Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(4):1531-1543. doi:10.7150/thno.36182 This issue Cite
Research Paper
Response to mTOR and PI3K inhibitors in enzalutamide-resistant luminal androgen receptor triple-negative breast cancer patient-derived xenografts
Florence Coussy1,2,3 ![]() , Marion Lavigne4, Leanne de Koning5, Rania El Botty2, Fariba Nemati2, Adnan Naguez2, Guillaume Bataillon4, Berengère Ouine5, Ahmed Dahmani2, Elodie Montaudon2, Pierre Painsec2, Sophie Chateau-Joubert6, Fuhrmann Laetitia4, Thibaut Larcher7, Sophie Vacher1, Walid Chemlali1, Adrien Briaux1, Samia Melaabi1, Anne Vincent Salomon4, Jean Marc Guinebretiere8, Ivan Bieche1,9*, Elisabetta Marangoni2*
, Marion Lavigne4, Leanne de Koning5, Rania El Botty2, Fariba Nemati2, Adnan Naguez2, Guillaume Bataillon4, Berengère Ouine5, Ahmed Dahmani2, Elodie Montaudon2, Pierre Painsec2, Sophie Chateau-Joubert6, Fuhrmann Laetitia4, Thibaut Larcher7, Sophie Vacher1, Walid Chemlali1, Adrien Briaux1, Samia Melaabi1, Anne Vincent Salomon4, Jean Marc Guinebretiere8, Ivan Bieche1,9*, Elisabetta Marangoni2*
1. Unit of Pharmacogenomics, Department of Genetics, Institut Curie, 26 rue d'Ulm, Paris, France
2. Laboratory of Preclinical Investigation, Department of Translational Research, Institut Curie Research Center, 26 rue d' Ulm, Paris, France
3. Department of Medical Oncology, Institut Curie, 26 rue d' Ulm, Paris, France
4. Department of Biopathology, Institut Curie, 26 rue d' Ulm, Paris, France
5. Translational Research Department, RPPA Platform, Institut Curie Research Center, 26 rue d'Ulm, Paris, France
6. BioPôle Alfort, National Veterinary School of Alfort, Maison Alfort, France
7. INRA, APEX-PAnTher, Oniris, Nantes, France
8. Department of Biopathology, Institut Curie, Hopital René Huguenin, Saint Cloud, France
9. U1016, Paris Descartes University, 4 avenue de l'observatoire, Paris, France.
*Contributed equally
Received 2019-4-29; Accepted 2019-10-18; Published 2020-1-1
Abstract

Luminal androgen receptor (LAR) breast cancer accounts for 10% of all triple-negative breast cancers (TNBC). Anti-androgen therapy for this subtype is in development, but yields only partial clinical benefits. In this study, we aimed to characterize the genomic alterations of LAR TNBC, to analyze activation of the PI3K signaling pathway and to compare the response to PI3K pathway inhibitors with that to anti-androgen therapy in patient-derived xenografts (PDX) of LAR TNBC.
Methods: Four LAR PDX models were identified, on the basis of their transcriptomic profiles, in a cohort of 57 PDX models of TNBC. The expression of AR-related genes, basal and luminal cytokeratins and EMT genes was analyzed by RT-PCR and IHC. AKT1 and PIK3CA mutations were identified by targeted NGS, and activation of the PI3K pathway was analyzed with a reverse-phase protein array. Three LAR PDXs with a PIK3CA or AKT1 mutation were treated with the AR inhibitor enzalutamide, a PI3K inhibitor, a dual PI3K-mTOR inhibitor and a mTORC1-mTORC2 inhibitor. Finally, we screened a clinical cohort of 329 TNBC for PIK3CA and AKT1 hotspot mutations.
Results: LAR TNBC PDXs were significantly enriched in PIK3CA and AKT1 mutations, and had higher levels of luminal-androgen-like gene expression and a higher PI3K pathway protein activation score than other TNBC subtypes. Immunohistochemistry analysis revealed strong expression of the luminal cytokeratin CK18 and AR in three LAR PDX models. We found that mTOR and PI3K inhibitors had marked antitumor activity in vivo in PDX harboring genomic alterations of PIK3CA and AKT1 genes that did not respond to the AR antagonist enzalutamide. PIK3CA mutations were detected in more than one third of AR+ TNBC from patients (38%), and only 10% of AR-negative TNBC.
Conclusion: Our results for PDX models of LAR TNBC resistant to enzalutamide indicate that PIK3CA and AKT1 are potential therapeutic targets.
Keywords: Triple-negative breast cancer, androgen receptor, luminal androgen receptor (LAR), genomic alteration, targeted therapy, PI3K pathway inhibitor.
Introduction
Triple-negative breast cancer (TNBC) accounts for about 10% of all breast cancers [1]. This disease is defined by the absence of estrogen receptor (ER) and progesterone receptor (PR) expression, and of human epidermal growth factor receptor 2 (ERBB2) overexpression [2]. TNBC generally has a poor prognosis, due to a lack of targeted therapy, in particular [3]. TNBC are highly heterogeneous in terms of their genomic and histological characteristics, and this may have hampered efforts to develop effective treatments. Various studies have addressed the issue of the heterogeneity of gene expression in TNBC [4, 5, 6]. Lehman et al. identified six classes of TNBC on the basis of transcriptomic analyses: basal-like1: BL1; basal-like2: BL2; immunomodulatory: IM; mesenchymal: M, mesenchymal stem-like: MSL; and luminal androgen receptor: LAR [5]. Each of these classes was characterized by alterations to specific pathways. The LAR subtype is the most differentiated subtype of TNBC and it accounts for about 10% of all TNBC (9% in the METABRIC cohort and 8.7% in the TCGA cohort) [7, 8]. This subtype is characterized by activation of the androgen receptor (AR) pathway, with expression of the genes encoding AR targets and coactivators. The prognosis of this subtype remains unclear, due in particular to differences in its classification on the basis of gene expression [7, 9] or histological descriptions [10]. Anti-androgen therapy is currently being developed for this subtype and has been shown to have definite, but only partial clinical benefits [11, 12].
The phosphatidylinositol 3-kinase (PI3K) signaling pathway is crucial for cell growth and survival. PIK3CA activating mutations and PTEN loss of expression may contribute to treatment resistance in breast cancer (BC). The LAR subtype, associated with the luminal phenotype, is enriched in PI3K pathway alterations [13]. However, no clinical data are available concerning the activity of PI3K inhibitors in this subtype.
PDX models are robust preclinical models for testing the suitability of genomic alterations for use as biomarkers and comparing responses to targeted therapy, as they conserve the molecular heterogeneity present in the patient [14] and are predictive of treatment response in clinical practice [15]. However, no PDX models of LAR TNBC have ever been described, possibly due to the low frequency of this subtype of breast cancer.
The objective of this study was to characterize the genomic and protein characteristics of LAR PDXs and to compare the efficacy of various therapies targeting the PI3K signaling pathway with that of AR inhibitors.
