Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(22):10186-10199. doi:10.7150/thno.46835 This issue Cite
Research Paper
Phorbol ester activates human mesenchymal stem cells to inhibit B cells and ameliorate lupus symptoms in MRL.Faslpr mice
Hong Kyung Lee1*, Hyung Sook Kim1*, Minji Pyo1, Eun Jae Park1, Sundong Jang1, Hye Won Jun1, Tae Yong Lee1,2, Kyung Suk Kim2, Sang-Cheol Bae3, Youngsoo Kim1, Jin Tae Hong1, Jaesuk Yun1 ![]() , Sang-Bae Han1
, Sang-Bae Han1 ![]()
1. College of Pharmacy, Chungbuk National University, Cheongju, Chungbuk 28160, Republic of Korea.
2. Bioengineering Institute, Corestem Inc., Gyeonggi 13486, Republic of Korea.
3. Hanyang University Hospital for Rheumatic Diseases, Seoul 04763, Republic of Korea.
*These authors contributed equally to this study.
Received 2020-4-9; Accepted 2020-8-4; Published 2020-8-13
Abstract

Rationale: Systemic lupus erythematosus (SLE) is a multi-organ autoimmune disease characterized by autoantibody production by hyper-activated B cells. Although mesenchymal stem cells (MSCs) ameliorate lupus symptoms by inhibiting T cells, whether they inhibit B cells has been controversial. Here we address this issue and reveal how to prime MSCs to inhibit B cells and improve the efficacy of MSCs in SLE.
Methods: We examined the effect of MSCs on purified B cells in vitro and the therapeutic efficacy of MSCs in lupus-prone MRL.Faslpr mice. We screened chemicals for their ability to activate MSCs to inhibit B cells.
Results: Mouse bone marrow-derived MSCs inhibited mouse B cells in a CXCL12-dependent manner, whereas human bone marrow-derived MSCs (hMSCs) did not inhibit human B (hB) cells. We used a chemical approach to overcome this hurdle and found that phorbol myristate acetate (PMA), phorbol 12,13-dibutyrate, and ingenol-3-angelate rendered hMSCs capable of inhibiting IgM production by hB cells. As to the mechanism, PMA-primed hMSCs attracted hB cells in a CXCL10-dependent manner and induced hB cell apoptosis in a PD-L1-dependent manner. Finally, we showed that PMA-primed hMSCs were better than naïve hMSCs at ameliorating SLE progression in MRL.Faslpr mice.
Conclusion: Taken together, our data demonstrate that phorbol esters might be good tool compounds to activate MSCs to inhibit B cells and suggest that our chemical approach might allow for improvements in the therapeutic efficacy of hMSCs in SLE.
Keywords: B cell, CXCL10, mesenchymal stem cell, PD-L1, phorbol ester, systemic lupus erythematosus
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by the production of autoantibodies to ubiquitous self-antigens [1]. Although various abnormalities of immune cells have been implicated in SLE pathogenesis, excessive B cell activation seems to play a crucial role [2]. Mesenchymal stem cells (MSCs) are multipotent adult stem cells that have emerged as a promising therapy for the treatment of SLE. Adoptive transfer of MSCs to lupus-prone MRL.Faslpr mice increased their survival and decreased anti-dsDNA antibody level and nephritis [3-9]. In clinical studies, MSCs improved renal functions and decreased autoantibody production [10-15]. As to the underlying mechanisms, MSCs inhibit T cell functions by producing such soluble mediators as IL-10, nitric oxide (NO), tumor growth factor (TGF)-β, prostaglandin E2 (PGE2), and indoleamine 2,3-dioxygenase (IDO) [16-18]. In addition, MSCs suppress T cell functions by CCL2- and Fas-dependent contact inhibition [19, 20]. MSCs also inhibit the functions of dendritic cells, neutrophils, and natural killer cells, but enhance those of regulatory T cells [21-24]. However, whether MSCs inhibit B cells has been controversial. Some studies have shown that MSCs inhibit proliferation, antibody and cytokine production, and migration of B cells [25-28]. But others reported that MSCs cannot inhibit the proliferation of and antibody production by B cells [29, 30], and even enhance B cell functions [23, 31]. Reportedly, MSCs indirectly inhibit B cell proliferation through the inhibition of T cells [29, 32].
Since B cells play a crucial role in SLE progression, it is essential to understand the effect of MSCs on B cells. In this study, we first investigated whether mouse and human MSCs inhibited B cells or not. We found that mouse MSCs (mMSCs) inhibited mouse B (mB) cells, but human MSCs (hMSCs) did not inhibit human B (hB) cells, suggesting a species difference in MSC effects on B cells. Then, we extended the scope of our study to find chemicals that could improve hMSC functions. We found that phorbol esters activated hMSCs to inhibit hB cells and enhanced the therapeutic activity of hMSCs in a SLE mouse model. We suggest here that a chemical approach might be useful to generate clinically useful hMSCs for the treatment of SLE patients.
Materials and Methods
Preparation of MSCs
hMSCs were generated from bone marrow (BM) cells aspirated from the posterior iliac crest of healthy donors. Mononuclear cells were collected by density gradient centrifugation (Ficoll-Paque; GE Healthcare Bio-Sciences AB, Uppsala, Sweden) and were cultured at 2 × 107 cells/T175 flask in CSBM-A06 medium (Corestem Inc., Gyeonggi, Korea) containing 10% fetal bovine serum (BD Biosciences, Franklin Lakes, NJ, USA), 2.5 mM L-glutamine, and penicillin/streptomycin (WelGene, Gyeonggi, Korea) in a 5% CO2 incubator at 37 °C. Medium was changed every 3-4 days and non-adherent cells were removed. Adherent cells were sub-cultured on day 10 or 11 (passage 1). hMSCs were used in experiments at passages 3-5 [33]. The surface marker profile of hMSCs was CD29+CD44+CD73+CD105+CD90+CD34-CD45-HLA-DR- (data not shown). All hMSC studies were approved by the Institutional Review Board of Hanyang University Hospital and were carried out in accordance with the approved guidelines. All participants provided written informed consent.
mMSCs were generated from the BM cells of tibiae and femurs of 6-8-week-old C3H/HeN mice (Orient Bio, Gyeonggi, Korea). Red blood cells were lysed with ACK buffer, and BM cells were cultured at 1 × 107 cells/well of a 6-well plate in α-MEM medium containing 10% fetal bovine serum, 2 mM L-glutamine, and penicillin/streptomycin in a 5% CO2 incubator at 37 °C. Medium was changed every 3 days and non-adherent cells were removed. Adherent cells were sub-cultured on day 10 or 11 (passage 1) and used in experiments at day 17-21 [33]. The surface marker profile of mMSCs was Sca-1+CD44+CD73+CD45-CD11b-CD11c-Gr-1-MHC-II- (data not shown). All animal studies were approved by the Chungbuk National University Animal Experimentation Ethics Committee and were carried out in accordance with the approved guidelines.
Priming of hMSCs with chemicals
hMSCs were treated with phorbol myristate acetate (PMA; 10 ng/mL) for 24 h, washed three times with medium, and used immediately in experiments. We also treated hMSCs with various chemicals for 24 h at the concentrations indicated in Table S1.
Preparation of B cells and mitogen assay
Human peripheral blood mononuclear cells were donated by the Chungbuk Red Cross Blood Center (Cheongju, Korea). Lymphocytes were isolated from these cells by density gradient centrifugation (Ficoll-Paque) [34]. hB cells were isolated from lymphocytes using a human B cell isolation kit (Miltenyi Biotec, Auburn, CA, USA). mB cells were purified from spleen cells of MRL.Faslpr mice by a negative depletion method using mouse B cell isolation kit (Miltenyi Biotec) [9]. Purity of hB and mB cells was typically >90%. B cells (1 × 105 cells/well) and MSCs (0.01-0.1 × 105 cells/well) were added in 200 µL to the wells of 96-well plates. CpG-oligodeoxynucleotide (ODN; 5 μg/mL) was used to activate hB cells, and lipopolysaccharide (LPS; 1 μg/mL) was used to activate mB cells. To measure B cell proliferation, cells were pulsed with [3H]-thymidine (113 Ci/nmol; NEN, Boston, MA, USA) at a concentration of 1 μCi/well for the last 18 h and were harvested on day 3 using an automated cell harvester (Inotech, Dottikon, Switzerland). The amount of [3H]-thymidine incorporated into the cells was measured using a Wallac Microbeta scintillation counter (Wallac, Turku, Finland) [3].
Time-lapse imaging and transwell assay
For time-lapse imaging, dishes were pre-warmed for 24 h in a 5% CO2 incubator at 37 °C. MSCs (70 µL of 0.3 × 106 cells/mL) were seeded into the left chamber and B cells (70 µL of 1 × 106 cells/mL) into the right chamber of culture-insert µ-Dish35mm culture dishes (ibidi GmbH, Martinsried, Germany). Cell-containing dishes were incubated for 3 h under the microscope and then inserts were carefully removed. Time-lapse imaging was performed with a Biostation IM-Q microscope (Nikon, Tokyo, Japan) equipped with a 10× magnification objective (numeric aperture 0.5) and an environmental chamber kept at 37 °C and 5% CO2. Images were acquired every 2 min for 12 h and were analyzed by using Imaris software 9.3.0 (Oxford Instruments plc, Abingdon, UK). The number of migrating B cells was counted [4].
