Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Design and synthesis of p-PROTAC
Characterization of biophysical...
Application of p-PROTAC
Future development
Conclusion and perspective
Abbreviations
Acknowledgements
References
Authors' Biographic
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(22):10141-10153. doi:10.7150/thno.46985 This issue Cite
Review
The peptide PROTAC modality: a novel strategy for targeted protein ubiquitination
Jinmei Jin1*, Ye Wu1*, Jinjiao Chen1,2*, Yiwen Shen1, Lijun Zhang1, Hong Zhang1, Lili Chen1, Hebao Yuan3, Hongzhuan Chen1,4, Weidong Zhang1 ![]() , Xin Luan1
, Xin Luan1 ![]()
1. Institute of Interdisciplinary Integrative Medicine Research, Shuguang Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai 201203, China.
2. Department of Pharmacology, School of Pharmacy, Fudan University, Shanghai 201203, China.
3. Department of Pharmaceutical Sciences, College of Pharmacy, University of Michigan, Ann Arbor, MI 48109 US.
4. Department of Pharmacology and Chemical Biology, Shanghai Universities Collaborative Innovation Center for Translational Medicine, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China.
*These authors contribute equally to this work.
Received 2020-4-14; Accepted 2020-7-30; Published 2020-8-8
Abstract

Despite dramatic advances in drug discovery over the decades, effective therapeutic strategies for cancers treatment are still in urgent demands. PROteolysis TArgeting Chimera (PROTAC), a novel therapeutic modality, has been vigorously promoted in preclinical and clinical applications. Unlike small molecule PROTAC, peptide PROTAC (p-PROTAC) with advantages of high specificity and low toxicity, while avoiding the limitations of shallow binding pockets through large interacting surfaces, provides promising substitutions for E3 ubiquitin ligase complex-mediated ubiquitination of “undruggable proteins”. It is worth noting that successful applications of p-PROTAC still have some obstacles, including low stability and poor membrane permeability. Hence, we highlight that p-PROTAC combined with cell-penetrating peptides, constrained conformation technique, and targeted delivery systems could be the future efforts for potential translational research.
Keywords: peptide PROTAC, ubiquitination, undruggable proteins, E3 ligase
Introduction
PROteolysis TArgeting Chimera (PROTAC) is an emerging class of therapeutic modality to induce the dynamic degradation of intracellular or nuclear protein of interest (POI), and it plays a significant role in solving drug resistance through the degradation of the entire pathogenic proteins without compensatory increase or mutation [1]. For instance, more than 80% of patients with chronic lymphocytic leukemia (CLL) developed C481S mutation after receiving the Bruton's tyrosine kinase (BTK) inhibitor ibrutinib, leading to acquired drug resistance [2]. It is gratifying that a series of PROTACs (MT-802, SJF620, and L18I) effectively degrade a variety of clinical BTK mutant proteins and overcome the resistance to ibrutinib induced by BTK mutations [2-4].
The core concept of PROTAC is that this bifunctional molecule binds the POI at one end while binding an E3 ligase at the other end, which forms a ternary complex to hijack the cellular ubiquitin-proteasome system (UPS) for proteasomal degradation of POI [3-5]. Compared with small molecule inhibitors, PROTAC modalities usually exhibit improved therapeutic effects due to their enhanced regulation of related fundamental signaling pathway and minimized drug resistance [6]. As a vital direction for novel drug discovery, the therapeutic strategies of PROTAC have been successfully applied to conditionally degrade plenty of POIs in vitro and in vivo, such as estrogen receptor (ER) [7], androgen receptor (AR) [8], bromodomain-containing protein 4 (BRD4) [9-11], anaplastic lymphoma kinase (ALK) [12, 13], Focal adhesion kinase (FAK) [14], cyclin dependent kinase 9 (CDK9) [15], BCR-ABL1 [16, 17], and cycle-related and expression-elevated protein in tumor (CREPT) [18]. The oral PROTACs ARV-110 for AR (ClinicalTrials.gov Identifier: NCT03888612) and ARV-471 for ER (ClinicalTrials.gov Identifier: NCT04072952) exhibited excellent therapeutic efficacies in preclinical studies, and are currently undergoing Phase I clinical trials for evaluation of their safety and tolerability. The preliminary clinical data of ARV-110 exhibited good safety and efficacy in patients with metastatic Castration-resistant Prostate Cancer [19].
However, most of the current PROTACs are using small molecules as targeting warheads, which heavily rely on the binding pockets of POI [20]. With the rapid development of structural biology, it is coming along more convenient to obtain the peptides with high affinity to POI epitopes [21, 22]. Therefore, designing PROTAC based on the specific peptides (p-PROTAC) is an emerging approach to realize the specific and effective degradation of POI, and extend the scope in regards to “undruggable” proteins targeting, while avoiding the restriction of shallow binding pockets through large interacting surfaces between POI and peptides [4, 23]. Compared with small molecule PROTACs, p-PROTACs have exhibited several unique advantages (Table 1). The previously reported peptide targeting warheads of p-PROTACs and E3 ubiquitin ligase-recruiting ligands so far were endogenous peptides with high safety and affinity. Those endogenous ligands have been regarded as ideal choices for drug development compared with other modalities (small molecules and antibodies) [24]. However, it is worth noting that most of the subsequent researches chose small molecule ligands due to the poor permeability and low stability of conventional peptides. Hence, the multifaceted understanding of p-PROTAC should be highly valued which can further promote its potential development.
Comparison of p-PROTAC and small molecule PROTAC
| p-PROTAC | small molecule PROTAC | |
|---|---|---|
| Targeting warhead | Peptides | Small molecules |
| Advantages | Ability to target "undruggable" POIs with specificity [23]; Resistance to mutation targets [21]; Simple design and synthesis process; Low toxicity and high-safety in vivo | High cellular permeability, High stability, and low cost [21] |
| Disadvantages | Poor cell membrane permeability [25]; Low stability [25];Few studies on their efficacy | Limitation of degradation of "undruggable" proteins [1]; Inability to target "undruggable" with large shallow surfaces [23];Severe side effects |
| Clinical trials | None | ARV-110, ARV-471 |
In this review, we summarized the proven approaches for design, synthesis, and application of p-PROTAC, while highlighting their unique characteristics and advantages. Regarding the current bottlenecks for clinical translation of p-PROTAC, we also especially focus on the potential efforts in establishing p-PROTAC platforms on the grounds of interdisciplinary technologies.
Design and synthesis of p-PROTAC
PROTAC is generally regarded as a bifunctional molecule that acts as a bridge between the POI and E3 ligase to induce the subsequent degradation of POI. p-PROTAC consists of following components, a peptide-based targeting warhead, a chemical linker, and a recruitment ligand for E3 ubiquitin ligase (Figure 1) [25-27]. In this part, we mainly summarized the design principles for the three components, the strategy to synthesize p-PROTAC, optimization of subsequent peptides, as well as providing theoretical basis for the subsequent use of p-PROTAC.
The schematic diagram of p-PROTAC. (A) With ATP, the enzyme E1 adheres to the Cys residue of the ubiquitin molecule. Then E1 transfers the activated ubiquitin molecule to the E3 ubiquitin ligase via the ubiquitin-binding enzyme E2. (B) Bifunctional p-PROTAC molecule binds to the POI with one end while the other end binding to an E3 ligase to form ternary complex. (C) Different types of E3 enzymes jointly recognize POI with polyubiquitinated modification to mediate degradation through ubiquitin proteasome, and then freed p-PROTAC can be recycled.
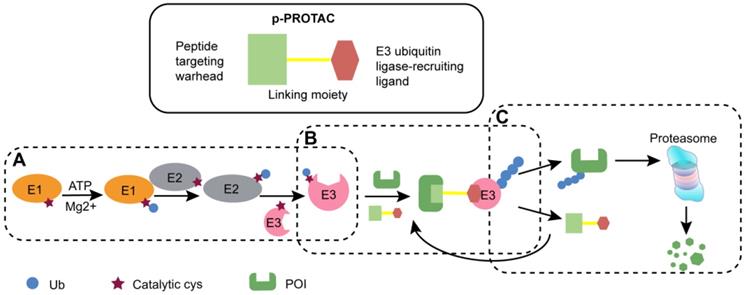
Design of p-PROTAC
Design of peptide targeting warhead
The appropriate choice of peptide targeting warheads has appreciable impacts on the binding affinity and spatial orientation of POI and E3 ligase, which affects the ubiquitination efficiency [28]. The obtainment of the targeting peptide is not a tedious trial-and-error process like small molecules which extremely relies on the database and virtual screening. Generally, based on the crystal structure of endogenous complex of POI and binding protein which reveals their key protein-protein interaction (PPI) motif and residues (Figure 2), the peptide targeting warheads, binding to the POI selectively, can be designed to disrupt the PPI. Then, the obtained sequence of protein epitope mimetic can be used as the leading candidate to synthesize targeting warheads [29]. To maximize the efficacy of protein epitope mimetics, the point mutation on the non-critical interacting residues can be further used to optimize peptides targeting warhead with high affinity for a specific POI [29].
