Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Drug combination strategies
Nanocarrier-based combination...
Perspectives
Conclusions
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(3):1355-1372. doi:10.7150/thno.38147 This issue Cite
Review
Nanocarrier-based drug combination therapy for glioblastoma
Mengnan Zhao1*, Demian van Straten2*, Marike L.D. Broekman3, Véronique Préat1 ![]() , Raymond M. Schiffelers2
, Raymond M. Schiffelers2 ![]()
1. Université catholique de Louvain, Louvain Drug Research Institute, Advanced Drug Delivery and Biomaterials, Avenue Mounier, 73, B1 73.12, 1200 Brussels, Belgium
2. Department of Clinical Chemistry and Haematology, University Medical Center Utrecht, Heidelberglaan 100, 3584 CX Utrecht, Netherlands
3. Department of Neurosurgery, Leiden University Medical Center, PO Box 9600, 2300 RC, Leiden, The Netherlands.
*Shared coauthorship
Received 2019-7-4; Accepted 2019-11-4; Published 2020-1-1
Abstract

The current achievements in treating glioblastoma (GBM) patients are not sufficient because many challenges exist, such as tumor heterogeneity, the blood brain barrier, glioma stem cells, drug efflux pumps and DNA damage repair mechanisms. Drug combination therapies have shown increasing benefits against those challenges. With the help of nanocarriers, enhancement of the efficacy and safety could be gained using synergistic combinations of different therapeutic agents. In this review, we will discuss the major issues for GBM treatment, the rationales of drug combinations with or without nanocarriers and the principle of enhanced permeability and retention effect involved in nanomedicine-based tumor targeting and promising nanodiagnostics or -therapeutics. We will also summarize the recent progress and discuss the clinical perspectives of nanocarrier-based combination therapies. The goal of this article was to provide better understanding and key considerations to develop new nanomedicine combinations and nanotheranostics options to fight against GBM.
Keywords: glioblastoma, nanomedicine, nanoparticles, local delivery, systemic delivery, EPR effect, theranostics
Introduction
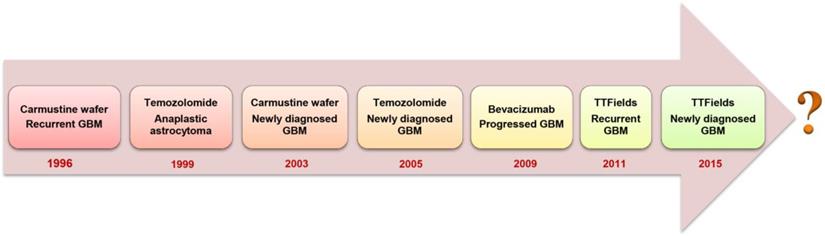
Glioblastoma multiforme (GBM) is a grade IV malignant glioma associated with very poor patient prognosis [1]. Currently, no curative treatment options exist and the 5-year survival of GBM-diagnosed patients remains lower than 6% [2]. Upon diagnosis, standard of care involves maximal surgical resection, followed by radiotherapy and concurrent chemotherapy with temozolomide (TMZ) [3]. Complete surgical resection is usually unachievable because GBM is only diagnosed when the patient develops symptoms, by which time the highly invasive tumor cells infiltrate to the crucial functional regions of the brain that control senses, actions and speech [4]. Although the continuous development of surgical imaging techniques allows increasingly more extensive surgical resections, there is always the need to balance between aggressive removal of tumor tissue whilst maintaining brain function and protecting the life quality of patients [1]. A median survival of 12.1 months can be obtained through addition of focal irradiation [3]. However, radiation is associated with cognitive impairment, DNA lesions and other severe systemic side effects [5]. The anti-angiogenic drug bevacizumab was approved by the U.S. Food and Drug Administration (FDA) to treat recurrent GBM that has progressed after prior therapy. The addition of bevacizumab to concomitant chemo-radiotherapy for newly diagnosed GBM showed prolonged progression-free survival (PFS) but failed to show an overall survival (OS) benefit in phase III trials [6, 7]. Optune®, a device that creates low-intensity and alternating tumor-treating fields, also obtained approval from the FDA as treatment together with TMZ for newly diagnosed GBM in 2015 [8]. Despite these multidisciplinary therapies, most patients still develop tumor recurrence within 1 to 2 years of diagnosis. Patients may then undergo repeated resection, different chemotherapies, bevacizumab therapy or additional radiotherapy. Unfortunately, there is limited evidence showing that those treatments can increase the survival time. Thus, regarding the unmet medical needs for GBM patients, the fight against GBM is far from over (Figure 1).
Evolution of FDA-approved GBM treatment approaches.
The general concerns associated with chemotherapy include the blood brain barrier, complicated tumor heterogeneity, glioma stem cells, DNA damage repair mechanisms and drug efflux pumps (Table 1 and Graphical abstract) [9, 10].
Rationale of nanocarrier-based combination therapy against GBM
| Issues with single drug treatment | Advantages of combination therapy | Advantages of nanocarrier-based drug delivery | Advantages of nanocarrier-based combination therapy |
|---|---|---|---|
| • Tumor heterogeneity • DNA damage repair • Glioma stem cells • Efflux pump • Dose-limiting toxicity | • Combination of drugs with different mechanisms of action • Combination with anti-GSC drug • Combination with efflux-pump inhibitor • Combination with MGMT inhibitor • Combination of drugs with non-overlapping toxicity | • Drug encapsulation and solubilization • Drug protection • Increase of cellular uptake of nanoparticles via endocytosis • Targeted delivery • Controlled/sustained release kinetics • Improvement of drug half-lives • Facilitate diagnostic or theranostic agents | • Combination of drug with different properties (solubility, BBB permeability, pharmacokinetics) • Ensure the synergistic drug ratio • Ensure the colocalization of drugs into tumor site • Facilitate sequential drug exposures |
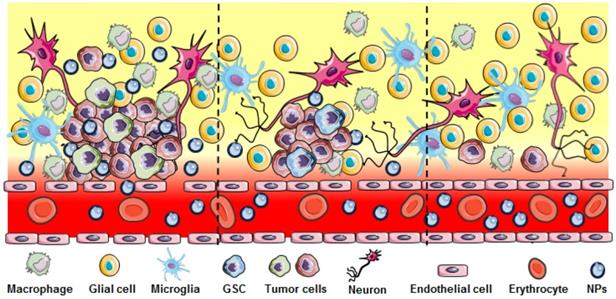
The blood brain barrier (BBB) is a particularly formidable challenge in developing therapeutics for brain tumors. Brain microvascular endothelial cells, pericytes, astrocytes, tight junctions, neurons and the basement membrane together form a physical barrier to protect the brain and maintain a well-defined intracranial environment [11]. Although several specialized transport systems mediate the entry of essential substances such as nucleosides, glucose, amino acids, hormones and receptor-mediated endocytosis via specific proteins (e.g., transferrin and lactoferrin), the tight junctions prevent the passive penetration of hydrophilic molecules from the blood circulation to the brain. GBM therapeutics need to be able to cross this barrier and penetrate the brain to reach the tumor. However, only small lipophilic chemotherapeutic agents with a molecular weight less 400 Da and 8 hydrogen bonds can passively pass through the BBB [12]. TMZ is an orally administered alkylating agent that can be transported across the BBB and has remarkable distribution at the tumor site. However, TMZ-induced cytotoxic effects can be neutralized by various DNA repair mechanisms, re-enforcing the structural integrity of the methylated DNA bases before causing extensive tumor cell death. High-grade gliomas are characterized by disrupted and heterogeneous blood brain tumor barrier (BBTB) (Figure 2), while the tricky task in GBM treatment is reaching the residual tumor cells infiltrating to brain parenchyma where the BBTB is intact or less compromised, leading to an insufficient therapeutic effect through passive drug diffusion [13].
Heterogeneous disruption in GBM. Significant BBB breakdown seen in the bulk tumor region (left panel) allows nanoparticle extravasation. Regions with infiltrating GBM and GSC cells show less or no breakdown of the BBB (middle and right) preventing NPs or other therapeutics to reach these cells.
Another difficulty is found in the heterogeneity of GBM. Genomic research has shown that GBM contains many different cell types depending on their origin or subsequent genetic and epigenetic conversions [14]. Single-cell sequencing of five primary GBM showed inherently variable gene expression in diverse transcriptional programs associated with oncogenic signaling, hypoxia, proliferation and the complement/immune response [15].
This genetic drift can result in self-renewing, tumorigenic glioblastoma stem cells (GSCs) that contribute to tumor initiation and therapeutic resistance [16]. Stem cell-like properties allow GSCs to differentiate into highly proliferating progenitor-like tumor cells or other differentiated tumor cells, which can be more resistant to radio- and chemotherapy than non GSC tumor cells both in vitro and in vivo. Consequently, populations of glioma stem cells remain alive after initial treatment and reinitiate tumor recurrence [10].
Multidrug resistance (MDR) presents another major barrier for chemotherapeutic drugs to get access to brain tumor cells effectively. Among the different mechanisms of MDR, ATP-binding cassette (ABC) transporter-mediated exocytosis was mostly noticed. The P-glycoprotein (P-gp) which is encoded by the human MDR1 gene has been mostly considered as the cause of anti-cancer drug resistance. MDR1 P-gp is present in the brain capillaries of the BBB, as well as in many other tissues. Many drugs exhibit significantly improved brain penetration when drug efflux transporters are inhibited [17].
Because of these challenges, combining drugs with different working mechanisms has gained great attention in recent years. The right combination of compounds could enhance efficacy by targeting these issues in a synergistic or additive manner. However, the efficiency of many chemotherapeutic agents is also limited by their dose-related toxicities. As the BBB shields the brain from most systemically administrated compounds, high doses are given to achieve intracranial therapeutic drug levels. Increasing the dose of a specific anticancer drug will inevitably lead to significant toxicity. Many GBM chemotherapeutic drugs have demonstrated off-target toxicity at the doses needed to reach an intracranial effect. For example, TMZ is associated with lymphopenia, thrombocytopenia and neutropenia [18] and bevacizumab is frequently associated with hypertension, leukopenia, non-central nervous system hemorrhage and thromboembolic events [19]. Thus, combining drugs with non-overlapping toxicities and reducing the dose of each single drug may be a better choice.
With growing investigation of the tumor microenvironment and by unravelling biological and molecular pathways, increasingly more potential drug combinations are emerging. However, just combining cytotoxic compounds does not address the problems associated with poor drug distribution at the desired tumor site. Different approaches have been raised to defeat unfavorable drug distribution in the brain [20]. Among these, nanotechnology-based drug delivery is a promising strategy to enhance chemotherapy efficiency. Various nanocarriers have been investigated for drug delivery in central nervous system (CNS) tumors, such as polymeric nanoparticles, liposomes, and lipid nanocapsules. The correct nanocarrier could enhance the solubility of hydrophobic drugs, prolong compound circulation times and provide sustained drug release, improving therapeutic efficacy and safety [21, 22]. They can be administered locally or systemically, with the potential benefit of the enhanced permeation and retention (EPR) effect.
In this review, we will discuss and address the advantages of drug combinations with or without nanocarriers, nanomedicine-based tumor targeting strategies, current preclinical drug combinations, and promising nanotherapeutics and nanodiagnostics. The aim of this review was to highlight the importance and potential of drug combinations and give a comprehensive understanding of different combination strategies for GBM therapy.