For transwell assay, B cells (1 × 106 cells/mL) were stained with 5-chloromethylfluorescein diacetate (CMFDA; 2 µM) at 37 °C for 15 min and washed three times with serum-free medium. B cells (1 × 105 cells; 100 µL) were added to each upper well of transwell plates with a 5-µm insert (Corning, Corning, NY, USA). Various concentrations of chemokines or MSCs were added to the lower wells in 600 µL of complete RPMI 1640 medium. The number of B cells migrated to the lower well over 1.5 h was counted using a flow cytometer (FACSCalibur; BD Biosciences) [35]. In some experiments, B cells were pre-incubated with the CCR2 antagonist RS102895 (30 µg/mL, Sigma-Aldrich, St. Louis, MO, USA) [36] or CXCL12 antagonist AMD3100 (300 µg/mL, Sigma-Aldrich) [37] for 1 h.
Apoptosis assay
hB cells (1 × 106 cells) and hMSCs (0.1 × 106 cells) were added in 2 mL onto 35-mm culture dishes (BD Biosciences) and cultured for 24 h. hB cell apoptosis was determined in three ways. First, hB cells were stained with anti-CD19 antibody conjugated with APC and then with FITC-annexin and propidium iodide for 15 min (FITC-Annexin V Apoptosis Detection Kit, BD Biosciences). hB cells were analyzed using a flow cytometer (FACSCalibur) and the data were processed using Cell Quest Pro software (BD Biosciences) [38]. Second, hB cells were stained with anti-CD19 antibody conjugated with APC and then labeled with FITC-ApoStat (Intracellular Caspase Detection ApoStat kit, Bio-Techne, Minneapolis, MN, USA) for 1 h. hB cells were analyzed using a flow cytometer (FACSCalibur) and the data were processed using Cell Quest Pro software [39]. Third, CellEvent Caspase-3/7 Green ReadyProbes Reagent (Thermo Fisher Scientific, Carlsbad, CA, USA) was directly added to the co-culture of hB cells and hMSCs [40]. Time-lapse imaging was performed with a Biostation IM-Q microscope (Nikon) and images were acquired in two channels (phase contrast and green filter) every 10 min for 24 h [4]. Images were analyzed by using Imaris software 9.3.0. Green-fluorescent cells were considered apoptotic.
Lupus-prone MRL.Faslpr mouse model
MRL.MpJ-Tnfrsf6Faslpr/J (called MRL.Faslpr hereafter) mice lack Fas and spontaneously develop an SLE-like disease [20]. The onset and symptom severity in these mice depend on their genetic background. Female MRL.Faslpr mice die at an average age of 17 weeks and males at 22 weeks. Similar to SLE patients, MRL.Faslpr mice have a marked increase in anti-dsDNA antibodies in their blood and develop severe nephritis. Female MRL.Faslpr mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Mice were housed in specific pathogen-free conditions at 21-24 °C and 40-60% relative humidity under a 12 h light/dark cycle and randomized into 3 groups. Mice were injected intravenously with PBS (vehicle, n = 6), 4 × 104 naïve hMSCs/mouse (n = 6), or 4 × 104 PMA-hMSCs/mouse (n = 6) once at the age of 12 weeks. Survival rate and body weight were examined every 2 weeks. Serum were collected every 3 weeks and stored at -70 °C until used. The levels of anti-dsDNA IgG and total IgG in serum and the levels of protein in urine were measured by using ELISA kits purchased from Alpha Diagnostic International (San Antonio, TX, USA), eBioscience (San Diego, CA, USA), and Sigma-Aldrich, respectively, according to the manufacturers' instructions.
Immunohistochemistry (IHC)
Kidneys were isolated from the surviving MRL.Faslpr mice at 22 weeks of age, fixed with 4% formalin, and immersed in PBS [5]. After dehydration with ethanol and xylene, the tissues were embedded in paraffin and cut into 4-µm sections. After removing paraffin, sections were hydrated and heated in a microwave oven (650 W, 20 min) for antigen retrieval, after which endogenous peroxidase activity was blocked with 3% hydrogen peroxide. To detect immune cells in the kidney, sections were incubated with the following primary goat antibodies against mouse IgG (diluted 1:100; Jackson ImmunoResearch, West Grove, PA, USA), C3 complement (1:100; GeneTex, San Antonio, TX, USA), CD19 (1:100; BioLegend, San Diego, CA, USA), CD3 (1:100; Santa Cruz Biotechnology, Dallas, TX, USA), F4/80 (1:100; Santa Cruz Biotechnology), CD209b (1:100; Santa Cruz Biotechnology), and Foxp3 (1:50; Abcam, Cambridge, UK) at 4 °C overnight. Then all sections were incubated with secondary antibody (anti-goat IgG conjugated with horseradish peroxidase; Vector Laboratories, Burlingame, CA, USA) for 1 h at room temperature. Signals were developed with a two-component high-sensitivity diaminobenzidine chromogenic substrate (Vector Laboratories) for 10 min and the sections were counter-stained with hematoxylin. Stained area (%) was calculated with ImageJ software (NIH, Bethesda, MD, USA) as follows: IHC stained area (brown staining) / total area (brown + non-brown staining).
RNA interference
Small interfering RNAs (siRNAs) were purchased from Bioneer (Daejon, Korea). Their sequences are shown in Table S2. MSCs were transfected with 100 nM siRNAs using Lipofectamine RNAiMAX reagent (Thermo Fisher Scientific) following the manufacturer's protocol. Cells were incubated at 37 °C in a CO2 incubator for 48 h [41].
RT-PCR, ELISA, and nitric oxide (NO) assay
Total RNA was isolated from MSCs using TRIZOL Reagent (Thermo Fisher Scientific). RNA was quantified using a spectrophotometer and stored at -80 °C at a concentration of 1 mg/mL. cDNA was synthesized from 3 µg total RNA using an RT kit (Bioneer). PCR was used to examine the levels of mRNAs of chemokines, cytokines, and other proteins in spleen cells of MRL.Faslpr mice at 22 weeks of age. The primer sequences are shown in Table S3. PCR products were separated on 1% agarose gels and stained with 5 µg/mL ethidium bromide [33].
The levels of CCL2, CCL4, CCL5, CXCL10, CXCL12, IFN-γ, IL-10, PGE2, and TGF-β, accumulated in medium were measured by ELISA (Bio-Techne). The levels of IDO were measured by ELISA (BlueGene Biotech; Shanghai, China) [20]. The levels of IgM and IgG were determined by ELISA (Thermo Fisher Scientific). The level of nitrite accumulation in medium was used as an indicator NO production [43]. Briefly, the cell culture supernatants were mixed with an equal volume of Griess reagent (1% sulfanilamide, 0.1% naphthylethyl-enediamine dihydrochloride, and 2% phosphoric acid) and incubated at room temperature for 10 min. Nitrite concentrations were measured as optical density at 540 nm.
Western blotting
Cells were lysed in ice-cold cell lysis buffer (Cell Signaling Technology, Danvers, MA, USA; 20 mM Tris-HCl, pH 7.5; 150 mM NaCl; 1 mM Na2EDTA; 1 mM EGTA; 1% Triton X-100; 2.5 mM sodium pyrophosphate; 1 mM β-glycerophosphate; 1 mM Na3VO4; 1 µg/mL leupeptin). Proteins were separated by 10% SDS-PAGE and transferred to a PDVF membrane (MilliporeSigma, Burlington, MA, USA). Membrane was blocked with 5% skim milk in TBS/Tween-20 (TTBS) for 1 h and incubated with primary antibody in TTBS containing 5% BSA overnight. Antibodies against mouse or human CCR2, CCR5, CXCR3, CXCR4, GAPDH, IDO, PKC-α, PKC-δ, phospho-STAT1, and STAT1 were purchased from Cell Signaling Technology. After washing, membranes were incubated with horseradish peroxidase- conjugated secondary antibody and signals were detected by enhanced chemiluminescence (Amersham Pharmacia Biotech, Piscataway, NJ, USA) [33, 42].
Phenotyping
Cells were stained for 15 min at 4 °C with FITC-conjugated antibody against mouse CD4 or CD138 (BD Biosciences). Alternatively, cells were fixed using a Cytofix-Cytoperm Kit (BD Biosciences) and then stained with anti-Foxp3-APC or anti-IgG-APC antibody (eBiosciences, San Diego, CA, USA). Cells were analyzed using a flow cytometer (FACSCalibur) and the data were processed using Cell Quest Pro Software [44].
Statistical analysis
Data are presented as the mean ± SEM of at least three independent in vitro experiments performed in triplicate or six mice. To determine statistical significance, p-values were calculated using one-way ANOVA (GraphPad Software, San Diego, CA, USA).
