Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Origin of MCs
Regulation of MC...
MC Deficiency Models: Historical...
Proteomic and Genomic Expression...
Mediators and Other Compounds...
Mechanisms Contributing to MC...
Heterogeneity and Versatility of...
Potential Physiologic and...
Potential Role(s) of MCs as...
Potential Role of MCs as Repair...
Potential Role of MCs as Repair...
Possible Role of MCs in...
Role of MCs as Inflammatory...
Potential Role of MCs in Tumor...
Neoplastic Disorders of MCs and...
New Treatment Concepts:...
Concluding Remarks and Future...
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(23):10743-10768. doi:10.7150/thno.46719 This issue Cite
Review
Mast cells as a unique hematopoietic lineage and cell system: From Paul Ehrlich's visions to precision medicine concepts
Peter Valent1 ![]() , Cem Akin2, Karin Hartmann3, Gunnar Nilsson4, Andreas Reiter5, Olivier Hermine6, Karl Sotlar7, Wolfgang R. Sperr1, Luis Escribano8, Tracy I. George9, Hanneke C. Kluin-Nelemans10, Celalettin Ustun11, Massimo Triggiani12, Knut Brockow13, Jason Gotlib14, Alberto Orfao8, Petri T. Kovanen15, Emir Hadzijusufovic1,16, Irina Sadovnik1, Hans-Peter Horny17, Michel Arock18, Lawrence B. Schwartz19, K. Frank Austen20, Dean D. Metcalfe21, Stephen J. Galli22
, Cem Akin2, Karin Hartmann3, Gunnar Nilsson4, Andreas Reiter5, Olivier Hermine6, Karl Sotlar7, Wolfgang R. Sperr1, Luis Escribano8, Tracy I. George9, Hanneke C. Kluin-Nelemans10, Celalettin Ustun11, Massimo Triggiani12, Knut Brockow13, Jason Gotlib14, Alberto Orfao8, Petri T. Kovanen15, Emir Hadzijusufovic1,16, Irina Sadovnik1, Hans-Peter Horny17, Michel Arock18, Lawrence B. Schwartz19, K. Frank Austen20, Dean D. Metcalfe21, Stephen J. Galli22
1. Department of Medicine I, Division of Hematology & Hemostaseology and Ludwig Boltzmann Institute for Hematology and Oncology, Medical University of Vienna, Austria.
2. Division of Allergy and Clinical Immunology, University of Michigan, Ann Arbor, MI, USA.
3. Division of Allergy, Department of Dermatology, University of Basel, Basel, Switzerland.
4. Department of Medicine Solna & Mastocytosis Centre, Karolinska Institutet and Karolinska University Hospital, Stockholm, Sweden.
5. Department of Hematology and Oncology, University Hospital Mannheim, Heidelberg University, Mannheim, Germany.
6. Imagine Institute Université Paris Descartes, Sorbonne, Paris Cité, Centre national de référence des mastocytoses, Paris, France.
7. Institute of Pathology, Paracelsus Medical University Salzburg, Austria.
8. Servicio Central de Citometria (NUCLEUS), Centro de Investigacion del Cancer (IBMCC; CSIC/USAL and CIBERONC) and Department of Medicine, University of Salamanca, Spain.
9. Department of Pathology, University of Utah, Salt Lake City, UT, USA.
10. Department of Hematology, University Medical Center Groningen, University of Groningen, The Netherlands.
11. Division of Hematology-Oncology and Transplantation, Department of Medicine, University of Minnesota, Minneapolis, MN, USA.
12. Division of Allergy and Clinical Immunology, University of Salerno, Italy.
13. Department of Dermatology and Allergy Biederstein, Technical University of Munich, Germany.
14. Stanford Cancer Center, Stanford University School of Medicine, Stanford, CA, USA.
15. Wihuri Research Institute, Helsinki, Finland.
16. Department of Companion Animals and Horses, Small Animal Clinic, Internal Medicine, University of Veterinary Medicine Vienna, Austria.
17. Institute of Pathology, Ludwig Maximilian University, Munich, Germany.
18. INSERM UMRS1138, Centre de Recherche des Cordeliers, Paris, France.
19. Department of Internal Medicine, Division of Rheumatology, Allergy & Immunology, Virginia Commonwealth University, Richmond, VA, USA.
20. Division of Allergy and Immunology, Department of Medicine, Brigham and Women's Hospital, Harvard Medical School, Boston.
21. Laboratory of Allergic Diseases, NIAID, NIH, Bethesda, MD, USA.
22. Departments of Pathology and of Microbiology and Immunology, and the Sean N. Parker Center for Allergy and Asthma Research, Stanford University School of Medicine, Stanford, USA.
*This paper, reflecting both the more recent developments in the field and the presentations and discussions at a meeting held in 2015 to mark the 100th Anniversary of the death of Paul Ehrlich, is dedicated to the pioneering work and scientific achievements of this remarkable scientist.
Received 2020-4-6; Accepted 2020-8-6; Published 2020-8-29
Abstract

The origin and functions of mast cells (MCs) have been debated since their description by Paul Ehrlich in 1879. MCs have long been considered 'reactive bystanders' and 'amplifiers' in inflammatory processes, allergic reactions, and host responses to infectious diseases. However, knowledge about the origin, phenotypes and functions of MCs has increased substantially over the past 50 years. MCs are now known to be derived from multipotent hematopoietic progenitors, which, through a process of differentiation and maturation, form a unique hematopoietic lineage residing in multiple organs. In particular, MCs are distinguishable from basophils and other hematopoietic cells by their unique phenotype, origin(s), and spectrum of functions, both in innate and adaptive immune responses and in other settings. The concept of a unique MC lineage is further supported by the development of a distinct group of neoplasms, collectively referred to as mastocytosis, in which MC precursors expand as clonal cells. The clinical consequences of the expansion and/or activation of MCs are best established in mastocytosis and in allergic inflammation. However, MCs have also been implicated as important participants in a number of additional pathologic conditions and physiological processes. In this article, we review concepts regarding MC development, factors controlling MC expansion and activation, and some of the fundamental roles MCs may play in both health and disease. We also discuss new concepts for suppressing MC expansion and/or activation using molecularly-targeted drugs.
Keywords: histamine, IgE receptor, KIT, mast cell activation, mastocytosis, tryptase
Introduction
Mast cells (MCs) are tissue-resident, multifunctional effector cells of the innate immune system that can also substantially contribute to adaptive immune responses. Historically, MCs were described and originally named by Paul Ehrlich in 1879 based on their unique dye-staining properties [1]. Ehrlich also described the blood basophil and certain morphologic similarities between both cell types. However, the origin of MCs, and their roles in health and disease, remained mysteries for many decades. Two key clues in defining the functions of MCs were the demonstration that MCs contain histamine and can bind immunoglobulin E (IgE) [2-6]. Based on these observations and subsequent preclinical and clinical studies, MCs are now recognized as key effector cells in IgE-dependent allergic inflammation [3-7].
Today, the contributions of MCs to allergic and other inflammatory reactions are well established [3-11]. MCs express high-affinity receptors for IgE and produce numerous biologically active substances, some of which are stored in cytoplasmic granules for rapid release [2-4]. It is now known that MCs may be activated by many different agents. These include specific antigens (allergens) through allergen-specific IgE and IgE receptors, by certain cytokines, anaphylatoxins, certain neuropeptides, and exogenous toxins, and also by IgG immune complexes and complement, certain drugs, and products of bacteria or other pathogens [2-9, 11]. MCs activated by such mechanisms can release their granule-stored mediators, including histamine. In addition, activated MCs may synthesize and secrete many other types of mediators, such as lipid mediators, cytokines and chemokines, and thereby trigger local and systemic reactions [2-7]. Although MC activation is observed in a variety of inflammatory, infectious, and other reactive conditions, it has been best characterized during IgE-dependent allergic reactions.
Human MCs have the ability to re-granulate after degranulation and thereby participate in multiple cycles of activation with mediator secretion [8]. Moreover, certain MCs resident constitutively within tissues have a remarkably long life span, ranging from months to years, contrasting with the much shorter life span of basophils and other type of granulocytes [9-11]. Some MC populations, such as those induced in the gut of mice during infections with certain parasites, can rapidly expand and then drop back to approximately baseline levels upon resolution of the infection [9, 12, 13]. Thus, in addition to the long-term, relatively stable MC populations found in many tissues, shorter-lived populations of MCs also appear to exist. These MCs can expand and contract in particular sites in response to local processes, including parasite infections.
However, the rapid expansion of MC populations in multiple organs, including the skin, respiratory system and gastrointestinal tract, is also observed in mice, rats and cynomolgus monkeys injected with the major mast cell growth factor stem cell factor (SCF), also known as KIT ligand [9, 12-14]. Upon cessation of SCF injections, the numbers of these expanded MC populations can rapidly return toward baseline values [12-14]. These findings, along with the observations that mucosal MCs are absent in T cell-deficient mice, indicate that tissue MC populations may be responsive to changes in ambient levels of soluble SCF, or, perhaps, to changes in membrane-bound SCF or other cytokines.
In this article, we will discuss the progress that has been made over the last decades in addressing the origin and functions of MCs in health and disease, and regarding their neoplastic expansion. We will also discuss current diagnostic and therapeutic approaches to diseases involving MCs, particularly neoplastic disorders.
Origin of MCs
After their description and naming by Paul Ehrlich in 1879 [1], MCs were soon identified as tissue-resident cells in multiple organs, prompting discussions about their origin and functions. It was proposed initially that MCs were derived from blood basophils or from a local histiocytic progenitor. This debate lasted many decades, as no experimental model was available to resolve the issue. However, approximately 100 years after their discovery, the origin of MCs from transplantable hematopoietic stem cells was convincingly demonstrated. This was done in a series of elegant transfer-experiments (and other studies) in mice by Yukihiko Kitamura and his colleagues [15-18]. Later, the origin of MCs from transplantable hematopoietic stem cells was confirmed in humans [10]. In both species, MCs were found to develop from immature (CD34+) hematopoietic precursor cells when these were cultured in the presence of certain cytokines or on stromal cells [19-23].
Subsequent attempts evaluated whether MCs were directly derived from hematopoietic stem cells, from a later progenitor, or even from a mature leukocyte. However, all attempts to grow human MCs from highly purified blood monocytes or blood basophils failed, regardless of the cytokines or culture conditions employed [24]. Moreover, in colony assay experiments, human MCs were found to grow preferentially in pure MC colonies or in colonies containing MCs and basophil-like cells [24, 25]. Based on these and more recent observations a bi-committed MC/basophil progenitor cell has been postulated, but the existence of such a cell is still under debate. By contrast, some MCs were also detected in mixed colonies derived from pluripotent stem cells [24, 25]. Based on these observations, human MCs were considered to derive directly from pluripotent CD34+/KIT+ stem cells in various organ systems. Such multi-lineage and MC-committed CD34+ stem cells are detectable in the bone marrow (BM), peripheral blood (PB) and in the peripheral tissues in most extramedullary organs [9, 22-27].
Based on such observations, most CD34+/KIT+ MC progenitors are now considered to originate in the BM, to translocate from the BM into the PB, and to enter peripheral tissue sites after homing to various organs (Figure 1A) [9, 26, 27]. In addition, progenitors with the potential to generate MCs have been identified in the yolk sac [28, 29] and white adipose tissue of mice [30]. These latter observations have supported the assumption that BM-independent pools of self-renewing stem cells giving rise to MC progenitors may also develop in local tissue sites early during prenatal development; these cells then can be maintained (via the self-renewal capacity of local stem cells) throughout the lifetime of the organism (Figure 1B).
Development of human mast cells from stem and progenitor cells. A. Development of mast cells from uncommitted bone marrow-derived stem and progenitor cells. In adult humans, most hematopoietic stem cells (HSC) and mast cell-committed progenitor cells (cells depicted as containing only one cytoplasmic granule in this figure) are considered to originate from the bone marrow (BM) under physiologic (normal) conditions. After leaving the BM, these stem/progenitor cells are detectable in the peripheral blood and after homing to various tissues and exposure to stem cell factor (SCF) these cells undergo maturation and develop into mature mast cells. Note that in the BM, only a few HSC will become mast cell-committed. This is because, apart from SCF, other hematopoietic cytokines are also present and induce differentiation into other (non-mast cell) cell types. B. Stem and progenitor cell evolution and distribution during prenatal development. During embryonal and fetal development, primitive (immature) mesenchymal stem cells develop in the yolk sac and give rise to multipotent HSC. Both types of cells may be distributed through the blood stream and can invade other organ systems, including the liver, BM and skin (green arrows). Some of these cells may form a local pool of self-renewing HSC in extramedullary organs before birth and may be able to maintain local pools of HSC and mast cell progenitors via self-renewal in adulthood. After birth, the largest pool of self-renewing HSC is located in the BM. These HSC can leave the BM and can home to extramedullary organs (red colored arrows) where they can undergo mast cell differentiation (blue arrows). However, pools of self-renewing HSC and HSC-derived mast cell progenitors may also originate from yolk sac-derived stem cells and may later persist in adults. These cells develop into mast cells in local organs independent of the BM. There may be additional trafficking routes followed by certain subsets of HSC and mast cell progenitors homing from one to another organ system (dotted line-arrows); however, robust data supporting such HSC trafficking routes in humans are not available. Abbreviations: GI tract, gastrointestinal tract; LNs, lymph nodes.
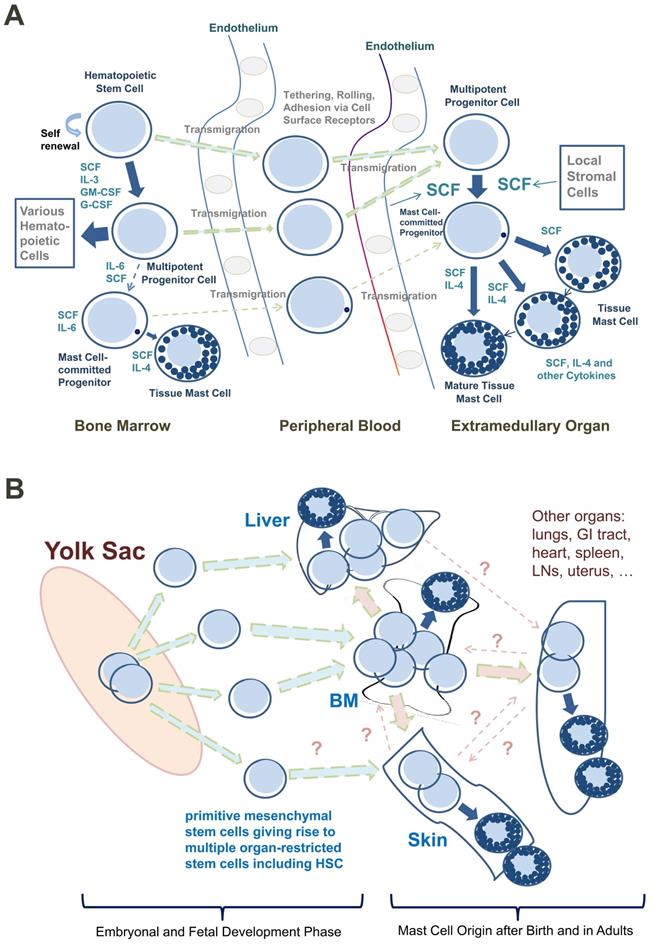
The phenotypic properties of MC-committed progenitor cells have been described in mice and humans [22, 26, 27, 31-37]. In humans, these cells express CD34, KIT (CD117), the interleukin-3 receptor (IL-3R) and CD13, but display only low, if any, amounts of FcεRI [31-37]. Later, during MC development, MC precursors stop expressing CD34 and the IL-3R alpha chain (CD123) and start expressing higher levels of FcεRI and KIT [27, 34-36]. Importantly, MC progenitors display a number of adhesion molecules through which these cells may enter peripheral (extramedullary) organs and tissues. For example, in the mouse, beta7 integrin mediates the adhesion needed for transendothelial movement of BM-derived MC progenitors into peripheral tissues [38].
Despite the many new insights into the origin and development of MCs, several questions remain. First, it is not clear whether human MCs also can be derived from pre-formed local pools of CD34+ stem cells within extramedullary organs. Such a local pool of CD34+ MC precursors might be established early in life or even during embryogenesis (Figure 1B). Another related question is whether MCs in local tissue sites can sometimes emerge from a bi-committed progenitor cell, giving rise to MCs and macrophages or MCs and other leukocytes. In other words, although the direct origin of MCs from blood-born mature monocytes has been formally excluded [24], it remains unknown whether some MCs originate from local bi-committed progenitor cells giving rise to MCs and macrophages at local tissue sites. In this regard, it is worth noting that MCs and macrophages share functional and phenotypic similarities and that advanced systemic mastocytosis (SM) is often accompanied by chronic myelomonocytic leukemia (CMML) [39-42]. On the other hand, no association between SM and histiocytic tumors has yet been described. Finally, while definitive information about the exact relationship between human mast cells and basophils remains to be ascertained, most available evidence is consistent with the conclusion that the mature basophil and mast cell lineages are distinct [24, 43, 44].
Regulation of MC Differentiation, Maturation and Survival
In the mouse, a number of interleukins (ILs), including IL-3, IL-4, IL-9 and IL-10, promote the development of MCs from their BM progenitor cells (Table 1) [9, 22, 45-48]. However, apart from the support provided by the IL-network, development of mouse MCs is also regulated by tissue-resident stromal cells [9, 20, 21]. In a series of pivotal studies, SCF was identified as a major player in stem cell- and MC development and survival in the mouse [9, 12, 26, 49, 50]. Deprivation of MC-cultures of SCF (or of SCF-expressing stromal cells) results in MC growth arrest and apoptosis [9, 26]. Correspondingly, Sl/Sld mice (now designated C57BL/6-KitlSl/Sl-d mice) lacking (functional) SCF and W/W-v mice (now designated WBB6F1-KitW/W-v mice) lacking a functional SCF receptor, KIT, exhibit a MC-deficient phenotype [9, 15-18, 26].
Effects of cytokines on growth and differentiation of mouse and human mast cells
| Effects on proliferation (p) or maturation (m) of: | ||||
|---|---|---|---|---|
| Mouse* | Human** | |||
| Cytokine | MC precursors | MCs | MC precursors | MCs |
| SCF | + (p,m) | + (p,m) | + (p,m) | + (m) |
| IL-3 | + (p,m) | + (p,m) | + (p) | - |
| IL-4 | + (p,m) | + (p,m) | +/- (p,m)*** | +/- (p,m)*** |
| IL-5 | + (p,m) | + (p,m) | - | - |
| IL-6 | + (p) | +/- (p,m) | + (p) | - |
| IL-9 | + (p,m) | + (p,m) | - | - |
| IL-10 | + (p,m) | + (p,m) | - | - |
| IL-11 | - | - | - | - |
| IL-12 | - | - | - | - |
| IL-13 | + (p,m) | + (p,m) | n.k. | +/- (p) |
| IL-15 | + (p,m) | + (p,m) | n.k. | n.k. |
| IL-33 | + (p) | + (p) | n.k. | + (m) |
| GM-CSF | + (p) | - | - | - |
| NGF | + (p,m) | + (m) | - | - |
Abbreviations: MC, mast cell; SCF, stem cell factor; (p), proliferation; (m), maturation; IL, interleukin; n.k., not known; GM-CSF, granulocyte-macrophage colony-stimulating factor; NGF, nerve growth factor. *In most studies mouse mast cell precursors were obtained from the bone marrow and studies on mouse mast cells refer to data obtained with IL-3-dependent bone marrow-derived mast cells. **Most studies on human MC precursors were performed using cord blood cell progenitors, and most studies on mature MCs were performed using isolated human lung mast cells, skin mast cells, or mast cells cultured from their cord blood progenitors using IL-3, SCF and IL-6. ***Under certain conditions, IL-4 can even induce apoptosis in MCs and MC precursor cells.
In 1990, human SCF was cloned and found to act as a ligand of human KIT [51]. Shortly thereafter, recombinant human SCF was found to induce the development of MCs from their (CD34+/KIT+) progenitor cells in vitro [52-54]. In stromal cells (e.g. fibroblasts and endothelial cells) SCF is often expressed in soluble and membrane-bound forms and can serve as a multifunctional MC regulator. In fact, membrane-bound SCF acts as an adhesion/homing receptor for KIT+ stem cells and MCs [9, 11, 26]. By contrast, soluble SCF, once released from stromal cells, acts as a MC chemoattractant and as an activation-inducing cytokine [55-62].
