Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(24):10957-10972. doi:10.7150/thno.49629 This issue Cite
Research Paper
Restoration of RNA helicase DDX5 suppresses hepatitis B virus (HBV) biosynthesis and Wnt signaling in HBV-related hepatocellular carcinoma
Saravana Kumar Kailasam Mani1,4*, Bingyu Yan2,4*, Zhibin Cui1,4, Jiazeng Sun1,4, Sagar Utturkar4, Adrien Foca5, Nadim Fares6, David Durantel5, Nadia Lanman4, Philippe Merle6, Majid Kazemian2,3,4 ![]() , Ourania Andrisani1,4
, Ourania Andrisani1,4 ![]()
1. Department of Basic Medical Sciences.
2. Department of Biochemistry.
3. Department of Computer Science.
4. Purdue Center for Cancer Research, Purdue University, West Lafayette, IN 47907.
5. Cancer Research Center of Lyon UMR Inserm 1052 - CNRS 5286.
6. Department of Hepatology, Hôpital de la Croix-Rousse, Hospices Civils de Lyon, Université Lyon 1, Lyon, France
*equal contribution
Received 2020-6-17; Accepted 2020-8-11; Published 2020-9-1
Abstract

Rationale: RNA helicase DDX5 is downregulated during hepatitis B virus (HBV) replication, and poor prognosis HBV-related hepatocellular carcinoma (HCC). The aim of this study is to determine the mechanism and significance of DDX5 downregulation for HBV-driven HCC, and identify biologics to prevent DDX5 downregulation.
Methods: Molecular approaches including immunoblotting, qRT-PCR, luciferase transfections, hepatosphere assays, Assay for Transposase-Accessible Chromatin sequencing (ATAC-seq), and RNA-seq were used with cellular models of HBV replication, HBV infection, and HBV-related liver tumors, as well as bioinformatic analyses of liver cancer cells from two independent cohorts.
Results: We demonstrate that HBV infection induces expression of the proto-oncogenic miR17~92 and miR106b~25 clusters which target the downregulation of DDX5. Increased expression of these miRNAs is also detected in HBV-driven HCCs exhibiting reduced DDX5 mRNA. Stable DDX5 knockdown (DDX5KD) in HBV replicating hepatocytes increased viral replication, and resulted in hepatosphere formation, drug resistance, Wnt activation, and pluripotency gene expression. ATAC-seq of DDX5KD compared to DDX5 wild-type (WT) cells identified accessible chromatin regions enriched in regulation of Wnt signaling genes. RNA-seq analysis comparing WT versus DDX5KD cells identified enhanced expression of multiple genes involved in Wnt pathway. Additionally, expression of Disheveled, DVL1, a key regulator of Wnt pathway activation, was significantly higher in liver cancer cells with low DDX5 expression, from two independent cohorts. Importantly, inhibitors (antagomirs) to miR17~92 and miR106b~25 restored DDX5 levels, reduced DVL1 expression, and suppressed both Wnt activation and viral replication.
Conclusion: DDX5 is a negative regulator of Wnt signaling and hepatocyte reprogramming in HCCs. Restoration of DDX5 levels by miR17~92 / miR106b~25 antagomirs in HBV-infected patients can be explored as both antitumor and antiviral strategy.
Keywords: Hepatitis B virus, Hepatocellular Carcinoma, RNA helicase DDX5, miR17~92/miR106b~25 & antagomirs, Wnt/β-catenin signaling.
Introduction
Hepatocellular carcinoma (HCC) is a leading type of primary cancer with increasing incidence globally [1]. Chronic Hepatitis B virus (HBV) infection remains one of the major etiologic factors in HCC pathogenesis [2]. Despite the HBV vaccine, the WHO estimates that globally 250 million people are chronically infected with HBV. Curative treatments for early stage HCC include liver resection, transplantation, or local ablation. However, high recurrence rates after resection compromise patient outcomes. In advanced stage HCC, multi-kinase inhibitors including sorafenib [3], regorafenib [4], cabozantinib [5], lenvatinib or anti-angiogenic monoclonal antibodies such as ramucirumab [6] offer only palliative benefits. Thus, a compelling need exists to determine key molecular drivers of HCC pathogenesis, in order to design effective, targeted therapies that suppress both virus biosynthesis and liver cancer pathogenesis.
A cellular mechanism hijacked by HBV that regulates both host and viral gene transcription [7] involves the chromatin modifying Polycomb Repressive Complex 2 (PRC2) complex [8] and RNA helicase DDX5 [9]. PRC2 represses transcription of genes by trimethylation of histone H3 on lysine 27 (H3K27me3). During HBV infection, viral covalently closed circular DNA (cccDNA) serving as template for viral transcription, assumes chromatin-like structure [10]. Histone modifications associated with the HBV cccDNA/minichromosome determine viral transcription and replication rate [11]. Our previous studies have shown that HBV replicating cells and HBV-related HCCs exhibit reduced PRC2 activity, resulting in de-repression of host PRC2 target genes [12-14]. Moreover, knockdown of the essential PRC2 subunit SUZ12 enhances HBV replication [12, 13], suggesting loss of PRC2-mediated gene repression is advantageous for viral growth. Loss of PRC2 function occurs by proteasomal degradation of SUZ12, dependent on cellular polo-like-kinase1 (PLK1) [15], a host pro-viral factor [16]. RNA helicases regulate a wide range of pathways [17]. For example, DDX5 interacts with SUZ12, contributing to enhanced SUZ12 protein stability [7]. Reduced DDX5 and SUZ12 protein levels correlate with enhanced viral transcription and replication, while in clinical samples, reduced DDX5 expression correlates with hepatocyte de-differentiation, expression of PRC2 target genes including EpCAM, a hepatic Cancer Stem Cell (hCSC) marker [18], and poor patient prognosis [7]. These observations suggest a role for DDX5 both in HBV replication and HBV-induced HCC.
In this study, we investigated how HBV infection mediates DDX5 downregulation, and the consequences of DDX5 downregulation for the infected hepatocyte. We show that HBV replication induces the expression of proto-oncogenic miR-17~92 and its paralog miR106b~25 [19] which directly target the three prime untranslated region (3'-UTR) of DDX5. miRNAs silence gene expression post-transcriptionally, regulating an array of biological processes, and linked to various diseased states including cancer [20]. miR17~92 is induced by the proto-oncogene c-Myc [21], and miR106b~25 is encoded within intron 13 of minichromosome maintenance complex component 7 (MCM7) [22]. Importantly, both miRNAs are upregulated in HBV-induced HCCs [19, 23], and their over-expression is associated with liver fibrosis, cirrhosis, and HCC [19, 24]. Biogenesis of mature miRNAs involves processing of primary miRNA (pri-miRNA) by the microprocessor complex [20]. DDX5 is a critical component of this complex, and importantly, genes involved in the microprocessor complex are haploinsufficient tumor suppressors [25, 26]. Herein, we show DDX5 downregulation imparts cancer stem cell properties to hepatocytes, including hepatosphere formation, resistance to sorafenib and cisplatin, expression of pluripotency genes, and activation of Wnt signaling. Antagomirs (inhibitors) to these miRNAs restore DDX5 levels in HBV replicating cells, suppressing Wnt pathway activation and virus biosynthesis, i.e., acting as both antitumor and antiviral agents.
Methods
Cell lines
Human HCC cell lines HepAD38 [27], HepG2 [7], HepG2-NTCP clone 7 [28], Huh7, and HepaRG cells [29] were grown as described. Cell lines were routinely tested for mycoplasma. HepAD38 cell line and its derivatives were authenticated by short tandem repeat (STR) analysis performed by ATCC.
Cell transfection and infection assays
HepG2 and HepAD38 cells (5x104 cells) were co-transfected with Renilla luciferase (25 ng), Luc-3'UTR-DDX5 (25 ng), and control (Ctrl) vectors or plasmid encoding miR106b~25 or miR17~92, using Lipofectamine 3000 (Life Technologies). In HepAD38 cells [27], HBV replication was induced by tetracycline removal 48 h prior to transfection. Luciferase activity was measured 48 h after transfection using Dual Luciferase Assay system as per manufacturer's protocol (Promega), and normalized to Renilla luciferase. Plasmids used are listed in Supporting Table S1. Infection assays of HepaRG and HepG2-NTCP cell lines were performed as described [28, 29], employing 100 HBV genome equivalents per cell.
Wnt reporter assay
HBV replicating HepAD38 cells (5x104 cells, day 3 of HBV replication) were co-transfected with TOPflash vector (25 ng) containing TCF-binding sites upstream of firefly luciferase, and Renilla luciferase vector (25 ng). Ctrl siRNA (40 nM) or DDX5 siRNA (40 nM) were co-transfected with Renilla and Firefly luciferase vectors using RNAiMax (Life Technologies). Luciferase activity was measured 48 h after transfection using Dual Luciferase Assay system as per manufacturer's protocol (Promega), and normalized to Renilla luciferase. Plasmids used are listed in Table S1.