Results
mMSCs inhibit mB cells in a CXCL12-dependent manner
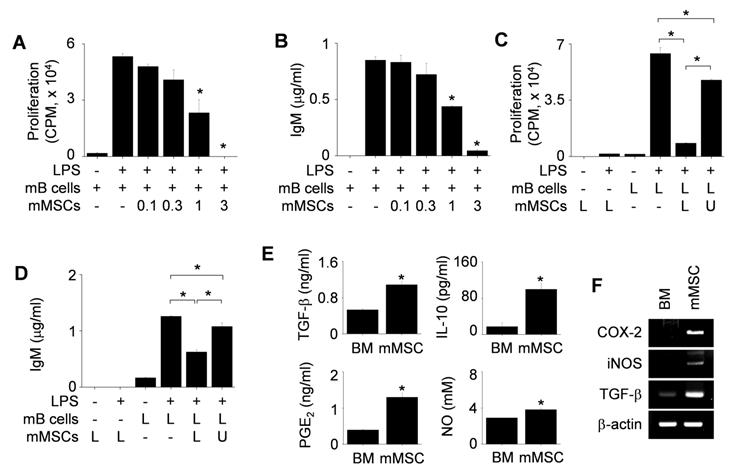
First, we used mMSCs generated from BM cells of C3H/HeN mice (H-2k) and splenic B cells isolated from lupus-prone MRL.Faslpr mice (H-2k). We found that mMSCs inhibited the proliferation of (Figure 1A) and IgM production (Figure 1B) by LPS-treated mB cells in a dose-dependent manner. mMSCs also inhibited the IgG production by mB cells activated with anti-CD40 antibody, IL-4, and IL-21 (Figure S1A). To assess whether mMSCs inhibit mB cells in a soluble factor- or contact-dependent manner, we used a transwell assay. When we added mMSCs and mB cells to the lower wells, thereby allowing cell-cell contact, mMSCs strongly inhibited mB cell proliferation (Figure 1C) and IgM production (Figure 1D). When we added mMSCs to the upper wells and mB cells to the lower wells, thereby preventing direct cell-cell contact, mMSCs inhibited mB cell functions much more weakly (Figure 1C-D). These data imply that mMSCs inhibit mB cell functions mainly in a contact-dependent manner and partially in a soluble factor-dependent manner. We confirmed that mMSCs produced the immunosuppressive soluble factors TGF-β, IL-10, PGE2, and NO (Figure 1E-F).
Effects of mMSCs on the proliferation of and IgM production by mB cells. (A-B) mMSCs (0.1-3 × 104 cells/well) and MRL.Faslpr mB cells (1 × 105 cells/well) were co-cultured for 72 h. LPS (1 μg/mL) was used to activate mB cells. The proliferation of and IgM production by mB cells were measured by the mitogen assay (A) and ELISA (B), respectively. (C-D) mMSCs (1 × 104 cells/well) were added to the upper (U) or lower (L) wells of transwell plates and mB cells (1 × 105 cells/well) to the L wells. After incubation with LPS for 72 h, the mitogen assay (C) and ELISA (D) were performed. (E) The levels of TGF-β, IL-10, and PGE2 accumulated in the culture medium of BM cells and mMSCs for 24 h were measured by ELISA. NO level was measured with Griess reagent. (F) Expression levels of COX-2, iNOS, and TGF-β mRNAs in BM cells and mMSCs were assessed by RT-PCR. *p < 0.01 (n = 3).
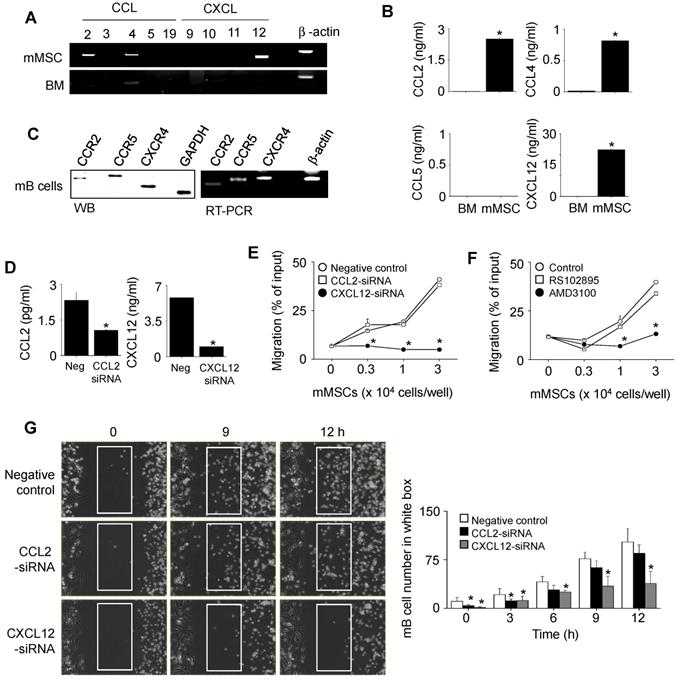
Next, we examined how mMSCs inhibited mB cell functions in a contact-dependent manner. To examine the role of chemokines, we assessed the expression profiles of chemokines in mMSCs and chemokine receptors in mB cells. mMSCs expressed mRNAs for CCL2, CCL4, and CXCL12 (Figure 2A), and the corresponding proteins were detectable by ELISA in conditioned medium (Figure 2B). We also confirmed that mB cells expressed mRNAs and proteins of CCR2, CCR5, and CXCR4 (Figure 2C). Then we used siRNAs to knock down CCL2 and CXCL12 in mMSCs (Figure 2D) and performed transwell assay to assess mB cell migration towards mMSCs at the population level. mB cells migrated well to mMSCs transfected with negative control or CCL2 siRNAs, but not to mMSCs transfected with CXCL12 siRNA (Figure 2E). mB cells treated with the CXCR4 antagonist AMD3100 showed little migration toward mMSCs, while mB cells treated with the CCR2 antagonist RS102895 migrated well to mMSCs (Figure 2F). To confirm these results, we used time-lapse imaging at the single-cell level. We placed mMSCs transfected with negative control siRNA, CCL2 siRNA, or CXCL12 on the left side of an imaging chamber and mB cells on the right side and acquired images every 2 min for 12 h (Movie S1). Representative images collected at 2-h intervals are shown in Figure 2G. The number of mB cells passing through the white box at each time point showed that negative control- and CCL2 siRNA-transfected mMSCs induced mB cell migration towards them, whereas CXCL12 siRNA-transfected mMSCs did not (Figure 2G). Overall, these data suggest that CXCL12 produced by mMSCs induces mB cell migration.
Effects of mMSCs on the migration of mB cells. (A) Expression levels of chemokine mRNAs in BM cells and mMSCs were assessed by RT-PCR. (B) The levels of CCL2, CCL4, CCL5, and CXCL12 accumulated in culture medium of BM cells and mMSCs for 24 h were measured by ELISA. (C) Expression levels of chemokine receptors and their mRNAs in mB cells were assessed by western blotting and RT-PCR, respectively. (D) mMSCs were transfected with negative (neg) control, CCL2, or CXCL12 siRNA for 48 h. The levels of CCL2 and CXCL12 accumulated in culture medium for 24 h were measured by ELISA. (E-F) CMFDA-labeled mB cells (1 × 105 cells/well) were added to the upper wells of transwell plates with a 5-μm insert. mMSCs (0.3-3 × 104 cells/well), which had been transfected with negative control, CCL2-siRNA, or CXCL12-siRNA, were added to the lower wells (E). CMFDA-labeled mB cells were pre-treated with dimethyl sulfoxide (0.1%, Control), CCR2 antagonist RS102895 (30 μg/mL), or CXCR4 antagonist AMD3100 (300 μg/mL) for 1 h, washed three times, and added to the upper wells. mMSCs (0.3-3 × 104 cells/well) were added to the lower wells (F). After 1.5 h, the number of CMFDA-labeled mB cells migrating to the lower well was determined. (G) For time-lapse imaging, mMSCs (70 μL of 0.3 × 106 cells/mL) were seeded into the left chamber and mB cells (70 μL of 1 × 106 cells/mL) into the right chamber of culture-insert μ-Dish35mm culture dishes. Images were acquired every 2 min for 12 h after 1-h pre-incubation. Representative photos are shown (n = 3). The numbers of mB cells passing through the white boxes are shown. *p < 0.01 (n = 3).
PMA-primed hMSCs inhibit hB cells in a CXCL10-dependent manner
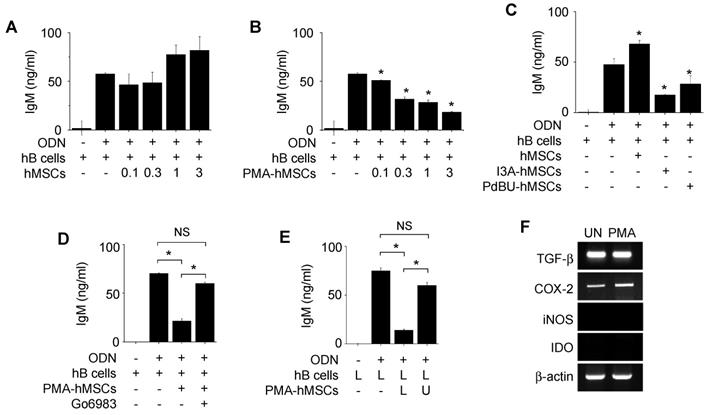
Next, we examined the effects of hMSCs generated from human BM cells on hB cells isolated from peripheral blood mononuclear cells of healthy donors. Unexpectedly, hMSCs did not inhibit IgM production by hB cells (Figure 3A). Thus, we screened a number of chemicals for their ability to activate hMSCs to inhibit hB cells (Table S1). Among these chemicals, three protein kinase C (PKC) activators (PMA, phorbol 12,13-dibutyrate, and ingenol 3-angelate) activated hMSCs so that they inhibited IgM production by ODN-treated hB cells (Figure 3B-C). PMA-primed hMSCs (called PMA-hMSCs hereafter) inhibited IgG production by hB cells activated with anti-CD40 antibody, IL-4, and IL-21 (Figure S1B). PKC inhibitor Go6983 abolished the ability of PMA-hMSCs to inhibit hB cells (Figure 3D). When we added PMA-hMSCs and hB cells together to the lower wells of transwell plates, PMA-hMSCs strongly inhibited hB cell IgM production, but, when we separated them, PMA-hMSCs did not (Figure 3E), suggesting that PMA-hMSCs inhibited hB cells mainly in a contact-dependent but not soluble factor-dependent manner. PMA did not increase the expression levels of immunosuppressive soluble factors, such as TGF-β, COX2, iNOS, or IDO, in hMSCs compared with naïve hMSCs (Figure 3F). PMA did not affect the phenotypes (Figure S2A), viability (Figure S2B), and proliferation (Figure S2C) of hMSCs. In addition, PMA did not affect the gene expression of IL-6 and IL-10 by hMSCs (Figure S2D).
