Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(25):11754-11774. doi:10.7150/thno.43163 This issue Cite
Research Paper
Cardiomyocyte-derived small extracellular vesicles can signal eNOS activation in cardiac microvascular endothelial cells to protect against Ischemia/Reperfusion injury
Guihao Chen1,2, Chuansheng Xu1, Thomas G. Gillette2, Tongyi Huang3, Peisen Huang1, Qing Li1, Xiangdong Li1, Qinfeng Li4, Yu Ning1, Ruijie Tang1, Cunrong Huang1, Yuyan Xiong1, Xiaqiu Tian1,5, Jun Xu1, Junyan Xu1, Liping Chang6, Cong Wei6,7, Chen Jin1, Joseph A. Hill2, Yuejin Yang1 ![]()
1. State Key Laboratory of Cardiovascular Disease, Department of Cardiology, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100037, China
2. Department of Internal Medicine (Cardiology), University of Texas Southwestern Medical Center, Dallas, TX 75390-8573, USA
3. Department of Medical Ultrasound, Institute of Diagnostic and Interventional Ultrasound, First Affiliated Hospital, Sun Yat-Sen University, Guangzhou, Guangdong 510080, China
4. Department of Cardiology, Biomedical Research (Therapy) Center, Sir Run Run Shaw Hospital, School of Medicine, Zhejiang University, Hangzhou 310020, China
5. Center for Cardiac Intensive Care, Beijing Anzhen Hospital, Capital Medical University, Beijing Institute of Heart, Lung and Blood Vessel Diseases, Beijing 100029, China
6. Yiling Hospital of Hebei Medical University, National Key Laboratory of Collateral Disease Research and Innovative Chinese Medicine, Shijiazhuang, Hebei 050035, China
7. National Key Laboratory of Collateral Disease Research and Innovative Chinese Medicine, Key Laboratory of State Administration of TCM (Cardio-Cerebral Vessel Collateral Disease), Shijiazhuang, Hebei 050035, China
Received 2019-12-17; Accepted 2020-8-12; Published 2020-9-23
Abstract

Rationale: The crosstalk between cardiac microvascular endothelial cells (CMECs) and cardiomyocytes (CMs) has emerged as a key component in the development of, and protection against, cardiac diseases. For example, activation of endothelial nitric oxide synthase (eNOS) in CMECs, by therapeutic strategies such as ischemic preconditioning, plays a critical role in the protection against myocardial ischemia/reperfusion (I/R) injury. However, much less is known about the signals produced by CMs that are able to regulate CMEC biology. Here we uncovered one such mechanism using Tongxinluo (TXL), a traditional Chinese medicine, that alleviates myocardial ischemia/reperfusion (I/R) injury by activating CMEC eNOS. The aim of our study is to identify the signals produced by CMs that can regulate CMEC biology during I/R.
Methods: Ex vivo, in vivo, and in vitro settings of ischemia-reperfusion were used in our study, with the protective signaling pathways activated in CMECs identified using genetic inhibition (p70s6k1 siRNA, miR-145-5p mimics, etc.), chemical inhibitors (the eNOS inhibitor, L-NNA, and the small extracellular vesicles (sEVs) inhibitor, GW4869) and Western blot analyses. TritonX-100 at a dose of 0.125% was utilized to inactivate the eNOS activity in endothelium to investigate the role of CMEC-derived eNOS in TXL-induced cardioprotection.
Results: We found that while CMEC-derived eNOS activity was required for the cardioprotection of TXL, activation of eNOS in CMECs by TXL did not occur directly. Instead, eNOS activation in CMECs required a crosstalk between CMs and CMECs through the uptake of CM-derived sEVs. We further demonstrate that TXL induced CM-sEVs contain increased levels of Long Intergenic Non-Protein Coding RNA, Regulator Of Reprogramming (Linc-ROR). Upon uptake into CMECs, linc-ROR downregulates its target miR-145-5p leading to activation of the eNOS pathway by facilitating the expression of p70s6k1 in these cells. The activation of CMEC-derived eNOS works to increase survival in both the CMECs and the CMs themselves.
Conclusions: These data uncover a mechanism by which the crosstalk between CMs and CMECs leads to the increased survival of the heart after I/R injury and point to a new therapeutic target for the blunting of myocardial I/R injury.
Keywords: Cardioprotection, tongxinluo, cardiomyocytes, endothelial cells, crosstalk
Introduction
Coronary heart disease is the leading cause of death worldwide [1]. Timely and successful reperfusion, such as primary percutaneous coronary intervention, is the most effective treatment for patients suffering from acute myocardial infarction (AMI) [2]. However, aside from the ischemic injury, restoration of blood flow to endangered cardiomyocytes per se can paradoxically result in injuries to the myocardium, a phenomenon called reperfusion injury. Therefore, it is theoretically feasible that the prognosis of patients with AMI will be further improved if reperfusion injury can be alleviated or even prevented [3]. Although numerous strategies such as ischemic preconditioning have been reported to exert robust protective effects in animal model of I/R injury in the past 30 years, the translation of these beneficial therapies into clinical practice has been disappointing [4]. Consequently, new therapeutics should still be tested to determine whether they can reduce I/R injury or can be used to elucidate the mechanisms of I/R injury.
In hearts, cardiac microvascular endothelial cells (CMECs), which are located in the capillaries, closely surround CMs, with a short distance between these two types of cells (approximately 1-2 μm) [5]. Anatomically, several CMECs border one cardiomyocyte and can be regarded as a working “perfusion unit”. In these “perfusion units”, the close proximity between CMs and CMECs guarantees the important role of the crosstalk between these two types of cells, in both physiological (normal cardiac development) and pathophysiological conditions (ischemia, remodelling and metabolic dysfunction) in the heart [6]. Accumulating evidence suggests that the crosstalk between CMECs and CMs may be instrumental in the progression of myocardial I/R injury and thus, is a new potential therapeutic target for mitigating myocardial I/R injury. For instance, endothelium-derived neuregulin has been demonstrated to protect CMs from I/R-induced injury [7], and sEVs secreted from endothelial cells after ischemic preconditioning can make CMs less vulnerable to injury induced by H/R (I/R) [8]. Furthermore, cardioprotection conferred by exercise training is dependent on the activation of eNOS in endothelium [9]. Although the role of endothelial cells in protecting CMs during I/R is clear, few studies have investigated whether signals produced by CMs are able to impact CMECs and thereby reduce myocardial I/R injury.
Early preconditioning, which is implemented before a prolonged period of ischemia, is a physiological adaptive phenomenon where brief episodes of ischemia interspersed with reperfusion (ischemic preconditioning), or organs are exposed to certain pharmacological agents (pharmacological preconditioning), such as isoflurane. The cardioprotective effect of early preconditioning has been confirmed in dozens of studies, and is proved to be largely mediated by the activation of eNOS pathway [10, 11]. eNOS is an enzyme primarily responsible for the generation of NO in the vascular endothelium [12]. In hearts, endothelial cells [13] and other non-endothelial cells such as cardiomyocytes [14] and fibroblasts [15] express eNOS. Our early studies have demonstrated that preconditioning the hearts with Tongxinluo (TXL), a traditional Chinese medicine, can protect the hearts from I/R injury by activating the eNOS pathway [16, 17]. However, it remains unknown that whether the infarct-sparing effect of TXL is mediated by eNOS in endothelial cells, and how TXL activates endothelial eNOS. When examining this question, we uncovered a mechanism whereby crosstalk between CMs and CMECs impact CMECs to reduce myocardial I/R injury.
We show that preconditioning the hearts with TXL leads to the release of small extracellular vesicles (sEVs) from CMs that then stimulate eNOS activation in CMECs. These studies suggest that targeting this cellular crosstalk may provide a novel approach for the reduction of myocardial I/R injury.
Materials and Methods
Preparation of TXL Solution for the In Vitro Experiments
A solution of TXL ultrafine powder (Lot Number: 071201; Shijiazhuang Yiling Pharmaceutical Co., Shijiazhuang, China) was made as described previously [18]. In brief, after TXL was dissolved in serum-free Dulbecco's modified Eagle's medium (DMEM; Life Technologies, Grand Island, NY, USA), the suspension was sonicated for 30 min and then centrifuged at 2,500 rpm for 15 min. A 0.22-µm filter was used to filter the supernatant to prepare a sterile TXL solution.
Animals
Male Sprague Dawley rats (220-250 g) were utilized in our experiments. Animal studies were performed in accordance with the “Guide for the Care and Use of Laboratory Animals” issued by the US National Institutes of Health (Bethesda, MD, USA, NIH Publication No. 85-23, revised 1996) and the “Regulation to the Care and Use of Experimental Animals” of the Beijing Council on Animal Care (1996). The experimental protocol was reviewed and approved by the Care of Experimental Animals Committee of Fuwai Hospital.
Establishment of an in vivo Myocardial I/R Injury Model and infarct size measurement
Male Sprague Dawley rats were anesthetized with sodium pentobarbital (50 mg/kg, intraperitoneally) before endotracheal intubation. I/R was induced by ligating the left anterior descending artery (LAD) for 45 min, followed by loosening the ligature for 3 hours, as described previously [19]. Rats were randomized to: (1) Sham group, in which LAD was encircled by a suture but not occluded, and rats were administered saline by gavage 1 h prior to I/R; (2) I/R group, in which rats were administered saline by gavage 1 h prior to I/R; (3) I/R + TXL group, in which rats were administered TXL dissolved in saline (0.4 g/kg, an equivalent dose to that used clinically in humans) by gavage 1 h prior to I/R; and (4) I/R + TXL + GW4869 (Selleck, Houston, TX, USA) group, in which the rats were administered GW4869 (1 mg/kg) intraperitoneally every 24 h for 3 days prior to I/R, as described previously [20, 21], in addition to receiving the same TXL treatment as the TXL group. GW4869 was dissolved in saline with 2.5% DMSO. The same volume of 2.5% DMSO saline buffer was injected as a vehicle control in groups where GW4869 was not given.
Myocardial infarct sizes were assessed as previously described [18]. Briefly, after reperfusion, the suture was re-tied to ligate the LAD again, and 1 mL 2% Evans Blue dye (Sigma, Darmstadt, Germany) was injected into the thoracic aorta to delineate the ischemic area (area-at-risk, AAR) and the non-ischemic area (Evans blue perfused region). Then the heart was harvested and stored at -80°C overnight. On the second day, frozen ventricles were cut into 5 or 6 slices and incubated in 1% 2, 3, 5-triphenyltetrazolium chloride (TTC, Amresco, USA) for 15 min at 37°C. After staining, slices were incubated in 10% formalin to increase the contrast between dyed and undyed tissues. Ischemic but viable tissue will be stained red and the infarcted area (IA) will be stained pale white. ImageJ software (National Institutes of Health) was used to measure the IA, AAR, and total cross-sectional heart area (TA). A percentage of AAR (IA/AAR) or TA (AAR/TA) was used to express the infarct size and ischemic area, respectively.
Detection of microcirculatory perfusion
To determine microcirculatory blood flow during myocardial (M) I/R, rats were anesthetized using pentobarbital sodium, intubated and ventilated with a positive-pressure respirator. After removing the thoracic muscle, the thoracic cage was opened via thoracotomy through the 4th intercostal space on both sides and the mid-sternum. The heart was then completely exposed by cutting 2-4 ribs on both sides and fixing the free anterior thorax wall towards the rat's head with a forcep. To induce myocardial ischemia, the LAD coronary artery was ligated with a 6-0 silk suture 2-3 mm from the origin. 45 minutes later, the ligature was removed to re-perfuse the ischemic myocardium. To monitor microcirculatory perfusion, hearts were scanned using laser Doppler flowmetry (PeriCam PSI System, Perimed, Sweden) at baseline (prior to ligation), and 0, 10, 20, 30 and 40 mins post-reperfusion. Left ventricular blood flow at the ischemic part was determined and analyzed blindly.
Langendorff model, study design and infarct size measurement
After being heparinized (3 IU/g BW) and anesthetized with sodium pentobarbital (50 mg/kg, intraperitoneally), rat hearts were rapidly removed (the procedure time from opening the chest to heart excision was less than 2 min). Then the aorta was immediately connected to a standard Langendorff apparatus and perfused with Krebs-Henseleit (K-H) buffer solution (pH 7.4: in mM: NaCl 118; KCl 4.7; CaCl2 2.0; MgSO4 1.2; NaHCO3 25; KH2PO4 1.2; glucose 11.1) gassed with 95% O2 and 5% CO2 during the perfusion, at a constant flow of 10 mL/min at 37°C. A pulmonary arteriotomy was performed to drain coronary effluent. To establish a global I/R model, the perfusion pump was switched off for 40 min to stop coronary flow and switched on for 60 min to re-establish perfusion.
Rats were randomized to: (1) I/R; (2) I/R +TXL; (3) I/R+TXL+TritonX-100; and (4) I/R+TXL+L-NNA. TXL (400 μg/mL) was added into the perfusion fluid before ischemia and throughout reperfusion in the presence or absence of L-NNA (0.1 mM) or TritonX-100 (0.125%). L-NNA is a competitive inhibitor with selectivity for NOS. TritonX-100 at a low dose, as demonstrated before [22], can be used to inactivate the eNOS activity and decrease NO synthesis in endothelium, without losing eNOS or denaturing eNOS. The Triton-induced membrane damage may lead to leakage of raw materials (both substrates such as L-arginine and cofactors such as BH4) necessary for NO generation through the plasma membrane [22]. To investigate whether the TXL-induced protective effect is dependent on the activation of eNOS in endothelium, a Triton X-100 (0.125 %) bolus solution was used to abolish eNOS-mediated NO production [9,23]. Triton-X 100 at a concentration of 0.25%, which is commonly used by other research groups [9,23], increased infarct size in our preliminary experiments. Therefore, a lower concentration of Triton-X 100, which had no impact on infarct size but significantly reduced NO production, was utilized in our study.