A toolbox of functional peptide PROTACs. (A) The design of peptide targeting warhead using chemical epitope targeting strategy (PDB code: 1QZ7). (B) The commonly used E3 ligase ligands. (C) Linkers-1 connecting POI and E3 ligase ligand. (D) Cell penetrating peptides used to increase membrane permeability. (E) Linkers-2 connecting POI and tag. (F) Functional tags used in p-PROTAC.
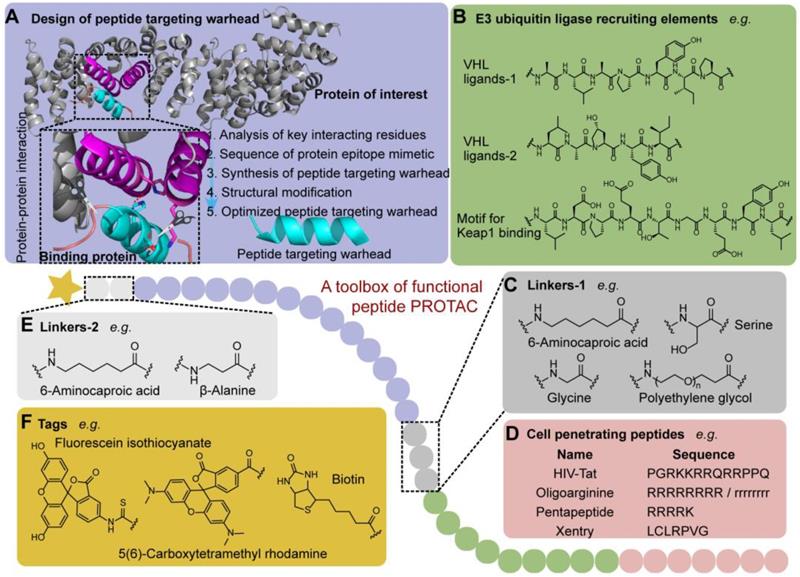
For example, using chemical epitope targeting strategy, Leduc et al. [29] proposed the peptide sequences that mimic binding helix of the coactivator domain with the ERα could function as ER antagonists. To maintain α-helical structure and specificity of protein epitope mimetic in short peptide, they reserved consensus pentapeptide motif, and developed a series of helix-stabilized cyclic peptides as selective inhibitors to bind ER tightly and selectively by regulating ER-coactivator interactions. Inspired by these peptidomimetic estrogen receptor modulators (PERMs), Li et al. [30] further used the N-terminal aspartic acid cross-linking strategy (terminal aspartic acid, TD) to design more stabilized peptide modulators (TD-PERMs) with good cell permeability. Then, they [31] conjugated TD-PERMs with the recruiting peptide of the Von Hippel-Lindau (VHL) E3 ligase to form a p-PROTAC molecule with enhanced biological activity compared to TD-PERMs.
In addition, the phage display and yeast display techniques can be used to develop targeting peptides with high affinity to POI [32, 33]. These techniques provide alternative tools to obtain peptide targeting warheads with highest frequency clone after several rounds of biopanning. More importantly, these techniques are independent of protein crystal complexes, and D-configuration peptide targeting warheads can be obtained via mirror-image phage display which can avoid easy degradation of L-peptides by proteolytic enzymes in vivo [34]. We expect that the peptide phage display techniques could be the future trends to design targeting peptides for p-PROTAC.
Exploitation of E3 ubiquitin ligase-recruiting ligand
Since ubiquitination tags guide the degradation caused by proteasomes which are hijacked by p-PROTAC to promote the degradation of POIs [35, 36], it is very important to rationally choose and design the recruiting moiety. There are many E3 ubiquitin ligases encoded in our bodies with specific degron recognition motifs [37], which provide huge theoretical possibility for PROTAC drug development. However, only less than 1% of E3 ligases, including Von Hippel-Lindau (VHL), Cereblon (CRBN), IAPs, Keap1, RNF4, RNF114, and MDM2, can be hijacked by PROTAC in vivo [38-40]. Heretofore, most of reported PROTACs have chosen VHL or CRBN as E3 ligase due to the existence of their specific and high affinity ligands [11, 41-43].
In the first reported PROTAC strategy, the researchers found that a phosphopeptide (DRHDSGLDSM) within IκBα, regulating the recruitment of Skp1-Cullin-F boxβ-TRCP (SCFβ-TRCP) in E3 ubiquitin ligase complexes, can be successfully used to induce the ubiquitination dependent degradation of POI [25]. In tumor sites, hypoxia-inducible factor-1α (HIF-1α) can be degraded through the VHL-mediated ubiquitination [44]. Therefore, the minimum recognition sequence (ALAPYIP) of HIF-1α has been chosen as an E3 ubiquitin ligase recruiting ligand to induce the degradation of HIF-1α in vivo [45, 46]. Afterwards, a series of peptides have been further optimized for VHL recruitment, including HIF-1α octapeptide and other five amino acids (LAP(OH)YI) [47]. So far, the E3 ubiquitin ligase recruiting ligands for VHL have also been widely applied to design p-PROTAC [18, 31, 33, 48, 49].
Owing to the convenient and easy synthesis, a series of small molecules with the E3 ubiquitin ligase-recruiting function could also be coupled with the targeting warheads to form the p-PROTAC. For example, three amine drugs (thalidomide, lenalidomide, and pomalidomide) have been identified as CRBN ligands [50].
Choice of linking moiety
Linking moiety has significant influences on the stability of the ternary complex and subsequent function [39, 51]. To maintain the delicate balance between the affinity and the spatial effect, the linking moiety should have an appropriate stereochemical structure and provide a suitable solvent-exposed position to connect the peptide targeting warhead with the E3 ubiquitin ligase-recruiting ligand [52]. So far, amino acids are the commonly used linking moieties in p-PROTAC, including aminohexanoic acid (Ahx), glycine, and serine [45, 53]. Polyethylene glycol (PEG) is another linking moiety regularly used to improve the hydrophilicity of p-PROTAC. For instance, VHL-recruiting PROTAC targeting ER showed optimal efficiency with 16 atoms chain length between E3 recognition moiety and warhead [54]. Interestingly, this PROTAC exhibited improved efficiency and affinity to ER due to the change of linker and connection mode in a separate study [55]. Similarly, a series of VHL-recruiting PROTACs targeting FAK exhibited different POI degradation potential due to the different length and constitution of linking moieties [14].
Synthesis strategy
The solid-phase synthesis strategies with Fmoc-protecting group have been widely used in p-PROTAC synthesis. During the process, the side chains of regular amino acids are protected by an unstable anti-acid protective group, while the α-amino group is protected by an unstable anti-base Fmoc-protective group [56]. The selection of resin is determined by the functional groups of C-terminal: the 2-cl-trt or Wang resin are suitable for the retain of C-terminus, while the Rink Amide-AM resin is suitable for the amination of C-terminus [57]. The final p-PROTAC products can be separated from the resin and further purified. Compared with the classic organic synthesis and segments linking of molecule, the amino acid condensation is more concise. In addition to the previously mentioned solid-phase synthesis method, the package of those three parts can also be performed with liquid-phase synthesis [25].
As the targeting warhead, peptides possess greater potential in structural modification compared with small molecules [31]. For example, the length, type, and the sequence of amino acids can be changed for specific secondary structures and physicochemical properties [58], and the progress of solid-phase synthesis technique further promotes automatic synthesis of peptides which accelerate additional layer of convenience for potential targeting peptide library and p-PROTAC fabrication.
In addition, the fluorescent tags (FITC and rhodamine) can be utilized to track the cell uptake process and fluorescence localization of p-PROTAC [56]. The fusion affinity tags (biotin, etc.) can help evaluate the targeting behavior of p-PROTAC by immunoprecipitation or Pull-down assays [54, 57, 59]. Non-radionuclide labeling, phosphopeptide synthesis, and other modification methods are also applied in peptide modification [49]. However, we must pay special attention to the modification position so that the binding affinity of p-PROTAC to POI and E3 ligase, as well as the stability of the ternary complex should not be affected.
