitumor M1-like TAMs, and therefore elicit a potent antitumor immune response, whilst inhibiting tumor growth. Similarly, Parayath et al. developed a CD44 targeting hyaluronic acid-poly(ethylenimine) (HA-PEI)-based nanoparticle for the delivery of miR-125b to peritoneal macrophages [160]. Overexpression of miR-125b in macrophages would promote M1-like TAMs activation and lead to inhibition of primary tumor growth and metastasis. In vivo results showed that there was a more than 6-fold increase in the ratio of M1 to M2 TAMs and 300-fold increase in the ratio of iNOS (M1 marker) to Arg-1 (M2 marker) in TAMs after treatment with HA-PEI-125b nanoparticles. All of the examples above indicated that successfully inducing M1-like TAMs polarization would enhance anti-cancer immunotherapy.
Summary of microRNA-based nanotherapeutics for cancer immunotherapy
| Cells | Nanocarriers | microRNA | Immunological effects | Ref. |
|---|---|---|---|---|
| T cells | Exosome-like nanovesicles | miR-150 Antagonist | T-cell regulation | [160] |
| TAMs | Layered double hydroxides NPs | miR-155 | Repolarize M2 to M1 | [161] |
| Lipid-coated NPs | miR-155 | Repolarize M2 to M1 | [158] | |
| CD44 coated HA-PEI based NPs | micR-125b | Reprogram TAMs into M1 | [159] | |
| DCs | Exosomes | miR-155 | Increase the expressions of MHC-II, CD86, CD40, and CD83, and promote the secretion of the IL12p70, IFN-gamma, and IL-10 | [162] |
| PEG-PLL-PLLeu polymeric NPs | miR-148a Antagonist | Reprogram DCs, reduce Treg cells and myeloid-derived suppressor cells | [163] | |
| NK cells | Exosomes | miR-186 | Promote NK activation | [164] |
3.3 Nanostructure-mediated mRNA delivery for immunotherapy
mRNA emerging as a cancer therapeutic has drawn increasing attention due to its multiple unique features[166], such as the absent risk of insertional mutagenesis, more consistent and predictable kinetics of protein expression, and relatively convenient in vitro synthesis. However, its poor stability (easily degraded by the nucleases) and propensity for immunostimulation have greatly hindered the in vivo application. Moreover, it very difficult for the larger mRNA molecules with negative charges to enter antigen-presenting cells (APCs) directly. Nanoparticle-based platforms with high cytosolic transportation and reduced renal filtration, could be emerging as a promising mRNA delivery tool to protect nucleic acids from enzymatic degradation and withstand multiple intracellular and extracellular barriers [35, 167]. Here, we will describe two examples to present the advances of mRNA nanomedicines in immunotherapy applications, including vaccination and cell engineering.
3.3.1 mRNA-based NPs for vaccination
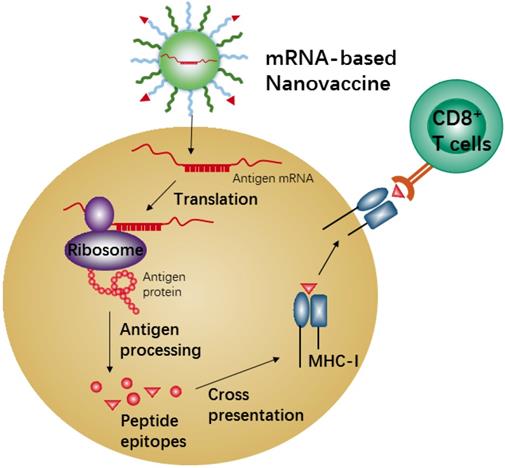
mRNA-based vaccines show a promising alternative due to the high potency, safe administration, and capacity for rapid development and low-cost manufacture[168, 169]. Recent nanotechnological advances have largely overcome the issues of in vivo mRNA delivery, and therefore mRNA-based nanovaccines have recently attracted increasing attention because of the promising results achieved in many anti-tumor vaccination studies in animal models [170, 171]. Meanwhile, the mRNA-based vaccine could effectively carry out antigen-encoding mRNA to antigen presenting cells (APCs) in vivo directly. Also, the mRNA-based vaccine is unlimited in molecular structures and number of tumor antigen proteins. When the nanocarriers efficiently deliver these antigen-encoding mRNAs into APCs, the mRNAs will be released and translated into tumor antigenic proteins in the cytoplasm of APCs, which are then processed into peptide epitopes for subsequent binding with the major histocompatibility complex (MHC) class I via cross-presentation pathway. The MHC-peptides are finally transferred to the cell surface of APCs for activation of CD8+ T cells, leading to corresponding anti-tumor immune responses (Figure 6). Efficient in vivo mRNA delivery and induction of strong cytotoxic T cell response are the key prerequisites for cancer immunotherapy by mRNA-based nanovaccines. There are so many advantages and disadvantages for in vivo application of mRNA-based nanovaccines, and many review articles have summarized the advertences of mRNA-based nanovaccines in cancer therapy. Here we just describe one example to present the mechanism of anti-tumor response by nanovaccine.
Schematic illustration of antigen cross-presentation by mRNA-based nanovaccine in APC.
A lipid nanovaccine with the loading of tumor-associated antigens mRNA (e.g., gp100 and TRP2) was designed by Oberli et al. [172]. In this study, the lipid nanoparticle that consisted of an ionizable lipid, a lipid-PEG, a phospholipid, cholesterol, and an additive was developed. The ionizable lipid was used for complexation with the negatively charged mRNA and also helped with cellular uptake. The results showed the NPs worked well for the delivery of mRNA to dendritic cells, macrophages, and neutrophils, and effectively led to strong activation of CD8+ T cells after a single immunization in the B16F10 melanoma model. Moreover, after treatment with these NPs, B16F10 melanoma tumor experienced shrinkage, and mice survival was significantly extended. The exciting result from this study demonstrated that the induction of cytotoxic T cell response by mRNA nanovaccine would be an excellent candidate in the application of cancer treatment.
In the nanovaccine field, the nanostructures with CpG oligodeoxynucleotides that act as immuno-adjuvants for inducing immunostimulatory responses are very attractive. CpG oligodeoxynucleotides are Toll-like Receptor 9 (TLR9) agonists, which are expressed in human B and plasmacytoid dendritic cells. Recent studies demonstrated CpG not only could induce apoptosis of tumor cells with high-expression of the TLR9 receptor, but also enhance NK cell activation and promote activation of anti-tumor immune response by combining with various of cytokine treatments [173, 174]. Therefore, CpG-based nanotherapeutics can only benefit from combination with strategies of immune checkpoint blockade.
3.3.2 mRNA-based NPs for T cell engineering
As a novel immunotherapy method, CAR-T therapy has achieved great success in treating patients with hematological malignancies [13], such as leukemia and lymphoma therapy. Currently, the most common techniques for the development of CAR-engineered T cells are using viral gene transduction by virus-mediated delivery. However, using viral vectors may lead to the potential insertional mutagenesis and genotoxicity for effector T cells[15]. Meanwhile, the feared side-effects would happen when virus vectors transduced cells [175]. Hence, more precise T cell manipulations are currently under investigation.
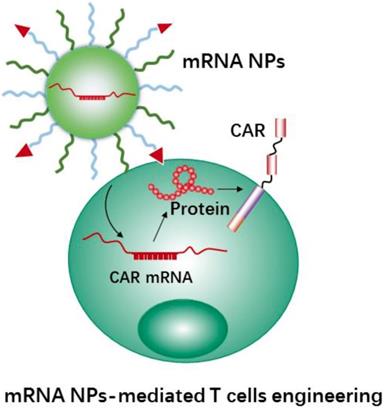
Using mRNA instead of DNA as T cell engineering therapeutics (Figure 7) is attractive due to the multiple unique features of mRNA [176, 177]. Meanwhile, the use of nanocarriers protect mRNA therapeutics from degradation and may help with cellular uptake and endosomal escape. In addition, nanocarriers provide further advantages for the engineering of immune cells, e.g., coating with specific ligands for increasing cell binding and cellular uptake, and carrying multiple RNA payloads. Moffett et al. developed a targeted nanocarrier for reprogramming T cells by delivery of several mRNAs to T cells [178]. The nanocarrier consisted of negatively charged polyglutamic acid (PGA), poly(β-amino ester) (PBAE) polymers and T cell targeting ligands (e.g., anti-CD3 and anti-CD8). In this study, the authors first used their nanocarrier to deliver FoxO1 (Forkhead box O1, a transcription factor to promote the generation and maintenance of memory T cells) mRNA to CAR T cells for overexpressing FoxO1. FoxO1 overexpression can bias CAR-T-cells toward a central memory phenotype and therefore improve the anti-tumor activity of CAR T cells. In addition, the authors developed mRNA nanocarriers to transiently express genome-editing proteins (CRISPR) for efficiently knocking out T cells receptors in CAR-programmed lymphocytes. The results demonstrated that the NPs could effectively transport mRNAs to targeted T cells, and subsequently led to the targeted T cells to express selected proteins. Next, we will describe CRISPR-Cas9 nanotechnology mediated cell engineering and its application in immunotherapy.