Materials and Methods
Patients
We analysed samples from 323 unilateral invasive non-metastatic triple-negative primary breast tumors excised from women managed at Institut Curie (Paris and Saint-Cloud, France) between 1980 and 2015 (Table S1). Most of the patients (67%) were diagnosed and treated after 2000. All patients admitted to our institution before 2007 were informed that their tumor samples might be used for scientific purposes and were given the opportunity to refuse such use. Since 2007, patients admitted to our institution also provide consent actively, by signing an informed consent form. Patients (mean age: 56 years, range: 28-91) met the following criteria: primary unilateral non-metastatic TNBC, with full clinical, histological and laboratory data and full follow-up at Institut Curie. Median follow-up was 7.8 years (range: 8 months to 36 years). Eighty-one patients developed metastases within 10 years.
Patient-derived xenografts
LAR PDX were identified in a recently described large cohort of TNBC PDX [16]. Clinical information for the four LAR patients is provided in table S2. The experimental protocol and animal housing complied with institutional guidelines, and with the requirements of the French Ethics Committee (Agreement B75-05-18, France). Three LAR PDX models with specific alterations were chosen for in vivo preclinical assays: HBCx-2 (AKT1 mutation), HBCx-31 (AKT1 mutation), HBCx-154 (PIK3CA mutation). A fourth model, HBCx-35, was lost after five passages in mice and was not used for experiments. These three models were treated five times per week with enzalutamide (50 mg/kg, once daily), five times per week with PF-04691502 (10 mg/kg, once daily) (MedChem Express®), three times per week with BAY80-6946 (14 mg/kg) (MedChem Express®), and five times per week with AZD2014 (15 mg/kg) (MedChem Express®). Time of sacrifice depending on treatment: BAY80-6946: 3h post treatment, PF-04691502: 1h post treatment, AZD2014: 4h post treatment.
Tumor growth was evaluated by measuring two perpendicular tumor diameters with calipers, twice weekly. Individual tumor volumes were calculated as follows: V=axb2/2, where “a” is the largest diameter, and “b” is the smallest diameter. For each tumor, volume is expressed relative to the initial volume, as relative tumor volume (RTV). Tumor growth inhibition (TGI) on treatment was assessed by calculating the ratio of the mean RTV (relative tumor volume) for the treated group to the mean RTV for the control group at the same time point. The statistical significance of TGI was assessed in a paired Student's t test comparing tumor volumes between the treated and control groups. p-values were considered statistically significant for *p < 0.05, **p < 0.01 and *** p < 0.001.
Transcriptomic data analysis
Transcriptomic profiling was performed with gene expression arrays on 57 PDX TNBC. The concentration and integrity/purity of each RNA sample were determined with the RNA 6000 LabChip kit (Agilent) and an Agilent 2100 bioanalyzer. Samples were hybridized with GeneChip Human 1.1 ST arrays in accordance with the manufacturer's (Affymetrix) recommendations, with the WT Expression Kit protocol (Life Technologies) and Affymetrix labeling and hybridization kits. The RMA normalization procedure was applied with the oligo package [17]. No additional human-mouse cross-hybridization filtering was applied, as our xenograft samples contained less than 5% mouse cells (determined by RT-PCR to quantify transcripts of the ubiquitously expressed TBP gene with specific mouse and human primer pairs), too small a proportion to affect the expression profiles obtained with HuGene1.0 arrays [18]. The TNBC molecular subtypes of the PDX were determined from gene expression data, with TNBCtype software developed by Chen et al. [19].
Somatic mutation analysis: We analyzed 57 triple-negative PDX by targeted NGS on 95 genes, selected from those most commonly mutated in breast cancer (>1%) and including potential therapeutic targets (Table S3). Specific NGS primers were designed based on the human reference genome (<1% of the total reads common to mice). NGS was performed on an Illumina HiSeq2500 sequencer. Reads were aligned with the BWA allowing up to 4% mismatches with the reference sequences. Only reads with a mapping quality of more than 20 were used for variant calling with the GATK unified genotyper. The genomic alterations detected included single-nucleotide variations of SMGs (i.e. base substitutions and short insertions/deletions) [20]. Genomic variants were annotated with data from the 1000 Genome and COSMIC databases [21]. Deleterious genomic alterations were defined as follows: (i) for oncogenes, we considered only mutations resulting in a gain of function (i.e. hotspot missense mutations, in-frame insertions/deletions/splicing variants reported to be oncogenic), (ii) for tumor suppressor genes (TSG), we considered only mutations resulting in a loss of function (i.e. biallelic truncating alterations (nonsense mutations, frameshift insertions/deletions/splicing) or monoallelic truncating alterations associated with heterozygous deletions detected by copy number analysis). Variants with a low allelic frequency (<5%) or low coverage (<100x) were excluded from the analysis. Genomic variants were validated biologically by comparison with the COSMIC, TumorPortal and cBioportal databases [6, 22].
Analysis of copy number alterations (SCNA)
PDX were profiled with Affymetrix genomics arrays (SNP 6.0 or Cytoscan HD array). Genome-wide copy number analysis was performed with Affymetrix SNP arrays, as previously described [23, 24]. SNP 6.0 or Cytoscan HD arrays were processed with 500 ng and 250 ng of gDNA, respectively, as the starting material, as recommended by the supplier. Raw data were normalized with Genotyping Console (SNP6.0 arrays) or Chromosome Analysis Suite (Cytoscan HD arrays). The focal amplification of oncogenes was defined as a log ratio>1.58 (6 copies per diploid genome) and a maximum size <10 megabases. Biallelic inactivation of TSG was defined as a homozygous deletion or truncating mutation associated with heterozygous deletion. Copy number alterations were compared with those of TCGA breast cancers in cBioPortal data [25, 26].
Screening for PIK3CA and AKT1 mutations in patients
PIK3CA mutations (exons 1, 2, 9, 20), and AKT1 (E17K, hotspot) were detected by sequencing cDNA fragments obtained by RT-PCR amplification. The exons from the two genes to be screened were chosen on the basis of the mutation frequency reported in COSMIC: Catalogue of Somatic Mutations in Cancer (cancer.sanger.ac.uk/). Screening was performed by high-resolution melting curve analysis on a LightCycler 480 (Roche Diagnostics, Penzberg, Germany) with LCGreen Plus + Melting Dye fluorescence (Biotech, Idaho Technology Inc., Salt Lake City, UT). Details of the primers and PCR conditions are available on request. The amplified products were sequenced with the BigDye Terminator kit on an ABI Prism 3130 automatic DNA sequencer (Applied Biosystems, Courtaboeuf, France) with a detection sensitivity of 5% mutated cells, and the sequences were compared with the corresponding cDNA reference sequences (PIK3CA NM_006218, AKT1 NM_005163). All the mutations detected were confirmed in a second independent sample testing run.
RT-qPCR in PDXs
Total RNA extraction and RT-PCR have been described elsewhere [27]. Expression of the TBP gene (GenBank accession no. NM_003194) encoding the TATA box-binding protein (a component of the DNA-binding protein complex TFIID) was quantified as an endogenous RNA control and each sample was normalized against its TBP mRNA content. Results, expressed as N-fold differences in target gene expression relative to the TBP gene and termed “Ntarget”, were determined as Ntarget = 2ΔCt sample, where the ΔCt value of the sample was determined by subtracting the mean Ct value of the target gene from the mean Ct value of the TBP gene [28].
