Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(3):1197-1212. doi:10.7150/thno.39320 This issue Cite
Research Paper
Smooth muscle SIRT1 reprograms endothelial cells to suppress angiogenesis after ischemia
Yong-Qing Dou1,4, Peng Kong1, Chang-Lin Li1, Hong-Xing Sun1, Wei-Wei Li1, Yuan Yu1, Lei Nie1, Li-Li Zhao1, Sui-Bing Miao1, Xiao-Kun Li1, Chen Dong1, Jin-Wen Zhang2, Yang Liu2, Xiao-Xia Huo3, Kui Chi2, Xiang Gao2, Ning Zhang1, Lin Weng1, Hongyuan Yang5, Fan Zhang1, Mei Han1 ![]()
1. Department of Biochemistry and Molecular Biology, College of Basic Medicine, Key Laboratory of Medical Biotechnology of Hebei Province, Hebei Medical University, Shijiazhuang, China.
2. Department of Vascular Surgery of Second Hospital, Hebei Medical University, Shijiazhuang, China.
3. Department of Gastroenterology of Second Hospital, Hebei Medical University, Shijiazhuang, China.
4. Key Laboratory of Integrative Medicine on Liver-kidney patterns of Hebei Province, College of Integrated Chinese and Western Medicine, Hebei University of Chinese Medicine, Shijiazhuang, China.
5. School of Biotechnology and Biomolecular Sciences, University of New South Wales, Sydney, Australia.
Received 2019-8-14; Accepted 2019-10-28; Published 2020-1-1
Abstract

Objective: Vascular smooth muscle cells (VSMCs) undergo the phenotypic changes from contractile to synthetic state during vascular remodeling after ischemia. SIRT1 protects against stress-induced vascular remodeling via maintaining VSMC differentiated phenotype. However, the effect of smooth muscle SIRT1 on the functions of endothelial cells (ECs) has not been well clarified. Here, we explored the role of smooth muscle SIRT1 in endothelial angiogenesis after ischemia and the underlying mechanisms.
Methods: We performed a femoral artery ligation model using VSMC specific human SIRT1 transgenic (SIRT1-Tg) and knockout (KO) mice. Angiogenesis was assessed in in vivo by quantification of the total number of capillaries, wound healing and matrigel plug assays, and in vitro ECs by tube formation, proliferation and migration assays. The interaction of HIF1α with circRNA was examined by using RNA immunoprecipitation, RNA pull-down and in situ hybridization assays.
Results: The blood flow recovery was significantly attenuated in SIRT1-Tg mice, and markedly improved in SIRT1-Tg mice treated with SIRT1 inhibitor EX527 and in SIRT1-KO mice. The density of capillaries significantly decreased in the ischemic gastrocnemius of SIRT1-Tg mice compared with SIRT1-KO and WT mice, with reduced expression of VEGFA, which resulted in decreased number of arterioles. We identified that the phenotypic switching of SIRT1-Tg VSMCs was attenuated in response to hypoxia, with high levels of contractile proteins and reduced expression of the synthetic markers and NG2, compared with SIRT1-KO and WT VSMCs. Mechanistically, SIRT1-Tg VSMCs inhibited endothelial angiogenic activity induced by hypoxia via the exosome cZFP609. The cZFP609 was delivered into ECs, and detained HIF1α in the cytoplasm via its interaction with HIF1α, thereby inhibiting VEGFA expression and endothelial angiogenic functions. Meantime, the high cZFP609 expression was observed in the plasma of the patients with atherosclerotic or diabetic lower extremity peripheral artery disease, associated with reduced ankle-brachial index. Knockdown of cZFP609 improved blood flow recovery after hindlimb ischemia in SIRT1-Tg mice.
Conclusions: Our findings demonstrate that SIRT1 may impair the plasticity of VSMCs. cZFP609 mediates VSMCs to reprogram endothelial functions, and serves as a valuable indicator to assess the prognosis and clinical outcomes of ischemic diseases.
Keywords: cZFP609, angiogenesis, vascular smooth muscle cells, ischemia, exosomes.
Introduction
The progressive occlusion in arterial conduits initiates a series of events during atherosclerotic or diabetic lower extremity peripheral artery disease (PAD), including pre-existing collateral arteries into functional conduit vessels proximally and ischemic angiogenesis distally [1]. Arteriole formation distally involves endothelial cell (EC) activation, basal membrane degradation, leukocyte invasion, and proliferation of vascular cells [2]. Vascular smooth muscle cells (VSMCs) undergo the phenotypic changes from contractile to synthetic state possibly enabling the mobilization, proliferation during arteriogenesis [2], and are capable to differentiate into pericytes to coat around capillaries during angiogenesis [3]. The cytokines and growth factors induce EC proliferation, and new capillary formation that occurs by sprouting or by longitudinal splitting (intussusception) of existing vessels [4, 5]. The ability of ECs to proliferate and to secrete growth factors, such as VEGF and PDGF, is crucial for the new vascular network development and subsequent arterialization via recruitment of mural cells.
The key factor regulating VEGF production in ischemic tissues is HIF1α. The regulation of HIF1α activity occurs in large part at post-translational modifications, resulting in its stabilization, nuclear translocation, DNA binding activity, and proper transcriptional activity [6, 7]. Noncoding RNAs provide novel post-transcriptional/-translation mechanisms of HIF1α regulation, and have been implicated in hypoxia signaling modulation [8, 9]. Despite recent advances in the field of angiogenesis, many details of this complex process have not been elucidated so far, especially about the roles (largely ignored) of VSMCs in the progression of angiogenesis after ischemia.
The silent information regulator 1 (SIRT1), a NAD+-dependent histone deacetylase, is highly expressed in the vasculature [10] and is an important modulator of cardiovascular functions in health and disease. Studies indicate that SIRT1 protects against stress-induced vascular remodeling [11, 12], such as neointimal formation [11], aortic stiffness [13], dissection [14] and atherosclerosis in mice [15, 16]. The activation of SIRT1 prevents senescence of vascular ECs and VSMCs [17, 18], suggesting a critical role of SIRT1 in vascular homeostasis and vascular diseases. In fact, SIRT1 prevents stress-induced vascular remodeling largely by maintaining the differentiated phenotype of VSMCs. It has been known that SIRT1 controls the angiogenic activity of ECs, and is required for ischemia-induced neovascularization [10]. Controversially, other reports have shown that SIRT1-mediated deacetylation inactivates HIF1α in hypoxic mouse tissues [7], and thus has negative effects on tumor growth and angiogenesis [7]. Reduced SIRT1 expression may be correlated with enhanced angiogenesis [19, 20]. Although there are extensive studies on the vascular protective effects of SIRT1, it is unclear whether continuous or excessive activation of SIRT1 is available for all atherosclerotic vascular diseases, such as atherosclerotic lower extremity PAD. Furthermore, the effect of smooth muscle SIRT1 on the endothelial angiogenic activity has not been well clarified.
In the present study, we performed a femoral artery ligation model using VSMC specific human SIRT1 transgenic (SIRT1-Tg) and SIRT1 knockout (SIRT1-KO) mice, and showed that SIRT1-Tg mice displayed delayed blood flow recovery and reduced density of capillaries and arterioles in the ischemic gastrocnemius. cZFP609 was expressed and released by SIRT1-Tg VSMCs via exosomes. Furthermore, the cZFP609 reprogramed ECs to suppress angiogenesis via blockade of HIF1α nuclear translocation and VEGFA expression in response to hypoxia. Thus, cZFP609 may act as a novel indicator to assess the prognosis and clinical outcomes of ischemic diseases.
Materials and Methods
All the data are available in the article and Supplementary Files, or available from the authors upon request.
Animals and ethics statement
All animal procedures conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication, 8th Edition, 2011) and were approved by the Institutional Animal Care and Use Committee of Hebei Medical University. VSMC specific SIRT1- transgenic (SIRT1-Tg) mice [11] and knockout (SIRT1-KO) mice [18] were kindly gifted by Dr. De-Pei Liu and Hou-Zao Chen of Chinese Academy of Medical Sciences and Peking Union Medical College, China. All mice were housed in a specific pathogen-free environment under a 12 h /12 h light-dark cycle and fed rodent diet ad libitum.
Hindlimb ischemia model
We used a previously described mice model of unilateral hindlimb ischemia [1]. In brief, the mice (male, 12-14 week-old) were anesthetized with a mixture of oxygen and 1.125% isoflurane. Left unilateral femoral artery occlusion was performed by double ligation of the left superficial femoral artery proximal and distal to the deep femoral artery. Animal numbers are stated with the different experimental results. A sham operation was performed on the contralateral right leg. At the same time, for activation or inhibition of SIRT1, the mice were treated with SIRT1 agonist RSV (2 mg/kg/d, J&K) or inhibitor EX527 (2 mg/kg/d, Cayman) by intraperitoneal injection. For intramuscular injection of AAV [21], 3 weeks before SIRT1-Tg mice were performed left unilateral femoral artery occlusion, left gastrocnemius muscles were injected with 1×1012 vg /mL AAV9-shRNA-cZFP609 (adeno-associated virus-9 short-hairpin RNA; shcZFP609, HANBIO) and AAV9-shRNA-NC (shNC, HANBIO) by multi-point injection, the injection volume was 10 μL per point, 5 points in total. On day 14 after surgery, mice were euthanized by injection of pentobarbital (80 mg/kg IP) [22], the gastrocnemius tissues were harvested and placed in FSC 22 Frozen Section Media (Leica, 3801480), and three random 10 μm frozen sections of the gastrocnemius muscle per animal were used for immunofluorescence analyses.