Drug combination strategies
Poor drug delivery, tumor heterogeneity and drug resistance pathways have prevented single compound therapies to show significant benefits for GBM patients [2]. Combining drugs could overcome some of the problems associated with GBM treatment. Ideally, drug combinations take advantage of each individual compound's strengths and weaknesses to improve efficacy, decrease toxicity and overcome drug resistance. It starts with the method of administration (systemic versus local), which can heavily influence these parameters and determines how each compound is delivered.
Systemic delivery
Drug delivery to GBM is notoriously difficult due to the inability of most drugs to cross the BBB and penetrate the tumor tissue. Only few systemically administrated drugs reach the tumor site in a therapeutic dose. Various approaches, such as chemical modification of chemotherapeutic drugs, BBB altering strategies and efflux transporter inhibitors, are being investigated to enhance the systemic delivery of potential anti-GBM drugs. For instance, drugs can be modified to a more lipophilic form by adding lipid groups to the polar ends of therapeutic molecules. A log P (octanol-water) value ranging from 1.5 to 2.5 of lipophilic analogs has better brain permeability [23]. However, this may also increase nonspecific uptake of the drug molecule by other tissues through the blood circulation.
Alternatively, hyperosmotic agents, bioactive molecules, surfactants and ultrasound or electromagnetic waves have been used to alter the permeability of the BBB. Yet, such approaches are often associated with risks such as possible tumor diffusion to the periphery, and exposure of the brain to neurotoxins.
In addition, inhibition of ABC efflux gene families can increase drug penetration into the brain without compromising the integrity of the tight junctions and endothelial layers. However, this approach will also reduce the efflux of potential neurotoxic compounds. In fact, many of these efflux pumps transporters could not be fully inhibited due to various reasons, including multifactorial multidrug resistance and genomically unstable tumor cells [24]. Thus, further investigations of various systemic delivery approaches through enhancing BBB permeability are still required to achieve a significant therapeutic effect for GBM treatment.
Local delivery
Local intracranial delivery not only overcomes BBB-associated drug delivery issues but also prevents systemic compound clearance and/or degradation and reduces systemic side effects. As such, much lower dosages are needed. Local drug delivery to the brain and further distribution within the brain can be mediated by simple diffusion using a reservoir-catheter system or positive pressure bulk flow via convection enhanced delivery. The Ommaya reservoir is a reservoir capsule connected to a catheter located in the lateral ventricle. The capsule is embedded under the scalp and is easily accessible for cerebrospinal fluid (CSF) aspiration or drug delivery directly into the ventricular CSF and relies on the flow of CSF to distribute chemotherapeutics, radioactive compounds, antibodies, viruses or cells throughout the brain [25].
Alternatively, drugs are administered to the brain continuously with a positive pressure bulk flow via convection-enhanced delivery (CED). The positive pressure drives convective local transport of therapeutic concentrations of anti-tumor drugs into the interstitial tumor space. This technique achieved higher drug concentrations in the targeted tumor tissue compared with diffusion-limited delivery [26]. Unfortunately, common side effects, such as edema, infection and backflow along the catheter have resulted in the limited application of CED to treat GBM [26].
Locally implanted (biodegradable) drug delivery depots have increasingly gained interest. Currently, the only FDA-approved biodegradable implant is the Gliadel® wafer for newly diagnosed malignant and recurrent GBM. However, the success of Gliadel® wafers is restricted by the limited penetration of the active compound, carmustine, into the brain tumor tissue. Moreover, use of the wafers has been associated with several adverse events, including intracranial infections, wafer migration, cerebral edema, CSF leakage and seizures [27].
Nevertheless, the concept of local delivery by implanting drug-releasing depots in the tumor resection cavity remains intriguing for the treatment of GBM. Films, foams and gels have been investigated for their use in local drug delivery. Particularly hydrogels have gained much attention in recent years. In general, hydrogels are injectable, biocompatible, biodegradable and mechanically comparable to soft tissue, making them attractive for intracranial implantation. They provide a versatile drug delivery system because they can be loaded with small-molecule drugs, biomacromolecules (such as DNA or protein) or cells. The gels can be engineered to tune the release of their contents in a timeframe ranging from hours up to several months [28]. A hydrogel composed of gemcitabine lipid nanocapsules obtained sustained drug release for at least 1 month and significantly delayed tumor recurrence and prolonged survival in a GBM resection mouse model [29]. This hydrogel was also able to co-deliver gemcitabine and paclitaxel (PTX). The drug combination was shown to be synergistic in different GBM cell lines [30]. Similarly, local treatment of GBM with a photopolymerizable hydrogel coloaded with PTX and TMZ suppressed tumor growth more efficiently than the single drugs in an orthotopic U87MG tumor resection model [31]. Taken together, local delivery appears to be an effective approach to improve chemotherapy-based treatment for GBM by increasing the local dose of chemotherapeutics and simultaneously reducing systemic side effects. Polymeric implants and hydrogels show great promise but need additional development and optimization before they can be translated to clinical practice [32].
Recent clinical trials of drug combinations for GBM treatment
Different drug combination strategies have been explored in clinical trials to tackle known drawbacks of GBM treatment. A non-exhaustive summary of combination therapy trials is presented in table 2 (Source: ClinicalTrials.gov). Several chemotherapeutics have been combined with anti-angiogenic drugs. A phase II clinical trial using bevacizumab and the topoisomerase inhibitor irinotecan showed a 6-month PFS rate of 50.3% compared to 42.6% with bevacizumab alone in recurrent GBM. The median overall survival times were 9.2 months and 8.7 month for combination therapy and bevacizumab alone respectively [33].
Recent clinical trials of drug combination for GBM treatment
| Drugs | Mechanism of action | Condition | Phase/Status | Major findings | Clinical trial ID |
|---|---|---|---|---|---|
| Bevacizumab; Irinotecan | Anti-VEGF antibody; Topoisomerase I inhibitor | Recurrent Gliomas | phase II/Completed in 2013 | No results found | NCT00921167 |
| O6-Benzylguanine; Temozolomide | O6-alkylguanine-DNA alkyltransferase inhibitor; Alkylating agent | Temozolomide- resistant malignant glioma | phase II/Completed in 2008 | No results found | NCT00613093 |
| Imatinib; Hydroxyurea | Tyrosine kinase inhibitor; ribonucleoside diphosphate reductase inhibitor | Recurrent/ progressive grade II low-grade Glioma | phase II/Completed in 2012 | Groups: patients with astrocytoma or oligodendroglioma; 12-month PFS:44% and 34% respectively | NCT00615927 |
| Cediranib; Lomustine | Tyrosine kinase; Alkylating agent | Recurrent GBM | Phase III/Completed in 2016 | Groups: patients received cediranib alone, lomustine alone or drug combination; PFS: 92, 125 and 82 days respectively | NCT00777153 |
| Erlotinib; Vorinostat; Temozolomide | Tyrosine kinase inhibitor; Histone deacetylase inhibitor; Alkylating agent | Recurrent GBM | Phase II/Terminated in 2014 (Unanticipated Toxicities) | No results found | NCT01110876 |
| Sorafenib; Temsirolimus | Tyrosine kinase inhibitor; mTOR inhibitor | Recurrent GBM | Phase I/II/Completed in 2013 | Groups: patients not undergoing surgery or received anti-VEGF therapy; 6-month PFS: 17% and 10% respectively | NCT00329719 |
| Bevacizumab; Sorafenib | Anti-VEGF antibody; Tyrosine protein kinases | Recurrent GBM | Phase II/Completed in 2014 | Groups: patients received sorafenib high dose or low dose; 6-month PFS: 26% and 17% respectively | NCT00621686 |
| Bevacizumab; Temsirolimus | Anti-VEGF antibody; mTOR inhibitor | Recurrent GBM | Phase II/Completed in 2010 | No results found | NCT00800917 |
| Erlotinib; Sirolimus | Tyrosine kinase inhibitor; mTOR inhibitor | Recurrent GBM | Phase II/Completed in 2009 | Group: patients received erlotinib and sirolimus; 6-month PFS: 3% | NCT00672243 |
| Vorinostat; Bortezomib | Deacetylase inhibitor; Proteasome inhibitor | Recurrent GBM | Phase II/ Completed in 2010 | Groups: patients not undergoing surgery or undergoing surgery; 6-month PFS: 0 and 29% respectively | NCT00641706 |
| Bevacizumab; Erlotinib | Anti-VEGF antibody; Tyrosine kinase inhibitor; | Recurrent GBM | Phase II/ Completed in 2010 | Groups: patients with grade III or grade IV malignant glioma; 6-month PFS:44% and 29% respectively | NCT00671970 |
| Temozolomide; SGT-53 | Alkylating agent; Liposome-p53 DNA | Recurrent GBM | Phase II/ Recruiting | No results found | NCT02340156 |
| Glasdegib; Temozolomide | Inhibits SHH pathway interfering with cancer stem cells and endothelial migration; Alkylating agent | Newly diagnosed GBM | Phase IB/II/ Recruiting | No results found | NCT03466450 |
| Bortezomib; Temozolomide | Deplete the MGMT enzyme; Alkylating agent | Recurrent GBM with unmethylated MGMT promoter | Phase IB/II/ Recruiting | No results found | NCT03643549 |
| Bevacizumab; Capecitabine | Anti-VEGF antibody; Target myeloid-derived suppressor cells | Recurrent GBM | Phase I/ Recruiting | No results found | NCT02669173 |
Another phase II clinical trial combined TMZ with an O6-methylguanine-DNA methyltransferase (MGMT inhibitor) O6-benzylguanine (O6-BG) to rebuild drug sensitivity in TMZ-resistant anaplastic glioma. Indeed, O6-BG was able to restore TMZ sensitivity in TMZ-resistant anaplastic glioma, but not in TMZ-resistant GBM [34].
More often than not, promising preclinical anti-tumor strategies disappoint in clinical trials due to various reasons. For example, a phase I/II trial to determine the efficacy of vorinostat + erlotinib versus vorinostat + erlotinib + TMZ in patients with recurrent GBM multiforme was terminated because of unanticipated toxicities (NCT01110876). A trial that combined the histone deacetylase inhibitor vorinostat and proteasome inhibitor bortezomib to treat recurrent GBM, reported that no patient achieved 6-month PFS and only one patient showed a partial response according to the Macdonald Criteria, probably due to brain delivery issues [35].
The underwhelming results of drug combinations in clinical trials compared to the encouraging in vitro results can be explained by several factors (table 1). In some cases, the ratio between the compounds is important for their combined efficacy. In vivo, the differences in drug distribution, metabolism and excretion for each single drug need to be considered and fine-tuned to realize the desired local concentrations. Dose-limiting toxicity and, although rarely reported, insufficient intracranial drug accumulation, seem to explain most of the disappointing trials. This appears to be surmountable as systemic toxicity and increased brain penetration of drugs can be addressed by alternative delivery strategies.
Nanocarrier-based combination therapy for GBM
Preclinical and clinical research has shown that various factors may compromise the efficacy of (combinations of) therapeutics, which has led to disappointing clinical outcomes (table 1). For GBM, the BBB seems to be the main dissonant. In recent years, nanomaterials have gained attention as they have the potential to overcome many of these hurdles, mask unfavorable characteristics of the active compounds and/or improve their efficacy. It is estimated that due to the BBB, 100% of large molecules and 98% of small molecules fail to sufficiently reach the brain to achieve therapeutic levels [36]. Nanoparticles (NPs) encapsulating these molecules can be tailored to enable specific transport of their payload to the brain or facilitate penetration through the BBB, thereby enabling encapsulated drugs for previously unreachable tumors such as GBM [37].