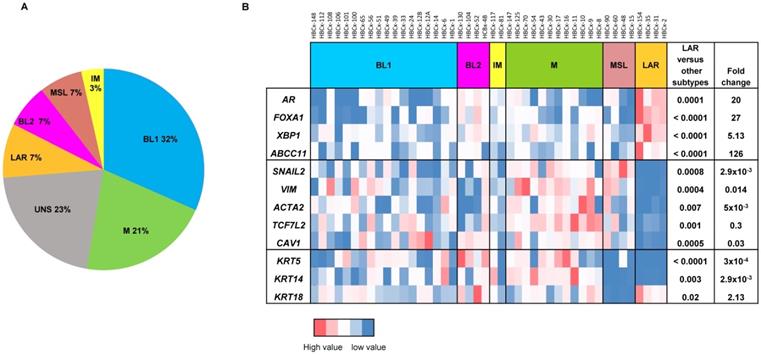
For the analysis of gene expression in PDX, mRNA levels were normalized to obtain a 'basal mRNA level' (smallest amount of mRNA quantifiable (Ct = 35)) equal to 1. We analyzed the expression of AR-related genes (AR, FOXA1, XBP1, ABCC11), genes involved in epithelial-mesenchymal transition (EMT) (SNAI2, VIM, ACTA2, TCF7L2, CAV1) and three cytokeratin genes (KRT5, KRT14, KRT18) in all TNBC PDX. The 13 PDX classified as unstable (UNS) in Lehmann's classification were excluded from the RT-PCR analysis shown in Figure 1B. Mann-Whitney tests were used to analyze gene expression differences between LAR and the other TNBC subtypes. Fold-changes in expression were calculated from the ratio of the mean value for LAR to the mean value for the subtypes.
Gene expression analysis of TNBC PDX. (A) Classification of PDX according to Lehmann's classification of TNBC (N=57). BL1: Basal-like1; BL2: Basal-like 2; IM: Immuno-modulatory, M:Mesenchymal; MSL: Mesenchymal Stem Like; LAR: Luminal Androgen Receptor; UNS: Unstable); (B) RT-PCR expression analysis of AR- related genes (AR, FOXA1, XBP1, ABCC11) , EMT genes (SNAIL2, VIM, ACTA2, TCF7L2, CAV1) and cytokeratin's KRT5, KRT14, KRT18 in TNBC PDX (n=44). Fold changes and p value are calculated to analyze gene expression in LAR PDX as compared to the other TNBC subtypes.
The expression of AR-related genes (AR, FOXA1, PIP, TFAP2B) and AR-induced genes (FN1, SERPINB5, S100P) was analyzed in treated xenografts.
Reverse-phase protein arrays (RPPA)
RPPA was performed as previously described [29] for 48 of the 57 TNBC PDX (9 PDX were established after the RPPA analysis). We calculated a PI3K pathway score with normalized data to assess pathway activation. Scores were obtained by calculating the sum for positive protein components (PI3K p110 subunit β, p-AKT1 (Ser473), p-AKT1 (Thr308), p-4E-BP1, p-p70-S6 kinase, p-S6 ribosomal protein; Cell Signaling Technology®) and subtracting the negative components of the pathway (PTEN, Cell Signaling Technology®). Eleven PDX classified as unstable (UNS) in Lehmann's classification were excluded from the analysis shown in Figure 3B.
Western blot analysis
Proteins were extracted from tumors in RIPA buffer (50 mM Tris HCL pH 8, 150 mM NaCl, 0.5% deoxycholic acid, 0.5% Triton), supplemented with protease and phosphatase inhibitors. Lysates were resolved by electrophoresis in 10% agarose gels. The resulting bands were transferred onto nitrocellulose membranes (Bio-Rad, Hercules, CA, USA), which were then probed with rabbit antibodies against AKT, p-AKT (Ser473), S6, p-S6, 4E-BP1, p-4E-BP1, PRAS, p-PRAS and GAPDH (Cell Signaling®). The membranes were washed and incubated with the appropriate horseradish peroxidase-conjugated affinity-purified goat anti-rabbit secondary antibodies (Jackson ImmunoResearch Laboratories, Inc., Interchim). Protein was quantified with Multi Gauge software and normalized against GAPDH levels.
Immunohistochemistry (IHC) analysis
TNBC PDX were fixed in 10% neutral buffered formalin, embedded in paraffin and stained with hematoxylin-eosin. TNBC PDX were included in TMAs in duplicate. Two cores were picked from each tumor paraffin block, with the Tissue-Tek Quick-Ray System from Sakura and a 6x10 matrix of the 2 mm core recipient block; 4 µm TMA sections were allowed to adhere to Superfrost Plus slides (MICROM, Walldorf, Germany). AR (Cell Signaling #5153, 1/400), CK5 (AbCam, ab52635, 1/400), CK8/18 (cloneE431-1, Invitrogen, 1/100), CK14 (Abcam, ab198167, 1/100), EGFR (Cell Signaling, #4267, 1/100), P-S6 (Cell Signaling ,#5364, 1/2000), and P-AKT (Ser 473,Cell Signaling, #4060, 1/50) antibodies were used. Slides incubated in parallel with pre-immune rabbit IgG were used as negative controls. Incubation and detection by the streptavidin-biotin-peroxidase complex method with DAB as the substrate were performed with a Ventana Medical System (ROCHE) DXT automat.
Statistical analysis
The proportions of the TNBC subgroup transcriptome were compared in Chi2 tests. The proportions of genomic alterations between PDX and TCGA were compared in Chi2 or Fisher's exact tests, as appropriate.
Metastasis-free survival (MFS) was determined as the interval between diagnosis and detection of the first distant metastasis. Overall survival (OS) was determined as the interval between diagnosis and death. Survival distribution was estimated by the Kaplan-Meier method.
Results
Establishment and characterization of LAR TNBC PDXs
Four PDX models of the LAR TNBC subtype were identified from a recently reported cohort of 57 TNBC PDX [16]. In this cohort, the Lehmann classification of TNBC subtypes was determined on the basis of transcriptomic profiles, with the TNBCtype tool. All Lehmanns' TNBC subtypes were represented: 32% BL1, 21% M, 7% LAR, 7% MSL, 7% BL2, 3% IM and 23% UNS (Figure 1A). We validated the correlation between transcriptomic classification and gene expression, by analyzing the expression of different sets of genes (AR-related genes, basal cytokeratin genes and EMT-related genes) by RT-PCR in 44 TNBC PDX (we excluded 13 PDX classified as unstable (UNS)). Figure 1B shows the heat map of normalized gene expression for the AR-related genes (AR, FOXA1, XBP1, ABCC11), cytokeratin genes (KRT5, KRT14, KRT18) and EMT genes (SNAI2, VIM, ACTA2, TCF7L2, and CAV1). The LAR subtype is characterized by significant overexpression of AR-related genes relative to the other TNBC subtypes. As expected, luminal cytokeratin KRT18 was overexpressed and the basal cytokeratins (KRT5, KRT14) were significantly underexpressed in the LAR subtype. Various genes associated with epithelial-mesenchymal transition (EMT: SNAI2, VIM, ACTA2, TCF7L2, and CAV1) were underexpressed in LAR PDX relative to the other TNBC subtypes.