Laser Doppler Perfusion Imaging
For laser Doppler perfusion imaging, the animals were anesthetized and measured under a Periscan PSI imager (Perimed, Sweden). The readings of 3 individual measurements per mouse were averaged for each time point. Measurements were performed just before and after the operation on day 0, 3, 7 and 14 with the last measurement constituting the main end point. Flow ratios of the left / right leg were compared between the treatment groups.
Human blood sample collection
Human blood sample collection was approved by the Human Ethics Committee, Hebei Medical University Second Hospital and the study was conducted in accordance with the principles of Good Clinical Practice and the Declaration of Helsinki. Written consent from participants or their guardians was obtained. Plasma samples were collected from the initial admission blood draws of those patients who attended the Department of Vascular surgery. Eligible patients were with lower extremity PAD, which were enrolled with an abnormal ankle-brachial index (ABI)≤0.80 at screening. Prior to blood collection, we carefully examined the medical history of control subjects and lower extremity PAD, which having one or more of the following criteria was excluded: prior surgery or trauma within one month prior to admission; autoimmune disease; severe infection; and malignancy. The present study recruited 19 patients with lower extremity PAD and 30 control subjects.
Wound healing assay
The male SIRT1-Tg, SIRT1-KO or WT mice (12-14 week-old) were anesthetized with a mixture of oxygen and 1.125% isoflurane. Dermal wound healing assays were done by creating in the back skin of 6 mm punch wounds without injuring the underlying muscle. Wound regions were photographed using a NIKON camera (D 7000) on day 0, 1, 3, 5, and 7. Wound area was calculated using Image J software. Wound sizes at different time points were expressed as percentage of the wound area on day 0.
In vivo Matrigel plug assay
Growth factor-reduced Matrigel (0.5 mL, Fisher Scientific) with or without VEGFA (100 ng/mL, Cell Signaling Technology) were injected subcutaneously into opposite iliac regions of SIRT1-Tg, SIRT1-KO or WT mice. After injection for 7 d, matrigel plugs were removed from the mice under anesthetization and used to EC invasion assay. Quantification of blood vessels was performed on three sections per plug to define CD31.
Immunofluorescence analysis
Immunofluorescence staining was performed on acetone-fixed EC monolayers or 10-μm-thick frozen sections. Sections were blocked using 5% normal goat serum in TBS for 30 min and then incubated with primary antibodies against CD31 (BD Biosciences), HIF1α (GeneTex) or α-SMA (Abcam) at 4 °C overnight, and isotype matched controls. Sections were washed 3 times with TBS and incubated with fluorescein-conjugated secondary antibodies (Alexa Fluor® 555 or Alexa Fluor 488, Invitrogen) for 1 h at room temperature. Nuclei were detected by DAPI (Antifade Mountant with DAPI, Thermofisher). Images were acquired using a fluorescence microscope (Olympus, Japan) or a Confocal Laser Scanning Microscope Systems (Leica). Digitized images were analyzed with Image J or software program LAS AF Lite. The total number of capillaries labeled with CD31 was counted on 5 random optical fields for each section. The relative tubule length and number of branches were expressed in capillaries per square millimeters.
Plasmid construction
To construct cZFP609 expression plasmids, mouse cZFP609 cDNA was synthesized by Sangon Biotech (Shanghai, China) and cloned into pcD-ciR vector (Geneseed Biotech, Guangzhou, China). The pcD-ciR vector contained a front circular frame and a back circular frame. The HIF1α luciferase reporter vector contained two tandem repeats of HIF1α binding site (ACGTG) of the mouse PDGFB gene, was inserted into pGL3-Promoter vector (Promega). Transfection was carried out using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
Cell culture and treatment
The VSMCs of WT, SIRT1-Tg and SIRT1-KO mice were isolated from aortas with 1% collagenase, and cultured in low glucose Dulbecco's modified Eagle's medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (FBS, Gibco). Passage 4 to 10 cells were used in the experiments. The primary ECs of mice were isolated from the lungs of 4-week-old C57BL/6J mice by two rounds of immunoselection with CD31 (BD Biosciences) and CD102 (BD Biosciences)-conjugated magnetic beads according to a previously described procedure [23], and cultured in Endothelial Cell Medium (ECM, Sciencell) containing 5% FBS and endothelial cell growth supplements (ECGS). Passage 5 to 10 cells were used in the experiments. Human umbilical vein endothelial cells (HUVECs) were obtained from Sciencell and cultured on gelatin-coated plates in ECM supplemented with 5% FBS and ECGS, and used to evaluate endothelial angiogenic functions. Before hypoxia, all of the above cells were incubated in serum-free medium for 24 h, and then under hypoxic (1% O2) or normoxic (21% O2) conditions for another 24 h. To block the degradation of HIF1α, the cells were pretreated with DMOG (1 mM, Sigma) for 4 h before the collection of cells. For activation or inhibition of SIRT1, the cells were pretreated with resveratrol (RSV, 25 μM, J&K) or EX527 (20 μM, Cayman) for 4 h before hypoxia. To examine the effect of SIRT1-Tg VSMCs on EC function, the ECs were incubated with the hypoxia-induced VSMC conditioned culture media for 24 h.
Cell proliferation and migration assays
ECs or HUVECs proliferation assay was performed as described in the product (2750, Millipore). The cells were labeled for 12 h with BrdU. OD readings were done at 450 nm.
The migration of ECs or HUVECs was evaluated by performing a cell-wounding assay. Cells grown to 100% confluence on glass slides were scraped off the slides with a cell scraper to create a 3-mm-wide wound and were then incubated at 37 ℃ for 24 h under hypoxia. The cells were fixed with methanol and stained with hexamethylpararosaniline. The migration activity of the cells was expressed as the number of cells that migrated into the wound area in each field.
In vitro tube formation assay
Cells were starved overnight in 0.5% FBS, and then detached with trypsin, seeded at a density of 30000 cells per well in a 48-well plate containing reduced-growth factor matrigel (BD Biosciences). Cells were in the conditioned culture media or in media containing 0.5% FBS, VEGFA (100 ng/mL). Tube length was quantified after 24 h by measuring the cumulative tube length in five random microscopic fields. The mean value of 10 cumulative total lengths per well represents an experimental point. The relative tubule length and number of branches were assessed using Image-Pro Plus.
Exosome Purification
The VSMCs were incubated in serum-free low glucose DMEM for 24 h. The conditioned medium was harvested, and centrifuged at 3,000 g for 15 min at 4℃. The supernatants were then passed through a 0.22 μm (Millipore) filter to remove cellular debris and large vesicles. The clarified medium was mixed with GSTM solution (Geneseed Biotech) and incubated overnight at 4℃, and then centrifuged twice at 5,000 g. The pelleted exosomes were resuspended in approximately 100 μL of PBS and subjected to subsequent experiments.
Small interfering RNA (siRNA) transfection
The siRNA duplexes targeting mouse cZFP609 (si-cZFP609), 5'- GUCUGAAAAGCAAUGAUGUTT-3' and 5'- ACAUCAUUGCUUUUCAGACTT-3' were obtained from GenePharma. Scrambled siRNA (si-Con) 5'-UUCUCCGAACGUGUCACGUTT-3' and 5'-ACGUGACACGUUCGGAGAATT-3' served as a negative control. The siRNAs were transiently transfected into VSMCs using Lipofectamine® RNAiMAX Transfection Reagent (Invitrogen) according to the manufacturer's protocol.
RNA isolation and quantitative reverse transcription-PCR (qRT-PCR)
Total RNAs from cell lysates were isolated using TRIzol reagent (Life Technologies). The nuclear and cytoplasmic fractions were extracted using Minute TM Cytoplasmic and Nuclear Extraction Kit (Invent Biotechnologies). To quantify the amount of mRNA and circRNA, cDNAs were synthesized using the M-MLV First Strand Kit (Life Technologies), and quantitative PCRs were performed using SYBR Green qPCR SuperMix-UDG (Life Technologies). For quantification, all RNA expression was normalized to the amount of Tubulin using the 2-ΔΔCt method.
Western blot analysis
RIPA buffer was used to lyse cells (50 mM Tris-Cl, pH 7.5, 1% NP-40, 0.5% Na-deoxycholate, 150 mM NaCl supplemented with complete proteinase inhibitor, Roche Applied Sciences) and mice muscles (50 mM Tris-Cl, pH 7.5, 1% NP-40, 0.5% Na-deoxycholate, 0.1% SDS, 1 mM EDTA, 150 mM NaCl supplemented with complete proteinase inhibitor). Equal amounts of protein (30~60 μg) were separated by 10% SDS-PAGE, and electrotransfered to a PVDF membrane. Membranes were blocked with 5% milk in TBS for 1 h at room temperature, and incubated with primary antibodies against HIF1α (GeneTex), VEGFA (Arigo), β-actin (Cell Signaling Technology), Lamin A/C (Cell Signaling Technology), Tubulin (Cell Signaling Technology), α-SMA, SM22α, MMP2, MMP9, OPN and NG2 at 4℃ overnight, and then with the HRP-conjugated secondary antibody (Abcam) for 1 h. The blots were evaluated with GE ImageQuant™ LAS 4000 detection system. The protein bands of interest were quantified using Image Pro Plus 6.0 software, and the integrated signal densities were normalized to β-actin or Tubulin (the loading control).