In line with these observations, administration of recombinant human SCF to humans resulted in the development of increased numbers of MCs at SCF injection sites [63, 64]. In addition, there was evidence for local activation of MCs, specifically reflected in wheal and flare reactions at the skin injections sites in these studies. Moreover, in two participants, systemic signs and symptoms suggestive of anaphylaxis occurred [63, 64]. All in all, SCF appears to be a master-regulator of human MCs, promoting the transmigration of circulating MC progenitors into tissues, their local development into mature MCs, the migration and survival of such mature MCs, and MC activation.
Other cytokines may also trigger MC development at certain stages of stem cell differentiation [9, 34, 45-48]. In a very early phase of human MC development, multi-lineage cytokines such as IL-3 may expand the pool of CD34+/KIT+ stem- and progenitor cells and thus facilitate human MC development (Table 1) [34-36, 52, 65]. A similar effect was also described for IL-6 [66-68]. Therefore, when applied in early phases of culture, both cytokines can promote SCF-induced MC development in vitro [34, 36, 52, 65-68]. Similar observations have been made in the mouse system. However, continuous application of IL-3 promotes the differentiation of basophils and other myeloid cells, while downregulating expression of KIT and counteracting differentiation of MCs [69, 70]. Correspondingly, late-stage MC progenitors and normal mature, resting human tissue MCs lack IL-3 receptors [34, 36, 71]. In these later phases of MC development, other cytokines, such as IL-4, may be involved in the differentiation and maturation of MCs [72-74]. In particular, IL-4 has been described as triggering expression of chymase, certain adhesion molecules, and high-affinity IgE receptors in human MC precursor cells [72-75]. Depending on the environment and organ, IL-4 may even promote the proliferation of maturing human MCs [75]. However, IL-4 has also been described as downregulating KIT expression and KIT-dependent differentiation of human MC precursor cells and causing apoptosis of human MC progenitors and of lung MCs that express only tryptase (MCT), unless IL-6 is also present [70, 76-78].
As mentioned, many different ILs and other cytokines can be involved in MC differentiation, maturation, and survival in the mouse. These cytokines can trigger the expression of multiple proteases in MCs and their progenitors, thus influencing the cells' phenotype, and/or can influence expression of molecules that enhance MC survival [12, 79-82]. It seems likely that all of these cytokines act in concert to promote MC differentiation and maturation in various tissues and organs [9, 11, 80, 81, 83]. Since cytokine-expression depends on the tissue-type and pathology of the affected organ, certain MC phenotypes appear to be tissue-dependent and pre-determined, for example by the cytokine-network characteristics of the underlying condition and/or pathology [7, 9, 11, 80, 81, 83]. In addition, MC phenotypes may change depending on the organ system and the local tissue microenvironment [9, 11, 83-87]. Table 1 provides an overview of the effects of cytokines on the development and maturation of mouse and human MCs.
The signal transduction pathways and effector molecules downstream of MC cytokine receptors have been analyzed extensively and a detailed description is beyond the scope of this review. However, a few important signaling nodes and effector molecules should be mentioned, as these are now also recognized as potential targets of therapy. One important target appears to be the PI3-kinase/AKT/mTOR pathway [88-91]. Disruption of this pathway is associated with a decreased ability of MC precursors to develop into mature MCs and also impairs MC activation [88-91]. The RAS/MEK/ERK pathway is also potentially important for MC development and function [92-94]. Finally, the JAK-STAT5 pathway has been implicated in KIT-dependent growth and survival of MCs [91, 95]. All three pathways may be highly active, particularly in neoplastic MCs exhibiting the transforming KIT mutant D816V. Depending on additional mutations (some of them directly affecting these pathways, like oncogenic RAS mutations), one, two, or all three of these pathways seem to play important roles in oncogenesis and drug resistance [91, 95].
Among several survival molecules acting downstream of AKT, mTOR, RAS or STAT5, members of the BCL2 family (such as MCL-1, BAX or BCL-xL, and several heat shock proteins, like heme oxygenase-1, HSP70 and HSP90) play critical roles in the survival of normal and/or neoplastic human MCs [96, 97]. Effects of such anti-apoptotic molecules may not only explain SCF-dependent survival of normal MCs, but also the accumulation of neoplastic MCs in patients with mastocytosis [96]. Indeed, MCs in such patients express excess amounts of these survival-promoting molecules, and pharmacologic inhibition of these survival molecules is associated with reduced survival and increased apoptosis in neoplastic MCs [96]. Table 2 provides a list of critical signaling and survival molecules relevant to KIT-dependent growth and survival in normal and neoplastic MCs.
Critical signaling and survival molecules relevant to KIT-dependent growth and survival in normal and neoplastic human mast cells
| Expressed in resting and/or activated human mast cells (MCs) | ||||
|---|---|---|---|---|
| Molecule | Resting MCs | FcεRI cross-linked MCs | SCF-activated MCs | KIT D816V+ neoplastic MCs |
| Phosphorylated (p) signaling molecules | ||||
| pERK | - | + | + | ++ |
| pAKT | - | + | + | ++ |
| pS6 | - | + | + | ++ |
| pSTAT5 | - | + | + | ++ |
| pBTK | - | + | +/- | +/- |
| Survival-related anti-apoptotic molecules | ||||
| BCL-2 | +/- | +/- | + | + |
| MCL-1 | - | - | +/- | ++ |
| BCL-xL | - | - | +/- | ++ |
| BIM | - | - | - | - |
| BAX | - | - | - | - |
| PUMA | - | - | - | - |
| NOXA | - | - | - | - |
| HO-1/HSP32 | - | - | +/- | ++ |
| HSP70 | - | - | + | ++ |
| HSP90 | - | - | + | ++ |
Abbreviations: MCs, mast cells; STAT5, signal transducer and activator of transcription 5; BTK, Bruton´s tyrosine kinase; HO-1, heme oxygenase 1.
Finally, a number of transcription factors are considered to be involved in the development and differentiation of mouse MCs, including, among others, GATA2, microphthalmia (mi) transcription factor (MITF), and STAT5 [98-102]. A role for STAT5 in the growth of neoplastic human MCs has also been postulated [91, 95]. Whether the transcription factors GATA2 and MITF also play a role in the development or function of normal and/or neoplastic human MCs remains unknown.
MC Deficiency Models: Historical Aspects and Recent Developments
As outlined above, the development and differentiation of human or mouse MCs from their uncommitted hematopoietic stem- and progenitor cells depends largely on the presence of a functional KIT receptor and its ligand, SCF. KIT is a tyrosine kinase receptor that acts as a facultative onco-protein and regulates the growth, differentiation and survival of germ cells, hematopoietic stem cells, MCs, interstitial (intestinal pacemaker) cells of Cajal (ICCs), and melanocytes. In mice, ´loss-of-function´ of KIT (for example in WBB6F1-KitW/W-v mice) results in macrocytic anemia, a profound MC deficiency, deficiency of germ cells, lack of ICCs, depigmentation of the skin, and failure of neutrophils to express inhibitory leukocyte immunoglobulin (Ig)-like receptor B4 [9, 15, 26, 103-105]. Apart from Kit-deficient mice, there are a number of additional MC deficiency models that implicate specific target pathways and molecules [106]. MC deficiency also may be due to a lack of functionally active SCF (also known as steel factor, SL), for example in Sl/Sld (C57BL/6-KitlSl/Sl-d) mice [16, 106]. These mice express only the extracellular, non-membrane associated, domain of SCF. Another model of MC deficiency is based on the lack of a functional MITF [106, 107]. MITF is involved in the regulation of expression of KIT in hematopoietic stem cells, MC progenitors and melanoblasts, and is itself regulated by KIT activity [106, 107]. In addition, MITF regulates the production of a number of mediators, including specific proteases in MCs and MC progenitor cells [108-113].
Initial studies investigating MC functions in vivo principally focused on MC-deficient mice whose MC deficiency was due to diminished function of KIT, such as in KitW/W-v mice, or decreased expression of KIT, such as in KitW-sh/W-sh mice. The MC deficiency of KitW-sh/W-sh mice is based on a large genetic inversion upstream of the c-kit locus which disrupts corin [114] and results in a profound MC deficiency as well as other phenotypic abnormalities, including increased levels of neutrophils and basophils [115, 116]. Both KitW/W-v mice and KitW-sh/W-sh mice develop tissue MC populations after adoptive transfer of MCs derived in vitro from the hematopoietic cells of the corresponding wild type (WT) mice or from other normal or genetically-altered mice of suitable strains, thus producing 'mast cell knock-in mice' [117]. After adoptive transfer, such in vitro-derived MC populations, which often consist of BM -derived cultured MCs (BMCMCs), gradually acquire multiple phenotypic characteristics which resemble those attributed to the native MC populations in the corresponding anatomical sites of WT mice [84-86, 117]. Accordingly, KitW/W-v mice and KitW-sh/W-sh mice engrafted with Kit WT MCs or MCs bearing specific genetic abnormalities have been used to investigate the functions of MCs and certain MC receptors or products in diverse biological responses and models of disease [115].
However, as detailed elsewhere [117-119], such experiments are subject to several admonitions, including: 1) the phenotypes and anatomical distribution of the adoptively-transferred MCs at diverse anatomical sites may not be identical to that of the corresponding native MC populations in WT mice, particularly after intravenous injection of such in vitro-derived MCs [117]; 2) the reality that a biological response in KitW/W-v or KitW-sh/W-sh mice can be “normalized” after the adoptive transfer of MCs to such mice indicates that MCs can express the detected function in the context of those mutant mice (which have multiple phenotypic abnormalities in addition to their profound MC deficiency), but does not by itself prove that MCs are necessary for the same functions in WT mice; and 3) the limited strain background of the KitW/W-v or KitW-sh/W-sh mice used for early studies of MC function may have revealed some MC roles that are strongly expressed in mice on the C57BL/6 or related backgrounds, but may be expressed weakly, if at all, on other strain backgrounds, such as BALB/c [119].
Given such concerns about the general utility of KitW/W-v or KitW-sh/W-sh mice for studies of MC function, several groups have attempted to generate transgenic mice that express MC deficiency by mechanisms independent of those reflecting abnormalities in KIT or its ligand. As reviewed elsewhere [115], a variety of mouse strains have now been developed which constitutively or inducibly lack, or are deficient in, some or all MC populations by KIT-independent mechanisms. While each of these more recent models of MC deficiency has abnormalities in addition to the MC deficiency (e.g., two of these strains have moderately reduced numbers of basophils [120, 121]), these non-MC abnormalities are less pronounced than those in KitW/W-v and KitW-sh/W-sh mice. The availability of these newer strains of MC-deficient mice should further advance our understanding of the roles of MCs in mammalian biology.
To date, there has been no description of humans with a genetically-determined MC deficiency. However, humans with a single loss-of-function mutation in KIT can exhibit the clinical picture of piebaldism, a rare autosomal dominant disorder characterized by symmetrical pigment defect reflecting a localized lack of melanocytes in the skin and lack of melanin in the hair shafts. This condition may present as a patch of white hair (poliosis) in the forehead and/or a patch of non-pigmented amelanotic skin (leukoderma) [122], a pigment phenotype much like that of mice with a single loss-of-function mutation in c-Kit [9, 15, 106]. Although the piebald phenotype also can reflect loss-of-function mutations in genes other than KIT (e.g., SNAI2 [123]), piebaldism was arguably the first genetic transmitted pathology recognized in recorded history by the ancient Greeks. In these patients, the ability of hematopoietic progenitor cells to differentiate into MCs in response to SCF may be slightly impaired (P.V., personal observation). However, based on the ´mosaic-pattern´ of the defect, no complete MC deficiency is found in these patients. Although some patients with AIDS or various genetically-determined immunodeficiencies may exhibit reduced numbers of MCs in the gastrointestinal mucosa [124], no other natural or clinical model of MC-deficiency has yet been identified in humans.
However, it has been described that patients with chronic myeloid leukemia (CML) receiving imatinib, a strong KIT inhibitor, for 2 or more years can develop a profound MC deficiency [125]. The long latency until MC deficiency occurs in these patients is best explained by the long time required by MC progenitors to develop into mature MCs and the long life-span (years) of some populations of mature MCs [9, 10, 26, 34, 36]. Given the evidence that the development of human tissue MCs from their progenitor cells is critically dependent on SCF and KIT, it is not unexpected that long-term treatment with strong inhibitors of KIT-activation can produce MC deficiency. The clinical relevance of such an induced MC-deficiency, however, remains to be determined.
Proteomic and Genomic Expression Profiling Confirms that MCs form a Unique Lineage within the Hematopoietic Cell System
It is generally acknowledged that MCs are distinct hematopoietic cells that display a unique profile of leukocyte differentiation antigens [40, 43]. Since tissue MCs display a number of myeloid determinants, these cells are commonly considered to be ´myeloid cells´ [40, 43]. Initially, protein profiling of human MCs was largely restricted to biochemical investigations and antibody-typing. Thus, between 1989 and 2005, extensive antibody-typing was performed to establish the phenotype of primary human tissue MCs obtained from various organs [40, 43, 126, 127]. In all organs tested, MCs were found to display substantial amounts of CD9 and KIT (CD117) as well as certain myeloid surface antigens, including CD13 and CD33. The phenotype of primary tissue MCs was also compared to that of blood basophils and blood monocytes. These studies revealed that many other myeloid cell surface structures, like CD11b, CD14, CD15, or CD35, otherwise displayed by basophils and/or monocytes, are not expressed, or expressed only at trace amounts, on human MCs [40, 43, 128].
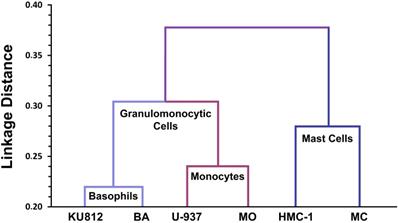
All in all, a detailed phenotypic comparison between MCs, monocytes and basophils, and their respective cell lines was consistent with the conclusion that MCs are neither related to basophils nor to blood monocytes (Figure 2) [128]. Later large-scale genomic studies, transcriptome analyses, and proteomic studies confirmed that the mRNA and protein expression profiles of MCs are unique and distinct within the family of hematopoietic cells [129-131]. Finally, KIT mutational studies in patients with systemic mastocytosis (SM) confirmed that MCs form a distinct cell lineage without a direct developmental relationship with basophils or other leukocytes [44].
Lineage relationships between human mast cells, basophils and monocytes based on CD antigen expression profiles. Human lung mast cells (MC), the human mast cell line HMC-1, normal blood basophils (BA), the human basophil cell line KU812, normal blood monocytes (MO) and the human monoblastic cell line U-937 were analyzed using a panel of 90 different CD antibodies provided by the Leukocyte Typing Workshops. Based on antibody-reactivities, linkage distance analyses were performed (Agis H, et al., Immunology. 1996; 87: 535-43). As expected, the linkage distance within the cell lineages examined (primary cells versus respective cell lines) is low. Primary basophils and monocytes and the respective lines were also found to be related phenotypically. By contrast, however, the phenotype of MC did not reveal a close relationship with BA or MO, neither in the cell line context nor in primary cells (primary MC vs primary BA). These data suggest that MC and BA form two separate (independent) hematopoietic cell lineages within the leukocyte family.
The notion that MCs form a separate hematopoietic cell lineage defined by specific cell surface antigens has major practical and clinical implications. First, the unique phenotype of MCs enables their detection and their enrichment from various tissues [43, 126, 127, 132]. Second, the counting and phenotyping of MCs critically contribute to the diagnostic work-up of patients with suspected SM [133-135]. In this context, it is noteworthy that the phenotype of neoplastic MCs in patients with SM is aberrant, recurrent, and unique. In particular, in contrast to MCs in normal or reactive tissues, MCs in SM usually display CD25 and often also CD2 and/or CD30 [133-136]. Currently, two of these antigens, CD2 and CD25, serve as minor diagnostic criteria for SM defined by the World Health Organization (WHO) [137-139]. The fact that these aberrantly expressed antigens are otherwise primarily expressed on lymphopoietic cells, has recently re-introduced a discussion about the nature and origin of normal and neoplastic MCs and, more importantly, re-enforced the concept that MCs form a unique hematopoietic cell lineage.
There are also additional antigens that are expressed aberrantly on neoplastic MCs, such as the IL-3 receptor alpha chain (CD123) [140]. Other cell surface antigens, including CD203c and the C5a receptor (C5aR) CD88, are usually expressed at higher levels on neoplastic MCs in SM compared to normal BM MCs [134, 141, 142]. Interestingly, some of these receptors are also expressed on 'reactive (activated) MCs' in various inflammatory conditions. For example, in patients with rheumatoid arthritis, MCs can express the C5aR CD88 [143]. Even CD25 may be detectable in reactive MCs in inflamed tissue sites (P.V., unpublished observation). Therefore, these surface antigens serve as minor, not major, criteria of SM. Table 3 provides a summary of relevant cell surface molecules expressed on normal, reactive and neoplastic MCs [43, 135, 144].
Cell surface antigens expressed on normal, reactive and neoplastic human mast cells
| Cell surface antigens expressed on normal, reactive and neoplastic human mast cells | ||||||
|---|---|---|---|---|---|---|
| Expressed on the surface of: | ||||||
| Antigen | CD | Lung MCs* | Skin MCs* | HMC-1 | MCs in ISM** | MCs in MCL** |
| LFA-2 | CD2 | - | - | +/- | +/- | -/+ |
| AMP-N | CD13 | + | + | + | + | + |
| LPS-R-r | CD14 | - | - | - | - | - |
| IL-2RA | CD25 | - | - | +/- | + | + |
| Ki-1 | CD30 | - | - | +/- | +/- | +/- |
| Siglec-3 | CD33 | + | + | + | + | + |
| HPCA-1 | CD34 | - | - | - | - | - |
| Leukosialin | CD43 | + | + | + | + | + |
| Hermes-R | CD44 | + | + | + | + | + |
| CLA | CD45 | + | + | + | + | + |
| ICAM-1 | CD54 | + | + | + | + | + |
| LFA-3 | CD58 | + | + | + | + | + |
| LAMP-3 | CD63 | + | + | + | + | + |
| C5aR | CD88 | -/+ | + | + | + | +/- |
| GM-CSFRA | CD116 | - | - | - | - | - |
| KIT | CD117 | + | + | + | + | + |
| IL-3RA | CD123 | - | - | - | - | +/- |
| L1CAM | CD171 | n.k. | + | +/- | n.k. | n.k. |
| ENPP3 | CD203c | +/- | +/- | +/- | + | + |
| FcεRI | n.c. | + | + | - | + | -/+ |
| MRGPRX2 | n.c. | - | + | - | n.k. | n.k. |
*Lung mast cells (MCs) from surgical tumor tissue samples and skin MCs from inflamed foreskin samples were examined. **Mast cells in patients with ISM and MCL usually express the KIT mutation D816V. All data were obtained from the available literature. Abbreviations: CD, cluster of differentiation; ISM, indolent systemic mastocytosis; MCL, mast cell leukemia; LFA-2, leukocyte function-associated antigen-2; AMP-N, aminopeptidase-N; IL-2RA, interleukin-2 receptor alpha chain; HPCA-1, human precursor cell antigen 1; LAMP-3, lysosomal associated membrane protein-3; C5aR, complement component 5a receptor; Ki-1, Kiel-antigen-1; CLA, common leukocyte antigen; ICAM-1, intercellular adhesion molecule-1; GM-CSFRA, granulocyte-macrophage colony-stimulating factor receptor alpha chain; ENPP3, ectonucleotide pyrophosphatase/phosphodiesterase 3; n.c., not (yet) clustered; n.k., not known.
Mediators and Other Compounds Produced by Tissue MCs
MCs contain many functionally defined and clinically relevant mediators in their cytoplasmic (secretory) granules [3, 4, 7, 145-152]. In human MCs, these include, among others, histamine, heparin, tryptase, carboxypeptidase, and chymase, which are stored in their metachromatic granules (Table 4). Other mediators, such as lipids (e.g. prostaglandin D2, leukotriene C4, platelet-activating factor, and sphingosine-1-phosphate), are primarily produced during MC activation. In general, MC-derived mediators are classified on the basis of their chemical structures and their specific biological functions, as well as their locations of storage or origin within MCs. Granule-derived mediators are often bound to heparin and many of them contribute to inflammatory (allergic) reactions [151-153]. Some MC-derived mediators, such as heparin, tissue type plasminogen activator (tPA), MC proteases and MC-derived cytokines (some of which appear to be pre-formed), are molecules characteristically associated with local tissue repair [154-159].