Infarct size was measured 1 h after reperfusion. The hearts were harvested and stored at -80°C overnight. The steps for TTC staining were the same as those described for measuring infarct size after in vivo I/R. ImageJ software (National Institutes of Health) was used to measure the IA and total cross-sectional heart area (TA). A percentage of TA (IA/TA) was used to express the infarct size.
Measurement of nitric oxide production
Total NO production in cardiac tissues was determined by measuring the concentration of nitrate and nitrite, the stable metabolites of NO, by the modified Griess reaction method, according to the manufacturer's instructions (Beyotime Institute of Biotechnology, Shanghai, China). Optical density was measured at 540 nm and the amount of nitrite in the tissue lysate was calculated using sodium nitrite as a reference standard.
Determination of CK-MB release in serum
Blood samples were collected from the abdominal aorta of rats using a 10 mL syringe 3 hours after reperfusion. Blood samples were collected and serum was separated by centrifugation and then kept at -80°C. The levels of CK-MB were measured using a commercially available Rat Elisa Assay kit with a microplate reader according to the manufacturer's instructions (Jiancheng Bioengineering Institute, Nanjing, China).
Cell Culture and Treatments
Human CMs isolated from the ventricles of human adult hearts were purchased from PromoCell (Heidelberg, Germany). Cells were grown in a monolayer to 80% confluence and then sub-cultured using Ready-to-Use Myocyte Growth Medium (PromoCell). Experiments with human CMs were performed at passages three to seven.
Human CMECs were purchased from ScienCell Research Laboratories (San Diego, CA, USA) and cultured according to the manufacturer's instructions. Cells were grown in endothelial cell medium (ScienCell Research Laboratories) containing 5% fetal bovine serum (ScienCell Research Laboratories), 1% endothelial cell growth supplement (ScienCell Research Laboratories), and 1% penicillin/streptomycin at 37°C with 5% CO2, and then sub-cultured at a ratio of 1:3 when reaching 90% confluence. Cells in passages three to seven were used in our experiments.
Human cardiac fibroblasts were bought from ScienCell Research Laboratories. Human cardiac fibroblasts between passages 3-7 at 70-80% confluence were used for experiments. Human cardiac fibroblasts were cultured on poly-L-lysine coated culture plates in fibroblast medium (ScienCell) containing 10% FBS, growth supplements and penicillin/streptomycin, according to the manufacturer's instructions.
For co-culture experiments, 120,000 CMs were seeded into 6-well plate inserts with 0.4 μm pores (Corning, New York, USA) in normal culture medium. CMs were allowed to attach for 24 h before different treatments such as TXL pretreatment (400 μg/mL, 1 h) or siRNA transfection, and then the inserts were placed into 6-well plates previously plated with CMECs.
For H/R experiments, cells were washed with warm PBS twice and then exposed to different treatments in serum-free DMEM medium. Then cells were kept in an airtight and hypoxic GENbox jar fitted with a catalyst (BioMérieux, Marcy l'Etoile, France) to remove oxygen and induce 18 h hypoxia, as previously described [24]. They were then placed under normal conditions for 2 h of reoxygenation. To ensure that the in vitro H/R model was successfully established, an anaerobic indicator dye (BioMérieux) was used to reflect the oxygen tension of the medium.
To investigate whether TXL protected CMECs via the eNOS pathway, CMECs were washed with phosphate buffered saline (PBS) and exposed to L-NNA (100 μM) [9] in serum-free DMEM for 30 min prior to hypoxia and throughout the H/R conditions in the absence or presence of TXL (800 μg/mL) [25].
For sEV inhibition, human CMs were treated in 6-well plate inserts (Corning) with a chemical sEV inhibitor, GW4869 [26,27] (10μm) (Sigma, USA) for 24 h. Then, they were co-treated with TXL (400 μg/mL) [18] and GW4869 for 1 h prior to hypoxia, and then exposed to GW4869 alone when subjected to H/R. Human CMs were treated with DMSO as the vehicle control.
For all the in vitro experiments, at least 3 independent batches of CMECs, CMs or cardiac fibroblasts were used.
Assessment of Morphological Changes
The chromatin dye Hoechst 33,342 was used to stain and assess cell nuclear condensation and fragmentation (Beyotime Institute of Biotechnology, China). Briefly, CMECs were fixed with paraformaldehyde (4%) for 30 min and then treated with 5 mg/mL Hoechst 33,342 for 30 min. Finally, stained cells were washed twice with PBS at room temperature and observed under a fluorescence microscope (Leica, Germany). Cells with fragmented and condensed nuclei were defined as dead cells.
Determination of Cell Death by flow Cytometry
The percentage of dead cells was evaluated using the Annexin V-FITC/PI Kit (Becton, Dickinson and Company, USA), following the manufacturer's instructions. Briefly, CMECs, after experimental treatments, were collected and resuspended in 100 µl 1× binding buffer. The cell suspension was incubated with Annexin V and propidium iodide (PI) at room temperature in the dark for 15 min. Next, the binding buffer was mixed with another 400 µl 1× binding buffer, and CMECs were harvested and analyzed using the FACS Calibur System (Becton-Dickinson). Viable CMECs were considered annexin V-/propidium iodide (PI)-, early apoptotic CMECs as annexin V+/PI- and late apoptotic CMECs and already dead CMECs as annexin V+/PI+. Cells of annexin V+/PI- or annexin V+/PI+ were added together to calculate the proportion of dead CMECs.
SEV isolation
SEVs were isolated by differential centrifugation described in detail elsewhere [28]. Briefly, to isolate sEVs derived from CMs that were untreated, pretreated with TXL, or treated with lincRNA-ROR siRNA or conditioned medium from cells pretreated with TXL pretreatment (1 h) and then stimulated by H/R (18 h/2 h) were harvested and centrifuged. Cells and debris were removed by differential centrifugation at 300 ×g (10 min) and 2000 ×g (20 min), respectively. Microvesicles were precipitated by centrifuging at 13,500 ×g (30 min) and discarded. Supernatants were then ultra-centrifuged at 120,000 ×g (70 min) to acquire a raw sEV pellet. The pellet was then rinsed with PBS followed by a second ultracentrifugation (120,000 ×g, 70 min). The sEV pellet was resuspended in PBS and stored at -80°C for the corresponding experiments. The whole procedure was undertaken under sterile conditions.
Transmission electron microscopy (TEM)
SEVs diluted in PBS were adsorbed to a 200-mesh formvar/carbon coated electron microscopy grid (Electron Microscopy Sciences, PA, USA) and incubated at room temperature for 5 min. Next, excessive PBS was removed and 0.75% uranyl acetate dihydrate was used to stain sEVs for 5 min, followed by rinsing the samples with PBS three times. After the excess liquid was removed, the grids were observed by TEM and images were recorded with a camera.
Fluorescence confocal microscopy for sEV uptake
To determine how fast sEVs were taken up by CMECs in vitro, isolated sEVs were labeled with fluorescent dye PKH26 using a Red Fluorescent Cell Linker Kit (Sigma-Aldrich) according to the manufacturer's instructions and rinsed with PBS followed by ultra-centrifuging twice to remove redundant dye. PKH26-labeled sEVs were incubated with CMECs for 1 or 18 h. CMECs were then rinsed twice with PBS, fixed with 4% paraformaldehyde for 15 min, rinsed twice with PBS and permeabilized with 0.1% Triton X-100 for 5 min. Thereafter, cells were blocked with 1% goat serum for 40 min and stained with phalloidine and DAPI at room temperature. Uptake of PKH26-labeled sEVs by CMECs was observed by confocal microscopy.
Real-Time RT-PCR
Total RNA from CMECs, human CMs or sEVs was extracted using Trizol, according to the manufacturer's protocol (Invitrogen). linc-ROR and GAPDH mRNAs were reverse transcribed to cDNA using an iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA) and then quantified by quantitative real-time RT-PCR with SYBR Green PCR Master Mix. MicroRNAs were reverse transcribed using a miScript II RT Kit (Qiagen, Valencia, CA, USA) and quantified by quantitative real-time RT-PCR using a miScript SYBR green PCR kit (Qiagen). qRT-PCR was performed on an ABI 7500 thermocycler (Applied Biosystems) for 40 cycles. The following PCR primers were used: GAPDH forward, 5′-GAAGGTGAAGGTCGGAGTCA-3′; reverse, 5′-GGAAGATGGTGATGGGATTTC-3′; linc-ROR forward, 5′-AGGAAGCCTGAGAGTTGGC-3′; reverse, 5′-CTCAGTGGGGAAGACTCCAG-3′. Specific primers for microRNAs (miR-497-5p, miR-145-5p, miR-128-3p, and U6) were purchased from Qiagen. The linc-ROR from CMECs or human CMs was quantified using the 2(-ΔΔCT) relative quantification method, using GAPDH as an internal control. Linc-ROR in sEVs was quantified using the 2(-ΔΔCT) relative quantification method, using ce-miR-39-3p as an external control, as previously described [29, 30]. MicroRNA levels were quantified by the 2(-ΔΔCT) relative quantification method, using U6 as an internal control.
Western Blotting
Proteins were extracted from CMECs, sEVs or myocardial tissue samples. The BCA assay (Beyotime Institute of Biotechnology, China) was used to measure protein concentrations. To assess protein levels in these samples, lysates containing 25μg protein were separated on NuPage 4%-12% Bis-Tris Gels (Novex, Life Technologies) and protein bands were transferred to nitrocellulose membranes using a dry electroblotting apparatus (Invitrogen). Then 5% non-fat dry milk was utilized to block the membranes. Membranes were incubated in solutions of primary antibodies overnight at 4°C. The primary antibodies used included eNOS (#32027; 1:1000) and phospho-eNOS (Ser 1177) (#9570; 1:1000), Calnexin (#2679S; 1:1000), all from Cell Signaling Technology. Other primary antibodies used were p70s6k1(ab32529; 1:5,000), phosphop70s6k1 (Thr389) (ab2571; 1:250), CD81(ab109201; 1:2000) and CD63 (ab134045; 1:2000), all from Abcam. After being incubated with primary antibodies, membranes were rinsed three times and incubated with appropriate secondary antibodies (1:5,000; Zhongshanjinqiao, China) on the second day. Then stained bands were visualized with a Chemiluminescence Detection Kit (Pierce) after washing membranes three times. Protein signals were normalized to GAPDH (ab181602; primary antibody at 1:5,000; Abcam, Cambridge, UK). ImageJ software was used to analyze the densitometry.
Cell Transfection
Small interfering RNA (siRNA) oligonucleotides against p70s6k1 (Santa Cruz Biotechnology), nonspecific control siRNA oligonucleotide (Santa Cruz Biotechnology), mimics of miR-145-5p (Thermo Fisher Scientific, Inc., Waltham, MA, USA) or the microRNA negative control (Thermo Fisher Scientific, Inc.) were transfected into CMECs using Lipofectamine™ 2000 (Thermo Fisher Scientific, Inc.) in Opti-MEM Reduced Serum Medium (Thermo Fisher Scientific, Inc.). siRNA against linc-ROR (Genepharma, Shanghai, China) and nonspecific control siRNA oligonucleotides (Genepharma, Shanghai, China) were transfected into human CMs with the above-mentioned reagents. Cells were used in the following experiments after 48 h transfection. The siRNA sequence for linc-ROR was 5'-GGAGAGGAAGCCTGAGAGT-3'. The other proprietary sequences of siRNAs and microRNAs were not divulged by the companies.
Statistical Analysis
Statistical analysis was conducted in the GraphPad Prism 8.0.1. Quantitative data are displayed as the means ± standard error of the mean (SEM). Continuous variables between two groups were compared by the Student t-test. In the experiment of microcirculatory perfusion, two-way analysis of variance (ANOVA) was used for data comparisons. Comparison in other experiments with more than two groups was performed by one-way ANOVA. Following either one-way or two-way ANOVA, post-hoc analysis by Tukey's test utilized for multiple comparisons. P < 0.05 was considered to show statistical significance.