Fluorescence in situ hybridization (FISH)
The ECs were washed in PBS and fixed in 4% paraformaldehyde for 10 min and permeabilized overnight in 70% ethanol. Then the cells were rehydrated for 10 min in 50% formamide and 2×SSC. In case of immunofluorescence, cells were blocked with 10% BSA in PBS for 2 h followed by incubation with HIF1α (GeneTex) (1:200) in PBS treated with DEPC (VETEC) at 4 ℃ overnight. After washing three times in PBS, cells were incubated with secondary antibody (Alexa Fluor 488, Invitrogen).
For FISH, the cells were incubated using specific probes of cZFP609. Hybridization was performed using fluorescence-labeled probes in hybridization buffer by incubation at 55 ℃ for 1 h. After stringent washing with SSC buffer, cell nuclei were counterstained with DAPI (Invitrogen). Images were acquired using a Confocal Laser Scanning Microscope Systems (Leica).
RNA immunoprecipitationn assay (RIP)
The ECs were washed in ice-cold PBS, lysed in lysis buffer (20 mM Tris-HCl, pH 7.0, 150 mM NaCl, 0.5% NP-40, 5 mM EDTA, with freshly added 1 mM DTT, 1 mM PMSF, and 2 U/μL RNase inhibitor), and then incubated with 5 μg the primary antibody at 4 ℃ for 2 h. 50 μL Dynabeads Protein G (Life technology) was added to each sample, and the mixtures were incubated at 4 ℃ for 4 h. The pellets were washed with PBS and resuspended in 1 mL TRizol Reagent (Invitrogen). The precipitated RNA in the aqueous solution was subject to qRT-PCR analysis to demonstrate the presence of the binding products using respective primers [24].
RNA pull-down assay
The ECs were washed in ice-cold phosphate-buffered saline, lysed in 500 μL lysis buffer (20 mM Tris-HCl, pH 7.0, 150 mM NaCl, 0.5% NP-40, 5 mM EDTA, with freshly added 1 mM DTT, 1 mM PMSF, and 2 U/μL RNase inhibitor), and then incubated with 3 μg biotinylated DNA oligo probes against endogenous or ectopically expressed cZFP609 at 4 ℃ for 2 h. A total of 50 μL Dynabeads™ MyOne™ Streptavidin C1 magnetic beads (Invitrogen) were added to each binding reaction and further incubated at 4 ℃ for 2 h. The beads were washed briefly with lysis buffer for three times. The bound proteins in the pull-down materials were analyzed by western blot [25].
Chromatin immunoprecipitation assay (ChIP)
The VSMCs or ECs were incubated at 20% or 1% O2 for 24 h and were fixed in 1% formaldehyde for 10 min to cross link proteins with DNA. The cross-linked chromatin was then prepared and sonicated to an average size of 400-600 bp. The samples were precleared with Dynabeads Protein G (Life technology) for 30 min at 4 ℃. The DNA fragments were immunoprecipitated overnight at 4 ℃ with the HIF1α (GeneTex) and normal rabbit IgG (Santa Cruz) antibodies. The precipitated DNA was recovered via phenol/chloroform extraction, and the HIF1α binding site (HBS) was amplified by qPCR. Each experiment was replicated at least three times.
Statistics
All statistical analyses were performed with the SPSS 21.0 software. The data are presented as means ± SEM. Two groups were compared by Student's T tests. Differences among groups were analyzed with one-way analysis of variance (ANOVA). Blood flow recovery imaging of the mouse foot and wound healing assay were analyzed with ANOVA of repeated measurement data. Spearman rank correlation test was performed to determine the relationship between human plasma cZNF609 level and ABI. Data were analyzed using IBM SPSS Statistics 21.0 (IBM Corporation, Armonk, NY, USA). For all statistical comparisons, P < 0.05 was considered significant.
Results
Blood flow recovery is delayed after hindlimb ischemia in SIRT1-Tg mice
To validate the effects of smooth muscle SIRT1 on angiogenesis [7, 10, 26], we first performed the hindlimb ischemia on VSMC specific SIRT1-Tg, SIRT1-KO and wild type (WT) mice by ligation of the left femoral artery. Using a laser Doppler blood flowmeter, we found that the blood flow recovery was significantly attenuated in SIRT1-Tg mice (Figure 1A-B). Conversely, the blood flow recovery was markedly improved, which displayed increased blood flow perfusion on day 3 after ischemia in SIRT1-KO mice (Figure. 1C-D). To determine the causal relationship between SIRT1 and blood flow perfusion, we examined the effect of the activation or inhibition of SIRT1 on blood flow recovery after ischemia. We showed that delayed blood flow recovery was markedly reversed in SIRT1-Tg mice treated with SIRT1 inhibitor EX527 compared with untreated control (Figure 1A-B). Conversely, SIRT1 agonist resveratrol (RSV) resulted in a reduced blood flow perfusion in the ischemic hindlimbs of WT mice, similar to SIRT1-Tg mice (Figure 1E-F). New capillary formation is an essential prerequisite for arteriole formation in ischemia distally. Then, we examined the capillary density in the ischemic gastrocnemius using immunofluorescence staining of the frozen tissue sections. We showed that the number of capillaries with CD31 positive ECs reduced in ischemic SIRT1-Tg tissues, accompanied by decrease in arteriole density (Figure 1G-I). In contrast, increased density of capillaries and arterioles was observed in the ischemic gastrocnemius of SIRT1-KO mice (Figure 1G-I). We speculated that the angiogenic dysfunction resulted in delayed blood flow recovery after ischemia in SIRT1-Tg mice.
Blood flow recovery is delayed after hindlimb ischemia in SIRT1-Tg mice. (A-F) Representative laser Doppler perfusion images at indicated time point after hindlimb ischemia. (A and B) WT, SIRT1-Tg and SIRT1-Tg mice treated with SIRTl inhibitor EX527 (n= 12). (C and D) WT and SIRT1-KO mice (n= 8 per group). (E and F) WT mice treated with DMSO or SIRTl agonist RSV after hindlimb ischemia (n= 8 per group), Laser Doppler perfusion at various time points was expressed as a ratio of flow between ischemic (L) and sham (R) limbs (L/R). (G) Representative immunofluorescence for CD31 (Scale bars = 100 μm) and α-SMA-positive cells (Scale bars = 20 μm) in the gastrocnemius tissue. (H) Immunostaining of CD31-positive cells represented the relative capillary density. (I) The density of arteriole was expressed as the quantity of arterioles per mm2. Data represent mean±SEM. Repeated Measures ANOVA, one-way ANOVA or student's t-test:*P<0.05, **P<0.01 versus the corresponding control.
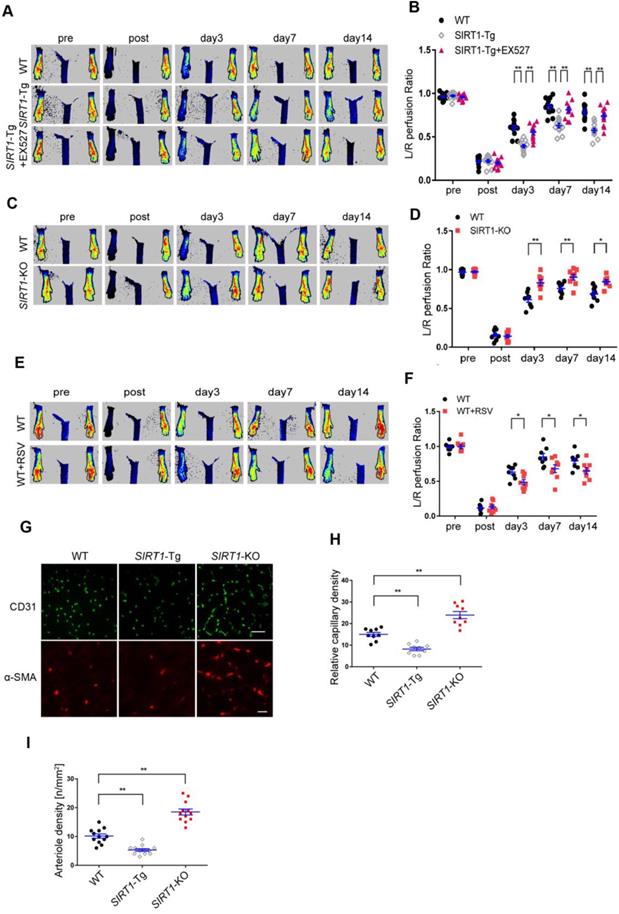
Angiogenesis is impaired in SIRT1-Tg mice
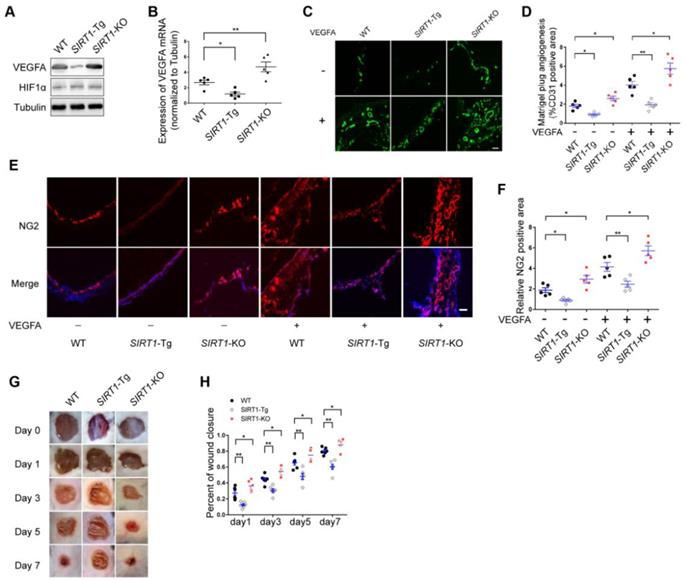
Arteriole formation depends on angiogenesis in ischemic distal region via mural cell and SMC coverage [3]. Angiogenesis is largely driven by VEGFA production in response to hypoxia [27]. To verify the angiogenic dysfunction in SIRT1-Tg mice, we first detected the expression of VEGFA in the ischemic gastrocnemius of SIRT1-Tg mice using Western blot and qRT-PCR. We showed decreased protein and mRNA level of VEGFA in SIRT1-Tg ischemic tissues (Figure 2A-B). Furthermore, this decrease was eliminated in SIRT1-KO mice under the same conditions. However, the expression of HIF1α was no difference in the ischemic tissues among WT, SIRT1-Tg and SIRT1-KO mice. To further confirm the inhibitory effect of smooth muscle SIRT1 on angiogenesis, we performed a matrigel plug implantation assay to evaluate angiogenesis in vivo. We found that the number of the capillaries and mural cells invaded into the matrigel with or without VEGFA was reduced in SIRT1-Tg mice, and increased in SIRT1-KO mice (Figure 2C-F). The inhibitory effect of smooth muscle SIRT1 on angiogenesis was also verified by a puncture wound model in SIRT1-Tg and -KO mice, which revealed a delayed wound closure in SIRT1-Tg mice (Figure 2G and H). These findings imply that VSMC SIRT1 may disturb angiogenesis and mural cell activation.