Selection of biologically relevant mediators that human mast cells produce and secrete
| Substance | Biologically relevant functions |
|---|---|
| Histamine | Vasodilation, vascular permeability, endothelial cell priming for leukocyte-rolling, neuroendocrine mediator, pro-inflammatory |
| Heparin | Co-factor of ATIII, of tPA and of tryptase, anti-inflammatory |
| PGD2 | Induces bronchoconstriction, activates endothelial cells, induces vasodilation and VEGF production, activates Th2 lymphocytes, eosinophils and basophils |
| Alpha tryptase | May promote mast cell activation |
| Beta tryptase | Fibrinogenolysis, mitogen for fibroblasts and endothelial cells, lipid-modifier, degrades VIP, endothelin, fibronectin, collagen, calcitonin gene‐related peptide, protease‐activated receptor 2, RANTES and eotaxin |
| Chymase | Lipid-modifier, degrades apolipoprotein B, VIP, fibronectin, vitronectin, bradykinin, HGF, SCF, C3a, and thrombin; induces smooth muscle cell and endothelial cell apoptosis, converts angiotensin (Ang) 1 to Ang 2, and big endothelin to endothelin, activates IL-1-beta and TGF-beta-1 |
| TNF-alpha | Endothelial cell and macrophage activation, induces apoptosis in smooth muscle cells and other perivascular cells, mediator of catabolic processes, tissue inflammation and tissue damage |
| IL-3 | Multipotent growth factor for myeloid cells, expands the pool of multilineage progenitor cells in the bone marrow, induces the differentiation of basophils, eosinophils, and macrophages, promotes activation of (primes) basophils and eosinophils |
| IL-8 | Induces chemotaxis (migration) and activation of granulocytes and monocytes, pro-inflammatory and angiogenic mediator |
| CCL2 | Induces leukocyte chemotaxis and activation |
| OSM | Promotes angiogenesis, fibrosis and tissue remodeling |
| VEGF | Promotes angiogenesis and vascular permeability |
| FGF | Promotes fibrosis and wound healing |
ATIII, anti-thrombin 3, tPA, tissue type plasminogen activator; PGD2, prostaglandin D2; VEGF, vascular endothelial growth factor; VIP, vasoactive intestinal peptide; RANTES, regulated on activation, normal T cell expressed and secreted; HGF, hepatocyte growth factor; SCF, stem cell factor; IL, interleukin; TGF, transforming growth factor; TNF, tumor necrosis factor; CCL2, CC-chemokine ligand 2; OSM, oncostatin M; VEGF, vascular endothelial growth factor; FGF, fibroblast growth factor.
In vitro-derived mouse MCs also can produce a wide variety of cytokines, chemokines and growth factors [11, 146-150] and some of these products also have been detected in isolated mouse peritoneal mast cells [11]. While some of these products apparently can be stored in certain MC granules, such as tumor necrosis factor (TNF) alpha or vascular permeability factor (VPF), also known as vascular endothelial growth factor (VEGF), the largest amounts of these substances are generally thought to be synthesized and directly secreted after MCs have been functionally activated [11, 146-150, 159, 160].
By contrast only a few cytokines and chemokines have been detectable in resting human MCs. In particular, normal human tissue MCs are considered to store some preformed TNF-alpha in their granules. However, normal human MCs do not express many other cytokines unless activated by external stimuli. By contrast, after IgE-dependent activation in vitro or exposure to activating cytokines such as SCF, human (and mouse) MCs can express and release several different cytokines, including interleukins (like IL-3) and chemokines [146-150, 160]. Most of these cytokines and chemokines are also produced in neoplastic MCs, where the activating KIT mutant D816V acts as a persistent stimulus for cytokine/chemokine synthesis. For example, KIT D816V-transformed neoplastic MCs display substantial amounts of oncostatin-M (OSM), IL-8 and the CC-chemokine ligand 2 (CCL2) also known as monocyte chemotactic protein 1 (MCP1), and CCL23 [161, 162]. However, other studies suggest that normal human MCs, either in the resting state or after activation via the FcεRI, do not generate multiple cytokines [129, 160, 163]. These observations highlight the need for additional research to ascertain the ability of normal vs. neoplastic human MCs to generate cytokines and chemokines under different conditions and states of activation.
Table 4 presents a summary of clinically relevant MC-derived mediators and cytokines. A comprehensive overview of all these molecules is beyond the scope of this article. However, it should be emphasized that several MC mediators, such as histamine, are clinically important as they contribute to the typical signs and symptoms of patients with IgE-dependent or independent allergies and patients with mastocytosis. Other mediators may be involved in tissue remodeling and repair processes, including fibrosis and angiogenesis [7, 152, 153, 161, 162]. Whether some of the specific signs and symptoms observed in advanced SM, such as weight loss (an established TNF effect) or fatigue, are triggered by MC-derived cytokines (e.g., during cytokine storm) is presently unknown.
Mechanisms Contributing to MC Mediator Release and Releasability
The ability of MCs and basophils to respond to IgE-dependent or -independent stimuli (i.e. the cells´ “releasability”) is determined by many factors. These include genetic and epigenetic effects, the type and dose of agonist (e.g., specific allergen recognized by the cells' FcεRI-bound IgE antibodies), the local micro-environment, the levels of ambient cytokines (which may either directly induce mediators secretion or modify the cell's responses to other stimuli), and the underlying disease pathology [164, 165]. The features and severity of reactions evoked by MC activation depend on additional factors, such as the type and amounts of mediators released and the number and location of reacting MCs (and other immune cells including basophils) involved in the response [166, 167]. When MC activation is substantial, this can result in systemic anaphylaxis and, in certain settings, the criteria for a MC activation syndrome (MCAS) may be fulfilled [168, 169]. However, MCs also can undergo activation chronically or locally in a number of inflammatory conditions without clinical signs or symptoms of systemic MC activation, so that MCAS criteria are not fulfilled [170].
In IgE-dependent allergic disorders, IgE-receptor cross-linking is usually the critical event that leads to mediator release [4-7]. Certain cytokines promote such IgE-dependent reactions. In humans, SCF augments MC IgE-dependent histamine release [60-62]. When applied at higher concentrations and/or for longer periods of time (>90 minutes at ≥100 ng/ml), SCF can directly induce histamine secretion in primary human tissue MCs [60-62]. Similar effects of SCF have been described in mice in vivo [57] and in humans injected subcutaneously with recombinant human SCF [63]. In those settings, SCF induced degranulation that resembled IgE-induced anaphylactic degranulation [57, 63]. However, it is worth noting that the activation of MCs by certain cytokines can differ substantially from anaphylactic degranulation. In particular, ultrastructural analyses have shown that the stimulation of MCs with certain cytokines results in piecemeal degranulation in which vesicles are thought to transport mediators to the cell surface in the absence of classical degranulation and in which the slow release of mediator substances follows. In contrast, IgE-dependent activation is usually followed by rapid degranulation and the release of stored MC mediators via the process of rapid compound exocytosis [171].
Apart from SCF and IgE-dependent stimuli, many other agonists also induce mediator release in human MCs. For example, C3a and C5a induce histamine release in human CD88+ skin MCs [172-174], and in a small subset of CD88+ lung MCs that express both tryptase and chymase (MCTC type) [175]. In many other extra-cutaneous organ systems, MCs usually lack CD88 and do not respond to C3a or C5a unless an inflammatory reaction is ongoing, such as in rheumatoid arthritis [143]. In the mouse, genetic and MC transfer studies indicate that C3a and C5a, acting via the C3aR and the C5aR respectively, can directly induce degranulation of skin MCs and that the endogenous production of such anaphylatoxins may also augment the intensity of IgE-dependent MC activation in the skin [176].
Several seminal review articles have discussed the various biochemical and signal transduction pathways that underlie IgE-receptor-mediated, cytokine-induced or C3a/C5a-mediated activation, degranulation, and mediator release in MCs [2-6, 9]. Figure S1 provides an overview of IgE-receptor-dependent and KIT-dependent signaling cascades. For more details, the reader is referred to the available literature. It is important to note that several of these activation-linked events and downstream signaling molecules are emerging novel targets of therapy for allergic (atopic) diseases, mastocytosis and MCAS.
Heterogeneity and Versatility of MCs
Several observations suggest that mouse and human MCs in various organs have heterogeneous phenotypic and functional properties [9, 79-81, 83, 87, 135, 143, 172-175, 177-179]. Despite earlier contributions by William Bate Hardy [179] and others, Lennart Enerbäck is often regarded as the first to provide a detailed description of MC heterogeneity in rodents [177]. He identified two distinct types of MCs in rat tissue sections: mucosal MCs (MMCs) and connective tissue-type MCs (CTMCs) [177]. Compared to CTMCs, MMCs are smaller, more variable in shape, and often hypo-granulated. CTMCs were also identified as a unique source of heparin. By contrast, rodent MMCs contain glycosaminoglycans of lower sulfation, no heparin, low amounts of histamine, and little or no serotonin [177]. A surprising observation was that special fixation procedures were necessary to detect MMCs in rat tissue sections. Later, MMCs and CTMCs were also detected in mice.
In both species, MMCs and CTMCs display specific patterns of MC proteases [180-182]. Notably, mouse MMCs preferentially contain mouse MC protease (mMCP)-1 and -2, whereas CTMCs express mMCP-4, -5, -6, and carboxypeptidase A [180-182]. In addition, it was found that the development of MMCs and CTMCs is controlled by distinct cytokines, including SCF (CTMCs), IL-3 (MMCs), and other MC-targeting interleukins [12, 46, 79-81, 83, 182]. CTMCs were also regarded as innate or constitutive MCs whereas MMCs were thought to represent an adaptive MC compartment, which is inducible by the tissue microenvironment in response to inflammatory processes [183].
Subsequent studies revealed that the phenotypes of MMCs and CTMCs are, at least in part, reversible in certain tissue locations and cytokine milieus, and that trans-differentiation between the two types of MCs may occur in physiologic and pathologic tissues [81, 83-87, 182]. In addition, based on the expression of various combinations of different MMCPs, MC heterogeneity is complex and cannot be fully understood based on a simple model predicting two major MC subsets (MMCs and CTMCs). Rather, increasing data suggest that MCs are functionally and phenotypically versatile cells. In fact, MCs may change their phenotype (e.g. protease-composition) rapidly depending on the cytokines to which these cells are exposed, the tissue microenvironment, the maturity of the responding MC population, and the underlying pathology [81, 83-87, 143, 182].
In humans, MC heterogeneity was first described on the basis of differential expression of neutral proteases in MCs derived from the skin and other organs. In particular, it has been reported that skin MCs express both tryptase and chymase in their secretory granules (MCTC), whereas most MCs in the lung and small intestinal mucosa display tryptase but not chymase (MCT) [178]. Attempts to relate MCTs to mouse MMCs and MCTCs to CTMCs were only partly successful, mostly because mixtures of MCTs and MCTCs were found in most extra-cutaneous tissue sites.
Human MCs can also be classified based on expression of certain surface receptors and responses to certain stimuli [143, 172-175]. For example, the C5aR CD88 is expressed abundantly on all MCs derived from skin (MCTCs) and on the small portion of lung MCs that are MCTCs, but not detectably on human lung MCTs [174, 175]. Expression of the C5aR on MCs may also depend on the underlying disease. For example, the C5aR CD88 is expressed on synovial MCs in rheumatoid arthritis but not on synovial MCs in patients with osteoarthritis [143]. Carboxypeptidase A3, which is usually expressed only in MCTCs in healthy tissues, is also expressed in chymase-deficient MCs in certain inflammatory sites, including eosinophilic esophagitis, severe asthma, exercise-induced bronchospasm and chronic sinusitis. Finally, opioid receptors, including the MRGPRX2 receptor, are expressed on human skin MCs, but not on MCs in other organs [163, 184].
To date, the factors and mechanisms underlying expression of CD88 and carboxy-peptidase A3 in MCs remain unknown. One hypothesis is that chronic cytokine exposure or continuous KIT activation triggered by a mutation leads to expression of CD88. In line with this hypothesis, MCs in patients with KIT D816V+ SM constitutively display CD88 [185]. Other markers are also expressed on both neoplastic MCs in SM and reactive MCs in inflamed tissues. One interesting surface marker-antigen is the alpha-chain of the IL-2 receptor (IL-2RA), CD25. This antigen is usually not expressed on MCs in healthy tissues. However, in pathologic tissues and chronic inflammatory reactions, MCs may display CD25 (P.V. unpublished observation). Again, the factors responsible for abnormal expression of CD25 on MCs remain unknown.
Another important observation is that MC antigens may be displayed differentially based on the cells´ stage of maturation. For example, the high affinity IgE receptor, FcεRI, is not expressed on very early MC precursors but appears later during MC maturation [34-36, 53, 67, 68]. Finally, certain cell surface antigens are rapidly expressed, or increase, upon IgE-dependent or cytokine-induced activation of MCs. Similarly, compared to resting MCs, IgE receptor-activated MCs express increased amounts of CD63 and CD203c [135, 142]. It has also been described that interferon-gamma induces the expression of HLA-DR and CD64 (FcγRI) on human MCs [186, 187]. Finally, in patients with mastocytosis, neoplastic MCs express various cell surface antigens, including CD25, CD30, CD32, CD64, CD88, CD123, CD203c, and HLA-DR in an augmented or aberrant manner [133-136, 138-142].
Potential Physiologic and Pathologic Role(s) of MCs
During the past few decades, several attempts have been made to decipher the possible physiologic roles of MCs, one of the last unresolved riddles about these cells. While no definitive solution to this quandary has been presented, a number of interesting possibilities have been suggested. One evolving hypothesis is that MCs are physiologically involved in orchestrating tissue repair processes, in a variety of settings, through which the evolution of certain pathologies can be avoided. This role would therefore be similar to that of neutrophils and macrophages in combatting bacterial and other infections. In fact, during the past 30 years, more and more data support the probability that MCs play an important role as defense and repair cells during various pathologic conditions and related biological processes. Another emerging concept is that MCs and their products are essentially involved in the innate, and acquired, defense against the lethality of animal-derived venoms, a concept that has recently been established experimentally in mice and may also apply to humans. These concepts, and the diverse roles MCs may play in the destructive and healing processes of various tissues, are discussed in the following paragraphs.
Potential Role(s) of MCs as Defenders against Microbes and Toxins
Several findings point to the possible roles of MCs as defenders in certain infectious diseases [188-194]. While a complete overview of all proposed functions of mast cells in infectious diseases is beyond the scope of the current article, we here discuss a few important concepts that relate to the role of MCs as possible contributors to host defense. One remarkable observation was that MC-deficient mice were more susceptible to certain fatal bacterial infections, such as peritoneal infection and septicemia following cecum ligation and puncture [189, 190]. Similar observations have been made in other models of bacterial infections [116, 188]. It had also been described that the protective 'MC-effect' depends on the rapid availability of MC-derived TNF-alpha, a cytokine known to augment the influx of phagocytes into inflamed tissue sites [189, 190]. Indeed, exposure of endothelial cells to MC-derived histamine and TNF-alpha promotes leukocyte rolling, leukocyte adhesion, and the consecutive transmigration of phagocytes. In the absence of such phagocyte-trafficking, any bacterial disease would become potentially life-threatening.
However, other studies performed in MC-deficient KitW/W-v and KitW-sh/W-sh mice suggested that MC-derived TNF-alpha may actually contribute to an adverse outcome in certain severe forms of cecal ligation and puncture [116]. More recent data suggest that, in the mouse, basophils represent a significant contributor to survival in a moderate form of cecal ligation and puncture, and that this role may at least in part depend on basophil production of TNF-alpha [195]. Other studies have shown that bacterial antigens directly induce the release of mediators, including TNF-alpha, from MCs [191-193]. However, it remains unknown how much TNF-alpha can be provided by human MCs in reactions such as these. A number of additional mechanisms may contribute to the ability of MCs in various organs to enhance resistance to bacterial infections [188, 194, 196, 197]. These mechanisms may include, among others, MC activation via Toll-like receptors (TLR)s [194] (although TLRs have not been identified on human skin MCs by all groups [163]), complement activation [196], and activation through endothelin-1 [198]. Moreover, both human and mouse MCs have been implicated in bacterial phagocytosis [194, 197].
On the other hand, several observations argue against a major role for human MCs as defenders against bacterial infections. First, human MCs are not capable of compensating a severe neutropenia to rescue septicemic patients, e.g. during high-dose chemotherapy (where MC usually survive). Second, in patients with imatinib-induced MC deficiency, no increase in the rate of total or severe bacterial infections has been reported [125]. These observations do not totally exclude the possibility that MCs contribute to the immunological defense system against bacteria or other microbes in humans. However, compared to other immune cells, in humans the contribution of MCs appears to be less important.
MCs have also been implicated in host defense against various parasites. For example, in mice, an intact MC system seems to be required for proper defense against certain helminth infections, such as infections by Strongyloides ratti, Strongyloides venezuelensis or Trichinella spiralis [199-201]. In addition, mouse MCs may contribute to host defense against primary cutaneous infections with Leishmania major [202].
A similar role of MCs as defenders against parasites in humans has not been established to date. In this regard, it is worth noting that the physiologic and pathogenetic role of MCs in host defense against pathogens may have changed during mammalian evolution. In mice and rats, MCs may be part of a strong defense-system against bacteria, helminths, and other pathogens. However, in humans, it is possible that these MC functions may no longer be as critically required to guarantee host survival. On the other hand, evidence favoring a protective role for MCs in response to Strongyloides stercoralis has been described in non-human primates [203].
Finally, MCs have been implicated in the etiology of several viral infections. In fact, a number of viruses have been reported to be capable of infecting human MCs, including the human immunodeficiency virus (HIV) [204-206]. MCs are unique among HIV-vulnerable cell types in that they appear to only be susceptible to infection with R5-tropic HIV for a brief period during their ontogeny when they are CD4+/CCR5+/CXCR4+ [204]. It also has been reported that IgE-FcεRI interactions enhance the susceptibility of these MC precursors to HIV [205]. As these infected MCs enter into tissues, mature, and persist, they appear to provide a long-lived, inducible, reservoir of persistent HIV in tissues. In vivo evidence supports this possibility, in that HIV-infected women have both circulating precursor MCs and placental tissue MCs that harbor inducible infectious HIV even after having been treated during their pregnancy with highly active antiretroviral therapy (HAART) [206]. Other viruses are also reported to infect human MCs. For example, the LAD2 MC line and primary human cord blood-derived MCs have been infected with human rhinoviruses and RSV [207, 208]. In addition, the corona virus receptor CD26 is expressed on human skin MCs [131].
MCs also may be able to contribute to immune defense mechanisms against viruses. For instance, rodent, monkey, and human MCs are able to detect dengue virus (DENV), a single-stranded, positive-sense RNA virus, which results in MC activation and degranulation [209]. This MC response has been linked to the MC-driven recruitment of natural killer and natural killer T cells. Similarly, human MCs can produce type I IFNs after exposure to double-stranded RNA and/or respiratory syncytial virus or reovirus type 1, the former via specific interactions with TLR-3 [208]. Such observations are consistent with the conclusion that MCs can contribute to innate immune responses to viral infections in part via the production of type I IFNs.
Recently, MCs and their products also have been implicated in the defense of mice against animal-derived venoms [210-216]. In fact, several findings in MC-deficient, MC-reconstituted, or MC chymase-deficient mice indicate that mouse MCs (and for certain venoms, mouse MC chymase), can enhance the survival of mice challenged for the first time with the venom of the honeybee, several snakes, a lizard, or two scorpions [210, 212, 215]. As a consequence, the venom-induced tissue damage and the resulting death of the host, are significantly ameliorated [210, 212, 215]. In the case of honeybee and Russell's viper venoms, IgE-dependent MC activation can further enhance resistance to the lethality of these venoms in mice [213, 215]. Moreover, zebra fish embryos were used to assess the toxicity of snake venoms treated with human MC proteases [217]. It was determined that human MC tryptase-ß, but not human MC chymase or CPA, could reduce the toxicity of the venoms of six poisonous snakes [217]. Taken together, this evidence indicates that MCs, MC proteases, and IgE may play important protective roles against the serious and indeed fatal, effects of animal-derived venoms. However, so far, no broadly applicable therapeutic approach has been developed using MC proteases.