Schematic representation of mRNA nanotherapeutics for T cell engineering. The nanocarriers delivery CAR mRNA to T cells, and induce T cells activation by expressing the CAR protein on the surface of T cells.
3.4 CRISPR-Cas9 nanotechnology for cell engineering
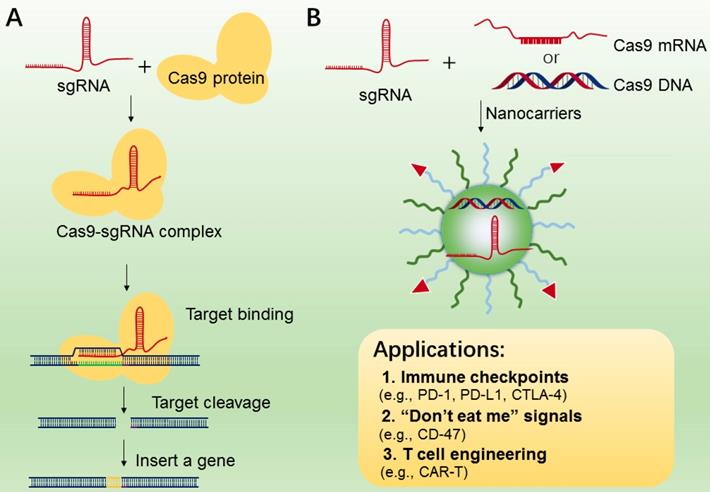
CRISPR/Cas9 system (clustered regularly interspaced short palindromic repeats) [178], is an RNA-guided DNA targeting technology, which has been widely applied to genome editing [180, 181], gene therapy [182] and manipulation of induced pluripotent stem cells (iPS) studies[183]. The CRISPR-Cas9 gene editing machinery is comprised of two essential components, i.e., sgRNA and DNA endonuclease Cas9 (Figure 8A). The Cas9 protein functions in locating and cleaving targeted DNA, and the guide RNA has a 5′ end that is complementary to the target DNA sequence. Only when two macromolecules are forming a complex, will the cleavage activity of Cas 9 begin to be triggered (Figure 8A). In cancer immunotherapy, CRISPR-Cas9-mediated genome editing is usually applied to knockout the genes that encode inhibitory receptor proteins of tumor or T cells, such as PD-1, PD-L1, and CTLA-4[184-186]. In 2016, a case of CRISPR-Cas9 application in a clinical trial of T cells engineering by deleting PD-1 was performed in China, and the result was promising[187], indicating the potential value of CRISPR-Cas9-mediated genome editing in immunotherapy.
A. The mechanism of CRISPR-Cas9 technology for gene editing. B. CRISPR-Cas 9 nanotherapeutics for cell engineering and the applications in cancer immunotherapy. The CRISPR-Cas 9 system (sgRNA with Cas9 mRNA or DNA or protein) was transported to tumor or immune cells by nanocarriers.
While CRISPR-Cas9 has been used as a particularly versatile and operationally simple tool for gene editing in many cell types, the effective delivery of CRISPR-Cas9 system into targeted T cells is still the common issue for efficient genome editing. In addition, off-target effects are also found in many CRISPR-Cas9-mediated gene-editing studies [188]. To reduce these effects, various nanocarriers have been developed as the delivery of CRISPR-Cas9 system[189-191], which may be applied to manipulations of immune cells (TAMs, B cells, and T cells) and tumor cells (Figure 8B). In addition, compared to the viral vectors with high immunogenicity, various types of lipid-based or polymer-based nanocarriers, which are actively targetable with low immunogenicity, showed exciting delivery efficiency[192, 193]. For example, Ray et al reported a nanomaterial platform based cationic arginine-coated gold nanoparticles[194], which delivered CRISPR-Cas9 gene editing machinery into macrophage cells for knockout expression of “don't eat me” signals (SIRP-α). The NPs based nanoplatform has shown ∼90% delivery efficiency as well as ∼30% gene editing efficiency. The in vitro experimental results also showed a 4-fold increase in the innate phagocytic capabilities of the macrophages by using this strategy to turn off the “don't eat me” signal on macrophages, indicating this strategy may be a promising tool for the development of “weaponized” TAMs for cancer immunotherapy. Similarly, Cheng et al. proposed a double emulsion method by complexing plasmids with stearyl polyethyleneimine (stPEI) as the core to form human serum albumin (HSA) (plasmid/stPEI/HSA) NPs for delivery of CRISPR/Cas9 [195]. The NPs could disrupt or silence the expression of PD-L1 by CRISPR/Cas9-mediated gene editing.
4. Conclusions and future perspectives
During the past few years, immunotherapy has shown to be one of the most promising therapeutic strategies and has resulted in a paradigm shift in the treatment of cancers [13, 112]. RNA-based therapeutics provide several advantages in immunomodulation and cancer vaccines, which will serve as a competitor and as a current immunotherapies partnership to achieve synergistic combinations for more efficient and personalized results [75, 169, 176, 196]. Compared to the conventional proteins, antibodies, and cell-based therapeutics (e.g., CAR-T), RNA-based therapeutics including siRNA, microRNA, and mRNA can either knock out or knock down targeted genes or upregulate expressions of specific proteins, and such features of RNA-based therapeutics cause high selectivity and low risk of off-target hitting. Meanwhile, RNA-based therapeutics have become much more diverse and broad in regulatory functions in immunotherapy than the other antibody or protein-based therapies. For example, RNA-based therapeutics could activate rapid and effective anti-cancer immune response by inducing immunogenic cell death of tumor cells, and thus may skip the requirement of deep tumor penetration-a significant hurdle faced by traditional cancer nanomedicines. Moreover, the development and production of RNA therapeutics are very convenient, rapid and cost-effective [197]. The success of pre-clinical studies has led to the initiation of clinical trials of different forms of RNA therapeutics for cancer immunotherapy (Table 4).