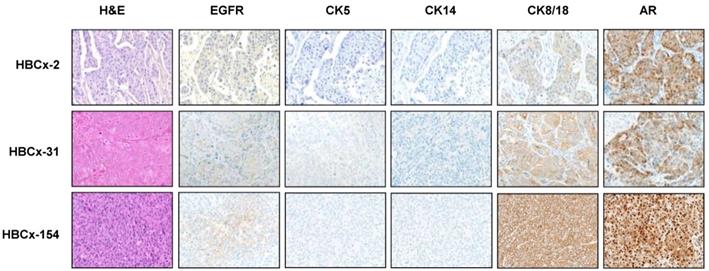
LAR TNBC are characterized by an apocrine morphology [30]. We therefore analyzed the histology of the LAR TNBC PDXs. Three cases, shown in Figure 2, presented typical apocrine differentiation. Tumor cells presented the characteristic nuclear and cytoplasmic features that must be present in more than 90% of tumor cells to confirm the morphological diagnosis of apocrine differentiation: (i) abundant (nuclear-cytoplasmic ratio = 1:2 or greater), finely granular eosinophilic cytoplasm (ii) round or pleomorphic nuclei (iii) prominent red nucleoli, often multiple (iv) sharply defined cell borders. On immunochemistry, tumor cells are usually positive for AR. AR was strongly expressed in HBCx-154 PDX (90%, +++), whereas weaker nuclear expression was observed in the HBCx-2 and HBCx-31 PDX. The three PDXs expressing the luminal cytokeratin CK18 and were negative for basal cytokeratins (CK5, CK14).
Morphological and immunohistochemistry analysis of 3 LAR TNBC PDX. Representative hematoxylin-eosin (H&E) stained sections and expression of EGFR, CK5, CK14, CK8/18 and AR (20X).
The PI3K signaling pathway is activated in the LAR subtype
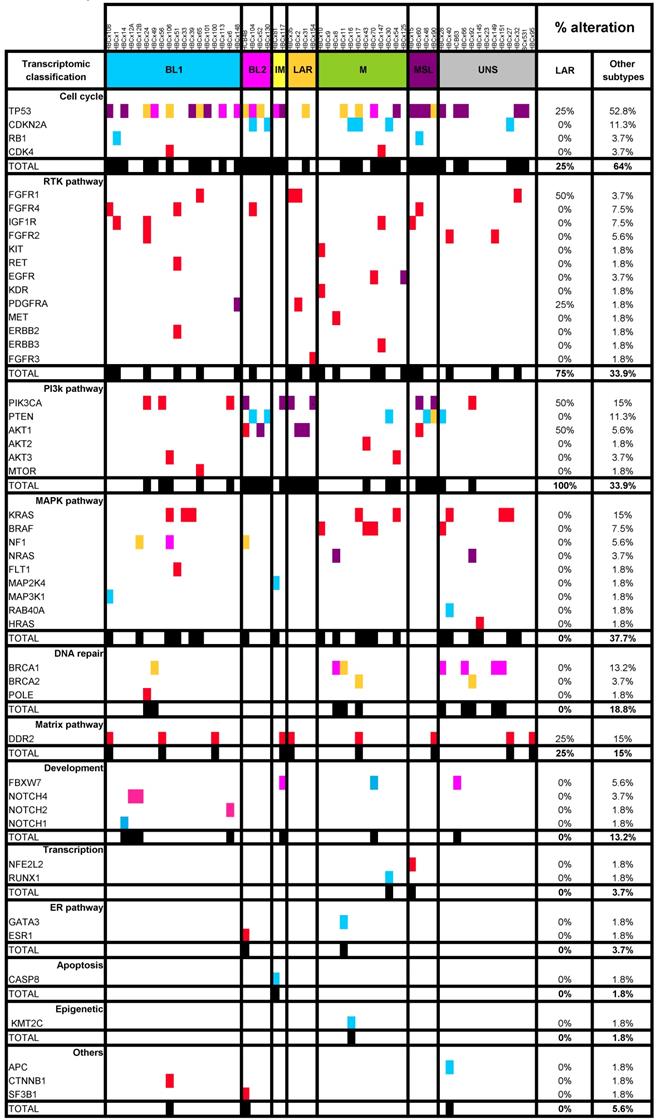
LAR TNBC are characterized by a high frequency of PIK3CA mutations [13]. We therefore compared the genomic profile of LAR PDX (n=4) with that of the other TNBC subtypes. According to targeted NGS (analysis of the 95 genes most frequently altered in breast cancer) and copy number analysis, LAR TNBC PDX had a genomic profile different from that of the other TNBC subtypes, with enrichment in PIK3CA and AKT1 mutations (100% versus 7,5%, p=0.0002, Fisher's exact test; LAR versus other subtypes), a lower frequency of TP53 mutations (25% for LAR and 52.8% in the other subtypes) and a high frequency of FGFR1 amplification. LAR models were also characterized by a lower frequency of genomic alterations in genes associated with the cycle cell pathway and an absence of alterations to the MAPK and DNA repair pathways (Table 1) (Figure 3A).
Representation of SMGs and SCNAs for 57 TNBC PDXs

Genomic alterations and activation of PIK3/AKT pathway in LAR PDX as compared to the other TNBC subtypes. (A) Percentage of genomic alterations (mutations, amplification and homozygous deletions) in TNBC PDX. (B) Heatmap representing PIK3CA, AKT1 and PTEN genomic alterations and expression of PI3Kp110, P-AKT, P-p70-S6K, P-S6 RP and PTEN proteins determined by RPPA analysis in the 37 TNBC PDX. RPPA activation score was determined by calculating the sum of the different protein components. (3 LAR models =HBCx-2, HBCx-31, HBCx-35)
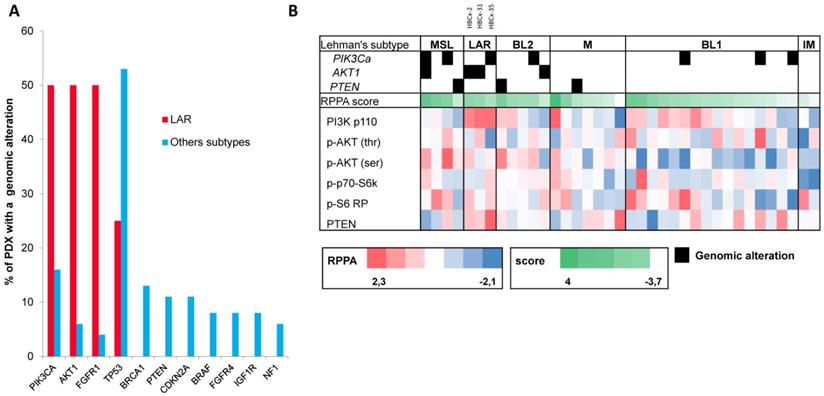
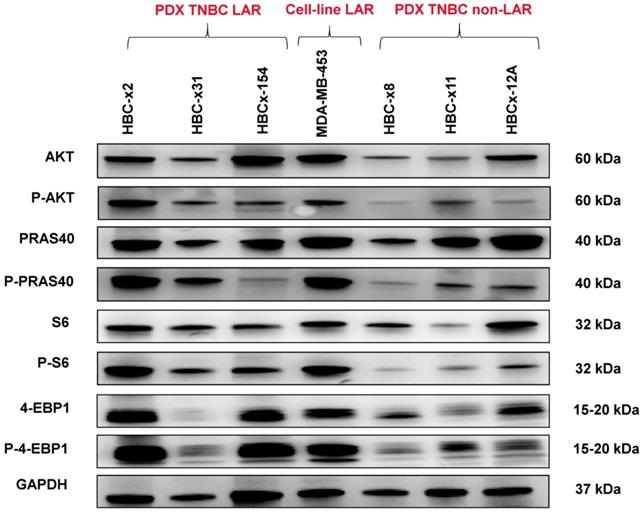
We analyzed the expression of the major effectors of the PI3K pathway (PI3-kinase, p-AKT, p-4E-BP1, p70-S6-kinase, p-S6RP, PTEN) by RPPA, to confirm the activation of this signalling pathway. The MSL and LAR subtypes had higher PI3K pathway activation scores: MSL TNBC are driven by low levels of PTEN protein and LAR TNBC, like other luminal breast cancers, are driven by high levels of PI3K protein (Figure 3B). A western blot analysis of P-AKT, PRAS, P-S6 and P 4EBP1 confirmed the activation of the PIK3 pathway in the three LAR TNBC PDX. Three TNBC PDX (HBCx-8, HBCx-11, HBCx-12A) with low levels of AR mRNA were used as negative controls (Figure 4 and S1) (mean Ct/TBP Hs vs. 35 for the 3 LAR PDX were 1776 versus 0.25 for the 3 negative controls).