Potential Role of MCs as Repair Cells in IgE-Dependent Allergic Reactions
A number of observations point to important 'repair-roles' of MCs during allergen-induced anaphylaxis [210, 218-220]. First, MC-derived proteases have been described as degrading several different allergens into inactive fragments [218, 219]. Moreover, MC-derived proteases have been reported to degrade IgE [220]. The functional consequence of this MC-derived protease activity would be a disruption of the allergen-induced tissue damage. All of these data point to important biological feedback mechanisms during human allergic reactions in which MCs and MC-derived products can help to limit the local and systemic reactions to allergens. In line with this concept, MC-derived proteases can also degrade a number of cytokines, chemokines, and vasoactive peptides into inactive fragments.
Potential Role of MCs as Repair Cells in Thromboembolic Disorders and Wound Healing
Several biochemical and functional studies in humans and other species suggest that MCs represent the major source of heparin [9, 154, 158, 221]. It has also been reported that MC-deficient KitW/Wv mice develop more severe thromboembolic events after India ink injection compared to their normal littermates, and that injection of heparin can rescue these mice from fatal thromboembolic events [222]. In addition, human MCs are a unique source of non-complexed tissue-type plasminogen activator (tPA) [157]. Whereas in many other cell types, tPA is expressed and released together with its natural inhibitors (PA inhibitors = PAIs) and in the absence of heparin, MCs produce and release un-complexed (active) tPA together with its stabilizer and activator, heparin [157]. In addition, it has been found that MCs accumulate in areas of thromboembolism, especially around thrombosed vessels [223], and that thrombin-activated endothelial cells express and release soluble SCF, which in turn attracts MCs (Figure S2) [224].
These observations support the hypothesis that MCs can act as major repair cells in thromboembolic disorders, and that in physiologic tissues MCs may even be integrated in a prophylactic repair system that counteracts thromboembolic events [157]. Other studies have shown that patients with clonal MC disorders may suffer from a bleeding tendency, and that in these patients, hyper-heparinemia and sometimes even hyperfibrinolysis (caused by elevated tPA) may be detected [225-228].
However, several questions remain. For example, it remains unknown which triggers cause liberation of heparin and tPA from MCs in patients with MC disorders. It also remains unknown whether MC-derived heparin and tPA can indeed prevent local thromboembolic events. Finally, no increase in thromboembolic events has been observed in CML patients receiving life-long imatinib, although these patients develop a MC deficiency [125]. This observation suggests that other cells and mechanisms can counteract thrombosis. One possibility is that basophils may counteract some of the effects of a MC deficiency.
MCs have also been implicated as repair cells in wound healing [155, 156, 229-232]. This hypothesis is based primarily on the observation that MCs can produce several important repair molecules, including heparin, proteases, and angiogenic cytokines, such as VEGF, fibroblast growth factor (FGF) and IL-6 [155, 156, 159, 229-231]. In addition, MCs represent a potentially rich source of oncostatin-M (OSM), IL-8 and CCL2 [161, 162]. In several experimental models, there is evidence that MCs indeed may contribute to wound healing [229-231]. However, an unequivocal role of MCs in wound healing could not be confirmed in all of the model systems examined [232, 233].
Possible Role of MCs in Atherosclerosis and related Cardiovascular Diseases
Several observations in experimental animals and in man suggest that MCs may play an important role in the evolution and progression of atherosclerosis [234-249]. Early studies revealed that activated MCs can support the generation of cholesterol-filled macrophages (foam cells) which is typical finding in atherosclerosis [234]. Mechanistically, the apolipoprotein B-100 (apoB-100) component of low-density lipoprotein (LDL) particles binds to the heparin proteoglycan component of exocytosed MC granules. Subsequently, MC granule-derived chymase can degrade apoB-100, thereby inducing fusion and tight binding of the LDL particles to the granules [236]. Ultimately, the LDL-loaded granules are phagocytosed by macrophages, thereby leading to cholesterol accumulation in these cells [234, 235]. MC granules can also facilitate uptake of LDL by smooth muscle cells and thereby promote their transformation into foam cells [238]. Chymase and tryptase were also later found to degrade the apolipoprotein A-I component of high-density lipoprotein (HDL) [243]. This renders the HDL particles unable to remove cholesterol from macrophages, thereby accelerating cholesterol accumulation and foam cell-formation in the arterial wall [243].
By immunohistochemistry, MCs containing tryptase and chymase have been detected in the normal intimal layer of healthy human aortas, as well as in the fatty streaks and vulnerable shoulder regions of atheromas in atherosclerotic aortas [237]. Moreover, studies in human coronary arteries revealed the accumulation of activated MCs at the actual site of erosion or rupture of the culprit coronary plaque that was responsible for an acute coronary syndrome or myocardial infarction [240, 241].
Mechanistic insights into the possible roles of MCs in plaque erosion and rupture have also been obtained from in vitro culture experiments. In these studies, human MC-derived proteases were found to degrade the pericellular matrix of endothelial cells and, together with TNF-alpha, induce their apoptotic death [245, 247]. This process is known to lead to de-endothelialization of the plaque and thus to plaque erosion. Moreover, MC-derived proteases can degrade the pericellular matrix of smooth muscle cells, with apoptotic cell death resulting from loss of matrix-derived (external) survival signals [242]. Loss of these collagen-producing cells may in turn result in further thinning of the collagenous cap of the plaque, thereby increasing the risk of plaque rupture [244]. Thus, MC-derived proteases may significantly contribute to the erosion and rupture of an atherosclerotic plaque, both by directly degrading pericellular matrices and by facilitating degradation of the extracellular matrix via activation of matrix metalloproteinases [246].
Finally, MCs and their products have been implicated in inflammatory processes that trigger and sustain atherogenesis [249]. All of these observations point to potentially important roles of arterial MCs and their inflammatory products in cholesterol accumulation in the vessel wall and in plaque vulnerability and thus in the progression of atherosclerosis. In addition, MC-engraftment of the meninges of genetically MC-deficient KitW/Wv mice has provided evidence that meningeal MCs can worsen stroke pathology in that species, perhaps in part through the release of IL-6 [250]. Supplemental Figure S3 shows some potential mechanisms by which activated MCs may contribute to various stages of atherogenesis.
However, several questions remain concerning the role of MCs in atherosclerosis. First, systemic disorders characterized by a marked chronic activation of MCs, or increases in MC numbers, such as atopic disorders, severe allergies or mastocytosis, are not known to be associated with an increased risk of atherosclerosis, myocardial infarct or stroke. In addition, multi-kinase inhibitors targeting KIT lead to MC deficiency [125] but cannot protect the individual from the development of atherosclerosis. Indeed, some of these kinase blockers (nilotinib and ponatinib) promote atherosclerosis and the occurrence of thromboembolic events [251]. On the other hand, the mechanisms through which these TKI promote atherosclerosis are not well understood and may involve several different cell types and molecules. Moreover, imatinib induces MC deficiency but does not promote the development of atherosclerosis in patients with CML [125].
MCs have also been proposed as providing some protective functions in the context of atherosclerosis. First, MCs provide heparin together with un-complexed (PAI-free) tPA [157], a 'master-cocktail´ that is used clinically to treat patients with acute arterial occlusion such as myocardial infarction or ischemic stroke. Moreover, heparin released from activated MCs in atherosclerotic lesions may restrict the development of a platelet-rich arterial thrombus on the de-endothelialized (eroded) surface of an atherosclerotic plaque. MCs also provide several additional repair molecules, such as FGF, VEGF, OSM, or IL-8 [155-159, 161, 162]. These MC-derived repair molecules may contribute to neo-angiogenesis and re-perfusion of ischemic organs (after infarction) as well as re-canalization of thrombosed vessels [157].
From these observations, the hypothesis has been raised that MCs may exert both pro-atherogenic and anti-atherogenic activities, as well as both pro- and anti-repair effects, during ongoing atherosclerosis. In reality, the net effect of MCs and MC-specific effector-molecules may largely depend on additional factors, including the involved vessel site and the stage of atherosclerosis. For example, the fibrinolytic and fibrinogenolytic activities of MC proteases (tPA, tryptase) may counteract atherosclerosis and related vascular events in small and large arteries, at both early and later stages of atherosclerosis. On the other hand, the effects of these molecules may also be fatal, for example when the thinned fibrous cap of an atheroma ruptures. Table 5 summarizes the potential harmful and protective effects of various MC products in atherosclerosis.
Potential contributions of mast cells (MCs) and their products to biological processes relevant to the development of atherosclerosis
| Effects | MC mediators involved |
|---|---|
| A. Proatherogenic effects | |
| Proinflammatory effects in the vessel wall | TNF-alpha and other MC-derived cytokines |
| Recruitment of leukocytes/phagocytes into the vessel wall | Histamine, MC-derived cytokines (TNF-alpha and others) and chemokines (CCL2, IL-8, others) |
| Modification and cellular uptake of LDL by macrophages and smooth muscle cells consecutive foam cell formation | Tryptase and chymase as well heparin |
| Modification of HDL molecules into pathologic species that are no longer capable of mobilizing cellular cholesterol | Tryptase and chymase |
| Endothelial cell and smooth muscle cell death with subsequent plaque rupture | MC proteases and TNF-alpha |
| Leakage and disruption of microvessels in neovascularized plaque areas | Histamine, tryptase and chymase |
| B. Protective effects and repair functions | |
| Thrombolysis: degradation of fibrin and fibrinogen | tPA (fibrin-degrader) and uPAR; Tryptase (fibrinogen-degrader); Heparin (co-factor for tPA, tryptase and anti-thrombin III) |
| Protection of endothelial cell layers and vessel wall integrity | Tryptase (endothelial cell survival) and angiogenic cytokines (VEGF, CCL-2, IL-8, others) |
| Neovascularization of hypoxic regions in atherosclerotic plaques | Angiogenic cytokines (VEGF, FGF) |
Abbreviations: tPA, tissue type plasminogen activator; VEGF, vascular endothelial cell growth factor; FGF, fibroblast growth factor.
Role of MCs as Inflammatory Effector Cells in Allergic and Immunologic Diseases
Previous reports have described the pivotal roles of MCs in IgE-dependent and IgE-independent inflammatory reactions [2-6]. Although a detailed review cannot be provided in this article, we should note that the key event in such reactions is the release of pre-formed granule-derived and newly generated pro-inflammatory and vasoactive mediators, cytokines, and chemokines [2-7, 145-150]. These mediators and cytokines are major triggers of immediate and late-phase reactions and thus of the clinical symptoms in allergic disorders. Depending on many factors, most of which regulate the releasability of MCs (and basophils), the reactions and symptoms range from mild to severe or even life-threatening [164-169].
However, as mentioned, MCs can also be involved in regulatory feed-back mechanisms that can help to limit allergic reactions and symptoms [210-212, 218-220]. Desensitization against IgE-dependent and IgE-independent stimuli also has been described [252-254]. Many of these mechanisms may also apply in patients with systemic mastocytosis (SM), where the excessive number of MCs results in an increased risk for life-threatening anaphylaxis in a subset of patients. However, many patients with SM maintain a clinically stable course without any mediator-related symptoms, so that the hypothesis has been raised that their neoplastic MCs may be less responsive to IgE-dependent and other stimuli through desensitization phenomena and other negative-feedback mechanisms [255].
On the other hand, many patients with SM suffer from recurrent systemic anaphylaxis or even MCAS [167-169]. In these patients, as well as in those with severe allergies (without SM), the regulatory mechanisms described above may have been bypassed or are too weak to keep the reaginic proinflammatory machinery under control. Recent data suggest that patients with severe allergy symptoms, or those with mastocytosis and severe symptoms, may have extra copies of their alpha tryptase gene, a genetic condition called hereditary alpha tryptasemia (HAT) [256, 257]. However, not all of these HAT patients have severe symptoms and in many cases, the exact mechanisms that cause severe anaphylaxis in SM and/or MCAS remain unknown. Regarding therapy, patients with severe MCAS are treated symptomatically with drugs targeting MC mediators or their receptors, such as histamine receptor blockers, MC-stabilizing drugs and other anti-allergic medications. In severe forms, antibodies targeting IgE, such as omalizumab, may be considered.
Until approximately 1980, MCs were primarily regarded as key effector cells of allergic reactions. However, since then MCs and their mediators have been implicated in various other inflammatory conditions and immunologic disorders [7, 9, 143, 150, 258-264]. For example, MCs have been described as contributing to pro-inflammatory reactions and processes in patients with various systemic auto-immune disorders [58, 143, 258-264]. While a complete overview of the MC´s potential involvement in these disorders is beyond the scope of this article, we wish to emphasize one of the most prominent and important examples, the impact of MCs on the pathogenesis and evolution of rheumatoid arthritis (RA).
Earlier studies had shown that MCs are detectable in the inflamed synovial tissue of the affected joints in patients with RA and that cytokines, such as TNF-alpha, which can be derived from MCs and other participants in these disorders, play a pivotal role in the pathogenesis of the disease [258-261]. TNF-alpha and other (potentially) MC-derived cytokines have also been implicated in the recruitment and activation of leukocytes into the inflamed joints in RA [258]. Subsequently, SCF produced by activated synovial fibroblasts and other local cells was described as attracting and activating MCs, thereby providing a mechanism through which MCs accumulate and release their cytokines in synovial areas in RA [59].
However, other MC agonists, such as C3a and C5a, as well as IgE and other immunoglobulins, may also be involved in MC recruitment and activation in RA [143]. Other studies have shown that MC-derived proteases play a crucial role in the pathogenesis of RA [262, 263]. For example, among several different effects on local effector cells of RA, MC-derived tryptase can augment the survival and activation of local (synovial) fibroblasts [263]. In addition, MC-derived proteases may augment autoimmune responses in RA [262].
The most critical MC-derived cytokine in RA appears to be TFN-alpha. Since this cytokine (which can be derived from MCs and other cell types) can activate synovial cells to produce collagenase, a key enzyme that can induce destruction of cartilage, antagonists of TNF-alpha have indeed been shown to have beneficial effects in this disease [265]. Other therapeutic attempts have aimed at the direct suppression of MC growth or MC activation in patients with RA [260]. However, to date, no robust therapeutic concept focusing solely on the MC compartment has been described in the context of RA or other autoimmune disorders. Also of note, recent work with both two types of MC-deficient mice and human synovium, has implicated MCs, and IgE/FcεRI and Syk, in the development of osteoarthritis [266].
Potential Role of MCs in Tumor Development and Tumor Surveillance
A number of different studies have pointed to several potential roles MCs may play in tumor development and/or tumor surveillance [267-271]. In fact, depending on the tumor type, tissue of origin, experimental model, and biologic context, MCs have been thought either to promote or inhibit the development of a neoplasm [267-271]. The mechanisms underlying this proposed dual role of MCs is not completely understood. However, MCs can produce a number of substances that may contribute to tumor or leukemia cell proliferation and survival (e.g., growth factors), tumor cell redistribution and metastasis (e.g., vasoactive substances, chemotactic factors, adhesion receptor-inducers) and/or modulation of the tumor-related microenvironment (e.g., angiogenesis-promoting compounds) [145-156, 159]. Many MC-related mechanisms and molecules may play a role in immune surveillance against tumor growth. These include, among others, TNF, perforin/cytolysin, proteases, and IgE-dependent processes that lead to tumor cell destruction [272, 273]. Unfortunately, most such observations have been made in experimental models, and the actual net impact of MCs during tumor development in patients suffering from various malignancies remains unknown. It is also worth noting that chronic MC activation is not known to be associated with a lower or higher risk of human malignancies.
Neoplastic Disorders of MCs and their Precursors
The pathologic accumulation of MCs in the skin of patients with urticaria pigmentosa (UP) was first described by Paul Gerson Unna in 1887. Later, the term mastocytosis was proposed, based on the stable clinical course of most patients and the belief that the MC accumulations were reactive rather than neoplastic in nature (the suffix 'osis' is usually employed to describe benign/reactive expansions of leukocytes). This hypothesis was supported by the observation that the cutaneous lesions of mastocytosis, when manifested in early childhood, often disappeared during or shortly after puberty [274].
In 1949, Ellis described the first case of systemic mastocytosis (SM) with involvement of internal organs [275]. From this and other similar cases it was appreciated that mastocytosis in adults is a persistent and neoplastic disease that may be complicated by organ involvement or even organ damage. At approximately the same time, the first cases of MC leukemia (MCL) were described. Since then, a bone marrow biopsy has been regarded as a basic standard in the diagnostic evaluation of patients with suspected SM.
A first comprehensive classification of mastocytosis was proposed by Karl Lennert and the Kiel group in 1979 [276]. In 1988, Travis et al. proposed a classification for systemic mast cell disease which introduced indolent, aggressive and associated hematologic disease categories [277]. In 1991, Dean Metcalfe proposed a first consensus classification of mastocytosis [278]. The clonal nature of SM was confirmed by demonstrating that the involved cells (MCs and their precursors) exhibit the transforming KIT mutation D816V [279-281]. This mutation had been described as one of two activating KIT mutations in the HMC-1 mast cell line that was derived from a patient with MCL [282, 283]. Shortly afterwards, KIT mutation analysis was introduced as a major diagnostic test in patients with suspected SM.
Between 1990 and 2000, specific morphologic, immunologic, and molecular features of neoplastic MCs were examined for their specificity and robustness. Based on such studies, criteria were proposed to make the diagnosis of SM and to support a related classification of mastocytosis [137]. This proposal serves as a basis of the WHO classification of mastocytosis [138, 139]. The most recent update of this classification divides the disease into cutaneous mastocytosis (CM), indolent SM (ISM), smoldering SM (SSM), SM with an associated hematologic neoplasm (SM-AHN), aggressive SM (ASM), MCL, and MC sarcoma (MCS) [138, 139]. We refer to the available literature for the details of the classification, and for the criteria of SM. However, it is essential to focus here on a few aspects related to the clinical impact of MCs in these disorders.
First, it is important to recognize that patients with SM may or may not present with severe mediator-related symptoms. Patients who are suffering from such symptoms and require continuous treatment with anti-mediator-type drugs are labeled as SMSY [169]. In severe cases, a MCAS is almost always detected in these patients [168, 169]. Others suffering from SM also have an underlying hypertryptasemia (HAT). Another important point is that in pediatric patients, in whom the disease was long believed to be a ´reactive´ (non-neoplastic) process, the disorder is now regarded as a neoplastic condition, because in most cases activating (somatic or germline) mutations in KIT are detectable [284, 285]. Remarkably, however, the presence of the D816V KIT mutation or other KIT mutations cannot predict the clinical course in pediatric CM. Rather, it is the presence of the typical (adult-like, i.e. small sized, monomorphic) form of the cutaneous lesions that helps predict persistence of pediatric mastocytosis into adulthood [286].
Another riddle regarding MCs in SM was the fact that the KIT D816V mutation is expressed in MCs in both indolent SM associated with a normal life-expectancy, as well as in MCs in advanced SM, including MCL where the survival time is usually very short [137-139, 279-281, 285]. In this regard, it is important to appreciate that KIT D816V acts as weak oncogene regarding proliferation but as a strong inducer of MC differentiation, maturation and survival, processes that are usually associated with reduced proliferation [287]. However, in a subset of patients with SM, KIT D816V can be detected in MCs, as well as in other hematopoietic lineages [288, 289]. Such multi-lineage involvement is typically found in smoldering SM and advanced SM and is of prognostic significance [290].
Moreover, several additional oncogenic mutations (apart from KIT mutations) have been detected and serve as new diagnostic and prognostic parameters in patients with advanced SM (SM-AHN, ASM or MCL) [291-295]. The current hypothesis is that these additional mutations (most commonly in: TET2, SRSF2, ASXL1, CBL, RUNX1, and RAS) act together with KIT D816V in advanced SM to trigger proliferation and survival of neoplastic MCs. The type and number of such additional mutations have a major impact on the prognosis (including overall survival and responses to therapy) of patients with SM [295]. For example, a particularly poor prognosis is found in patients who carry mutations in SRSF2, ASXL1, and RUNX1 (S/A/R-patients) [295]. It is also worth mentioning that most of these mutations are also found in other myeloid neoplasms (without SM) and that, among advanced SM patients, they are most frequently detected in SM-AHN.
Taken together, the unique features of SM and the cell-specific evolution of MC progenitors and other cell lineages in advanced SM provide additional evidence that MCs form a separate myeloid lineage within the hematopoietic cell system. Moreover, despite expression of certain ´lymphoid´ antigens on neoplastic MCs, the progression-patterns seen in patients with SM-AHN confirm that MCs are best regarded as myeloid cells. In fact, in almost all patients with SM-AHN, the AHN portion of the disease manifests as a myeloid neoplasm, but only rarely as a lymphoid malignancy [41, 137-139].