Current clinical studies of RNA-mediated immunotherapy for the treatment of cancer
| Targeting Cell | RNAs Encoding | Cancer Types | Status | ClinicalTrials.gov Identifier Number |
|---|---|---|---|---|
| T cells | MET scFv CAR | Malignant Melanoma, Breast Cancer | Early Phase 1 Recruiting | NCT03060356 |
| cMet CAR | Metastatic Breast Cancer; Triple Negative Breast Cancer | Phase 1 Completed | NCT01837602 | |
| Chimeric anti-mesothelin immunoreceptor SS1 | Pancreatic Cancer | Phase 1 Completed | NCT01897415 | |
| DCs | TAAs: NY-ESO-1, MAGEC1, MAGEC2, 5 T4, Survivin, and MUC1 | Lung Cancer | Phase 2 Recruiting | NCT03164772 |
| TAAs: PSA, PSCA, PSMA, STEAP1, PAP and MUC1 | Prostate Carcinoma | Phase 2 Completed | NCT02140138 | |
| Neo-Ag | Melanoma | Active No Recruiting | NCT02035956 | |
| Neo-Ag | Solid tumor | Phase 1 Recruiting | NCT03313778 | |
| Neo-Ag | Melanoma; Colon Cancer; Gastrointestinal Cancer; Genitourinary Cancer; Hepatocellular Cancer | Phase 2 Completed | NCT03480152 | |
| Three variant RNAs; p53, and Neo-Ag based on NGS screening | Breast Cancer (Triple Negative Breast Cancer) | Phase 1 Recruiting | NCT02316457 | |
| Carcinoembryonic antigen RNA | Colorectal Cancer; Metastatic Cancer | Phase 2 Completed | NCT00003433 | |
| Prostate specific antigen (PSA) | Prostate Cancer | Phase 2 Completed | NCT00004211 | |
| Carcinoembryonic antigen | Breast Cancer; Colorectal Cancer; Extrahepatic Bile Duct Cancer | Phase 1 Completed | NCT00004604 | |
| Total tumor RNA | Kidney Cancer | Phase 1 Completed | NCT00005816 | |
| Autologous tumor RNA | Melanoma | Phase 3 Recruiting | NCT01983748 | |
| TAAs: NYESO-1, MAGE-A3, tyrosinase, and TPTE | Melanoma | Phase 1 Recruiting | NCT02410733 | |
| siRNA: LMP2, LMP7, and MECL1; mRNA: MART-1, tyrosinase, gp100, and MAGE-3 | Melanoma | Phase 1 Completed | NCT00672542 | |
| Melan-A, Mage-A1, Mage-A3, Survivin, GP100 and Tyrosinase | Malignant Melanoma | Phase 1/2 Completed | NCT00204516 | |
| pp65-flLAMP | Glioblastoma | Active No recruiting | NCT03615404 |
Given the potentials of these RNA therapeutics, various nanoparticle-based delivery systems, such as lipid-based, polymeric, inorganic and bio-inspired nanomaterials have been explored extensively [49, 167, 198, 199]. These nanoparticle-based delivery systems not only carry a high dose of therapeutic payloads to targeted cells or tissues, but also show the same regulatory functions in immunotherapy with RNA therapeutics. In addition, the combination of RNA-mediated nano-immunotherapy with chem- or photodynamic therapies demonstrated promising results in an animal model[81, 106]. Meanwhile, combining RNA-mediated nanotherapy with current immunotherapies (e.g., anti-CTLA-4, anti-PD-1, and anti-PD-L1) is also a great opportunity to enhance cancer treatments [133]. However, there are still several issues need to be solved before such applications are translated to the clinic.
To begin with, some of the main barriers that hinder the stability and in vivo safety of the various nanomaterial carriers include cytotoxicity and undesirable immune stimulation [21, 200]. As mentioned previously, potential in vivo toxicity issues can arise from the use of cationic units in polymeric NPs, payload leakages from lipid-based nanostructures, and the non-biodegradability of inorganic NPs [20, 38]. In addition, the NPs may be recognized as a foreign substance, and thus easily excreted by renal/hepatic clearance or eliminated by the innate immune systems [198]. To address these issues, nanocarriers should be bio-compatible and capable of biodegradability by modulating the surface of the NPs with biomolecules or proper hydrophilic materials such as PEG.
Secondly, nanocarriers must increase cell targeting and cell internalization. For example, for targeting tumor cells to silence PD-L1 by RNA-based NPs, it needs to target as many of them as possible, thus requiring high accumulation and deep tumor penetration. To increase cellular uptake and thus increase the effectiveness of RNA delivery, NPs could be coated with targeting ligands, which would increase the chance of binding to the targeted cells. In addition, effective on-demand RNA delivery and release would become feasible through the incorporation of backbones of that are sensitive to different stimuli, either endogenous (e.g., pH, enzyme, and redox) or exogenous (e.g., temperature, electricity, light, magnetic force, ultrasound)[201, 202]. Such a feature of nanocarriers would enable optimal spatial and temporal release of RNA payloads. Despite many nanovesicles infiltrating the targeted cells through the endocytic pathway, they must still overcome the barriers that endosomes and lysosomes pose before achieving the ultimate goal of cytosolic release of RNA therapeutics [203]. Generally, nanomaterials are transported into the endosome, which then fuses with lysosomes and ultimately destroys the RNA cargoes. Some specific targeting peptides (e.g., HA) or surfactants can be used in the preparation of nanocarriers to enable the endosomal/lysosomal escape via the proton sponge effect or membrane lysis. It should be noted that many stimuli-responsive nanomaterials had potential dose-dependent cytotoxicity. This cytotoxicity was due to the stimuli-responsive motif often being limited by insufficient biocompatibility or the need for bio-degradability [204]. Meanwhile, the complexity of the architectural design and difficulties in the synthesis of these NPs are likely to hamper their clinical translation. Therefore, the benefit-to-risk ratio for the development of responsive NPs needs to be balanced, and the issues related to the responsive characteristics would eventually need to be solved.
In summary, the goal of the current review article was to summarize and highlight the RNA-based nanotherapeutics for the immunomodulation and enhancement of cancer immunotherapy. Given the convenience and importance of RNA therapeutics in cancer immunotherapy, using nanomaterials for effectively delivery of RNA to a targeted tumor or immune cells for triggering anti-tumor immune response have already produced some exciting results in the treatment of cancer. Meanwhile, by combining RNA-mediated immunomodulation with ICB immunotherapy, chemotherapy, and photodynamic therapy, synthesized effects have been noted and a bright future for the clinical use of cancer treatment awaits. Although the RNA delivery nanoplatforms for clinical applications is still challenging, some issues need to be solved before this nanotherapeutics translated from the bench to the bedside. We believe that the RNA-mediated nano-immunotherapy has great potential to overcome some of these shortcomings and has the opportunity to be used for cancer treatment in the future.
Abbreviations
siRNA: small interfering RNA; mRNA: messenger RNA; ICB: immune checkpoint blockade; CAR-T: chimeric antigen receptor T-cell immunotherapy; RISC: RNA-induced silencing complex; AGO2: Argonaute-2; pri-microRNA: primary microRNA; pre-microRNA: precursor microRNA; UTR: untranslated region; ORF: open reading frame; NPs: nanoparticles; EPR: enhanced permeability and retention effect; PLGA: poly(lactic-co-glycolic acid); PEI: polyethyleneimine; PEG: polyethylene glycol; PVA: polyvinyl alcohol; PLA: poly-l-lactic acid; MSNs: mesoporous silica nanoparticles; CNTs: carbon nanotubes; QDs: quantum dots; PCL-PEG: polycaprolactone-polyethyleneglycol; PCL-PEI: polycaprolactone-polyethylenimine; FA-PEI: folic acid- polyethylenimine; PDPA: poly(2-(diisopropylamino)ethyl methacrylate; PEG-PLL-PLLeu: poly(ethylene glycol)-b-poly(l-lysine)-b-poly(l-leucine); PEG /MT/PC NPs: polyethylene glycol (PEG)/mannose doubly modified trimethyl chitosan ( MT)/poly (allylamine hydrochloride) (PC)-based nanoparticles (NPs); HA: hyaluronic acid; AuNPs: gold nanoparticles; OVA: Ovalbumin; TME: tumor microenvironment; HCC: hepatocellular carcinoma cells; TGF-β: transforming growth factor-β; VEGF: vascular endothelial growth factor; IL-6: interleukin-6; CXCL5: C-X-C motif chemokine 5; PD-1: programmed death receptor-1; PD-L1: programmed death receptor-1 ligand; CTLA-4: cytotoxic T-lymphocyte-associated protein 4; TIM-3: mucin-containing protein 3; CSF1: colony stimulating factor 1; CSF-1R: colony stimulating factor-1 receptor; IFN-γ: interferon γ; TNF-α: tumor necrosis factor α; Tregs: regulatory T cells; Th cells: T helper cells; DCs: dendritic cells; TAMs: Tumor-Associated Macrophages; NK: natural killer cells; MDSCs: myeloid derived suppressor cells; APCs: antigen-presenting cells; MHC: major histocompatibility complex; SOCS1: suppressor of cytokine signaling 1; IDO: indoleamine 2,3-dioxygenase; STAT3: signal transducer and activator of transcription-3; ARG1: arginase-1; TLR: Toll-like receptors; NOS: nitric oxide synthase; FoxO1: Forkhead box O1; CRISPR: clustered regularly interspaced short palindromic repeat; MHC: major histocompatibility complex.