Effects of PMA-hMSCs on IgM production by hB cells. (A) hMSCs (0.1-3 × 103 cells/well) and hB cells (1 × 105 cells/well) were co-cultured for 72 h. (B) hMSCs were treated with PMA (10 ng/mL) for 24 h and washed three times with medium. PMA-treated hMSCs (PMA-hMSCs; 0.1-3 × 103 cells/well) and hB cells (1 × 105 cells/well) were co-cultured for 72 h. (C) hMSCs were treated with ingenol-3-acetate (I3A, 10 µg/mL) or phorbol 12,13-dibutyrate (PdBU, 10 µg/mL) for 24 h and washed three times with medium. Chemical-treated hMSCs (1 × 103 cells/well) were co-cultured with hB cells (1 × 105 cells/well) for 72 h. (D) hMSCs were treated with PMA in the presence or absence of the PKC inhibitor Go6983 (1 μg/mL) for 24 h. PMA-hMSCs were washed three times with medium. PMA-hMSCs (1 × 103 cells/well) were co-cultured with hB cells (1 × 105 cells/well) for 72 h. (E) PMA-hMSCs (1 × 103 cells/well) were added to the lower wells and hB cells (1 × 105 cells/well) to the upper wells of transwell plates with a 5-μm insert. CpG-oligodeoxynucleotide (ODN, 5 μg/mL) was used to activate hB cells. IgM production by hB cells was measured by ELISA (A-E). (F) Total RNA was isolated from chemically untreated hMSCs (UN) or PMA-treated hMSCs (PMA). Gene expression levels were assessed by RT-PCR. *p < 0.01 (n = 3).
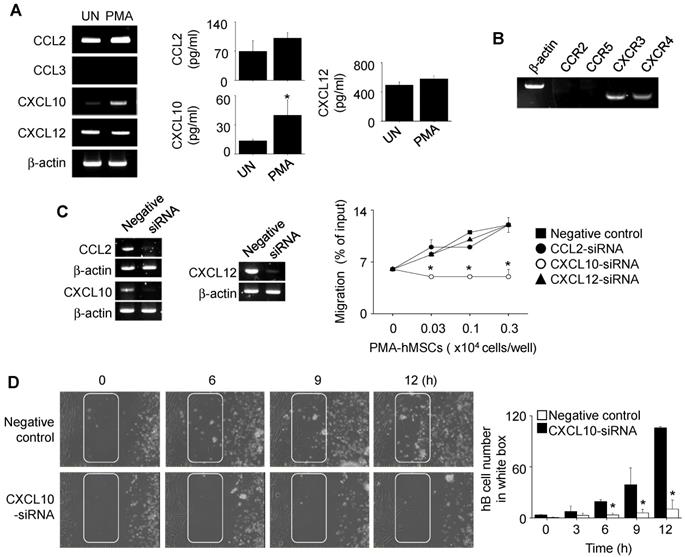
Next, we investigated how PMA-hMSCs inhibited hB cell functions in a contact-dependent manner. PMA increased the expression of CXCL10, but not of CCL2, CCL3, or CXCL12, by hMSCs (Figure 4A). hB cells expressed the CXCL10 receptor CXCR3 (Figure 4B). We decreased the expression of CCL2, CXCL10, and CXCL12 in PMA-hMSCs with specific siRNAs and used them in transwell assay. PMA-hMSCs transfected with CXCL10 siRNA did not induce hB cell migration, but other PMA-hMSCs attracted hB cells (Figure 4C). These results were confirmed by time-lapse imaging (Movie S2). Representative images collected at 2-h intervals are shown in Figure 4D. The number of hB cells passing through the white box at each time point showed that CXCL10 siRNA-transfected PMA-hMSCs did not induce hB cell migration (Figure 4D). Overall, these data suggest that PMA-hMSCs use CXCL10 to attract hB cells, unlike mMSCs, which use CXCL12.
Effects of PMA on the migration of hMSCs. (A) hMSCs were activated with PMA for 24 h. Chemokine expression levels were measured by RT-PCR and ELISA. UN, untreated. (B) Expression levels of chemokine receptors in hB cells were assessed by RT-PCR. (C) PMA-hMSCs were transfected with negative-control, CCL2, CXCL10, or CXCL12 siRNA for 48 h, and chemokine expression levels were analyzed by RT-PCR. PMA-hMSCs (0.03-0.3 × 104 cells/well) were added to the lower wells and CMFDA-labeled hB cells (1 × 105 cells/well) to the upper wells of transwell plates with a 5-μm insert. After 1.5 h, the number of CMFDA-labeled hB cells migrating to the lower well was determined. (D) For time-lapse imaging, hMSCs (70 μL of 0.3 × 106 cells/mL) were seeded into the left chamber and hB cells (70 μL of 1 × 106 cells/mL) into the right chamber of culture-insert μ-Dish35mm culture dishes. Images were acquired every 2 min for 12 h. Representative photos are shown. The numbers of hB cells passing through the white boxes are shown. *p < 0.01 (n = 3).
PMA-hMSCs inhibit hB cells in a PD-L1-dependent manner
Next, we examined how PMA-hMSCs directly inhibit hB cell functions after contact. PMA strongly increased the expression of PD-L1 and weakly that of PD-L2 and FasL by hMSCs (Figure 5A). Anti-PD-L1 blocking antibody abolished the inhibitory capacity of PMA-hMSCs, but anti-PD-L2 and anti-FasL antibodies did not (Figure 5B). PMA-hMSCs transfected with PD-L1 siRNA did not inhibit B cells, but PMA-hMSCs transfected with FasL siRNA inhibited hB cells well (Figure 5C).
Effects of PMA-hMSCs on the viability of hB cells. (A) hMSCs were activated with PMA for 24 h. Expression levels of the death ligands PD-L1, PD-L2, and FasL were measured by RT-PCR and flow cytometric analysis. UN, untreated. (B-C) PMA-hMSCs (1 × 103 cells/well) were co-cultured with hB cells (1 × 105 cells/well) for 72 h in the presence of blocking antibodies against PD-L1, PD-L2, or FasL (B). PMA-hMSCs, which had been transfected with PD-L1 or FasL siRNA, were co-cultured with hB cells (C). ODN (5 μg/mL) was used to activate hB cells. IgM production by hB cells was measured by ELISA assay. (D-F) PMA-hMSCs (0.1 × 106 cells), which had been transfected with PD-L1 siRNA, were cultured with hB cells (1 × 106 cells) in 35-mm culture dishes for 24 h. hB cells were stained with anti-CD19-APC and then stained with FITC-Annexin V and propidium iodide (PI). Cells were analyzed using a flow cytometer (D). hB cells were stained with anti-CD19-APC and Intracellular Caspase Detection ApoStat kit and analyzed using a flow cytometer (E). CellEvent Caspase-3/7 Green ReadyProbes Reagent was added to the culture of hB cells and hMSCs, and the cells were imaged every 10 min for 24 h with a Biostation IM-Q microscope (Nikon). Green fluorescent cells were considered apoptotic (F). *p < 0.01 (n = 3).
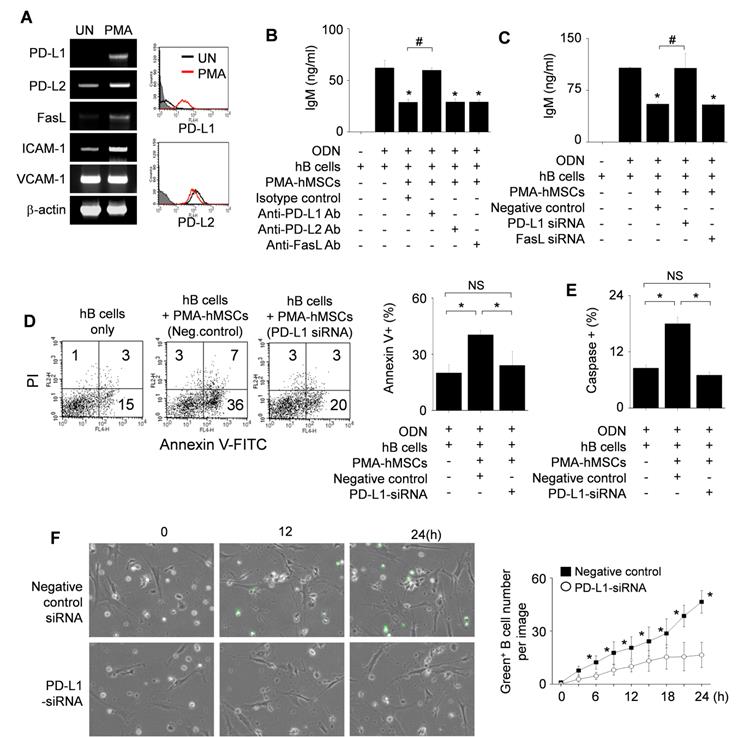
Then, we examined whether PD-L1-expressing PMA-hMSCs can induce apoptosis of hB cells in three ways. First, we stained hB cells with annexin V and propidium iodide. PMA-hMSCs markedly increased the proportion of annexin V-positive apoptotic hB cells, but PMA-hMSCs transfected with PD-L1 siRNA did so only weakly (Figure 5D). Second, we labeled hB cells with FITC-VAD-FMK (ApoStat), a cell-permeable fluorescent pan-caspase probe. PMA-hMSCs increased pan-caspase activity in hB cells, but PMA-hMSCs transfected with PD-L1 siRNA did not (Figure 5E). Third, we labeled hB cells with the DEVD peptide conjugated to a nucleic acid-binding dye (CellEvent Caspase-3/7 Green ReadyProbes Reagent). Upon activation of caspase-3/7 in apoptotic cells, DEVD is cleaved and the free dye binds DNA, generating bright green fluorescence. By using time-lapse imaging, we showed that caspase-3/7 activity was higher in PMA-hMSCs than in PMA-hMSCs transfected with PD-L1 siRNA (Movie S3 and Figure 5F). Overall, these data suggest that PMA-hMSCs activates caspases in a PD-L1-dependent manner, followed by hB cell apoptosis.