Sphere assay
HBV replicating HepAD38 cells (1x103) were seeded in ultra-low attachment 6-well plates (Corning). Cisplatin (10 μM) and Sorafenib (2.5 μM) were replaced every 3 days for 2 weeks, using sphere media containing DMEM/F12 (90% v/v), Penicillin/Streptomycin (1% v/v), G418 50 mg/mL (0.8% v/v), Fibroblast Growth factor 100 ng/μL (0.02% v/v), B27 (1X), and Epidermal growth factor 100 ng/μL (0.02% v/v).
Cell viability assay
HBV replicating HepAD38 cells (1x104) seeded in 96-well plates were treated with cisplatin (40 μM), sorafenib (7.5 μM), or DMSO for 24 h (day 5 of HBV replication). Growth inhibition was measured at 490 nm by CellTiter 96 AQueous One Solution Cell Proliferation assay (Promega). 100% viability refers to A490 value of DMSO-treated cells. Background absorbance was measured from wells containing media and MTS without cells.
Immunoblot analysis and Immunofluorescence microscopy
Methods are described in detail in Supplementary Material section. Antibodies employed are listed in Table S2.
RNA extraction and qRT-PCR
Detailed methods are described in Supplementary Material section; primer sequences are listed in Table S3, and reagents, chemical inhibitors and kits in Table S4.
RNA-seq analysis
HepAD38 cells, wild type (WT) and DDX5 knockdown (KD5) grown +/- tetracycline for 10 days to induce HBV replication [27]. Sorafenib (2.5 μM) treatment was for three days prior to harvesting. Three independent biological replicates were prepared for RNA isolation and RNA sequencing. Total RNA submitted to Purdue Genomics Core Facility for quality assessment and next-generation sequencing. Paired-end 2x50 bp sequencing performed using a HiSeq2500 system (Illumina). Data quality control performed using FastQC v0.11.8. The RNA expression level in each library estimated by “rsem-calculate-expression” procedure in RSEM v1.3.112 using default parameters except “--bowtie-n 1 -bowtie-m 100 -seed-length 28 --paired-end”. The bowtie index required by RSEM software generated by “rsem-prepare-reference” on all RefSeq genes, obtained from UCSC table browser on April 2017. EdgeR v3.24.013 package used to normalize gene expression among all libraries and identify differentially expressed genes among samples with following constraints: fold change > 1.5, FDR < 0.05 and TPM > 1. Gene set enrichment analysis (GSEA) performed using GSEA software [30].
ATAC-seq analysis
Two independent biological replicates of WT HepAD38 and DDX5 knockdown (KD5) cells used for ATAC-seq analyses. Protocol and method of ATAC-seq data analysis described in Supplementary Material section.
Data availability
All sequencing data are available through the NCBI Gene Expression Omnibus (GEO) database (accession number GSE131257).
Chromatin immunoprecipitation (ChIP)
ChIP assays were performed using Millipore ChIP Assay Kit (catalog no.: 17-295). Antibodies used listed in Table S2. ChIP primers used are described in [21].
Statistical analysis
One-way ANOVA with Sidak's multiple comparison test with single pooled variance was performed using GraphPad Prism version 5.0, comparing mean of each sample to mean of control (Figure 1B, 1D). Two-way ANOVA with Sidaks's multiple comparison test was performed comparing: (i) mean of each microRNA in infected to uninfected samples (Figure 2A) and (Figure S2A), (ii) mean of each cell line to mean of WT cells (Figure 3C), (Figure 5A), and (Figure 5C), (iii) mean of each siRNA to control siRNA (Figure 5A and B), and (iv) mean of each inhibitor to mean of control inhibitor (Figure 8C and D). Two tailed t-test with Welch's correction was used to determine significance in Figure 2D. Results were considered statistically significant if p < 0.05.
DDX5 is target of miR106b~25 and miR17~92 in HBV replicating cells. (A) Luc-3'UTR-DDX5 containing the WT 3'UTR, indicated deletions Δ1, Δ2, Δ3, and site directed changes of nucleotides 129-135 (mut-Δ3). (B) Transient transfections of Luc-3'UTR-DDX5 co-transfected with Renilla luciferase, and plasmids expressing miR106b~25 or miR-17~92 in HepAD38 cells without (-) and with (+) HBV replication by tetracycline removal for 2 days. (C) Immunoblots of DDX5 and HBc following transfection of plasmids expressing miR106b~25 or miR-17~92, in HepAD38 cells with HBV replication for 2 days. Relative intensity (rel. int.) quantified vs. actin using ImageJ software. (D) Luc-3'UTR-DDX5 containing the WT 3'UTR, indicated deletions Δ1, Δ2, Δ3, and site directed changes of nucleotides 129-135 (mut-Δ3), co-transfected with Renilla luciferase in HepAD38 cells with (+) or without (-) HBV replication for 2 days (n=3). * P < 0.05; Error bars indicate Mean ± SEM. (E) WT and mut-Δ3 Luc-3'UTR-DDX5 co-transfected with Renilla luciferase and 10 nM each of indicated miRNA inhibitors (antogomirs) or 50 nM control inhibitor (Ctrli) in HepAD38 cells with (+) or without (-) HBV replication for 2 days (n=3). * P < 0.05; Error bars indicate Mean ± SEM.
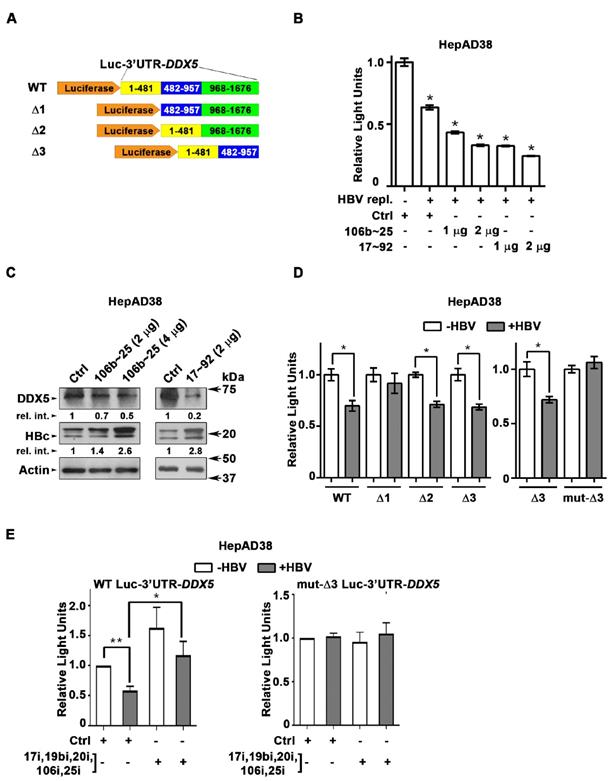
HBV replication induces expression of miR106b~25 and miR17~92. (A) qRT-PCR of indicated miRNAs in HepAD38 cells without (-) and with (+) HBV replication for 5 days (n=3). (B) ChIP assays in HepAD38 cells with (+) or without (-) HBV replication for 5 days, using c-Myc antibody and primer sets 1-3 spanning c-Myc binding sites [21]. IgG was negative control. (n=3). Schematic representation of genomic interval encompassing the miR17~92 cluster. RT-PCR amplicons represented by numbered lines. (C) Immunoblots of c-Myc and MCM7 using lysates from HepAD38 cells with (+) or without (-) HBV replication for 7days. Quantification by ImageJ software of three independent experiments. (D) qRT-PCR of indicated miRNAs, and DDX5 in HBV-related HCC tumor vs. peritumor samples. Tumor samples were grouped according to Mani et al, [14] into Group III vs. Rest (refer also to Figure S2C). * P < 0.05; Error bars indicate Mean ± SEM.
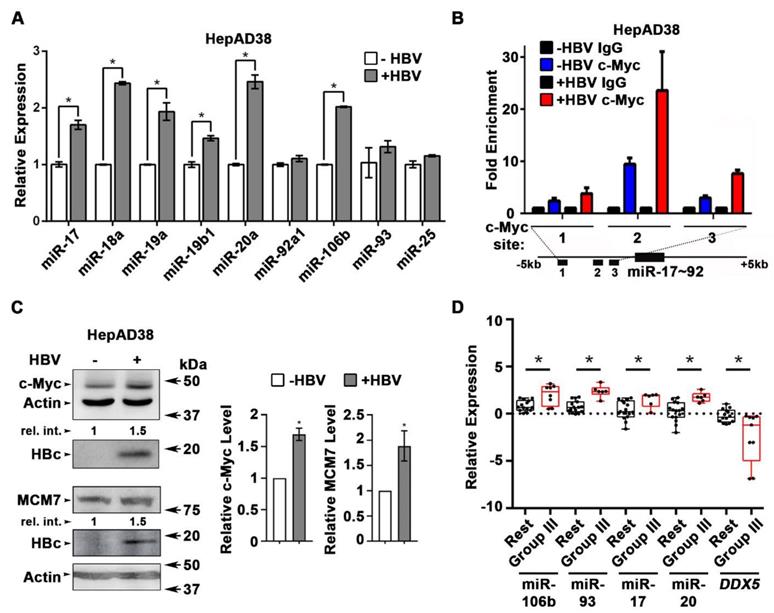
DDX5 knockdown confers stem cell-like properties. (A) Immunoblots of DDX5 and HBc in WT and DDX5 knockdown (KD2, KD3 and KD5) HepAD38 cells, and in WT HepAD38 cells following transient transfection of two different siRNAs for DDX5 or control siRNA (Ctrl), without (-) and with (+) HBV replication for 5 days. Panels shown below the immunoblots are cumulative quantification of three independent biological replicates. (B) HBV replicating WT, KD2, KD3 and KD5 HepAD38 cells grown for 14 days in hepatic sphere media using ultra-low attachment plates, with cisplatin (10 μM) or sorafenib (2.5 μM). Shown is a representative assay of three independent biological replicates. (C) Proliferation (MTS) assays performed with WT, KD2, KD3 and KD5 HepAD38 cells grown with (+) HBV replication for 5 days. Cells were treated with cisplatin (40 μM), sorafenib (7.5 μM) or DMSO for 24 h (n=3). *P < 0.05; Error bars indicate Mean ± SEM.