Characterization of biophysical properties
According to the construction features of p-PROTAC ternary complexes, the following biophysical properties mainly include the determination of conformation, binding affinity with POI, cell membrane permeability, and the characterization of therapeutic effects. More importantly, the ubiquitin-proteasome-dependent degradation process of POI should to be verified carefully. Herein, we present all the techniques used to evaluate these characteristics of p-PROTAC.
Conformation
In the p-PROTACs, peptides with an α-helical structure and rich positive charge possess better cell membrane permeability [60, 61]. The peptides containing an α-helix exhibit the typical absorption peaks at 208 nm and 222 nm in circular dichroism spectrum. At given concentration of the peptides, the α-helix degree of the peptides can be evaluated through the absorption signal strength of the circular dichroism spectrum at 222 nm, which could be utilized to compare the cell membrane permeability and stability of different p-PROTACs. In a recent study, a facile N-terminal aspartic acid cross-linking strategy (TD strategy) was invented to construct p-PROTAC with α-helix [31]. Using circular dichroism spectroscopy, they successfully evaluated the helicity of these peptides and proved that the obtained p-PROTAC with α-helix from TD strategy exhibited better cell penetration than linear ones [31].
Target binding affinity
Fluorescence polarization (FP), isothermal titration calorimetry (ITC), surface plasmon resonance (SPR), microscale thermophoresis (MST), and co-immunoprecipitation (Co-IP) techniques have been widely used to determine the affinity of polypeptides with POI. Even better, the FP, ITC, SPR, and MST can provide the binding constant (Kd) between them. The characteristics of those binding affinity analysis methods have been listed in Table 2.
Characteristics of different binding affinity analysis methods
| FP [64, 74] | ITC [75] | SPR [66, 76] | MST [67, 69] | Co-IP [70, 77] | |
|---|---|---|---|---|---|
| Measuring range | pM ~ mM | nM ~μM | pM ~ mM | pM ~ mM | − |
| Protein fixation | No | No | Yes | No | Yes |
| Sample consumption | Low | High | Low | Minimal | − |
| Sample number | 1 | 1 | 1 | 1 ~ 16 | 1 |
| Applicable sample | Proteins; Peptides; Small molecules | Proteins; Peptides; Small molecules | Proteins; Peptides; Small molecules; Cell lysates; Culture medium Viruses, et al. | Proteins; Serum; Cell lysates; Culture medium | Cell lysates; Culture medium |
| Sensitivity | High | High | High | High | Low |
| Time-consuming | Hours | Hours | Hours | Minutes | Days |
| Fluorescent labeling | Yes | Not required | Not required | Yes | No |
| Advantages | Low sample purity requirements; Real-time monitoring | Low sample purity requirements; Easy to use; Sample could be reused after the test | Real-time monitoring; Wide range of samples; Classic approach | Fast and efficient; Easy to use; Close to the natural testing environment; Reaction system in solution. | Close to the natural testing environment; Commonly used for in vivo assay |
| Disadvantages | Expensive equipment; | Large sample consumption; Kinetics cannot be determined | Non-specific binding; Expensive equipment | Non-specific binding; Kinetics cannot be determined | Low affinity and instant PPI are difficult to detect; the error in predicting the target protein could lead to the failure |
In the FP assays, the small molecules can be used as fluorophores to detect the formation of complexes from the increased fluorescence polarization for assessing the strength of PPI [62-64]. For p-PROTAC strategy, FP is often used to detect the affinity between a polypeptide fragment and POI [31, 48]. Previous research found that FP assay could be used to detect the affinity of the fluorescein isothiocyanate (FITC)-labeled peptides with the ERα ligand binding domain, providing varying affinity parameters between different PERM and ERα [31].
ITC is a well-established thermodynamic dependent technique that determines a series of thermodynamic parameters for bimolecular interactions, including binding constants, molar binding enthalpy (ΔH°), the binding reaction entropy (ΔS°), and the like [65]. When constructing p-PROTAC's degradation ability on intracellular Tau, the results of ITC experiments exhibited that peptide 1 retained its binding affinity to Keap1 and Tau, with Kd values of 22.8 nM and 763 nM, respectively [26]. However, the ITC method also has some obstacles for biological samples evaluation, such as low sensitivity and relatively large number of samples required for sufficient thermal signal.
SPR biosensor technology is widely used because of its rapid selection of fragments with binding affinity. It enables researchers to quantify the binding properties of a lead compound to its target based on affinity, specificity, and association/dissociation rate [66]. Due to the directly measured interaction between peptides and POI, the kinetic process of binding and the kinetic data of the interaction can be easily obtained. It should be noted that the modes of surface-immobilized binding partners may affect the molecular dynamics, thereby artificially altering the binding event.
MST is a unique physical principle of thermophoresis dependent technique to quantify the interaction between biomolecules [67]. Laudably, the sample consumption during the process is much smaller and time saving than previously mentioned methods. Moreover, MST used a label-free method which can avoid anthropogenic influence and increase authenticity [68, 69]. In a previous study, the MST technology has been successfully used to detect the affinity of p-PROTAC with gradient dilutions of purified CREPT proteins (Kd ≈ 0.34 μM ± 0.11) [18].
Co-IP has recently become one of the most popular assays in PPI research field, and it is often combined with previously mentioned techniques to determine whether two target proteins are bound in vivo [70-72]. In the Co-IP experiment, the assay begins with the preparation of cell or tissue lysate in an appropriate lysis buffer, and then the POI in the lysate is captured using a specific antibody and precipitated along with its binding proteins with resin [73]. It is worth noting that Co-IP cannot be used to detect relatively weak affinity or transient PPI, and the wrong choices of target protein can also lead to the failure of this assay.
Cell membrane permeability
The cell membrane permeability of fluorescein-labeled p-PROTAC can be assessed using flow cytometry and confocal microscopy. For instance, the fluorescence intensity could be measured to quantify the p-PROTAC within cell membrane using flow cytometry. The specific subcellular localization of FITC or carboxytetramethylrhod-amine (TAMRA) labeled p-PROTAC can be further visualized through co-localization with confocal microscopy [26, 31].
Bioactivities
In the previous studies, the bioactivities of p-PROTAC were mainly evaluated in the molecular and cellular levels. In the molecular level, western blotting (WB) and quantitative polymerase chain reaction (qPCR) are often used to access the degradation ability of p-PROTAC on POI and related signaling pathway. For example, WB analysis confirmed the degradation of ERα in T47D cells and Tau degradation in multiple cell lines after p-PROTACs treatment, respectively [26, 31]. In addition, the mRNA level of pS2, a gene transcriptionally regulated by ERα, was further measured with qPCR analysis for the subsequent downstream effects [31]. On the cellular level, the cell viability, cell proliferation, and cell apoptosis are commonly tested to evaluate the efficacy of p-PROTAC [31].
Ubiquitin-proteasome system-dependent degradation
Once the p-PROTAC ternary complex is successfully formed, ubiquitin proteins will be recruited to POI to initiate the degradation of the pathogenic proteins through the UPS. MG132, the proteasome inhibitor, is often used to verify the specificity of the UPS-dependent degradation caused by p-PROTAC. The previously reported p-PROTACs have exhibited the ability to reduce the expression of POI, such as ERα [31], Tau protein [26], and CREPT [18], which can be antagonized by MG132. Moreover, VHL, an E3 ligase ligand, is also used to verify the specificity of VHL-recruitment in p-PROTAC strategy through preventing recruitment of E3 ligase. The most typical example is that c-Met levels could be rescued after treatment with 50-fold excess free VHL ligand following the PROTAC-7 treatment [78]. These functional studies suggested that the degradation through ubiquitin proteasome is the main mechanism of p-PROTAC.
Application of p-PROTAC
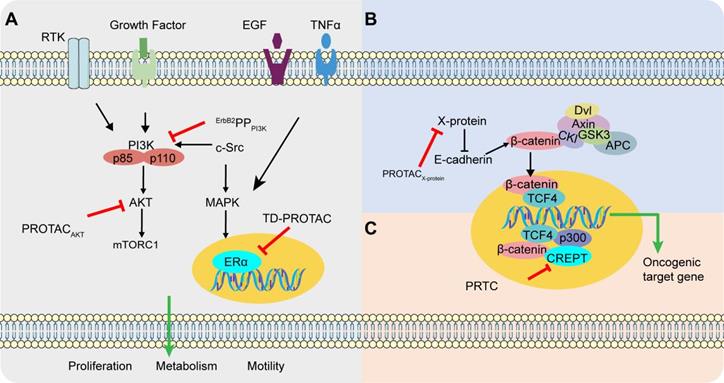
The interface of most protein-protein complexes is usually hydrophobic, relatively flat, and lacking deep docking pockets [79]. Compared with small molecules, peptide-based modulators exhibit greater potential to regulate PPI because of their large contact surface area and easy modification [40, 80]. Therefore, p-PROTAC is a more attracting strategy to eliminate the intracellular pathogenic proteins through UPS dependent degradation cascade. Several p-PROTACs have already been successfully used in some cancers and neurodegenerative disease with the specific POIs, including ERα, PI3K, AKT, CREPT, X-protein, Tau-protein, and FRS2α, which are summarized in Table 3. The main signaling pathways targeted by p-PROTACs for tumor treatment are illustrated in Figure 3.