Angiogenesis is impaired in SIRT1-Tg mice. (A) Western blot of VEGFA, HIF1α and (B) qRT-PCR for the expression of VEGFA in gastrocnemius tissues after 14 days of hindlimb ischemia (n=5). (C) Representative images of CD31 immunostaining in Matrigel plugs containing VEGFA or not implanted in mice (n=5 mice per group). (D) Quantification of angiogenesis was expressed as CD31-positive area for each section in randomly acquired images. Scale bars = 100 μm. (E) Representative images of NG2 immunostaining of Matrigel plugs containing VEGFA or not implanted in mice (n=5 mice per group). Nuclei were stained with DAPI in blue. (F) Relative NG2 positive areas show the number of vessels. Scale bars = 100 μm. (G) Representative images of wound healing at various time points. (H) Wound closure at different time points were expressed as percentage of the wound area on day 0 (n=5 mice per group). Data represent mean±SEM. Student's t-test, one-way ANOVA or repeated Measures ANOVA: *P<0.05, **P<0.01 versus the corresponding control.
The exosomes of SIRT1-Tg VSMCs inhibit the endothelial angiogenic function in vitro
To examine the phenotypic state of SIRT1-Tg VSMCs in response to hypoxia, we first detected the expression of the phenotypic markers in the cells. We showed that there were lower expression of the synthetic markers OPN, MMP2 and MMP9, and higher level of contractile markers α-SMA and SM22α in SIRT1-Tg VSMCs exposed to hypoxia, compared with WT and SIRT1-KO VSMCs that displayed increased expression of synthetic markers (Figure 3A), suggesting that SIRT1-Tg VSMCs were a differentiated phenotype even under hypoxic conditions. Importantly, the expression of NG2 almost disappeared in SIRT1-Tg VSMCs exposed to hypoxia, implying that the plasticity of the VSMCs was impaired to some extent, which might be associated with reduced mural cell coverage. Cells are able to release exosomes into their environment and thereby have an additional form of communication to influence target cell behavior over long distances [28]. To find out how SIRT1-Tg VSMCs lead to angiogenic dysfunction observed in the model, we isolated the exosomes from the conditioned media of WT, SIRT1-Tg and SIRT1-KO VSMCs to treat human and mouse ECs, respectively. Using BrdU incorporation, we showed that the exosomes from SIRT1-Tg VSMCs significantly reduced the proliferation activity of the ECs from the two species in response to hypoxia (Figure 3B), consistent with the results observed in the ischemic gastrocnemius of SIRT1-Tg mice. Migration of ECs is of central importance to angiogenesis. We performed a cell-wounding assay, and showed that the migration of the two ECs was attenuated following incubation with the SIRT1-Tg exosomes compared with WT control (Figure 3C-D). However, the exosomes of SIRT1-KO VSMCs increased the migration activity of the ECs. Using an in vitro tube formation assay, we found that relative tube length and number of branches were significantly reduced in SIRT1-Tg exosome group and increased in the cells treated with SIRT1-KO exosomes, compared with WT control (Figure 3E-G). The ability of ECs to secrete growth factors, such as VEGF, is crucial for the new vascular network development via induction of proliferation and migration. We performed qRT-PCR of VEGFA, and showed decreased VEGFA expression in ECs treated by the exosomes of SIRT1-Tg VSMCs in response to hypoxia (Figure 3H), consistent with the results observed in the model. In addition, this reduction was reversed in ECs treated by the exosomes of SIRT1-KO VSMCs, in which the level of VEGFA mRNA was 1.9-fold higher than that of WT control (Figure 3H).
The exosomes of SIRT1-Tg VSMCs inhibit the endothelial angiogenic functions in vitro. (A) VSMCs from WT, SIRT1-Tg or SIRT1-KO mice were incubated under hypoxia for 24 h. Western blot of OPN, MMP2, MMP9, α-SMA, SM22α and NG2. (B-J) HUVECs or mouse ECs were incubated with the hypoxia-induced VSMC exosomes (Exo) for 24 h and exposed to hypoxia. (B) The relative activity of proliferation by BrdU incorporation. (C and D) The relative activity of migration using a cell-wounding assay. (E) Representative images of tube formation. Scale bars = 200 μm. Relative tube length (F) and number of branches (G) were quantified by measuring the cumulative tube length and branches. (H) qRT-PCR of VEGFA expression in mouse ECs. (I) Immunofluorescent confocal microscopy of HIF1α nuclear translocation in the ECs. Scale bars =100 μm. (J) The quantification of the nuclear-to-cytosol ratio of HIF1α protein in ECs (n=15). Bar graphs show mean±SEM. Student's t-test or one-way ANOVA: *P<0.05, **P<0.01 versus the corresponding control.
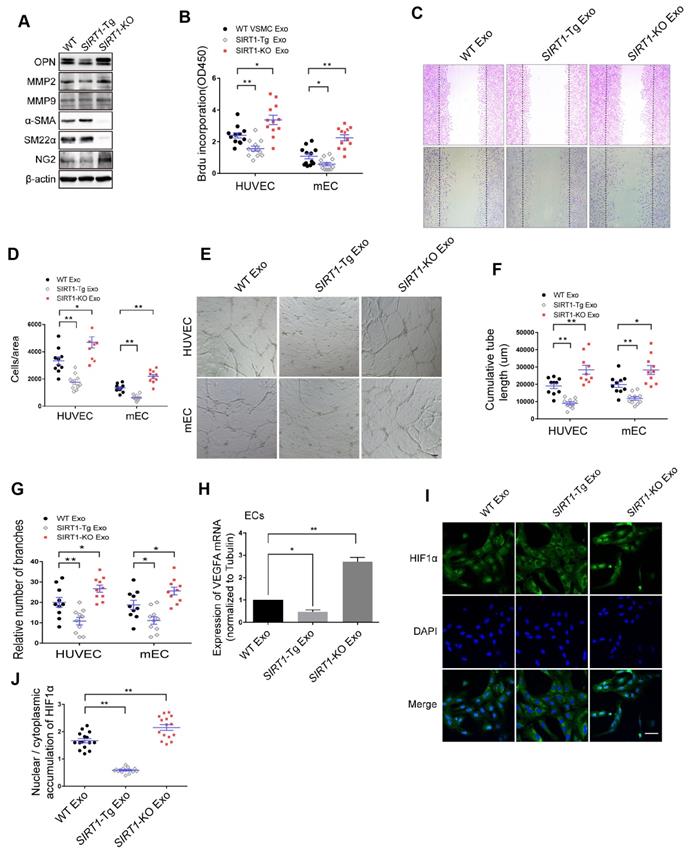
HIF1α nuclear translocation is a key step for activating expression of the target genes including VEGFA. We examined hypoxia-induced HIF1α nuclear translocation in ECs treated with the exosomes of WT, SIRT1-Tg and SIRT1-KO VSMCs using immunofluorescence staining by anti-HIF1α antibody. We found an almost disappeared nuclear HIF1α expression in ECs treated with the exosomes of SIRT1-Tg VSMCs under hypoxia (Figure 3I). In contrast, HIF1α was mainly accumulated in the nucleus of ECs treated with exosomes of SIRT1-KO cells (Figure 3I-J). These findings imply that some components of SIRT1-Tg exosomes may be delivered into ECs and inhibit VEGFA expression via sequestrating HIF1α in the cytoplasm.