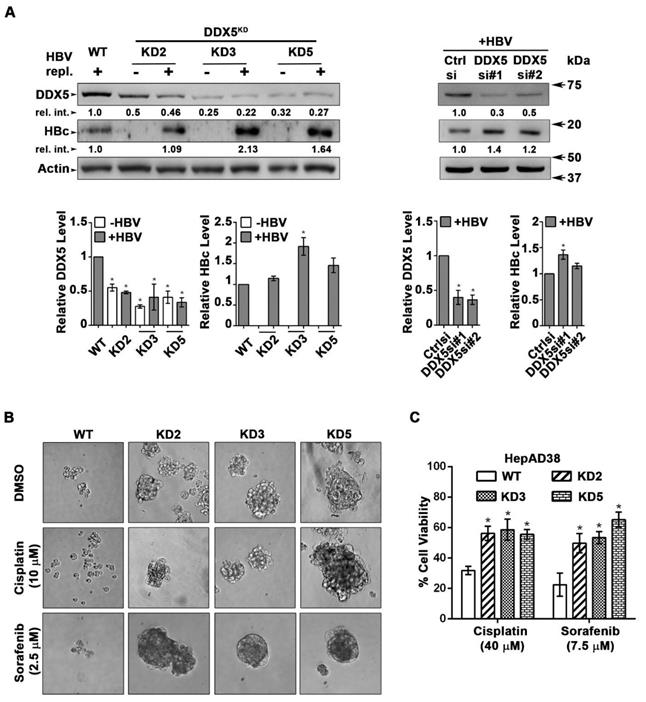
Downregulation of DDX5 activates Wnt signaling. (A-B) Transient co-transfections of TOPFlash and Renilla luciferase reporters in HepAD38 cells (WT, KD2, KD3, KD5 and miRO/E) (A), and Huh7 and HepaRG cells (B), transfected with DDX5si#1, DDX5si#2 or negative control siRNA (Ctrlsi). Luciferase activity from three independent assays, measured on day 5 of HBV replication. (C) qRT-PCR of OCT4 NANOG, SOX2 expression in WT and DDX5KD HepAD38 hepatospheres with (+) HBV replication for 14 days. (D) WT, KD2, KD3 and KD5 HepAD38 cells grown for 14 days in hepatic sphere media using ultra-low attachment plates, in presence of Wnt inhibitors ICG-001 (10 μM) or XAV-939 (10 μM). Shown is a representative assay (left) and quantification of DDX5KD hepatospheres (right) from three independent biological replicates. *P < 0.05; Error bars indicate Mean ± SEM.
Antagomirs restore DDX5 in HBV replicating hepatocytes. (A) Immunoblots of DDX5, LKB1 and PTEN in HepAD38 cells with (+) HBV replication for 5 days. Cells were transfected for 24 h with 50 nM of indicated miRNA inhibitors/antagomirs (106i, 19b1i, 17i, 20ai) or control inhibitor (Ctrli), on day 4 of HBV replication. Cumulative immunoblot quantification of three independent biological replicates for DDX5 shown on right panel, and for LKB1 and PTEN in Figure S6B. *P < 0.05; Error bars indicate Mean ± SEM. (B) Immunofluorescence confocal microscopy of HBc, DDX5 and β-catenin in HepAD38 cells without (-) or with (+) HBV replication for 5 days. Fluorescent antagomir for miR-19b1 (miR-19b1i-FAM, 50 nM) transfected on day 4 of HBV replication. White arrows indicate membrane localization of β-catenin. A representative assay from three independent biological replicates. Right panel, quantification of fluorescence intensities from 400 cells (mean gray value per μm2), employing ImageJ software. *P < 0.05; Error bars indicate Mean ± SEM. (C - D) HepG2-NTCP cells infected with 100 HBV genome equivalents/cell for 5 and 7 days. Infected cells were transfected with antagomirs of indicated miRNAs or Ctrli (50 nM each) 24 h prior to cell harvesting. (C) qRT-PCR of HBc RNA, HBV pgRNA, total HBV RNA and (D) qRT-PCR of DVL1 mRNA. Expression values calculated relative to Ctrli at 5 and 7 days p.i. using ΔΔCt method (n=3). *P < 0.05; Error bars indicate Mean ± SEM. (E) Immunofluorescence confocal microscopy of HBc in HepG2-NTCP cells at 7 day p.i., infected with 100 HBV genome equivalents per cell. Antagomir miR-19b1i-FAM (50 nM) transfected on day 6 p.i. A representative assay from three independent biological replicates.
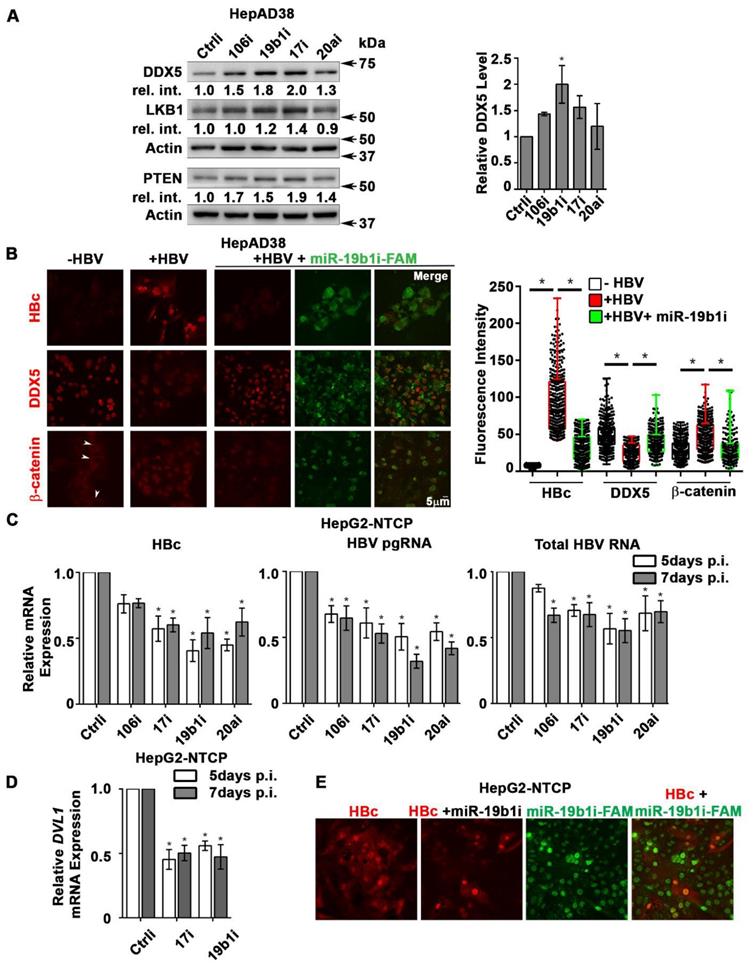
Results
DDX5 downregulation in HBV replicating cells by miR106b~25 and miR17~92
DDX5 is downregulated during HBV replication, and reduced levels of DDX5 in HBV-related HCCs tended towards poor patient prognosis [7]. Employing the target prediction algorithm TargetScan, a highly conserved seed sequence for miR17~92 and its paralog miR106b~25 [19, 31] was identified in 3' UTR of DDX5, located at nucleotides 129-135 (Figure S1A). To test whether miR106b~25 and miR17~92 downregulate DDX5, expression vectors encoding each of these miRNAs were co-transfected with expression vectors encoding the Firefly luciferase gene fused to 3'UTR of DDX5 (Luc-3'UTR-DDX5), and a Renilla luciferase vector for normalization of luciferase activity (Figure 1A). Overexpression of either miR106b~25 or miR17~92 in HepG2 cells reduced Firefly luciferase activity, indicating a functional seed sequence in 3'UTR of DDX5 (Figure S1B). Furthermore, transfection of Luc-3'UTR-DDX5 vector in HepAD38 cells exhibited reduced luciferase activity upon induction of HBV replication, consistent with upregulation of both miRNAs in HBV-induced HCCs [19, 23] and their role in targeting DDX5. Overexpression of miR106b~25 or miR17~92 clusters further reduced luciferase activity (Figure 1B). Importantly, overexpression of these miRNAs in HBV replicating HepAD38 cells [27] reduced protein level of endogenous DDX5, while levels of viral core antigen (HBc) were increased (Figure 1C), suggesting loss of DDX5 is advantageous to viral biosynthesis. Deletion analyses and site directed mutagenesis of 3'UTR of DDX5 confirmed the presence of miRNA seed sequence at nucleotides 129-135 (mut-Δ3), analyzed in the context of HBV replication (Figure 1D). Inhibitors (antagomirs) for miR106b~25 and miR17~92 transfected in HBV replicating cells reversed the reduction in luciferase activity from the WT Luc-3'UTR-DDX5, without an effect on the mut-Δ3 construct lacking the conserved seed sequence (Figure 1E). Together, these results indicate that HBV replication downregulates DDX5 mRNA via induction of miR106b~25 and miR17~92.