Results
Indirect activation of eNOS in CMECs can induce cardioprotection after I/R
Activation of eNOS is often required by beneficial agents to confer protection on endothelial cells under pathological conditions [31, 32]. For instance, the anti-apoptotic effect of sphingosine 1-phosphate on endothelial cells stressed with ethanol is dependent on the increased activity of this enzyme [31]. As we previously showed that TXL could protect CMECs from H/R-induced injury in vitro [25], and more importantly, the infarct-sparing effect of TXL in the in-vivo model of I/R required the activation of eNOS [16, 17], we first hypothesized that TXL was able to directly activate the eNOS pathway in CMECs, leading to the reduction of H/R-induced CMEC death. To test this hypothesis, we exposed CMECs to a model of acute simulated ischemia/reperfusion injury (18 h hypoxia followed by 2 h reoxygenation), as previously described [18]. As shown in Figure S1A-B, TXL had few effect (10.45 ± 0.92% vs 9.10 ± 1.01% in the Normal group, P > 0.05) on the viability of CMECs under normal conditions. Consistent with our previous research, TXL slightly inhibited the cell death of CMECs under the H/R conditions (33.43 ± 1.45% vs 42.53 ± 1.49% in the H/R group, P < 0.05) (Figure 1A, C), as detected by flow cytometry assay. Surprisingly, L-NNA, an inhibitor of eNOS, failed to inhibit the protective effect of TXL (34.30 ± 2.11% vs 33.43 ± 1.45% in the TXL group, P > 0.05) (Figure 1A, C), despite its efficient inhibition of eNOS activity (0.40 ± 0.09 vs 1.62 ± 0.07 in the TXL group, P < 0.05) (Figure 1E-F). The above findings were further confirmed by Hoechst 33342 staining (38.16 ± 1.44% vs 40.6 ± 3.68% in the TXL group, P > 0.05) (Figure 1B, D) and the CCK-8 assay (81.05 ± 1.43% vs 78.54 ± 1.51% in the TXL group, P > 0.05) (Figure S2). Importantly, eNOS was not activated by TXL in these conditions (Figure 1E-F). These results suggested that the protective effect of TXL on I/R injury is not through a direct activation of the eNOS pathway in CMECs.
Indirect activation of eNOSin CMECs can induce cardioprotectionafter I/R.A) Representative scatter diagram of the death of CMECs in flow cytometry assay (n=4). B) Representative images of Hoechst 33342 nucleic acid staining for CMECs; white arrows point to dead cells (Bar=100 μm, n=3). C-D) Quantitative analysis for dead cells in flow cytometry assay and Hoechst staining, respectively. E-F) The p-eNOSand t-eNOSwere detected by Western Blot for the in vitro experiments (n=3). G) A schematic representation of the experimental protocol for the Langendorffmodel. H) Representative images of TTC staining in each group (n = 5, 5-6 slices per heart). The red-stained areas indicate viable tissue, and the pale white areas indicate infarct tissue. I) Quantitative analysis for infarct size in each group. J) NO production in cardiac tissues for each group (Griess reaction method (n=5). K-L) The p-eNOSand t-eNOSwere detected by Western Blot for the ex-vivohearts (n=5). *P<0.05 vs. Control; #P <0.05 vs. TXL. Control: H/R or I/R; All data are mean ±SEM. Statistical analysis was performed with one-way ANOVA followed by Tukey's test.
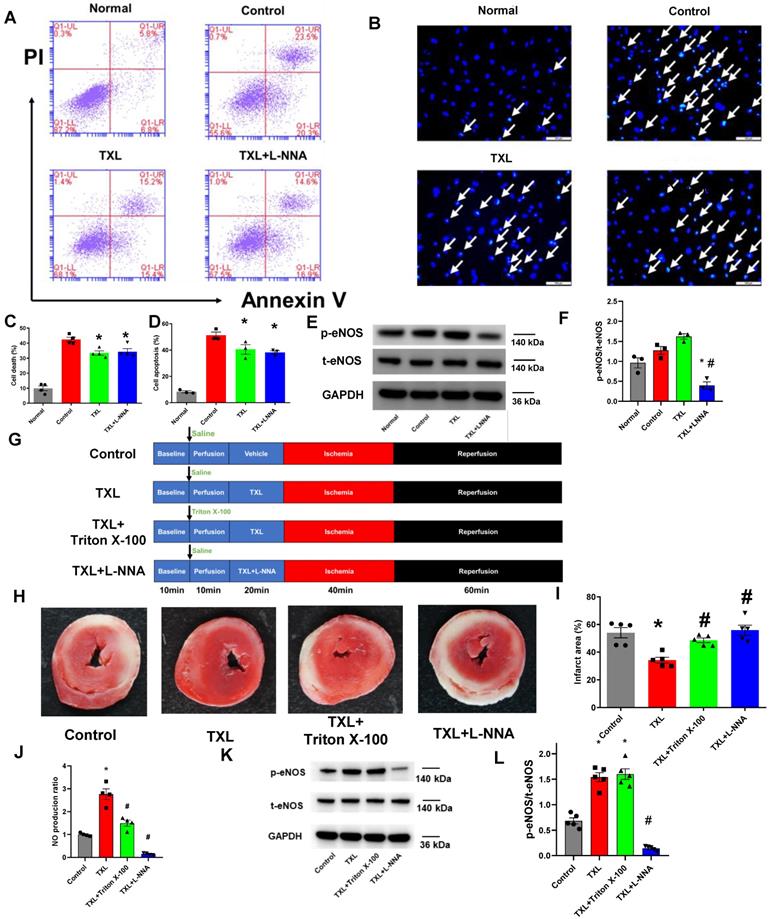
Our earlier work demonstrated a key role for eNOS in the cardioprotective effects of TXL [16, 17]. Therefore, we next asked if the eNOS activation observed in TXL-treated hearts was from CMECs and more importantly, if this CMEC derived eNOS was required for the cardioprotective effects of TXL. To address this question, we utilized the Langendorff model and exposed rat hearts to I/R injury (Figure 1G). As expected and shown in Figure 1H-I, TXL treatment significantly decreased the infarct size after I/R (34.14 ± 2.14% vs 54.02 ± 3.69% in the Control group, P < 0.05). Treatment with L-NNA, a competitive inhibitor of nitric oxide synthase, was effective in repressing the phosphorylation of eNOS (0.14 ± 0.02 vs 1.54 ± 0.09 in the TXL group, P < 0.05) (Figure 1K-L). Repression of eNOS activity in this manner completely abrogated the cardioprotective benefits of TXL treatment (55.84 ± 3.65% vs 34.14 ± 2.14% in the TXL group, P < 0.05) (Figure 1H-I). As shown in Figure S3, compared with the Control group, TXL group did not display an increase in p-nNOS/t-nNOS or the expression of iNOS, which ruled out the possibility that L-NNA took effects in our ex-vivo study by inhibiting the activity of nNOS or iNOS. In addition, we found that L-NNA treatment alone had no significant impact on infarct size (Figure S4A-B). Previously, low levels of Triton-X 100 pretreatment has been shown to be effective in abolishing coronary endothelial eNOS-mediated NO production with negligible effects on myocardial function and infarct size [22, 23]. We therefore pretreated the hearts with Triton-X 100 under these conditions and examined the impact on infarct size and TXL cardioprotection. Triton-X 100 pretreatment alone did not significantly change the infarct size after I/R (57.13 ± 4.26% vs 54.02 ± 3.69% in the Control group, P > 0.05, Figure S4A-B). In stark contrast, Triton-X 100 pretreatment dramatically inhibited the cardioprotective effects of TXL (48.54 ± 1.67% vs 34.14 ± 2.14% in the TXL group, P < 0.05) (Figure 1H-I). As shown in Figure 1J, Triton-X 100 pretreatment significantly reduced the level of NO in cardiac tissues (1.50 ± 0.13 vs 2.76 ± 0.24 in the TXL group, P < 0.05), indicating that it efficiently disrupted the function of eNOS in the coronary endothelium. Importantly, as shown in Figure 1K-L, Triton-X 100 inhibits NO without affecting the increased phosphorylation of eNOS induced by TXL (1.60 ± 0.10 vs 1.54 ± 0.09 in the TXL group, P > 0.05), demonstrating that the Triton-X treatment is not merely disrupting TXL uptake into cells. The correlation of decreased cardioprotection with decreased NO production are in agreement with our previous studies that TXL attenuated I/R injury in an eNOS-dependent pathway in vivo [16,17]. However, these data further suggest that the cardioprotection induced by TXL in ex vivo hearts were dependent on the activation of eNOS in cardiac endothelial cells. Taken together, the data from these experiments suggested that cardioprotection, induced by certain agents such as TXL, can result from the indirect activation of eNOS in CMECs during I/R.
Evidence of eNOS activation in CMECs through a cardiomyocyte-dependent paracrine pathway
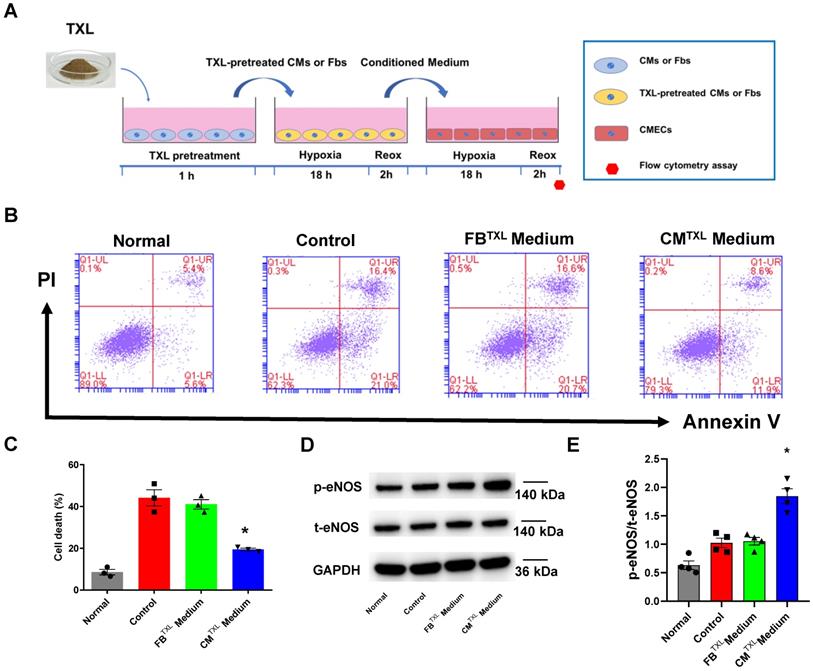
Our data suggests that the activation of endothelial eNOS is required by TXL to exert its infarct-sparing effect in the ex vivo model (Figure 1H-I). In addition, our in vitro experiments demonstrated that TXL did not directly activate eNOS in endothelial cells (Figure 1E-F). Furthermore, our ex vivo results (Figure 1H-I) rule out the possibility from our earlier in vivo studies that TXL conferred its cardioprotective effect via a systemic effect [16, 17]. Taken together, we hypothesized that TXL promotes crosstalk between endothelial and non-endothelial cells in hearts that promotes cell survival during I/R. Because fibroblasts (approximately 62.6% in rats, and approximately 26.1% in mice) and cardiomyocytes (approximately 26.4% in rats, and approximately 55.9% in mice) are the other two main types of cells in hearts [33], we used their conditioned medium to treat CMECs under H/R conditions (Figure 2A). In this experiment, we pretreated fibroblasts and cardiomyocytes with TXL for 1 h and then subjected them to H/R, after which their conditioned medium was collected (FbTXL medium and CMTXL medium) and used to treat CMECs when exposed to H/R. As shown in Figure 2B-C, compared with the Control group, the group with CMTXL medium had significantly fewer numbers of dead cells (19.43 ± 0.61% vs 44.1 ± 3.90% in the Control group, P < 0.05), while no difference was observed between the Control and FbTXL medium groups (41.00 ± 2.26% vs 44.1 ± 3.90% in the Control group, P > 0.05). These findings were confirmed in the CCK-8 assay, showing that CMTXL medium, rather than FbTXL medium, could protect CMECs from H/R-induced injury (Figure S5). Of note, conditioned medium from Fb or CM without TXL pretreatment had few effects on the cell death of CMECs (44.1 ± 3.90% vs 42.77 ± 3.09% in the Fb medium group and 39.63 ± 2.96% in the CM medium group, P > 0.05) (Figure S6A-B) during H/R, supporting the model in which TXL promotes crosstalk between these cell types to increase survival. Furthermore, CMTXL medium significantly increased the phosphorylation of eNOS in CMECs (1.84 ± 0.13 vs 1.03 ± 0.08 in the Control group, P < 0.05) (Figure 2D-E), suggesting that TXL treatment not only induces CMs to release a paracrine factor increasing endothelial cell survival after I/R, but also stimulates eNOS activity in these cells leading to an increase in cardioprotection.
Evidence of eNOSactivation in CMECs through a cardiomyocyte-dependent paracrine pathway. A) Schematic of the experimental protocol for investigating the effects of conditioned medium of CMs or Fbson CMECs. B-C) Dead CMECs in flow cytometry assay (n=3). D-E) The p-eNOSand t-eNOSwere detected by Western Blot (n=4). *P<0.05 vs. Control. CM: cardiomyocyte; CMTXL: TXL-pretreated cardiomyocyte; Fb: fibroblast; FbTXL: TXL-pretreated fibroblast; Reox: reoxygenation. All data are mean ±SEM. Statistical analysis was performed with one-way ANOVA followed by Tukey's test.