Smooth muscle exosome cZFP609 attenuates hypoxia-induced VEGFA expression in ECs
Non-coding RNAs carried by exosomes can be functionally transferred to recipient cells and subsequently regulate gene expression [29]. The latest study leads to the identification of 7770 circRNAs in human and mouse VSMCs that shared similar circRNA signatures [30]. To determine whether reduced nuclear HIF1α expression in ECs is mediated by circRNAs of SIRT1-Tg exosomes, we screened a set of the latest reported circRNAs involved in angiogenesis or regulated by hypoxia as candidate sponge RNAs and assessed their potentially interaction with HIF1α by RIP assay using anti-HIF1α antibody [31, 32]. We showed that HIF1α markedly recruited cZFP609 in ECs treated with the exosomes of SIRT1-Tg VSMCs under hypoxia (Figure 4A). Furthermore, the exosomes of SIRT1-Tg VSMCs knocked down for cZFP609 increased the HIF1α nuclear translocation induced by hypoxia in ECs (Figure S1).The level of cZFP609 expression was higher in SIRT1-Tg VSMCs and especially its exosomes under hypoxia (Figure 4B-C). Conversely, SIRT1-KO and WT VSMCs displayed lower expression of cZFP609 under the same conditions. However, the level of cZFP609 was decreased in ECs under hypoxia compared with normoxia state (Figure 4D), and significantly increased following incubation with the exosomes from SIRT1-Tg VSMCs exposed to hypoxia (Figure 4E). Moreover, this increase was abolished in ECs treated with the exosomes from the cZFP609 specific siRNA-treated SIRT1-Tg VSMCs, accompanied by an increased expression of VEGFA with unchanged ZFP609 mRNA (Figure 4F-G), suggesting that cZFP609 is generated by SIRT1-Tg VSMCs and delivered to ECs.
Smooth muscle exosome cZFP609 attenuates hypoxia-induced VEGFA expression in ECs. (A-G and I) qRT-PCR. VSMCs or ECs were exposed to hypoxia for 24 h, except if stated otherwise. (A) RIP assay were performed using HIF1α antibodies in ECs treated with the exosomes of SIRT1-Tg VSMCs under hypoxia. (B and C) cZFP609 expression in VSMCs (B) and the matched exosomes (C). (D) cZFP609 expression in the ECs. (E) cZFP609 expression in the ECs incubated in the VSMC exosome. (F) cZFP609 expression in SIRT1-Tg VSMCs transfected with siScr or sicZFP609. (G) cZFP609 and VEGFA mRNA expression in the ECs treated with the exosome from the cZFP609 siRNA-transfected SIRT1-Tg VSMCs. (H) The sequence of cZFP609 was obtained from Sanger sequencing. (I) cZFP609 and VEGFA mRNA expression. The ECs were transfected with vector and cZFP609 for 24 h. VEGFA protein expression were detected by Western blot (J). (A-G and I) Bar graphs show mean±SEM from 3 independent experiments (n=3). Student's t-test or one-way ANOVA: *P<0.05, **P<0.01 versus the corresponding control.
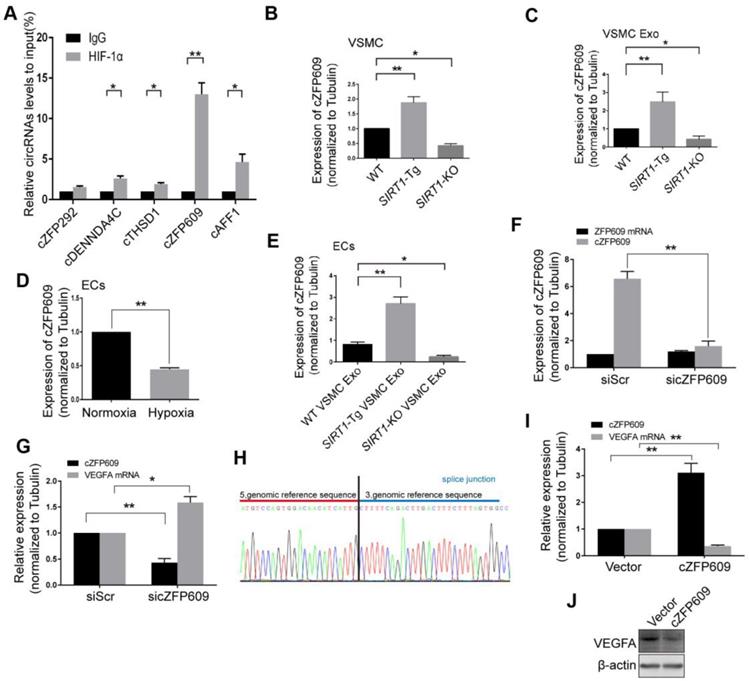
To further confirm the inhibitory effect of cZFP609 on VEGFA expression, we construct cZFP609 expression plasmid. Mouse cZFP609 cDNA was synthesized and cloned into pcD-ciR vector (Figure 4H). We showed that overexpression of cZFP609 resulted in reduced expression of VEGFA in ECs compared with vehicle control (Figure 4I-J). These data indicate that the exosome cZFP609 of SIRT1-Tg VSMCs attenuates hypoxia-induced VEGFA expression in ECs.
Knockdown of cZFP609 improves endothelial angiogenic functions
Hypoxia-induced angiogenesis is achieved through VEGFA-driven proliferation and migration of ECs [33]. To verify the direct effect of cZFP609 on endothelial angiogenic functions, the expressing vector was transfected into ECs to overexpress cZFP609, and proliferation of ECs was determined by BrdU incorporation. We showed that overexpression of cZFP609 inhibited the proliferation of ECs in response to hypoxia (Figure 5A). BrdU incorporation was decreased to ~40% of that of the vehicle control. We then performed cell-wounding assay following overexpression of cZFP609 in ECs. The migration was attenuated in cZFP609-overexpressed ECs, consistent with reduced proliferative activity (Figure 5B-C). The tube formation assay is a preferred protocol to identify inhibitors or stimulators of the angiogenic activity of ECs in vitro [34]. Using this protocol, we measured the effect of cZFP609 on the tubule formation in a quantifiable manner. Overexpression of cZFP609 resulted in shortened relative tube length (Figure 5D-E), accompanied with reduced relative number of branches in ECs transfected with cZFP609 vector compared with vehicle control (Figure 5F). To validate whether cZFP609 is a target to improve angiogenesis, the matrigel was premixed with siScr or sicZFP609, and was injected subcutaneously into SIRT1-Tg mice to knockdown of endogenous cZFP609 expression. We showed that the angiogenic dysfunction was reversed by knockdown of cZFP609 in in vivo matrigel plug assay (Figure 5G). Furthermore, the number of the capillaries invaded into the matrigel was significantly increased in the matrigel plugs with sicZFP609, accompanied with the density of NG2-positive cells (Figure 5G-J). qRT-PCR displayed that the expression level of CD31 and VEGFA mRNAs was elevated in the matrigel with sicZFP609 compared with the control (Figure 5K).These findings indicate that knockdown of cZFP609 improves endothelial angiogenic functions via promoting VEGFA expression in response to hypoxia.
cZFP609 inhibits the endothelial angiogenic functions. (A-F) The mouse ECs were transfected with vector or cZFP609 for 24 h, and then treated by hypoxia for 24 h. (A) Proliferation of ECs was determined by BrdU incorporation. Bar graphs show the relative activity of proliferation. (B and C) The migration of cells was evaluated by a cell-wounding assay. The number of cells in the wounded area shows the relative activity of migration. (D-F) Representative images of tube formation. Relative tube length and number of branches were quantified by measuring the cumulative tube length and branches. Scale bars = 200 μm. (G-K) The matrigel was premixed with siScr or sicZFP609, and was injected subcutaneously into SIRT1-Tg mice. (G) Bright field image of angiogenesis (upper lane) and representative images of CD31 (lower lane) and NG2 (I) immunostaining in matrigel plugs. Nuclei were stained with DAPI in blue. Scale bars = 100 μm. Angiogenesis in matrigel plugs was quantified by measuring CD31 (H) and NG2 positive area (J). (K) qRT-PCRs of CD31 mRNA, VEGFA mRNA and cZFP609 expression in matrigel plugs (n=5 mice per group). (A, C, E, F, H, J and K) Bar graphs show mean±SEM. Student's t-test: *P<0.05, **P<0.01 versus the corresponding control.