HBV replication induces expression of miR106b~25 and miR17~92
Next, we quantified the expression of individual members of miR17~92 and miR106b~25 clusters in HBV replicating HepAD38 cells (Figure 2A), and HBV infected HepaRG cells (Figure S2A). Five out of six members of miR17~92 cluster and one out of three members of miR106b~25 cluster were significantly induced in HBV replicating HepAD38 cells (Figure 2A), while two members of miR17~92 were induced more than two-fold in HBV infected HepaRG cells (Figure S2A). To understand how HBV infection increased miR17~92 expression, we focused on the miR17~92 promoter region which contains three binding sites for c-Myc, often overexpressed in liver cancer and a key regulator of miR17~92 [21]. Two out of these three sites (i.e. sites #2 and #3) exhibited further increased c-Myc occupancy in HBV replicating HepAD38 cells (Figure 2B), consistent with induction of miR17~92 members (Figure 2A), and the increased c-Myc expression detected by immunoblots (Figure 2C). miR106b~25 is encoded by intron13 of the MCM7 gene and when MCM7 is overexpressed, it also increases expression of miR106b~25 [22]. To determine whether MCM7 is overexpressed, we quantified MCM7 protein levels in lysates from HBV replicating HepAD38 cells (Figure 2C). Approximately 2-fold increased MCM7 protein level was observed, in agreement with the well-established activation of cellular mitogenic pathways by the viral HBx protein [32, 33], supporting the increased expression of miR106b~25 in HBV replicating cells (Figure 2A). In addition to DDX5 (Figure 1), the increased expression of these miRNAs also downregulates other known cellular targets, including PTEN [22] and LKB1 [34]. Indeed, HBV replicating cells display reduced PTEN and LKB1 levels, which regulate AKT phosphorylation (Figure S2B), and AMPK activity [35], respectively. Importantly both of these pathways are known to exert a positive effect on HBV replication [36-38]. Overall, these data indicate that HBV replication induces expression of miR106b~25 and miR17~92.
To determine whether miRNA-mediated downregulation of DDX5 is clinically relevant, the level of these miRNAs was quantified in HBV-related HCCs. Our previous study identified a hepatic cancer stem cell (hCSC)-like gene signature to be associated with tumor recurrence after surgery in HBV-related HCCs [14]. In that study (14), we denoted liver tumors expressing the hCSC-like gene signature as “Group III”, while tumors that did not express the hCSC-like signature [14] were referred to as the “Rest” (Figure S2C). Herein, we quantified miRNA expression in the same HBV-related HCCs. Significantly, “Group III” HBV-related HCCs, in comparison to the “Rest”, exhibited increased expression of miR106b~25 and miR17~92 and reduced levels of DDX5 mRNA (Figure 2D). Considering that miRNAs act by reducing either mRNA stability or translation of their targets, these clinical data strongly suggest an inverse correlation between high expression of miR106b~25 and miR17~92 vs. reduced DDX5 levels (Figure S2D), and, in turn, tumor aggressiveness.
DDX5 knockdown induces viral biosynthesis and confers cancer stem cell-like properties to hepatocytes
We next studied the functional significance of DDX5 loss to viral biosynthesis, and hepatosphere formation. We derived three stable DDX5 knockdown cell lines (DDX5KD) from HepAD38 cells, referred to as KD2, KD3 and KD5 (Figure 3A) using three different shRNAs. To assess whether DDX5 downregulation affected viral replication, we quantified HBc levels by immunoblots. HBc levels were increased after DDX5 knockdown, both in the stable cell lines (Figure 3A) and after transient transfection of DDX5 siRNAs #1 and #2 (Figure 3A). These results also agree with the increased HBc levels observed upon overexpression of miR106b~25 and miR17~92 in HBV replicating HepAD38 cells (Figure 1C). Collectively, these data suggest that DDX5 acts as a host restriction factor for HBV biosynthesis.
Since DDX5 was shown to act as a roadblock to pluripotency [39], we examined whether loss of DDX5 promotes a stem cell-like phenotype in HBV replicating hepatocytes. WT HepAD38 cells failed to form hepatospheres in ultra-low attachment plates. By contrast, DDX5KD cells (KD2, KD3, and KD5) formed robust hepatospheres that survived treatment with chemotherapeutic drugs cisplatin (10 μM) and sorafenib (2.5 μM) (Figure 3B). Furthermore, proliferation assays demonstrated reduced sensitivity to cisplatin and sorafenib upon DDX5 downregulation (Figure 3C and Figure S3), a characteristic feature of cancer stem cells (CSCs).
To determine pathways contributing to the observed stemness characteristics, we performed the Assay for Transposase-Accessible Chromatin sequencing (ATAC-seq) and compared the chromatin accessibility of WT vs. DDX5KD cells (Figure 4A and Table S5). Interestingly, 376 loci became more accessible in DDX5KD cells, while 186 loci became less accessible. Next, we performed pathway analysis of the genes neighboring these accessible regions. We found that regulation of Wnt signaling pathway was the fifth-most enriched pathway among all biological pathways assessed (Figure 4B). Examples of Wnt pathway genes exhibiting changes in chromatin accessibility are shown in Figure 4C, suggesting DDX5 may regulate Wnt signaling by affecting chromatin accessibility of genes involved in Wnt signaling.
DDX5 downregulation alters chromatin accessibility near genes associated with Wnt pathway. (A) Heatmap showing signal intensity of each differential ATAC peak (n=2 biological replicates) and clustering of peaks into two groups, more accessible group (top) and less accessible group (bottom) in DDX5KD cells. (B) Pathway analysis of genes neighboring the top group with representative examples of open chromatin regions (OCRs) (C) Highlighted with arrow are OCRs corresponding to ATAC peaks in heatmap.
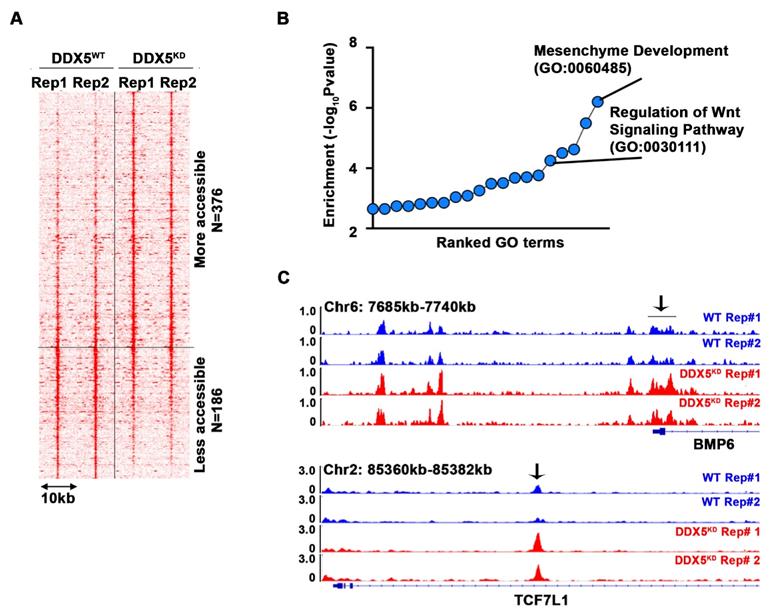
DDX5 downregulation activates Wnt signaling in liver cancer cell lines
Based on the ATAC-seq results (Figure 4), we examined activation of Wnt/β-catenin signaling, one of the key pathways regulating stemness [40, 41]. We performed luciferase assays to assess whether Wnt signaling was upregulated upon reduction of DDX5. Wnt-responsive TOPFlash reporter, containing LEF/TCF binding sites upstream of Firefly luciferase, was co-transfected with control Renilla luciferase into WT HepAD38 cells, HepAD38 cells overexpressing miR106b~25 and miR17~92 (miRO/E, Figure S4A-B), and stably or transiently DDX5 knockdown cells. Increased luciferase activity, i.e. increased Wnt/β-catenin pathway activation, was observed upon DDX5 downregulation by stable overexpression of miR106b~25 and miR17~92 (miRO/E), stable (KD2, KD3, and KD5) and transient (siRNAs #1 and #2) knockdown of DDX5 (Figure 5A). Furthermore, transient (siRNAs #1 and #2) knockdown of DDX5 in Huh7 and HepaRG cells also increased luciferase expression from the TOPFlash reporter, supporting this mechanism is functional in other liver cancer cell lines (Figure 5B). Interestingly, hepatospheres of HBV replicating DDX5 knockdown cells exhibited higher expression of pluripotency genes, determined by qRT-PCR (Figure 5C) and immunoblotting of NANOG, SOX2, OCT4 and hCSC marker CD44 (Figure S4C). Wnt inhibitors, ICG-001 [42] and XAV-939 [43], targeting different steps of Wnt signaling, suppressed hepatosphere formation, thereby linking Wnt pathway activation to the hCSC phenotype (Figure 5D, left panel). Quantification of hepatosphere formation shows statistically increased hepatosphere formation in DDX5KD cells (Figure 5D, right panel). Taken together, these data indicate that DDX5 maintains hepatocyte differentiation, and DDX5 downregulation promotes hepatocyte deprogramming via activation of Wnt signaling.