CM-derived sEVs mediate the eNOS activation in CMECs during H/R in a trans-well co-culture system
Our data suggest that treatment of CMs with TXL results in the release of a factor(s) that decreases endothelial cell death during I/R. SEVs, previously named as exosomes [34], are nanosized particles released into the extracellular space by various cell types. They contain substances such as proteins, lipids and nucleic acids (mRNA, long non-coding RNA, etc) and have an important role in cell-to-cell communication [35]. Close contact between CMs and CMECs suggests that sEVs can mediate the crosstalk between these two types of cells. Indeed, one study demonstrated that cardiomyocytes mediated anti-angiogenesis via the sEV transfer of miR-320 into endothelial cells in type 2 diabetic rats [36]. Furthermore, sEVs released from endothelial cells have been suggested, in some cases, to protect cardiomyocytes from H/R-induced injury [8]. Taken together, we hypothesized that TXL-pretreated cardiomyocytes protect endothelial cells exposed to H/R by releasing sEVs. To test this hypothesis, a trans-well co-culture system was introduced into our experiments (Figure 3A). As expected, being co-cultured with CMTXL, instead of CM (37.47 ± 0.64% in the CM group vs 40.03 ± 2.06% in the Control group, P > 0.05) (Figure S7), inhibited the cell death of CMECs during H/R (19.17 ± 0.74% vs 38.13 ± 0.63% in the Control group, P < 0.05) (Figure 3B-C), validating the results of the above experiments (Figure 2B-C). However, this beneficial effect was totally abolished when GW4869, an sEV release inhibitor, was used to treat CMTXL (37.73 ± 0.55% vs 38.13 ± 0.63% in the Control group, P > 0.05) (Figure 3B-C) , suggesting a requirement for secreted sEVs in the CMTXL mediated protection of CMECs during H/R. GW4869 alone did not affect the death of CMECs (37.87 ± 0.59% vs 38.13 ± 0.63% in the Control group, P > 0.05) (Figure S8A-B) or CM (21.53 ± 1.25% in the CMTXL group vs 20.23 ± 1.11% in the CMTXL+GW4869 group, P > 0.05) (Figure S9A-B) under H/R conditions. sEVs released from H/R-treated CMTXL (CMTXL-sEVs) were also isolated and incubated with CMECs. CMTXL-sEVs were able to alleviate the H/R-induced injury of CMECs (21.10 ± 1.15% vs 38.13 ± 0.63% in the Control group, P < 0.05) (Figure 3B-C), while CM-sEV (Figure S7) or CMTXL-medium depleted of sEVs (Figure S10) did not affect the death of CMECs under H/R. Additionally, sEVs derived from H/R-treated NRVMTXL (NRVMTXL-sEVs; NRVM refers to neonatal rat ventricular myocyte) were isolated and used to treat rat aortic endothelial cells. Consistently, NRVMTXL-sEVs significantly inhibited the cell death of rat aortic endothelial cells exposed to H/R (28.27 ± 1.13% vs 44.90 ± 2.38% in the Control group, P < 0.05) (Figure S11A-B).
CM-derived sEVsmediate the eNOSactivation in CMECs during H/R in a trans-well co-culture system.A) Schematic of the protocol for co-culture experiments. B-C) Dead CMECs in flow cytometry assay (n=3). D-E) The p-eNOSand t-eNOSwere detected by Western Blot (n=4). F) NO production in CMECs for each group (Griess reaction method, n=4). G) Western blots confirmed the presence of sEVmarker in sEVsderived from TXL-pretreated CM after H/R (n=4). H) Electron micrographs of purified sEVsfrom TXL-pretreated CM after H/R. (Bar: 200 nm; n=3). I) Representative confocal images showing that red fluorescence dye PKH26 labelled sEVsfrom TXL-pretreated CM after H/R were endocytosed by CMECs after 1 hour and 18 hours incubation. *P<0.05 vs. Control; #P <0.05 vs. CMTXL. sEVs: small extracellular vesicles; CM: cardiomyocyte; CMTXL: TXL-pretreated cardiomyocyte; CMTXL-sEV: sEVsderived from CMTXL. All data are mean ±SEM. Statistical analysis was performed with one-way ANOVA followed by Tukey's test.
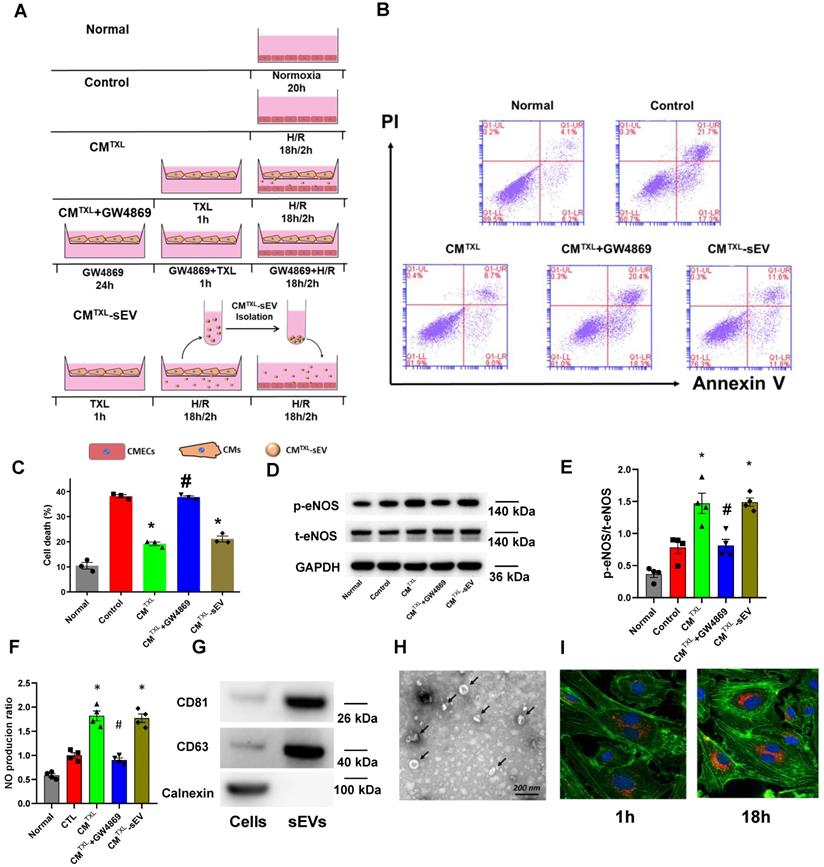
While TXL did not induce direct CMEC protection against H/R through eNOS activation, this activation of eNOS by TXL is critical to the cardio-protective benefits of the treatment. We therefore investigated the requirement of eNOS activity in the CMTXL derived protection of CMECs to H/R. Surprisingly, under these conditions, eNOS was necessary for the protective effect of CMTXL-sEVs on CMECs to H/R (Figure S12A-B). As shown in Figure 3D-E, compared with the Control group, CMECs in the CMTXL group had a significantly higher ratio of p-eNOS/t-eNOS (1.47 ± 0.16 vs 0.78 ± 0.10 in the Control group, P < 0.05). This trend was reversed when GW4869 was introduced to inhibit the release of sEVs from CMTXL (0.81 ± 0.10 vs 0.78 ± 0.10 in the Control group, P > 0.05) (Figure 3D-E). The treatment of CMECs with CMTXL-sEVs also facilitates the phosphorylation of eNOS compared with the Control group (1.49 ± 0.07 vs 0.78 ± 0.10 in the Control group, P < 0.05) (Figure 3D-E). Our further experiments (Figure 3F) showed that CMECs in the CMTXL group had more NO production (1.82 ± 0.10 vs 1.00 ± 0.05 in the Control group, P < 0.05), compared with the Control group. However, this effect could be offset by GW4869 (0.90 ± 0.04 vs 1.00 ± 0.05 in the Control group, P < 0.05) (Figure 3F). Additionally, treating CMECs with CMTXL-sEVs also augmented the production of NO (1.77 ± 0.09 vs 1.00 ± 0.05 in the Control group, P < 0.05) (Figure 3F), compared with the Control group. These data show that CM-derived sEVs are necessary and sufficient for the CMTXL activation of eNOS in the CMECs.
To further support the above findings, western blotting, transmission electron microscopy (TEM) and nanoparticle tracking analysis (NTA) were used to characterize sEVs derived from CMTXL. Western blot analyses demonstrated that CMTXL-sEVs had a high expression of the sEV specific markers, CD81 and CD63, while the marker of cell lysate, Calnexin, could not be detected (Figure 3G). TXL pretreatment had no impact on CM's ability to release sEVs, as demonstrated by the amount of these markers (Figure S13A) in the conditioned medium from CM or CMTXL and shown in NTA (Figure S13B). CMTXL pretreated with GW4869 secreted much less sEVs compared with the group without GW4869 pretreatment, as shown by the dramatical reduction of CD81 and CD63 in the conditioned medium of CMTXL (Figure S14A), suggesting the efficacy of GW4869 in our conditions to inhibit sEV release. TEM imaging analysis showed that CMTXL-sEVs had a cup-shaped structure and were approximately 100 nm in diameter (Figure 3H). To assess the internalization of sEVs by CMECs, CMTXL-sEVs were pre-labeled with PKH26 (a red fluorescent dye that stains the cell membrane) and cultured with CMECs. Confocal images showed that CMECs had internalized many CMTXL-sEVs 1 h after incubation and had taken up a significantly higher amount of stained CMTXL-sEVs by 18 h (Figure 3I), indicating CMECs internalize sEVs rapidly. PKH26 dye was also used to label conditioned medium from CMTXL pretreated with GW4869, and there was no transfer of dye into CMECs, supporting the inhibition efficacy of GW4969 again (Figure S14B).
The improvement of cardiac microcirculation and infarct-sparing effect induced by TXL is mediated by sEVs
To investigate if TXL could reduce the death of CMECs during I/R and thus improve cardiac microcirculation, we then used laser Doppler flowmetry to record cardiac microvascular perfusion after the release of coronary artery ligation. As hypothesized, the TXL group displayed a significant improvement in cardiac microcirculation after I/R, compared with the control group (Figure 4A-B).
The improvement of cardiac microcirculation and infarct-sparing effect induced by TXL is mediated by sEVs.A-B) Representative perfusion images for myocardial microcirculatory perfusion (percentage relative to baseline) measured by laser Doppler flow during myocardial I/R (n=6-8), and the statistical graph were shown, respectively. C) Representative images for myocardial infraction size assessed by Evans blue/TTC double staining (n=7-8). D-E) Quantitative analysis for infarct size and area at risk after I/R, respectively. F) Serum CK-MB activity in each group (n=5-6). G-H) The p-eNOSand t-eNOSwere detected by Western Blot (n=4). * P<0.05 TXL vs. Control; # P <0.05 TXL + GW4869 vs TXL. AAR: area at risk; IA: infarct area; TA: total area. All data are mean ±SEM. Statistical analysis was performed with two-way (microcirculatory perfusion) or one-way (other experiments) ANOVA followed by Tukey's test.
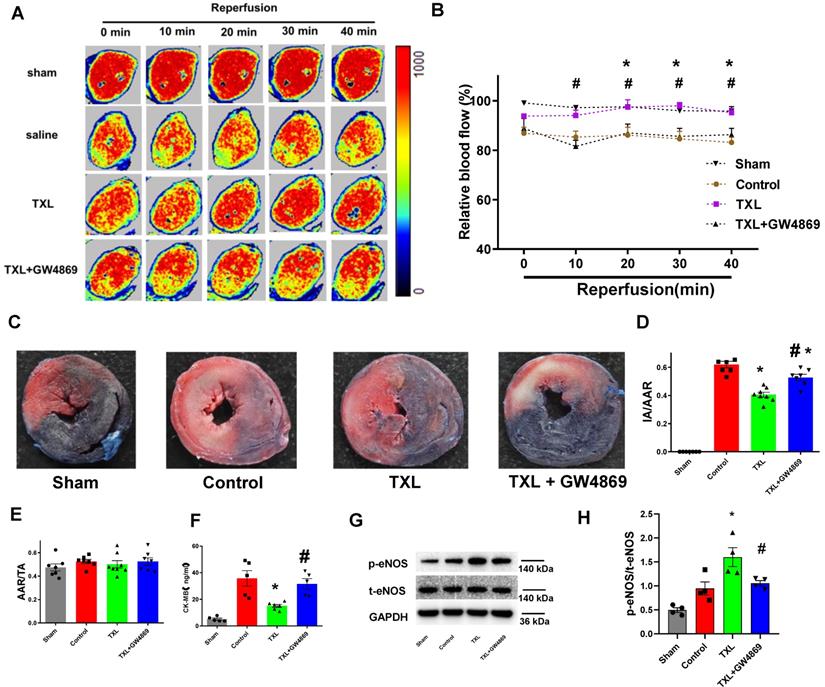
To examine the requirement of sEVs in the in vivo protective effect induced by TXL, GW4869 was used to inhibit the secretion of sEVs in vivo. As shown in Figure 4A-B, GW4869 could offset the protective effects of TXL on cardiac microvascular perfusion.