New Treatment Concepts: Targeting of MC Growth and/or Activation
During the past 3 decades, several different treatment approaches have been established for MC proliferative disorders and for conditions defined by MC activation. Whereas initial attempts to block MC growth in patients with advanced SM were based on treatment with conventional anti-neoplastic drugs, like interferon-alpha or cladribine [296, 297], more recent approaches are based on using validated drug targets [138, 298-302]. Considering the possibility of inducing MC deficiency by blocking MC development from their progenitors, the clinically most relevant targets identified so far are WT KIT (normal MCs) and the KIT mutant D816V that acts as a driver of MC development and differentiation in CM and SM [255, 283, 284, 287]. As mentioned before, imatinib is a strong KIT inhibitor and can induce an almost complete MC deficiency when administered continuously [125]. MC-depletion in subjects treated with imatinib takes about 2 years [125]. Application of such KIT inhibitors also may be of interest in the context of MC activation, atopic disorders or severe allergies. Indeed, a recent double-blind, placebo-controlled study in a small number of patients with severe asthma indicates that imatinib may be useful in this setting, although it is not clear to what extent the benefit observed was due to effects on MCs as opposed to other actions of the drug [298, 299].
However, the KIT D816V mutation confers imatinib resistance on neoplastic MCs [300, 301]. Therefore, novel KIT tyrosine kinase inhibitors have been developed for testing in preclinical and clinical studies. One of these drugs is midostaurin (PKC412), which blocks the kinase activity of wild type KIT, KIT D816V and several other clinically relevant KIT mutants [300, 301]. In addition, in contrast to most other KIT blockers, midostaurin inhibits SYK activation and several other IgE-receptor downstream targets; midostaurin thereby blocks MC activation and IgE-dependent mediator secretion in MCs and basophils [302].
In a first pilot patient with advanced SM, midostaurin was found to block the expansion of KIT D816V-mutated MCs [303]. More recently, midostaurin was administered to patients with advanced SM in a global, single arm, phase II trial. In this study, midostaurin was found to induce major and often durable clinical responses in many cases with advanced SM, including MCL [304]. Remarkably, many patients not only developed meaningful hematologic responses but also major responses in mediator-related symptoms and a substantial increase in their quality of life [304]. The latter effect of the drug may reflect its ability to suppress mediator release in MCs [302]. Based on these data, midostaurin was approved by the US Food and Drug Administration and the European Medicines Agency in 2017 for all subtypes of advanced SM [305]. In addition, midostaurin may be an interesting drug for patients with non-advanced SM in whom mediator-induced symptoms cannot be controlled by conventional anti-mediator type drugs.
In addition to midostaurin, several other KIT-targeting drugs have been tested in the context of advanced SM [301]. Whereas some of them, like imatinib or masitinib, are unable to suppress the kinase activity of mutant KIT (especially D816V), others, like avapritinib or ripretinib, are potent inhibitors of KIT D816V and can block the growth of neoplastic progenitor cells [306]. As noted above, all of these inhibitors are candidates for evaluation in the context of allergic/atopic disorders and various immunologic or rheumatologic diseases where MC activation is thought to play an important role [298, 307]. However, in these non-neoplastic diseases, the use of these agents must be carefully balanced against their side effects. It should also be kept in mind that some of the clinical effects and side effects of the 'KIT-targeting' drugs may reflect actions on KIT expressed by cells other than MCs or on tyrosine kinases other than KIT. Other drugs act on various signaling or effector molecules downstream of KIT and/or downstream of the IgE receptor [308, 309]. The clinical efficacy of many of these agents is currently being explored in clinical trials. Finally, for patients with severe allergies and MCAS, and for those who are suffering from severe allergies, MCAS and/or additional aggravating MC conditions (like HAT and/or mastocytosis), application of antibodies removing IgE, such as omalizumab or ligelizumab, or targeting MC tryptase, may represent promising additional approaches [310-314]. Finally, a number of inhibitory receptors, such as Siglec-7, Siglec-8, or CD300a, have been identified on human MCs and are currently being validated for their role as therapeutic target sites [315-318].
Concluding Remarks and Future Perspectives
Since their description and naming by Paul Ehrlich, MCs have been the subject of intensive research and have fascinated generations of scientists. Some of the riddles concerning the origin and functions of MCs have now been solved. MCs are hematopoietic cells derived from CD34+ stem cells, develop and survive under the influence of SCF, and can undergo neoplastic transformation based on acquiring gain-of-function mutations in the SCF receptor (KIT) gene and other critical target genes. Moreover, some of the mechanisms underlying MC activation in various disease states and related syndromes have been defined. Also, a variety of diagnostic markers and drug targets have been identified and have been successfully validated, and several drugs targeting MC expansion, MC activation, or both have been translated from preclinical testing into clinical applications. In advanced MC disorders, KIT appears to be a most promising target. Finally, diagnostic criteria for MC disorders, including mastocytosis and mast cell activation syndromes, have been established.
However, many questions remain. For example, there is still debate about the possible roles of MCs in healthy tissues. Notably, the long-term MC-deficiency induced by persistent treatment with imatinib in humans has not yet been associated with a particular adverse phenotype. Also, in several pathologic conditions, it remains unknown whether MC activation may be useful, e.g., by enhancing repair processes, is harmful and leads to tissue damage, or, depending on the circumstances, may contribute to either outcome. Finally, it remains unknown which KIT-independent molecular pathways and molecules play the most decisive role in the progression of mast cell neoplasms. We trust that future MC research will provide answers to such questions, leading to a better understanding of MC biology in health and disease, as well as to the development of new and better diagnostics and better treatments for patients suffering from MCAS, advanced MC neoplasms or other MC-dependent pathologies.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank all members of the mastocytosis research team of the Medical University of Vienna for their support in the project and for their assistance in the organization of the Working Conference on Mast Cells and Mastocytosis in Vienna in August 2015.
Authors' Contributions
All co-authors listed participated actively as faculty members in the Workshop “Mast Cells and Mastocytosis” in the Paul Ehrlich memorial meeting in Vienna in 2015. Each co-author contributed essentially to the faculty discussion as well as to the conception and drafting of the paper. All co-authors approved the final version of the document.
Funding and Support
P.V. was supported by the Austrian Science Fund (FWF), grants F4701-B28, F4704-B28 and P32470-B. DDM is supported by the Division of Intramural Research, NIAID. SJG's research is supported by the NIH, USA, the US-Israel Binational Foundation, and the Department of Pathology and the Sean N. Parker Center for Allergy and Asthma Research at Stanford University, Stanford, CA. Finally, we thank 4 anonymous reviewers for helpful comments and suggestions.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ehrlich P. Beiträge zur Kenntniss der granulirten Bindegewebszellen und der eosinophilen Leukocythen. Arch Anat Phys. 1879; 166-9.
2. Ishizaka T, Ishizaka K. Activation of mast cells for mediator release through IgE receptors. Prog Allergy. 1984;34:188-235
3. Metcalfe DD. Mast cells and mastocytosis. Blood. 2008;112:946-56
4. Galli SJ, Tsai M. IgE and mast cells in allergic disease. Nat Med. 2012;18:693-704
5. Ishizaka T, Conrad DH. Binding characteristics of human IgE receptors and initial triggering events in human mast cells for histamine release. Monogr Allergy. 1983;18:14-24
6. Nadler MJ, Matthews SA, Turner H, Kinet JP. Signal transduction by the high-affinity immunoglobulin E receptor Fc epsilon RI: coupling form to function. Adv Immunol. 2000;76:325-55
7. Theoharides TC, Valent P, Akin C. Mast Cells, Mastocytosis, and Related Disorders. N Engl J Med. 2015;373:163-72
8. Dvorak AM, Schleimer RP, Schulman ES, Lichtenstein LM. Human mast cells use conservation and condensation mechanisms during recovery from degranulation. In vitro studies with mast cells purified from human lungs. Lab Invest. 1986;54:663-78
9. Galli SJ. New insights into "the riddle of the mast cells": microenvironmental regulation of mast cell development and phenotypic heterogeneity. Lab Invest. 1990;62:5-33
10. Foedinger M, Fritsch G, Winkler K, Emminger W, Mitterbauer G, Gadner H. et al. Origin of human mast cells: development from transplanted hematopoietic stem cells after allogeneic bone marrow transplantation. Blood. 1994;84:2954-9
11. Galli SJ, Kalesnikoff J, Grimbaldeston MA, Piliponsky AM, Williams CM, Tsai M. Mast cells as "tunable" effector and immunoregulatory cells: recent advances. Annu Rev Immunol. 2005;23:749-86
12. Tsai M, Shih LS, Newlands GF, Takeishi T, Langley KE, Zsebo KM. et al. The rat c-kit ligand, stem cell factor, induces the development of connective tissue-type and mucosal mast cells in vivo. Analysis by anatomical distribution, histochemistry, and protease phenotype. J Exp Med. 1991;174:125-31
13. Galli SJ, Iemura A, Garlick DS, Gamba-Vitalo C, Zsebo KM, Andrews RG. Reversible expansion of primate mast cell populations in vivo by stem cell factor. J Clin Invest. 1993;91:148-52
14. Maurer M, Galli SJ. Lack of significant skin inflammation during elimination by apoptosis of large numbers of mouse cutaneous mast cells after cessation of treatment with stem cell factor. Lab Invest. 2004;84:1593-602
15. Kitamura Y, Go S, Hatanaka K. Decrease of mast cells in W/Wv mice and their increase by bone marrow transplantation. Blood. 1978;52:447-52
16. Kitamura Y, Go S. Decreased production of mast cells in S1/S1d anemic mice. Blood. 1979;53:492-7
17. Kitamura Y, Matsuda H, Hatanaka K. Clonal nature of mast-cell clusters formed in W/Wv mice after bone marrow transplantation. Nature. 1979;281:154-5
18. Kitamura Y, Yokoyama M, Matsuda H, Ohno T, Mori KJ. Spleen colony-forming cell as common precursor for tissue mast cells and granulocytes. Nature. 1981;291:159-60
19. Horton MA, O'Brien HA. Characterization of human mast cells in long-term culture. Blood. 1983;62:1251-60
20. Levi-Schaffer F, Dayton ET, Austen KF, Hein A, Caulfield JP, Gravallese PM. et al. Mouse bone marrow-derived mast cells cocultured with fibroblasts. Morphology and stimulation-induced release of histamine, leukotriene B4, leukotriene C4, and prostaglandin D2. J Immunol. 1987;139:3431-41
21. Fujita J, Nakayama H, Onoue H, Kanakura Y, Nakano T, Asai H. et al. Fibroblast-dependent growth of mouse mast cells in vitro: duplication of mast cell depletion in mutant mice of W/Wv genotype. J Cell Physiol. 1988;134:78-84
22. Jarboe DL, Marshall JS, Randolph TR, Kukolja A, Huff TF. The mast cell-committed progenitor. I. Description of a cell capable of IL-3-independent proliferation and differentiation without contact with fibroblasts. J Immunol. 1989;142:2405-17
23. Kirshenbaum AS, Kessler SW, Goff JP, Metcalfe DD. Demonstration of the origin of human mast cells from CD34+ bone marrow progenitor cells. J Immunol. 1991;146:1410-5
24. Agis H, Willheim M, Sperr WR, Wilfing A, Kroemer E, Kabrna E. et al. Monocytes do not make mast cells when cultured in the presence of SCF. Characterization of the circulating mast cell progenitor as a c-kit+, CD34+, Ly-, CD14-, CD17-, colony-forming cell. J Immunol. 1993;151:4221-7
25. Kempuraj D, Saito H, Kaneko A, Fukagawa K, Nakayama M, Toru H. et al. Characterization of mast cell-committed progenitors present in human umbilical cord blood. Blood. 1999;93:3338-46
26. Galli SJ, Tsai M, Wershil BK. The c-kit receptor, stem cell factor, and mast cells. What each is teaching us about the others. Am J Pathol. 1993;142:965-74
27. Valent P. The riddle of the mast cell: kit(CD117)-ligand as the missing link? Immunol Today. 1994;15:111-4
28. Sonoda T, Hayashi C, Kitamura Y. Presence of mast cell precursors in the yolk sac of mice. Dev Biol. 1983;97:89-94
29. Tsai FY, Orkin SH. Transcription factor GATA-2 is required for proliferation/survival of early hematopoietic cells and mast cell formation, but not for erythroid and myeloid terminal differentiation. Blood. 1997;89:3636-43
30. Poglio S, De Toni-Costes F, Arnaud E, Laharrague P, Espinosa E, Casteilla L. et al. Adipose tissue as a dedicated reservoir of functional mast cell progenitors. Stem Cells. 2010;28:2065-72
31. Dahlin JS, Malinovschi A, Oehrvik H, Sandelin M, Janson C, Alving K. et al. Lin- CD34hi CD117int/hi FcepsilonRI+ cells in human blood constitute a rare population of mast cell progenitors. Blood. 2016;127:383-91
32. Rodewald HR, Dessing M, Dvorak AM, Galli SJ. Identification of a committed precursor for the mast cell lineage. Science. 1996;271:818-22
33. Chen CC, Grimbaldeston MA, Tsai M, Weissman IL, Galli SJ. Identification of mast cell progenitors in adult mice. Proc Natl Acad Sci U S A. 2005;102:11408-13
34. Ochi H, Hirani WM, Yuan Q, Friend DS, Austen KF, Boyce JA. T helper cell type 2 cytokine-mediated comitogenic responses and CCR3 expression during differentiation of human mast cells in vitro. J Exp Med. 1999;190:267-80
35. Kirshenbaum AS, Goff JP, Semere T, Foster B, Scott LM, Metcalfe DD. Demonstration that human mast cells arise from a progenitor cell population that is CD34(+), c-kit(+), and expresses aminopeptidase N (CD13). Blood. 1999;94:2333-42
36. Schernthaner GH, Hauswirth AW, Baghestanian M, Agis H, Ghannadan M, Worda C. et al. Detection of differentiation- and activation-linked cell surface antigens on cultured mast cell progenitors. Allergy. 2005;60:1248-55
37. Maaninka K, Lappalainen J, Kovanen PT. Human mast cells arise from a common circulating progenitor. J Allergy Clin Immunol. 2013;132:463-9 e3
38. Gurish MF, Tao H, Abonia JP, Arya A, Friend DS, Parker CM. et al. Intestinal mast cell progenitors require CD49dbeta7 (alpha4beta7 integrin) for tissue-specific homing. J Exp Med. 2001;194:1243-52
39. Horny HP, Ruck P, Xiao JC, Kaiserling E. Immunoreactivity of normal and neoplastic human tissue mast cells with macrophage-associated antibodies, with special reference to the recently developed monoclonal antibody PG-M1. Hum Pathol. 1993;24:355-8
40. Valent P, Ashman LK, Hinterberger W, Eckersberger F, Majdic O, Lechner K. et al. Mast cell typing: demonstration of a distinct hematopoietic cell type and evidence for immunophenotypic relationship to mononuclear phagocytes. Blood. 1989;73:1778-85
41. Sperr WR, Horny HP, Lechner K, Valent P. Clinical and biologic diversity of leukemias occurring in patients with mastocytosis. Leuk Lymphoma. 2000;37:473-86
42. Sotlar K, Colak S, Bache A, Berezowska S, Krokowski M, Bueltmann B. et al. Variable presence of KITD816V in clonal haematological non-mast cell lineage diseases associated with systemic mastocytosis (SM-AHNMD). J Pathol. 2010;220:586-95
43. Valent P, Bettelheim P. Cell surface structures on human basophils and mast cells: biochemical and functional characterization. Adv Immunol. 1992;52:333-423
44. Kocabas CN, Yavuz AS, Lipsky PE, Metcalfe DD, Akin C. Analysis of the lineage relationship between mast cells and basophils using the c-kit D816V mutation as a biologic signature. J Allergy Clin Immunol. 2005;115:1155-61
45. Ihle JN, Keller J, Oroszlan S, Henderson LE, Copeland TD, Fitch F. et al. Biologic properties of homogeneous interleukin 3. I. Demonstration of WEHI-3 growth factor activity, mast cell growth factor activity, p cell-stimulating factor activity, colony-stimulating factor activity, and histamine-producing cell-stimulating factor activity. J Immunol. 1983;131:282-7
46. Hamaguchi Y, Kanakura Y, Fujita J, Takeda S, Nakano T, Tarui S. et al. Interleukin 4 as an essential factor for in vitro clonal growth of murine connective tissue-type mast cells. J Exp Med. 1987;165:268-73
47. Thompson-Snipes L, Dhar V, Bond MW, Mosmann TR, Moore KW, Rennick DM. Interleukin 10: a novel stimulatory factor for mast cells and their progenitors. J Exp Med. 1991;173:507-10
48. Shelburne CP, Ryan JJ. The role of Th2 cytokines in mast cell homeostasis. Immunol Rev. 2001;179:82-93
49. Zsebo KM, Williams DA, Geissler EN, Broudy VC, Martin FH, Atkins HL. et al. Stem cell factor is encoded at the Sl locus of the mouse and is the ligand for the c-kit tyrosine kinase receptor. Cell. 1990;63:213-24
50. Tsai M, Takeishi T, Thompson H, Langley KE, Zsebo KM, Metcalfe DD. et al. Induction of mast cell proliferation, maturation, and heparin synthesis by the rat c-kit ligand, stem cell factor. Proc Natl Acad Sci U S A. 1991;88:6382-6
51. Martin FH, Suggs SV, Langley KE, Lu HS, Ting J, Okino KH. et al. Primary structure and functional expression of rat and human stem cell factor DNAs. Cell. 1990;63:203-11
52. Kirshenbaum AS, Goff JP, Kessler SW, Mican JM, Zsebo KM, Metcalfe DD. Effect of IL-3 and stem cell factor on the appearance of human basophils and mast cells from CD34+ pluripotent progenitor cells. J Immunol. 1992;148:772-7
53. Valent P, Spanbloechl E, Sperr WR, Sillaber C, Zsebo KM, Agis H. et al. Induction of differentiation of human mast cells from bone marrow and peripheral blood mononuclear cells by recombinant human stem cell factor/kit-ligand in long-term culture. Blood. 1992;80:2237-45