Depending on the materials, nanoformulations are able to load hydrophilic and hydrophobic drugs, ensure sustained drug release and enhance the half-life of the drug in the circulation. For example, the half-life of TMZ was enhanced to 13.4 h compared to 1.8 h of the free drug by encapsulation in a chitosan-based nanoparticle [38]. Improving compound solubility, stability and reducing systemic toxicity are major goals in designing such formulations. Several FDA approved nanoformulations (e.g. Abraxane®, Doxil®, DaunoXome®) mainly reduce toxicity of the parent compound and thereby improve its therapeutic index. Small interfering RNAs (siRNA) show great therapeutic promise but have disappointing clinical relevance due to stability and delivery issues. The only currently FDA approved siRNA therapeutic is Patisiran®, which is based on a lipid NP formulation that improves siRNA stability.
Two decades have passed since the first NP-based cancer treatment was approved by the FDA, with an increasing number of clinical trials now ongoing, including many for GBM. A variety of systems based on polymers (micelles, dendrimers), inorganic materials (iron, silica, gold) and/or lipids (liposomes, solid lipid nanocarriers) have been investigated for drug delivery to the brain. The therapeutic drugs can be loaded in these particles by encapsulation, covalent linking or surface adsorption [37]. Depending on the design of the drug delivery system, the drugs are either passively or actively targeted to the tumor to exert their effect.
Passive targeting
Passive tumor targeting is based on the observation that certain sized particles tend to accumulate in tumor tissue much more than they do in healthy tissues. This is known as the enhanced permeability and retention effect (EPR) effect, which was first described by Matsumura and Maeda in 1986 [39]. EPR is based on aberrant pathophysiological characteristics of tumors. The presence of abnormal, fenestrated vasculature and a lack of proper lymphatic drainage results in the extravasation of NPs and reduced lymphatic clearance [40, 41] .
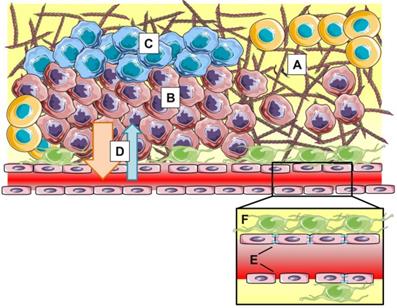
However, within the last couple of years, more and more researchers have realized that the EPR effect is highly heterogeneous both intra- and intertumorally, varies during tumor development and does not always hold up in clinical settings (Figure 3). Moreover, the magnitude of the EPR effect as seen in rodent models fails to translate to the clinic. Human tumors are drastically different from preclinical tumor models in many critical aspects such as: (i) heterogeneity or lack of fenestrations in the tumor endothelium, (ii) presence of acidic and hypoxic areas (iii) lower or heterogeneous pericyte and basement membrane coverage, (iv) and high interstitial fluid pressure (IFP) induced by dense extracellular matrix, explaining the difficulty in translating the EPR effect from bench to bedside [42].
The EPR effect is influenced by stromal parameters such as dense extracellular matrix (A), hypercellularity (B), hypoxia (C) and high interstitial fluid pressure (D). At blood vessel level (insert), heterogeneity in vascular permeability, tight junction expression (E) and pericyte coverage (F) result in varying clinical manifestations of EPR.
Interestingly, GBM is characterized by robust endothelial proliferation resulting in tortuous, disorganized and highly permeable vasculature [43, 44]. Such excessive neovascularization also affects BBB integrity which can be visualized by magnetic resonance imaging (MRI). Contrast agents such as gadolinium do not cross a healthy BBB, but in pathologies such as GBM, hypervascularization causes physical disruptions of the BBB that allow leakage of gadolinium into the tumor tissue, making the tumor visible on T1-weighted MRI. Gadolinium-enhanced areas form the golden standard in GBM diagnosis and provide guidance for surgical resection. Thus, theoretically, intravenously administered nanocarriers could exploit this phenomenon in GBM. Yet, it is evidently not this simple in the clinic.
First of all, when comparing different imaging modalities, it becomes clear that T1-weighted MRI alone does not visualize the entire tumor. Beyond the contrast-enhancing region, essentially all of GBM show non-enhancing edema on T2-weighted or fluid attenuation inversion recovery imaging [45]. These areas have impaired fluid regulation yet do not accumulate contrast agent, suggesting that the BBB is intact. Additionally, the impaired fluid regulation leads to high interstitial fluid pressure which compromises transvascular transport of molecules. It is suggested that the invasive tumor cells and tumor associated stromal cells in this peritumoral brain zone drive 90% of all recurrences [46, 47]. These cells are not or barely reached by passively targeted drugs or NPs due to their location behind an intact BBB. This should be considered when designing new therapies and drug delivery systems (DDSs), as relying solely on the EPR effect may be insufficient to target these cells [24, 44, 46].
Other key pathological features of GBM are hypoxic areas surrounded by hypercellular rings of actively migrating tumor cells [48]. These hypoxic zones, arising from inadequate vascularization, cannot be adequately reached by NPs or therapeutics in general. Moreover, hypoxia gives rise to a group of so-called pseudopalisading tumor cells that are highly migratory, pro-angiogenic, therapy resistant and show decreased proliferation [48-50].
Due to the differences in EPR effect between and within tumors, approaches to still take advantage of it may vary. In certain situations, one might benefit from tumor vasculature normalization [51-53], while in others, increasing vascular leakiness or opening up the BBB will improve treatment [54, 55]. Non-steroidal anti-inflammatory drugs (NSAID) such as COX-inhibitors are able to reduce BBB disruption in neuro-inflammation [56] and prevent protein extravasation in glioma [57]. COX-2 inhibition normalized the tumor microenvironment including its vasculature and improved the penetration and accumulation of micelles (22 nm) rather than bigger (100 nm) NPs in a solid tumor mouse model [58]. Similarly, reducing the leakiness of tumor vasculature by blocking the VEGF receptor, improved the delivery of nanoparticles in a size dependent manner in a mammary mouse model [59]. In contrast, the A2A adenosine receptor agonist lexiscan is approved by FDA for myocardial perfusion imaging. Lexiscan has the ability to transiently open the BBB and enhance the permeability of NPs to the brain [60]. Integrating lexiscan in NPs improved its BBB traversing properties and increased the efficacy of encapsulated therapeutics in an orthotopic glioma mouse model [61]. Focused ultrasound (FUS) in combination with circulating microbubbles has been demonstrated to be an effective approach to locally increase BBB permeability. The delivery of PTX-liposomes to mice brain tissue could be effectively improved by pulsed FUS sonication resulting in a two-fold higher local drug concentration and improved survival in an intracranial mouse GBM model [62].
The extent of EPR in a tumor could be tested prior to treatment using imaging modalities. In a preclinical study, the relative blood volume was found to correlate with the tumor accumulation of polymeric drug carriers. With the help of contrast-enhanced ultrasound imaging, this vascular parameter proved useful in predicting EPR-mediated tumor accumulation of NPs and could possibly be used to preselect patients eligible for nanotherapeutic treatment [63]. The size of NPs would affect their tumor distribution as well. In one study, 64Cu-labeled long-circulating particles of different size were systemically administered and tracked via MRI and positron-emission tomography (PET) in an intracranial rat GBM model. It was demonstrated that 7 hours after systemic injection, the uptake of 20 nm NPs is significantly greater than those of 110 nm. It also showed that PET/MRI co-registration of brain images may contribute to the monitoring of disease progression and determining what drug delivery approach is feasible [64].
The high variability of the EPR effect is considered as one of the major challenges for translating nanomedicine into the clinic. This issue can be addressed by indirect EPR imaging, utilizing companion diagnostics or developing systems with both diagnostic and therapeutic properties (theranostics). Indirect EPR imaging can non-invasively visualize and quantify the key EPR-determining parameters of the tumor vasculature, while theranostics or companion diagnostics can give insight in NPs distribution and tumor responses.
Tumor immune microenvironment and nanocarrier-based drug delivery
Many cancers are preceded by infections or (chronic) inflammation and it is increasingly clear that the immune system plays a central role not only in cancer development but also in tumor progression [65]. In GBM, the tumor immune microenvironment heavily influences progression, invasion, metabolic reprogramming and therapy resistance, mostly orchestrated by the present immune cells [66]. Peripheral monocytes are attracted by tumor secreted factors, infiltrate the brain, differentiate to macrophages and together with the residential microglia develop a class of cells called tumor associated microglia/macrophages (TAM) [67]. They can represent up to 50% of the GBM mass and high TAM density has been correlated with glioma grade [68] and poor prognosis [69]. GBM cells produce and secrete chemoattractants and signaling molecules to create an environment that drives TAM towards a predominantly immunosuppressive and tumor supportive (TAM2), rather than an immune-stimulatory and anti-tumor phenotype (TAM1) [70]. Immunosuppressive TAM promote tissue remodeling and angiogenesis, thereby driving GBM progression [67]. Additionally, TAM further drive angiogenesis via the secretion of pro-angiogenic factors such as vascular endothelial growth factor (VEGF) and CXC-chemokine ligand 2, paving the way for hypervascularity as seen in GBM [71]. Modulation of the TAM polarization has been emerging as a new therapeutic target for GBM. Polarization from TAM2 towards TAM1 not only activates the cytotoxic T cells but also induces the secretion of antitumor cytokines. Zhao et al. developed an albumin-based biomimetic NPs with transferrin receptor-binding peptide T12 and mannose as targeting ligands for codelivery of the disulfiram/copper complex and the macrophage modulator regorafenib. The T12 peptide can enhance BBB permeability and glioma cell uptake. The mannose ligand can bind to mannose receptors on TAM2. This system efficiently inhibited the glioma cell proliferation, successfully induced the protumor TAM2 towards antitumor TAM1 and triggered macrophage-directed anti-glioma immunotherapy via TAM, regulatory T cells, CD8+ T cells and cytokines [72].
The inflammatory response observed in GBM results in increased vascular permeability as well. In turn, this will not only allow for extravasation of immune cells, but also of therapeutics including NPs. Furthermore, extracellular matrix (ECM) remodeling can influence NPs transport through the interstitial space [73]. Second, in addition to modulation of the physical tumor environment (e.g. ECM and vasculature), TAM can also directly contribute to drug retention. Macrophages protect the body by specializing in phagocytosing pathogens, cellular debris and foreign materials. It can be expected that the bulk of NPs that are taken up by the tumor ends up in these cells [74]. Indeed, most NPs end up in TAM, even when they only represent 1% of tumor mass [75]. Elegant studies by Miller et al. show that NPs predominantly accumulate in tumor associated immune cells rather than tumor cells. They used fluorescently labeled polymeric NP (100 nm) together with magnetic NP (20 nm) in several tumor mouse models to predict NP tumor accumulation and treatment outcome. It appeared that NP distribution was mainly determined by vascularization and permeability at early time points, but at later time points by cellular uptake. Both NPs were mostly taken up by host phagocytes (> 90%). High local TAM counts correlated with increased NP accumulation and as such proved an important component of the EPR effect [76]. Interestingly, the same group showed that NP accumulating TAM can function as a drug depot, which slowly releasing encapsulated therapeutics to surrounding (tumor) cells. Depleting TAM numbers reduced NP accumulation and treatment efficacy, illustrating a role for TAM in tumor drug retention [77]. Furthermore, it was observed that phagocytes, after ingestion of NPs, can move across the vascular wall and carry the NP to the extravascular space. In vitro studies show monocyte mediated transfer of NP over an endothelial monolayer [78]. In vivo, TAM with ingested NPs has been observed to cross the BBB and migrate to (distant) tumors. As such, NPs were shuttled between contralateral CNS tumors via migrating TAMs [79]. Thus, TAMs are able to modulate the tumor microenvironment thereby enhancing vascular and interstitial permeability, while their phagocytic nature drives NP uptake and prolongs retention in the tumor. Since TAMs are key regulators of several EPR driving mechanisms, it seems natural to investigate ways to take advantage and try to improve drug delivery via these cells.