Further experiments demonstrated that, the TXL group had a significantly decreased myocardial infarct size after I/R compared with the control group (40.51 ± 1.75% vs 61.89 ± 2.29% in the Control group, P < 0.05) (Figure 4C-E) , which was consistent with our published research [16-18]. However, GW4869 pretreatment reduced the beneficial effect of TXL (52.64 ± 2.27% vs 61.89 ± 2.29% in the Control group, P > 0.05) (Figure 4C-E). GW4869 did not on its own have any impact on infarct size under these conditions (61.73 ± 2.49% vs 59.57 ± 2.22% in the Control group, P > 0.05) (Figure S15A-C). Serum CK-MB levels, a marker of cardiac cell death, was reduced in the TXL treated group compared to the untreated control after I/R (15.06 ± 1.16% vs 35.69 ± 5.89% in the Control group, P < 0.05) (Figure 4F). Consistent with the infarct size, no difference in serum CK-MB levels was observed between the control and TXL+GW4869 groups (31.59 ± 3.91% vs 35.69 ± 5.89% in the Control group, P < 0.05) (Figure 4F). And compared with the control group, the phosphorylation of eNOS was statistically increased in the TXL treated group (1.60 ± 0.20 vs 0.95 ± 0.14 in the Control group, P < 0.05) (Figure 4G-H); however, this effect was counteracted by GW4869 (1.05 ± 0.05 vs 0.95 ± 0.14 in the Control group, P > 0.05) (Figure 4G-H). We then isolated the adult rat CMECs after myocardial I/R (Figure S16A-B), demonstrating that CMECs in the TXL-pretreated hearts had increased eNOS activity (1.73 ± 0.24% vs 0.81 ± 0.12% in the Control group, P < 0.05), which was offset by the treatment of GW4869 (0.80 ± 0.18% vs 0.81 ± 0.12% in the Control group, P > 0.05). These data support a model by which TXL-induced sEVs drive crosstalk between CMs and endothelial cells increasing eNOS production and cell survival after I/R.
CM-derived sEVs mediate the eNOS activation in CMECs through the p70s6k1 pathway
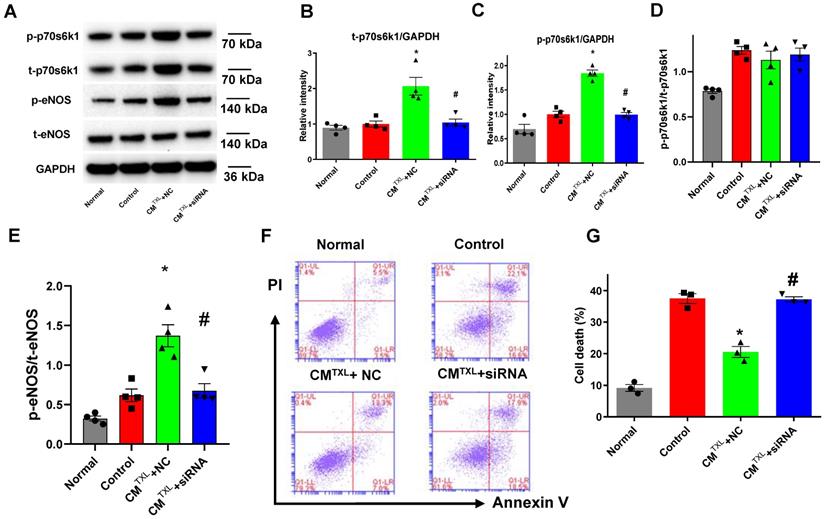
Previous studies have shown that cysteine-rich, angiogenic inducer 61 or neuropeptide Y activate the eNOS pathway by increasing the phosphorylation of p70s6k [37,38]. Furthermore, our recent study reported that TXL attenuated I/R injury by upregulating the steady-state level of p70s6k1 and increasing p-p70s6k1 [18]. Consequently, we hypothesized that CMTXL-sEVs protect CMECs from H/R injury by activating the p70s6k1/eNOS pathway. As shown in Figure 5A-D, compared with the Control group, TXLTXL+NC group displayed higher levels of both t-p70s6k1 (2.07 ± 0.25 in CMTXL + NC group vs 1.00 ± 0.08 in the Control group, P < 0.05) and p-p70s6k1 (1.84 ± 0.07 in CMTXL + NC group vs 1.00 ± 0.06 in the Control group, P < 0.05), with no significant difference in the ratio of p-p70s6k1/t-p70s6k1 (1.13 ± 0.10 in CMTXL + NC group vs 1.24 ± 0.04 in the Control group, P > 0.05) being found between these two groups. Consistently, similar results could be observed in the adult rat CMECs isolated after myocardial I/R (Figure S16A, C-E). These data indicated that the increased p-p70s6k1 level in the CMTXL+NC group was dependent on the augmentation of its steady-state level. In addition, transfection of siRNA against p70s6k1 into CMECs reversed the increasing trend of t-p70s6k1 levels (1.04 ± 0.10 in CMTXL + siRNA group vs 1.00 ± 0.08 in the Control group, P > 0.05) (Figure 5A-B) induced by co-culture with CMTXL under H/R conditions; thus, the level of p-p70s6k1 (0.99 ± 0.04 in CMTXL + siRNA group vs 1.00 ± 0.06 in the Control group, P > 0.05) (Figure 5A, C) in CMECs was unchanged when co-cultured with CMTXL. As hypothesized, the decreased level of p-p70s6k1, caused by the reduction in the expression of t-p70s6k1, inhibited the activation of eNOS (0.67 ± 0.09 in CMTXL + siRNA group vs 0.62 ± 0.08 in the Control group, P > 0.05) (Figure 5A, E) in CMECs induced by co-culture with CMTXL, indicating that the increased phosphorylation of p70s6k1 was required for activation of the eNOS pathway under our conditions. Because of the above-mentioned effects induced by p70s6k1 siRNA, no difference in the percentage of dead CMECs was observed between the CMTXL+siRNA and Control groups (37.20 ± 0.85% in CMTXL + siRNA group vs 37.47 ± 1.54% in the Control group, P > 0.05) (Figure 5F-G). The siRNA against p70s6k1 alone did not increase the cell death of CMECs during H/R (36.27 ± 1.17% vs 37.47 ± 1.54% in the Control group, P > 0.05), as shown in Figure S17. The above findings were further confirmed with a second siRNA construct against p70s6k1 (Figure S18). Therefore, we concluded that CMTXL reduced the H/R-induced injury of CMECs by promoting the expression of the p70s6k1, and a subsequent increase in the level of p-p70s6k1, thus activating eNOS pathway.
CM-derived sEVsmediate the eNOSactivation in CMECs through the p70s6k1 pathway. A-E) The levels of p-70s6k1, t-p70s6k1, p-eNOSand t-eNOSwere detected by Western Blot (n=4). F-G) Dead CMECs in flow cytometry assay (n=3). * P<0.05 vs. Control; # P <0.05 vs CMTXL+NC. sEVs: small extracellular vesicles; CM: cardiomyocyte; CMTXL: TXL-pretreated CMs; CMTXL+siRNA group: CMECs were transfected with p70s6k1 siRNA before being co-cultured with CMTXL and then exposed to H/R; CMTXL+NC group: CMECs were transfected with negative control of siRNA before being co-cultured with CMTXL and then exposed to H/R. All data are mean ±SEM. Statistical analysis was performed with one-way ANOVA followed by Tukey's test.
CM-derived sEVs mediate eNOS activation by increasing p70s6k1 in CMECs through reduction of miR-145-5p
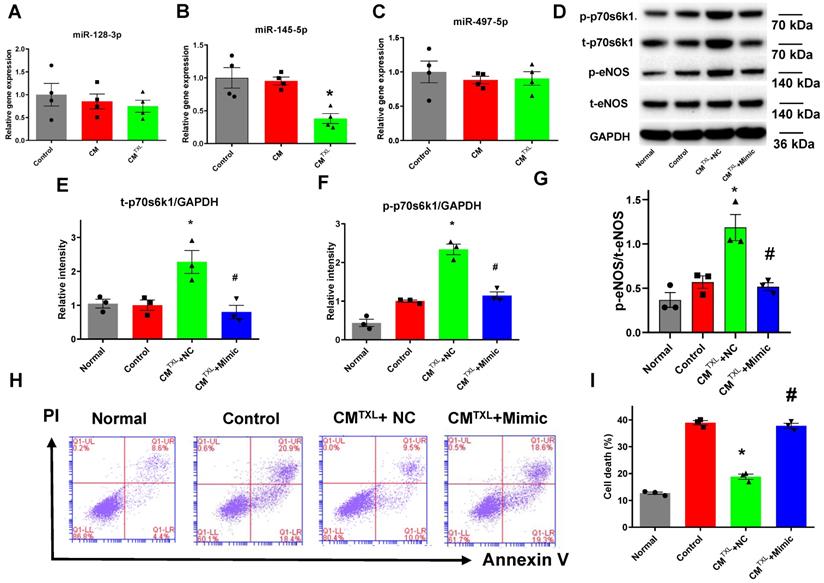
Our previous research demonstrated that treating CMs with TXL promoted the expression of p70s6k1 via the inhibition of miR-128-3p [18]. Therefore, we measured the level of miR-128-3p in CMECs co-cultured with or without CMTXL in the co-culture system after being exposed to H/R. Surprisingly, no change in the miR-128-3p level was observed in CMECs co-cultured with CMTXL after H/R, compared with CMECs co-cultured with or without CM (0.75 ± 0.13% vs 1.00 ± 0.25% in the Control group and 0.85 ± 0.16% in the CM group, P > 0.05) (Figure 6A). Next, we measured the levels of two other microRNAs, miR-145-5p [39] and miR-497-5p [40], which were reported to target the mRNA of p70s6k1. No change was observed in miR-497-5p levels under any of the conditions (Figure 6C). However, CMECs co-cultured with CMTXL had a lower level of miR-145-5p as compared with cells co-cultured with or without CM (0.38 ± 0.07% vs 1.00 ± 0.16% in the Control group and 0.95 ± 0.06% in the CM group, P < 0.05) (Figure 6B), confirmed by the in vivo results that the level of miR-145-5p was lower in the rat adult CMEC isolated from the TXL-pretreated hearts (0.32 ± 0.04% vs 1.00 ± 0.11% in the Control group, P < 0.05) (Figure S16F) after myocardial I/R. To investigate whether the downregulation of miR-145-5p was necessary in the beneficial effects of TXL against H/R-induced cell death, miR-145-5p mimics were utilized. CMECs were transfected with miR-145-5p mimics or control, and then co-cultured with CMTXL when subjected to H/R. Western blotting analysis demonstrated that miR-145-5p mimics downregulated the expression of p70s6k1 (0.80 ± 0.20 in CMTXL + Mimic group vs 2.28 ± 0.19 in the CMTXL +NC group, P < 0.05) (Figure 6D-E) and thus decreased its phosphorylation (1.14 ± 0.10 in CMTXL + Mimic group vs 2.34 ± 0.14 in the CMTXL +NC group, P < 0.05) (Figure 6D, F), with a significant reduced eNOS activity (0.52 ± 0.04 in CMTXL + Mimic group vs 1.19 ± 0.15 in the CMTXL +NC group, P < 0.05) (Figure 6D, G). As a consequence of the decrease in the p-eNOS level in CMECs during H/R, being co-cultured with CMTXL did not reduce cell death in CMECs (37.77 ± 0.98% in CMTXL + Mimic group vs 38.90 ± 0.83% in the Control group, P > 0.05) (Figure 6H-I), indicating that the upregulation of miR-145-5p abolished the beneficial effects of CMTXL on CMECs. As shown in Figure S19, transfection of miR-145-5p mimics into CMECs had negligible effects on the cell death of CMECs after H/R (37.57 ± 1.15% vs 35.33 ± 1.00% in the Control group, P > 0.05). These results suggested that the decrease of miR-145-5p in CMECs mediated the protective effect of CMTXL on CMECs during H/R.
CM-derived sEVsmediate eNOSactivation by increasing p70s6k1 in CMECs through reduction of miR-145-5p. A-C) The level of microRNAs targeting the p70s6k1 mRNA in CMECs (n = 4). D-G) The levels of p-70s6k1, t-p70s6k1, p-eNOSand t-eNOSwere detected by Western Blot (n=3). H-I) Dead CMECs in flow cytometry assay (n=3). * P<0.05 vs. Control; # P <0.05 vs. CMTXL+NC. sEVs: small extracellular vesicles; CM: cardiomyocyte; CMTXL: TXL-pretreated CMs; CMTXL+Mimic group: CMECs were transfected with miR-145-5p mimics before being co-cultured with CMTXL and then exposed to H/R; CMTXL+NC: CMECs were transfected with negative control of mimics before being co-cultured with CMTXL and then exposed to H/R. All data are mean ±SEM. Statistical analysis was performed with one-way ANOVA followed by Tukey's test.