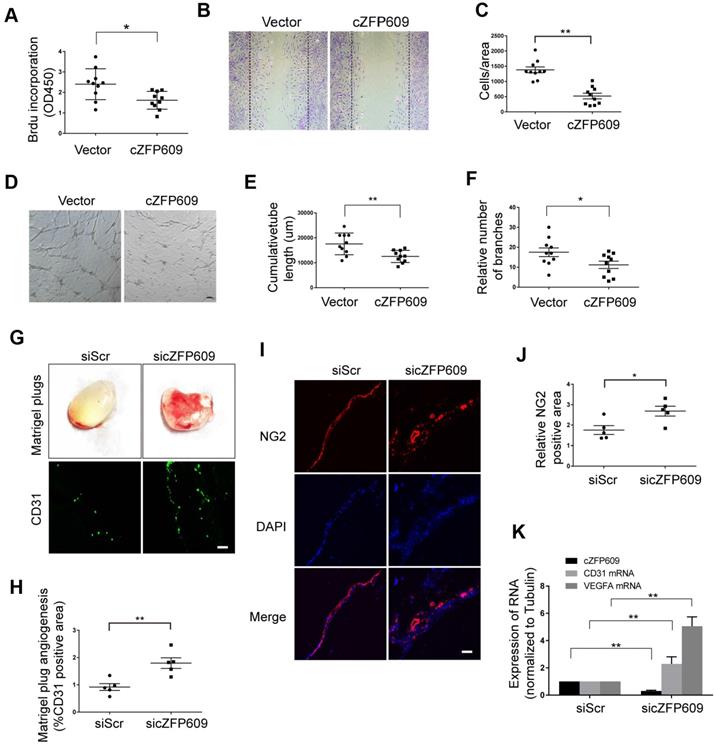
cZFP609 blocks hypoxia-induced HIF1α nuclear translocation in ECs
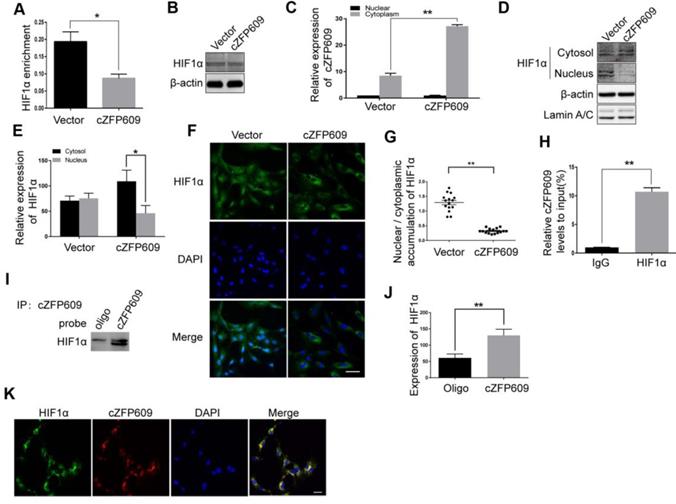
HIF1α binding to the promoter region is essential for hypoxia-induced transcription of VEGFA gene. To validate the causal relationship between decrease in expression of VEGFA and nuclear HIF1α activity in ECs overexpressing cZFP609, we first performed ChIP assay using a HIF1α antibody to detect the binding activity between HIF1α and the promoter. We showed that overexpression of cZFP609 significantly decreased the binding of HIF1α to the promoter of VEGFA gene (Figure 6A) with unchanged expression of HIF1α in ECs (Figure 6B). We then isolated nuclear and cytosolic fractions from ECs to examine the distribution of cZFP609 in ECs using qRT-PCR. We found that cZFP609 was mainly localized in the cytoplasm, and significantly increased in the cZFP609-transfected ECs (Figure 6C). Using Western blot and immunofluorescence staining, we found that the distribution of HIF1α in the nuclei fraction decreased in ECs overexpressing cZFP609 in response to hypoxia (Figure 6D-E), and hypoxia-induced nuclear translocation of HIF1α disappeared under the same conditions (Figure 6F-G). To confirm that cZFP609 inhibits the nuclear translocation of HIF1α, we performed RIP and RNA pull-down assays, respectively. We demonstrated that cZFP609 was retrieved by HIF1α antibody (Figure 6H), and HIF1α protein was also retrieved by cZFP609 probe in ECs overexpressing cZFP609 (Figure 6I-J). In addition, the cZFP609 binding to HIF1α was further verified by fluorescence in situ hybridization (FISH) assay, which displayed the co-localization of cZFP609 and HIF1α in the cytoplasm of ECs (Figure 6K). Taken together, our data demonstrate that cZFP609, as a sponge, detains HIF1α in the cytoplasm, resulting in suppression of hypoxia-induced VEGFA expression.
cZFP609 interacts with and blocks HIF1α nuclear translocation in ECs. The mouse ECs were transfected with vector or cZFP609 for 24 h and then incubated under hypoxia for 24 h. (A) ChIP assay for VEGFA gene promoter region in the ECs using HIF1α antibody. (B) Western blot analysis of HIF1α in the ECs. (C) qRT-PCRs for cZFP609 expression in the nucleus and cytoplasm of the ECs. (D and E) Western blot for HIF1α expression in the ECs. (F) Immunofluorescent confocal microscopy of HIF1α nuclear translocation in the ECs. Scale bars =100 μm. (G) The quantification of the nuclear-to-cytosol ratio of HIF1α protein in ECs (n=15). (H) RIP assay was performed using HIF1α antibodies in the ECs. qRT-PCR was used to detect pulled-down cZFP609. (I and J) The cytoplasm was extracted in ECs incubated under hypoxia for 24 h. RNA pull-down assay was performed using the probe. Western blot was used to validate the interactions between cZFP609 and HIF1α. (K) Confocal FISH images of colocalization between HIF1α and cZFP609 in the ECs. Scale bars=50 μm. Bar graphs show mean±SEM from 3 independent experiments (n=3). Student's t-test: *P<0.05, **P<0.01 versus the corresponding control.
cZFP609 is negatively correlated with blood flow perfusion after ischemia
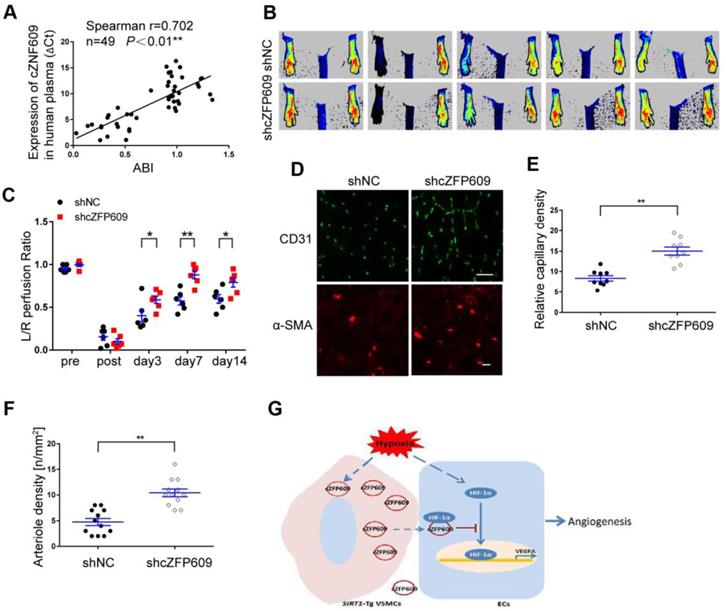
To explore the clinical value of cZNF609 in assessment of blood flow perfusion during tissue ischemia, we measured the plasma level of cZNF609 (cZFP609 in mouse or cZNF609 in human) in patients with atherosclerotic or diabetic lower extremity PAD (n=19). We showed that the plasma level of cZNF609 was significantly higher in the patients than that in normal individuals (n=30), and correlated with reduced ankle-brachial index (ABI), which reflected reduced blood flow in lower extremity (Figure 7A, Table S2). To explore whether inhibition of cZFP609 expression improves blood flow perfusion after ischemia, AAV9-shcZFP609 (1×1012 vg /mL) was injected into the gastrocnemius of SIRT1-Tg mice followed by the femoral artery ligation [21]. We observed that knockdown of cZFP609 promoted early appearance of blood flow recovery (Figure 7B-C), and increased number of capillaries and arterioles in the ischemic tissues of SIRT1-Tg mice compared with the control (Figure 7D-F). These data indicate that plasma cZNF609 level can reflect the severity of lower extremity PAD in patients to some extent.
cZFP609 negatively regulates blood flow perfusion. (A) Plasma samples were collected from the patients with PAD (n = 19) and control subjects (n = 30). The plasma cZNF609 was determined by qPCR. ABI was calculated by measuring the ankle and the brachial systolic blood pressure using PeriFlux System 5000 (Perimed AB, Datavagen 9A, 175 43 Jarfalla, Sweden). Spearman's correlation analysis was performed. (B) Representative laser Doppler perfusion images at indicated time point after hindlimb ischemia. SIRT1-Tg mice treated with shcZFP609 or shNC (n= 6). (C) Laser Doppler perfusion at various time points was expressed as a ratio of flow between ischemic (L) and sham (R) limbs (L/R). (D) Immunofluorescence for CD31 (Scale bars = 100 μm) and α-SMA-positive cells (Scale bars = 20 μm) in the gastrocnemius tissue. (E) Immunostaining of CD31-positive cells represented the relative capillary density. (F) The density of arteriole was expressed as the quantity of arterioles per mm2. Data represent mean±SEM. Repeated Measures ANOVA or student's t-test: *P<0.05, **P<0.01 versus the corresponding control. (G) A working model for SIRT1-Tg VSMCs to impair angiogenesis via inhibition of HIF1α.