PMA-hMSCs are more efficient than naïve hMSCs in ameliorating lupus progression in MRL.Faslpr mice
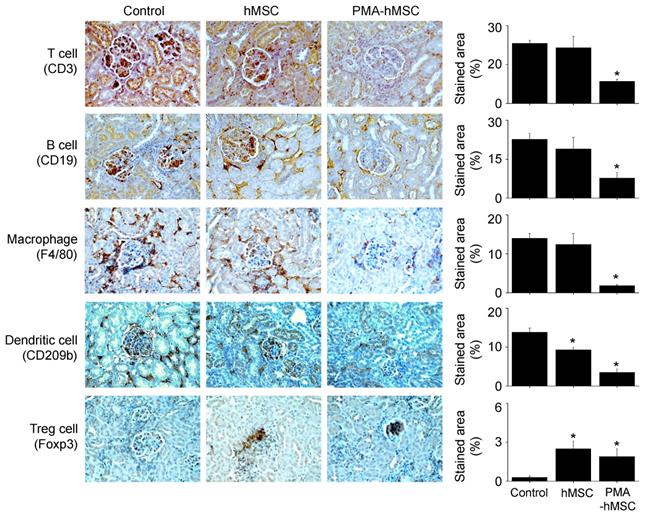
Finally, we examined the therapeutic activity of PMA-hMSCs in lupus-prone MRL.Faslpr mice. Our preliminary experiments showed that hMSCs at 4 × 106 cells/injection completely prevented lupus progression in MRL.Faslpr mice. To compare the efficacy of hMSCs and PMA-hMSCs, we therefore injected lower numbers of hMSCs and PMA-hMSCs (4 × 104 cells/injection). All mice (n = 6) that received PMA-hMSCs survived up to 30 weeks of age, which was much longer than control and hMSC-injected groups (Figure 6A). Regardless of PMA priming, hMSCs did not affect body weight (Figure 6B), and no untoward effects were noted. In another experiment, when 50% of control mice survived (22 weeks of age), we sacrificed the surviving mice. The serum levels of anti-dsDNA (Figure 6C) and total IgG (Figure 6D) antibodies and the levels of protein in urine (Figure 6E) were much lower in PMA-hMSC-treated mice than in the control and hMSC-treated mice. The deposition of IgG and C3 complement in the kidney was significantly decreased in PMA-hMSC-treated mice in comparison with the control and hMSC-treated mice (Figure 6F). The expression of all inflammatory cytokines tested (IL-1β, IL-12, IFN-γ, and TNF-α) in the spleen was also lower in PMA-hMSC-treated mice than in the control and hMSC-treated mice (Figure 6G). The frequency of Foxp3-expressing CD4+ Treg cells was higher and that of IgG-producing CD138+ plasma cells was lower in the spleens of PMA-hMSC-treated mice than in those of the control and hMSC-treated mice (Figure 6H). The infiltration of T cells, B cells, macrophages, and dendritic cells into the kidney was significantly decreased in PMA-hMSC-treated mice in comparison with the control and hMSC-treated mice (Figure 7). In contrast, the infiltration of Treg cells into the kidney was significantly increased in PMA-hMSC-treated mice in comparison with the control mice (Figure 7). Overall, our data suggest that PMA-hMSCs ameliorate the development of SLE-like disease in MRL.Faslpr mice more effectively than do naïve hMSCs.
In vivo efficacy of PMA-hMSCs in MRL.Faslpr mice. (A-B) MRL.Faslpr mice were intravenously injected with PBS (control), naïve hMSCs (4 × 104 cells /injection), or PMA-hMSCs (4 × 104 cells/injection) once at the age of 12 weeks. Survival was measured every week (A) and body weight (B) every 2 weeks up to 30 weeks of age. *p < 0.01 (n = 6). (C-H) Injections were performed as in (A). Surviving mice were sacrificed at the age of 22 weeks. The serum levels of anti-dsDNA IgG (C) and total IgG (D) were measured every 3 weeks. Proteinurea levels were measured at the age of 22 weeks (E). Kidney sections were stained with antibodies against IgG and C3 complement (F). Total RNA was isolated from spleen cells and the expression levels of inflammatory cytokine genes (IL-1β, IL-12, IFN-γ, and TNF-α) were examined by RT-PCR (G). The ratios of Foxp3-expressing CD4+ Treg cells (CD4+Foxp3int) and IgG-producing CD138+ plasma cells (CD138+IgGint) in the spleen were measured by flow cytometry (H). *p < 0.01 (n = 6).
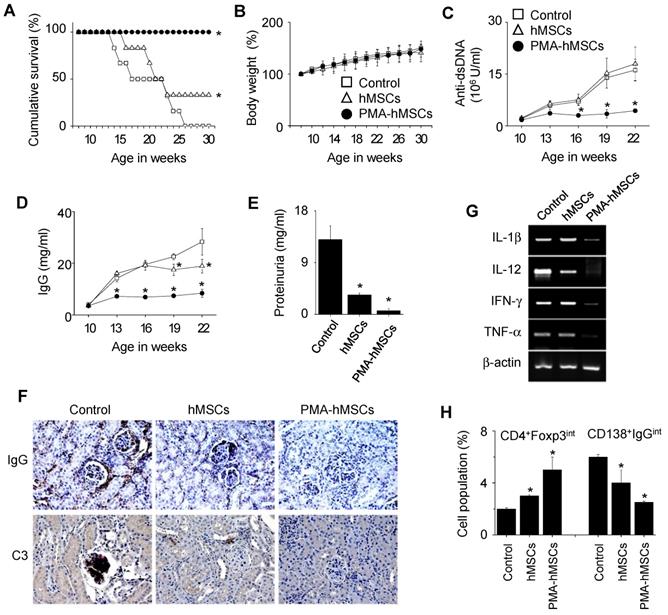
Immunohistochemical analysis. Kidney sections used in Figure 6F were stained with antibodies against CD3 (T cells), CD19 (B cells), F4/80 (macrophages), CD209b (dendritic cells), or Foxp3 (Treg cells).
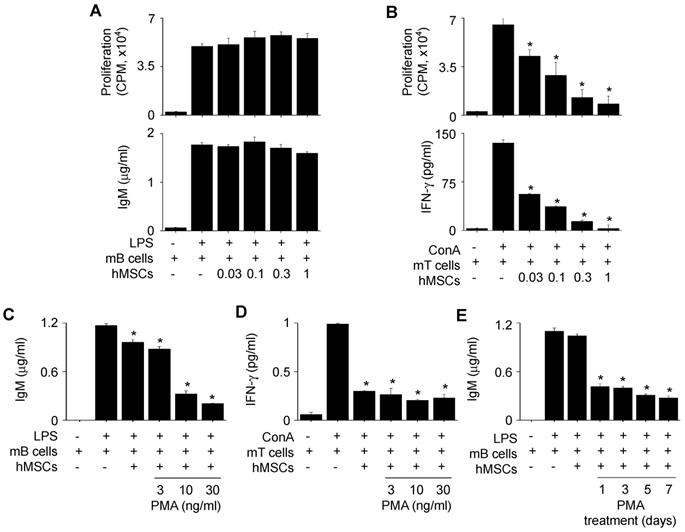
Since we injected PMA-hMSCs into the xenogeneic MRL.Faslpr mice, we examined whether PMA-hMSCs inhibited mB cells. hMSCs did not inhibit the proliferation of or IgM production by mB cells from MRL.Faslpr mice (Figure 8A). However, hMSCs inhibited well the proliferation of and IFN-γ production by concanavalin A (ConA)-activated T cells from the same mice (Figure 8B). PMA-hMSCs inhibited both IgM production by mB cells (Figure 8C) and IFN-γ production by mouse T cells (Figure 8D). We found that 1-day priming of hMSCs with PMA was enough to activate hMSCs to inhibit mB cells (Figure 8E). We also proved that mMSCs, hMSCs, and PMA-hMSCs inhibited IFN-γ production by T cells activated with anti-CD3 and anti-CD28 antibody, which mimicked TCR-dependent T cell activation (Figure S1C-D). Overall, our data suggest that the improved efficacy of PMA-hMSCs in MRL.Faslpr mice might be due to the dual inhibition of T and B cells.
Effects of PMA-hMSCs on xenogeneic mB cells. (A) hMSCs (0.03-1 × 104 cells/well) and MRL.Faslpr mB cells (1 × 105 cells/well) were co-cultured for 72 h. LPS (1 μg/mL) was used to activate mB cells. The proliferation of and IgM production by mB cells were measured by the mitogen assay and ELISA, respectively. (B) hMSCs (0.03-1 × 104 cells/well) and MRL.Faslpr mT cells (1 × 105 cells/well) were co-cultured for 72 h. Concanavalin A (ConA, 1 μg/mL) was used to activate mT cells. The proliferation of and IFN-γ production by mT cells were measured by the mitogen assay and ELISA, respectively. (C-D) hMSCs (1 × 104 cells/well) were activated with PMA (3-30 ng/mL) for 24 h and then co-cultured with mB cells (1 × 105 cells/well) (C) or mT cells (1 × 105 cells/well) (D). (E) hMSCs were activated with PMA (10 ng/mL) for 1 to 7 days and then co-cultured with mB cells. *p < 0.01 (n = 3).