Western Blot analysis of different PI3K/AKT/mTOR pathway components in the 3 LAR PDX, a LAR cell line (MDA-MB 453) and 3 non LAR TNBC PDX models.
Efficacy of PI3K pathway inhibitors in 3 LAR TNBC PDX
We assessed the dependence of LAR PDX tumor growth on AR signaling by the in vivo efficacy of enzalutamide, an androgen receptor inhibitor approved for the treatment of prostatic cancer. Surprisingly, enzalutamide was completely ineffective in the three PDX (TGI:-28% for HBCx-2 and 27% for HBCx-31 and HBCx-154) (Figure 5). We investigated the possible association between enzalutamide treatment and a decrease in AR-targeted gene expression, by analysing the expression of various AR-related genes and AR-inducible genes in control and enzalutamide-treated xenografts, by RT-PCR. The expression of AR-related and AR-inducible genes in treated xenografts was similar to that in controls (Figure S2).
In vivo response to targeted therapies and chemotherapy in LAR PDX. (A) In vivo response to enzalutamide, AZD2014 (dual mTORC1 and C2 inhibitor), BAY80-6946 (PI3K inhibitor) , PF-04691502 (dual PI3K and mTOR) in the 3 LAR PDX. mean +/- SD. (B) Response to AC (Adriamycin + cyclophosphamide) in the HBCx-2 and HBCx-31 PDX. (C) response to AC and to the combination of BAY80-6946 + enzalutamide in the HBCx-154 PDX.
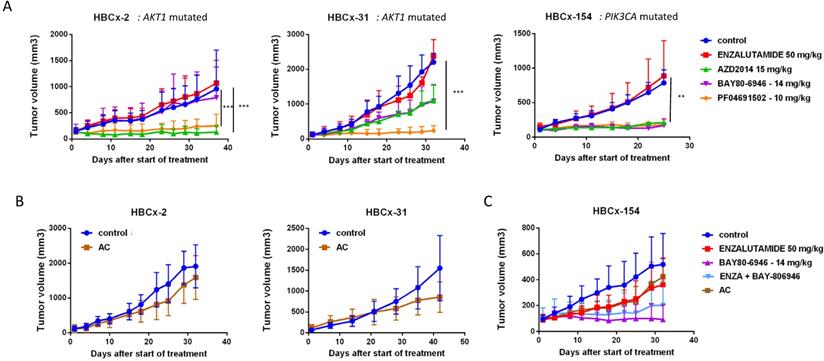
LAR TNBC PDX are characterized by AKT1 or PIK3CA mutations (associated with the activation of PI3K protein signaling). We therefore compared three different PI3K pathway inhibitors (BAY80-6946, a specific inhibitor of the PI3K p110α subunit; PF-04691502, a dual inhibitor of PI3K and mTOR; and AZD2014, an mTORC1 and mTORC2 inhibitor) with enzalutamide. HBCx-2 and HBCx-31 harbored an AKT1 hotspot mutation (p.E17K), whereas HBCx-154 harbored two PIK3CA mutations (p.N345K and p.H1048L). In the PIK3CA mutant model (HBCx-154), treatment with the three PI3K pathway inhibitors resulted in a significant inhibition of tumor growth (TGI= 80% for BAY-80-6946, p=0.006, 72% for PF-04691502 and AZD2014, p=0.02 and p=0.01, respectively). PI3K pathway inhibitors did not decrease tumor growth in the two AKT1-mutated models, but HBCx-2 PDX responded to the mTOR inhibitor AZD2014 (TGI=86%, p=0.0001) and the dual inhibitor (mTOR and PIK3Ca) (TGI =70%, p=0.001). The second AKT1-mutated model (HBCx-31) presented a significant response only to the dual inhibitor (TGI=81%, p=0.02). We also tested a combination of adriamycin and cyclophosphamide [AC] (standard chemotherapy used in breast cancer treatment) in the three models: all three PDX were resistant to AC (Figure 5 B and C). Previous studies in cell line models of LAR breast cancer have reported increased antitumor activity of PI3K inhibitors used in combination with AR inhibitors [13]. We therefore tested the combination of BAY-80-6946 and enzalutamide in the HBCx-154 PDX, the PIK3CA-mutated PDX with the strongest nuclear AR expression. Combination treatment resulted in no greater antitumor activity than observed with BAY-80-6946 alone (Figure 5C).
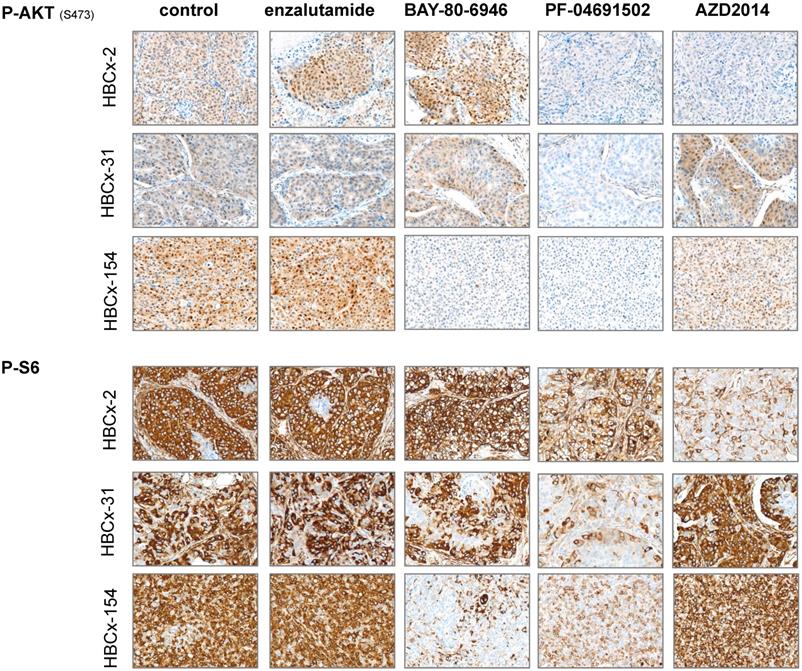
We analyzed pathway inhibition in treated tumors, by performing an IHC analysis of P-AKT and P-S6 on tumors harvested from control and treated xenografts. We confirmed the lower levels of P-AKT and P-S6 expression (i) in the 3 models after PF-04691502 treatment, (ii) in HBCx-154 after BAY-80-6946 treatment and (iii) in HBCx-2 after AZD2014 treatment (Figure 6).