Discussion
The results of the present study reveal that VSMC SIRT1 negatively regulates angiogenesis, resulting in a delayed blood flow recovery following hindlimb ischemia, similar to results from RSV administrate. Mechanically, the VSMCs-derived cZFP609 could be delivered to ECs by exosomes, and attenuated endothelial angiogenic function via blockade of HIF1α nuclear translocation and inhibition of VEGFA expression in response to hypoxia (Figure 7G). Higher cZFP609 expression was observed in the plasma, and associated with reduced ABI in the patients with atherosclerotic or diabetic lower extremity PAD. Thus, cZFP609 may be a novel biomarker and potential therapeutic target in treatment of ischemic diseases.
The restoration of hindlimb perfusion after ischemia is dependent on angiogenesis as well as capillary arterialization. VEGFA plays a central role in development and postnatal angiogenesis, and the source of the growth factor and the stimulus for its production include ischemic tissues, infiltrating monocytes/macrophages and the blood vessels themselves. The key factor regulating VEGFA production in ischemic tissues is HIF1α. HIF1α levels in turn are controlled in a highly complex manner by several regulators [35]. The recent study showed that the activation of the endothelial NF-κB cascade can lead not only to local production of VEGF but also to accumulation of monocytes/macrophages due to increased expression of adhesion molecules leading to further increase in local accumulation of VEGFA [27]. Furthermore, activated ECs induce the expression of PDGF-AA, PDGF-BB, and TGF- β in VSMCs, and the shear stress modulates VSMC migration, apoptosis, proliferation, and gene expressions in an EC-dependent manner [36, 37]. According to a series of research results, activation of ECs and accumulation of monocytes/macrophages are considered the principal regulatory mechanisms for angiogenesis after ischemia. However, there is almost no attention to the regulating role of VSMCs on this process. In the present study, we first demonstrated that VSMCs actively reprogrammed the angiogenic functions of ECs using SIRT1-overexpressed VSMCs in response to hypoxia in vivo and in vitro. We showed that the blood flow recovery was delayed, and angiogenesis was impaired in SIRT1-Tg mice after ischemia. Our findings suggest that VSMCs may be independently associated with angiogenesis rather than not only involved in arteriogenesis.
The remarkable plasticity of the VSMCs, particularly their ability to change phenotype from the contractile to the synthetic in response to hypoxia, makes the proliferation possible through a HIF1α-dependent mechanism [38]. VSMC phenotypic changes are typical for arteriogenesis [2]. The activation of SIRT1 in VSMCs prevents neointimal formation, hypertension and atherosclerosis [11, 16, 39]. We showed that overexpression of SIRT1 modulated VSMCs to be over-differentiated phenotype, which maintained higher expression of the contractile proteins and reduced synthetic markers in response to hypoxia, suggesting that SIRT1 might attenuate the phenotypic plasticity of VSMCs. The non-adaptive phenotype of VSMCs may result in generation of the abnormal signal component and lack of growth factors, and then disturb the angiogenic functions of ECs. It illustrates well the importance of balanced VSMC phenotype plasticity for normal vascular function.
SIRT1 is highly expressed in the vasculature during blood vessel growth. It's well known that SIRT1 controls the angiogenic activity of ECs via deacetylation of FoxO1 and uniquely regulates angiogenesis signaling [10]. Loss of SIRT1 function blocks sprouting angiogenesis and branching morphogenesis of ECs with consequent down-regulation of genes involved in blood vessel development and vascular remodeling that is essential for postnatal blood vessel development. The inactivation of SIRT1 causes reduced vascular branching and density in vivo [26]. However, other study revealed SIRT1 has negative effects on angiogenesis via deacetylating and inactivating HIF1α [7]. Moreover, SIRT1 is transcriptionally downregulated during hypoxia to modulate cellular adaptation to hypoxia by targeting HIF1α [40]. In the present study, we confirmed that overexpression of SIRT1 in VSMCs inhibited the angiogenic response in vivo and in vitro via exosome cZFP609-mediated intercellular communication, whatever the role of SIRT1 of ECs. Our findings provide evidence that VSMCs may reprogram angiogenesis, and targeting VSMCs may operate the balance between positive and negative mechanisms of angiogenesis in a cell-specific manner.
circRNAs are a novel class of non-coding RNAs that form a covalently closed continuous loop, and are conserved and stable [31]. circRNAs are specifically expressed in a cell type or developmental stage, indicating that circRNAs may play important roles in many physiological and pathophysiological processes. It has been demonstrated that circRNAs regulate gene expression by acting as miRNA sponges, RNA-binding protein sequestering agents, or nuclear transcriptional regulators [41]. The recent studies suggest that circRNAs are involved in angiogenesis or are regulated by hypoxia, such as cFoxo3, cZNF292, cZNF609, cITCH, cTHSD1 [31, 32, 42]. Silencing cZNF609 increases EC migration and tube formation, and protects EC against oxidative stress and hypoxia stress [32]. In the present study, we showed that the expression of cZFP609 was significantly increased in SIRT1-Tg VSMCs and released via the exosomes. The exosome cZFP609 was delivered into ECs that expressed low cZFP609, and acted as HIF1α sequestering agent to block its nuclear translocation, led to a decreased HIF1α-driven VEGFA gene expression in ECs. VSMC-derived cZFP609 disturbed hypoxia-induced reprogramming of growth signals, and impaired endothelial angiogenic functions. Furthermore, knockdown of cZFP609 effectively improved the growth of capillaries and promoted blood flow recovery after ischemia in vivo. Additionally, the plasma level of cZNF609 was negatively correlated with the ankle-brachial index in patients with atherosclerotic or diabetic lower extremity PAD, suggesting the potential clinical value of cZNF609 in assessment of blood flow perfusion in tissue ischemia. Collectively, these findings provide strong support for cZFP609, as an intercellular messenger, to mediate VSMCs to reprogram angiogenesis of ECs, which may be an additional mechanism of HIF1α post-translation regulation.
However, several questions in understanding the production and function of cZFP609 in VSMC reprogramming angiogenesis still remain to be clarified. First, SIRT1 maintains the differentiated phenotype of VSMCs, whether cZFP609 attenuates the plasticity of VSMCs in response to hypoxia, and the mechanism underlying SIRT1 mediating cZFP609 formation, are unclear. Second, we have demonstrated that SIRT1 inhibits vascular inflammation via suppressing the activation of NF-κB and the expression of pro-inflammatory factors in VSMCs [24, 43], and however, whether SIRT1 inhibiting NF-κB signaling limits accumulation of monocytes/macrophages due to decreased expression of adhesion molecules further leading to decrease in local level of VEGFA, a crucial pathological process involved in angiogenesis after ischemia, thereby impairing angiogenesis after ischemia, also remains unknown. Third, a recent study demonstrated that cZFP609 can be translated into a protein in a splicing-dependent and cap-independent manner [44], further investigation is required to determine whether its translated protein is involved in regulation of angiogenesis.
In summary, our study, for the first time, demonstrate that VSMC cZFP609 reprograms ECs to attenuate angiogenesis after ischemia via inhibition of HIF1α activation in SIRT1-dependent manner. Our findings provide not only novel insight into the molecular mechanism and cellular regulatory network in angiogenesis after ischemia, but also new valuable indicator to assess the prognosis and clinical outcomes of ischemic diseases.
Abbreviations
AAV: adeno-associated virus; ABI: ankle-brachial index; CD31: cluster of differentiation 31; ChIP: chromatin immunoprecipitation; EC: endothelial cell; FISH: fluorescence in situ hybridization; HIF: hypoxia-inducible factor; HUVEC: human umbilical vein endothelial cell; PCR: polymerase chain reaction; PDGF: platelet-derived growth factor; RIP: RNA immunoprecipitationn assay; SIRT1: silent information regulator 1; SIRT1-Tg: SIRT1-transgenic; VEGF: vascular endothelial growth factor; VSMC: vascular smooth muscle cell; WT: wild-type.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank Dr. D. P. Liu and H. Z. Chen for the SIRT1-VSMC-specific transgenic (SIRT1-Tg) mice and SIRT1-VSMC-specific knockout (SIRT1-KO) mice. We thank Y. Y. Zhang helpful discussions and Y. Zhang for technical contributions to this project. This work was supported by the National Natural Science Foundation of China 91739301, 91849102 and 31471092 (to M.H.), and 31872788 (to F.Z.); the Key Natural Science Foundation Projects of Hebei Province H2019206028 (to M.H.); Science and Technology Project of Hebei Province (172777199 to Y.-Q. Dou).