SEV transfer of linc-ROR from CMs activates the eNOS pathway by promoting the expression of p70s6k1 in CMECs
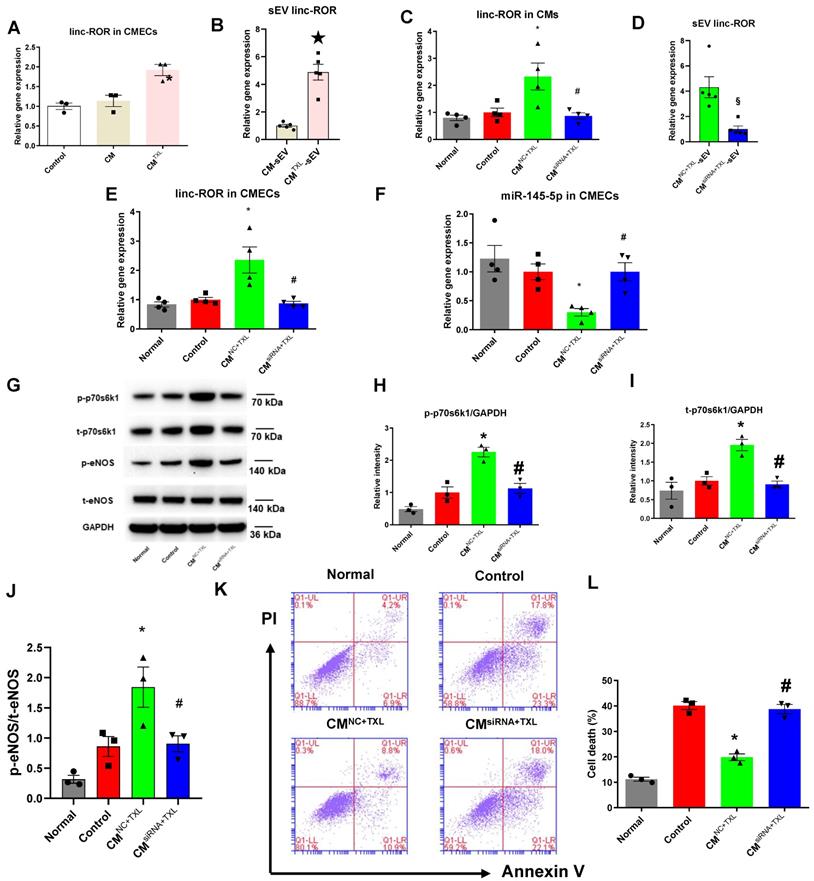
SEVs transfer cargos such as proteins and RNAs (including mRNAs, lncRNAs and microRNAs). Therefore, we hypothesized that the sEVs released from CMTXL shuttled a protective substance (or substances) from CMTXL to CMECs during H/R. Competing endogenous RNAs (ceRNAs) regulate other RNA transcripts by functioning as a kind of “sponge” for microRNAs, sequestering these microRNAs from their normal targets [41, 42]. lncRNAs are important members of the ceRNA family and have been frequently reported to regulate gene expression by sequestering microRNAs [41]. A recent study reported that sEV transfer of one such lncRNA, linc-ROR, promoted cell survival among hepatocellular cancer cells during hypoxia by targeting and downregulating miR-145-5p [43]. Therefore, we measured the level of linc-ROR in CMECs co-cultured with CMTXL under H/R conditions and found a significant augmentation of linc-ROR compared with those cultured with or without CMs (1.92 ± 0.14% vs 1.00 ± 0.08% in the Control group and 1.14 ± 0.14% in the CM group, P < 0.05) (Figure 7A). Furthermore, CMTXL-sEVs contained more linc-ROR than CM-sEVs (4.88 ± 0.57% in the CMTXL-sEV group vs 1.00 ± 0.10% in the CM-sEV group, P < 0.05) (Figure 7B). Similarly, we found that TXL pretreatment could increase the level of linc-ROR (1.96 ± 0.15% vs 1.00 ± 0.11% in the Control group, P < 0.05) (Figure S16G) in the adult rat CMECs isolated from the hearts having been stressed by I/R, and GW4869 treatment could reverse this trend (0.83 ± 0.06% vs 1.00 ± 0.11% in the Control group, P > 0.05) (Figure S16G). To determine whether linc-ROR mediates the protective effect of CMTXL-sEVs on CMECs during H/R, linc-ROR siRNA was used to silence linc-ROR in CMs before being pretreated with TXL (CMsiRNA+TXL) and exposed to H/R. As shown in Figure 7C, transfection of linc-ROR siRNA into CMs significantly decreased the level of linc-ROR in CMs (0.87 ± 0.11% in the CMsiRNA+TXL group vs 2.33 ± 0.50% in the CMNC+TXL group, P < 0.05), leading to a reduction of the level of linc-ROR in sEVs derived from CMTXL(1.00 ± 0.25% in the CMsiRNA+TXL-sEV group vs 4.31 ± 0.83% in the CM NC+TXL-sEV group, P < 0.05) (Figure 7D). The reduced level of sEV linc-ROR from CMTXL led to the unchanged levels of linc-ROR in CMECs when co-cultured with CMTXL (0.87 ± 0.07% in the CMsiRNA+TXL group vs 1.00 ± 0.08% in the Control group, P > 0.05) (Figure 7E). As a result, the level of miR-145-5p in CMECs co-cultured with CMsiRNA+TXL were similar to that in CMECs of the Control group (1.00 ± 0.16% in the CMsiRNA+TXL group vs 1.00 ± 0.14% in the Control group, P > 0.05) (Figure 7F). Correspondingly, the increase of p-p70s6k1 (1.13 ± 0.16 in the CMsiRNA+TXL group vs 1.00 ± 0.17 in the Control group, P > 0.05) (Figure 7G-H), t-p70s6k1 (0.91 ± 0.09 in the CMsiRNA+TXL group vs 1.00 ± 0.11 in the Control group, P > 0.05) (Figure 7G, I) and the ratio of p-eNOS/t-eNOS (0.91 ± 0.13 in the CMsiRNA+TXL group vs 0.86 ± 0.17 in the Control group, P > 0.05) (Figure 7G, J) in CMECs co-cultured with CMTXL was prevented once CMs were transfected with linc-ROR siRNA before being pretreated with TXL. As a consequence of all these changes induced by the transfection of linc-ROR siRNA into CMs, the percentage of dead CMECs in the CMsiRNA+TXL group was similar to that in the Control group (38.73 ± 1.78% in the CMsiRNA+TXL group vs 40.13 ± 1.55% in the Control group, P > 0.05) (Figure 7K-L). Furthermore, coculturing CMECs with CMs transfected with siRNA against linc-ROR did not affect the death percentage of CMECs under H/R conditions (37.83 ± 1.58% vs 40.13 ± 1.55% in the Control group, P > 0.05) (Figure S20A-B). These data demonstrated that CMTXL transferred linc-ROR to CMECs via sEVs, which protected CMECs during H/R.
SEV transfer of linc-ROR from CMs activates the eNOSpathway by promoting the expression of p70s6k1 in CMECs. A) The level of linc-ROR in CMECs co-cultured with or without CM (or CMTXL) (n=3). B) The level of linc-ROR in sEVsderived from TXL-pretreated and non-TXL-pretreated CMs after H/R (n=5). C-K) CMs were transfected with siRNA against linc-ROR or siRNA negative control as indicated, and then pretreated with TXL for 1 h before being co-cultured with CMECs in the co-culture system under normal or H/R condition. RNA and proteins were isolated for RT-PCR or WB. C) The level of linc-ROR in CMs (n=4). D) The level of linc-ROR in sEVsfrom CMs (n=5). E) The level of linc-ROR in CMECs (n=4). F) The level of miR-145-5p in CMECs (n=4). G-J) The levels of p-70s6k1, t-p70s6k1, p-eNOSand t-eNOSwere detected by Western Blot (n=3). K-L) Dead CMECs in flow cytometry assay (n=3). * P<0.05 vs. Control; ☆P <0.05 vs. CM-sEV; # P <0.05 vs. CMNC+TXL; §P<0.05 vs. CMNC+TXL-sEV. sEV: small extracellular vesicles; CM: cardiomyocyte; CMNC+TXL: CMs having been transfected with negative control of siRNA and then pretreated with TXL; CMsiRNA+TXL: CMs having been transfected with linc-ROR siRNA and then pretreated with TXL; CMNC+TXL group: CMECs were co-cultured with CMNC+TXL and subjected to H/R; CMsiRNA+TXL group: CMECs were co-cultured with CMsiRNA+TXL and subjected to H/R. All data are mean ±SEM. Statistical analysis was performed with one-way ANOVA followed by Tukey's test for multiple comparison or unpaired Student T-test for two-group comparison
Discussion
The crosstalk between CMECs and CMs has emerged as a key player in heart development and during the onset and progression of heart diseases [6]. Anatomically, CMECs closely surround CMs to form “perfusion units”, with a small distance between these two types of cells (approximately 1-2 μm) [5, 44]. The close proximity between CMs and CMECs allow them to communicate with each other in a paracrine manner, or in some certain scenarios, even through direct cell-to-cell contact [6]. Despite an increasing number of studies showing that endothelial cells can protect CMs during I/R [7, 8, 9], whether CMs are capable of sending messengers to CMECs and thus blunting myocardial I/R injury remains unknown. Here, we showed that cardiomyocyte-derived sEVs can enhance eNOS activity in CMECs to protect against I/R injury. Our study demonstrated that CMEC derived eNOS has an indispensable role in the protection induced by TXL against I/R injury. This beneficial effect is mediated in part by the transfer of sEV linc-ROR from CMTXL to CMECs. The transfer of linc-ROR to CMECs decreased miR-145-5p levels. Downregulation of miR-145-5p increased the expression of p70s6k1 and augmented its phosphorylation, thereby stimulating the eNOS pathway and promoting NO production, improving microcirculation and attenuating I/R injury. To the best of our knowledge, this is the first study demonstrating that CMs, stimulated by agents such as TXL, protect CMECs via sEVs during H/R (I/R).
In hearts, CMECs outnumber CMs by 3:1, despite the mass ratio of CMEC-to-CM being approximately 0.04 [45]. CMECs have a key role in the microcirculation by providing nutrition and oxygen, and they also release protective substances to impact CMs under certain conditions. It was reported that the dysfunction/death of CMECs leads to no-reflow phenomena after reperfusion [46], and that neuregulin secreted by CMECs protected CMs from I/R injury [7], demonstrating the important role of CMECs in I/R injury. Furthermore, CMECs are more vulnerable to I/R, during which the apoptosis of CMECs precedes that of CMs [47]. Therefore, protecting CMECs during I/R might be a method to reduce I/R injury. Indeed, our data suggest a reciprocal benefit whereby CM-derived sEVs can activate eNOS in CMECs, thus protecting CMECs, as well as CMs, from I/R injury.
Tongxinluo (TXL) is a traditional Chinese medicine that was registered with the China State Food and Drug Administration (CFDA) in 1996 [18]. The major ingredients of TXL are extracted from Radix ginseng, Buthus martensi, Hirudo, Eupolyphaga seusteleophaga, Scolopendra subspinipes, Periostracum cicadae, Radix paeoniae rubra, Semen ziziphispinosae, Lignum dalbergiae odoriferae, Lignum santali albi, and Borneolum syntheticum [48]. Its active constituents include peoniflorin, ginsenoside Rg1, ginsenoside Rb1, jujuboside A, jujuboside B, isoborneol and borneol [49]. Although our previous studies suggested that TXL exerted its cardioprotective effects by activating the eNOS pathway [16, 17], it remained unclear whether CMEC derived eNOS was involved in the TXL-induced cardioprotection. In the present study, disrupting the function of eNOS in CMECs with Triton X-100 largely offset the cardioprotective effect of TXL, indicating that CMEC eNOS is required by TXL to exert its cardioprotective effects. As Triton X-100 is a detergent, the risk exists that it may completely destroy endothelial cell function and microvascular vessel structure, stopping TXL from going through microvascular vessels to impact cardiomyocytes. However, in our study, we found that Triton-X 100 at a concentration of 0.125 % was able to reduce NO production, without affecting the increased phosphorylation of eNOS induced by TXL, suggesting cellular processes were still occurring. The fact that TXL could still facilitate eNOS phosphorylation in the ex vivo hearts treated with Triton-X 100 indicates that Triton-X 100 could cross the microvascular vessels and impact cardiomyocytes, as TXL alone could not directly activate eNOS in CMECs. If cardiac endothelium had been completely destroyed by Triton-X 100 and had not allowed TXL to go through it and reach cardiomyocytes, the p-eNOS could not have been increased in the whole hearts. This was consistent with the results from other groups that low levels of Triton-X 100 pretreatment was effective in abolishing coronary endothelial eNOS-mediated NO production with negligible effects on myocardial function and infarct size [9, 23, 50]. The indispensable role of CMEC eNOS in cardioprotection has also been suggested before. For example, ECs rather than CMs had a key role in eNOS-dependent cardioprotection induced by exercise training [9]. Additionally, our results are consistent with another study reporting that eNOS in ECs plays a critical role in ischemic preconditioning-induced cardioprotection [23].
ENOS is a membrane protein mainly found in plasma membrane caveolae or on the Golgi apparatus [51]. The activity of eNOS can be regulated by a variety of posttranslational modifications, such as acylation, nitrosylation and phosphorylation. Among these, phosphorylation is the major way that influences eNOS enzyme activity [51]. The biological effects of eNOS activation also depend on whether eNOS is coupled or not [52]. When the dihydrobiopetrin (BH2) to tetrahydrobiopetrin (BH4) ratio is increased, eNOS is not coupled to L-arginine oxidation and therefore produces superoxide (SO). SO is subsequently converted to hydrogen peroxide, leading to a burst production of ROS [53]. Whereas the coupled eNOS generates NO in the presence of BH4 by promoting the reduction of oxygen to L-arginine oxidation. It is worth noting that the increase of NO in cells or tissues does not necessarily mean beneficial effects in pathological conditions such as ischemia or I/R [53]. High concentrations of NO can cause cell death [54, 55] and depress contractile function [56], while small concentrations of NO, which can be induced by certain treatments such as antioxidants [57], improve ventricular function [58] and confer protection on the hearts during I/R [59]. Here, we found that TXL could alleviate I/R injury by activating eNOS and thus increasing the level of NO, whereas this TXL-induced protective effects were totally offset by L-NNA, an eNOS inhibitor that inhibited eNOS activity and reduced NO generation. These data suggest that, the augmented eNOS phosphorylation and NO level, which were induced by TXL, could protect the hearts from I/R injury.