Transcriptomic analyses define dysregulated Wnt signaling in DDX5 knockdown cells
To quantify the global effects of DDX5 knockdown on global gene expression in hepatocytes, we compared the transcriptome of WT vs. DDX5KD HepAD38 cells as a function of HBV replication, and sorafenib treatment using RNA-seq. Three comparisons were performed, namely, DDX5KD vs. WT cells in the absence of HBV replication (-HBV), DDX5KD vs. WT cells in the presence of HBV replication (+HBV), and DDX5KD HBV replicating cells with or without sorafenib treatment. Nearly 1000 genes were differentially expressed between DDX5KD vs. WT cells (Fold change >1.5; FDR<0.05; Figure 6A-B and Table S6). Interestingly, sorafenib treatment exerted a highly pronounced effect on the number of both upregulated and downregulated genes (Figure 6B). Importantly, DDX5 downregulation, irrespective of HBV replication, increased EpCAM expression (Figure 6C) among other genes, which is a known hepatic cancer stem cell marker [18] and a Wnt/β-catenin signaling target gene [44]. To further investigate the effect of DDX5 downregulation on Wnt/β-catenin signaling pathway, we asked whether expression of Wnt receptor genes differed between WT vs. DDX5KD cells, using Gene Set Enrichment Analysis (GSEA) [30]. We found that DDX5KD cells have higher expression of several genes involved in Wnt receptor signaling, irrespective of HBV replication, consistent with activation of the Wnt pathway in DDX5KD cells (Figure 5A and B). For example, Wnt receptors (frizzled/FZD) were among the upregulated genes in DDX5KD cells (Figure 6D), in agreement with earlier studies linking Wnt receptor (FZD7) overexpression to Wnt pathway activation in HCCs [45].
Transcriptomic analyses define dysregulated Wnt signaling in DDX5 knockdown cells. (A) Heat map of differentially expressed genes between DDX5KD vs. WT HBV replicating cells for 10 days. RNA-seq samples are from three independent biological replicates. (B) Differentially expressed genes in three indicated comparisons: DDX5KD vs. WT cells in the absence of HBV replication (-HBV), DDX5KD vs. WT cells in the presence of HBV replication (+HBV), and DDX5KD HBV replicating cells with or without sorafenib treatment. (C) Scatter plot showing mean gene expression values (n=3) in three, above mentioned comparisons. Differentially expressed genes are highlighted in orange (FDR<0.05) and grey (FDR>0.05) (D) GSEA for Wnt-activated receptor activity (GO: 0042813), comparing DDX5KD vs. WT cells in the absence of HBV replication (left panel), DDX5KD vs. WT cells in the presence (+) of HBV replication (middle panel), and DDX5KD HBV replicating cells with or without sorafenib treatment (right panel). (E) qRT-PCR validation of indicated genes using RNA isolated from HepAD38 cells, comparing DDX5KD vs. WT cells in the absence (-) or presence (+) of HBV replication for 5 days, and with sorafenib (2.5 µM) treatment for 3 days. Expression values calculated for WT -HBV vs. DDX5 -HBV; WT +HBV vs. DDX5 +HBV and DDX5+HBV vs. DDX5+HBV+Sorafenib, using ΔΔCt method. (n=3). (F) Immunoblot of indicated proteins in WT and DDX5KD HepAD38 cells, with HBV replication for 5 days. A representative assay from three biologic replicates. Quantification is shown in Fig. S5.
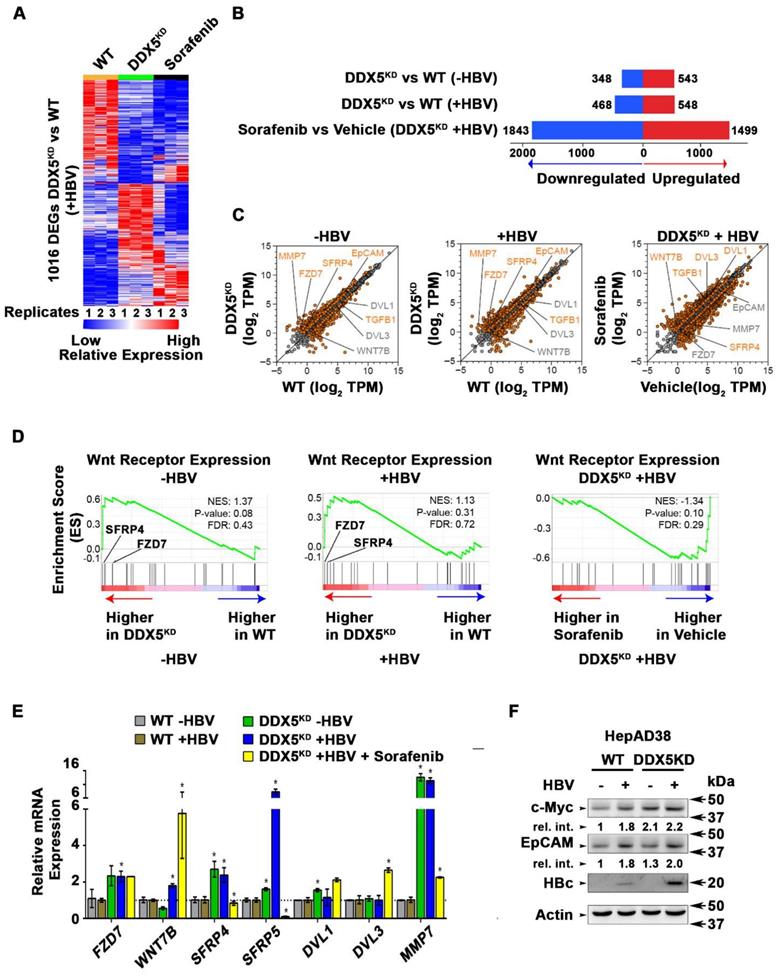
The expression of select genes involved in Wnt/β-catenin signaling was validated by qRT-PCR (Figure 6E). Specifically, in DDX5KD cells expression of FZD7, SFRP4, SFRP5, DVL1 and MMP7 increased irrespective of HBV replication (Figure 6E). Sorafenib treatment of HBV replicating DDX5KD cells increased expression of WNT7B ligand, while significantly suppressing expression of SFRP4 and SFRP5 (Figure 6E), the negative regulators of Wnt signaling [46]. These observations, together with the results of Figure 5 (A and B) demonstrate that downregulation of DDX5 is a key event leading to activation of Wnt signaling. To further confirm these findings, we examined by immunoblots protein levels of known Wnt regulated genes, including EpCAM and c-Myc (Figure 6F and Figure S5). We observe increased protein levels of both c-Myc and EpCAM in HBV replicating DDX5KD cells.
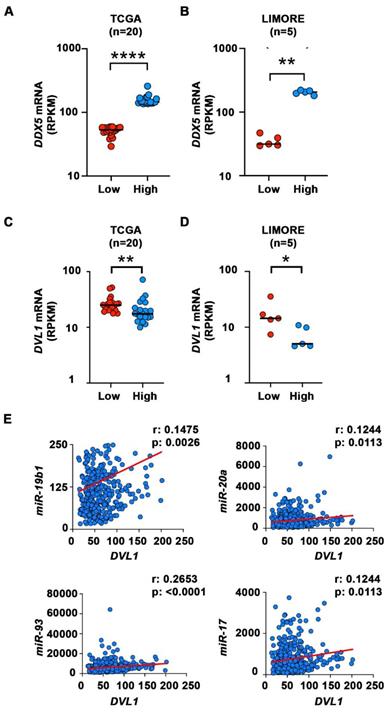
Elevated mRNA expression of DVL1 in liver cancer cells with low DDX5 mRNA
We next analyzed transcriptomic data of HCCs from The Cancer Genome Atlas (TCGA) [47] and Liver cancer Model Repository (LIMORE) cell line databases [48]. We divided the samples into two groups, those with the lowest and those with the highest expression of DDX5 mRNA (Figure 7A-B). We then compared the expression of several genes from Figure 6E that are involved in activation of Wnt signaling in these two groups. Among these genes, DVL1, a key positive regulator of Wnt signaling [49-51], was significantly different between the two groups in both TCGA and LIMORE (Figure 7 C-D). Interestingly, samples with the lowest expression of DDX5 exhibited statistically higher expression of DVL1. This result is consistent with the observed increased expression of DVL1 (Figure 6E) and the activation of Wnt signaling (Figures 4-6) in the DDX5KD cell lines. To further link DVL1 expression to the mechanism that mediates DDX5 downregulation, we performed correlation analysis between miR-19b1, miR-93, miR-20a, miR-17 and DVL1 expression across all HCC samples in TCGA. DVL1 mRNA expression positively correlates with all four miRNAs (Figure 7E).