Immunohistochemistry analysis of 3 LAR TNBC PDX: Analysis of p-AKT and p-S6 before and after treatments with enzalutamide, BAY-80-6946, PF-04691502 and AZD2014 (X20)
Prognostic value of PIK3Ca mutation
We analyzed PIK3CA and AKT1 hotspot mutations in a large cohort of TNBC with no distant metastasis at diagnosis, treated at the Institut Curie, to confirm the high frequency of PIK3CA and AKT1 mutations in AR+ TNBC tumors. Transcriptomic analysis is not routinely performed in clinical practice. We therefore focused on 21 TNBC positive by IHC for AR (AR+)(>10%) from a cohort of 323 TNBC. All of these cases were characterized by an apocrine morphology. PIK3CA mutations were strongly correlated with AR+ TNBC (38% (8/21) versus 10% (30/302) in other subtypes; Chi2 =0.001), but AKT1 mutations were not correlated with AR+ TNBC (4.7% (1/21) versus 3.3% in other subtypes, Chi2 =0.5) probably due to the small size of the sample of tumors presenting these alterations. On univariate analysis, PIK3CA mutations were associated with a poorer prognosis in the overall population of TNBC, for both metastasis-free survival (MFS, p=0.01) and overall survival (OS, p=0.0016) (Figure S3), but these associations were not significant on multivariate analysis. Details of the prognostic factors identified are provided in Table S4. In the population of patients with apocrine tumors, the association was close to significance, but the small number of patients made it impossible to draw robust conclusions (n=21; MFS: p=0.06 and OS: p=0.07) (Figure S3).
Discussion
Luminal androgen receptor-positive (LAR) tumors are a rare subtype of TNBC, accounting for 9% of our TNBC PDX cohort [7, 8]. LAR is the most highly differentiated subtype of TNBC. Gene ontology analyses reveal an enrichment of LAR TNBC in hormonally regulated pathways. Lehmann et al. found that AR mRNA levels were much higher in LAR TNBC than in the other TNBC subtypes, and that tumors of the LAR group expressed numerous downstream AR targets and coactivators [7]. Our findings confirm the strong expression of AR-related genes in the LAR subtype, associated with low levels of EMT gene expression (these genes being more strongly expressed by the M and MSL subtypes), consistent with the luminal properties of these tumors.
The role of AR in carcinogenesis has been studied more extensively in prostate cancer, in which there is compelling evidence for a crucial role of this pathway, due to its effects on prostate cell proliferation and differentiation. Furthermore, in clinical practice, androgen inhibitors constitute the cornerstone of medical treatment for prostatic cancer [32]. The role of AR in breast cancer is complex and depends on the signaling pathways activated simultaneously [33]. The lack of efficient targeted therapies and the poor prognosis of TNBC has led to the development of AR-targeted therapies in TNBC with AR expression.
Moreover, chemoresistance is much more frequent in LAR than in the other subtypes, as demonstrated by Masuda, who reported a low rate of pCR after primary chemotherapy in LAR TNBC [34], highlighting the need for new therapies for this subtype of TNBC.
Little is known about the genomic features of the LAR subtype. In the TCGA cohort, LAR TNBC displayed a higher proportion of PIK3CA mutations than other subtypes (46.2% versus 4.5%, p<0.0001) [5]. PIK3CA mutations are associated with AR protein levels [35]. We report specific features of this subtype in terms of genomic alterations and the activation of signaling pathways. Mutations of the PIK3CA and AKT1 genes are frequent in LAR PDX (100%; 4/4 PDX models) and AR+ TNBC (38%; 8/21 patients for PIK3CA). The PIK3 pathway is the only major pathway altered in the LAR subtype, contrasting with the situation in other subtypes and opening up new possibilities for effective treatment. As in luminal breast cancer, this activation involves PIK3CA and AKT1 mutations to a greater extent than PTEN loss (which is more frequently reported in TNBC in general). No specific data are available concerning the levels of PI3K pathway proteins in the LAR subtype. An analysis of the proteins present in 105 breast cancers from the TCGA cohort confirmed that PI3K was expressed in luminal A and B breast cancers, but also in TNBC [36]. We analyzed the activation of the PI3K pathway in the various TNBC subtypes, confirming its correlation with genomic alterations.
The aim of this study was, therefore, to compare PI3K pathway inhibitors with an AR antagonist in chemoresistant LAR PDX models. We describe here the first LAR PDX models used to test the efficacy of targeted therapies in this TNBC subtype. Indeed, few data are available for LAR models: only cell lines or CDX (cell line-derived xenografts) have been described [5, 13].
Our three LAR PDX models were resistant to the AR inhibitor enzalutamide and to standard chemotherapy for breast cancer (combination of anthracyclines and cyclophosphamide). Lehman et al. described the effects of the AR inhibitor bicalutamide in LAR cell lines [13]. Three clinical trials have tested androgen inhibitors in TNBC and have reported a clinical benefit of 19% to 29% [11, 12]. The Advanced Breast Cancer (ABC4) guidelines propose androgen inhibitors as a treatment option for AR+ TNBC [37]. Despite the potential utility of AR inhibitors in the treatment of LAR tumors, the clinical benefits of such treatment are modest and unmet clinical needs remain for LAR TNBC.
Our PDX models displaying PI3K pathway activation responded strongly to PI3K inhibitors, suggesting that PI3KCA and AKT1 mutations drove the proliferation of these tumors. As expected, BAY-80-6946, a specific inhibitor of PI3K, was effective in the PIK3CA-mutated model, but not in the two AKT1-mutated PDX [38]. The PI3K-mTOR dual inhibitor (PF-04691502) strongly decreased tumor growth in the three LAR PDXs. This dual inhibitor abolished the PI3K pathway more completely, by inhibiting all catalytic isoforms of PI3K as well as mTORC1 and mTORC2 [39]. AZD2014 was effective in two models (one with a PIK3CA mutation and one with a AKT1 mutation). AZD2014 inhibited mTORC1 and mTORC2 and abolished the feedback loops related to the inhibition of mTORC1 alone, thereby increasing the efficacy of a specific mTORC1 inhibitor [40]. In HBCx-154 PDX (PIK3CA-mutated) the 3 compounds had similar anti-tumor activities, however inhibition of P-AKT and P-S6 was higher in xenografts treated by BAY-80-6946 and PF-04691502 compounds, suggesting that in this tumor inhibition of PI3K is necessary to inhibit PI3K-Akt signaling pathway.