Discussion
Two approaches have been proposed to improve the functions of MSCs. One is MSC priming with cytokines and growth factors. IFN-γ, TNF-α, IL-17, and IL-1β are well known to enhance the immunosuppressive properties of MSCs by upregulating the secretion of IDO, PGE2, and TGF-β [45-49]. The second approach is MSC priming with chemicals, which might be more cost effective than priming with cytokines. Valproic acid, sphingosine-1-phosphate, 5-aza-2'-deoxycytidine, dimethyloxalylglycine, 2-chloro-N6-cyclopentyl-adenosine, rapamycin, and all-trans retinoic acid enhance the differentiation, proliferation, and tissue-homing ability of MSCs [50-55]. In this study, we investigated in detail the effects of chemicals on the ability of MSCs to suppress the functions of B cells and found that PMA improves the suppressive activity of hMSCs on hB cells. Compared to naïve hMSCs, PMA-hMSCs strongly inhibited IgM production by hB cells.
The first question addressed in this study is how PMA-hMSCs inhibit B cells. It is well documented that MSCs can inhibit T cells through both contact- and soluble factor-dependent mechanisms [16-18, 24, 56, 57]. However, PMA-hMSCs inhibit hB cells mainly in a contact-dependent manner, but not through soluble factors. When those cells were separated in our transwell assay, PMA-hMSCs did not affect hB cells, but allowing their contact resulted in PMA-hMSCs strongly and significantly inhibiting hB cells.
The second question is how PMA-hMSCs contact hB cells. Our data suggest an important role of CXCL10 produced by adherent PMA-hMSCs in migration of non-adherent hB cells. It is well known that serum levels of CXCL10 are increased in SLE patients and are strongly correlated with disease activity [58]. In lupus-prone MRL/lpr mice, the number of CXCR3-positive plasma B cells is increased in the secondary lymphoid organs during the development of lupus nephritis [59]. Importantly, the nephritic kidney shows high expression of CXCL10 [60], which increases the migration of CXCR3-positive plasma B cells from the secondary lymphoid organs into the inflamed kidney. Our data that PMA-hMSCs transfected with CXCL10 siRNA did not attract B cells suggest an additional role of CXCL10 in that this chemokine optimizes the interaction between MSCs and B cells, which might happen in the nephritic kidney. CCL2 and CXCL12 appear not to be involved in PMA-hMSC-induced attraction of hB cells. Two interesting aspects are worth noting. First, mMSCs use a different chemokine, CXCL12, to induce mB cell migration. CXCL12 siRNA-transfected mMSCs were unable to induce mB cell migration, and a CXCR4 antagonist blocked mB cell migration to mMSCs. Second, MSCs use CCL2 to attract T cells. CCL2-deficient MSCs can neither attract T cells nor inhibit T cell functions [4]. MSCs use CCL2 for the recruitment and subsequent contact-dependent inhibition of inflammatory Th17 cells and for the homing of Tregs and myeloid-derived suppressor cells to the damaged organs in models of experimental autoimmune encephalomyelitis and experimental autoimmune uveitis [61, 62]. Overall, our data suggest that MSCs use different chemokines to attract T and B cells in a species-dependent manner.
The next question is how PMA-hMSCs inhibit hB cells after contact. Our data suggest a key role for PD-L1. PD-1 is broadly expressed on B cells and other immune cells, including T cells [63]. Although PD-1 interacts with PD-L1 and PD-L2, PD-L1 might act as the primary ligand of PD-1 [64]. Unlike PD-L2, PD-L1 induces a considerable conformational change of PD-1 and is widely expressed on most cell types, including dendritic cells, macrophages, endothelial cells, and placenta cells [64, 65]. PD-L1 has been extensively studied as a negative regulator of the immune response that enables tumor cells to escape T cell immunity [66, 67], and as an inducer of peripheral tolerance to prevent autoimmune diseases [66, 68]. MSCs highly express PD-L1 upon activation with IFN-γ or TNF-α, although naïve MSCs express it very weakly [64, 69, 70]. IFN-γ treated MSCs inhibit cytokine production, proliferation, and differentiation of T cells and induce T cell apoptosis in a PD-L1-dependent manner, although some discrepancies are reported depending on the experimental conditions [63, 64, 69, 71-73]. MSCs inhibit B cell antibody production in a PD-1-dependent contact-inhibition manner [69]. In agreement with these data, our data show that PMA-hMSCs inhibit hB cell functions in a PD-L1-dependent manner, which was proved by siRNA and blocking-antibody experiments.
The implications of our study are limited by several caveats. First, we did not clarify the signaling in PMA-hMSCs. PMA activates PKCs and subsequently activates several transcription factors, such as NF-AT and NF-κB [74, 75], which are able to increase the mRNA expression of CXCL10 and PD-L1 [66, 76, 77]. It is also unclear how long PKC activation in hMSCs is maintained after washing out PMA. Our preliminary experiments demonstrated that the amount of PKCs in the cytosol of hMSCs decreased from 1 h and PKC almost disappeared 24 h after the onset of PMA treatment (Figure S3A). After removing PMA, the amount of PKCs in the cytosol of hMSCs was maintained at low level up to 3 days and increased from 4 days (Figure S3B). The gene expression of PD-L1 in hMSCs was maintained up to 2 days and decreased from 3 days after washing out PMA (Figure S3C). Further studies to delineate the detailed signaling cascades in PMA-hMSCs are needed, because elucidation of the underlying mechanism would enable us to develop a strategy for the generation of MSCs with potent immunosuppressive activities. Second, we did not study the negative role of PD-1 signaling in B cell activation. Although much has been learned about the inhibitory mechanisms of PD-1 signaling in T cell activation, little is known about how PD-1 inhibits B cell activation [71]. Thus, further studies are required to reveal the detailed inhibitory mechanisms of PD-1 signaling in B cell activation. Third, our experimental system, which uses MSCs and purified B cells, is not identical to in vivo conditions, since the interaction between MSCs and B cells is extremely complicated and is likely affected by many different factors in vivo. However, our system might be optimal for investigating their interaction because it is easily controllable and can be expected to yield reproducible data since there are few interfering factors [78].
Despite these shortcomings, the results of this study provide several insights into the mechanisms of B cell inhibition by MSCs. First, mMSCs inhibit mB cells, but hMSCs do not inhibit hB cells, which suggests a species difference in MSC effects on B cells. Second, phorbol ester priming of hMSCs can increase their immunosuppressive capacity against hB cells. Third, PMA-hMSCs inhibit hB cells via unique mechanisms using CXCL10 and PD-L1. Fourth, in comparison with naïve hMSCs, PMA-hMSCs have better therapeutic efficacy in vivo in an SLE mouse model. Most importantly, our data provide a clue on how to generate hMSCs effective for the treatment of patients with SLE and other autoimmune diseases characterized by excessive B cell activation.
Abbreviations
BM: bone marrow; ConA: concanavalin A; IDO: indoleamine 2,3-dioxygenase; I3A: ingenol 3-angelate; LPS: lipopolysaccharide; MSCs: mesenchymal stem cells; NO: nitric oxide; ODN: oligodeoxynucleotide; PdBU: phorbol 12,13-dibutyrate; PGE2: prostaglandin E2; PI: propidium iodide; PMA: phorbol myristate acetate; siRNAs: small interfering RNAs; SLE: systemic lupus erythematosus; TGF-β: tumor growth factor-beta.
Supplementary Material
Supplementary figures and tables.
Supplementary movie S1.
Supplementary movie S2.
Supplementary movie S3.
Acknowledgements
This study was supported by grants funded by the National Research Foundation of Korea (2017R1A5A2015541, 2017M3A9B4050336, and 2020R1A2C2004200).
Author Contributions
Study conception and design: KS Kim, SC Bae, J Yun, and SB Han. Acquisition of data: HK Lee, HS Kim, MJ Pyo, EJ Park, S Jang, and TY Lee. Analysis and interpretation of data: Y Kim, JT Hong, J Yun, and SB Han. Drafting the article: J Yun and SB Han.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Liu Z, Davidson A. Taming lupus-a new understanding of pathogenesis is leading to clinical advances. Nat Med. 2012;18:871-82
2. Morel L. Immunometabolism in systemic lupus erythematosus. Nat Rev Rheumatol. 2017;13:280-90
3. Lee HK, Kim KH, Kim HS, Kim JS, Lee JH, Ji A. et al. Effect of a Combination of Prednisone or Mycophenolate Mofetil and Mesenchymal Stem Cells on Lupus Symptoms in MRL.Fas(lpr) Mice. Stem Cells Int. 2018;2018:4273107
4. Lee HK, Kim HS, Kim JS, Kim YG, Park KH, Lee JH. et al. CCL2 deficient mesenchymal stem cells fail to establish long-lasting contact with T cells and no longer ameliorate lupus symptoms. Sci Rep. 2017;7:41258
5. Lee JH, Lee HK, Kim HS, Kim JS, Ji AY, Lee JS. et al. CXCR3-deficient mesenchymal stem cells fail to infiltrate into the nephritic kidney and do not ameliorate lupus symptoms in MRL. Fas(lpr) mice. Lupus. 2018;27:1854-9
6. Zhou T, Liao C, Li HY, Lin W, Lin S, Zhong H. Efficacy of mesenchymal stem cells in animal models of lupus nephritis: a meta-analysis. Stem Cell Res Ther. 2020;11:48
7. Dang J, Xu Z, Xu A, Liu Y, Fu Q, Wang J. et al. Human gingiva-derived mesenchymal stem cells are therapeutic in lupus nephritis through targeting of CD39(-)CD73 signaling pathway. J Autoimmun. 2020: 102491.