Another point that might influence the immune microenvironment-based therapy for GBM is the immunosuppressive effects of chemotherapy. Indeed, systemic chemotherapy might suppress the bone marrow and consequently impact the amount and activation state of immune cells [80]. Nevertheless, some promising results have been seen when giving local chemotherapy. It is exciting to note that local chemotherapy was able to potentiate anti- programmed death 1 (PD-1)-mediated antitumor immune response through increased percentage of dendritic cells, greater antigen presentation and further clonal activation of tumor-specific T cell responses [81]. Additionally, it was shown that cancer cells may undergo bona fide immunogenic cell death (ICD) after exposure to some chemotherapeutics that are currently used in clinic. This process generates specific changes in cell surface structures and releases soluble mediators, e.g. ATP, calreticulin, high-mobility group box 1(HMGB1) and chemokine ligand 10 (CL10), allowing dendritic cells to recognize the dying cell and initiate an anti-tumor immune response to clear tumor cells [82]. Further investigations may focus on nanoimmunotherapy in combination with the optimized dosing of chemotherapeutic drugs.
Active targeting
When passive targeting is insufficient, active targeting of NPs can facilitate their transport over the BBB. With polymeric NPs this is often achieved by modifying their surface with surfactants or BBB- or glioma-specific ligands. For example, doxorubicin (DOX) loaded poly butyl-cyanoacrylate NPs were more effective when coated with the surfactant P80 and increased the survival time of GBM tumor bearing rats compared to the non-coated group [83]. Additionally, conjugation of the BBB ligand transferrin to PTX loaded poly(lactic-co-glycolic acid (PLGA)-NPs showed significant enhancement of cellular uptake and cytotoxicity on C6 rat glioma cell line, in comparison with the non-conjugated PLGA NPs, by taking advantage of receptor mediated endocytosis (RME) [84]. Similarly, targeting polymeric micelles grafted with cyclic Arg-Gly-Asp (cRGD), a ligand with selective affinity for the αvβ3 and αvβ5 integrins that are overexpressed on tumor vasculature and tumor cells, vastly enhanced micelle uptake in tumor in an orthotopic mouse GBM model. Compared to specific targeted micelles, RGD-micelles loaded with oxiplatin significantly reduced tumor growth [85].
Lipid NPs, liposomes in particular, are also widely investigated in the drug delivery field. Liposomes can encapsulate hydrophilic compounds in the aqueous compartment and hydrophobic compounds in the bilayer making it a versatile delivery vehicle. Covering the surface of these particles (or NPs in general) with polyethylene glycol (PEG) neutralizes surface charge but thereby ensures prolonged circulation and increases the EPR effect. Radiotherapy supplemented with PEGylated liposomal DOX (Caelyx®) resulted in a significantly higher intratumoral concentration of DOX than normal brain tissue in GBM patients [86]. Positively charged liposomes have been shown to penetrate BBB by electrostatic interaction with polyanions on the BBB, which leads to adsorptive-mediated endocytosis [87]. Alternatively, the liposomal surface can be decorated with targeting ligands to facilitate BBB transport via RME and promote tumor specific uptake. Transferrin conjugated liposomes increased brain delivery of 5-fluorouracil by 13 times compared to non-conjugated liposomes [88]. In another study, interleukin-13-grafted liposomes significantly enhanced the cytotoxicity and tumor accumulation of DOX in comparison with the free drug on a subcutaneous mouse glioma model [89].
Lipid nanocarriers (LNCs) are also able to load both hydrophilic and hydrophobic molecules in an aqueous core by formation of reversed micelles [90]. Similar to liposomes, LNCs may also be coated with PEG or grafted with ligands to ensure efficient brain drug delivery [91]. One LNC component, the non-ionic surfactant HS15, was indicated as a key element in producing a P-gp suppressing effect which could aid retaining the delivered therapeutic in the brain [92].
Sometimes NPs are made by organic-inorganic hybrid materials as the magnetic and optical features of metallic NPs can be used to actively target to tumor site and monitor delivery non-invasively. In an U87MG orthotopic tumor model, magnetic targeting treatment with superparamagnetic iron oxide (SPIO) and PTX coloaded PLGA NPs significantly enhanced the median survival time compared with the passive targeting treatment group [93]. The tunable small size of gold NPs makes them good candidates as carriers for delivering cargoes across the BBB and targeting brain tumors. Transactivator of transcription (TAT) peptide-targeted multifunctional gold NPs were able to efficiently cross the BBB in an intracranial GBM mouse model and deliver anticancer drug DOX and gadolinium contrast agents to brain tumor tissues [94]. Chlorotoxin (CTX) has been shown to be a specific target and efficacious in blocking the glioma Cl channel activity. In one publication, a 131I-labeded CTX-functionalized polyethylenimine-entrapped gold nanoparticles as a multifunctional glioma-targeting nanoprobe was generated. After incubation with the NPs, C6 cells displayed much stronger fluorescence intensities than those treated with negative controls under the same conditions by confocal imaging. This CTX-loaded NP could also act as a nanoprobe for the targeted single photon emission computed tomography (SPECT)/ computed tomography (CT) imaging of glioma cells and in a subcutaneous tumor model [95]. In another study, the PLGA-gold hybrid NPs were loaded with docetaxel and targeted using angiopep-2. Targeting the NPs improved the efficacy of docetaxel due to improved delivery, while the gold allowed X-ray imaging of the accumulated NPs in a xenograft mouse GBM model. Moreover, thermal therapy could be applied by exposing the gold NPs to an 808 nm laser, which further increased therapeutic efficacy through local heating [96].
Overall, NPs show great potential in preclinical studies. They often improve accumulation of therapeutic compounds via passive targeting, while active targeting might be employed to increase this accumulation in hard to reach tumors such as GBM. Imaging (whether direct or indirect) can give an indication of the potential of certain tumors to be treated with NP formulations, thereby preselecting patients and possibly improving treatment success.
Nanocarrier-loaded with diagnostic or theranostic agents
Nanocarrier formulations additionally offer the opportunity to incorporate imaging features facilitating diagnostic as well as monitoring features. Accurate diagnosis is essential for adequate cancer treatment. For GBM, imaging is pivotal as the alternative diagnostics (e.g. biopsies or surgery) are highly invasive. As such, imaging is crucial for (preliminary) tumor characterization and localization, planning of surgical strategies and monitoring of treatment response.
Generally, maximal gross resection is correlated with increased survival [97, 98]. Maximum safe resection is executed based on preoperative imaging combined with intraoperative image-guided surgery. Contrast enhanced MRI is the mainstay imaging technique of GBM, sometimes supported with PET or CT. They are invaluable yet suffer from drawbacks such as limited sensitivity (tumor vs healthy tissue), discriminative power (pseudoprogression vs progression), low anatomical information, hazardous radiation and contrast agent delivery issues (as discussed earlier in this review) [99, 100]. As such, there is a constant search to improve technologies and methodologies to push neuroimaging forward. In recent years more advanced techniques such as dynamic susceptibility contrast (DSC)-MRI [101], dynamic contrast enhanced (DEC)-MRI [102] and amino acid PET [100] allow for more accurate imaging of GBM as well as monitoring treatment response.
Where conventional contrast enhancers and tracers can only enter the brain via compromised and leaky vasculature, amino acid linked PET tracers can transverse the intact BBB [103]. Subsequent tumor accumulation of these tracers occurs due to the increased metabolic demands, hyper vascularity and overexpression of specific transporters in neoplastic tissue [104]. Similar to amino acids, NPs can help to cross the BBB and improve tumor localization and simultaneously offer a platform for additional imaging probes or other molecules such as targeting ligands. Moreover, nanoformulations can increase tracer circulation times which proved beneficial for their clinical relevance [105]. It is therefore of no surprise there is growing interest in designing diagnostic NPs and further improving brain tumor imaging [106].
Coupling the integrin targeting ligand RGD to PET tracers increased their tumor specificity in mouse models [107] and allowed more accurate tumor to background distinction and treatment response monitoring [108, 109]. Tumor localization of RGD coupled-tracers was also seen in patients but seemed to be hampered by intact BBB and partial tumor volume issues [110]. Nevertheless, RGD-tracers were able to detect GBM lesions and predict treatment response to chemo-radiotherapy in patients [111]. Similarly, a gastrin-releasing peptide receptor targeted gadolinium tracer was conjugated with the near infrared fluorophore IRDye800CW forming a dual modality PET/near infrared (NIR) tracer which allowed intra-operational NIR image-guided resection in human GBM patients [112].
Preclinically, novel materials and approaches are being investigated such as quantum dots (QDs), metallic NPs, protein conjugates and polymeric particles [113]. QDs are semi-conducting nanocrystals (2-10 nm) with superior optical properties such as strong resistance to photobleaching, broad excitation spectra with narrow emission spectra, and long fluorescent lifetime [114]. Therefore they hold great promise in the field of fluorescent imaging of tumors. QDs functionalized with a tumor penetrating peptide showed increased localization to intracranial GBM tumor tissue compared to healthy brain tissue in mice, mostly due to EPR effect. The QDs could be visualized in the tumor tissue via fluorescence imaging [115]. Alternatively, smaller 5.74 nm cationic carbon dots emitting red light were able to cross the intact rat BBB, allowing early diagnosis contrary to contrast agents. The dots accumulated in orthotopic glioma with high tumor to background ratio allowing accurate tumor delineation [116]. cRGD functionalized QDs allowed intravital fluorescent imaging of the tumor for prolonged periods of time in an intracranial GBM mouse model, with down to single cell level fluorescence imaging ex vivo [117]. These dots could potentially allow real-time imaging and visualization of residual tumor for the surgeon or aid in cell identification and localization in biopsies. NIR and (infrared) IR probes have deeper tissue penetration than fluorescent imaging but might still be insufficient for deep seated tumors. One way to increase the imaging possibilities of fluorescent probes beyond the optical diffusion limits is photoacoustic (PA) imaging. This technique takes advantage of the sound waves generated by particles absorbing light, which can be converted into high resolution structural images. Ge et al. designed a carbon-based QD that could emit red light. Intravenously injected dots accumulated in xenograft HeLa tumors via EPR and allowed in vivo fluorescent as well as PA imaging. Moreover, the dots could be used as photothermal inducers, as a large percentage of the absorbed energy is converted to heat, facilitating thermal ablation of tumor tissue [118]. The FDA approved fluorescent dye indocyanine green (ICG) has been used in a similar fashion in a theranostic particle for deep seated GBM. Molybdenum disulfide NPs where used to passively target ICG to intracranial glioma in an orthotopic mouse model. Using PA imaging it was possible to identify tumor mass up to 3.5 mm below the scalp [119].