Despite the lack of enzalutamide activity, we hypothesized that a combination of enzalutamide and a PI3K inhibitor would promote tumor regression, as a synergistic effect has been described in an LAR cell line [13]. However, the combination of enzalutamide and BAY80-6946 in the PIK3CA-mutated PDX model with the highest levels of AR expression did not result in greater antitumor activity than BAY80-6946 monotherapy. This finding is not consistent with Lehmann's results showing a pan-PI3K inhibitor and a dual PI3K/mTOR inhibitor to be more effective in LAR CDX than bicalutamide in two cell-line models: bicalutamide slowed growth at the limit of significance, but the PI3K inhibitor appeared to be more effective both alone and in combination with bicalutamide [13], and several clinical trials of combination therapy for AR+ TNBC are currently underway (NCT02457910, NCT03207529).
Our LAR models present primary resistance to enzalutamide, which can be explained partly by the low levels of nuclear AR expression in the HBCx-2 and HBCx-31 models, although the AR present seems to be active, as shown by the high levels of AR-related gene expression. The presence of oncogenic activating mutations associated with PI3K pathway activation and the sensitivity to PIK3CA/mTOR inhibitors observed suggest that AKT1 and PIK3CA mutations are the main drivers of cell proliferation in these tumors. Alternatively, enzalutamide may not target AR in these models, as suggested by the absence of inhibition of AR-dependent gene expression after enzalutamide therapy. However, the dose used in our study (50 mg/kg/day) has been shown to be effective in the cell line-derived xenograft model of ER breast cancer [41] and in xenograft models of prostate cancer [42]. Moreover, we can speculate that the engraftment of androgen-dependent TNBC may be compromised in female nude mice, in which androgen production may be insufficient to support AR-dependent tumor growth. Our results show that the combination of PI3K and AR inhibitors does not increase sensitivity. Reciprocal feedback inhibition of AR by PI3K signaling in prostatic cancer has been reported, with the inhibition of PI3K leading to the derepression and activation of AR target genes, and the inhibition of AR leading to reciprocal PI3K pathway activation [43]. However, in the HBCx-154 model, PI3K pathway inhibition did not result in changes in AR-related gene expression (data not shown). PI3K inhibition thus appears to be insufficient to reverse AR resistance, but other mechanisms of resistance have been reported, including NFkB activation, MYC gain, and mitochondrial reprogramming [44]. Further investigations, possibly involving enzalutamide-resistant TNBC cell lines, are required, to investigate the molecular mechanisms underlying the crosstalk between PIK3 signaling and enzalutamide resistance.
In clinical practice, AKT1 inhibitors will probably be developed for the treatment of tumors of the LAR subtype, and interesting results have already been obtained for combinations of these agents with chemotherapy in TNBC [45].
The prognostic impact of AR in TNBC patients remains a matter of debate. Lehmann described a tendency towards a poorer prognosis for LAR (distant metastasis-free survival) [7]. By contrast, Masuda reported a tendency towards better survival for LAR after primary chemotherapy [34]. In a meta-analysis of TNBC patients, AR expression was also found to be associated with a better prognosis [35]. Furthermore, in breast cancer, PIK3CA mutation is significantly associated with longer invasive disease-free survival (p = 0.043), but not with longer distant DFS or OS [46]. The prognostic value of these parameters appears to have been demonstrated more clearly for estrogen receptor-positive breast cancer [47]. Multivariate analysis of our TNBC cohort did not confirm the prognostic value of PIK3CA mutation.
In conclusion, our results provide a robust rationale for screening for PIK3CA and AKT1 mutations in AR-TNBC patients and for treating selected patients with PI3K/AKT/mTOR pathway inhibitors.
Abbreviations
AR: Androgen receptor; BL1: Basal-like1; BL2: Basal-like2; CDX: Cell line-derived xenografts; CK: Cytokeratin; EMT: Epithelial-mesenchymal transition; ER: Estrogen receptor; ERBB2: Human epidermal growth factor receptor 2; IM: Immunomodulatory; LAR: Luminal androgen receptor; M: Mesenchymal; MFS: Metastasis-free survival; MSL: Mesenchymal stem-Like; OS: Overall survival; PDX: Patient-derived xenografts; PI3K: Phosphatidylinositol 3-kinase; PR: Progesterone receptor; RPPA: Reverse-phase protein arrays; RTV: Relative tumor volume; TGI: Tumor growth inhibition; TNBC: Triple-negative breast cancers.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank the animal platform of the Institut Curie and Julie Sappa of Alex Edelman and Associates for her help in correcting the English version of the article. We thank Jean-Luc Servely for technical assistance in immunohistochemistry staining. We thank the Translational Research Department and SIRIC2 for funding.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kwan ML, Kushi LH, Weltzien E, Maring B, Kutner SE, Fulton RS. et al. Epidemiology of breast cancer subtypes in two prospective cohort studies of breast cancer survivors. Breast Cancer Res. 2009;11:R31
2. Morris GJ, Naidu S, Topham AK, Guiles F, Xu Y, McCue P. et al. Differences in breast carcinoma characteristics in newly diagnosed African-American and Caucasian patients: a single-institution compilation compared with the National Cancer Institute's Surveillance, Epidemiology, and End Results database. Cancer. 2007;110:876-84
3. Aydiner A, Sen F, Tambas M, Ciftci R, Eralp Y, Saip P. et al. Metaplastic breast carcinoma versus triple-negative breast cancer: survival and response to treatment. Medicine (Baltimore). 2015;94:e2341
4. Burstein MD, Tsimelzon A, Poage GM, Covington KR, Contreras A, Fuqua SA. et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin Cancer Res. 2015;21:1688-98
5. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y. et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750-67
6. Bonsang-Kitzis H, Sadacca B, Hamy-Petit AS, Moarii M, Pinheiro A, Laurent C. et al. Biological network-driven gene selection identifies a stromal immune module as a key determinant of triple-negative breast carcinoma prognosis. Oncoimmunology. 2016;5:e1061176
7. Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61-70
8. Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA. et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016;7:11479
9. Lehmann-Che J, Hamy AS, Porcher R, Barritault M, Bouhidel F, Habuellelah H. et al. Molecular apocrine breast cancers are aggressive estrogen receptor negative tumors overexpressing either HER2 or GCDFP15. Breast Cancer Res. 2013;15:R37
10. Wang C, Pan B, Zhu H, Zhou Y, Mao F, Lin Y. et al. Prognostic value of androgen receptor in triple negative breast cancer: A meta-analysis. Oncotarget. 2016;7:46482-91
11. Gucalp A, Tolaney S, Isakoff SJ, Ingle JN, Liu MC, Carey LA. et al. Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic Breast Cancer. Clin Cancer Res. 2013;19:5505-12