8. Liu F, Chen H, Chen T, Lau CS, Yu FX, Chen K. et al. Immunotherapeutic effects of allogeneic mesenchymal stem cells on systemic lupus erythematosus. Lupus. 2020;29:872-83
9. Lee HK, Kim EY, Kim HS, Park EJ, Lee HJ, Lee TY. et al. Effect of Human Mesenchymal Stem Cells on Xenogeneic T and B Cells Isolated from Lupus-Prone MRL.Fas (lpr) Mice. Stem Cells Int. 2020;2020:5617192
10. Wang D, Zhang H, Liang J, Li X, Feng X, Wang H. et al. Allogeneic mesenchymal stem cell transplantation in severe and refractory systemic lupus erythematosus: 4 years of experience. Cell Transplant. 2013;22:2267-77
11. Li X, Wang D, Liang J, Zhang H, Sun L. Mesenchymal SCT ameliorates refractory cytopenia in patients with systemic lupus erythematosus. Bone Marrow Transplant. 2013;48:544-50
12. Gu F, Wang D, Zhang H, Feng X, Gilkeson GS, Shi S. et al. Allogeneic mesenchymal stem cell transplantation for lupus nephritis patients refractory to conventional therapy. Clin Rheumatol. 2014;33:1611-9
13. Zhou T, Li HY, Liao C, Lin W, Lin S. Clinical Efficacy and Safety of Mesenchymal Stem Cells for Systemic Lupus Erythematosus. Stem Cells Int. 2020;2020:6518508
14. Wen L, Labopin M, Badoglio M, Wang D, Sun L, Farge-Bancel D. Prognostic Factors for Clinical Response in Systemic Lupus Erythematosus Patients Treated by Allogeneic Mesenchymal Stem Cells. Stem Cells Int. 2019;2019:7061408
15. Liang J, Zhang H, Hua B, Wang H, Lu L, Shi S. et al. Allogenic mesenchymal stem cells transplantation in refractory systemic lupus erythematosus: a pilot clinical study. Ann Rheum Dis. 2010;69:1423-9
16. de Castro LL, Lopes-Pacheco M, Weiss DJ, Cruz FF, Rocco PRM. Current understanding of the immunosuppressive properties of mesenchymal stromal cells. J Mol Med (Berl). 2019;97:605-18
17. Shi Y, Wang Y, Li Q, Liu K, Hou J, Shao C. et al. Immunoregulatory mechanisms of mesenchymal stem and stromal cells in inflammatory diseases. Nat Rev Nephrol. 2018;14:493-507
18. Fan XL, Zhang Y, Li X, Fu QL. Mechanisms underlying the protective effects of mesenchymal stem cell-based therapy. Cell Mol Life Sci. 2020;77:2771-94
19. Akiyama K, Chen C, Wang D, Xu X, Qu C, Yamaza T. et al. Mesenchymal-stem-cell-induced immunoregulation involves FAS-ligand-/FAS-mediated T cell apoptosis. Cell Stem Cell. 2012;10:544-55
20. Kim JS, Kim YG, Lee HK, Park EJ, Kim B, Kang JS. et al. Cytokine-induced killer cells hunt individual cancer cells in droves in a mouse model. Cancer Immunol Immunother. 2017;66:193-202
21. Hackstein H, Tschipakow I, Bein G, Nold P, Brendel C, Baal N. Contact-dependent abrogation of bone marrow-derived plasmacytoid dendritic cell differentiation by murine mesenchymal stem cells. Biochem Biophys Res Commun. 2016;476:15-20
22. Salami F, Tavassoli A, Mehrzad J, Parham A. Immunomodulatory effects of mesenchymal stem cells on leukocytes with emphasis on neutrophils. Immunobiology. 2018;223:786-91
23. Cras A, Farge D, Carmoi T, Lataillade JJ, Wang DD, Sun L. Update on mesenchymal stem cell-based therapy in lupus and scleroderma. Arthritis Res Ther. 2015;17:301
24. Pers YM, Quentin J, Feirreira R, Espinoza F, Abdellaoui N, Erkilic N. et al. Injection of Adipose-Derived Stromal Cells in the Knee of Patients with Severe Osteoarthritis has a Systemic Effect and Promotes an Anti-Inflammatory Phenotype of Circulating Immune Cells. Theranostics. 2018;8:5519-28
25. Comoli P, Ginevri F, Maccario R, Avanzini MA, Marconi M, Groff A. et al. Human mesenchymal stem cells inhibit antibody production induced in vitro by allostimulation. Nephrol Dial Transplant. 2008;23:1196-202
26. Corcione A, Benvenuto F, Ferretti E, Giunti D, Cappiello V, Cazzanti F. et al. Human mesenchymal stem cells modulate B-cell functions. Blood. 2006;107:367-72
27. Che N, Li X, Zhang L, Liu R, Chen H, Gao X. et al. Impaired B cell inhibition by lupus bone marrow mesenchymal stem cells is caused by reduced CCL2 expression. J Immunol. 2014;193:5306-14
28. Chen X, Cai C, Xu D, Liu Q, Zheng S, Liu L. et al. Human Mesenchymal Stem Cell-Treated Regulatory CD23(+)CD43(+) B Cells Alleviate Intestinal Inflammation. Theranostics. 2019;9:4633-47
29. Rosado MM, Bernardo ME, Scarsella M, Conforti A, Giorda E, Biagini S. et al. Inhibition of B-cell proliferation and antibody production by mesenchymal stromal cells is mediated by T cells. Stem Cells Dev. 2015;24:93-103
30. Krampera M, Cosmi L, Angeli R, Pasini A, Liotta F, Andreini A. et al. Role for interferon-gamma in the immunomodulatory activity of human bone marrow mesenchymal stem cells. Stem Cells. 2006;24:386-98
31. Ji YR, Yang ZX, Han ZB, Meng L, Liang L, Feng XM. et al. Mesenchymal stem cells support proliferation and terminal differentiation of B cells. Cell Physiol Biochem. 2012;30:1526-37
32. Xu J. Therapeutic Applications of Mesenchymal Stem Cells for Systemic Lupus Erythematosus. Adv Exp Med Biol. 2018;1089:73-85
33. Kim HS, Lee JS, Lee HK, Park EJ, Jeon HW, Kang YJ. et al. Mesenchymal Stem Cells Ameliorate Renal Inflammation in Adriamycin-induced Nephropathy. Immune Netw. 2019;19:e36
34. Kim JW, Lee J, Hong SM, Lee J, Cho ML, Park SH. Circulating CCR7(lo)PD-1(hi) Follicular Helper T Cells Indicate Disease Activity and Glandular Inflammation in Patients with Primary Sjogren's Syndrome. Immune Netw. 2019;19:e26
35. Kim J, Kim JS, Lee HK, Kim HS, Park EJ, Choi JE. et al. CXCR3-deficient natural killer cells fail to migrate to B16F10 melanoma cells. Int Immunopharmacol. 2018;63:66-73
36. Ishikawa M, Ito H, Kitaori T, Murata K, Shibuya H, Furu M. et al. MCP/CCR2 signaling is essential for recruitment of mesenchymal progenitor cells during the early phase of fracture healing. PLoS One. 2014;9:e104954
37. Zhao FY, Cheng TY, Yang L, Huang YH, Li C, Han JZ. et al. G-CSF Inhibits Pulmonary Fibrosis by Promoting BMSC Homing to the Lungs via SDF-1/CXCR4 Chemotaxis. Sci Rep. 2020;10:10515
38. Kim SN, Lee HJ, Jeon MS, Yi T, Song SU. Galectin-9 is Involved in Immunosuppression Mediated by Human Bone Marrow-derived Clonal Mesenchymal Stem Cells. Immune Netw. 2015;15:241-51
39. Larsen SE, Voss K, Laing ED, Snow AL. Differential cytokine withdrawal-induced death sensitivity of effector T cells derived from distinct human CD8(+) memory subsets. Cell Death Discov. 2017;3:17031
40. Huang TC, Lee JF, Chen JY. Pardaxin, an antimicrobial peptide, triggers caspase-dependent and ROS-mediated apoptosis in HT-1080 cells. Mar Drugs. 2011;9:1995-2009
41. Kim JS, Shin BR, Lee HK, Lee JH, Kim KH, Choi JE. et al. Cd226(-/-) natural killer cells fail to establish stable contacts with cancer cells and show impaired control of tumor metastasis in vivo. Oncoimmunology. 2017;6:e1338994
42. Yeon JT, Kim KJ, Son YJ, Park SJ, Kim SH. Idelalisib inhibits osteoclast differentiation and pre-osteoclast migration by blocking the PI3Kdelta-Akt-c-Fos/NFATc1 signaling cascade. Arch Pharm Res. 2019;42:712-21
43. Jo SH, Kim ME, Cho JH, Lee Y, Lee J, Park YD. et al. Hesperetin inhibits neuroinflammation on microglia by suppressing inflammatory cytokines and MAPK pathways. Arch Pharm Res. 2019;42:695-703
44. Kim D, Koh J, Ko JS, Kim HY, Lee H, Chung DH. Ubiquitin E3 Ligase Pellino-1 Inhibits IL-10-mediated M2c Polarization of Macrophages, Thereby Suppressing Tumor Growth. Immune Netw. 2019;19:e32
45. Noronha NC, Mizukami A, Caliari-Oliveira C, Cominal JG, Rocha JLM, Covas DT. et al. Priming approaches to improve the efficacy of mesenchymal stromal cell-based therapies. Stem Cell Res Ther. 2019;10:131