Successful application of p-PROTACs
| Name [Ref.] | Sequence | Target | Warhead | E3 ligase | Linker | Cancer |
|---|---|---|---|---|---|---|
| PROTACAKT [33] | 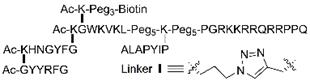 | Akt | tri_a | VHL | PEG5 | Ovarian cancer |
| TD-PROTAC [31] | 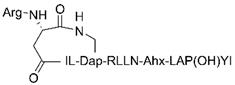 | ERα | PERMs | VHL | Ahx | Breast cancer |
| ErbB2PPPI3K [49] | GPGGDYAAMGACPASEQGYEEMRA-PEG3-ALAPYIP-(D-R)8 | PI3K | ErbB2 peptide | VHL | PEG3 | Breast cancer; Ovarian cancer |
| PRTC [18] | KRRRR-VRALKQKYEELKKEK; ESLVDK-Ahx-LAP(OH)YI. | CREPT | CREPT ligand | VHL | Ahx | Pancreatic cancer |
| PROTACX-protein [48] | (D-R)8-LCLRPVGAESRGRPVS; GPFG-GMLAPYIPM. | X-protein | oligomerization peptide | VHL | − | HBV-induced HCC |
| TrKAPPFRS2α [49] | IENPQYFSDA-Ahx2-ALAPYIP-(D-R)8 | FRS2α | TrKA phosphorylation site | VHL | Ahx2 | Neuroma (PC12) |
| PROTAC Tau-protein [26] | YQQYQDATADEQG-GSGS-LDPETGEYL-(D-R)8 | Tau-protein | Sequence targeting Tau | Keap1 | GSGS | Neurodegenerative disease |
Different pathways in cancer targeted by p-PROTAC. (A) ErbB2PPPI3K and PROTACAKT inhibit breast tumor growth and promote apoptosis through the classic PI3K/AKT signaling pathway. c-Src promotes the transcription of ERα in the nucleus through the MAPK signaling pathway. The degradation of ERα by TD-PROTAC inhibits the transcription of downstream target genes and mediates the killing effect on ERα-positive breast cancer. (B) Reduced expression of E-cadherin is closely related to disruption of cell-cell contacts and enhanced cancer cell invasion. When E-cadherin expression is suppressed, β-catenin is released and translocated to the nucleus, which can be antagonized by PROTACX-protein. (C) PRTC proficiently inhibits cell proliferation and motility in pancreatic by inhibiting transcription initiation upon Wnt signaling as well as downstream target gene cyclin D1 thus inducing cycle arrest and DNA damage.
Breast cancer
The ErbB2/ErbB3/PI3K signal transduction pathway plays an important role in mitosis and inhibition of apoptosis [49]. Overexpression of ErbB2 in breast and ovarian cancers leads to the formation and phosphorylation of heterodimers, thereby activating the downstream signal transduction [81, 82]. In addition, the ErbB3 phosphorylation recruits the lipid kinase PI3K through the SH2 domain of the PI3K regulatory subunit. And the PI3K activation stimulates the survival-promoting kinase Akt (protein kinase B), which inactivates the pro-apoptotic mechanism in tumor cells [49]. The successful inhibition of ErbB3/PI3K signaling pathway is emerging as a promising anti-tumor strategy. Recently, a PI3K-targeted phosphor-PROTAC (ErbB2PPPI3K) has been successfully fabricated to degrade PI3K in breast cancer cell lines MDA-MB-175 and MDA-MB-231 through the inherent affinity of peptide targeting warheads with SH2 domain of PI3K [65], as the first reported p-PROTAC with confirmed in vivo activity and no acquired drug-resistant mutations compared with erlotinib and gefitinib [49, 83].
ERα is often overexpressed in breast cancer cells, and promotes estrogen-dependent cell proliferation [84]. As the classic methods for inhibiting ERα, modulation of the conformational state of ERα with various unnatural ligands often causes drug resistance in breast cancer patients [85-87]. In the previous reported PROTAC, Zhao et al. [30] used the N-terminal aspartic acid to promote formation of helical structures to enhance the stability and cell permeability, which helped the TD strategy-based PROTAC to form a complex with E3 ubiquitin ligase for effective degradation of ERα and inhibition of estrogen-positive breast cancer cell [31]. It is worth noting that TD strategy-based p-PTOTAC targeted the different sites of the POI compared with the small molecules PROTAC, and this study proved the successful application of stabilized peptides based PROTAC and its prospects.
Ovarian cancer
When investigating the effect of p-PROTAC ErbB2PPPI3K, authors found that ErbB2PPPI3K could knock out PI3K in two separate ovarian cancer cell lines OVCAR8 and SKOV3 in vitro and inhibit tumor growth in OVCAR8 xenograft mouse model [49]. In addition, compared with small molecule inhibitor of PI3K (LY294002), the p-PROTAC at dose of 10 mg/kg/day (maximum tolerated dose, MTD) through intraperitoneal injection exhibited lower toxicity [49]. In another study, a p-PROTAC (CPP-tria-PR) was prepared to degrade Akt protein in vitro which is also closely related with the occurrence of ovarian cancer [33].
HBV-induced hepatocellular carcinoma (HCC)
The X-protein of hepatitis B virus (HBV) is essential for viral infection and promotes HBV-induced HCC [88-90]. A p-PROTAC has been successfully fabricated with an oligomerization peptide, to recruite the E3 ubiquitin ligase for UPS-dependent degradation of X-protein [48].
Pancreatic cancer
Associated with RNA polymerase II which could induce chromatin loops' formation and activation thus promoting cyclin D1 transcription [91], CREPT is identified as a novel oncogene which is highly expressed in pancreatic cancer [18, 86, 87, 91]. Based on this, a cell-permeable p-PROTAC (PRTC), using a peptide binding key in the CCT domain, was fabricated to degrade the CREPT in pancreatic cancer cells [18]. These results showed that PRTC is effective in degradation of CREPT proteins, as well as the inhibition effect of tumorigenesis in the tumor bearing xenograft mice without obvious side-effects [18].
Neurodegenerative diseases
Microtubule-associated protein Tau is clearly clustered in neurodegenerative diseases, which is an ideal target for the treatment of neurodegenerative diseases [92-94]. Potential anti-Tau therapies, including kinase inhibition, inhibition of Tau aggregation, or microtubules stabilization, have been discontinued because of the toxicity and/or lack of efficacy [95]. In the previous study, a p-PROTAC composed of a peptide from β-tubulin recruiting Tau and a peptide as a strong binder of Keap1 through a short linker has been fabricated. By recruiting Tau to the Keap1-Cul3 ubiquitin ligase complex, the protein Tau was finally degraded through proteasome in several Tau over-expressed cell lines such as SH-SY5Y, Neuro-2a, and PC-12 cells in a concentration-dependent and time-dependent manner [26]. Unfortunately, the blood-brain barrier (BBB) permeability of this p-PROTAC was not evaluated.
Future development
PROTAC technology has achieved notable advances in drug discovery, and several PROTAC molecules are currently in clinical trials [96]. There are still many challenges for the future development of PROTAC, including off-target toxicity caused by non-specific degradation of POI in normal cells, major obstacle of pharmacokinetic (PK) evaluation for PROTAC, limited choices of E3 ligases for PROTAC design, and restricted applications for only intracellular proteins but not extracellular proteins, including membrane proteins and secreted proteins. In addition, as one vital form of PROTAC, p-PROTAC further broadens the applicable targets in “undruggable proteins” with potential high affinity (Table 1) [3, 25, 31]. Nevertheless, p-PROTACs also have their own limitations, including poor structural stability, easy degradation, and poor transmembrane capacity. To fully address those shortcomings, we propose the following possible solutions as shown in Table 4, which may be used to tackle the weakness of p-PROTACs thus forwarding their potential clinical translation.