In our study, we found a novel pathway of crosstalk between CMs and CMECs during I/R, whereby the indirect activation of eNOS in CMECs is enough to induce cardioprotection. Previously, we have shown that TXL activated eNOS in the heart [16, 17]. However, in the present study our in vitro experiments showed that TXL had a mild but significant protection against H/R-induced injury of CMECs without significantly facilitating the phosphorylation of eNOS, and that an inhibitor of eNOS, L-NNA, failed to abrogate this beneficial effect of TXL. This suggested that TXL treatment on its own does not directly activate eNOS in CMECs. In contrast, the CMEC-CM co-culture model demonstrated that CMECs cultured with CMTXL had a strong protection against H/R injury, dependent of the enhanced eNOS activity. Importantly, the effects were reversed if CM were treated with GW4869, which blocks the generation of sEVs. The role of sEVs in mediating the beneficial effect of TXL was further validated by the result that CMECs treated with CMTXL-sEVs had significantly higher levels of p-eNOS and fewer dead cells. Finally, in endothelium-permeabilized hearts, where brief low levels of Triton X-100 were used to abolish eNOS-mediated NO generation in endothelial cells, the infarct-limiting effect of TXL was largely offset. These data support a model where crosstalk between the preconditioned-CMs and CMECs result in reciprocal protection against I/R injury.
SEVs are double-layer extracellular vesicles between 30-100 nm in size [35]. They can be secreted by most cells and have been found in most body fluids, including saliva, urine and plasma [60]. These nanosized particles are considered to transfer proteins, microRNA and long non-coding RNA cargos through the bloodstream and other body fluids, protecting them from enzymatic degradation [35]. The amounts and contents of sEVs vary depending on the type of parent cell stimuli (e.g. alcohol [61], hypoxia [62], hypoxia/reoxygenation [61]). Regarding intercellular communication, sEVs impact target cells in different ways, such as directly transferring their biochemical contents to target cells after being taken up [35]. In the “perfusion units”, CMECs and CMs are adjacent to each other anatomically in a heart, therefore sEVs released from these two types of cells can significantly influence one another because of the relatively high local concentration of sEVs [5, 44]. Indeed, many studies have demonstrated that sEVs can mediate crosstalk between CMECs and CMs under certain conditions [36, 63]. Of note, sEVs released from endothelial cells treated with ischemic preconditioning protected cardiomyocytes under H/R conditions [8]. In our study, using GW4869, we found it was CMTXL-sEVs that exerted the protective effects of CMTXL on CMECs during H/R. Although GW4869 is an inhibitor of neutral sphingomyelinase as well as sEV biogenesis, GW4869 on its own had no effect on cell death of CMECs and CMs under our conditions, suggesting against an off-target effect induced by inhibiting the neutral sphingomyelinase. In isolation this experiment alone would not be enough to show that sEVs are involved in the phenotype. Taking this into consideration, we isolated sEVs from CMTXL and demonstrated that these vesicles were capable of protecting endothelial cells from H/R injury. Furthermore, we found that sEV-depleted conditioned medium from CMTXL didn't affect the cell death of CMECs during H/R. Another point that needs attention is that the cardioprotective effect of TXL didn't entirely depend on the CMTXL-sEVs, as in vivo administration of GW4869 largely attenuated, but did not completely block the infarct-sparing effect of TXL. This may be due to the direct protection of TXL on CMECs [25] and CMs [18] during H/R (I/R). In summary, our findings may contribute to deeper insights into the crosstalk between CMs and CMECs during I/R.
P70s6k is a member of the AGC family and is regulated by the mammalian target of rapamycin (mTOR) pathway [64]. It was demonstrated that p70s6k plays important roles in various biological processes, such as protein synthesis, glucose homeostasis, and cell survival [65]. Previous studies reported that the p70s6k protein is involved in the cardioprotective RISK pathway and might be phosphorylated by the RISK-related proteins Erk and Akt [66]. There are two subtypes of p70S6K, p70S6K1 and p70S6K2 [67], with the latter being detected at very low levels in adult hearts [68]. eNOS has a cardioprotective role during I/R [9, 23] and although facilitating p70s6k phosphorylation with cysteine-rich, angiogenic inducer 61 or neuropeptide Y activated the eNOS pathway [37, 38], it remains unclear whether activating the p70s6k1/eNOS pathway during I/R is beneficial. Our current and previous studies demonstrated that activation of the eNOS pathway was indispensable for TXL-induced cardioprotective effects [16, 17]. In this study, we found that CMECs co-cultured with CMTXL had significantly higher levels of steady-state p70s6k1 and consequently, its phosphorylated form. Furthermore, knocking down p70s6k1 prevented the increased phosphorylation of eNOS induced by TXL in CMECs during H/R, with the inhibitory effects on cell death being abrogated. This indicated that p70s6k1 is required for the phosphorylation of eNOS in cardioprotection induced by certain agents such as TXL. To the best of our knowledge, this is the first study to unravel the role of the p70s6k1/eNOS pathway in I/R injury.
MicroRNAs, a family of small non-coding single-stranded RNAs, are emerging as robust molecules that modulate the post-transcriptional expression of genes [69]. In our CMEC-CM co-culture model, the upregulation of steady-state levels of p70s6k1 in CMECs co-cultured with CMTXL was correlated with the reduction of miR-145-5p, which targets and represses the translation of p70s6k1 mRNA [43]. Our further experiments showed that transfecting CMECs with miR-145-5p mimics prevented the upregulation of p70s6k1 in CMECs co-cultured with CMTXL. Following the inhibition of the p70s6k1/eNOS pathway, CMTXL no longer protected CMECs from H/R-induced injury in the co-culture system. Therefore, the downregulation of miR-145-5p mediated the beneficial effects of CMTXL on CMECs. This finding is partly consistent with a recent study where suppression of miR-145-5p attenuated acute cerebral ischemia/reperfusion injury in rats [70]. Two other studies also showed that the overexpression (inhibition) of miR-145-5p promoted (repressed) the apoptosis of hepatocellular carcinoma cells in vitro [43, 71]. Previously, we showed that treating CMs with TXL directly could reduce the level of miR-128-3p in CMs after H/R [18]. However, in the current study, we found that the expression of miR-145-5p in CMECs, but not miR-128-3p, was decreased after CMECs took up CMTXL-sEVs (rich in linc-ROR) under the condition of H/R. As the effects of these two studies resulted from two different treatments (linc-ROR-rich CMTXL-sEVs or TXL alone), it is understandable that the trends in the change of miR-128-3p were not the same in these two studies.
lncRNAs are a type of RNA, defined as transcripts with lengths exceeding 200 nucleotides that are not translated into proteins [72]. lncRNAs have important roles in remodeling chromatin and genome architecture, RNA stabilization and transcription regulation, including enhancer-associated activity [73]. lncRNA can function as ceRNAs that sequester specific miRNAs from their mRNA targets, thereby increasing the expression of target mRNAs [73]. In the current study, the downregulation of miR-145-5p in CMECs co-cultured with CMTXL was attributable to the increased level of linc-ROR, a lncRNA transferred from CMTXL via sEVs. This can explain the phenomenon that the beneficial effects of CMTXL-sEVs disappeared after excessive miR-145-5p mimics were transfected into CMECs to overwhelm the binding capacity of linc-ROR. Our finding is in line with a prior study, where the sEV transfer of linc-ROR promoted the survival of liver cancer cells during hypoxic stress via the downregulation of miR-145-5p [43]. Furthermore, linc-ROR was recently been reported to alleviate hypoxia-induced damage by downregulating miR-145-5p in H9c2 cells [74]. Another study reported that linc-ROR attenuated cobalt chloride-induced hypoxia injury by inhibiting miR-145-5p [75].
Our study uncovers a new pathway of crosstalk in regulating cardioprotection during I/R injury. It adds to a growing literature that demonstrates the co-dependency of the “perfusion unit” cells in the heart in response to injury and stress. The major limitation of our study is that, further work will need to be done to understand the mechanism responsible for the increase of sEV linc-ROR from CMTXL during I/R and whether this occurs through the transcription of this gene in CMs or, enhanced packaging of linc-ROR from CMs to sEVs. Another limitation is that mice with cardiomyocyte-specific knockout of linc-ROR (or endothelium-specific knockout of miR-145-5p) were not used in our study to validate the therapeutic values of our study. However, it is clear from this study and others that the emerging role for crosstalk between CMs and CMECs, in physiological adaptations as well as pathological responses, has strong potential for therapeutic targeting.
Abbreviations
AAR: area-at-risk; AMI: acute myocardial infarction; CeRNAs: competing endogenous RNAs; CMECs: cardiac microvascular endothelial cells; CMs: cardiomyocytes; eNOS: endothelial nitric oxide synthase; H/R: hypoxia/reoxygenation; IA: infarcted area; I/R: ischemia/reperfusion; LAD: left anterior descending artery; Linc-ROR: long intergenic non-protein coding RNA, regulator of reprogramming; PBS: phosphate buffered saline; PI: propidium iodide; siRNA: small interfering RNA; TEM: transmission electron microscopy; TTC: 2, 3, 5-triphenyltetrazolium chloride; TXL: tongxinluo.
Supplementary Material
Supplementary figures.
Acknowledgements
This study was supported by grants from the CAMS Innovation Fund for Medical Sciences (CIFMS) (2016-I2M-1-009), the National Key Research and Development Program of China (No. 2017YFC1700503), the National Basic Research Program (973 Program) of China (No.2012CB518602), the National Natural Science Foundation of China (No. 81370223, No. 81573957 and No. 81874461). We thank Shuo Gao from State Key Laboratory of Cardiovascular Disease, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College for providing relevant suggestions on doing experiments related to flow cytometry.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Li Y, Xiong Z, Yan W, Gao E, Cheng H, Wu G. et al. Branched chain amino acids exacerbate myocardial ischemia/reperfusion vulnerability via enhancing GCN2/ATF6/PPAR-alpha pathway-dependent fatty acid oxidation. Theranostics. 2020;10:5623-40
2. Anderson JL, Morrow DA. Acute Myocardial Infarction. N Engl J Med. 2017;376:2053-64
3. Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121-35
4. Davidson SM, Ferdinandy P, Andreadou I, Botker HE, Heusch G, Ibanez B. et al. Multitarget Strategies to Reduce Myocardial Ischemia/Reperfusion Injury: JACC Review Topic of the Week. J Am Coll Cardiol. 2019;73:89-99
5. Brutsaert DL. Cardiac endothelial-myocardial signaling: its role in cardiac growth, contractile performance, and rhythmicity. Physiol Rev. 2003;83:59-115
6. Colliva A, Braga L, Giacca M, Zacchigna S. Endothelial cell-cardiomyocyte crosstalk in heart development and disease. J Physiol. 2019
7. Hedhli N, Huang Q, Kalinowski A, Palmeri M, Hu X, Russell RR. et al. Endothelium-derived neuregulin protects the heart against ischemic injury. Circulation. 2011;123:2254-62
8. Davidson SM, Riquelme JA, Zheng Y, Vicencio JM, Lavandero S, Yellon DM. Endothelial cells release cardioprotective exosomes that may contribute to ischaemic preconditioning. Sci Rep. 2018;8:15885
9. Farah C, Nascimento A, Bolea G, Meyer G, Gayrard S, Lacampagne A. et al. Key role of endothelium in the eNOS-dependent cardioprotection with exercise training. J Mol Cell Cardiol. 2017;102:26-30
10. Lamberts RR, Onderwater G, Hamdani N, Vreden MJ, Steenhuisen J, Eringa EC. et al. Reactive oxygen species-induced stimulation of 5'AMP-activated protein kinase mediates sevoflurane-induced cardioprotection. Circulation. 2009;120:S10-5