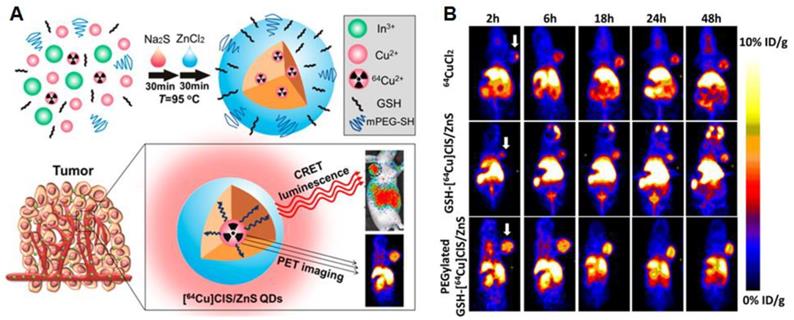
Alternatively, QDs can be coupled to other imaging agents such as radioactive tracers to allow deeper tissue imaging. PEGylated radioactive QDs were made using metal chlorides and 64CuCl2. These particles could be imaged using PET scans in a xenograft GBM mouse model. The dots were self-luminescent via cerenkov resonance energy transfer, making it possible to visualize tumors via luminescence imaging as the EPR effect resulted in tumor accumulation of the PEGylated particles [120] (Figure 4A and B). Radiolabeled carbon based dots (C-dots) with cRGD targeting showed promising tumor localization and imaging possibilities together with favorable pharmacokinetics and dynamics in human melanoma patients with metastases. The C-dots could be visualized accurately via PET imaging and showed promising fluorescence imaging opportunities in an earlier mouse study [121].
(A) and (B): Radioactive [64Cu]CLS/ZnS QDs as PET/self-illuminating luminescence imaging agents show promising in vivo visualization possibilities. Adapted with permission from [120], copyright 2014 American chemical society.
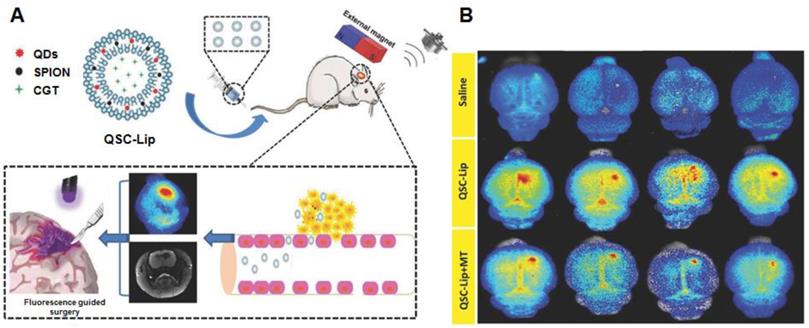
As mentioned in the section of active targeting, superparamagnetic iron oxide nanoparticles (SPIONs) are extensively investigated as standalone theranostic particles as well. The magnetic iron core can be visualized via MRI allowing tracking of the particles and simultaneously present a way of directing the particles toward their goal via magnetic fields. A second biocompatible layer reduces toxicity and extends circulation [122]. By including QDs in a liposome loaded with SPION, a dual-imaging platform was created that simultaneously functions as a carrier for therapeutics. The liposomes could be directed to the tumor via magnetic targeting, assisted by ultrasound-targeted microbubble destruction of the BBB. They were visualized by MRI (SPION) and fluorescent imaging (using the QD). The fluorescent signal was able to improve gross resection while the loaded cinglitide could inhibit tumor growth [123] (Figure 5A and B). Locally, SPIONs can be used to induce hyperthermia. Aminosilane-coated SPIONs are approved in Europe under the name NanoTherm® for hyperthermal treatment of primary and recurrent GBM. The particles are injected intratumorally, followed by applying alternating magnetic fields to produce cell killing heat. Combined with radiotherapy, NanoTherm® prolonged survival in 66 patients with recurrent GBM [124]. Similarly, by coating the cavity wall with magnetic NPs after tumor resection, hyperthermal treatment of residual tumor was possible. Combined with radiotherapy this approach showed a prolonged anti-tumor immune response with some patients achieving long lasting stable disease [125]. A hybrid NP, consisting of a magnetic iron core coated with a carbon shell with photoluminescent properties, was used to image and treat mice bearing C6 GBM tumors. The particles could be visualized with fluorescent imaging as well as MRI and by using a NIR, photothermal treatment significantly inhibited tumor growth in tumor-bearing mice [126].
(A) and (B) Using an exogenous magnetic field to target liposomes loaded with multiple imaging agents and therapeutic drugs to an intracranial tumor. The integrated QDs can be used for fluorescence guided resection. Adapted with permission from [123], copyright 2018 WILEY‐VCH Verlag GmbH & Co. KGaA, Weinheim.
As more and more information is gathered on the presence of different cellular compositions in different tumor regions, subtype specific diagnostic/ theranostic NPs might be used to elucidate the tumor make up and provide tailored therapy [46].
Considerations for nanocarrier-based drug combination therapy for GBM
Due to differences in BBB permeability, drug stability and pharmacokinetics, therapeutic efficacy may be disappointing when simply administering two or more theoretically synergistic compounds (e.g. Bortezomib and HDAC; O6-BG and TMZ) [127, 128]. Careful planning is needed to make sure both compounds arrive at the target site with desired concentration. Alternatively, encapsulating multiple agents in a single nanocarrier ensures both agents will travel and reach their destination together. A folate-targeted poly (ε-caprolactone) (PCL) and linear poly (ethylene imine) (PEI) cationic delivery system was capable of co-encapsulating BCL-2 siRNA and DOX. Blocking the production of BCL-2 effectively inhibited the anti-apoptotic response and sensitized C6 cells to DOX treatment, demonstrating the synergistic effect of the DOX and BCL-siRNA when they are delivered simultaneously [129]. By tweaking the release kinetics of the individual compounds, it is possible to control drug ratios and achieve improved therapeutic effects. By optimizing PTX: TMZ ratios within a single NP, therapeutic efficacy could be improved compared to single drug NPs or free drug combinations against GBM cell lines in vitro. Intravenous administration of NPs encapsulating the compounds in their optimal ratio significantly inhibited tumor growth in a U87 subcutaneous model in vivo [130].
To find optimized ratios, different approaches to define the combination effect need to be conducted before the nanoformulations. The most well-known median-effect-based method described by Chou and Talalay could be helpful to analyze the drug combination effect, which provides strict quantitative standards to establish drug combination therapy by research institutions, drug inspection bureaus and pharmaceutical factories [131]. As for the nanocarrier design, drug coloaded in same nanocarrier might be superior at cellular levels as synergistic drugs can be both released in the same cell. However, a few factors such as the partition coefficient, the molecular weight and the synergistic ratio of the payloads have to be considered. Thus, free drug form or drug encapsulated in different NPs seems to be more flexible for combination regimen design.
Besides the optimal ratios, the timing and order in which drugs are administered can have significant effects on the treatment outcome. For instance, the complex tumor microenvironment can greatly impair drug distribution within the tumor [132]. Improved drug distribution and efficacy can be achieved by administering the drugs in a specific order, thereby “priming” the microenvironment. Patients who were treated with the antiangiogenic drug cediranib together with radio-chemotherapy achieved improved PFS and OS when they showed improved tumor perfusion because of the vasculature normalization induced by the anti-angiogenic kinase inhibitor given first [133]. Whether the improved survival is due to increased efficacy of the secondary treatment, the additional effect of the anti-angiogenic therapy or both remains unclear [134]. However, several clinical studies observed that bevacuzimab only provided survival benefits when combined with radio-chemotherapy [53]. This suggests that the anti-angiogenic drug alone was not primarily responsible for the therapeutic effect and was more important in a supportive role to improve drug distribution with possible additional effects on interstitial fluid pressure and hypoxia, two factors that influence both drug distribution and patient survival [53]. Another approach addresses the movement of (nano-) particles in the dense tumor interstitial space. Inducing cellular apoptosis can open up this interstitial space, thereby improving tumor penetration and transfection by siRNA NPs [135]. Similar results were achieved by attacking structural ECM molecules. Pretreating mice with losartan, a drug against hypertension that additionally acts as an anti-fibrotic agent, reduced collagen I levels and improved the distribution and efficacy of an oncolytic virus and Doxil® (both ~100 nm) in several tumor models [73].
The timing and order of treatments can also significantly affect the way tumors respond to certain insults on a cellular level, a factor that often seems overlooked. Knocking down epidermal growth factor receptor (EGFR) signaling in triple-negative breast cancer cells markedly increased their sensitivity to subsequent DOX treatment. Reverse order or simultaneous administration had no such effect and could even be inhibitory, emphasizing the importance of timing rather than just co-administration [136]. Inhibiting the Wnt/β‐catenin signaling pathway with aspirin, sensitized glioma cells to TMZ. Co-loading aspirin with TMZ in a PLGA microsphere significantly increased tumor inhibition compared particles loaded with TMZ alone in a xenograft mouse model [137]. Similarly, a wildtype p53 gene plasmid was loaded in a transferrin targeted liposome (SGT-53) which could sensitize human glioma cells to TMZ. Moreover, pretreatment of TMZ resistant tumors with SGT-53 reversed TMZ resistance, while co-delivery of SGT-53 and TMZ postponed the development of TMZ resistance in an intracranial mouse GBM model [138]. Alternatively, a stapled peptide was designed against the p53 inhibitors MDMX and MDM2 which was loaded in an RGD-targeted micelle. The micelles could reach an intracranial tumor and exert potent p53-dependent anti-proliferative activity. In addition, activating p53 sensitized the tumors for subsequent TMZ treatment [139].
Intelligent designs of NPs [140, 141] or more elaborate DDSs [142] can facilitate such sequential drug exposures and might improve the therapeutic efficacy of compound combinations. For instance, a 1,2-dioleoyl-sn-glycero-3-phosphate (DOPA) and poly L-lactide (PLA)-PEG NPs system loaded with erlotinib and DOX was found to facilitate an optimal order of administration for these two drugs, which is according to the fact that basal A type of triple negative breast cancer cells can be sensitized to DNA-damaging agents after EGFR signaling is suppressed [141]. Thus, it could be possible for nanocarriers to not only codeliver different therapeutic compounds with different physicochemical properties, but also sequentially release them in a desired order.
Perspectives
As single drug therapies often seem insufficient for the treatment GBM, the focus has shifted to combination therapies. It is important to find combinations that act synergistically and preferably on different targets as tumor heterogeneity and emergence of resistance pathways need to be considered. Genetic profiling of tumors can help in finding the most promising approach. Incorporating established therapies in combination approaches will ensure faster translation to the clinic while adding compounds for novel targets such as the tumor immune microenvironment has shown promising results. However, simply administering two compounds systemically will most likely result in underwhelming results. Brain delivery of most compounds has proven challenging and seems to be insufficiently acknowledged as few clinical trials investigate or report brain concentrations of the used compounds.
Intracranial delivery of therapeutics is promising but still needs significant optimization before it is sufficiently practical to be implemented in standard care. Systemic delivery is predominantly hampered by the BBB. There is increasing interest in nanoformulations for the delivery of drugs to the brain and has shown promising results in preclinical (small) animal studies. The ideal design of a nanosized delivery system depends predominantly on the encapsulated molecules, the preferred release profile of each compound (simultaneous or time staggered) and which cells are targeted. Nevertheless, certain characteristics seem to be universally beneficial. PEGylating the particle and keeping the size around 20-75nm ensures prolonged circulation and at the same time allows sufficient tissue penetration. Factors such as the partition coefficient, the molecular weight and the synergistic ratio of the payloads have to be considered as well. Although nanocarrier based brain delivery shows promising results in preclinical in vivo studies, results fail to translate to the clinic. This is partly due to too simplistic (murine) models that don't represent the complex human tumor heterogeneity.