Strategies used to improve p-PROTACs
| CPPs [26, 48, 49, 66] | Constrained conformation [31] | Target delivery | |
|---|---|---|---|
| Tools | TAT; poly-D-arginine; Xentry | α-helical conformation of peptides | Nanocarriers |
| Application | PROTACTau-protein; TrkAPPFRS2α; ErbB2PPPI3K; PROTACX-protein; PRTC | TD-PROTAC | − |
| Advantages | Increased permeability | Increased permeability and stability | Precise treatment |
Utilization of cell-penetrating peptides
The cell-penetrating peptides (CPPs), including HIV-1 TAT peptide (YGRKKRRQRRR) and poly-D-arginine (RRRRRRRR), have already been used to effectively penetrate cell membrane through endocytosis or direct penetration for delivery of many types of cargo, including peptides, proteins, oligonucleotides, and drug molecules. Studies confirmed that these CPPs can also be used for p-PROTACs [49, 97-99]. In previous study, two different CPP, poly-D-arginine and TAT peptide, have been used to enhance the Keap1-dependent p-PROTACs for degradation of Tau-protein [26]. Through the comparison of EGFP-labelled series of modified p-PROTAC, the poly-D-arginine conjugated p-PROTAC possess the best membrane permeability in SH-SY5Y cells. The similar result can be seen in the poly-D-arginine modified phosphoPROTAC TrkAPPFRS2α and ErbB2PPPI3K with increased inhibitory effects on cells [49]. In addition, a new short CPP-Xentry (LCLRPVG), an N-terminal region of the X-protein from hepatitis B virus, has been successfully used in the synthesis of p-PROTAC for X-protein [100].
Constrained conformation
Besides the cell membrane penetration, there are still some other barriers for improvement of p-PROTAC drug-like characteristics [101]. One vital issue is the retention time of the protein epitope mimetic conformation, especially for the short peptides. It is conformational flexibility that causes poor stability and cell permeability of peptides, thereby making intracellular targets inaccessible by originally fragile and hydrophilic peptides [30]. Hence, conformational constraint of peptides with unnatural backbone structure provides promising method to solve the previous problems [102-103]. Illuminated by nucleation strategies, Li et al. [30] introduced unnatural amino acid 2,3-diaminopropionic acid into PERMs motif backbone to lock its helical conformation. TD strategy-based p-PROTAC (TD-PROTAC) exhibited higher helicity, binding affinities, better stability, and cell permeability than linear PROTACs [31]. Another widely applied conformation-constrained strategy for peptide is all hydrocarbon stapled peptide technology developed by Verdine et al [104]. This strategy has successfully pushed mimetic peptide-ALRN-6924 as p53-MDM2/MDMX inhibitor into Phase II trial (Clinical Trials.gov Identifier: NCT02264613). Our group has also previously used stapled peptides as PPI inhibitor to regulate Axin-β-catenin interaction with better structural stability, protease resistance and stronger efficacy of Axin (469-482) than linear peptides [102]. On this basis, we subsequently reported a new p-PROTAC with stapled technology (xStAx-VHLL) that effectively inhibited Wnt-dependent intestinal cancer in mice and the survival of colorectal cancer patient-derived organoids through degradation of β-catenin [105]. These findings strongly corroborate that constrained conformation exhibits the ability to promote the drug-like property of p-PROTAC.
Targeted delivery
Another key issue is non-specific degradation of POI in normal cells. In this pursuit, the safe biodegradable delivery systems should be proposed for the target delivery of p-PROTAC, such as nanocarriers which have been used to deliver protein or peptide drugs [106-107]. While protecting the stability of cargoes, nanomedicine can significantly increase the bioavailability, circulation time, and tissue-specific targeting through surface modification, which can further improve the efficacy [108]. For example, PEG conjugated nanoparticle can escape from monocyte phagocytic system uptake, prolonging blood residence time and circulation [109-110]. It is also well known that tumor tissues often have enhanced permeability and retention effect (EPR) due to the uncontrolled angiogenesis process and leaky vascular structure [111], and numerous evidences proved the successful utilization of EPR effects for nanomedicine accumulation in tumor sites [112]. These findings suggest that the combination of nanotechnology with p-PROTAC strategy may help to solve the difficulties in targeted delivery and specific accumulation of p-PROTAC in tumor sites. For instance, Aronson et al [111] loaded a cytotoxic anti-cancer peptide into the lipid nanoparticles, and this DDS successfully facilitated the selective damage in cancer cells while avoiding off-target effects in normal tissues. Similarly, a serum albumin-coated boehmite nano-DDS for bee venom peptide melittin has also been used to reduce the hemolysis and enhance the cytotoxic effects compared to the free drug [114].
Conclusion and perspective
The booming p-PROTAC technology offers superior advantages over traditional peptide drugs with auxiliary action mechanism and synergetic therapeutic effect. It also provides great opportunity to drive the further development of existing peptides, especially for PPI inhibitors. At the same time, increasing crystal structure identifications of pathogenic proteins help to accelerate the discovery of specific peptides, making “undruggable” targets potentially accessible by mimetic peptides. So far, a series of studies have proved the successful application of p-PROTAC for POI degradation through ubiquitination.
It is worth noting that, there are still many challenges and limitations for p-PROTACs which could be partially solved through interdisciplinary collaboration including structure biology, chemistry, nanotechnology, and pharmacology. For example, with the emergence of conformational constraint strategy, stabilized peptide-based PROTAC exhibited improved activities, stability, and cell permeability compared to the linear ones. Moreover, we expect that multifunctional drug delivery systems (DDS) can be regarded as novel approaches to improve therapeutic utility of p-PROTAC or achieve combination therapy. Meanwhile, the development of novel preclinical tumor models, including patient-derived organoids (PDO) and patient-derived xenografts (PDX), provides tools to better evaluate the efficacy and side effects of p-PROTACs [105, 115]. However, the compositional complexity of p-PROTACs makes it being drug-like more challenge than simple peptides drugs, including the increased technical difficulties in evaluation of drug pharmacokinetics, stability, safety, and especially the unique catalytic cycle manner for p-PROTACs. To better forwarding the future clinical translation of p-PROTACs, experiences learned from the encouraging clinical trials of small molecule PROTACs, including ARV-471 and ARV-110, should be valued.
In general, p-PROTAC technology provides an attractive platform to obtain leading candidates for potential translational research. We expect that the emerging p-PRTOACs will become leading PROTAC modality in the advancement of novel drug discovery.
Abbreviations
PROTAC: PROteolysis TArgeting Chimera; p-PROTAC: peptide PROTAC; POI: protein of interest; CLL: chronic lymphocytic leukemia; BTK: Bruton's tyrosine kinase; UPS: ubiquitin-proteasome system; ER: estrogen receptor; AR: androgen receptor; BRD4: bromodomain-containing protein 4; ALK: anaplastic lymphoma kinase; CDK9: cyclin dependent kinase 9; CREPT: cycle-related and expression-elevated protein; PPI: protein-protein interaction; PERMs: peptidomimetic estrogen receptor modulators; TD: terminal aspartic acid; VHL: Von Hippel-Lindau; CRBN: cereblon; SCFβ-TRCP: Skp1-Cullin-F boxβ-TRCP; HIF-1α: hypoxia-inducible factor-1α; Ahx: aminohexanoic acid; PEG: polyethylene glycol; TD strategy: N-terminal aspartic acid cross-linking strategy; FP: fluorescence polarization; ITC: isothermal titration calorimetry; SPR: surface plasmon resonance; MST: microscale thermophoresis; Co-IP: co-immunoprecipitation; FITC: fluorescein isothiocyanate; ΔH°: molar binding enthalpy; ΔS°: binding reaction entropy; TAMRA: carboxytetramethylrhod-amine; WB: western blotting; qPCR: quantitative polymerase chain reaction; ErbB2PPPI3K: PI3K-targeted phosphor-PROTAC; MTD: maximum tolerated dose; HCC: hepatocellular carcinoma; HBV: hepatitis B virus; BBB: blood-brain barrier; CPPs: cell-penetrating peptides; TD-PROTAC: TD strategy-based p-PROTAC; EPR: enhanced permeability and retention effect; DDS: drug delivery systems; PDO: patient-derived organoids; PDX: patient-derived xenografts.