46. de Witte SF, Franquesa M, Baan CC, Hoogduijn MJ. Toward Development of iMesenchymal Stem Cells for Immunomodulatory Therapy. Front Immunol. 2015;6:648
47. Prasanna SJ, Gopalakrishnan D, Shankar SR, Vasandan AB. Pro-inflammatory cytokines, IFNgamma and TNFalpha, influence immune properties of human bone marrow and Wharton jelly mesenchymal stem cells differentially. PLoS One. 2010;5:e9016
48. Sivanathan KN, Rojas-Canales D, Grey ST, Gronthos S, Coates PT. Transcriptome Profiling of IL-17A Preactivated Mesenchymal Stem Cells: A Comparative Study to Unmodified and IFN-gamma Modified Mesenchymal Stem Cells. Stem Cells Int. 2017;2017:1025820
49. Luz-Crawford P, Espinosa-Carrasco G, Ipseiz N, Contreras R, Tejedor G, Medina DA. et al. Gilz-Activin A as a Novel Signaling Axis Orchestrating Mesenchymal Stem Cell and Th17 Cell Interplay. Theranostics. 2018;8:846-59
50. Lim J, Lee S, Ju H, Kim Y, Heo J, Lee HY. et al. Valproic acid enforces the priming effect of sphingosine-1 phosphate on human mesenchymal stem cells. Int J Mol Med. 2017;40:739-47
51. Xu R, Chen W, Zhang Z, Qiu Y, Wang Y, Zhang B. et al. Integrated data analysis identifies potential inducers and pathways during the endothelial differentiation of bone-marrow stromal cells by DNA methyltransferase inhibitor, 5-aza-2'-deoxycytidine. Gene. 2018;657:9-18
52. Liu XB, Wang JA, Ji XY, Yu SP, Wei L. Preconditioning of bone marrow mesenchymal stem cells by prolyl hydroxylase inhibition enhances cell survival and angiogenesis in vitro and after transplantation into the ischemic heart of rats. Stem Cell Res Ther. 2014;5:111
53. D'Alimonte I, Nargi E, Lannutti A, Marchisio M, Pierdomenico L, Costanzo G. et al. Adenosine A1 receptor stimulation enhances osteogenic differentiation of human dental pulp-derived mesenchymal stem cells via WNT signaling. Stem Cell Res. 2013;11:611-24
54. Wang B, Lin Y, Hu Y, Shan W, Liu S, Xu Y. et al. mTOR inhibition improves the immunomodulatory properties of human bone marrow mesenchymal stem cells by inducing COX-2 and PGE2. Stem Cell Res Ther. 2017;8:292
55. Pourjafar M, Saidijam M, Mansouri K, Ghasemibasir H, Karimi Dermani F, Najafi R. All-trans retinoic acid preconditioning enhances proliferation, angiogenesis and migration of mesenchymal stem cell in vitro and enhances wound repair in vivo. Cell Prolif. 2017 50
56. Cosenza S, Toupet K, Maumus M, Luz-Crawford P, Blanc-Brude O, Jorgensen C. et al. Mesenchymal stem cells-derived exosomes are more immunosuppressive than microparticles in inflammatory arthritis. Theranostics. 2018;8:1399-410
57. Monguio-Tortajada M, Roura S, Galvez-Monton C, Pujal JM, Aran G, Sanjurjo L. et al. Nanosized UCMSC-derived extracellular vesicles but not conditioned medium exclusively inhibit the inflammatory response of stimulated T cells: implications for nanomedicine. Theranostics. 2017;7:270-84
58. Lit LC, Wong CK, Tam LS, Li EK, Lam CW. Raised plasma concentration and ex vivo production of inflammatory chemokines in patients with systemic lupus erythematosus. Ann Rheum Dis. 2006;65:209-15
59. Lacotte S, Decossas M, Le Coz C, Brun S, Muller S, Dumortier H. Early differentiated CD138(high) MHCII+ IgG+ plasma cells express CXCR3 and localize into inflamed kidneys of lupus mice. PLoS One. 2013;8:e58140
60. Teramoto K, Negoro N, Kitamoto K, Iwai T, Iwao H, Okamura M. et al. Microarray analysis of glomerular gene expression in murine lupus nephritis. J Pharmacol Sci. 2008;106:56-67
61. Rafei M, Hsieh J, Fortier S, Li M, Yuan S, Birman E. et al. Mesenchymal stromal cell-derived CCL2 suppresses plasma cell immunoglobulin production via STAT3 inactivation and PAX5 induction. Blood. 2008;112:4991-8
62. Lee HJ, Ko JH, Jeong HJ, Ko AY, Kim MK, Wee WR. et al. Mesenchymal stem/stromal cells protect against autoimmunity via CCL2-dependent recruitment of myeloid-derived suppressor cells. J Immunol. 2015;194:3634-45
63. Cho KA, Park M, Kim YH, Ryu KH, Woo SY. Poly I:C primes the suppressive function of human palatine tonsil-derived MSCs against Th17 differentiation by increasing PD-L1 expression. Immunobiology. 2017;222:394-8
64. Davies LC, Heldring N, Kadri N, Le Blanc K. Mesenchymal Stromal Cell Secretion of Programmed Death-1 Ligands Regulates T Cell Mediated Immunosuppression. Stem Cells. 2017;35:766-76
65. Ghiotto M, Gauthier L, Serriari N, Pastor S, Truneh A, Nunes JA. et al. PD-L1 and PD-L2 differ in their molecular mechanisms of interaction with PD-1. Int Immunol. 2010;22:651-60
66. Salmaninejad A, Valilou SF, Shabgah AG, Aslani S, Alimardani M, Pasdar A. et al. PD-1/PD-L1 pathway: Basic biology and role in cancer immunotherapy. J Cell Physiol. 2019;234:16824-37
67. Ohaegbulam KC, Assal A, Lazar-Molnar E, Yao Y, Zang X. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol Med. 2015;21:24-33
68. Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219-42
69. Schena F, Gambini C, Gregorio A, Mosconi M, Reverberi D, Gattorno M. et al. Interferon-gamma-dependent inhibition of B cell activation by bone marrow-derived mesenchymal stem cells in a murine model of systemic lupus erythematosus. Arthritis Rheum. 2010;62:2776-86
70. Liu Y, Jing H, Kou X, Chen C, Liu D, Jin Y. et al. PD-1 is required to maintain stem cell properties in human dental pulp stem cells. Cell Death Differ. 2018;25:1350-60
71. Zhou K, Guo S, Tong S, Sun Q, Li F, Zhang X. et al. Immunosuppression of Human Adipose-Derived Stem Cells on T Cell Subsets via the Reduction of NF-kappaB Activation Mediated by PD-L1/PD-1 and Gal-9/TIM-3 Pathways. Stem Cells Dev. 2018;27:1191-202
72. Luz-Crawford P, Noel D, Fernandez X, Khoury M, Figueroa F, Carrion F. et al. Mesenchymal stem cells repress Th17 molecular program through the PD-1 pathway. PLoS One. 2012;7:e45272
73. Yan Z, Zhuansun Y, Liu G, Chen R, Li J, Ran P. Mesenchymal stem cells suppress T cells by inducing apoptosis and through PD-1/B7-H1 interactions. Immunol Lett. 2014;162:248-55
74. Seo HH, Lee CY, Lee J, Lim S, Choi E, Park JC. et al. The role of nuclear factor of activated T cells during phorbol myristate acetate-induced cardiac differentiation of mesenchymal stem cells. Stem Cell Res Ther. 2016;7:90
75. Chang W, Lim S, Song BW, Lee CY, Park MS, Chung YA. et al. Phorbol myristate acetate differentiates human adipose-derived mesenchymal stem cells into functional cardiogenic cells. Biochem Biophys Res Commun. 2012;424:740-6
76. Vazirinejad R, Ahmadi Z, Kazemi Arababadi M, Hassanshahi G, Kennedy D. The biological functions, structure and sources of CXCL10 and its outstanding part in the pathophysiology of multiple sclerosis. Neuroimmunomodulation. 2014;21:322-30
77. Staron MM, Gray SM, Marshall HD, Parish IA, Chen JH, Perry CJ. et al. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8(+) T cells during chronic infection. Immunity. 2014;41:802-14
78. Fan L, Hu C, Chen J, Cen P, Wang J, Li L. Interaction between Mesenchymal Stem Cells and B-Cells. Int J Mol Sci. 2016 17
Author contact
![]() Corresponding authors: Jaesuk Yun, PhD and Sang-Bae Han, PhD, College of Pharmacy, Chungbuk National University, 194-21, Osongsaengmyung-1, Heungdeok, Cheongju, Chungbuk 28160, Republic of Korea. Phone: 82-43-261-2815; Fax: 82-43-268-2732; E-mails: jyunac.kr and shanac.kr.
Corresponding authors: Jaesuk Yun, PhD and Sang-Bae Han, PhD, College of Pharmacy, Chungbuk National University, 194-21, Osongsaengmyung-1, Heungdeok, Cheongju, Chungbuk 28160, Republic of Korea. Phone: 82-43-261-2815; Fax: 82-43-268-2732; E-mails: jyunac.kr and shanac.kr.