54. Irani AM, Nilsson G, Miettinen U, Craig SS, Ashman LK, Ishizaka T. et al. Recombinant human stem cell factor stimulates differentiation of mast cells from dispersed human fetal liver cells. Blood. 1992;80:3009-21
55. Meininger CJ, Yano H, Rottapel R, Bernstein A, Zsebo KM, Zetter BR. The c-kit receptor ligand functions as a mast cell chemoattractant. Blood. 1992;79:958-63
56. Nilsson G, Butterfield JH, Nilsson K, Siegbahn A. Stem cell factor is a chemotactic factor for human mast cells. J Immunol. 1994;153:3717-23
57. Wershil BK, Tsai M, Geissler EN, Zsebo KM, Galli SJ. The rat c-kit ligand, stem cell factor, induces c-kit receptor-dependent mouse mast cell activation in vivo. Evidence that signaling through the c-kit receptor can induce expression of cellular function. J Exp Med. 1992;175:245-55
58. Baghestanian M, Agis H, Bevec D, Bankl HC, Hofbauer R, Kress HG. et al. Stem cell factor-induced downregulation of c-kit in human lung mast cells and HMC-1 mast cells. Exp Hematol. 1996;24:1377-86
59. Kiener HP, Hofbauer R, Tohidast-Akrad M, Walchshofer S, Redlich K, Bitzan P. et al. Tumor necrosis factor alpha promotes the expression of stem cell factor in synovial fibroblasts and their capacity to induce mast cell chemotaxis. Arthritis Rheum. 2000;43:164-74
60. Columbo M, Horowitz EM, Botana LM, MacGlashan DW Jr, Bochner BS, Gillis S. et al. The human recombinant c-kit receptor ligand, rhSCF, induces mediator release from human cutaneous mast cells and enhances IgE-dependent mediator release from both skin mast cells and peripheral blood basophils. J Immunol. 1992;149:599-608
61. Bischoff SC, Dahinden CA. c-kit ligand: a unique potentiator of mediator release by human lung mast cells. J Exp Med. 1992;175:237-44
62. Sperr WR, Czerwenka K, Mundigler G, Mueller MR, Semper H, Klappacher G. et al. Specific activation of human mast cells by the ligand for c-kit: comparison between lung, uterus and heart mast cells. Int Arch Allergy Immunol. 1993;102:170-5
63. Costa JJ, Demetri GD, Harrist TJ, Dvorak AM, Hayes DF, Merica EA. et al. Recombinant human stem cell factor (kit ligand) promotes human mast cell and melanocyte hyperplasia and functional activation in vivo. J Exp Med. 1996;183:2681-6
64. Dvorak AM, Costa JJ, Monahan-Earley RA, Fox P, Galli SJ. Ultrastructural analysis of human skin biopsy specimens from patients receiving recombinant human stem cell factor: subcutaneous injection of rhSCF induces dermal mast cell degranulation and granulocyte recruitment at the injection site. J Allergy Clin Immunol. 1998;101:793-806
65. Durand B, Migliaccio G, Yee NS, Eddleman K, Huima-Byron T, Migliaccio AR. et al. Long-term generation of human mast cells in serum-free cultures of CD34+ cord blood cells stimulated with stem cell factor and interleukin-3. Blood. 1994;84:3667-74
66. Tsujino Y, Wada H, Misawa M, Kai S, Hara H. Effects of mast cell growth factor, interleukin-3, and interleukin-6 on human primitive hematopoietic progenitors from bone marrow and cord blood. Exp Hematol. 1993;21:1379-86
67. Saito H, Ebisawa M, Tachimoto H, Shichijo M, Fukagawa K, Matsumoto K. et al. Selective growth of human mast cells induced by Steel factor, IL-6, and prostaglandin E2 from cord blood mononuclear cells. J Immunol. 1996;157:343-50
68. Yanagida M, Fukamachi H, Ohgami K, Kuwaki T, Ishii H, Uzumaki H. et al. Effects of T-helper 2-type cytokines, interleukin-3 (IL-3), IL-4, IL-5, and IL-6 on the survival of cultured human mast cells. Blood. 1995;86:3705-14
69. Shimizu Y, Matsumoto K, Okayama Y, Sakai K, Maeno T, Suga T. et al. Interleukin-3 does not affect the differentiation of mast cells derived from human bone marrow progenitors. Immunol Invest. 2008;37:1-17
70. Sillaber C, Sperr WR, Agis H, Spanbloechl E, Lechner K, Valent P. Inhibition of stem cell factor dependent formation of human mast cells by interleukin-3 and interleukin-4. Int Arch Allergy Immunol. 1994;105:264-8
71. Valent P, Besemer J, Sillaber C, Butterfield JH, Eher R, Majdic O. et al. Failure to detect IL-3-binding sites on human mast cells. J Immunol. 1990;145:3432-7
72. Xia HZ, Du Z, Craig S, Klisch G, Noben-Trauth N, Kochan JP. et al. Effect of recombinant human IL-4 on tryptase, chymase, and Fc epsilon receptor type I expression in recombinant human stem cell factor-dependent fetal liver-derived human mast cells. J Immunol. 1997;159:2911-21
73. Toru H, Eguchi M, Matsumoto R, Yanagida M, Yata J, Nakahata T. Interleukin-4 promotes the development of tryptase and chymase double-positive human mast cells accompanied by cell maturation. Blood. 1998;91:187-95
74. Yamaguchi M, Sayama K, Yano K, Lantz CS, Noben-Trauth N, Ra C. et al. IgE enhances Fc epsilon receptor I expression and IgE-dependent release of histamine and lipid mediators from human umbilical cord blood-derived mast cells: synergistic effect of IL-4 and IgE on human mast cell Fc epsilon receptor I expression and mediator release. J Immunol. 1999;162:5455-65
75. Bischoff SC, Sellge G, Lorentz A, Sebald W, Raab R, Manns MP. IL-4 enhances proliferation and mediator release in mature human mast cells. Proc Natl Acad Sci U S A. 1999;96:8080-5
76. Nilsson G, Miettinen U, Ishizaka T, Ashman LK, Irani AM, Schwartz LB. Interleukin-4 inhibits the expression of Kit and tryptase during stem cell factor-dependent development of human mast cells from fetal liver cells. Blood. 1994;84:1519-27
77. Oskeritzian CA, Wang Z, Kochan JP, Grimes M, Du Z, Chang HW. et al. Recombinant human (rh)IL-4-mediated apoptosis and recombinant human IL-6-mediated protection of recombinant human stem cell factor-dependent human mast cells derived from cord blood mononuclear cell progenitors. J Immunol. 1999;163:5105-15
78. Oskeritzian CA, Zhao W, Pozez AL, Cohen NM, Grimes M, Schwartz LB. Neutralizing endogenous IL-6 renders mast cells of the MCT type from lung, but not the MCTC type from skin and lung, susceptible to human recombinant IL-4-induced apoptosis. J Immunol. 2004;172:593-600
79. Ghildyal N, McNeil HP, Gurish MF, Austen KF, Stevens RL. Transcriptional regulation of the mucosal mast cell-specific protease gene, MMCP-2, by interleukin 10 and interleukin 3. J Biol Chem. 1992;267:8473-7
80. Gurish MF, Ghildyal N, McNeil HP, Austen KF, Gillis S, Stevens RL. Differential expression of secretory granule proteases in mouse mast cells exposed to interleukin 3 and c-kit ligand. J Exp Med. 1992;175:1003-12
81. Levi-Schaffer F, Austen KF, Gravallese PM, Stevens RL. Coculture of interleukin 3-dependent mouse mast cells with fibroblasts results in a phenotypic change of the mast cells. Proc Natl Acad Sci U S A. 1986;83:6485-8
82. Zheng PY, Geng XR, Hong JY, Yang G, Liu JQ, Mo LH. et al. Regulating Bcl2L12 expression in mast cells inhibits food allergy. Theranostics. 2019;9:4982-92
83. Austen KF, Boyce JA. Mast cell lineage development and phenotypic regulation. Leuk Res. 2001;25:511-8
84. Sonoda S, Sonoda T, Nakano T, Kanayama Y, Kanakura Y, Asai H. et al. Development of mucosal mast cells after injection of a single connective tissue-type mast cell in the stomach mucosa of genetically mast cell-deficient W/Wv mice. J Immunol. 1986;137:1319-22
85. Otsu K, Nakano T, Kanakura Y, Asai H, Katz HR, Austen KF. et al. Phenotypic changes of bone marrow-derived mast cells after intraperitoneal transfer into W/Wv mice that are genetically deficient in mast cells. J Exp Med. 1987;165:615-27
86. Kanakura Y, Thompson H, Nakano T, Yamamura T, Asai H, Kitamura Y. et al. Multiple bidirectional alterations of phenotype and changes in proliferative potential during the in vitro and in vivo passage of clonal mast cell populations derived from mouse peritoneal mast cells. Blood. 1988;72:877-85
87. Friend DS, Ghildyal N, Austen KF, Gurish MF, Matsumoto R, Stevens RL. Mast cells that reside at different locations in the jejunum of mice infected with Trichinella spiralis exhibit sequential changes in their granule ultrastructure and chymase phenotype. J Cell Biol. 1996;135:279-90
88. Serve H, Yee NS, Stella G, Sepp-Lorenzino L, Tan JC, Besmer P. Differential roles of PI3-kinase and Kit tyrosine 821 in Kit receptor-mediated proliferation, survival and cell adhesion in mast cells. EMBO J. 1995;14:473-83
89. Timokhina I, Kissel H, Stella G, Besmer P. Kit signaling through PI 3-kinase and Src kinase pathways: an essential role for Rac1 and JNK activation in mast cell proliferation. EMBO J. 1998;17:6250-62
90. Fukao T, Yamada T, Tanabe M, Terauchi Y, Ota T, Takayama T. et al. Selective loss of gastrointestinal mast cells and impaired immunity in PI3K-deficient mice. Nat Immunol. 2002;3:295-304
91. Harir N, Boudot C, Friedbichler K, Sonneck K, Kondo R, Martin-Lanneree S. et al. Oncogenic Kit controls neoplastic mast cell growth through a Stat5/PI3-kinase signaling cascade. Blood. 2008;112:2463-73
92. Moeller C, Alfredsson J, Engstroem M, Wootz H, Xiang Z, Lennartsson J. et al. Stem cell factor promotes mast cell survival via inactivation of FOXO3a-mediated transcriptional induction and MEK-regulated phosphorylation of the proapoptotic protein Bim. Blood. 2005;106:1330-6
93. Takayama G, Ohtani M, Minowa A, Matsuda S, Koyasu S. Class I PI3K-mediated Akt and ERK signals play a critical role in FcepsilonRI-induced degranulation in mast cells. Int Immunol. 2013;25:215-20
94. Hundley TR, Gilfillan AM, Tkaczyk C, Andrade MV, Metcalfe DD, Beaven MA. Kit and FcepsilonRI mediate unique and convergent signals for release of inflammatory mediators from human mast cells. Blood. 2004;104:2410-7
95. Baumgartner C, Cerny-Reiterer S, Sonneck K, Mayerhofer M, Gleixner KV, Fritz R. et al. Expression of activated STAT5 in neoplastic mast cells in systemic mastocytosis: subcellular distribution and role of the transforming oncoprotein KIT D816V. Am J Pathol. 2009;175:2416-29
96. Peter B, Cerny-Reiterer S, Hadzijusufovic E, Schuch K, Stefanzl G, Eisenwort G. et al. The pan-Bcl-2 blocker obatoclax promotes the expression of Puma, Noxa, and Bim mRNA and induces apoptosis in neoplastic mast cells. J Leukoc Biol. 2014;95:95-104
97. Maurer M, Tsai M, Metz M, Fish S, Korsmeyer SJ, Galli SJ. A role for Bax in the regulation of apoptosis in mouse mast cells. J Invest Dermatol. 2000;114:1205-6
98. Pullen NA, Barnstein BO, Falanga YT, Wang Z, Suzuki R, Tamang TD. et al. Novel mechanism for Fc{epsilon}RI-mediated signal transducer and activator of transcription 5 (STAT5) tyrosine phosphorylation and the selective influence of STAT5B over mast cell cytokine production. J Biol Chem. 2012;287:2045-54
99. Ohneda K, Ohmori S, Yamamoto M. Mouse Tryptase Gene Expression is Coordinately Regulated by GATA1 and GATA2 in Bone Marrow-Derived Mast Cells. Int J Mol Sci. 2019;20:4603
100. Li Y, Qi X, Liu B, Huang H. The STAT5-GATA2 pathway is critical in basophil and mast cell differentiation and maintenance. J Immunol. 2015;194:4328-38
101. Li Y, Liu B, Harmacek L, Long Z, Liang J, Lukin K. et al. The transcription factors GATA2 and microphthalmia-associated transcription factor regulate Hdc gene expression in mast cells and are required for IgE/mast cell-mediated anaphylaxis. J Allergy Clin Immunol. 2018;142:1173-84
102. Morii E, Ogihara H, Oboki K, Kataoka TR, Maeyama K, Fisher DE. et al. Effect of a large deletion of the basic domain of mi transcription factor on differentiation of mast cells. Blood. 2001;98:2577-9
103. Fekete E, Little CC, Cloudman AM. Some Effects of the Gene W(Dominant Spotting) in Mice. Proc Natl Acad Sci U S A. 1941;27:114-7
104. Geissler EN, McFarland EC, Russell ES. Analysis of pleiotropism at the dominant white-spotting (W) locus of the house mouse: a description of ten new W alleles. Genetics. 1981;97:337-61
105. Zhou JS, Xing W, Friend DS, Austen KF, Katz HR. Mast cell deficiency in Kit(W-sh) mice does not impair antibody-mediated arthritis. J Exp Med. 2007;204:2797-802
106. Kitamura Y, Morii E, Ogihara H, Jippo T, Ito A. Mutant mice: a useful tool for studying the development of mast cells. Int Arch Allergy Immunol. 2001;124:16-9
107. Kitamura Y, Morii E, Jippo T, Ito A. mi-transcription factor as a regulator of mast cell differentiation. Int J Hematol. 2000;71:197-202
108. Tsujimura T, Morii E, Nozaki M, Hashimoto K, Moriyama Y, Takebayashi K. et al. Involvement of transcription factor encoded by the mi locus in the expression of c-kit receptor tyrosine kinase in cultured mast cells of mice. Blood. 1996;88:1225-33
109. Ito A, Morii E, Kim DK, Kataoka TR, Jippo T, Maeyama K. et al. Inhibitory effect of the transcription factor encoded by the mi mutant allele in cultured mast cells of mice. Blood. 1999;93:1189-96
110. Kim DK, Morii E, Ogihara H, Lee YM, Jippo T, Adachi S. et al. Different effect of various mutant MITF encoded by mi, Mior, or Miwh allele on phenotype of murine mast cells. Blood. 1999;93:4179-86
111. Adachi S, Morii E, Kim D, Ogihara H, Jippo T, Ito A. et al. Involvement of mi-transcription factor in expression of alpha-melanocyte-stimulating hormone receptor in cultured mast cells of mice. J Immunol. 2000;164:855-60
112. Ogihara H, Morii E, Kim DK, Oboki K, Kitamura Y. Inhibitory effect of the transcription factor encoded by the mutant mi microphthalmia allele on transactivation of mouse mast cell protease 7 gene. Blood. 2001;97:645-51
113. Kitamura Y, Morii E, Jippo T, Ito A. Regulation of mast cell phenotype by MITF. Int Arch Allergy Immunol. 2002;127:106-9
114. Nigrovic PA, Gray DH, Jones T, Hallgren J, Kuo FC, Chaletzky B. et al. Genetic inversion in mast cell-deficient (Wsh) mice interrupts corin and manifests as hematopoietic and cardiac aberrancy. Am J Pathol. 2008;173:1693-701
115. Reber LL, Marichal T, Galli SJ. New models for analyzing mast cell functions in vivo. Trends Immunol. 2012;33:613-25
116. Piliponsky AM, Chen CC, Grimbaldeston MA, Burns-Guydish SM, Hardy J, Kalesnikoff J. et al. Mast cell-derived TNF can exacerbate mortality during severe bacterial infections in C57BL/6-KitW-sh/W-sh mice. Am J Pathol. 2010;176:926-38
117. Grimbaldeston MA, Chen CC, Piliponsky AM, Tsai M, Tam SY, Galli SJ. Mast cell-deficient W-sash c-kit mutant Kit W-sh/W-sh mice as a model for investigating mast cell biology in vivo. Am J Pathol. 2005;167:835-48
118. Rodewald HR, Feyerabend TB. Widespread immunological functions of mast cells: fact or fiction? Immunity. 2012;37:13-24
119. Becker M, Reuter S, Friedrich P, Doener F, Michel A, Bopp T. et al. Genetic variation determines mast cell functions in experimental asthma. J Immunol. 2011;186:7225-31
120. Feyerabend TB, Weiser A, Tietz A, Stassen M, Harris N, Kopf M. et al. Cre-mediated cell ablation contests mast cell contribution in models of antibody- and T cell-mediated autoimmunity. Immunity. 2011;35:832-44
121. Lilla JN, Chen CC, Mukai K, BenBarak MJ, Franco CB, Kalesnikoff J. et al. Reduced mast cell and basophil numbers and function in Cpa3-Cre; Mcl-1fl/fl mice. Blood. 2011;118:6930-8
122. Giebel LB, Spritz RA. Mutation of the KIT (mast/stem cell growth factor receptor) protooncogene in human piebaldism. Proc Natl Acad Sci U S A. 1991;88:8696-9
123. Sanchez-Martin M, Perez-Losada J, Rodriguez-Garcia A, Gonzalez-Sanchez B, Korf BR, Kuster W. et al. Deletion of the SLUG (SNAI2) gene results in human piebaldism. Am J Med Genet A. 2003;122A:125-32
124. Irani AM, Craig SS, DeBlois G, Elson CO, Schechter NM, Schwartz LB. Deficiency of the tryptase-positive, chymase-negative mast cell type in gastrointestinal mucosa of patients with defective T lymphocyte function. J Immunol. 1987;138:4381-6
125. Cerny-Reiterer S, Rabenhorst A, Stefanzl G, Herndlhofer S, Hoermann G, Muellauer L. et al. Long-term treatment with imatinib results in profound mast cell deficiency in Ph+ chronic myeloid leukemia. Oncotarget. 2015;6:3071-84
126. Sperr WR, Bankl HC, Mundigler G, Klappacher G, Grossschmidt K, Agis H. et al. The human cardiac mast cell: localization, isolation, phenotype, and functional characterization. Blood. 1994;84:3876-84
127. Ghannadan M, Baghestanian M, Wimazal F, Eisenmenger M, Latal D, Karguel G. et al. Phenotypic characterization of human skin mast cells by combined staining with toluidine blue and CD antibodies. J Invest Dermatol. 1998;111:689-95
128. Agis H, Fuereder W, Bankl HC, Kundi M, Sperr WR, Willheim M. et al. Comparative immunophenotypic analysis of human mast cells, blood basophils and monocytes. Immunology. 1996;87:535-43
129. Saito H, Nakajima T, Matsumoto K. Human mast cell transcriptome project. Int Arch Allergy Immunol. 2001;125:1-8
130. Dwyer DF, Barrett NA, Austen KF, Immunological Genome Project C. Expression profiling of constitutive mast cells reveals a unique identity within the immune system. Nat Immunol. 2016;17:878-87
131. Gschwandtner M, Paulitschke V, Mildner M, Brunner PM, Hacker S, Eisenwort G. et al. Proteome analysis identifies L1CAM/CD171 and DPP4/CD26 as novel markers of human skin mast cells. Allergy. 2017;72:85-97
132. Willheim M, Agis H, Sperr WR, Koeller M, Bankl HC, Kiener H. et al. Purification of human basophils and mast cells by multistep separation technique and mAb to CDw17 and CD117/c-kit. J Immunol Methods. 1995;182:115-29
133. Escribano L, Orfao A, Diaz-Agustin B, Villarrubia J, Cervero C, Lopez A. et al. Indolent systemic mast cell disease in adults: immunophenotypic characterization of bone marrow mast cells and its diagnostic implications. Blood. 1998;91:2731-6
134. Escribano L, Diaz-Agustin B, Bellas C, Navalon R, Nunez R, Sperr WR. et al. Utility of flow cytometric analysis of mast cells in the diagnosis and classification of adult mastocytosis. Leuk Res. 2001;25:563-70
135. Valent P, Cerny-Reiterer S, Herrmann H, Mirkina I, George TI, Sotlar K. et al. Phenotypic heterogeneity, novel diagnostic markers, and target expression profiles in normal and neoplastic human mast cells. Best Pract Res Clin Haematol. 2010;23:369-78
136. Valent P, Sotlar K, Horny HP. Aberrant expression of CD30 in aggressive systemic mastocytosis and mast cell leukemia: a differential diagnosis to consider in aggressive hematopoietic CD30-positive neoplasms. Leuk Lymphoma. 2011;52:740-4
137. Valent P, Horny HP, Escribano L, Longley BJ, Li CY, Schwartz LB. et al. Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res. 2001;25:603-25