Acknowledgements
Funding
This work was supported by funds from the National Natural Science Foundation of China (No. 81903654), Program for Professor of Special Appointment (Young Eastern Scholar) at Shanghai Institutions of Higher Learning (QD2018035), “Chenguang Program” of Education Commission of Shanghai Municipality (18CG46), and Shanghai Sailing Program (19YF1449400), Shanghai Engineering Research Center for the Preparation of Bioactive Natural Products (16DZ2280200), the National Key Research and Development Program of China (2017YFC1700200), National Key Subject of Drug Innovation (2019ZX09201005-007), National key R & D program for key research project of modernization of traditional Chinese medicine (2019YFC1711602), National Science and Technology Major Project of China (2019ZX09201004-003-010).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Salami J, Crews CM. Waste disposal-An attractive strategy for cancer therapy. Science. 2017;355:1163-7
2. Buhimschi AD, Armstrong HA, Toure M, Jaime-Figueroa S, Chen TL, Lehman AM. et al. Targeting the C481S Ibrutinib-Resistance Mutation in Bruton's Tyrosine Kinase Using PROTAC-Mediated Degradation. Biochemistry. 2018;57:3564-75
3. Lai AC, Crews CM. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov. 2017;16:101-14
4. Cromm PM, Crews CM. Targeted Protein Degradation: from Chemical Biology to Drug Discovery. Cell Chem Biol. 2017;24:1181-90
5. Neklesa TK, Winkler JD, Crews CM. Targeted Protein Degradation by PROTACs. Pharmacol Ther. 2017;174:138-44
6. Burslem GM, Crews CM. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell. 2020;181:102-14
7. Hu J, Hu B, Wang M, Xu F, Miao B, Yang C. et al. Discovery of ERD-308 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Estrogen Receptor (ER). J Med Chem. 2019;62:1420-42
8. Han X, Wang C, Qin C, Xiang W, Fernandez-Salas E, Yang C. et al. Discovery of ARD-69 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Androgen Receptor (AR) for the treatment of prostate cancer. J Med Chem. 2019;62:941-64
9. Zhang F, Wu Z, Chen P, Zhang J, Wang T, Zhou J. et al. Discovery of a new class of PROTAC BRD4 degraders based on a dihydroquinazolinone derivative and lenalidomide/pomalidomide. Bioorg Med Chem. 2020;28:115228
10. Raina K, Lu J, Qian Y, Altieri M, Gordon D, Rossi AM. et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc Natl Acad Sci U S A. 2016;113:7124-9
11. Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S. et al. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348:1376-81
12. Zhang C, Han XR, Yang X, Jiang B, Liu J, Xiong Y. et al. Proteolysis Targeting Chimeras (PROTACs) of Anaplastic Lymphoma Kinase (ALK). Eur J Med Chem. 2018;151:304-14
13. Kang CH, Lee DH, Lee CO, Du Ha J, Park CH, Hwang JY. Induced protein degradation of anaplastic lymphoma kinase (ALK) by proteolysis targeting chimera (PROTAC). Biochem Biophys Res Commun. 2018;505:542-7
14. Cromm PM, Samarasinghe K, Hines J, Crews CM. Addressing Kinase-Independent Functions of Fak via PROTAC-Mediated Degradation. J Am Chem Soc. 2018;140:17019-26
15. Olson CM, Jiang B, Erb MA, Liang Y, Doctor ZM, Zhang Z. et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat Chem Biol. 2018;14:163-70
16. Burslem GM, Schultz AR, Bondeson DP, Eide CA, Savage SS, Druker BJ. et al. Targeting BCR-ABL1 in Chronic Myeloid Leukemia by PROTAC-Mediated Targeted Protein Degradation. Cancer Res. 2019;79:4744-53
17. Lai AC, Toure M, Hellerschmied D, Salami J, Jaime-Figueroa S, Ko E. et al. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew Chem Int Ed Engl. 2016;55:807-10
18. Ma D, Zou Y, Chu Y, Liu Z, Liu G, Chu J. et al. A cell-permeable peptide-based PROTAC against the oncoprotein CREPT proficiently inhibits pancreatic cancer. Theranostics. 2020;10:3708-21
19. Proof-of-Concept with PROTACs in Prostate Cancer. Cancer Discov. 2020; doi: 10.1158/2159-8290.CD-NB2020-054. Epub ahead of print.
20. Burslem GM, Crews CM. Small-Molecule Modulation of Protein Homeostasis. Chem Rev. 2017;117:11269-301
21. Pelay-Gimeno M, Glas A, Koch O, Grossmann TN. Structure-Based Design of Inhibitors of Protein-Protein Interactions: Mimicking Peptide Binding Epitopes. Angew Chem Int Ed Engl. 2015;54:8896-927
22. Milroy LG, Grossmann TN, Hennig S, Brunsveld L, Ottmann C. Modulators of protein-protein interactions. Chem Rev. 2014;114:4695-748
23. Lazo JS, Sharlow ER. Drugging Undruggable Molecular Cancer Targets. Annu Rev Pharmacol Toxicol. 2016;56:23-40
24. Kaspar AA, Reichert JM. Future directions for peptide therapeutics development. Drug Discov Today. 2013;18:807-17
25. Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2001;98:8554-9
26. Toure M, Crews CM. Small-Molecule PROTACS: New Approaches to Protein Degradation. Angew Chem Int Ed Engl. 2016;55:1966-73
27. Lu M, Liu T, Jiao Q, Ji J, Tao M, Liu Y. et al. Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiquitination-proteasome degradation pathway. Eur J Med Chem. 2018;146:251-9
28. Huang HT, Dobrovolsky D, Paulk J, Yang G, Weisberg EL, Doctor ZM. et al. A Chemoproteomic Approach to Query the Degradable Kinome Using a Multi-kinase Degrader. Cell Chem Biol. 2018;25:88-99
29. Leduc AM, Trent JO, Wittliff JL, Bramlett KS, Briggs SL, Chirgadze NY. et al. Helix-stabilized cyclic peptides as selective inhibitors of steroid receptor-coactivator interactions. Proc Natl Acad Sci U S A. 2003;100:11273-8
30. Zhao H, Liu QS, Geng H, Tian Y, Cheng M, Jiang YH. et al. Crosslinked Aspartic Acids as Helix-Nucleating Templates. Angew Chem Int Ed Engl. 2016;55:12088-93
31. Jiang Y, Deng Q, Zhao H, Xie M, Chen L, Yin F. et al. Development of Stabilized Peptide-Based PROTACs against Estrogen Receptor α. ACS Chem Biol. 2018;13:628-35
32. Crew AP, Raina K, Dong H, Qian Y, Wang J, Vigil D. et al. Identification and Characterization of Von Hippel-Lindau-Recruiting Proteolysis Targeting Chimeras (PROTACs) of TANK-Binding Kinase 1. J Med Chem. 2018;61:583-98
33. Henning RK, Varghese JO, Das S, Nag A, Tang G, Tang K. et al. Degradation of Akt using protein-catalyzed capture agents. J Pept Sci. 2016;22:196-200
34. Chang HN, Liu BY, Qi YK, Zhou Y, Chen YP, Pan KM. et al. Blocking of the PD-1/PD-L1 Interaction by a D-Peptide Antagonist for Cancer Immunotherapy. Angew Chem Int Ed Engl. 2015;54:11760-4
35. Hershko A, Ciechanover A. The ubiquitin system for protein degradation. Annu Rev Biochem. 1992;61:761-807
36. Hershko A, Heller H, Elias S, Ciechanover A. Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J Biol Chem. 1983;258:8206-14
37. Lucas X, Ciulli A. Recognition of substrate degrons by E3 ubiquitin ligases and modulation by small-molecule mimicry strategies. Curr Opin Struct Biol. 2017;44:101-10
38. Paiva SL, Crews CM. Targeted protein degradation: elements of PROTAC design. Curr Opin Chem Biol. 2019;50:111-9
39. Bondeson DP, Smith BE, Burslem GM, Buhimschi AD, Hines J, Jaime-Figueroa S. et al. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem Biol. 2018;25:78-87
40. Hu K, Yin F, Yu M, Sun C, Li J, Liang Y. et al. In-Tether Chiral Center Induced Helical Peptide Modulators Target p53-MDM2/MDMX and Inhibit Tumor Growth in Stem-Like Cancer Cell. Theranostics. 2017;7:4566-76
41. Bondeson DP, Mares A, Smith IE, Ko E, Campos S, Miah AH. et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol. 2015;11:611-7
42. Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K. et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem Biol. 2015;22:755-63
43. Zengerle M, Chan KH, Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem Biol. 2015;10:1770-7
44. Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M. et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464-8
45. Schneekloth JJ, Fonseca FN, Koldobskiy M, Mandal A, Deshaies R, Sakamoto K. et al. Chemical genetic control of protein levels: selective in vivo targeted degradation. J Am Chem Soc. 2004;126:3748-54