11. Liu Y, Paterson M, Baumgardt SL, Irwin MG, Xia Z, Bosnjak ZJ. et al. Vascular endothelial growth factor regulation of endothelial nitric oxide synthase phosphorylation is involved in isoflurane cardiac preconditioning. Cardiovasc Res. 2019;115:168-78
12. Fish JE, Marsden PA. Endothelial nitric oxide synthase: insight into cell-specific gene regulation in the vascular endothelium. Cell Mol Life Sci. 2006;63:144-62
13. Li Y, Hu XX, Fu L, Chen J, Lu LH, Liu X. et al. Time Window Is Important for Adenosine Preventing Cold-induced Injury to the Endothelium. J Cardiovasc Pharmacol. 2017;69:382-8
14. Symons JD, Hu P, Yang Y, Wang X, Zhang QJ, Wende AR. et al. Knockout of insulin receptors in cardiomyocytes attenuates coronary arterial dysfunction induced by pressure overload. Am J Physiol Heart Circ Physiol. 2011;300:H374-81
15. Chen J, Shearer GC, Chen Q, Healy CL, Beyer AJ, Nareddy VB. et al. Omega-3 fatty acids prevent pressure overload-induced cardiac fibrosis through activation of cyclic GMP/protein kinase G signaling in cardiac fibroblasts. Circulation. 2011;123:584-93
16. Cheng YT, Yang YJ, Zhang HT, Qian HY, Zhao JL, Meng XM. et al. Pretreatment with Tongxinluo protects porcine myocardium from ischaemia/reperfusion injury through a nitric oxide related mechanism. Chin Med J (Engl). 2009;122:1529-38
17. Li XD, Yang YJ, Geng YJ, Jin C, Hu FH, Zhao JL. et al. Tongxinluo reduces myocardial no-reflow and ischemia-reperfusion injury by stimulating the phosphorylation of eNOS via the PKA pathway. Am J Physiol Heart Circ Physiol. 2010;299:H1255-61
18. Chen GH, Xu CS, Zhang J, Li Q, Cui HH, Li XD. et al. Inhibition of miR-128-3p by Tongxinluo Protects Human Cardiomyocytes from Ischemia/reperfusion Injury via Upregulation of p70s6k1/p-p70s6k1. Front Pharmacol. 2017;8:775
19. Kang B, Hong J, Xiao J, Zhu X, Ni X, Zhang Y. et al. Involvement of miR-1 in the protective effect of hydrogen sulfide against cardiomyocyte apoptosis induced by ischemia/reperfusion. Mol Biol Rep. 2014;41:6845-53
20. Unal B, Ozcan F, Tuzcu H, Kirac E, Elpek GO, Aslan M. Inhibition of neutral sphingomyelinase decreases elevated levels of nitrative and oxidative stress markers in liver ischemia-reperfusion injury. Redox Rep. 2017;22:147-59
21. Wang X, Gu H, Huang W, Peng J, Li Y, Yang L. et al. Hsp20-Mediated Activation of Exosome Biogenesis in Cardiomyocytes Improves Cardiac Function and Angiogenesis in Diabetic Mice. Diabetes. 2016;65:3111-28
22. Giraldez RR, Panda A, Zweier JL. Endothelial dysfunction does not require loss of endothelial nitric oxide synthase. Am J Physiol Heart Circ Physiol. 2000;278:H2020-7
23. Yang C, Talukder MA, Varadharaj S, Velayutham M, Zweier JL. Early ischaemic preconditioning requires Akt- and PKA-mediated activation of eNOS via serine1176 phosphorylation. Cardiovasc Res. 2013;97:33-43
24. Chen J, Baydoun AR, Xu R, Deng L, Liu X, Zhu W. et al. Lysophosphatidic acid protects mesenchymal stem cells against hypoxia and serum deprivation-induced apoptosis. Stem Cells. 2008;26:135-45
25. Cui H, Li X, Li N, Qi K, Li Q, Jin C. et al. Induction of autophagy by Tongxinluo through the MEK/ERK pathway protects human cardiac microvascular endothelial cells from hypoxia/reoxygenation injury. J Cardiovasc Pharmacol. 2014;64:180-90
26. Bang C, Batkai S, Dangwal S, Gupta SK, Foinquinos A, Holzmann A. et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J Clin Invest. 2014;124:2136-46
27. Luo H, Li X, Li T, Zhao L, He J, Zha L. et al. microRNA-423-3p exosomes derived from cardiac fibroblasts mediates the cardioprotective effects of ischaemic post-conditioning. Cardiovasc Res. 2019;115:1189-204
28. Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006 Chapter 3: 3-22
29. Jin X, Chen Y, Chen H, Fei S, Chen D, Cai X. et al. Evaluation of Tumor-Derived Exosomal miRNA as Potential Diagnostic Biomarkers for Early-Stage Non-Small Cell Lung Cancer Using Next-Generation Sequencing. Clin Cancer Res. 2017;23:5311-9
30. Kroh EM, Parkin RK, Mitchell PS, Tewari M. Analysis of circulating microRNA biomarkers in plasma and serum using quantitative reverse transcription-PCR (qRT-PCR). Methods. 2010;50:298-301
31. Zheng DM, Kitamura T, Ikejima K, Enomoto N, Yamashina S, Suzuki S. et al. Sphingosine 1-phosphate protects rat liver sinusoidal endothelial cells from ethanol-induced apoptosis: Role of intracellular calcium and nitric oxide. Hepatology. 2006;44:1278-87
32. Watanabe T, Suzuki J, Yamawaki H, Sharma VK, Sheu SS, Berk BC. Losartan metabolite EXP3179 activates Akt and endothelial nitric oxide synthase via vascular endothelial growth factor receptor-2 in endothelial cells: angiotensin II type 1 receptor-independent effects of EXP3179. Circulation. 2005;112:1798-805
33. Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am J Physiol Heart Circ Physiol. 2007;293:H1883-91
34. Thery C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R. et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7:1535750
35. Chen GH, Xu J, Yang YJ. Exosomes: promising sacks for treating ischemic heart disease? Am J Physiol Heart Circ Physiol. 2017;313:H508-23
36. Wang X, Huang W, Liu G, Cai W, Millard RW, Wang Y. et al. Cardiomyocytes mediate anti-angiogenesis in type 2 diabetic rats through the exosomal transfer of miR-320 into endothelial cells. J Mol Cell Cardiol. 2014;74:139-50
37. Cheng PW, Wu AT, Lu PJ, Yang YC, Ho WY, Lin HC. et al. Central hypotensive effects of neuropeptide Y are modulated by endothelial nitric oxide synthase after activation by ribosomal protein S6 kinase. Br J Pharmacol. 2012;167:1148-60
38. Hwang S, Lee HJ, Kim G, Won KJ, Park YS, Jo I. CCN1 acutely increases nitric oxide production via integrin alphavbeta3-Akt-S6K-phosphorylation of endothelial nitric oxide synthase at the serine 1177 signaling axis. Free Radic Biol Med. 2015;89:229-40
39. Xu Q, Liu LZ, Qian X, Chen Q, Jiang Y, Li D. et al. MiR-145 directly targets p70S6K1 in cancer cells to inhibit tumor growth and angiogenesis. Nucleic Acids Res. 2012;40:761-74
40. Xu S, Fu GB, Tao Z, OuYang J, Kong F, Jiang BH. et al. MiR-497 decreases cisplatin resistance in ovarian cancer cells by targeting mTOR/P70S6K1. Oncotarget. 2015;6:26457-71
41. Kartha RV, Subramanian S. Competing endogenous RNAs (ceRNAs): new entrants to the intricacies of gene regulation. Front Genet. 2014;5:8
42. Leisegang MS. LET's sponge: How the lncRNA PFL promotes cardiac fibrosis. Theranostics. 2018;8:874-7
43. Takahashi K, Yan IK, Haga H, Patel T. Modulation of hypoxia-signaling pathways by extracellular linc-RoR. J Cell Sci. 2014;127:1585-94
44. Leucker TM, Bienengraeber M, Muravyeva M, Baotic I, Weihrauch D, Brzezinska AK. et al. Endothelial-cardiomyocyte crosstalk enhances pharmacological cardioprotection. J Mol Cell Cardiol. 2011;51:803-11
45. Leucker TM, Bienengraeber M, Muravyeva M, Baotic I, Weihrauch D, Brzezinska AK. et al. Endothelial-cardiomyocyte crosstalk enhances pharmacological cardioprotection. J Mol Cell Cardiol. 2011;51:803-11
46. Heusch G. The Coronary Circulation as a Target of Cardioprotection. Circ Res. 2016;118:1643-58
47. Scarabelli T, Stephanou A, Rayment N, Pasini E, Comini L, Curello S. et al. Apoptosis of endothelial cells precedes myocyte cell apoptosis in ischemia/reperfusion injury. Circulation. 2001;104:253-6
48. Karalliedde LD, Kappagoda CT. The challenge of traditional Chinese medicines for allopathic practitioners. Am J Physiol Heart Circ Physiol. 2009;297:H1967-9
49. Chen WQ, Zhong L, Zhang L, Ji XP, Zhao YX, Zhang C. et al. Chinese medicine tongxinluo significantly lowers serum lipid levels and stabilizes vulnerable plaques in a rabbit model. J Ethnopharmacol. 2009;124:103-10
50. Leucker TM, Ge ZD, Procknow J, Liu Y, Shi Y, Bienengraeber M. et al. Impairment of endothelial-myocardial interaction increases the susceptibility of cardiomyocytes to ischemia/reperfusion injury. Plos One. 2013;8:e70088
51. Heiss EH, Dirsch VM. Regulation of eNOS enzyme activity by posttranslational modification. Curr Pharm Des. 2014;20:3503-13
52. Chen Q, Kim EE, Elio K, Zambrano C, Krass S, Teng JC. et al. The role of tetrahydrobiopterin and dihydrobiopterin in ischemia/reperfusion injury when given at reperfusion. Adv Pharmacol Sci. 2010;2010:963914
53. Heusch G. Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ Res. 2015;116:674-99
54. Taimor G, Hofstaetter B, Piper HM. Apoptosis induction by nitric oxide in adult cardiomyocytes via cGMP-signaling and its impairment after simulated ischemia. Cardiovasc Res. 2000;45:588-94
55. Hofstaetter B, Taimor G, Inserte J, Garcia-Dorado D, Piper HM. Inhibition of apoptotic responses after ischemic stress in isolated hearts and cardiomyocytes. Basic Res Cardiol. 2002;97:479-88
56. Heinzel FR, Gres P, Boengler K, Duschin A, Konietzka I, Rassaf T. et al. Inducible nitric oxide synthase expression and cardiomyocyte dysfunction during sustained moderate ischemia in pigs. Circ Res. 2008;103:1120-7
57. Wang T, Mao X, Li H, Qiao S, Xu A, Wang J. et al. N-Acetylcysteine and allopurinol up-regulated the Jak/STAT3 and PI3K/Akt pathways via adiponectin and attenuated myocardial postischemic injury in diabetes. Free Radic Biol Med. 2013;63:291-303
58. Rassaf T, Poll LW, Brouzos P, Lauer T, Totzeck M, Kleinbongard P. et al. Positive effects of nitric oxide on left ventricular function in humans. Eur Heart J. 2006;27:1699-705
59. Rassaf T, Totzeck M, Hendgen-Cotta UB, Shiva S, Heusch G, Kelm M. Circulating nitrite contributes to cardioprotection by remote ischemic preconditioning. Circ Res. 2014;114:1601-10
60. Yellon DM, Davidson SM. Exosomes: nanoparticles involved in cardioprotection? Circ Res. 2014;114:325-32
61. Malik ZA, Kott KS, Poe AJ, Kuo T, Chen L, Ferrara KW. et al. Cardiac myocyte exosomes: stability, HSP60, and proteomics. Am J Physiol Heart Circ Physiol. 2013;304:H954-65
62. Zhu LP, Tian T, Wang JY, He JN, Chen T, Pan M. et al. Hypoxia-elicited mesenchymal stem cell-derived exosomes facilitates cardiac repair through miR-125b-mediated prevention of cell death in myocardial infarction. Theranostics. 2018;8:6163-77
63. Garcia NA, Moncayo-Arlandi J, Sepulveda P, Diez-Juan A. Cardiomyocyte exosomes regulate glycolytic flux in endothelium by direct transfer of GLUT transporters and glycolytic enzymes. Cardiovasc Res. 2016;109:397-408
64. Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010;11:9-22
65. Fenton TR, Gout IT. Functions and regulation of the 70kDa ribosomal S6 kinases. Int J Biochem Cell Biol. 2011;43:47-59
66. Heusch G. Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ Res. 2015;116:674-99
67. Shima H, Pende M, Chen Y, Fumagalli S, Thomas G, Kozma SC. Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. Embo J. 1998;17:6649-59
68. Tseng YT, Yano N, Rojan A, Stabila JP, McGonnigal BG, Ianus V. et al. Ontogeny of phosphoinositide 3-kinase signaling in developing heart: effect of acute beta-adrenergic stimulation. Am J Physiol Heart Circ Physiol. 2005;289:H1834-42
69. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281-97
70. Xie X, Peng L, Zhu J, Zhou Y, Li L, Chen Y. et al. miR-145-5p/Nurr1/TNF-alpha Signaling-Induced Microglia Activation Regulates Neuron Injury of Acute Cerebral Ischemic/Reperfusion in Rats. Front Mol Neurosci. 2017;10:383
71. Wang B, Dong W, Li X. miR-145-5p Acts as a Novel Tumor Suppressor in Hepatocellular Carcinoma Through Targeting RAB18. Technol Cancer Res Treat. 2019;18:1078117837
72. Perkel JM. Visiting "noncodarnia". Biotechniques. 2013;54:301 303-4
73. Ransohoff JD, Wei Y, Khavari PA. The functions and unique features of long intergenic non-coding RNA. Nat Rev Mol Cell Biol. 2018;19:143-57
74. Wang P, Yuan Y. LncRNA-ROR alleviates hypoxia-triggered damages by downregulating miR-145 in rat cardiomyocytes H9c2 cells. J Cell Physiol. 2019
75. Ge H, Liu J, Liu F, Sun Y, Yang R. Long non-coding RNA ROR mitigates cobalt chloride-induced hypoxia injury through regulation of miR-145. Artif Cells Nanomed Biotechnol. 2019;47:2221-9
Author contact
![]() Corresponding author: Yuejin Yang, MD, PhD. E-mail: yangyuejinorg or yangyjfwcom. Postal address: State Key Laboratory of Cardiovascular Disease, Department of Cardiology, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100037, China. Phone number: 86-10-88398760
Corresponding author: Yuejin Yang, MD, PhD. E-mail: yangyuejinorg or yangyjfwcom. Postal address: State Key Laboratory of Cardiovascular Disease, Department of Cardiology, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100037, China. Phone number: 86-10-88398760