138. Valent P, Akin C, Metcalfe DD. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood. 2017;129:1420-7
139. Horny HP, Akin C, Arber D, Peterson LA, Tefferi A, Metcalfe DD. et al. Mastocytosis. In: World Health Organization (WHO) Classification of Tumours. Pathology & Genetics. Tumours of Haematopoietic and Lymphoid Tissues. Eds: Swerdlow, SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Arber DA, Hasserjian RP, Le Beau MM, Orazi A, Siebert R. Lyon, France: IARC Press. 2017:62-9
140. Pardanani A, Lasho T, Chen D, Kimlinger TK, Finke C, Zblewski D. et al. Aberrant expression of CD123 (interleukin-3 receptor-alpha) on neoplastic mast cells. Leukemia. 2015;29:1605-8
141. Teodosio C, Garcia-Montero AC, Jara-Acevedo M, Sanchez-Munoz L, Alvarez-Twose I, Nunez R. et al. Mast cells from different molecular and prognostic subtypes of systemic mastocytosis display distinct immunophenotypes. J Allergy Clin Immunol. 2010;125:719-26 26 e1-26 e4
142. Hauswirth AW, Escribano L, Prados A, Nunez R, Mirkina I, Kneidinger M. et al. CD203c is overexpressed on neoplastic mast cells in systemic mastocytosis and is upregulated upon IgE receptor cross-linking. Int J Immunopathol Pharmacol. 2008;21:797-806
143. Kiener HP, Baghestanian M, Dominkus M, Walchshofer S, Ghannadan M, Willheim M. et al. Expression of the C5a receptor (CD88) on synovial mast cells in patients with rheumatoid arthritis. Arthritis Rheum. 1998;41:233-45
144. Teodosio C, Mayado A, Sanchez-Munoz L, Morgado JM, Jara-Acevedo M, Alvarez-Twose I. et al. The immunophenotype of mast cells and its utility in the diagnostic work-up of systemic mastocytosis. J Leukoc Biol. 2015;97:49-59
145. Schwartz LB. Mediators of human mast cells and human mast cell subsets. Ann Allergy. 1987;58:226-35
146. Wodnar-Filipowicz A, Heusser CH, Moroni C. Production of the haemopoietic growth factors GM-CSF and interleukin-3 by mast cells in response to IgE receptor-mediated activation. Nature. 1989;339:150-2
147. Burd PR, Rogers HW, Gordon JR, Martin CA, Jayaraman S, Wilson SD. et al. Interleukin 3-dependent and -independent mast cells stimulated with IgE and antigen express multiple cytokines. J Exp Med. 1989;170:245-57
148. Plaut M, Pierce JH, Watson CJ, Hanley-Hyde J, Nordan RP, Paul WE. Mast cell lines produce lymphokines in response to cross-linkage of Fc epsilon RI or to calcium ionophores. Nature. 1989;339:64-7
149. Gordon JR, Galli SJ. Mast cells as a source of both preformed and immunologically inducible TNF-alpha/cachectin. Nature. 1990;346:274-6
150. Galli SJ, Gordon JR, Wershil BK. Mast cell cytokines in allergy and inflammation. Agents Actions Suppl. 1993;43:209-20
151. Schwartz LB, Bradford TR. Regulation of tryptase from human lung mast cells by heparin. Stabilization of the active tetramer. J Biol Chem. 1986;261:7372-9
152. Stevens RL, Adachi R. Protease-proteoglycan complexes of mouse and human mast cells and importance of their beta-tryptase-heparin complexes in inflammation and innate immunity. Immunol Rev. 2007;217:155-67
153. Castells M. Mast cell mediators in allergic inflammation and mastocytosis. Immunol Allergy Clin North Am. 2006;26:465-85
154. Metcalfe DD, Lewis RA, Silbert JE, Rosenberg RD, Wasserman SI, Austen KF. Isolation and characterization of heparin from human lung. J Clin Invest. 1979;64:1537-43
155. Reed JA, Albino AP, McNutt NS. Human cutaneous mast cells express basic fibroblast growth factor. Lab Invest. 1995;72:215-22
156. Artuc M, Hermes B, Steckelings UM, Gruetzkau A, Henz BM. Mast cells and their mediators in cutaneous wound healing-active participants or innocent bystanders? Exp Dermatol. 1999;8:1-16
157. Sillaber C, Baghestanian M, Bevec D, Willheim M, Agis H, Kapiotis S. et al. The mast cell as site of tissue-type plasminogen activator expression and fibrinolysis. J Immunol. 1999;162:1032-41
158. Lassila R, Jouppila A. Mast cell-derived heparin proteoglycans as a model for a local antithrombotic. Semin Thromb Hemost. 2014;40:837-44
159. Boesiger J, Tsai M, Maurer M, Yamaguchi M, Brown LF, Claffey KP. et al. Mast cells can secrete vascular permeability factor/ vascular endothelial cell growth factor and exhibit enhanced release after immunoglobulin E-dependent upregulation of fc epsilon receptor I expression. J Exp Med. 1998;188:1135-45
160. Baghestanian M, Hofbauer R, Kiener HP, Bankl HC, Wimazal F, Willheim M. et al. The c-kit ligand stem cell factor and anti-IgE promote expression of monocyte chemoattractant protein-1 in human lung mast cells. Blood. 1997;90:4438-49
161. Hoermann G, Cerny-Reiterer S, Perne A, Klauser M, Hoetzenecker K, Klein K. et al. Identification of oncostatin M as a STAT5-dependent mediator of bone marrow remodeling in KIT D816V-positive systemic mastocytosis. Am J Pathol. 2011;178:2344-56
162. Teodosio C, Garcia-Montero AC, Jara-Acevedo M, Sanchez-Munoz L, Pedreira CE, Alvarez-Twose I. et al. Gene expression profile of highly purified bone marrow mast cells in systemic mastocytosis. J Allergy Clin Immunol. 2013;131:1213-24 24 e1-4
163. Plum T, Wang X, Rettel M, Krijgsveld J, Feyerabend TB, Rodewald HR. Human Mast Cell Proteome Reveals Unique Lineage, Putative Functions, and Structural Basis for Cell Ablation. Immunity. 2020;52:404-16 e5
164. Marone G, Poto S, Celestino D, Bonini S. Human basophil releasability. III. Genetic control of human basophil releasability. J Immunol. 1986;137:3588-92
165. Kepley CL, Youssef L, Andrews RP, Wilson BS, Oliver JM. Syk deficiency in nonreleaser basophils. J Allergy Clin Immunol. 1999;104:279-84
166. Bonadonna P, Perbellini O, Passalacqua G, Caruso B, Colarossi S, Dal Fior D. et al. Clonal mast cell disorders in patients with systemic reactions to Hymenoptera stings and increased serum tryptase levels. J Allergy Clin Immunol. 2009;123:680-6
167. Matito A, Alvarez-Twose I, Morgado JM, Sanchez-Munoz L, Orfao A, Escribano L. Anaphylaxis as a clinical manifestation of clonal mast cell disorders. Curr Allergy Asthma Rep. 2014;14:450
168. Akin C, Valent P, Metcalfe DD. Mast cell activation syndrome: Proposed diagnostic criteria. J Allergy Clin Immunol. 2010;126:1099-104 e4
169. Valent P, Akin C, Arock M, Brockow K, Butterfield JH, Carter MC. et al. Definitions, criteria and global classification of mast cell disorders with special reference to mast cell activation syndromes: a consensus proposal. Int Arch Allergy Immunol. 2012;157:215-25
170. Valent P, Akin C, Bonadonna P, Hartmann K, Brockow K, Niedoszytko M. et al. Proposed Diagnostic Algorithm for Patients with Suspected Mast Cell Activation Syndrome. J Allergy Clin Immunol Pract. 2019;7:1125-33 e1
171. Dvorak AM. Basophils and mast cells: piecemeal degranulation in situ and ex vivo: a possible mechanism for cytokine-induced function in disease. Immunol Ser. 1992;57:169-271
172. Lawrence ID, Warner JA, Cohan VL, Hubbard WC, Kagey-Sobotka A, Lichtenstein LM. Purification and characterization of human skin mast cells. Evidence for human mast cell heterogeneity. J Immunol. 1987;139:3062-9
173. el-Lati SG, Dahinden CA, Church MK. Complement peptides C3a- and C5a-induced mediator release from dissociated human skin mast cells. J Invest Dermatol. 1994;102:803-6
174. Fuereder W, Agis H, Willheim M, Bankl HC, Maier U, Kishi K. et al. Differential expression of complement receptors on human basophils and mast cells. Evidence for mast cell heterogeneity and CD88/C5aR expression on skin mast cells. J Immunol. 1995;155:3152-60
175. Oskeritzian CA, Zhao W, Min HK, Xia HZ, Pozez A, Kiev J. et al. Surface CD88 functionally distinguishes the MCTC from the MCT type of human lung mast cell. J Allergy Clin Immunol. 2005;115:1162-8
176. Schafer B, Piliponsky AM, Oka T, Song CH, Gerard NP, Gerard C. et al. Mast cell anaphylatoxin receptor expression can enhance IgE-dependent skin inflammation in mice. J Allergy Clin Immunol. 2013;131:541-8 e1-9
177. Enerbaeck L. Mast cells in rat gastrointestinal mucosa. 2. Dye-binding and metachromatic properties. Acta Pathol Microbiol Scand. 1966;66:303-12
178. Irani AA, Schechter NM, Craig SS, DeBlois G, Schwartz LB. Two types of human mast cells that have distinct neutral protease compositions. Proc Natl Acad Sci U S A. 1986;83:4464-8
179. Crivellato E, Ribatti D. The fundamental contribution of William Bate Hardy to shape the concept of mast cell heterogeneity. Br J Haematol. 2010;150:152-7
180. Befus AD, Chin B, Pick J, Evans S, Osborn S, Forstrom J. Proteinases of rat mast cells. Peritoneal but not intestinal mucosal mast cells express mast cell proteinase 5 and carboxypeptidase A. J Immunol. 1995;155:4406-11
181. Pejler G, Abrink M, Ringvall M, Wernersson S. Mast cell proteases. Adv Immunol. 2007;95:167-255
182. Kitamura Y. Heterogeneity of mast cells and phenotypic change between subpopulations. Annu Rev Immunol. 1989;7:59-76
183. Bankova LG, Dwyer DF, Liu AY, Austen KF, Gurish MF. Maturation of mast cell progenitors to mucosal mast cells during allergic pulmonary inflammation in mice. Mucosal Immunol. 2015;8:596-606
184. McNeil BD, Pundir P, Meeker S, Han L, Undem BJ, Kulka M. et al. Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature. 2015;519:237-41
185. Nunez-Lopez R, Escribano L, Schernthaner GH, Prados A, Rodriguez-Gonzalez R, Diaz-Agustin B. et al. Overexpression of complement receptors and related antigens on the surface of bone marrow mast cells in patients with systemic mastocytosis. Br J Haematol. 2003;120:257-65
186. Okayama Y, Kirshenbaum AS, Metcalfe DD. Expression of a functional high-affinity IgG receptor, Fc gamma RI, on human mast cells: Up-regulation by IFN-gamma. J Immunol. 2000;164:4332-9
187. Lotfi-Emran S, Ward BR, Le QT, Pozez AL, Manjili MH, Woodfolk JA. et al. Human mast cells present antigen to autologous CD4(+) T cells. J Allergy Clin Immunol. 2018;141:311-21 e10
188. Galli SJ, Tsai M. Mast cells in allergy and infection: versatile effector and regulatory cells in innate and adaptive immunity. Eur J Immunol. 2010;40:1843-51
189. Echtenacher B, Maennel DN, Hueltner L. Critical protective role of mast cells in a model of acute septic peritonitis. Nature. 1996;381:75-7
190. Malaviya R, Ikeda T, Ross E, Abraham SN. Mast cell modulation of neutrophil influx and bacterial clearance at sites of infection through TNF-alpha. Nature. 1996;381:77-80
191. Malaviya R, Ikeda T, Abraham SN, Malaviya R. Contribution of mast cells to bacterial clearance and their proliferation during experimental cystitis induced by type 1 fimbriated E. coli. Immunol Lett. 2004;91:103-11
192. Dreskin SC, Abraham SN. Production of TNF-alpha by murine bone marrow derived mast cells activated by the bacterial fimbrial protein, FimH. Clin Immunol. 1999;90:420-4
193. Cruse G, Fernandes VE, de Salort J, Pankhania D, Marinas MS, Brewin H. et al. Human lung mast cells mediate pneumococcal cell death in response to activation by pneumolysin. J Immunol. 2010;184:7108-15
194. Hofmann AM, Abraham SN. New roles for mast cells in modulating allergic reactions and immunity against pathogens. Curr Opin Immunol. 2009;21:679-86
195. Piliponsky AM, Shubin NJ, Lahiri AK, Truong P, Clauson M, Niino K. et al. Basophil-derived tumor necrosis factor can enhance survival in a sepsis model in mice. Nat Immunol. 2019;20:129-40
196. Prodeus AP, Zhou X, Maurer M, Galli SJ, Carroll MC. Impaired mast cell-dependent natural immunity in complement C3-deficient mice. Nature. 1997;390:172-5
197. Kulka M, Fukuishi N, Rottem M, Mekori YA, Metcalfe DD. Mast cells, which interact with Escherichia coli, up-regulate genes associated with innate immunity and become less responsive to Fc(epsilon)RI-mediated activation. J Leukoc Biol. 2006;79:339-50
198. Piliponsky AM, Chen CC, Nishimura T, Metz M, Rios EJ, Dobner PR. et al. Neurotensin increases mortality and mast cells reduce neurotensin levels in a mouse model of sepsis. Nat Med. 2008;14:392-8
199. Abe T, Nawa Y. Reconstitution of mucosal mast cells in W/WV mice by adoptive transfer of bone marrow-derived cultured mast cells and its effects on the protective capacity to Strongyloides ratti-infection. Parasite Immunol. 1987;9:31-8
200. Lantz CS, Boesiger J, Song CH, Mach N, Kobayashi T, Mulligan RC. et al. Role for interleukin-3 in mast-cell and basophil development and in immunity to parasites. Nature. 1998;392:90-3
201. Newlands GF, Miller HR, MacKellar A, Galli SJ. Stem cell factor contributes to intestinal mucosal mast cell hyperplasia in rats infected with Nippostrongylus brasiliensis or Trichinella spiralis, but anti-stem cell factor treatment decreases parasite egg production during N brasiliensis infection. Blood. 1995;86:1968-76
202. Maurer M, Lopez Kostka S, Siebenhaar F, Moelle K, Metz M, Knop J. et al. Skin mast cells control T cell-dependent host defense in Leishmania major infections. FASEB J. 2006;20:2460-7
203. Barrett KE, Neva FA, Gam AA, Cicmanec J, London WT, Phillips JM. et al. The immune response to nematode parasites: modulation of mast cell numbers and function during Strongyloides stercoralis infections in nonhuman primates. Am J Trop Med Hyg. 1988;38:574-81
204. Bannert N, Farzan M, Friend DS, Ochi H, Price KS, Sodroski J. et al. Human Mast cell progenitors can be infected by macrophagetropic human immunodeficiency virus type 1 and retain virus with maturation in vitro. J Virol. 2001;75:10808-14
205. Sundstrom JB, Hair GA, Ansari AA, Secor WE, Gilfillan AM, Metcalfe DD. et al. IgE-FcepsilonRI interactions determine HIV coreceptor usage and susceptibility to infection during ontogeny of mast cells. J Immunol. 2009;182:6401-9
206. Sundstrom JB, Ellis JE, Hair GA, Kirshenbaum AS, Metcalfe DD, Yi H. et al. Human tissue mast cells are an inducible reservoir of persistent HIV infection. Blood. 2007;109:5293-300
207. Akoto C, Davies DE, Swindle EJ. Mast cells are permissive for rhinovirus replication: potential implications for asthma exacerbations. Clin Exp Allergy. 2017;47:351-60
208. Kulka M, Alexopoulou L, Flavell RA, Metcalfe DD. Activation of mast cells by double-stranded RNA: evidence for activation through Toll-like receptor 3. J Allergy Clin Immunol. 2004;114:174-82
209. St John AL, Rathore AP, Yap H, Ng ML, Metcalfe DD, Vasudevan SG. et al. Immune surveillance by mast cells during dengue infection promotes natural killer (NK) and NKT-cell recruitment and viral clearance. Proc Natl Acad Sci U S A. 2011;108:9190-5
210. Metz M, Piliponsky AM, Chen CC, Lammel V, Abrink M, Pejler G. et al. Mast cells can enhance resistance to snake and honeybee venoms. Science. 2006;313:526-30
211. Schneider LA, Schlenner SM, Feyerabend TB, Wunderlin M, Rodewald HR. Molecular mechanism of mast cell mediated innate defense against endothelin and snake venom sarafotoxin. J Exp Med. 2007;204:2629-39
212. Akahoshi M, Song CH, Piliponsky AM, Metz M, Guzzetta A, Abrink M. et al. Mast cell chymase reduces the toxicity of Gila monster venom, scorpion venom, and vasoactive intestinal polypeptide in mice. J Clin Invest. 2011;121:4180-91
213. Marichal T, Starkl P, Reber LL, Kalesnikoff J, Oettgen HC, Tsai M. et al. A beneficial role for immunoglobulin E in host defense against honeybee venom. Immunity. 2013;39:963-75
214. Palm NW, Rosenstein RK, Yu S, Schenten DD, Florsheim E, Medzhitov R. Bee venom phospholipase A2 induces a primary type 2 response that is dependent on the receptor ST2 and confers protective immunity. Immunity. 2013;39:976-85
215. Starkl P, Marichal T, Gaudenzio N, Reber LL, Sibilano R, Tsai M. et al. IgE antibodies, FcepsilonRIalpha, and IgE-mediated local anaphylaxis can limit snake venom toxicity. J Allergy Clin Immunol. 2016;137:246-57 e11
216. Galli SJ, Starkl P, Marichal T, Tsai M. Mast cells and IgE in defense against venoms: Possible "good side" of allergy? Allergol Int. 2016;65:3-15
217. Anderson E, Stavenhagen K, Kolarich D, Sommerhoff CP, Maurer M, Metz M. Human Mast Cell Tryptase Is a Potential Treatment for Snakebite Envenoming Across Multiple Snake Species. Front Immunol. 2018;9:1532
218. Rauter I, Krauth MT, Flicker S, Gieras A, Westritschnig K, Vrtala S. et al. Allergen cleavage by effector cell-derived proteases regulates allergic inflammation. FASEB J. 2006;20:967-9
219. Mellon MB, Frank BT, Fang KC. Mast cell alpha-chymase reduces IgE recognition of birch pollen profilin by cleaving antibody-binding epitopes. J Immunol. 2002;168:290-7
220. Rauter I, Krauth MT, Westritschnig K, Horak F, Flicker S, Gieras A. et al. Mast cell-derived proteases control allergic inflammation through cleavage of IgE. J Allergy Clin Immunol. 2008;121:197-202
221. Hill M. Secretion of heparin by mast cells. Nature. 1957;180:654-5
222. Kitamura Y, Taguchi T, Yokoyama M, Inoue M, Yamatodani A, Asano H. et al. Higher susceptibility of mast-cell-deficient W/WV mutant mice to brain thromboembolism and mortality caused by intravenous injection of India ink. Am J Pathol. 1986;122:469-80
223. Bankl HC, Radaszkiewicz T, Klappacher GW, Glogar D, Sperr WR, Grossschmidt K. et al. Increase and redistribution of cardiac mast cells in auricular thrombosis. Possible role of kit ligand. Circulation. 1995;91:275-83
224. Baghestanian M, Hofbauer R, Kress HG, Wojta J, Fabry A, Binder BR. et al. Thrombin augments vascular cell-dependent migration of human mast cells: role of MGF. Thromb Haemost. 1997;77:577-84
225. Nenci GG, Berrettini M, Parise P, Agnelli G. Persistent spontaneous heparinaemia in systemic mastocytosis. Folia Haematol Int Mag Klin Morphol Blutforsch. 1982;109:453-63
226. Fanning SF, White MJ, Douglas JB, Rowell JA. Systemic heparin-like activity in mastocytosis. Aust N Z J Med. 1992;22:170-1
227. Sucker C, Mansmann G, Steiner S, Gattermann N, Schmitt-Graeff A, Loncar R. et al. Fatal bleeding due to a heparin-like anticoagulant in a 37-year-old woman suffering from systemic mastocytosis. Clin Appl Thromb Hemost. 2008;14:360-4
228. Wimazal F, Sperr WR, Horny HP, Carroll V, Binder BR, Fonatsch C. et al. Hyperfibrinolysis in a case of myelodysplastic syndrome with leukemic spread of mast cells. Am J Hematol. 1999;61:66-77
229. Wulff BC, Parent AE, Meleski MA, DiPietro LA, Schrementi ME, Wilgus TA. Mast cells contribute to scar formation during fetal wound healing. J Invest Dermatol. 2012;132:458-65