Acknowledgements
This work was supported by Prostate Cancer Foundation Young Investigator Award and CA200900 (J.S.). Y.-X. Lin thanks the China Postdoctoral Science Foundation (2018M633260, 2018T110918) and Guangdong Natural Science Foundation (2018A030310098) for financial support, and S. Blake thanks the Tufts Center for Stem Diversity and the Harvard Yes for Cure Program.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Adams BD, Parsons C, Walker L, Zhang WC, Slack FJ. Targeting noncoding RNAs in disease. J Clin Invest. 2017;127:761-71
2. Chakraborty C, Sharma AR, Sharma G, Doss CGP, Lee SS. Therapeutic miRNA and siRNA: moving from bench to clinic as next generation medicine. Mol The Nucleic Acids. 2017;8:132-43
3. Kaczmarek JC, Kowalski PS, Anderson DG. Advances in the delivery of RNA therapeutics: from concept to clinical reality. Genome Med. 2017;9:60
4. Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16:203-21
5. Zhao BXS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2017;18:31-42
6. Mirzaei H, Masoudifar A, Sahebkar A, Zare N, Nahand JS, Rashidi B. et al. MicroRNA: a novel target of curcumin in cancer therapy. J Cell Physiol. 2018;233:3004-15
7. Rosenblum D, Joshi N, Tao W, Karp JM, Peer D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat Commun. 2018;9:1410
8. Zhou ZX, Liu XR, Zhu DC, Wang Y, Zhang Z, Zhou XF. et al. Nonviral cancer gene therapy: delivery cascade and vector nanoproperty integration. Adv Drug Deliv Rev. 2017;115:115-54
9. Iyer AK, Duan ZF, Amiji MM. Nanodelivery systems for nucleic acid therapeutics in drug resistant tumors. Mol Pharm. 2014;11:2511-26
10. Chen BL, Dai WB, He B, Zhang H, Wang XQ, Wang YG. et al. Current multistage drug delivery systems based on the tumor microenvironment. Theranostics. 2017;7:538-58
11. Hajj KA, Whitehead KA. Tools for translation: non-viral materials for therapeutic mRNA delivery. Nat Rev Mater. 2017;2:17056
12. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252-64
13. June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359:1361-5
14. Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Clin Pract Oncol. 2016;13:273-90
15. Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol The Oncolytics. 2016;3:16011
16. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL. et al. Chimeric antigen receptor T-cell therapy-assessment and management of toxicities. Nat Clin Pract Oncol. 2018;15:47-62
17. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R. et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134-44
18. Aagaard L, Rossi JJ. RNAi therapeutics: principles, prospects and challenges. Adv Drug Deliv Rev. 2007;59:75-86
19. Li ZH, Rana TM. Therapeutic targeting of microRNAs: current status and future challenges. Nat Rev Drug Discov. 2014;13:622-38
20. Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65:36-48
21. Guo X, Huang L. Recent advances in nonviral vectors for gene delivery. Acc Chem Res. 2012;45:971-9
22. Samal SK, Dash M, Van Vlierberghe S, Kaplan DL, Chiellini E, Van Blitterswijk C. et al. Cationic polymers and their therapeutic potential. Chem Soc Rev. 2012;41:7147-94
23. Zhang SB, Zhao B, Jiang HM, Wang B, Ma BC. Cationic lipids and polymers mediated vectors for delivery of siRNA. J Control Release. 2007;123:1-10
24. Lin YX, Gao YJ, Wang Y, Qiao ZY, Fan G, Qiao SL. et al. pH-sensitive polymeric nanoparticles with gold(I) compound payloads synergistically induce cancer cell death through modulation of autophagy. Mol Pharm. 2015;12:2869-78
25. Lin YX, Wang Y, Qiao SL, An HW, Zhang RX, Qiao ZY. et al. pH-sensitive polymeric nanoparticles modulate autophagic effect via lysosome impairment. Small. 2016;12:2921-31
26. Wang Y, Lin YX, Qiao ZY, An HW, Qiao SL, Wang L. et al. Self-assembled autophagy-inducing polymeric nanoparticles for breast cancer interference in-vivo. Adv Mater. 2015;27:2627-34
27. Loh XJ, Lee TC, Dou QQ, Deen GR. Utilising inorganic nanocarriers for gene delivery. Biomater Sci. 2016;4:70-86
28. Shen JL, Zhang W, Qi RG, Mao ZW, Shen HF. Engineering functional inorganic-organic hybrid systems: advances in siRNA therapeutics. Chem Soc Rev. 2018;47:1969-95
29. Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21-and 22-nucleotide RNAs. Genes Dev. 2001;15:188-200
30. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215-33
31. Tan L, Sun X. Recent advances in mRNA vaccine delivery. Nano Res. 2018;11:5338-54
32. Lundqvist A, Noffz G, Pavlenko M, Saeboe-Larssen S, Fong T, Maitland N. et al. Nonviral and viral gene transfer into different subsets of human dendritic cells yield comparable efficiency of transfection. J Immunother. 2002;25:445-54
33. Van Tendeloo VFI, Ponsaerts P, Lardon F, Nijs G, Lenjou M, Van Broeckhoven C. et al. Highly efficient gene delivery by mRNA electroporation in human hematopoietic cells: superiority to lipofection and passive pulsing of mRNA and to electroporation of plasmid cDNA for tumor antigen loading of dendritic cells. Blood. 2001;98:49-56
34. Matsui A, Uchida S, Ishii T, Itaka K, Kataoka K. Messenger RNA-based therapeutics for the treatment of apoptosis-associated diseases. Sci Rep. 2015;5:15810
35. Xiong QQ, Lee GY, Ding JX, Li WL, Shi JJ. Biomedical applications of mRNA nanomedicine. Nano Res. 2018;11:5281-309
36. Pecot CV, Calin GA, Coleman RL, Lopez-Berestein G, Sood AK. RNA interference in the clinic: challenges and future directions. Nat Rev Cancer. 2011;11:59-67
37. Mura S, Nicolas J, Couvreur P. Stimuli-responsive nanocarriers for drug delivery. Nat Mater. 2013;12:991-1003
38. Gomes-da-Silva LC, Fonseca NA, Moura V, de Lima MCP, Simoes S, Moreira JN. Lipid-based nanoparticles for siRNA delivery in cancer therapy: paradigms and challenges. Acc Chem Res. 2012;45:1163-71
39. Li WJ, Szoka FC. Lipid-based nanoparticles for nucleic acid delivery. Pharm Res. 2007;24:438-49
40. Tseng YC, Mozumdar S, Huang L. Lipid-based systemic delivery of siRNA. Adv Drug Deliv Rev. 2009;61:721-31
41. Jayaraman M, Ansell SM, Mui BL, Tam YK, Chen J, Du X. et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew Chem Int Ed. 2012;51:8529-33
42. Sato Y, Hashiba K, Sasaki K, Maeki M, Tokeshi M, Harashima H. Understanding structure-activity relationships of pH-sensitive cationic lipids facilitates the rational identification of promising lipid nanoparticles for delivering siRNAs in vivo. J Control Release. 2019;295:140-52
43. Shim MS, Kwon YJ. Stimuli-responsive polymers and nanomaterials for gene delivery and imaging applications. Adv Drug Deliv Rev. 2012;64:1046-58
44. Tanner P, Baumann P, Enea R, Onaca O, Palivan C, Meier W. Polymeric vesicles: from drug carriers to nanoreactors and artificial organelles. Acc Chem Res. 2011;44:1039-49
45. Dahlman JE, Barnes C, Khan OF, Thiriot A, Jhunjunwala S, Shaw TE. et al. In vivo endothelial siRNA delivery using polymeric nanoparticles with low molecular weight. Nat Nanotechnol. 2014;9:648-55
46. Kumar A, Kumar V. Biotemplated inorganic nanostructures: supramolecular directed nanosystems of semiconductor(s)/metal(s) mediated by nucleic acids and their properties. Chem Rev. 2014;114:7044-78