Elevated mRNA expression of DVL1 in liver cells with low DDX5 mRNA. (A-B) Dot plots showing expression of DDX5 (A-B) and DVL1 (C-D) mRNA in samples with lowest DDX5 and samples with highest expression of DDX5 in TCGA (n=20) (A and C) and LIMORE (B and D) datasets. Median highlighted. *: P < 0.05; **: P < 0.01; ****: P < 0.0001. (E) Scatter plots show Spearman correlation between indicated miRNAs and DVL1 for all liver cancer samples in TCGA.
miRNA inhibitors (antagomirs) restore DDX5 in HBV replicating hepatocytes
DDX5 acts as a host cell restriction factor for HBV replication (Figures 1C and 3A), a barrier to hepatocyte dedifferentiation, and a negative regulator of Wnt signaling (Figures 4 and 5). We have also shown that overexpression of miR106b~25 and miR17~92 downregulate DDX5 (Figure 1). However, whether miRNA inhibitors can prevent DDX5 downregulation and associated phenotypes is unclear. To investigate the effect of the miRNA inhibitors (antagomirs), we targeted specific members of miR106b~25 and miR17~92 families. Tumor suppressors LKB1 [34] and PTEN [22], well-described targets of miR17~92 and miR106b~25 clusters, respectively, were downregulated in HBV replicating cells (supporting Figure S6A). Transfection of indicated antagomirs restored DDX5 as well as tumor suppressors LKB1 and PTEN in HBV replicating cells (Figure 8A and Figure S6B), suggesting these antagomirs have antitumor potential.
Next, we analyzed the antagomir effect on HBV replication, employing immunofluorescence microscopy of HBc. Co-transfection of combination of antagomirs for miR-106b, miR-17, miR-20a, and miR-19b1 reduced HBc immunostaining by nearly 50% in HepAD38 cells (Figure S6C). Accordingly, we designed antagomirs to miR-19b1 and miR-17 conjugated to a fluorophore (FAM). HepAD38 cells were transfected with miR-19b1i-FAM (Figure 8B) or miR-17i-FAM (Figure S6D) on day3 of HBV replication. Inhibitor miR-19b1i-FAM reduced HBc immunofluorescence in HBV replicating cells, while the signal for DDX5 increased. Interestingly, miR-19b1i-FAM also reduced nuclear immunostaining of β-catenin in HBV replicating HepAD38 cells (Figure 8B), indicating that restoring DDX5 suppressed Wnt/β-catenin pathway activation. Quantification of the fluorescence intensity demonstrates that the observed effects by miR-19b1i-FAM (Figure 8B) and miR-17i-FAM (Figure S6D) are statistically significant.
We then utilized the infection model of HepG2-NTCP cells to verify effects of miR106b~25 and miR17~92 antagomirs on virus biosynthesis. HepG2-NTCP cells were infected with 100 HBV genome equivalents per cell; on days 4 and 6 post infection (p.i.), HBV infected cells were transfected for 24 h with antagomirs for miR-106b, miR-17, miR-20a, and miR-19b1 or control inhibitor (Ctrli). Quantification of viral RNA on days 5 and 7 p.i. demonstrated significant reduction in the expression of HBc mRNA, pre-genomic RNA (pgRNA), and total HBV RNA in infected cells transfected with the indicated antagomirs (Figure 8C). Interestingly, antagomir transfected samples also exhibited reduced expression of DVL1 mRNA (Figure 8D), supporting the role of DDX5 as a negative regulator of Wnt signaling. We also monitored HBV biosynthesis in HBV infected HepG2-NTCP cells by fluorescence microscopy for HBc, as a function of transfection of miR-19b1i-FAM (Figure 8E) and miR-17i-FAM (Figure S6E). On day 7 p.i., HBV infected cells with strong HBc immunofluorescence (red) exhibited low fluorescence due to miR-19b1i-FAM (green); conversely, cells with strong green fluorescence exhibited weak immunostaining for HBc (Figure 8D). Similar results were obtained with miR-17i-FAM (Figure S6E). Taken together, these results demonstrate that antagomirs for miR106b~25 and miR17~92 restore DDX5 levels in HBV infected cells, attenuating Wnt pathway activation and HBV replication.
Discussion
In this study, we investigated the mechanism by which HBV infection downregulates the RNA helicase DDX5, and the significance of this downregulation to HCC pathogenesis. RNA helicases are involved in all aspects of RNA metabolism, from transcription, epigenetic regulation, miRNA processing, to mRNA splicing, decay, and translation [9, 17]. As a RNA helicase, DDX5 acts as a pro-viral host factor in biosynthesis of several RNA viruses, including HIV and HCV [52]. By contrast, DDX5 has antiviral function in myxoma virus biosynthesis [53] and HBV biosynthesis [7]. In our earlier studies, we have observed that DDX5 protein levels decrease in HBV replicating/infected hepatocytes [7]. Moreover, DDX5 knockdown in HBV infected HepG2-NTCP cells increased viral transcription, while the level of H3K27me3, a transcriptionally silencing histone modification associated with cccDNA, was reduced [7]. Since DDX5 interacts with the silencing PRC2 complex [7], we interpret these results to mean that DDX5 is involved in epigenetically regulating chromatin modifications of the viral minichromosome. Further studies are needed to determine the cellular context of this regulation.
Transcriptomic and functional analyses reveal that downregulation of DDX5 in HepAD38 hepatocytes results in activation of Wnt/β-catenin signaling (Figures 4-7), a pathway involved in reprogramming of hepatocytes towards a hCSC phenotype in HCCs [18, 44, 54]. In addition, the Wnt/β-catenin pathway is an important regulator of adult liver size, liver regeneration, and metabolic zonation [55, 56]. Whether DDX5 has a role in regulation of normal liver size, regeneration, and liver zonation is presently unknown. Importantly, recent studies have demonstrated the role of epigenetic mechanisms involving the PRC2 complex in liver regeneration [57].
In this study, we show that downregulation of DDX5 during the course of HBV infection is mediated by miR106b~25 [22] and miR17~92 [21] (Figure 1). These miRNAs are upregulated during HBV replication (Figure 2) and in HBV-related HCCs [23]. However, their role in HBV replication and virus-mediated hepatocarcinogenesis has been unknown. These miRNAs suppress pro-apoptotic functions of TGF-β pathway [31], downregulate tumor suppressors PTEN [22] and LKB1 [34], and have important roles in maintenance of pluripotency, progenitor cell growth, and regulation of normal development [58]. Interestingly, as we demonstrate herein, HBV-induced miR106b~25 and miR17~92 target the same seed sequence in 3'UTR of DDX5 and downregulate DDX5 during HBV replication (Figure 1). The increased expression of MCM7 encoding miR106b~25 [22] and c-Myc-driven transcription of miR17~92 [21], occurring during HBV replication, offer mechanistic insights into the observed induction of these miRNAs (Figure 2). Regarding the question of how HBV replication increases expression of these two miRNA clusters, it is well established that the viral HBx protein functions as an activator of cellular mitogenic pathways [32, 33, 59].
HBV-related HCCs display upregulated expression of these miRNAs [23]. The highest induction of miR106b~25 and miR17~92 is observed in HBV-related HCCs that display the lowest DDX5 mRNA level (Figure 2D). Importantly, HBV-related HCCs with low DDX5 mRNA belong to “Group III” tumors that express the hCSC-like gene signature, associated with poor patient prognosis [14]. The expression levels of miR106b~25, miR17~92 and DDX5 in these two groups of tumors, namely, “Group III” vs. the “Rest” (Figure S2C), display statistically significant differences (Figure 2D), suggesting a link of DDX5 loss to a hCSC-like phenotype. Mechanistically, loss of function studies of DDX5 demonstrate that DDX5 is multifunctional, having a role in viral biosynthesis (Figure 3A), in drug resistance, and hepatocyte stemness (Figure 3B and C). Specifically, HBV replicating DDX5KD cells form hepatospheres when grown in low attachment plates, are resistant to cisplatin and sorafenib, and express elevated levels of pluripotency genes and hCSC marker CD44 (Figure S4C). Moreover, loss of DDX5 either by overexpression of miR106b~25 and miR17~92, stable knockdown of DDX5, or DDX5 siRNA transfection, activate Wnt/β-catenin signaling in various liver cancer cell lines (Figure 5).