46. Hon WC, Wilson MI, Harlos K, Claridge TD, Schofield CJ, Pugh CW. et al. Structural basis for the recognition of hydroxyproline in HIF-1 alpha by pVHL. Nature. 2002;417:975-8
47. Bargagna-Mohan P, Baek SH, Lee H, Kim K, Mohan R. Use of PROTACS as molecular probes of angiogenesis. Bioorg Med Chem Lett. 2005;15:2724-7
48. Montrose K, Krissansen GW. Design of a PROTAC that antagonizes and destroys the cancer-forming X-protein of the hepatitis B virus. Biochem Biophys Res Commun. 2014;453:735-40
49. Hines J, Gough JD, Corson TW, Crews CM. Posttranslational protein knockdown coupled to receptor tyrosine kinase activation with phosphoPROTACs. Proc Natl Acad Sci U S A. 2013;110:8942-7
50. Philip P Chamberlain, Brian E Cathers. Cereblon modulators: Low molecular weight inducers of protein degradation. Drug Discov Today Technol. 2019;31:29-34
51. Khan S, Zhang X, Lv D, Zhang Q, He Y, Zhang P. et al. A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nat Med. 2019;25:1938-47
52. Smith BE, Wang SL, Jaime-Figueroa S, Harbin A, Wang J, Hamman BD. et al. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat Commun. 2019;10:131
53. Rodriguez-Gonzalez A, Cyrus K, Salcius M, Kim K, Crews CM, Deshaies RJ. et al. Targeting steroid hormone receptors for ubiquitination and degradation in breast and prostate cancer. Oncogene. 2008;27:7201-11
54. Cyrus K, Wehenkel M, Choi EY, Han HJ, Lee H, Swanson H. et al. Impact of linker length on the activity of PROTACs. Mol Biosyst. 2011;7:359-64
55. Cyrus K, Wehenkel M, Choi EY, Lee H, Swanson H, Kim KB. Jostling for position: optimizing linker location in the design of estrogen receptor-targeting PROTACs. ChemMedChem. 2010;5:979-85
56. Redman JE, Ghadiri MR. Synthesis of photoactive p-azidotetrafluorophenylalanine containing peptide by solid-phase Fmoc methodology. Org Lett. 2002;4:4467-9
57. Gao S, Guo Y, Li H, Fang G. Chemical synthesis and applications of stapled peptides. Prog Chem. 2014;26:100-9
58. Eskandari S, Guerin T, Toth I, Stephenson RJ. Recent advances in self-assembled peptides: Implications for targeted drug delivery and vaccine engineering. Adv Drug Deliv Rev. 2017;110-111:169-87
59. Sakamoto KM, Kim KB, Verma R, Ransick A, Stein B, Crews CM. et al. Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol Cell Proteomics. 2003;2:1350-8
60. Cronican JJ, Thompson DB, Beier KT, McNaughton BR, Cepko CL, Liu DR. Potent delivery of functional proteins into Mammalian cells in vitro and in vivo using a supercharged protein. ACS Chem Biol. 2010;5:747-52
61. McNaughton BR, Cronican JJ, Thompson DB, Liu DR. Mammalian cell penetration, siRNA transfection, and DNA transfection by supercharged proteins. Proc Natl Acad Sci U S A. 2009;106:6111-6
62. Marlowe T, Alvarado C, Rivera A, Lenzo F, Nott R, Bondugji D. et al. Development of a High-Throughput Fluorescence Polarization Assay to Detect Inhibitors of the FAK-Paxillin Interaction. Slas Discov. 2020;25:21-32
63. Zhou W, Li Y, Song J, Li C. Fluorescence polarization assay for the identification and evaluation of inhibitors at YAP-TEAD protein-protein interface 3. Anal Biochem. 2019;586:113413
64. Raines RT. Fluorescence polarization assay to quantify protein-protein interactions: an update. Methods Mol Biol. 2015;1278:323-7
65. Baranauskiene L, Kuo TC, Chen WY, Matulis D. Isothermal titration calorimetry for characterization of recombinant proteins. Curr Opin Biotechnol. 2019;55:9-15
66. Olaru A, Bala C, Jaffrezic-Renault N, Aboul-Enein HY. Surface plasmon resonance (SPR) biosensors in pharmaceutical analysis. Crit Rev Anal Chem. 2015;45:97-105
67. Jerabek-Willemsen M, Wienken CJ, Braun D, Baaske P, Duhr S. Molecular interaction studies using microscale thermophoresis. Assay Drug Dev Technol. 2011;9:342-53
68. Seidel SA, Dijkman PM, Lea WA, van den Bogaart G, Jerabek-Willemsen M, Lazic A. et al. Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods. 2013;59:301-15
69. Wienken CJ, Baaske P, Rothbauer U, Braun D, Duhr S. Protein-binding assays in biological liquids using microscale thermophoresis. Nat Commun. 2010;1:100
70. Lin JS, Lai EM. Protein-Protein Interactions: Co-Immunoprecipitation. Methods Mol Biol. 2017;1615:211-9
71. Li X, Zhong L, Wang Z, Chen H, Liao D, Zhang R. et al. Phosphorylation of IRS4 by CK1gamma2 promotes its degradation by CHIP through the ubiquitin/lysosome pathway. Theranostics. 2018;8:3643-53
72. Huang Y, Hu K, Zhang S, Dong X, Yin Z, Meng R. et al. S6K1 phosphorylation-dependent degradation of Mxi1 by beta-Trcp ubiquitin ligase promotes Myc activation and radioresistance in lung cancer. Theranostics. 2018;8:1286-300
73. Lee C. Coimmunoprecipitation assay. Methods Mol Biol. 2007;362:401-6
74. Zhao C, Zhang L, Ni Y. Development of Fluorescence PolarizationinLife Sciences. Progress in Modern Biomedicine. 2010;10:3154-6
75. Ward WH, Holdgate GA. Isothermal titration calorimetry in drug discovery. Prog Med Chem. 2001;38:309-76
76. Piliarik M, Vaisocherova H, Homola J. Surface plasmon resonance biosensing. Methods Mol Biol. 2009;503:65-88
77. Tang Z, Takahashi Y. Analysis of Protein-Protein Interaction by Co-IP in Human Cells. Methods Mol Biol. 2018;1794:289-96
78. Burslem GM, Smith BE, Lai AC, Jaime-Figueroa S, McQuaid DC, Bondeson DP. et al. The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chem Biol. 2018;25:67-77
79. Tsomaia N. Peptide therapeutics: targeting the undruggable space. Eur J Med Chem. 2015;94:459-70
80. Ivanov AA, Khuri FR, Fu H. Targeting protein-protein interactions as an anticancer strategy. Trends Pharmacol Sci. 2013;34:393-400
81. Stern DF. ERBB3/HER3 and ERBB2/HER2 duet in mammary development and breast cancer. J Mammary Gland Biol Neoplasia. 2008;13:215-23
82. Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CR, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci U S A. 2003;100:8933-8
83. Jiao Q, Bi L, Ren Y, Song S, Wang Q, Wang YS. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol Cancer. 2018;17:36
84. Holst F, Stahl PR, Ruiz C, Hellwinkel O, Jehan Z, Wendland M. et al. Estrogen receptor alpha (ESR1) gene amplification is frequent in breast cancer. Nat Genet. 2007;39:655-60
85. Mohseni M, Cidado J, Croessmann S, Cravero K, Cimino-Mathews A, Wong HY. et al. MACROD2 overexpression mediates estrogen independent growth and tamoxifen resistance in breast cancers. Proc Natl Acad Sci U S A. 2014;111:17606-11
86. Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009;9:631-43
87. Peng J, Sengupta S, Jordan VC. Potential of selective estrogen receptor modulators as treatments and preventives of breast cancer. Anticancer Agents Med Chem. 2009;9:481-99
88. Zhang C, Liu P, Zhang C. Hepatitis B virus X protein upregulates alpha-fetoprotein to promote hepatocellular carcinoma by targeting miR-1236 and miR-329. J Cell Biochem. 2020;121:2489-99
89. Yen CJ, Yang ST, Chen RY, Huang W, Chayama K, Lee MH. et al. Hepatitis B virus X protein (HBx) enhances centrosomal P4.1-associated protein (CPAP) expression to promote hepatocarcinogenesis. J Biomed Sci. 2019;26:44
90. Yang Z, Li J, Feng G, Wang Y, Yang G, Liu Y. et al. Hepatitis B virus X protein enhances hepatocarcinogenesis by depressing the targeting of NUSAP1 mRNA by miR-18b. Cancer Biol Med. 2019;16:276-87
91. Lu D, Wu Y, Wang Y, Ren F, Wang D, Su F. et al. CREPT accelerates tumorigenesis by regulating the transcription of cell-cycle-related genes. Cancer Cell. 2012;21:92-104
92. Akoury E, Pickhardt M, Gajda M, Biernat J, Mandelkow E, Zweckstetter M. Mechanistic basis of phenothiazine-driven inhibition of Tau aggregation. Angew Chem Int Ed Engl. 2013;52:3511-5
93. Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S. et al. Tau reduction prevents Abeta-induced defects in axonal transport. Science. 2010;330:198
94. Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8:663-72
95. Congdon EE, Sigurdsson EM. Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol. 2018;14:399-415