47. Lin G, Mi P, Chu CC, Zhang J, Liu G. Inorganic nanocarriers overcoming multidrug resistance for cancer theranostics. Adv Sci. 2016;3:1600134
48. Yoo JW, Irvine DJ, Discher DE, Mitragotri S. Bio-inspired, bioengineered and biomimetic drug delivery carriers. Nat Rev Drug Discov. 2011;10:521-35
49. Li X, Liang QM, Zhang W, Li YL, Ye JD, Zhao FJ. et al. Bio-inspired bioactive glasses for efficient microRNA and drug delivery. J Mater Chem B. 2017;5:6376-84
50. Abd E, Khalil IA, Elewa YHA, Kusumoto K, Sato Y, Shobaki N. et al. Lung-endothelium-targeted nanoparticles based on a pH-sensitive lipid and the gala peptide enable robust gene silencing and the regression of metastatic lung cancer. Adv Funct Mater. 2019;29:1807677
51. Singha K, Namgung R, Kim WJ. Polymers in small-interfering RNA delivery. Nucleic Acid Ther. 2011;21:133-47
52. Usman A, Zia KM, Zuber M, Tabasum S, Rehman S, Zia F. Chitin and chitosan based polyurethanes: a review of recent advances and prospective biomedical applications. Int J Biol Macromol. 2016;86:630-45
53. Mano JF, Silva GA, Azevedo HS, Malafaya PB, Sousa RA, Silva SS. et al. Natural origin biodegradable systems in tissue engineering and regenerative medicine: present status and some moving trends. J Royal Soc Interface. 2007;4:999-1030
54. Houacine C, Yousaf SS, Khan I, Khurana RK, Singh KK. Potential of natural biomaterials in nano-scale drug delivery. Curr Pharm Des. 2018;24:5188-206
55. Islam MA, Xu YJ, Tao W, Ubellacker JM, Lim M, Aum D. et al. Restoration of tumour-growth suppression in vivo via systemic nanoparticle-mediated delivery of PTEN mRNA. Nat Biomed Eng. 2018;2:850-64
56. Liu YL, Ji XY, Tong WWL, Askhatova D, Yang TY, Cheng HW. et al. Engineering multifunctional RNAi nanomedicine to concurrently target cancer hallmarks for combinatorial therapy. Angew Chem Int Ed. 2018;57:1510-3
57. Shi JJ, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17:20-37
58. Xu XD, Wu J, Liu SS, Saw PE, Tao W, Li YJ. et al. Redox-responsive nanoparticle-mediated systemic RNAi for effective cancer therapy. Small. 2018;14:1802565
59. Xu XD, Wu J, Liu YL, Saw PE, Tao W, Yu M. et al. Multifunctional envelope-type siRNA delivery nanoparticle platform for prostate cancer therapy. ACS Nano. 2017;11:2618-27
60. Hom C, Lu J, Liong M, Luo HZ, Li ZX, Zink JI. et al. Mesoporous silica nanoparticles facilitate delivery of siRNA to shutdown signaling pathways in mammalian cells. Small. 2010;6:1185-90
61. Li X, Chen YJ, Wang MQ, Ma YJ, Xia WL, Gu HC. A mesoporous silica nanoparticle-PEI-fusogenic peptide system for siRNA delivery in cancer therapy. Biomaterials. 2013;34:1391-401
62. Popat A, Hartono SB, Stahr F, Liu J, Qiao SZ, Lu GQ. Mesoporous silica nanoparticles for bioadsorption, enzyme immobilisation, and delivery carriers. Nanoscale. 2011;3:2801-18
63. Lu Q, Moore JM, Huang G, Mount AS, Rao AM, Larcom LL. et al. RNA polymer translocation with single-walled carbon nanotubes. Nano Lett. 2004;4:2473-7
64. Lee T, Yagati AK, Pi FM, Sharma A, Choi JW, Guo PX. Construction of RNA-quantum dot chimera for nanoscale resistive biomemory application. ACS Nano. 2015;9:6675-82
65. He CB, Lu KD, Liu DM, Lin WB. Nanoscale metal-organic frameworks for the co-delivery of cisplatin and pooled siRNAs to enhance therapeutic efficacy in drug-resistant ovarian cancer cells. J Am Chem Soc. 2014;136:5181-4
66. Samanta A, Banerjee S, Liu Y. DNA nanotechnology for nanophotonic applications. Nanoscale. 2015;7:2210-20
67. Stulz E. DNA architectonics: towards the next generation of bio-inspired materials. Chem A Eur J. 2012;18:4456-69
68. Johnsen KB, Gudbergsson JM, Skov MN, Pilgaard L, Moos T, Duroux M. A comprehensive overview of exosomes as drug delivery vehicles-endogenous nanocarriers for targeted cancer therapy. BBA Rev Cancer. 2014;1846:75-87
69. Van den Boorn JG, Dassler J, Coch C, Schlee M, Hartmann G. Exosomes as nucleic acid nanocarriers. Adv Drug Deliv Rev. 2013;65:331-5
70. Evans BC, Nelson CE, Yu SS, Beavers KR, Kim AJ, Li HM. et al. Ex vivo red blood cell hemolysis assay for the evaluation of pH-responsive endosomolytic agents for cytosolic delivery of biomacromolecular drugs. Jove J Vis Exp. 2013 e50166
71. Zhang P, Liu G, Chen X. Nanobiotechnology: cell membrane-based delivery systems. Nano today. 2017;13:7-9
72. Sato V, Bolzenius JK, Eteleeb AM, Su X, Maher CA, Sehn JK. et al. CD4(+) T cells induce rejection of urothelial tumors after immune checkpoint blockade. JCI Insight. 2018;3:e121062
73. Aleku M, Schulz P, Keil O, Santel A, Schaeper U, Dieckhoff B. et al. Atu027, a liposomal small interfering RNA formulation targeting protein kinase n3, inhibits cancer progression. Cancer Res. 2008;68:9788-98
74. Anfossi S, Babayan A, Pantel K, Calin GA. Clinical utility of circulating non-coding RNAs-an update. Nat Clin Pract Oncol. 2018;15:541-63
75. Ghafouri-Fard S, Ghafouri-Fard S. siRNA and cancer immunotherapy. Immunotherapy. 2012;4:907-17
76. Conde J, Arnold CE, Tian FR, Artzi N. RNAi nanomaterials targeting immune cells as an anti-tumor therapy: the missing link in cancer treatment? Mater Today. 2016;19:29-43
77. Freeley M, Long A. Advances in siRNA delivery to T-cells: potential clinical applications for inflammatory disease, cancer and infection. Biochem J. 2013;455:133-47
78. Tang X, Rao JD, Yin S, Wei JJ, Xia CY, Li M. et al. PD-L1 knockdown via hybrid micelle promotes paclitaxel induced cancer-immunity cycle for melanoma treatment. Eur J Pharm Sci. 2019;127:161-74
79. Teo PY, Yang C, Whilding LM, Parente-Pereira AC, Maher J, George AJT. et al. Ovarian cancer immunotherapy using PD-L1 siRNA targeted delivery from folic acid-functionalized polyethylenimine: strategies to enhance T cell killing. Adv Healthcare Mater. 2015;4:1180-9
80. Dai LL, Li K, Li MH, Zhao XJ, Luo Z, Lu L. et al. Size/charge changeable acidity-responsive micelleplex for photodynamic-improved PD-L1 immunotherapy with enhanced tumor penetration. Adv Funct Mater. 2018;28:1707249
81. Wang DG, Wang TT, Liu JP, Yu HJ, Jiao S, Feng B. et al. Acid-activatable versatile micelleplexes for PD-L1 blockade enhanced cancer photodynamic immunotherapy. Nano Lett. 2016;16:5503-13
82. Gulla SK, Kotcherlakota R, Nimushakavi S, Nimmu NV, Khalid S, Patra CR. et al. Au-CGKRK nanoconjugates for combating cancer through T-cell-driven therapeutic RNA interference. ACS Omega. 2018;3:8663-76
83. Xu ZH, Wang YH, Zhang L, Huang L. Nanoparticle-delivered transforming growth factor-beta siRNA enhances vaccination against advanced melanoma by modifying tumor microenvironment. ACS Nano. 2014;8:3636-45
84. Qiao CM, Yang J, Shen Q, Liu RY, Li YH, Shi YJ. et al. Traceable nanoparticles with dual targeting and ROS response for RNAi-based immunochemotherapy of intracranial glioblastoma treatment. Adv Mater. 2018;30:1705054
85. Wang YH, Xu ZH, Guo ST, Zhang L, Sharma A, Robertson GP. et al. Intravenous delivery of siRNA targeting CD47 effectively inhibits melanoma tumor growth and lung metastasis. Mol Ther. 2013;21:1919-29