12. Bonnefoi H, Grellety T, Tredan O, Saghatchian M, Dalenc F, Mailliez A. et al. A phase II trial of abiraterone acetate plus prednisone in patients with triple-negative androgen receptor positive locally advanced or metastatic breast cancer (UCBG 12-1). Ann Oncol. 2016;27:812-8
13. Lehmann BD, Bauer JA, Schafer JM, Pendleton CS, Tang L, Johnson KC. et al. PIK3CA mutations in androgen receptor-positive triple negative breast cancer confer sensitivity to the combination of PI3K and androgen receptor inhibitors. Breast Cancer Res. 2014;16:406
14. Bruna A, Rueda OM, Greenwood W, Batra AS, Callari M, Batra RN. et al. A biobank of breast cancer explants with preserved intra-tumor heterogeneity to screen anticancer compounds. Cell. 2016;167:260-74 e22
15. Izumchenko E, Paz K, Ciznadija D, Sloma I, Katz A, Vasquez-Dunddel D. et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann Oncol. 2017;28:2595-605
16. Coussy F, de Koning L, Lavigne M, Bernard V, Ouine B, Boulai A. et al. A large collection of integrated genomically characterized patient-derived xenografts highlighting the heterogeneity of triple-negative breast cancer. Int J Cancer. 2019;145:1902-1912
17. Carvalho BS, Louis TA, Irizarry RA. Quantifying uncertainty in genotype calls. Bioinformatics. 2009;26:242-9
18. Samuels AL, Peeva VK, Papa RA, Firth MJ, Francis RW, Beesley AH. et al. Validation of a mouse xenograft model system for gene expression analysis of human acute lymphoblastic leukaemia. BMC Genomics. 2010;11:256
19. Chen X, Li J, Gray WH, Lehmann BD, Bauer JA, Shyr Y. et al. TNBCtype: a subtyping tool for triple-negative breast cancer. Cancer Inform. 2012;11:147-56
20. Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J. et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023-31
21. Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J. et al. The Catalogue of Somatic Mutations in Cancer (COSMIC). Curr Protoc Hum Genet. 2008 Chapter 10: Unit 10 1
22. Hidalgo M, Amant F, Biankin AV, Budinska E, Byrne AT, Caldas C. et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014;4:998-1013
23. Laurent C, Gentien D, Piperno-Neumann S, Nemati F, Nicolas A, Tesson B. et al. Patient-derived xenografts recapitulate molecular features of human uveal melanomas. Mol Oncol. 2013;7:625-36
24. Crepin R, Gentien D, Duche A, Rapinat A, Reyes C, Nemati F. et al. Nanobodies against surface biomarkers enable the analysis of tumor genetic heterogeneity in uveal melanoma patient-derived xenografts. Pigment Cell Melanoma Res. 2017;30:317-27
25. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1
26. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401-4
27. Bieche I, Parfait B, Le Doussal V, Olivi M, Rio MC, Lidereau R. et al. Identification of CGA as a novel estrogen receptor-responsive gene in breast cancer: an outstanding candidate marker to predict the response to endocrine therapy. Cancer Res. 2001;61:1652-8
28. Bieche I, Onody P, Laurendeau I, Olivi M, Vidaud D, Lidereau R. et al. Real-time reverse transcription-PCR assay for future management of ERBB2-based clinical applications. Clin Chem. 1999;45:1148-56
29. Rondeau S, Vacher S, De Koning L, Briaux A, Schnitzler A, Chemlali W. et al. ATM has a major role in the double-strand break repair pathway dysregulation in sporadic breast carcinomas and is an independent prognostic marker at both mRNA and protein levels. Br J Cancer. 2015;112:1059-66
30. O'Malley FP, Bane A. An update on apocrine lesions of the breast. Histopathology. 2008;52:3-10
31. Cornford P, Bellmunt J, Bolla M, Briers E, De Santis M, Gross T. et al. EAU-ESTRO-SIOG Guidelines on Prostate Cancer. Part II: Treatment of relapsing, metastatic, and castration-resistant prostate cancer. Eur Urol. 2017;71:630-42
32. Litwin MS, Tan HJ. The diagnosis and treatment of prostate cancer: A Review. JAMA. 2017;317:2532-42
33. Gerratana L, Basile D, Buono G, De Placido S, Giuliano M, Minichillo S. et al. Androgen receptor in triple negative breast cancer: A potential target for the targetless subtype. Cancer Treat Rev. 2018;68:102-10
34. Masuda H, Baggerly KA, Wang Y, Zhang Y, Gonzalez-Angulo AM, Meric-Bernstam F. et al. Differential response to neoadjuvant chemotherapy among 7 triple-negative breast cancer molecular subtypes. Clin Cancer Res. 2013;19:5533-40
35. Gonzalez-Angulo AM, Stemke-Hale K, Palla SL, Carey M, Agarwal R, Meric-Berstam F. et al. Androgen receptor levels and association with PIK3CA mutations and prognosis in breast cancer. Clin Cancer Res. 2009;15:2472-8
36. Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P. et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534:55-62
37. Cardoso F, Senkus E, Costa A, Papadopoulos E, Aapro M, Andre F. et al. 4th ESO-ESMO International Consensus Guidelines for Advanced Breast Cancer (ABC 4). Ann Oncol. 2018;29:1634-57
38. Xu YC, Wang X, Chen Y, Chen SM, Yang XY, Sun YM. et al. Integration of receptor tyrosine kinases determines sensitivity to PI3Kα-selective inhibitors in breast cancer. Theranostics. 2017;23:974-986
39. Markman B, Tabernero J, Krop I, Shapiro GI, Siu L, Chen LC. et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of the oral phosphatidylinositol-3-kinase and mTOR inhibitor BGT226 in patients with advanced solid tumors. Ann Oncol. 2012;23:2399-408
40. Pike KG, Malagu K, Hummersone MG, Menear KA, Duggan HM, Gomez S. et al. Optimization of potent and selective dual mTORC1 and mTORC2 inhibitors: the discovery of AZD8055 and AZD2014. Bioorg Med Chem Lett. 2013;23:1212-6
41. Barton VN, D'Amato NC, Gordon MA, Lind HT, Spoelstra NS, Babbs BL. et al. Multiple molecular subtypes of triple-negative breast cancer critically rely on androgen receptor and respond to enzalutamide in vivo. Mol Cancer Ther. 2015;14:769-78
42. Guerrero J, Alfaro IE, Gomez F, Protter AA, Bernales S. Enzalutamide, an androgen receptor signaling inhibitor, induces tumor regression in a mouse model of castration-resistant prostate cancer. Prostate. 2013;73:1291-305
43. Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S. et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19(5):575-586
44. Armstrong AJ, Halabi S, Healy P, Alumkal JJ, Winters C, Kephart J. et al. Phase II trial of the PI3 kinase inhibitor buparlisib (BKM- 120) with or without enzalutamide in men with metastatic castration resistant prostate cancer. Eur J Cancer. 2017;81:228-36
45. Kim SB, Dent R, Im SA, Espié M, Blau S, Tan AR. et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicenter, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017;18:1360-1372
46. Zardavas D, Te Marvelde L, Milne RL, Fumagalli D, Fountzilas G, Kotoula V. et al. Tumor PIK3CA Genotype and Prognosis in Early-Stage Breast Cancer: A Pooled Analysis of Individual Patient Data. J Clin Oncol. 2018;36:981-90
47. Pang B, Cheng S, Sun SP, An C, Liu ZY, Feng X. et al. Prognostic role of PIK3CA mutations and their association with hormone receptor expression in breast cancer:a meta-analysis. Sci Rep. 2014;4:6255
Author contact
![]() Corresponding author: Florence Coussy. E-mail: florence.coussyfr; Phone: + 33 1 56245841; Fax: + 33 1 53 10 26 65.
Corresponding author: Florence Coussy. E-mail: florence.coussyfr; Phone: + 33 1 56245841; Fax: + 33 1 53 10 26 65.