In order to have a more accurate prediction of clinical outcome of novel therapeutic strategies, an ideal mouse glioma model should be orthotopic and highly reproducible with predictable tumor growth, bear a genetic similarity to human glioma, show cellular heterogeneity and angiogenic-like growth [143]. Patient derived GBM cells can mimic the invasiveness and infiltration behavior of human GBM. Nevertheless, the inability to fully restate the genetic heterogeneity and phenotype of spontaneously occurring human brain tumors in a foreign microenvironment is still a major limitation [144]. Mostly, genetically engineered mouse models (GEMMs) have used combinations of the tumor suppressor p53 and/or Rb knock-down and the activation of pro-survival RTK and Ras signaling to allow for de novo tumor formation. With GEMMs, it could be possible to investigate the function of tight junction proteins, transporters, or ECM components in BBB development and biology as well. On the other hand, GEMMs also failed to recapitulate the intratumoral heterogeneity seen in patients. Other limitations like expensive breeding and low tumor penetrance have to be considered [145]. Canine spontaneous brain tumors are valuable model systems to evaluate the clinical translation of different brain tumor therapeutic strategies for many reasons. The prevalence of malignant gliomas among dogs is comparable to that in humans. The gross pathological, microscopic and immune-histochemical features are similar between human and canine spontaneous brain tumors. The immunologic features associated with human and canine brain tumors also share similarities, such as the tumor infiltration of macrophages or T cells and PD-1 immunoinhibitory mechanisms. Those features are less frequently observed or sometimes absent in rodent human brain tumor xenograft models. Therefore, those models could be considered as a clinical relevant tumor model for investigations of novel drug delivery systems [146].
Additionally, EPR based passive brain targeting of NPs as seen in animal models fails to translate to the clinic. Active targeting is often suggested to improve tumor accumulation yet it is most likely more favorable to pre-select for patients that are suited for NP based treatment. Novel imaging modalities as well as the rise of nanotheranostics will provide the opportunity to pre-select patients with appropriate tumor characteristics such as high tumor perfusion and large relative blood volumes. Additionally, nanotheranostics will provide a way to monitor drug delivery and possibly visualize treatment outcome.
The preclinical success of immunotherapy of GBM has not been replicated in human clinical trials. Both vaccines as single therapy and immune check point inhibitors have led to disappointing clinical results [147, 148]. Future immune-based strategies will be focused on combination of different immunotherapeutic approaches to reduce immunosuppression and/or enhance immune response with other modalities.
Conclusions
GBM is a complicated cancer that involves various sophisticated molecular pathways, gene mutations, and tumor microenvironments. Despite plentiful investigations, an unmet medical need persists for the treatment of invasive GBM. Here we have described the current strategies for nanomedicine-based drug combination therapies. With increasing knowledge of GBM molecular pathways, increasingly more intelligent drug combination strategies will be developed. Nanocarriers will be designed to improve delivery and allow targeting of different pathways within the tumor microenvironment. Future approaches will exploit different nanocarrier-based combinations, the most promising being immunotherapy and nanotheranostics, to enhance the therapeutic benefits for GBM. With the broad knowledge and efforts of researchers, nanocarrier-based combination therapies are expanding the way for success in the battle for GBM treatment.
Acknowledgements
This study was supported by a grant from the Fondation contre le Cancer (Belgium) and grant 15204 in the Technology for Oncology Partnership between Applied and Engineering Sciences of the Netherlands Organization for Scientific Research and the Dutch Cancer Society.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Young RM, Jamshidi A, Davis G, Sherman JH. Current trends in the surgical management and treatment of adult glioblastoma. Ann Transl Med. 2015;3:121
2. Shergalis A, Bankhead A, Luesakul U, Muangsin N, Neamati N. Current challenges and opportunities in treating glioblastoma. Pharmacol Rev. 2018;70:412-45
3. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987-96
4. Davis ME. Glioblastoma: overview of disease and treatment. Clin J Oncol Nurs. 2016;20:S2-S8
5. Ozdemir-Kaynak E, Qutub AA, Yesil-Celiktas O. Advances in glioblastoma multiforme treatment: new models for nanoparticle therapy. Front Physiol. 2018;9:170
6. Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R. et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014;370:709-22
7. Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA. et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370:699-708
8. Stupp R, Taillibert S, Kanner AA. et al. Maintenance therapy with tumor-treating fields plus temozolomide vs temozolomide alone for glioblastoma: A randomized clinical trial. JAMA. 2015;314:2535-43
9. Ganipineni LP, Danhier F, Préat V. Drug delivery challenges and future of chemotherapeutic nanomedicine for glioblastoma treatment. J Control Release. 2018;281:42-57
10. Haar CP, Hebbar P, Wallace GC, Das A, Vandergrift WA, Smith JA. et al. Drug resistance in glioblastoma: a mini review. Neurochem Res. 2012;37:1192-200
11. Dong X. Current strategies for brain drug delivery. Theranostics. 2018;8:1481-93
12. Pardridge WM. Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab. 2012;32:1959-72
13. Wolburg H, Noell S, Fallier-Becker P, Mack AF, Wolburg-Buchholz K. The disturbed blood-brain barrier in human glioblastoma. Mol Aspects Med. 2012;33:579-89
14. Osuka S, Van Meir EG. Overcoming therapeutic resistance in glioblastoma: the way forward. J Clin Invest. 2017;127:415-26
15. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H. et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396-401
16. Safa AR, Saadatzadeh MR, Cohen-Gadol AA, Pollok KE, Bijangi-Vishehsaraei K. Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs. Genes & Diseases. 2015;2:152-63
17. Li Y, Yuan H, Yang K, Xu W, Tang W, Li X. The structure and functions of P-glycoprotein. Curr Med Chem. 2010;17:786-800
18. Perry JR, Laperriere N, O'callaghan CJ, Brandes AA, Menten J, Phillips C. et al. Short-course radiation plus temozolomide in elderly patients with glioblastoma. N Engl J Med. 2017;376:1027-37
19. Diaz RJ, Ali S, Qadir MG, De La Fuente MI, Ivan ME, Komotar RJ. The role of bevacizumab in the treatment of glioblastoma. J Neurooncol. 2017;133:455-67
20. Alam MI, Beg S, Samad A, Baboota S, Kohli K, Ali J. et al. Strategy for effective brain drug delivery. Eur J Pharm Sci. 2010;40:385-403
21. Davis ME, Chen Z, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nanoscience and technology: a collection of reviews from nature journals: World Scientific. 2010:239-50
22. Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751
23. Lu CT, Zhao YZ, Wong HL, Cai J, Peng L, Tian XQ. Current approaches to enhance CNS delivery of drugs across the brain barriers. Int J Nanomedicine. 2014;9:2241
24. Van Tellingen O, Yetkin-Arik B, De Gooijer M, Wesseling P, Wurdinger T, De Vries H. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist Updat. 2015;19:1-12
25. Ommaya AK. Subcutaneous reservoir and pump for sterile access to ventricular cerebrospinal fluid. Lancet. 1963;2:983-4
26. Vogelbaum MA, Aghi MK. Convection-enhanced delivery for the treatment of glioblastoma. Neuro Oncol. 2015;17:ii3-ii8
27. De Bonis P, Anile C, Pompucci A, Fiorentino A, Balducci M, Chiesa S. et al. Safety and efficacy of Gliadel wafers for newly diagnosed and recurrent glioblastoma. Acta Neurochir (Wien). 2012;154:1371-8
28. Buwalda SJ, Vermonden T, Hennink WE. Hydrogels for therapeutic delivery: current developments and future directions. Biomacromolecules. 2017;18:316-30
29. Bastiancich C, Bianco J, Vanvarenberg K, Ucakar B, Joudiou N, Gallez B. et al. Injectable nanomedicine hydrogel for local chemotherapy of glioblastoma after surgical resection. J Control Release. 2017;264:45-54
30. Bastiancich C, Bozzato E, Luyten U, Danhier F, Bastiat G, Préat V. Drug combination using an injectable nanomedicine hydrogel for glioblastoma treatment. Int J Pharm. 2019;559:220-7
31. Zhao M, Bozzato E, Joudiou N, Ghiassinejad S, Danhier F, Gallez B. et al. Codelivery of paclitaxel and temozolomide through a photopolymerizable hydrogel prevents glioblastoma recurrence after surgical resection. J Control Release. 2019;309:72-81
32. Bastiancich C, Danhier P, Préat V, Danhier F. Anticancer drug-loaded hydrogels as drug delivery systems for the local treatment of glioblastoma. J Control Release. 2016;243:29-42
33. Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE. et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J clin oncol. 2009;27:4733-40
34. Quinn JA, Jiang SX, Reardon DA, Desjardins A, Vredenburgh JJ, Rich JN. et al. Phase II trial of temozolomide plus o6-benzylguanine in adults with recurrent, temozolomide-resistant malignant glioma. J clin Oncol. 2009;27:1262
35. Friday BB, Anderson SK, Buckner J, Yu C, Giannini C, Geoffroy F. et al. Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: a north central cancer treatment group study. Neuro Oncol. 2011;14:215-21
36. Pardridge WM. The blood-brain barrier: bottleneck in brain drug development. NeuroRx. 2005;2:3-14
37. Karim R, Palazzo C, Evrard B, Piel G. Nanocarriers for the treatment of glioblastoma multiforme: current state-of-the-art. J Control Release. 2016;227:23-37
38. Fang C, Wang K, Stephen ZR, Mu Q, Kievit FM, Chiu DT. et al. Temozolomide nanoparticles for targeted glioblastoma therapy. ACS Appl Mater Interfaces. 2015;7:6674-82
39. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1999;46:6387-92
40. Moghimi SM, Hunter AC, Murray JC. Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol Rev. 2001;53:283-318
41. Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Del Rev. 2011;63:136-51
42. Danhier F. To exploit the tumor microenvironment: Since the EPR effect fails in the clinic, what is the future of nanomedicine? J Control Release. 2016;244:108-21
43. Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in brain tumours. Nat Rev Neurosci. 2007;8:610-22
44. D'Alessio A, Proietti G, Sica G, Scicchitano BM. Pathological and molecular features of glioblastoma and its peritumoral tissue. Cancers (Basel). 2019;11:469
45. Sarkaria JN, Hu LS, Parney IF, Pafundi DH, Brinkmann DH, Laack NN. et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro Oncol. 2018;20:184-91
46. Lemée JM, Clavreul A, Menei P. Intratumoral heterogeneity in glioblastoma: don't forget the peritumoral brain zone. Neuro Oncol. 2015;17:1322-32
47. Lemée JM, Clavreul A, Aubry M, Com E, de Tayrac M, Eliat PA. et al. Characterizing the peritumoral brain zone in glioblastoma: a multidisciplinary analysis. J Neurooncol. 2015;122:53-61
48. Rong Y, Durden DL, Van Meir EG, Brat DJ. 'Pseudopalisading' necrosis in glioblastoma: a familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J Neuropathol Exp Neurol. 2006;65:529-39
49. Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer. 2004;4:437-47
50. Huang WJ, Chen WW, Zhang X. Glioblastoma multiforme: effect of hypoxia and hypoxia inducible factors on therapeutic approaches. Oncol Lett. 2016;12:2283-8
51. Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ, Jain RK. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004;64:3731-6
52. Chauhan VP, Stylianopoulos T, Martin JD, Popovic Z, Chen O, Kamoun WS. et al. Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nat Nanotechnol. 2012;7:383-8
53. Jain RK. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J Clin Oncol. 2013;31:2205-18
54. Maeda H. The link between infection and cancer: tumor vasculature, free radicals, and drug delivery to tumors via the EPR effect. Cancer Sci. 2013;104:779-89
55. Golombek SK, May J-N, Theek B, Appold L, Drude N, Kiessling F. et al. Tumor targeting via EPR: strategies to enhance patient responses. Adv Drug Del Rev. 2018;130:17-38
56. Candelario-Jalil E, Taheri S, Yang Y, Sood R, Grossetete M, Estrada EY. et al. Cyclooxygenase inhibition limits blood-brain barrier disruption following intracerebral injection of tumor necrosis factor-alpha in the rat. J Pharmacol Exp Ther. 2007;323:488-98
57. Reichman HR, Farrell CL, Del Maestro RF. Effects of steroids and nonsteroid anti-inflammatory agents on vascular permeability in a rat glioma model. J Neurosurg. 1986;65:233-7
58. Zhang B, Hu Y, Pang Z. Modulating the tumor microenvironment to enhance tumor nanomedicine delivery. Front Pharmacol. 2017;8:952
59. Chauhan VP, Stylianopoulos T, Martin JD, Popović Z, Chen O, Kamoun WS. et al. Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nat Nanotechnol. 2012;7:383
60. Han L, Kong DK, Zheng M-q, Murikinati S, Ma C, Yuan P. et al. Increased nanoparticle delivery to brain tumors by autocatalytic priming for improved treatment and imaging. ACS Nano. 2016;10:4209-18
61. Zou Y, Liu Y, Yang Z, Zhang D, Lu Y, Zheng M. et al. Effective and targeted human orthotopic glioblastoma xenograft therapy via a multifunctional biomimetic nanomedicine. Adv Mater. 2018;30:1803717
62. Shen Y, Pi Z, Yan F, Yeh C-K, Zeng X, Diao X. et al. Enhanced delivery of paclitaxel liposomes using focused ultrasound with microbubbles for treating nude mice bearing intracranial glioblastoma xenografts. Int J Nanomedicine. 2017;12:5613
63. Theek B, Gremse F, Kunjachan S, Fokong S, Pola R, Pechar M. et al. Characterizing EPR-mediated passive drug targeting using contrast-enhanced functional ultrasound imaging. J Control Release. 2014;182:83-9
64. Seo JW, Ang J, Mahakian LM, Tam S, Fite B, Ingham ES. et al. Self-assembled 20-nm (64)Cu-micelles enhance accumulation in rat glioblastoma. J Control Release. 2015;220:51-60
65. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860-7
66. Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia. 2011;59:1169-80
67. Broekman ML, Maas SLN, Abels ER, Mempel TR, Krichevsky AM, Breakefield XO. Multidimensional communication in the microenvirons of glioblastoma. Nat Rev Neurol. 2018;14:482-95
68. Prosniak M, Harshyne LA, Andrews DW, Kenyon LC, Bedelbaeva K, Apanasovich TV. et al. Glioma grade is associated with the accumulation and activity of cells bearing M2 monocyte markers. Clin Cancer Res. 2013;19:3776-86
69. Sorensen MD, Dahlrot RH, Boldt HB, Hansen S, Kristensen BW. Tumour-associated microglia/macrophages predict poor prognosis in high-grade gliomas and correlate with an aggressive tumour subtype. Neuropathol Appl Neurobiol. 2018;44:185-206
70. Zhang J, Sarkar S, Cua R, Zhou Y, Hader W, Yong VW. A dialog between glioma and microglia that promotes tumor invasiveness through the CCL2/CCR2/interleukin-6 axis. Carcinogenesis. 2012;33:312-9
71. Brandenburg S, Muller A, Turkowski K, Radev YT, Rot S, Schmidt C. et al. Resident microglia rather than peripheral macrophages promote vascularization in brain tumors and are source of alternative pro-angiogenic factors. Acta Neuropathol. 2016;131:365-78
72. Zhao P, Wang Y, Kang X, Wu A, Yin W, Tang Y. et al. Dual-targeting biomimetic delivery for anti-glioma activity via remodeling the tumor microenvironment and directing macrophage-mediated immunotherapy. Chem Sci. 2018;9:2674-89
73. Diop-Frimpong B, Chauhan VP, Krane S, Boucher Y, Jain RK. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. PNAS. 2011;108:2909-14
74. Gustafson HH, Holt-Casper D, Grainger DW, Ghandehari H. Nanoparticle uptake: the phagocyte problem. Nano Today. 2015;10:487-510
75. Roode LE, Brighton H, Bo T, Perry JL, Parrott MC, Kersey F. et al. Subtumoral analysis of PRINT nanoparticle distribution reveals targeting variation based on cellular and particle properties. Nanomedicine. 2016;12:1053-62
76. Miller MA, Gadde S, Pfirschke C, Engblom C, Sprachman MM, Kohler RH. et al. Predicting therapeutic nanomedicine efficacy using a companion magnetic resonance imaging nanoparticle. Sci Transl Med. 2015;7:314ra183
77. Miller MA, Zheng YR, Gadde S, Pfirschke C, Zope H, Engblom C. et al. Tumour-associated macrophages act as a slow-release reservoir of nano-therapeutic Pt(IV) pro-drug. Nat Commun. 2015;6:8692
78. Moore TL, Hauser D, Gruber T, Rothen-Rutishauser B, Lattuada M, Petri-Fink A. et al. Cellular shuttles: monocytes/macrophages exhibit transendothelial transport of nanoparticles under physiological flow. ACS Appl Mater Interfaces. 2017;9:18501-11
79. Alizadeh D, Zhang L, Hwang J, Schluep T, Badie B. Tumor-associated macrophages are predominant carriers of cyclodextrin-based nanoparticles into gliomas. Nanomedicine. 2010;6:382-90
80. Lombardi G, Rumiato E, Bertorelle R, Saggioro D, Farina P, Della Puppa A. et al. Clinical and genetic factors associated with severe hematological toxicity in glioblastoma patients during radiation plus temozolomide treatment. J Clin Oncol. 2015;38:514-9
81. Mathios D, Kim JE, Mangraviti A, Phallen J, Park C-K, Jackson CM. et al. Anti-PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci Transl Med. 2016;8:370ra180
82. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97
83. Steiniger SC, Kreuter J, Khalansky AS, Skidan IN, Bobruskin AI, Smirnova ZS. et al. Chemotherapy of glioblastoma in rats using doxorubicin-loaded nanoparticles. Int J Cancer. 2004;109:759-67
84. Shah N, Chaudhari K, Dantuluri P, Murthy R, Das S. Paclitaxel-loaded PLGA nanoparticles surface modified with transferrin and Pluronic® P85, an in vitro cell line and in vivo biodistribution studies on rat model. J Drug Target. 2009;17:533-42
85. Miura Y, Takenaka T, Toh K, Wu S, Nishihara H, Kano MR. et al. Cyclic RGD-linked polymeric micelles for targeted delivery of platinum anticancer drugs to glioblastoma through the blood-brain tumor barrier. ACS Nano. 2013;7:8583-92
86. Koukourakis M, Koukouraki S, Fezoulidis I, Kelekis N, Kyrias G, Archimandritis S. et al. High intratumoural accumulation of stealth® liposomal doxorubicin (Caelyx®) in glioblastomas and in metastatic brain tumours. Br J Cancer. 2000;83:1281
87. Vieira DB, Gamarra LF. Getting into the brain: liposome-based strategies for effective drug delivery across the blood-brain barrier. Int J Nanomedicine. 2016;11:5381-414
88. Soni V, Kohli DV, Jain SK. Transferrin coupled liposomes as drug delivery carriers for brain targeting of 5-florouracil. J Drug Target. 2005;13:245-50
89. Madhankumar A, Slagle-Webb B, Mintz A, Sheehan JM, Connor JR. Interleukin-13 receptor-targeted nanovesicles are a potential therapy for glioblastoma multiforme. Mol Cancer Ther. 2006;5:3162-9
90. Anton N, Mojzisova H, Porcher E, Benoit JP, Saulnier P. Reverse micelle-loaded lipid nano-emulsions: new technology for nano-encapsulation of hydrophilic materials. Int J Pharm. 2010;398:204-9
91. Aparicio-Blanco J, Torres-Suarez AI. Glioblastoma multiforme and lipid nanocapsules: a review. J Biomed Nanotechnol. 2015;11:1283-311
92. Garcion E, Lamprecht A, Heurtault B, Paillard A, Aubert-Pouessel A, Denizot B. et al. A new generation of anticancer, drug-loaded, colloidal vectors reverses multidrug resistance in glioma and reduces tumor progression in rats. Mol Cancer Ther. 2006;5:1710-22
93. Ganipineni LP, Ucakar B, Joudiou N, Bianco J, Danhier P, Zhao M. et al. Magnetic targeting of paclitaxel-loaded poly (lactic-co-glycolic acid)-based nanoparticles for the treatment of glioblastoma. Int J Nanomedicine. 2018;13:4509
94. Cheng Y, Dai Q, Morshed RA, Fan X, Wegscheid ML, Wainwright DA. et al. Blood-brain barrier permeable gold nanoparticles: an efficient delivery platform for enhanced malignant glioma therapy and imaging. Small. 2014;10:5137-50
95. Zhao L, Li Y, Zhu J, Sun N, Song N, Xing Y. et al. Chlorotoxin peptide-functionalized polyethylenimine-entrapped gold nanoparticles for glioma SPECT/CT imaging and radionuclide therapy. J Nanobiotechnology. 2019;17:30
96. Hao Y, Zhang B, Zheng C, Ji R, Ren X, Guo F. et al. The tumor-targeting core-shell structured DTX-loaded PLGA@Au nanoparticles for chemo-photothermal therapy and X-ray imaging. J Control Release. 2015;220:545-55
97. Brown TJ, Brennan MC, Li M, Church EW, Brandmeir NJ, Rakszawski KL. et al. Association of the extent of resection with survival in glioblastoma: a systematic review and meta-analysis. JAMA oncology. 2016;2:1460-9
98. Trifiletti DM, Alonso C, Grover S, Fadul CE, Sheehan JP, Showalter TN. Prognostic implications of extent of resection in glioblastoma: analysis from a large database. World Neurosurg. 2017;103:330-40
99. Sarkaria JN, Hu LS, Parney IF, Pafundi DH, Brinkmann DH, Laack NN. et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro Oncol. 2017;20:184-91