86. Wu JM, Li Z, Yang ZP, Guo L, Zhang Y, Deng HH. et al. A glutamine-rich carrier efficiently delivers anti-CD47 siRNA driven by a "glutamine trap" to inhibit lung cancer cell growth. Mol Pharm. 2018;15:3032-45
87. Jadidi-Niaragh F, Atyabi F, Rastegari A, Kheshtchin N, Arab S, Hassannia H. et al. CD73 specific siRNA loaded chitosan lactate nanoparticles potentiate the antitumor effect of a dendritic cell vaccine in 4T1 breast cancer bearing mice. J Control Release. 2017;246:46-59
88. Matsuda A, Ishiguro K, Yan IK, Patel T. Extracellular vesicle-based therapeutic targeting of β-catenin to modulate anticancer immune responses in hepatocellular cancer. Hepatol Commun. 2019;3:525-41
89. Wu YH, Gu WY, Li L, Chen C, Xu ZP. Enhancing PD-1 gene silence in T lymphocytes by comparing the delivery performance of two inorganic nanoparticle platforms. Nanomaterials. 2019;9:159
90. Li SY, Liu Y, Xu CF, Shen S, Sun R, Du XJ. et al. Restoring anti-tumor functions of T cells via nanoparticle-mediated immune checkpoint modulation. J Control Release. 2016;231:17-28
91. Jiang Y, Hardie J, Liu YC, Ray M, Luo X, Das R. et al. Nanocapsule-mediated cytosolic siRNA delivery for anti-inflammatory treatment. J Control Release. 2018;283:235-40
92. Conde J, Bao CC, Tan YQ, Cui DX, Edelman ER, Azevedo HS. et al. Dual targeted immunotherapy via in vivo delivery of biohybrid RNAi-peptide nanoparticles to tumor-associated macrophages and cancer cells. Adv Funct Mater. 2015;25:4183-94
93. Qian Y, Qiao S, Dai YF, Xu GQ, Dai BL, Lu LS. et al. Molecular-targeted immunotherapeutic strategy for melanoma via dual-targeting nanoparticles delivering small interfering RNA to tumor-associated macrophages. ACS Nano. 2017;11:9536-49
94. Zhang YJ, Fu JM, Shi YM, Peng SS, Cai Y, Zhan XL. et al. A new cancer immunotherapy via simultaneous DC-mobilization and DC-targeted IDO gene silencing using an immune-stimulatory nanosystem. Int J Cancer. 2018;143:2039-52
95. Zhang YJ, Song N, Fu JM, Liu YL, Zhan XL, Peng SS. et al. Synergic therapy of melanoma using GNRs-MUA-PEI/siIDO2-FA through targeted gene silencing and plasmonic photothermia. RSC Adv. 2016;6:77577-89
96. Roeven MWH, Hobo W, Van der Voort R, Fredrix H, Norde WJ, Teijgeler K. et al. Efficient nontoxic delivery of PD-L1 and PD-L2 siRNA into dendritic cell vaccines using the cationic lipid SAINT-18. J Immunother. 2015;38:145-54
97. Cubillos-Ruiz JR, Engle X, Scarlett UK, Martinez D, Barber A, Elgueta R. et al. Polyethylenimine-based siRNA nanocomplexes reprogram tumor-associated dendritic cells via TLR5 to elicit therapeutic antitumor immunity. J Clin Invest. 2009;119:2231-44
98. Liu DQ, Lu S, Zhang LX, Ji M, Liu SY, Wang SW. et al. An indoleamine 2, 3-dioxygenase siRNA nanoparticle-coated and Trp2-displayed recombinant yeast vaccine inhibits melanoma tumor growth in mice. J Control Release. 2018;273:1-12
99. Luo ZC, Wang C, Yi HQ, Li P, Pan H, Liu LL. et al. Nanovaccine loaded with poly I:C and STAT3 siRNA robustly elicits anti-tumor immune responses through modulating tumor-associated dendritic cells in vivo. Biomaterials. 2015;38:50-60
100. Heo MB, Lim YT. Programmed nanoparticles for combined immunomodulation, antigen presentation and tracking of immunotherapeutic cells. Biomaterials. 2014;35:590-600
101. Warashina S, Nakamura T, Sato Y, Fujiwara Y, Hyodo M, Hatakeyama H. et al. A lipid nanoparticle for the efficient delivery of siRNA to dendritic cells. J Control Release. 2016;225:183-91
102. Heo MB, Cho MY, Lim YT. Polymer nanoparticles for enhanced immune response: combined delivery of tumor antigen and small interference RNA for immunosuppressive gene to dendritic cells. Acta Biomater. 2014;10:2169-76
103. Warashina S, Nakamura T, Harashima H. A20 silencing by lipid envelope-type nanoparticles enhances the efficiency of lipopolysaccharide-activated dendritic cells. Biol Pharm Bull. 2011;34:1348-51
104. Leuschner F, Dutta P, Gorbatov R, Novobrantseva TI, Donahoe JS, Courties G. et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat Biotechnol. 2011;29:1005-10
105. Song YD, Tang C, Yin CH. Combination antitumor immunotherapy with VEGF and PIGF siRNA via systemic delivery of multi-functionalized nanoparticles to tumor-associated macrophages and breast cancer cells. Biomaterials. 2018;185:117-32
106. Van Woensel M, Mathivet T, Wauthoz N, Rosiere R, Garg AD, Agostinis P. et al. Sensitization of glioblastoma tumor micro-environment to chemo-and immunotherapy by Galectin-1 intranasal knock-down strategy. Sci Rep. 2017;7:1217
107. Berraondo P, Sanmamed MF, Ochoa MC, Etxeberria I, Aznar MA, Perez-Gracia JL. et al. Cytokines in clinical cancer immunotherapy. Br J Cancer. 2019;120:6-15
108. Kalathil SG, Thanavala Y. High immunosuppressive burden in cancer patients: a major hurdle for cancer immunotherapy. Cancer Immunol Immunother. 2016;65:813-9
109. Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology. 2013;138:105-15
110. Munn DH, Bronte V. Immune suppressive mechanisms in the tumor microenvironment. Curr Opin Immunol. 2016;39:1-6
111. Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267-96
112. Byun DJ, Wolchok JD, Rosenberg LM, Girotra M. Cancer immunotherapy-immune checkpoint blockade and associated endocrinopathies. Nat Rev Endocrinol. 2017;13:195-207
113. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27:109-18
114. Vasievich EA, Huang L. The suppressive tumor microenvironment: a challenge in cancer immunotherapy. Mol Pharm. 2011;8:635-41
115. Wang Y, Lin YX, Qiao SL, Wang J, Wang H. Progress in tumor-associated macrophages: from bench to bedside. Adv Biosyst. 2019;3:1800232
116. Wang Y, Lin YX, Qiao SL, An HW, Ma Y, Qiao ZY. et al. Polymeric nanoparticles promote macrophage reversal from M2 to M1 phenotypes in the tumor microenvironment. Biomaterials. 2017;112:153-63
117. Lehmann B, Biburger M, Brueckner C, Ipsen-Escobedo A, Gordan S, Lehmann C. et al. Tumor location determines tissue-specific recruitment of tumor-associated macrophages and antibody-dependent immunotherapy response. Sci Immunol. 2017;2:eaah6413
118. Frankenberger C, Rabe D, Bainer R, Sankarasharma D, Chada K, Krausz T. et al. Metastasis suppressors regulate the tumor microenvironment by blocking recruitment of prometastatic tumor-associated macrophages. Cancer Res. 2015;75:4063-73
119. Guo Q, Jin Z, Yuan Y, Liu R, Xu T, Wei H. et al. New mechanisms of tumor-associated macrophages on promoting tumor progression: recent research advances and potential targets for tumor immunotherapy. J Immunol Res. 2016;2016:9720912
120. Sioud M. Releasing the immune system brakes using siRNAs enhances cancer immunotherapy. Cancers. 2019;11:176
121. Conroy H, Galvin KC, Higgins SC, Mills KHG. Gene silencing of TGF-beta 1 enhances antitumor immunity induced with a dendritic cell vaccine by reducing tumor-associated regulatory T cells. Cancer Immunol Immunother. 2012;61:425-31
122. Kim JH, Kang TH, Noh KH, Bae HC, Ahn YH, Lee YH. et al. Blocking the immunosuppressive axis with small interfering RNA targeting interleukin (IL)-10 receptor enhances dendritic cell-based vaccine potency. Clin Exp Immunol. 2011;165:180-9
