Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Epigenetic regulation
Epigenetic alterations in...
Epigenetic impact on targeted...
Epigenetic impact on...
Epigenetics in uveal melanomas...
Epigenetic modifications and...
Conclusion
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(4):1777-1797. doi:10.7150/thno.36218 This issue Cite
Review
The “ART” of Epigenetics in Melanoma: From histone “Alterations, to Resistance and Therapies”
Thomas Strub1,2, ![]() , Robert Ballotti1,2, Corine Bertolotto1,2
, Robert Ballotti1,2, Corine Bertolotto1,2
1. Université Nice Côte d'Azur, Inserm, C3M, France
2. Biology and pathologies of melanocytes, Equipe labellisée ARC 2019, C3M, team 1, France
Received 2019-4-30; Accepted 2019-11-14; Published 2020-1-1
Abstract

Malignant melanoma is the most deadly form of skin cancer. It originates from melanocytic cells and can also arise at other body sites. Early diagnosis and appropriate medical care offer excellent prognosis with up to 5-year survival rate in more than 95% of all patients. However, long-term survival rate for metastatic melanoma patients remains at only 5%. Indeed, malignant melanoma is known for its notorious resistance to most current therapies and is characterized by both genetic and epigenetic alterations. In cutaneous melanoma (CM), genetic alterations have been implicated in drug resistance, yet the main cause of this resistance seems to be non-genetic in nature with a change in transcription programs within cell subpopulations. This change can adapt and escape targeted therapy and immunotherapy cytotoxic effects favoring relapse.
Because they are reversible in nature, epigenetic changes are a growing focus in cancer research aiming to prevent or revert the drug resistance with current therapies. As such, the field of epigenetic therapeutics is among the most active area of preclinical and clinical research with effects of many classes of epigenetic drugs being investigated. Here, we review the multiplicity of epigenetic alterations, mainly histone alterations and chromatin remodeling in both cutaneous and uveal melanomas, opening opportunities for further research in the field and providing clues to specifically control these modifications. We also discuss how epigenetic dysregulations may be exploited to achieve clinical benefits for the patients, the limitations of these therapies, and recent data exploring this potential through combinatorial epigenetic and traditional therapeutic approaches.
Keywords: melanoma, epigenetics, drug resistance, targeted therapy, immunotherapy
Introduction
Incidence of cutaneous malignant melanoma is rising steadily. Its therapeutic management is a real challenge for oncologists as it is amongst the solid malignancies most refractory to conventional cancer therapies [1]. Recently, our improved understanding of the molecular mechanisms underlying cutaneous melanoma (CM) biology has led to improved treatments for advanced CM, which includes targeting the MAPK signaling pathway which dramatically improved patient outcome. More recently, the use of immune checkpoint inhibitors have shown to be effective in almost a third of all patients [2]. Moreover, improved overall survival outcomes were observed with targeted therapies in patients with BRAFV600 mutant unresectable stage III or stage IV melanoma, with up to 70% of patients who responded according to Response Evaluation Criteria in Solid Tumors (RECIST) along with tumor size reduction in 95% of patients in phase 3 randomized clinical trials [3-6]. Unfortunately, these results are either transient or limited to restricted subsets of patients due to intrinsic or acquired resistance. What is certain is that both intrinsic and acquired resistances can be driven by genetic and epigenetic alterations underlying gene expression changes in genetically identical cells. Epigenetic reprogramming rewires metabolic and signaling networks, thereby driving the emergence of tumor cell subpopulations with distinct behavior and altered antigenic landscape [7]. This intratumor heterogeneity drives new resistance mechanisms to escape drug cytotoxicity or surveying by the immune system, enabling tumor regrowth and disease relapse. As the side effects are often severe and can be life-threatening, alternative therapies must be explored. To advance in this field, we must understand the mechanisms of resistance in order to identify novel targets and therapeutic approaches for more effective and long-lasting treatments for patients.
In this review, we introduce the major advances in CM treatments and summarize recent discoveries of epigenetic influences. We focus on histone modifications, chromatin remodeling and histone variants in metastatic CM, followed by their role in resistance to therapy, and discuss why they are important therapeutic targets. We also discuss epigenetic changes, which are now receiving attention in metastatic uveal melanoma (UM), for which therapeutic intervention remains extremely limited. Additional important epigenetic events such as DNA methylation and non-coding RNAs are beyond the aim of this review. These processes will be briefly discussed and we refer the reader to other reviews [8,9].
Epigenetic regulation
Recent advances in deciphering the mechanisms of melanoma progression underlined a critical role for epigenetic alterations, thereby turning both academic and medical attention towards the application of epigenetics to melanoma detection and therapeutics.