The transcriptomic studies comparing the mRNA expression profile of DDX5KD cells, as a function of HBV replication and sorafenib treatment, further support the role of DDX5 as an upstream regulator of Wnt pathway activation. The RNA-seq analysis identified deregulated expression of several FZD receptors, as well as regulators of Wnt signaling, upon DDX5 knockdown (Figures 6 and 7). The deregulated expression of Wnt pathway genes including FZD7, DVL1, SFRP4, and SFRP5, found in our study, agrees with similarly deregulated expression of FZD7 and SFRP5 observed in HBV-related HCCs [60], and the upregulated expression of DVL1 in all HCC types [51]. Significantly, upregulated expression of DVL1, a key activator of Wnt signaling [49, 50], correlates with reduced DDX5 expression in HCCs and HCC-derived LIMORE cell lines (Figure 7). Furthermore, downregulation of DDX5 alters chromatin accessibility, enhancing chromatin accessibility near genes of the Wnt pathway (Figure 4). Thus, based on the well-established role of Wnt pathway activation in cellular reprogramming and pluripotency [41], our studies provide a mechanistic link of DDX5 loss to hepatocyte reprogramming/stemness in HCC.
The mechanism by which DDX5 effects chromatin changes likely involves binding to noncoding RNAs [9], and interaction with epigenetic complexes including PRC2 [7]. Further studies are required to address this mechanism and the role of DDX5 in regulating HBV biosynthesis. DDX5 could regulate viral transcription from cccDNA [7] and/or translation of viral transcripts. Similarly, how DDX5 regulates stemness is incompletely understood. DDX5 was shown to act as a roadblock of somatic cell reprogramming by processing miR-125b, which in turn represses RING1 and YY1 Binding Protein (RYBP), a known inducer of pluripotency-associated genes [39]. We also reported earlier that DDX5 is a positive regulator of PRC2 stability [7], and loss of PRC2 function during HBV replication leads to activation of Wnt signaling and re-expression of a hCSC-like gene signature [14]. Hence, restoring DDX5 expression in chronically infected hepatocytes could suppress re-expression of the hCSC-like phenotype, providing therapeutic benefit. Considering this important role of DDX5, antagomirs against miR106b~25 and miR17~92 restore DDX5 levels. Significantly, these antagomirs exert both antiviral effects reducing expression of HBV pgRNA and HBc, as well as anti-tumor effects restoring tumor suppressor PTEN and LKB1 (Figure 8). Additionally, antagomir-mediated rescue of DDX5 reversed Wnt/β-catenin activation in HBV replicating cells, determined by loss of nuclear localization of β-catenin and reduction in DVL1 expression (Figure 8).
Conclusion
The results reported herein identify DDX5 as a promising therapeutic target, and also suggest that antagomirs against miR106b~25 and miR17~92 can be explored as therapeutic strategy to suppress DDX5 downregulation. This strategy of therapeutic antagomirs to restore DDX5 levels in HBV infected hepatocytes targets multiple pathways important for HCC, namely, (i) inhibition of HBV replication/biosynthesis (ii) rescue of tumor suppressor genes, and (iii) repression of Wnt signaling. Several miRNA-targeted therapeutic delivery strategies have reached clinical development [61], including lipid-based nanoparticle formulations [62]. Hepatocyte-specific deliveries include miRNA-conjugation to cholesterol [63, 64], and N-acetyl-glucosamine (GalNac) which binds with high affinity to the asialoglycoprotein receptor expressed in hepatocytes [61, 65]. Recent studies have also developed folate-linked miRNAs targeting folate receptor overexpressing cancer cells [66]. Ongoing studies are investigating folate receptor expression in HBV infected hepatocytes. Thus, several promising approaches are available to explore antagomir-mediated restoration of DDX5 in chronically infected HBV patients.
Abbreviations
DDX5: DEAD-box helicase 5/p68; HBV: hepatitis B virus; HCC: hepatocellular carcinoma; HBc: hepatitis B virus core antigen; DVL1: Disheveled 1; miR: microRNA; ATAC-seq: assay for transposase-accessible chromatin sequencing; GSEA: gene set enrichment analysis; FDR: false discovery rate; qRT-PCR: quantitative real-time polymerase chain reaction; ChIP: Chromatin immunoprecipitation; TCGA: the cancer genome atlas; LIMORE: liver cancer model repository; PRC2: polycomb repressive complex 2; EpCAM: epithelial cell adhesion molecule; MCM7: minichromosome maintenance complex; PTEN: phosphatase and tensin homologue; LKB1: serine/threonine protein kinase 11; FZD7: frizzled class receptor 7; SFRP: secreted frizzled-related protein; TCF7L1: transcription factor 7-like1.
Supplementary Material
Supplementary figures and tables.
Supplementary table S5, S6.
Acknowledgements
The authors thank the French National Biological Resources Centre for frozen human liver tissues, obtained following approved consent from the French Liver Tumor Network Scientific Committee. The French Liver Tumor Network is funded by the Institut National de la Santé et de la Recherche Médicale (INSERM) and the Agence Nationale de la Recherche (ANR). The authors also thank Dr. Adam Zlotnick for generously providing the HBc antibody and Dr. RL Hullinger for manuscript editing.
Funding
This work was supported by NIH grant DK044533 to OA and 5K22HL125593 to MK. Shared Resources (Computational Genomics Facility) supported by NIH grant P30CA023168, Pilot grants (Phase I and II) supported by the Purdue Center for Cancer Research to OA, the Walther Cancer Foundation, and NIH/NCRR RR025761.
Author contributions
SKKM performed experiments, analyzed data and wrote the manuscript; ZC and JS performed experiments and analyzed data; AF and DD performed HepaRG and PHH infections and prepared materials. SU and NAL performed RNA-seq and TCGA analyses; NF and PM provided RNA samples from human HBV-related HCCs. BY performed bioinformatics analyses of Fig. 4, 6& 7; MK directed all bioinformatics analyses, wrote related sections and edited manuscript; OA directed the studies, analyzed data, and wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557-76
2. Beasley RP, Hwang LY, Lin CC, Chien CS. Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22 707 men in Taiwan. Lancet. 1981;2:1129-33
3. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF. et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378-90
4. Bruix J, Qin S, Merle P, Granito A, Huang YH, Bodoky G. et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389:56-66
5. Abou-Alfa GK, Meyer T, Cheng AL, El-Khoueiry AB, Rimassa L, Ryoo BY. et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N Engl J Med. 2018;379:54-63
6. Zhu AX, Kang YK, Yen CJ, Finn RS, Galle PR, Llovet JM. et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased alpha-fetoprotein concentrations (REACH-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019;20:282-96
7. Zhang H, Xing Z, Mani SK, Bancel B, Durantel D, Zoulim F. et al. RNA helicase DEAD box protein 5 regulates Polycomb repressive complex 2/Hox transcript antisense intergenic RNA function in hepatitis B virus infection and hepatocarcinogenesis. Hepatology. 2016;64:1033-48
8. Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343-9
9. Jarmoskaite I, Russell R. RNA helicase proteins as chaperones and remodelers. Ann Rev Biochem. 2014;83:697-725
10. Seeger C, Mason WS. Molecular biology of hepatitis B virus infection. Virology. 2015 479-480C: 672-86
11. Pollicino T, Belloni L, Raffa G, Pediconi N, Squadrito G, Raimondo G. et al. Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound H3 and H4 histones. Gastroenterology. 2006;130:823-37
12. Wang WH, Studach LL, Andrisani OM. Proteins ZNF198 and SUZ12 are down-regulated in hepatitis B virus (HBV) X protein-mediated hepatocyte transformation and in HBV replication. Hepatology. 2011;53:1137-47
13. Studach LL, Menne S, Cairo S, Buendia MA, Hullinger RL, Lefrançois L. et al. Subset of Suz12/PRC2 target genes is activated during hepatitis B virus replication and liver carcinogenesis associated with HBV X protein. Hepatology. 2012;56:1240-51
14. Mani SK, Zhang H, Diab A, Pascuzzi PE, Lefrancois L, Fares N. et al. EpCAM-regulated intramembrane proteolysis induces a cancer stem cell-like gene signature in hepatitis B virus-infected hepatocytes. J Hepatol. 2016;65:888-98
15. Zhang H, Diab A, Fan H, Mani SK, Hullinger R, Merle P. et al. PLK1 and HOTAIR Accelerate Proteasomal Degradation of SUZ12 and ZNF198 during Hepatitis B Virus-Induced Liver Carcinogenesis. Cancer Res. 2015;75:2363-74