123. Van der Waart AB, Fredrix H, Van der Voort R, Schaap N, Hobo W, Dolstra H. siRNA silencing of PD-1 ligands on dendritic cell vaccines boosts the expansion of minor histocompatibility antigen-specific CD8(+) T cells in NOD/SCID/IL2Rg(null) mice. Cancer Immunol Immunother. 2015;64:645-54
124. Zheng XF, Koropatnick J, Chen D, Velenosi T, Ling H, Zhang XS. et al. Silencing IDO in dendritic cells: a novel approach to enhance cancer immunotherapy in a murine breast cancer model. Int J Cancer. 2013;132:967-77
125. Aarts CEM, Kuijpers TW. Neutrophils as myeloid-derived suppressor cells. Eur J Clin Investig. 2018;48:e12989
126. Martin F, Apetoh L, Ghiringhelli F. Role of myeloid-derived suppressor cells in tumor immunotherapy. Immunotherapy. 2012;4:43-57
127. Yu JP, Wang Y, Yan F, Zhang P, Li H, Zhao H. et al. Noncanonical NF-kappa B activation mediates STAT3-stimulated IDO upregulation in myeloid-derived suppressor cells in breast cancer. J Immunol. 2014;193:2574-86
128. Zhang J, Xu XF, Shi M, Chen Y, Yu DH, Zhao CY. et al. CD13(hi) neutrophil-like myeloid-derived suppressor cells exert immune suppression through arginase 1 expression in pancreatic ductal adenocarcinoma. Oncoimmunology. 2017;6:e1258504
129. Sumida K, Ohno Y, Ohtake J, Kaneumi S, Kishikawa T, Takahashi N. et al. IL-11 induces differentiation of myeloid-derived suppressor cells through activation of STAT3 signalling pathway. Sci Rep. 2015;5:13650
130. Ibanez-Vea M, Zuazo M, Gato M, Arasanz H, Fernandez-Hinojal G, Escors D. et al. Myeloid-derived suppressor cells in the tumor microenvironment: current knowledge and future perspectives. Arch Immunol Ther Exp. 2018;66:113-23
131. Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D. et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res. 2016;76:5671-82
132. He W, Liang P, Guo GX, Huang Z, Niu YM, Dong L. et al. Re-polarizing myeloid-derived suppressor cells (MDSCs) with cationic polymers for cancer immunotherapy. Sci Rep. 2016;6:24506
133. Highfill SL, Cui YZ, Giles AJ, Smith JP, Zhang H, Morse E. et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med. 2014;6:237ra67
134. Liu YJ, Wei GW, Cheng WA, Dong ZY, Sun H, Lee VY. et al. Targeting myeloid-derived suppressor cells for cancer immunotherapy. Cancer Immunol Immunother. 2018;67:1181-95
135. Tuettenberg A, Steinbrink K, Schuppan D. Myeloid cells as orchestrators of the tumor microenvironment: novel targets for nanoparticular cancer therapy. Nanomed. 2016;11:2735-51
136. Denaro N, Merlano M, Nigro C. Long noncoding RNAs as regulators of cancer immunity. Mol Oncol. 2018;13:61-73
137. Garzon R, Fabbri M, Cimmino A, Calin GA, Croce CM. MicroRNA expression and function in cancer. Trends Mol Med. 2006;12:580-7
138. Kent OA, Mendell JT. A small piece in the cancer puzzle: microRNAs as tumor suppressors and oncogenes. Oncogene. 2006;25:6188-96
139. Zhang BH, Pan XP, Cobb GP, Anderson TA. microRNAs as oncogenes and tumor suppressors. Dev Biol. 2007;302:1-12
140. Hosseinahli N, Aghapour M, Duijf PHG, Baradaran B. Treating cancer with microRNA replacement therapy: a literature review. J Cell Physiol. 2018;233:5574-88
141. Lu LF, Liston A. MicroRNA in the immune system, microRNA as an immune system. Immunology. 2009;127:291-8
142. Grenda A, Krawczyk P. New dancing couple: PD-L1 and microRNA. Scand J Immunol. 2017;86:130-4
143. Wang QS, Lin W, Tang XQ, Li SH, Guo LB, Lin Y. et al. The roles of microRNAs in regulating the expression of PD-1/PD-L1 immune checkpoint. Int J Mol Sci. 2017;18:2540
144. Xu SH, Tao Z, Hai B, Liang HG, Shi Y, Wang T. et al. miR-424(322) reverses chemoresistance via T-cell immune response activation by blocking the PD-L1 immune checkpoint. Nat Commun. 2016;7:11406
145. Zhu JJ, Chen LX, Zou LT, Yang PP, Wu RR, Mao Y. et al. MiR-20b,-21, and-130b inhibit PTEN expression resulting in B7-H1 over-expression in advanced colorectal cancer. Hum Immunol. 2014;75:348-53
146. He X, Tang R, Sun Y, Wang YG, Zhen KY, Zhang DM. et al. MicroR-146 blocks the activation of M1 macrophage by targeting signal transducer and activator of transcription 1 in hepatic schistosomiasis. Ebiomedicine. 2016;13:339-47
147. Wang Z, Brandt S, Medeiros A, Wang SJ, Wu H, Dent A. et al. MicroRNA 21 is a homeostatic regulator of macrophage polarization and prevents prostaglandin E-2-mediated M2 generation. Plos One. 2015;10:e0115855
148. Shen R, Wang Y, Wang CX, Yin M, Liu HL, Chen JP. et al. miRNA-155 mediates TAM resistance by modulating SOCS6-STAT3 signalling pathway in breast cancer. Am J Transl Res. 2015;7:2115-26
149. Squadrito ML, Etzrodt M, De Palma M, Pittet MJ. MicroRNA-mediated control of macrophages and its implications for cancer. Trends Immunol. 2013;34:350-9
150. Leong JW, Sullivan RP, Fehniger TA. microRNA management of NK-cell developmental and functional programs. Eur J Immunol. 2014;44:2862-8
151. Liu XG, Zhan ZZ, Xu L, Ma F, Li D, Guo ZH. et al. MicroRNA-148/152 impair innate response and antigen presentation of TLR-triggered dendritic cells by targeting CaMKII alpha. J Immunol. 2010;185:7244-51
152. Liang X, Liu Y, Mei SY, Zhang MM, Xin JX, Zhang Y. et al. MicroRNA-22 impairs anti-tumor ability of dendritic cells by targeting p38. Plos One. 2015;10:e0121510
153. Min SP, Liang X, Zhang MM, Zhang Y, Mei SY, Liu JZ. et al. Multiple tumor-associated microRNAs modulate the survival and longevity of dendritic cells by targeting YWHAZ and Bcl2 signaling pathways. J Immunol. 2013;190:2437-46
154. Jeker LT, Bluestone JA. MicroRNA regulation of T-cell differentiation and function. Immunol Rev. 2013;253:65-81
155. Kroesen BJ, Teteloshvili N, Smigielska-Czepiel K, Brouwer E, Boots AMH, van den Berg A. et al. Immuno-miRs: critical regulators of T-cell development, function and ageing. Immunology. 2015;144:1-10
156. Zhang J, Cheng Y, Cui W, Li MX, Li B, Guo L. MicroRNA-155 modulates Th1 and Th17 cell differentiation and is associated with multiple sclerosis and experimental autoimmune encephalomyelitis. J Neuroimmunol. 2014;266:56-63
157. Du CS, Liu C, Kang JH, Zhao GX, Ye ZQ, Huang SC. et al. MicroRNA miR-326 regulates T-H-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat Immunol. 2009;10:1252-9
158. Kang SG, Liu WH, Lu PW, Jin HY, Lim HW, Shepherd J. et al. MicroRNAs of the miR-17 similar to 92 family are critical regulators of T-FH differentiation. Nat Immunol. 2013;14:849-57
159. Zang XL, Zhang XX, Zhao XL, Hu HY, Qiao MX, Deng YH. et al. Targeted delivery of miRNA 155 to tumor associated macrophages for tumor immunotherapy. Mol Pharm. 2019;16:1714-22
160. Parayath NN, Parikh A, Amiji MM. Repolarization of tumor-associated macrophages in a genetically engineered nonsmall cell lung cancer model by intraperitoneal administration of hyaluronic acid-based nanoparticles encapsulating microRNA-125b. Nano Lett. 2018;18:3571-9
161. Bryniarski K, Ptak W, Jayakumar A, Puellmann K, Caplan MJ, Chairoungdua A. et al. Antigen-specific, antibody-coated, exosome-like nanovesicles deliver suppressor T-cell microRNA-150 to effector T cells to inhibit contact sensitivity. J Allergy Clin Immunol. 2013;132:170-81