96. Wang Y, Jiang X, Feng F, Liu W, Sun H. Degradation of proteins by PROTACs and other strategies. Acta Pharm Sin B. 2020;10:207-38
97. He Y, Li F, Huang Y. Smart Cell-Penetrating Peptide-Based Techniques for Intracellular Delivery of Therapeutic Macromolecules. Adv Protein Chem Struct Biol. 2018;112:183-220
98. Zou L, Peng Q, Wang P, Zhou B. Progress in Research and Application of HIV-1 TAT-Derived Cell-Penetrating Peptide. J Membr Biol. 2017;250:115-22
99. Guidotti G, Brambilla L, Rossi D. Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol Sci. 2017;38:406-24
100. Montrose K, Yang Y, Sun X, Wiles S, Krissansen GW. Xentry, a new class of cell-penetrating peptide uniquely equipped for delivery of drugs. Sci Rep. 2013;3:1661
101. Jochim AL, Arora PS. Systematic analysis of helical protein interfaces reveals targets for synthetic inhibitors. ACS Chem Biol. 2010;5:919-23
102. Wu Y, Li Y, Li X, Zou Y, Liao H, Liu L. et al. A novel peptide stapling strategy enables the retention of ring-closing amino acid side chains for the Wnt/β-catenin signalling pathway. Chem Sci. 2017;8:7368-73
103. Hilinski GJ, Kim YW, Hong J, Kutchukian PS, Crenshaw CM, Berkovitch SS. et al. Stitched α-helical peptides via bis ring-closing metathesis. J Am Chem Soc. 2014;136:12314-22
104. Schafmeister CE, Po J, Verdine GL. An All-Hydrocarbon Cross-Linking System for Enhancing the Helicity and Metabolic Stability of Peptides. J Am Chem Soc. 2000;122:5891-2
105. Liao H, Li X, Zhao L, Wang Y, Wang X, Wu Y. et al. A PROTAC peptide induces durable β-catenin degradation and suppresses Wnt-dependent intestinal cancer. Cell Discov. 2020;6:35
106. Zhao L, Li N, Wang K, Shi C, Zhang L, Luan Y. A review of polypeptide-based polymersomes. Biomaterials. 2014;35:1284-301
107. Urban P, Valle-Delgado JJ, Moles E, Marques J, Diez C, Fernandez-Busquets X. Nanotools for the delivery of antimicrobial peptides. Curr Drug Targets. 2012;13:1158-72
108. Katz E, Willner I. Integrated nanoparticle-biomolecule hybrid systems: synthesis, properties, and applications. Angew Chem Int Ed Engl. 2004;43:6042-108
109. Kajiwara E, Kawano K, Hattori Y, Fukushima M, Hayashi K, Maitani Y. Long-circulating liposome-encapsulated ganciclovir enhances the efficacy of HSV-TK suicide gene therapy. J Control Release. 2007;120:104-10
110. Blume G, Cevc G. Liposomes for the sustained drug release in vivo. Biochim Biophys Acta. 1990;1029:91-7
111. Park J, Choi Y, Chang H, Um W, Ryu JH, Kwon IC. Alliance with EPR Effect: Combined Strategies to Improve the EPR Effect in the Tumor Microenvironment. Theranostics. 2019;9:8073-90
112. Greish K. Enhanced permeability and retention (EPR) effect for anticancer nanomedicine drug targeting. Methods Mol Biol. 2010;624:25-37
113. Aronson MR, Simonson AW, Orchard LM, Llinás M, Medina SH. Lipopeptisomes: Anticancer peptide-assembled particles for fusolytic oncotherapy. Acta Biomater. 2018;80:269-77
114. Liu H, Hu Y, Sun Y, Wan C, Zhang Z, Dai X. et al. Co-delivery of Bee Venom Melittin and a Photosensitizer with an Organic-Inorganic Hybrid Nanocarrier for Photodynamic Therapy and Immunotherapy. ACS Nano. 2019;13:12638-52
115. Lee SH, Hu W, Matulay JT, Silva MV, Owczarek TB, Kim K. et al. Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell. 2018;173:515-28
Authors' Biographic
 Associate Prof. Xin Luan received his B.Sc. in Pharmaceutics from China Pharmaceutical University (2011). He was awarded his Ph.D. degree in Pharmacology at Shanghai Jiao Tong University (2016), under the supervision of Prof. Hong-Zhuan Chen. He subsequently worked for two years at the University of Michigan under the guidance of Prof. Duxin Sun. His current appointment is Associate Professor at the Shanghai University of Traditional Chinese Medicine (SHUTCM). He is currently working on anti-tumor pharmacology and delivery systems of natural products.
Associate Prof. Xin Luan received his B.Sc. in Pharmaceutics from China Pharmaceutical University (2011). He was awarded his Ph.D. degree in Pharmacology at Shanghai Jiao Tong University (2016), under the supervision of Prof. Hong-Zhuan Chen. He subsequently worked for two years at the University of Michigan under the guidance of Prof. Duxin Sun. His current appointment is Associate Professor at the Shanghai University of Traditional Chinese Medicine (SHUTCM). He is currently working on anti-tumor pharmacology and delivery systems of natural products.
 Prof. Wei-Dong Zhang obtained his B.Sc. and Master degree in Natural Medicinal Chemistry from Second Military Medical University in 1988 and 1991, respectively. He received his Ph.D. in Natural Medicinal Chemistry from Shanghai Institute of Pharmaceutical Industry (1998), under the supervision of Professor Hui-Ting Li. He is currently a professor of the SHUTCM and Second Military Medical University. His research mainly focuses on Chinese medicine formula, isolation, structural identification and modification, total synthesis, and structure-activity relationship of bioactive natural products.
Prof. Wei-Dong Zhang obtained his B.Sc. and Master degree in Natural Medicinal Chemistry from Second Military Medical University in 1988 and 1991, respectively. He received his Ph.D. in Natural Medicinal Chemistry from Shanghai Institute of Pharmaceutical Industry (1998), under the supervision of Professor Hui-Ting Li. He is currently a professor of the SHUTCM and Second Military Medical University. His research mainly focuses on Chinese medicine formula, isolation, structural identification and modification, total synthesis, and structure-activity relationship of bioactive natural products.
 Jinmei Jin obtained her Master's degree (2019) in institute of Chinese Materia Medica at SHUTCM. She is currently a Ph.D. student under the supervision of Prof. Hong-Zhuan Chen and Xin Luan. Her current research focuses on the synthesis of peptide PROTAC and related oncotherapy.
Jinmei Jin obtained her Master's degree (2019) in institute of Chinese Materia Medica at SHUTCM. She is currently a Ph.D. student under the supervision of Prof. Hong-Zhuan Chen and Xin Luan. Her current research focuses on the synthesis of peptide PROTAC and related oncotherapy.
 Ye Wu obtained his B.Sc. in Pharmaceutics (2013) and Master degree in Pathology and Pathophysiology (2017) at Chengdu Medical College. He is currently pursuing his Ph.D. studies at SHUTCM, under the supervision of Prof. Wei-Dong Zhang and Xin Luan. His research mainly focuses on the conformational constraint and targeted delivery of natural cytotoxic peptides.
Ye Wu obtained his B.Sc. in Pharmaceutics (2013) and Master degree in Pathology and Pathophysiology (2017) at Chengdu Medical College. He is currently pursuing his Ph.D. studies at SHUTCM, under the supervision of Prof. Wei-Dong Zhang and Xin Luan. His research mainly focuses on the conformational constraint and targeted delivery of natural cytotoxic peptides.
 Jinjiao Chen obtained her bachelor's degree from the School of Life Science and Medicine, Dalian University of Technology in 2015. She is currently studying as a combined training student of Fudan University and SHUTCM, under the guidance of Prof. Xue-Mei Zhang and Xin Luan. Her research focuses on the development of anti-tumor proteolysis targeting chimera compounds.
Jinjiao Chen obtained her bachelor's degree from the School of Life Science and Medicine, Dalian University of Technology in 2015. She is currently studying as a combined training student of Fudan University and SHUTCM, under the guidance of Prof. Xue-Mei Zhang and Xin Luan. Her research focuses on the development of anti-tumor proteolysis targeting chimera compounds.
Author contact
![]() Corresponding authors: E-mail: luanxinedu.cn (X.L.), wdzhangycom (W.D.Z); Tel.: +86(021)-51322720.
Corresponding authors: E-mail: luanxinedu.cn (X.L.), wdzhangycom (W.D.Z); Tel.: +86(021)-51322720.