230. Douaiher J, Succar J, Lancerotto L, Gurish MF, Orgill DP, Hamilton MJ. et al. Development of mast cells and importance of their tryptase and chymase serine proteases in inflammation and wound healing. Adv Immunol. 2014;122:211-52
231. Younan G, Suber F, Xing W, Shi T, Kunori Y, Abrink M. et al. The inflammatory response after an epidermal burn depends on the activities of mouse mast cell proteases 4 and 5. J Immunol. 2010;185:7681-90
232. Antsiferova M, Martin C, Huber M, Feyerabend TB, Forster A, Hartmann K. et al. Mast cells are dispensable for normal and activin-promoted wound healing and skin carcinogenesis. J Immunol. 2013;191:6147-55
233. Nauta AC, Grova M, Montoro DT, Zimmermann A, Tsai M, Gurtner GC. et al. Evidence that mast cells are not required for healing of splinted cutaneous excisional wounds in mice. PLoS One. 2013;8:e59167
234. Kokkonen JO, Kovanen PT. Stimulation of mast cells leads to cholesterol accumulation in macrophages in vitro by a mast cell granule-mediated uptake of low density lipoprotein. Proc Natl Acad Sci U S A. 1987;84:2287-91
235. Kovanen PT. Mast cell granule-mediated uptake of low density lipoproteins by macrophages: a novel carrier mechanism leading to the formation of foam cells. Ann Med. 1991;23:551-9
236. Kovanen PT, Kokkonen JO. Modification of low density lipoproteins by secretory granules of rat serosal mast cells. J Biol Chem. 1991;266:4430-6
237. Kaartinen M, Penttila A, Kovanen PT. Mast cells of two types differing in neutral protease composition in the human aortic intima. Demonstration of tryptase- and tryptase/chymase-containing mast cells in normal intimas, fatty streaks, and the shoulder region of atheromas. Arterioscler Thromb. 1994;14:966-72
238. Wang Y, Lindstedt KA, Kovanen PT. Mast cell granule remnants carry LDL into smooth muscle cells of the synthetic phenotype and induce their conversion into foam cells. Arterioscler Thromb Vasc Biol. 1995;15:801-10
239. Ma H, Kovanen PT. IgE-dependent generation of foam cells: an immune mechanism involving degranulation of sensitized mast cells with resultant uptake of LDL by macrophages. Arterioscler Thromb Vasc Biol. 1995;15:811-9
240. Kovanen PT, Kaartinen M, Paavonen T. Infiltrates of activated mast cells at the site of coronary atheromatous erosion or rupture in myocardial infarction. Circulation. 1995;92:1084-8
241. Kaartinen M, van der Wal AC, van der Loos CM, Piek JJ, Koch KT, Becker AE. et al. Mast cell infiltration in acute coronary syndromes: implications for plaque rupture. J Am Coll Cardiol. 1998;32:606-12
242. Leskinen M, Wang Y, Leszczynski D, Lindstedt KA, Kovanen PT. Mast cell chymase induces apoptosis of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2001;21:516-22
243. Lee M, Sommerhoff CP, von Eckardstein A, Zettl F, Fritz H, Kovanen PT. Mast cell tryptase degrades HDL and blocks its function as an acceptor of cellular cholesterol. Arterioscler Thromb Vasc Biol. 2002;22:2086-91
244. Leskinen MJ, Kovanen PT, Lindstedt KA. Regulation of smooth muscle cell growth, function and death in vitro by activated mast cells-a potential mechanism for the weakening and rupture of atherosclerotic plaques. Biochem Pharmacol. 2003;66:1493-8
245. Mayranpaa MI, Heikkila HM, Lindstedt KA, Walls AF, Kovanen PT. Desquamation of human coronary artery endothelium by human mast cell proteases: implications for plaque erosion. Coron Artery Dis. 2006;17:611-21
246. Kovanen PT. Mast cells and degradation of pericellular and extracellular matrices: potential contributions to erosion, rupture and intraplaque haemorrhage of atherosclerotic plaques. Biochem Soc Trans. 2007;35:857-61
247. Heikkila HM, Latti S, Leskinen MJ, Hakala JK, Kovanen PT, Lindstedt KA. Activated mast cells induce endothelial cell apoptosis by a combined action of chymase and tumor necrosis factor-alpha. Arterioscler Thromb Vasc Biol. 2008;28:309-14
248. Kovanen PT. Mast cells in atherogenesis: actions and reactions. Curr Atheroscler Rep. 2009;11:214-9
249. Heikkila HM, Trosien J, Metso J, Jauhiainen M, Pentikainen MO, Kovanen PT. et al. Mast cells promote atherosclerosis by inducing both an atherogenic lipid profile and vascular inflammation. J Cell Biochem. 2010;109:615-23
250. Arac A, Grimbaldeston MA, Nepomuceno AR, Olayiwola O, Pereira MP, Nishiyama Y. et al. Evidence that meningeal mast cells can worsen stroke pathology in mice. Am J Pathol. 2014;184:2493-504
251. Valent P, Hadzijusufovic E, Schernthaner GH, Wolf D, Rea D, le Coutre P. Vascular safety issues in CML patients treated with BCR/ABL1 kinase inhibitors. Blood. 2015;125:901-6
252. Kagey-Sobotka A, MacGlashan DW, Lichtenstein LM. Role of receptor aggregation in triggering IgE-mediated reactions. Fed Proc. 1982;41:12-6
253. MacGlashan D Jr, Lavens-Phillips S, Katsushi M. IgE-mediated desensitization in human basophils and mast cells. Front Biosci. 1998;3:d746-56
254. Uermoesi C, Zabel F, Manolova V, Bauer M, Beerli RR, Senti G. et al. IgG-mediated down-regulation of IgE bound to mast cells: a potential novel mechanism of allergen-specific desensitization. Allergy. 2014;69:338-47
255. Saleh R, Wedeh G, Herrmann H, Bibi S, Cerny-Reiterer S, Sadovnik I. et al. A new human mast cell line expressing a functional IgE receptor converts to tumorigenic growth by KIT D816V transfection. Blood. 2014;124:111-20
256. Lyons JJ, Yu X, Hughes JD, Le QT, Jamil A, Bai Y. et al. Elevated basal serum tryptase identifies a multisystem disorder associated with increased TPSAB1 copy number. Nat Genet. 2016;48:1564-9
257. Lyons JJ. Hereditary Alpha Tryptasemia: Genotyping and Associated Clinical Features. Immunol Allergy Clin North Am. 2018;38:483-95
258. Woolley DE. The mast cell in inflammatory arthritis. N Engl J Med. 2003;348:1709-11
259. Nigrovic PA, Binstadt BA, Monach PA, Johnsen A, Gurish M, Iwakura Y. et al. Mast cells contribute to initiation of autoantibody-mediated arthritis via IL-1. Proc Natl Acad Sci U S A. 2007;104:2325-30
260. Eklund KK. Mast cells in the pathogenesis of rheumatic diseases and as potential targets for anti-rheumatic therapy. Immunol Rev. 2007;217:38-52
261. Sandler C, Lindstedt KA, Joutsiniemi S, Lappalainen J, Juutilainen T, Kolah J. et al. Selective activation of mast cells in rheumatoid synovial tissue results in production of TNF-alpha, IL-1beta and IL-1Ra. Inflamm Res. 2007;56:230-9
262. Shin K, Nigrovic PA, Crish J, Boilard E, McNeil HP, Larabee KS. et al. Mast cells contribute to autoimmune inflammatory arthritis via their tryptase/heparin complexes. J Immunol. 2009;182:647-56
263. Sawamukai N, Yukawa S, Saito K, Nakayamada S, Kambayashi T, Tanaka Y. Mast cell-derived tryptase inhibits apoptosis of human rheumatoid synovial fibroblasts via rho-mediated signaling. Arthritis Rheum. 2010;62:952-9
264. Lee H, Kashiwakura J, Matsuda A, Watanabe Y, Sakamoto-Sasaki T, Matsumoto K. et al. Activation of human synovial mast cells from rheumatoid arthritis or osteoarthritis patients in response to aggregated IgG through Fcgamma receptor I and Fcgamma receptor II. Arthritis Rheum. 2013;65:109-19
265. Atzeni F, Doria A, Carrabba M, Turiel M, Sarzi-Puttini P. Potential target of infliximab in autoimmune and inflammatory diseases. Autoimmun Rev. 2007;6:529-36
266. Wang Q, Lepus CM, Raghu H, Reber LL, Tsai MM, Wong HH. et al. IgE-mediated mast cell activation promotes inflammation and cartilage destruction in osteoarthritis. Elife. 2019;8:e39905
267. Jensen-Jarolim E, Achatz G, Turner MC, Karagiannis S, Legrand F, Capron M. et al. AllergoOncology: the role of IgE-mediated allergy in cancer. Allergy. 2008;63:1255-66
268. Ribatti D, Crivellato E. The controversial role of mast cells in tumor growth. Int Rev Cell Mol Biol. 2009;275:89-131
269. Rabenhorst A, Schlaak M, Heukamp LC, Forster A, Theurich S, von Bergwelt-Baildon M. et al. Mast cells play a protumorigenic role in primary cutaneous lymphoma. Blood. 2012;120:2042-54
270. Marichal T, Tsai M, Galli SJ. Mast cells: potential positive and negative roles in tumor biology. Cancer Immunol Res. 2013;1:269-79
271. Choi H, Na KJ. Integrative analysis of imaging and transcriptomic data of the immune landscape associated with tumor metabolism in lung adenocarcinoma: Clinical and prognostic implications. Theranostics. 2018;8:1956-65
272. Tharp MD, Kasper C, Thiele D, Charley MR, Kennerly DA, Sullivan TJ. Studies of connective tissue mast cell-mediated cytotoxicity. J Invest Dermatol. 1989;93:423-8
273. Gallagher SJ, Marshall JS, Hoskin DW. Human mast cells induce caspase-independent DNA fragmentation in leukemic T cells. Oncol Rep. 2003;10:1019-23
274. Caplan RM. The natural course of urticaria pigmentosa. Analysis and follow-up of 112 cases. Arch Dermatol. 1963;87:146-57
275. Ellis JM. Urticaria pigmentosa; a report of a case with autopsy. Arch Pathol (Chic). 1949;48:426-35
276. Lennert K, Parwaresch MR. Mast cells and mast cell neoplasia: a review. Histopathology. 1979;3:349-65
277. Travis WD, Li CY, Bergstralh EJ, Yam LT, Swee RG. Systemic mast cell disease. Analysis of 58 cases and literature review. Medicine (Baltimore). 1988;67:345-68
278. Metcalfe DD. Classification and diagnosis of mastocytosis: current status. J Invest Dermatol. 1991;96:2S-4S
279. Nagata H, Worobec AS, Oh CK, Chowdhury BA, Tannenbaum S, Suzuki Y. et al. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc Natl Acad Sci U S A. 1995;92:10560-4
280. Longley BJ, Tyrrell L, Lu SZ, Ma YS, Langley K, Ding TG. et al. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet. 1996;12:312-4
281. Fritsche-Polanz R, Jordan JH, Feix A, Sperr WR, Sunder-Plassmann G, Valent P. et al. Mutation analysis of C-KIT in patients with myelodysplastic syndromes without mastocytosis and cases of systemic mastocytosis. Br J Haematol. 2001;113:357-64
282. Butterfield JH, Weiler D, Dewald G, Gleich GJ. Establishment of an immature mast cell line from a patient with mast cell leukemia. Leuk Res. 1988;12:345-55
283. Furitsu T, Tsujimura T, Tono T, Ikeda H, Kitayama H, Koshimizu U. et al. Identification of mutations in the coding sequence of the proto-oncogene c-kit in a human mast cell leukemia cell line causing ligand-independent activation of c-kit product. J Clin Invest. 1993;92:1736-44
284. Bodemer C, Hermine O, Palmerini F, Yang Y, Grandpeix-Guyodo C, Leventhal PS. et al. Pediatric mastocytosis is a clonal disease associated with D816V and other activating c-KIT mutations. J Invest Dermatol. 2010;130:804-15
285. Arock M, Sotlar K, Akin C, Broesby-Olsen S, Hoermann G, Escribano L. et al. KIT mutation analysis in mast cell neoplasms: recommendations of the European Competence Network on Mastocytosis. Leukemia. 2015;29:1223-32
286. Hartmann K, Escribano L, Grattan C, Brockow K, Carter MC, Alvarez-Twose I. et al. Cutaneous manifestations in patients with mastocytosis: Consensus report of the European Competence Network on Mastocytosis; the American Academy of Allergy, Asthma & Immunology; and the European Academy of Allergology and Clinical Immunology. J Allergy Clin Immunol. 2016;137:35-45
287. Mayerhofer M, Gleixner KV, Hoelbl A, Florian S, Hoermann G, Aichberger KJ. et al. Unique effects of KIT D816V in BaF3 cells: induction of cluster formation, histamine synthesis, and early mast cell differentiation antigens. J Immunol. 2008;180:5466-76
288. Akin C, Kirshenbaum AS, Semere T, Worobec AS, Scott LM, Metcalfe DD. Analysis of the surface expression of c-kit and occurrence of the c-kit Asp816Val activating mutation in T cells, B cells, and myelomonocytic cells in patients with mastocytosis. Exp Hematol. 2000;28:140-7
289. Yavuz AS, Lipsky PE, Yavuz S, Metcalfe DD, Akin C. Evidence for the involvement of a hematopoietic progenitor cell in systemic mastocytosis from single-cell analysis of mutations in the c-kit gene. Blood. 2002;100:661-5
290. Munoz-Gonzalez JI, Alvarez-Twose I, Jara-Acevedo M, Henriques A, Vinas E, Prieto C. et al. Frequency and prognostic impact of KIT and other genetic variants in indolent systemic mastocytosis. Blood. 2019;134:456-68
291. Tefferi A, Levine RL, Lim KH, Abdel-Wahab O, Lasho TL, Patel J. et al. Frequent TET2 mutations in systemic mastocytosis: clinical, KITD816V and FIP1L1-PDGFRA correlates. Leukemia. 2009;23:900-4
292. Wilson TM, Maric I, Simakova O, Bai Y, Chan EC, Olivares N. et al. Clonal analysis of NRAS activating mutations in KIT-D816V systemic mastocytosis. Haematologica. 2011;96:459-63
293. Schwaab J, Schnittger S, Sotlar K, Walz C, Fabarius A, Pfirrmann M. et al. Comprehensive mutational profiling in advanced systemic mastocytosis. Blood. 2013;122:2460-6
294. Damaj G, Joris M, Chandesris O, Hanssens K, Soucie E, Canioni D. et al. ASXL1 but not TET2 mutations adversely impact overall survival of patients suffering systemic mastocytosis with associated clonal hematologic non-mast-cell diseases. PLoS One. 2014;9:e85362
295. Jawhar M, Schwaab J, Schnittger S, Meggendorfer M, Pfirrmann M, Sotlar K. et al. Additional mutations in SRSF2, ASXL1 and/or RUNX1 identify a high-risk group of patients with KIT D816V(+) advanced systemic mastocytosis. Leukemia. 2016;30:136-43
296. Kluin-Nelemans HC, Jansen JH, Breukelman H, Wolthers BG, Kluin PM, Kroon HM. et al. Response to interferon alfa-2b in a patient with systemic mastocytosis. N Engl J Med. 1992;326:619-23
297. Kluin-Nelemans HC, Oldhoff JM, Van Doormaal JJ, Van 't Wout JW, Verhoef G, Gerrits WB. et al. Cladribine therapy for systemic mastocytosis. Blood. 2003;102:4270-6
298. Cahill KN, Katz HR, Cui J, Lai J, Kazani S, Crosby-Thompson A. et al. KIT Inhibition by Imatinib in Patients with Severe Refractory Asthma. N Engl J Med. 2017;376:1911-20
299. Galli SJ. Mast Cells and KIT as Potential Therapeutic Targets in Severe Asthma. N Engl J Med. 2017;376:1983-4
300. Gleixner KV, Mayerhofer M, Aichberger KJ, Derdak S, Sonneck K, Boehm A. et al. PKC412 inhibits in vitro growth of neoplastic human mast cells expressing the D816V-mutated variant of KIT: comparison with AMN107, imatinib, and cladribine (2CdA) and evaluation of cooperative drug effects. Blood. 2006;107:752-9
301. Ustun C, DeRemer DL, Akin C. Tyrosine kinase inhibitors in the treatment of systemic mastocytosis. Leuk Res. 2011;35:1143-52
302. Krauth MT, Mirkina I, Herrmann H, Baumgartner C, Kneidinger M, Valent P. Midostaurin (PKC412) inhibits immunoglobulin E-dependent activation and mediator release in human blood basophils and mast cells. Clin Exp Allergy. 2009;39:1711-20
303. Gotlib J, Berube C, Growney JD, Chen CC, George TI, Williams C. et al. Activity of the tyrosine kinase inhibitor PKC412 in a patient with mast cell leukemia with the D816V KIT mutation. Blood. 2005;106:2865-70
304. Gotlib J, Kluin-Nelemans HC, George TI, Akin C, Sotlar K, Hermine O. et al. Efficacy and Safety of Midostaurin in Advanced Systemic Mastocytosis. N Engl J Med. 2016;374:2530-41
305. Kasamon YL, Ko CW, Subramaniam S, Ma L, Yang Y, Nie L. et al. FDA Approval Summary: Midostaurin for the Treatment of Advanced Systemic Mastocytosis. Oncologist. 2018;23:1511-9
306. Lubke J, Naumann N, Kluger S, Schwaab J, Metzgeroth G, Evans E. et al. Inhibitory effects of midostaurin and avapritinib on myeloid progenitors derived from patients with KIT D816V positive advanced systemic mastocytosis. Leukemia. 2019;33:1195-205
307. Eklund KK, Joensuu H. Treatment of rheumatoid arthritis with imatinib mesylate: clinical improvement in three refractory cases. Ann Med. 2003;35:362-7
308. Smiljkovic D, Blatt K, Stefanzl G, Dorofeeva Y, Skrabs C, Focke-Tejkl M. et al. BTK inhibition is a potent approach to block IgE-mediated histamine release in human basophils. Allergy. 2017;72:1666-76
309. Dispenza MC, Pongracic JA, Singh AM, Bochner BS. Short-term ibrutinib therapy suppresses skin test responses and eliminates IgE-mediated basophil activation in adults with peanut or tree nut allergy. J Allergy Clin Immunol. 2018;141:1914-6 e7
310. Jendoubi F, Gaudenzio N, Gallini A, Negretto M, Paul C, Bulai Livideanu C. Omalizumab in the treatment of adult patients with mastocytosis: A systematic review. Clin Exp Allergy. 2020;50:654-61
311. Lemal R, Fouquet G, Terriou L, Vaes M, Livideanu CB, Frenzel L. et al. Omalizumab Therapy for Mast Cell-Mediator Symptoms in Patients with ISM, CM, MMAS, and MCAS. J Allergy Clin Immunol Pract. 2019;7:2387-95 e3
312. Maurer M, Gimenez-Arnau AM, Sussman G, Metz M, Baker DR, Bauer A. et al. Ligelizumab for Chronic Spontaneous Urticaria. N Engl J Med. 2019;381:1321-32
313. Gasser P, Tarchevskaya SS, Guntern P, Brigger D, Ruppli R, Zbaren N. et al. The mechanistic and functional profile of the therapeutic anti-IgE antibody ligelizumab differs from omalizumab. Nat Commun. 2020;11:165
314. Maun HR, Jackman JK, Choy DF, Loyet KM, Staton TL, Jia G. et al. An Allosteric Anti-tryptase Antibody for the Treatment of Mast Cell-Mediated Severe Asthma. Cell. 2019;179:417-31 e19
315. Wang Y, Nakahashi-Oda C, Okayama Y, Shibuya A. Autonomous regulation of IgE-mediated mast cell degranulation and immediate hypersensitivity reaction by an inhibitory receptor CD300a. J Allergy Clin Immunol. 2019;144:323-7 e7
316. Landolina N, Zaffran I, Smiljkovic D, Serrano-Candelas E, Schmiedel D, Friedman S. et al. Activation of Siglec-7 results in inhibition of in vitro and in vivo growth of human mast cell leukemia cells. Pharmacol Res. 2020;158:104682
317. Youngblood BA, Brock EC, Leung J, Falahati R, Bochner BS, Rasmussen HS. et al. Siglec-8 antibody reduces eosinophils and mast cells in a transgenic mouse model of eosinophilic gastroenteritis. JCI Insight. 2019;4:e126219
318. Bochner BS. "Siglec"ting the allergic response for therapeutic targeting. Glycobiology. 2016;26:546-52
Author contact
![]() Corresponding author: Peter Valent, M.D. Department of Internal Medicine I, Division of Hematology and Hemostaseology and Ludwig Boltzmann Institute for Hematology and Oncology, Medical University of Vienna, Vienna, Austria. Phone: 43 1 40400 60850; Fax: 43 1 40400 40300; E-mail: peter.valentac.at.
Corresponding author: Peter Valent, M.D. Department of Internal Medicine I, Division of Hematology and Hemostaseology and Ludwig Boltzmann Institute for Hematology and Oncology, Medical University of Vienna, Vienna, Austria. Phone: 43 1 40400 60850; Fax: 43 1 40400 40300; E-mail: peter.valentac.at.