162. Yang LN, Sun J, Liu Q, Zhu RR, Yang QN, Hua JH. et al. Synergetic functional nanocomposites enhance immunotherapy in solid tumors by remodeling the immunoenvironment. Adv Sci. 2019;6:1802012
163. Asadirad A, Hashemi SM, Baghaei K, Ghanbarian H, Mortaz E, Zali MR. et al. Phenotypical and functional evaluation of dendritic cells after exosomal delivery of miRNA-155. Life Sci. 2019;219:152-62
164. Liu LL, Yi HQ, Wang C, He HM, Li P, Pan H. et al. Integrated nanovaccine with microrNA-148a inhibition reprograms tumor-associated dendritic cells by modulating miR-148a/DNMT1/SOCS1 Axis. J Immunol. 2016;197:1231-41
165. Neviani P, Wise PM, Murtadha M, Liu CW, Wu CH, Jong AY. et al. Natural killer-derived exosomal miR-186 inhibits neuroblastoma growth and immune escape mechanisms. Cancer Res. 2019;79:1151-64
166. Yamamoto A, Kormann M, Rosenecker J, Rudolph C. Current prospects for mRNA gene delivery. Eur J Pharm Biopharm. 2009;71:484-9
167. Islam MA, Reesor EKG, Xu YJ, Zope HR, Zetter BR, Shi JJ. Biomaterials for mRNA delivery. Biomater Sci. 2015;3:1519-33
168. Maruggi G, Zhang CL, Li JW, Ulmer JB, Yu D. mRNA as a transformative technology for vaccine development to control infectious diseases. Mol Ther. 2019;27:757-72
169. Pardi N, Hogan MJ, Porter FW, Weissman D. mRNA vaccines-a new era in vaccinology. Nat Rev Drug Discov. 2018;17:261-79
170. Reichmuth AM, Oberli MA, Jaklenec A, Langer R, Blankschtein D. mRNA vaccine delivery using lipid nanoparticles. Ther Deliv. 2016;7:319-34
171. Weiss R, Scheiblhofer S, Roesler E, Weinberger E, Thalhamer J. mRNA vaccination as a safe approach for specific protection from type I allergy. Expert Rev Vaccines. 2012;11:55-67
172. Oberli MA, Reichmuth AM, Dorkin JR, Mitchell MJ, Fenton OS, Jaklenec A. et al. Lipid nanoparticle assisted mRNA delivery for potent cancer immunotherapy. Nano Lett. 2017;17:1326-35
173. Krishnamachari Y, Salem AK. Innovative strategies for co-delivering antigens and CpG oligonucleotides. Adv Drug Deliv Rev. 2009;61:205-17
174. Wilson KD, De Jong SD, Tam YK. Lipid-based delivery of CpG oligonucleotides enhances immunotherapeutic efficacy. Adv Drug Deliv Rev. 2009;61:233-42
175. Stauss HJ, Morris EC. Immunotherapy with gene-modified T cells: limiting side effects provides new challenges. Gene Ther. 2013;20:1029
176. Schlake T, Thess A, Thran M, Jordan I. mRNA as novel technology for passive immunotherapy. Cell Mol Life Sci. 2019;76:301-28
177. Wang ZJ, Liu WH, Shi JY, Chen N, Fan CH. Nanoscale delivery systems for cancer immunotherapy. Mater Hor. 2018;5:344-62
178. Moffett HF, Coon ME, Radtke S, Stephan SB, McKnight L, Lambert A. et al. Hit-and-run programming of therapeutic cytoreagents using mRNA nanocarriers. Nat Commun. 2017;8:389
179. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281-308
180. Doudna JA, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096
181. Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262-78
182. Gori JL, Hsu PD, Maeder ML, Shen S, Welstead GG, Bumcrot D. Delivery and specificity of CRISPR/Cas9 genome editing technologies for human gene therapy. Hum Gene Ther. 2015;26:443-51
183. Li HL, Fujimoto N, Sasakawa N, Shirai S, Ohkame T, Sakuma T. et al. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Rep. 2015;4:143-54
184. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJC, Hamieh M, Cunanan KM. et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543:113-17
185. Ren JT, Liu XJ, Fang CY, Jiang SG, June CH, Zhao YB. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res. 2017;23:2255-66
186. Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA. et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. 2017;7:737
187. Lu Y, Xue JX, Deng T, Zhou XJ, Yu K, Huang MJ. A phase I trial of PD-1 deficient engineered T cells with CRISPR/Cas9 in patients with advanced non-small cell lung cancer. J Clin Oncol. 2018;36:3050
188. Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK. et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31:822
189. Wang HX, Li M, Lee CM, Chakraborty S, Kim HW, Bao G. et al. CRISPR/Cas9-based genome editing for disease modeling and therapy: challenges and opportunities for nonviral delivery. Chem Rev. 2017;117:9874-906
190. Sun W, Ji W, Hall JM, Hu Q, Wang C, Beisel CL. et al. Self-assembled DNA nanoclews for the efficient delivery of CRISPR-Cas9 for genome editing. Angew Chem Int Ed. 2015;54:12029-33
191. Sau S, Alsaab HO, Bhise K, Alzhrani R, Nabil G, Iyer AK. Multifunctional nanoparticles for cancer immunotherapy: a groundbreaking approach for reprogramming malfunctioned tumor environment. J Control Release. 2018;274:24-34
192. Miller JB, Zhang S, Kos P, Xiong H, Zhou K, Perelman SS. et al. Non-viral CRISPR/cas gene editing in vitro and in vivo enabled by synthetic nanoparticle co-delivery of Cas9 mRNA and sgRNA. Angew Chem Int Ed. 2017;56:1059-63
193. Liu C, Zhang L, Liu H, Cheng K. Delivery strategies of the CRISPR-Cas9 gene-editing system for therapeutic applications. J Control Release. 2017;266:17-26
194. Ray M, Lee Y-W, Hardie J, Mout R, Yeşilbag Tonga G, Farkas ME. et al. CRISPRed macrophages for cell-based cancer immunotherapy. Bioconjug Chem. 2018;29:445-50
195. Cheng W-J, Chen L-C, Ho H-O, Lin H-L, Sheu M-T. Stearyl polyethylenimine complexed with plasmids as the core of human serum albumin nanoparticles noncovalently bound to CRISPR/Cas9 plasmids or siRNA for disrupting or silencing PD-L1 expression for immunotherapy. Int J Nanomedicine. 2018;13:7079-94
196. Kohnken R, Mishra A. Micrornas in cutaneous T-cell lymphoma: the future of therapy. J Invest Dermatol. 2019;139:528-34
197. Shin H, Park SJ, Yim Y, Kim J, Choi C, Won C. et al. Recent advances in RNA therapeutics and RNA delivery systems based on nanoparticles. Adv Ther. 2018;1:1800065
198. Kanasty R, Dorkin JR, Vegas A, Anderson D. Delivery materials for siRNA therapeutics. Nat Mater. 2013;12:967-77
199. Kowalski P, Rudra A, Miao L, Anderson D. Delivering the messenger: advances in technologies for therapeutic mRNA delivery. Mol Ther. 2019;27:710-28
200. Xue HY, Liu SM, Wong HL. Nanotoxicity: a key obstacle to clinical translation of siRNA-based nanomedicine. Nanomed. 2014;9:295-312
201. Eloy JO, Petrilli R, Lopez RFV, Lee RJ. Stimuli-responsive nanoparticles for siRNA delivery. Curr Pharm Des. 2015;21:4131-44
202. Grzelczak M, Liz-Marzan LM, Klajn R. Stimuli-responsive self-assembly of nanoparticles. Chem Soc Rev. 2019;48:1342-61
203. Guo S, Huang L. Nanoparticles escaping res and endosome: challenges for siRNA delivery for cancer therapy. J Nanomater. 2011:742895
204. Liu X, Yang Y, Urban MW. Stimuli-responsive polymeric nanoparticles. Macromol Rapid Commun. 2017;38:1700030
Author contact
![]() Corresponding author: E-mail: meilin7sysu.edu.cn (L. Mei), wanghaocn (H. Wang), jshiharvard.edu (J. Shi).
Corresponding author: E-mail: meilin7sysu.edu.cn (L. Mei), wanghaocn (H. Wang), jshiharvard.edu (J. Shi).