16. Diab A, Foca A, Fusil F, Lahlali T, Jalaguier P, Amirache F. et al. Polo-like-kinase 1 is a proviral host factor for hepatitis B virus replication. Hepatology. 2017;66:1750-65
17. Jankowsky E, Fairman ME. RNA helicases-one fold for many functions. Curr Opin Struct Biol. 2007;17:316-24
18. Yamashita T, Ji J, Budhu A, Forgues M, Yang W, Wang HY. et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136:1012-24
19. Tan W, Li Y, Lim SG, Tan TM. miR-106b-25/miR-17-92 clusters: polycistrons with oncogenic roles in hepatocellular carcinoma. World J Gastroenterol. 2014;20:5962-72
20. Adams BD, Kasinski AL, Slack FJ. Aberrant regulation and function of microRNAs in cancer. Curr Biol. 2014;24:R762-76
21. O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839-43
22. Poliseno L, Salmena L, Riccardi L, Fornari A, Song MS, Hobbs RM. et al. Identification of the miR-106b~25 microRNA cluster as a proto-oncogenic PTEN-targeting intron that cooperates with its host gene MCM7 in transformation. Sci Signal. 2010;3:ra29
23. Connolly E, Melegari M, Landgraf P, Tchaikovskaya T, Tennant BC, Slagle BL. et al. Elevated expression of the miR-17-92 polycistron and miR-21 in hepadnavirus-associated hepatocellular carcinoma contributes to the malignant phenotype. Am J Pathol. 2008;173:856-64
24. Pineau P, Volinia S, McJunkin K, Marchio A, Battiston C, Terris B. et al. miR-221 overexpression contributes to liver tumorigenesis. Proc Natl Acad Sci U S A. 2010;107:264-9
25. Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N. et al. The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432:235-40
26. Zhang L, Li LX, Zhou JX, Harris PC, Calvet JP, Li X. RNA helicase p68 inhibits the transcription and post-transcription of Pkd1 in ADPKD. Theranostics. 2020;10:8281-97
27. Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT. et al. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother. 1997;41:1715-20
28. Michailidis E, Pabon J, Xiang K, Park P, Ramanan V, Hoffmann HH. et al. A robust cell culture system supporting the complete life cycle of hepatitis B virus. Sci Rep. 2017;7:16616
29. Lucifora J, Durantel D, Belloni L, Barraud L, Villet S, Vincent IE. et al. Initiation of hepatitis B virus genome replication and production of infectious virus following delivery in HepG2 cells by novel recombinant baculovirus vector. J Gen Vir. 2008;89:1819-28
30. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545-50
31. Petrocca F, Vecchione A, Croce CM. Emerging role of miR-106b-25/miR-17-92 clusters in the control of transforming growth factor beta signaling. Cancer Res. 2008;68:8191-4
32. Rakotomalala L, Studach L, Wang WH, Gregori G, Hullinger RL, Andrisani O. Hepatitis B virus X protein increases the Cdt1-to-geminin ratio inducing DNA re-replication and polyploidy. J Biol Chem. 2008;283:28729-40
33. Bouchard MJ, Schneider RJ. The enigmatic X gene of hepatitis B virus. J Virol. 2004;78:12725-34
34. Izreig S, Samborska B, Johnson RM, Sergushichev A, Ma EH, Lussier C. et al. The miR-17 approximately 92 microRNA Cluster Is a Global Regulator of Tumor Metabolism. Cell Rep. 2016;16:1915-28
35. Ciccarese F, Zulato E, Indraccolo S. LKB1/AMPK Pathway and Drug Response in Cancer: A Therapeutic Perspective. Oxid Med Cell Longev. 2019;2019:8730816
36. Rawat S, Bouchard MJ. The hepatitis B virus (HBV) HBx protein activates AKT to simultaneously regulate HBV replication and hepatocyte survival. J Virol. 2015;89:999-1012
37. Xie N, Yuan K, Zhou L, Wang K, Chen HN, Lei Y. et al. PRKAA/AMPK restricts HBV replication through promotion of autophagic degradation. Autophagy. 2016;12:1507-20
38. Li W, Cao F, Li J, Wang Z, Ren Y, Liang Z. et al. Simvastatin exerts anti-hepatitis B virus activity by inhibiting expression of minichromosome maintenance protein 7 in HepG2.2.15 cells. Mol Med Rep. 2016;14:5334-42
39. Li H, Lai P, Jia J, Song Y, Xia Q, Huang K. et al. RNA Helicase DDX5 Inhibits Reprogramming to Pluripotency by miRNA-Based Repression of RYBP and its PRC1-Dependent and -Independent Functions. Cell stem cell. 2017;20:571
40. Atlasi Y, Looijenga L, Fodde R. Cancer stem cells, pluripotency, and cellular heterogeneity: a WNTer perspective. Curr Top Dev Biol. 2014;107:373-404
41. de Jaime-Soguero A, Abreu de Oliveira WA, Lluis F. The Pleiotropic Effects of the Canonical Wnt Pathway in Early Development and Pluripotency. Genes (Basel). 2018 9
42. Higuchi Y, Nguyen C, Yasuda SY, McMillan M, Hasegawa K, Kahn M. Specific Direct Small Molecule p300/beta-Catenin Antagonists Maintain Stem Cell Potency. Curr Mol Pharmacol. 2016;9:272-9
43. Dregalla RC, Zhou J, Idate RR, Battaglia CL, Liber HL, Bailey SM. Regulatory roles of tankyrase 1 at telomeres and in DNA repair: suppression of T-SCE and stabilization of DNA-PKcs. Aging (Albany NY). 2010;2:691-708
44. Yamashita T, Budhu A, Forgues M, Wang XW. Activation of hepatic stem cell marker EpCAM by Wnt-beta-catenin signaling in hepatocellular carcinoma. Cancer Res. 2007;67:10831-9
45. Merle P, de la Monte S, Kim M, Herrmann M, Tanaka S, Von Dem Bussche A. et al. Functional consequences of frizzled-7 receptor overexpression in human hepatocellular carcinoma. Gastroenterology. 2004;127:1110-22
46. Chan KK, Lo RC. Deregulation of Frizzled Receptors in Hepatocellular Carcinoma. Int J Mol Sci. 2018 19
47. Cancer Genome Atlas Research Network. Electronic address wbe, Cancer Genome Atlas Research N. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell. 2017;169:1327-41 e23
48. Qiu Z, Li H, Zhang Z, Zhu Z, He S, Wang X. et al. A Pharmacogenomic Landscape in Human Liver Cancers. Cancer Cell. 2019;36:179-93 e11
49. Funato Y, Michiue T, Asashima M, Miki H. The thioredoxin-related redox-regulating protein nucleoredoxin inhibits Wnt-beta-catenin signalling through dishevelled. Nat Cell Biol. 2006;8:501-8
50. Lee YN, Gao Y, Wang HY. Differential mediation of the Wnt canonical pathway by mammalian Dishevelleds-1, -2, and -3. Cell Signal. 2008;20:443-52
51. Song J, Xie C, Jiang L, Wu G, Zhu J, Zhang S. et al. Transcription factor AP-4 promotes tumorigenic capability and activates the Wnt/beta-catenin pathway in hepatocellular carcinoma. Theranostics. 2018;8:3571-83
52. Cheng W, Chen G, Jia H, He X, Jing Z. DDX5 RNA Helicases: Emerging Roles in Viral Infection. Int J Mol Sci. 2018 19
53. Rahman MM, Bagdassarian E, Ali MAM, McFadden G. Identification of host DEAD-box RNA helicases that regulate cellular tropism of oncolytic Myxoma virus in human cancer cells. Sci Rep. 2017;7:15710
54. Yamashita T, Nault JC. Stemness of liver cancer: From hepatitis B virus to Wnt activation. J Hepatol. 2016;65:873-5
55. Leibing T, Geraud C, Augustin I, Boutros M, Augustin HG, Okun JG. et al. Angiocrine Wnt signaling controls liver growth and metabolic maturation in mice. Hepatology. 2018;68:707-22
56. Yang J, Mowry LE, Nejak-Bowen KN, Okabe H, Diegel CR, Lang RA. et al. beta-catenin signaling in murine liver zonation and regeneration: a Wnt-Wnt situation!. Hepatology. 2014;60:964-76
57. Wang S, Zhang C, Hasson D, Desai A, SenBanerjee S, Magnani E. et al. Epigenetic Compensation Promotes Liver Regeneration. Dev Cell. 2019;50:43-56.e6
58. Vidigal JA, Ventura A. Embryonic stem cell miRNAs and their roles in development and disease. Sem Cancer Biol. 2012;22:428-36
59. Slagle BL, Andrisani OM, Bouchard MJ, Lee CG, Ou JH, Siddiqui A. Technical standards for hepatitis B virus X protein (HBx) research. Hepatology. 2015;61:1416-24
60. Bengochea A, de Souza MM, Lefrançois L, Le Roux E, Galy O, Chemin I. et al. Common dysregulation of Wnt/Frizzled receptor elements in human hepatocellular carcinoma. Br J Cancer. 2008;99:143
61. Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16:203-22
62. Ickenstein LM, Garidel P. Lipid-based nanoparticle formulations for small molecules and RNA drugs. Expert Opin Drug Deliv. 2019;16:1205-26
63. Park JK, Kogure T, Nuovo GJ, Jiang J, He L, Kim JH. et al. miR-221 silencing blocks hepatocellular carcinoma and promotes survival. Cancer Res. 2011;71:7608-16
64. Trajkovski M, Hausser J, Soutschek J, Bhat B, Akin A, Zavolan M. et al. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature. 2011;474:649-53
65. Weingärtner A, Bethge L, Weiss L, Sternberger M, Lindholm MW. Less Is More: Novel Hepatocyte-Targeted siRNA Conjugates for Treatment of Liver-Related Disorders. Mol Ther Nucleic Acids. 2020;21:242-50
66. Orellana EA, Tenneti S, Rangasamy L, Lyle LT, Low PS, Kasinski AL. FolamiRs: Ligand-targeted, vehicle-free delivery of microRNAs for the treatment of cancer. Sci Transl Med. 2017 9
Author contact
![]() Corresponding author: kazemianedu, andrisaoedu
Corresponding author: kazemianedu, andrisaoedu