100. Galldiks N, Law I, Pope WB, Arbizu J, Langen K-J. The use of amino acid PET and conventional MRI for monitoring of brain tumor therapy. NeuroImage Clin. 2017;13:386-94
101. Boxerman JL, Shiroishi MS, Ellingson BM, Pope WB. Dynamic susceptibility contrast MR imaging in glioma: review of current clinical practice. Magn Reson Imaging Clin. 2016;24:649-70
102. Bergamino M, Bonzano L, Levrero F, Mancardi G, Roccatagliata L. A review of technical aspects of T1-weighted dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI) in human brain tumors. Phys Med. 2014;30:635-43
103. Smits A, Baumert BG. The clinical value of PET with amino acid tracers for gliomas WHO grade II. Int J Mol Imaging. 2011. 2011 Doi:10.1155/2011/372509
104. Haining Z, Kawai N, Miyake K, Okada M, Okubo S, Zhang X. et al. Relation of LAT1/4F2hc expression with pathological grade, proliferation and angiogenesis in human gliomas. BMC clinical pathology. 2012;12:4
105. Verduin M, Compter I, Steijvers D, Postma AA, Eekers DB, Anten MM. et al. Noninvasive glioblastoma testing: multimodal approach to monitoring and predicting treatment response. Dis Markers. 2018. 2018 Doi:10.1155/2018/2908609
106. Wu X, Yang H, Yang W, Chen X, Gao J, Gong X. et al. Nanoparticle-based diagnostic and therapeutic systems for brain tumors. J Mater Chem B. 2019;7:4734-50
107. Isal S, Pierson J, Imbert L, Clement A, Collet C, Pinel S. et al. PET imaging of 68 Ga-NODAGA-RGD, as compared with 18 F-fluorodeoxyglucose, in experimental rodent models of engrafted glioblastoma. EJNMMI research. 2018;8:51
108. Novy Z, Stepankova J, Hola M, Flasarova D, Popper M, Petrik M. Preclinical evaluation of radiolabeled peptides for PET imaging of glioblastoma multiforme. Molecules. 2019;24:2496
109. Provost C, Prignon A, Rozenblum-Beddok L, Bruyer Q, Dumont S, Merabtene F. et al. Comparison and evaluation of two RGD peptides labelled with (68)Ga or (18)F for PET imaging of angiogenesis in animal models of human glioblastoma or lung carcinoma. Oncotarget. 2018;9:19307-16
110. Schnell O, Krebs B, Carlsen J, Miederer I, Goetz C, Goldbrunner RH. et al. Imaging of integrin αvβ3 expression in patients with malignant glioma by [18F] Galacto-RGD positron emission tomography. Neuro Oncol. 2009;11:861-70
111. Zhang H, Liu N, Gao S, Hu X, Zhao W, Tao R. et al. Can an 18F-ALF-NOTA-PRGD2 PET/CT scan predict treatment sensitivity to concurrent chemoradiotherapy in patients with newly diagnosed glioblastoma? J Nucl Med. 2016;57:524-9
112. Li D, Zhang J, Chi C, Xiao X, Wang J, Lang L. et al. First-in-human study of PET and optical dual-modality image-guided surgery in glioblastoma using 68Ga-IRDye800CW-BBN. Theranostics. 2018;8:2508
113. Tang W, Fan W, Lau J, Deng L, Shen Z, Chen X. Emerging blood-brain-barrier-crossing nanotechnology for brain cancer theranostics. Chem Soc Rev. 2019;48:2967-3014
114. Pericleous P, Gazouli M, Lyberopoulou A, Rizos S, Nikiteas N, Efstathopoulos EP. Quantum dots hold promise for early cancer imaging and detection. Int J Cancer. 2012;131:519-28
115. Gao L, Zhao X, Wang J, Wang Y, Yu L, Peng H. et al. Multiple functionalized carbon quantum dots for targeting glioma and tissue imaging. Opt Mater. 2018;75:764-9
116. Liu Y, Liu J, Zhang J, Li X, Lin F, Zhou N. et al. Noninvasive brain tumor imaging using red emissive carbonized polymer dots across the blood-brain barrier. ACS Omega. 2018;3:7888-96
117. Liu L, Miao Q, Liang G. Quantum dots as multifunctional materials for tumor imaging and therapy. Materials (Basel). 2013;6:483-99
118. Ge J, Jia Q, Liu W, Guo L, Liu Q, Lan M. et al. Red-emissive carbon dots for fluorescent, photoacoustic, and thermal theranostics in living mice. Adv Mater. 2015;27:4169-77
119. Liu C, Chen J, Zhu Y, Gong X, Zheng R, Chen N. et al. Highly sensitive MoS2-indocyanine green hybrid for photoacoustic imaging of orthotopic brain glioma at deep site. Nano-Micro Lett. 2018;10:48
120. Guo W, Sun X, Jacobson O, Yan X, Min K, Srivatsan A. et al. Intrinsically radioactive [64Cu]CuInS/ZnS quantum dots for PET and optical imaging: improved radiochemical stability and controllable cerenkov luminescence. ACS Nano. 2015;9:488-95
121. Phillips E, Penate-Medina O, Zanzonico PB, Carvajal RD, Mohan P, Ye Y. et al. Clinical translation of an ultrasmall inorganic optical-PET imaging nanoparticle probe. Sci Transl Med. 2014;6:260ra149
122. Gobbo OL, Sjaastad K, Radomski MW, Volkov Y, Prina-Mello A. Magnetic nanoparticles in cancer theranostics. Theranostics. 2015;5:1249
123. Xu HL, Yang JJ, ZhuGe DL, Lin MT, Zhu QY, Jin BH. et al. Glioma-targeted delivery of a theranostic liposome integrated with quantum dots, superparamagnetic iron oxide, and cilengitide for dual-imaging guiding cancer surgery. Adv Healthc Mater. 2018;7:1701130
124. Maier-Hauff K, Ulrich F, Nestler D, Niehoff H, Wust P, Thiesen B. et al. Efficacy and safety of intratumoral thermotherapy using magnetic iron-oxide nanoparticles combined with external beam radiotherapy on patients with recurrent glioblastoma multiforme. J Neurooncol. 2011;103:317-24
125. Grauer O, Jaber M, Hess K, Weckesser M, Schwindt W, Maring S. et al. Combined intracavitary thermotherapy with iron oxide nanoparticles and radiotherapy as local treatment modality in recurrent glioblastoma patients. J Neurooncol. 2019;141:83-94
126. Wang H, Mu Q, Revia R, Wang K, Tian B, Lin G. et al. Iron oxide-carbon core-shell nanoparticles for dual-modal imaging-guided photothermal therapy. J Control Release. 2018;289:70-8
127. Asklund T, Kvarnbrink S, Holmlund C, Wibom C, Bergenheim T, Henriksson R. et al. Synergistic killing of glioblastoma stem-like cells by bortezomib and HDAC inhibitors. Anticancer Res. 2012;32:2407-13
128. Quinn JA, Jiang SX, Reardon DA, Desjardins A, Vredenburgh JJ, Rich JN. et al. Phase I trial of temozolomide plus O6-benzylguanine 5-day regimen with recurrent malignant glioma. Neuro Oncol. 2009;11:556-61
129. Cheng D, Cao N, Chen J, Yu X, Shuai X. Multifunctional nanocarrier mediated co-delivery of doxorubicin and siRNA for synergistic enhancement of glioma apoptosis in rat. Biomaterials. 2012;33:1170-9
130. Xu Y, Shen M, Li Y, Sun Y, Teng Y, Wang Y. et al. The synergic antitumor effects of paclitaxel and temozolomide co-loaded in mPEG-PLGA nanoparticles on glioblastoma cells. Oncotarget. 2016;7:20890-901
131. Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440-6
132. Khawar IA, Kim JH, Kuh H-J. Improving drug delivery to solid tumors: priming the tumor microenvironment. J Control Release. 2015;201:78-89
133. Batchelor TT, Gerstner ER, Emblem KE, Duda DG, Kalpathy-Cramer J, Snuderl M. et al. Improved tumor oxygenation and survival in glioblastoma patients who show increased blood perfusion after cediranib and chemoradiation. PNAS. 2013;110:19059-64
134. Sorensen AG, Emblem KE, Polaskova P, Jennings D, Kim H, Ancukiewicz M. et al. Increased survival of glioblastoma patients who respond to antiangiogenic therapy with elevated blood perfusion. Cancer Res. 2012;72:402-7
135. Wong HL, Shen Z, Lu Z, Wientjes MG, Au JL-S. Paclitaxel tumor-priming enhances siRNA delivery and transfection in 3-dimensional tumor cultures. Mol Pharm. 2011;8:833-40
136. Lee MJ, Ye AS, Gardino AK, Heijink AM, Sorger PK, MacBeath G. et al. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell. 2012;149:780-94
137. Shi ZD, Qian XM, Liu CY, Han L, Zhang KL, Chen LY. et al. Aspirin-/TMZ-coloaded microspheres exert synergistic antiglioma efficacy via inhibition of β-catenin transactivation. CNS Neurosci Ther. 2013;19:98-108
138. Kim SS, Rait A, Kim E, Pirollo KF, Nishida M, Farkas N. et al. A nanoparticle carrying the p53 gene targets tumors including cancer stem cells, sensitizes glioblastoma to chemotherapy and improves survival. ACS Nano. 2014;8:5494-514
139. Chen X, Tai L, Gao J, Qian J, Zhang M, Li B. et al. A stapled peptide antagonist of MDM2 carried by polymeric micelles sensitizes glioblastoma to temozolomide treatment through p53 activation. J Control Release. 2015;218:29-35
140. Morton SW, Lee MJ, Deng ZJ, Dreaden EC, Siouve E, Shopsowitz KE. et al. A nanoparticle-based combination chemotherapy delivery system for enhanced tumor killing by dynamic rewiring of signaling pathways. Sci Signal. 2014;7:ra44
141. Zhou Z, Kennell C, Jafari M, Lee JY, Ruiz-Torres SJ, Waltz SE. et al. Sequential delivery of erlotinib and doxorubicin for enhanced triple negative breast cancer treatment using polymeric nanoparticle. Int J Pharm. 2017;530:300-7
142. Majumder P, Baxa U, Walsh STR, Schneider JP. Design of a multicompartment hydrogel that facilitates time-resolved delivery of combination therapy and synergized killing of glioblastoma. Angew Chem Int Ed Engl. 2018;57:15040-4
143. Stylli SS, Luwor RB, Ware TM, Tan F, Kaye AH. Mouse models of glioma. J Clin Neurosci. 2015;22:619-26
144. Huszthy PC, Daphu I, Niclou SP, Stieber D, Nigro JM, Sakariassen PØ. et al. In vivo models of primary brain tumors: pitfalls and perspectives. Neuro Oncol. 2012;14:979-93
145. Simeonova I, Huillard E. In vivo models of brain tumors: roles of genetically engineered mouse models in understanding tumor biology and use in preclinical studies. Cell Mol Life Sci. 2014;71:4007-26
146. Arami H, Patel CB, Madsen SJ, Dickinson PJ, Davis RM, Zeng Y. et al. Nanomedicine for spontaneous brain tumors: a companion clinical trial. ACS nano. 2019;13:2858-69
147. Young JS, Dayani F, Morshed RA, Okada H, Aghi MK. Immunotherapy for high grade gliomas: a clinical update and practical considerations for neurosurgeons. World Neurosurg. 2019:124 397-409
148. Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol. 2018;15:422
Author contact
![]() Corresponding authors: Véronique Préat, Université catholique de Louvain, Louvain Drug Research Institute, Advanced Drug Delivery and Biomaterials, Avenue Mounier, 73, B1 73.12, 1200 Brussels, Belgium. veronique.preatbe OR Raymond M. Schiffelers, Clinical Chemistry and Haematology, University Medical Center Utrecht, G03.550, P.O. Box 85500, 3508 GA Utrecht, The Netherlands. r.schiffelersnl
Corresponding authors: Véronique Préat, Université catholique de Louvain, Louvain Drug Research Institute, Advanced Drug Delivery and Biomaterials, Avenue Mounier, 73, B1 73.12, 1200 Brussels, Belgium. veronique.preatbe OR Raymond M. Schiffelers, Clinical Chemistry and Haematology, University Medical Center Utrecht, G03.550, P.O. Box 85500, 3508 GA Utrecht, The Netherlands. r.schiffelersnl