Author contributions
Yong-Qing Dou conducted experiments, analyzed data, and wrote the manuscript. Peng Kong conducted in vivo experiments, analyzed data. Wei-Wei Li, Chang-Lin Li, Yuan Yu, Lei Nie, Li-Li Zhao and Sui-Bing Miao performed in vitro experiments. Xiao-Kun Li and Chen Dong conducted in vivo experiments. Chang-Lin Li, Xiao-Kun Li, Jin-Wen Zhang, Yang Liu, Xiao-Xia Huo, Kui Chi and Xiang Gao prepared hindlimb ischemia model of mice. Hongyuan Yang and Fan Zhang discussed the project. Mei Han supervised the project, analyzed data, and wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Limbourg A, Korff T, Napp LC, Schaper W, Drexler H, Limbourg FP. Evaluation of postnatal arteriogenesis and angiogenesis in a mouse model of hind-limb ischemia. Nat Protoc. 2009;4:1737-46
2. Cai W, Schaper W. Mechanisms of arteriogenesis. Acta Biochim Biophys Sin. 2008;40:681-92
3. Nicosia RF, Villaschi S. Rat aortic smooth muscle cells become pericytes during angiogenesis in vitro. Lab Invest. 1995;73:658-66
4. Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:873-87
5. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298-307
6. Richard DE, Berra E, Gothie E, Roux D, Pouyssegur J. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (HIF-1alpha) and enhance the transcriptional activity of HIF-1. J Biol Chem. 1999;274:32631-7
7. Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol Cell. 2010;38:864-78
8. Kenji T, Yan IK, Hiroaki H, Tushar P. Modulation of hypoxia-signaling pathways by extracellular linc-RoR. J Cell Sci. 2014;127:1585-94
9. Maistrovski Y, Biggar KK, Storey KB. HIF-1α regulation in mammalian hibernators: role of non-coding RNA in HIF-1α control during torpor in ground squirrels and bats. J Comp Physiol B. 2012;182:849-59
10. Potente M, Ghaeni L, Baldessari D, Mostoslavsky R, Rossig L, Dequiedt F. et al. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev. 2007;21:2644-58
11. Li L, Zhang HN, Chen HZ, Gao P, Zhu LH, Li HL. et al. SIRT1 acts as a modulator of neointima formation following vascular injury in mice. Circ Res. 2011;108:1180-9
12. Ma S, Motevalli SM, Chen J, Xu MQ, Wang Y, Feng J. et al. Precise theranostic nanomedicines for inhibiting vulnerable atherosclerotic plaque progression through regulation of vascular smooth muscle cell phenotype switching. Theranostics. 2018;8:3693-706
13. Cooper LL, Woodard T, Sigurdsson S, Buchem MAV, Torjesen AA, Inker LA. et al. Cerebrovascular damage mediates relations between aortic stiffness and memory. Hypertension. 2016;67:176-82
14. Fry JL, Shiraishi Y, Turcotte R, Yu X, Yuan ZG, Akiki R. et al. Vascular smooth muscle Sirtuin-1 protects against aortic dissection during angiotensin II-induced hypertension. J Am Heart Assoc. 2015:4
15. Stein S, Lohmann C, Schäfer N, Hofmann J, Rohrer L, Besler C. et al. SIRT1 decreases Lox-1-mediated foam cell formation in atherogenesis. Eur Heart J. 2010;31:2301-9
16. Gorenne I, Kumar S, Gray K, Figg N, Yu H, Mercer J. et al. Vascular smooth muscle cell Sirtuin 1 protects against DNA damage and inhibits atherosclerosis. Circulation. 2013;127:386-96
17. Zu Y, Liu L, Lee MY, Xu C, Liang Y, Man RY. et al. SIRT1 promotes proliferation and prevents senescence through targeting LKB1 in primary porcine aortic endothelial cells. Circ Res. 2010;106:1384-93
18. Chen HZ, Wang F, Gao P, Pei JF, Liu Y, Xu TT. et al. Age-associated Sirtuin 1 reduction in vascular smooth muscle links vascular senescence and inflammation to abdominal aortic aneurysm. Circ Res. 2016;119:1076-88
19. Marcu R, Kotha S, Zhi Z, Qin W, Neeley CK, Wang RK. et al. The mitochondrial permeability transition pore regulates endothelial bioenergetics and angiogenesis. Circ Res. 2015;116:1336-45
20. Brooks CL, Gu W. How does SIRT1 affect metabolism, senescence and cancer? Nat Rev Cancer. 2009;9:123-8
21. Tasfaout H, Lionello VM, Kretz C, Koebel P, Messaddeq N, Bitz D. et al. Single intramuscular injection of AAV-shRNA reduces DNM2 and prevents myotubular myopathy in mice. Mol Ther. 2018;26:1082-92
22. Xue T, Siu CW, Lieu DK, Lau CP, Tse HF, Li RA. Mechanistic role of I(f) revealed by induction of ventricular automaticity by somatic gene transfer of gating-engineered pacemaker (HCN) channels. Circulation. 2007;115:1839-50
23. Nie L, Guo X, Esmailzadeh L, Zhang J, Asadi A, Collinge M. et al. Transmembrane protein ESDN promotes endothelial VEGF signaling and regulates angiogenesis. J Clin Invest. 2013;123:5082-97
24. Kong P, Yu Y, Wang L, Dou YQ, Zhang XH, Cui Y. et al. circ-Sirt1 controls NF-kappaB activation via sequence-specific interaction and enhancement of SIRT1 expression by binding to miR-132/212 in vascular smooth muscle cells. Nucleic Acids Res. 2019;47:3580-93
25. Zeng Y, Du WW, Wu Y, Yang Z, Awan FM, Li X. et al. A circular RNA binds to and activates AKT phosphorylation and nuclear localization reducing apoptosis and enhancing cardiac repair. Theranostics. 2017;7:3842-55
26. Guarani V, Deflorian G, Franco CA, Kruger M, Phng LK, Bentley K. et al. Acetylation-dependent regulation of endothelial Notch signalling by the SIRT1 deacetylase. Nature. 2011;473:234-8
27. Tirziu D, Jaba IM, Yu P, Larrivée B, Coon BG, Cristofaro B. et al. Endothelial nuclear factor-κB-dependent regulation of arteriogenesis and branching. Circulation. 2012;126:2589-600
28. Yu S. et al. Tumor exosomes inhibit differentiation of bone marrow dendritic cells. J Immunol. 2007;178:6867-75
29. Pegtel DM, Cosmopoulos K, Thorley-Lawson DA, van Eijndhoven MA, Hopmans ES, Lindenberg JL. et al. Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci U S A. 2010;107:6328-33
30. Hall IF, Climent M, Quintavalle M, Farina FM, Schorn T, Zani S. et al. Circ_Lrp6, a circular RNA enriched in vascular smooth muscle cells, acts as a sponge regulating miRNA-145 function. Circ Res. 2019;124:498-510
31. Boeckel JN, Jae N, Heumuller AW, Chen W, Boon RA, Stellos K. et al. Identification and characterization of hypoxia-regulated endothelial circular RNA. Circ Res. 2015;117:884-90
32. Liu C, Yao MD, Li CP, Shan K, Yang H, Wang JJ. et al. Silencing of circular RNA-ZNF609 ameliorates vascular endothelial dysfunction. Theranostics. 2017;7:2863-77
33. Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843-5
34. Arnaoutova I, Kleinman HK. In vitro angiogenesis: endothelial cell tube formation on gelled basement membrane extract. Nat Protoc. 2010;5:628-35
35. Semenza Gregg L. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399-408
36. Heydarkhan-Hagvall S, Helenius G, Johansson BR, Li JY, Mattsson E, Risberg B. Co-culture of endothelial cells and smooth muscle cells affects gene expression of angiogenic factors. J Cell Biochem. 2003;89:1250-9
37. Qi YX, Jiang J, Jiang XH, Wang XD, Ji SY, Han Y. et al. PDGF-BB and TGF-{beta}1 on cross-talk between endothelial and smooth muscle cells in vascular remodeling induced by low shear stress. Proc Natl Acad Sci U S A. 2011;108:1908-13
38. Schultz K, Fanburg BL, Beasley D. Hypoxia and hypoxia-inducible factor-1α promote growth factor-induced proliferation of human vascular smooth muscle cells. Am J Physiol. 2006;290:2528-34
39. Gao P, Xu TT, Lu J, Li L, Xu J, Hao DL. et al. Overexpression of SIRT1 in vascular smooth muscle cells attenuates angiotensin II-induced vascular remodeling and hypertension in mice. J Mol Med (Berl). 2014;92:347-57
40. Zhang Q, Wang SY, Fleuriel C, Leprince D, Rocheleau JV, Piston DW. et al. Metabolic regulation of SIRT1 transcription via a HIC1:CtBP corepressor complex. Proc Natl Acad Sci U S A. 2007;104:829-33
41. Salzman J. Circular RNA Expression: Its Potential Regulation and Function. Trends Genet. 2016;32:309-16
42. Guo W, Zhang J, Zhang D, Cao S, Li G, Zhang S. et al. Polymorphisms and expression pattern of circular RNA circ-ITCH contributes to the carcinogenesis of hepatocellular carcinoma. Oncotarget. 2017;8:48169-77
43. Shu YN, Dong LH, Li H, Pei QQ, Miao SB, Zhang F. et al. CKII-SIRT1-SM22alpha loop evokes a self-limited inflammatory response in vascular smooth muscle cells. Cardiovasc Res. 2017;113:1198-207
44. Legnini I, Timoteo GD, Rossi F, Morlando M, Briganti F, Sthandier O. et al. Circ-ZNF609 Is a circular RNA that can be translated and functions in myogenesis. Mol Cell. 2017;66:22-37
Author contact
![]() Corresponding author: Mei Han, Department of Biochemistry and Molecular Biology, College of Basic Medicine, Hebei Medical University, No. 361 Zhongshan East Road, Shijiazhuang, Hebei, China. Phone: 86-311-86265557; E-mail: hanmeiedu.cn
Corresponding author: Mei Han, Department of Biochemistry and Molecular Biology, College of Basic Medicine, Hebei Medical University, No. 361 Zhongshan East Road, Shijiazhuang, Hebei, China. Phone: 86-311-86265557; E-mail: hanmeiedu.cn