Chromatin structure and histone modifications
By remodeling the chromatin structure, epigenetics co-operate with transcription factors and the translational machinery in fine-tuning gene expression [10]. Chromatin is the physiological state of the eukaryotic genome, in which DNA is packaged with its intimately associated proteins, the majority of which are histones. The nucleosome is the basic repeating unit of chromatin, which consists of 146 base pairs of DNA wrapped around an octamer of histone proteins: two of each histones H2A, H2B, H3 and H4 and/or their variants [11,12]. While chromatin is a highly organized structure, changes in its structure are essential for regulation of key cellular processes and therefore must be dynamic. Changes include post-translational modifications (affecting histones N-terminal tails, such as acetylation (ac), methylation (me), ubiquitylation, phosphorylation, sumoylation or glycosylation), ATP-dependent chromatin remodeling, and the incorporation of specialized histone variants into chromatin [11-14]. As a biological consequence, the genome can be partitioned into distinct domains, such as euchromatin (where DNA is “open” allowing transcription) and heterochromatin (where DNA is “closed” preventing transcription). This dynamic process is driven by the activity of specific cellular enzymes, for example, histone methyltransferases (HMTs), histone demethylases and histone acetyl transferases (HATs)/histone deacetylases (HDACs) for determining the status of histone methylation and acetylation, respectively. The balance of these histone modifications orchestrates the above mentioned states by modifying the charges of the nucleosomal structure by respectively decreasing or increasing the histone-DNA interactions and therefore modulation of transcriptional activation and repression [15].
Histone methylation comes in different forms. Indeed, histone lysine (K) can be mono-, di- or tri-methylated and defines different regulatory regions according to their methylation status. For example, H3K4me1 was the first histone modification connected with distal regulatory regions, called enhancers, whereas H3K4me3 is enriched at active promoters [16]. Of note, these regulatory elements are known to play a key role in regulating expression of genes important for maintaining cell identity and disease [17]. In addition to its status, the methylation site on the histone tail is also critical for diverse functions; H3K79me2 or H3K36me3 are mainly found where active transcription takes place whereas H3K9me3 or H3K27me3 are linked to transcriptional repression [13]. On the other hand, the histone acetylation state is also considered as a recruitment platform for transcription factors such as for bromodomain-containing proteins [15].
Another player in chromatin remodeling processes is the SWI/SNF complex (also known as BAF complex). This large multi-subunit complex uses the energy of ATP hydrolysis to remodel and evict nucleosomes at gene promoter, impacting the recruitment of regulators and therefore transcription regulation. This complex contains more than 15 members including an ATPase (BRG1, or BRM also known as SMARCA4 or SMARCA2 respectively) and a DNA binding domain subunit (ARID1A, ARID1B or ARID2) [18]. Finally, with their sequences and structural variations from the canonical histones, histone variants can replace their counterparts within the nucleosome [12]. Histone variants can have temporal and tissue-specific expression and affect a variety of DNA-templated processes.
Thus, by disrupting chromatin contact or by affecting the recruitment of nonhistone proteins to chromatin, all the above-mentioned reversible modifications orchestrated by “writers, readers and erasers” (enzymes that add, bind or remove chemical modifications to histones) influence many fundamental biological processes [19]. Strikingly, it has become evident in the last decade that the epigenetic landscape contains a unique ability to regulate key cellular and developmental processes [13,20,21], and that its deregulation may contribute to melanoma initiation, progression and drug resistance that will be discussed hereafter.
Epigenetic alterations in melanoma development and pathogenesis
Despite the unquestionable importance of oncogene activation and/or tumor suppressor inactivation in melanoma tumor burden, a growing body of evidence suggests that modifications in the epigenetic landscape drives the alteration of transcriptional programs that are tightly associated with the development of melanoma pathogenesis.
Regulation of chromatin in its various active states is largely controlled through post-translational modifications (PTMs) of the core histone proteins mediated by histone writers, erasers, and readers. Epigenetic regulations in melanoma, especially through these histone modifications, are gaining more and more attention. To begin with, insight into the importance of histone modifications in melanoma development emerged due to the fact that nevi, which are benign melanocytic lesions, mostly carry the oncogenic BRAFV600E mutated form but rarely become malignant melanoma. This indicates that additional events are necessary to initiate melanoma. Patton et al., developed the first animal model of a BRAFV600E driven melanoma using a transgenic zebrafish model expressing the human BRAFV600E under the control of the mitfa promoter. They showed that in a p53 deficient background, only a fraction of zebrafish develop melanoma tumors [22]. As only a subpopulation of genetically identical cells promote melanoma, this fact highlights the importance of additional molecular events beyond genetic alterations. To assess this, the same group developed a p53/BRAF/crestin: EGFP zebrafish model. The crestin gene first marks the neural crest progenitors during embryonic development but importantly, it is re-expressed specifically in melanoma tumors in adult zebrafish allowing them to track melanoma lesions at the time of their initiation [23]. Relevant in the scope of this review, they found H3K27ac super-enhancer marks (enhancer cluster regions) at the sox10 locus, which plays a key role in neural crest formation and melanomagenesis, suggesting an epigenetic mechanism to increase SOX10 expression leading to the reemergence of the neural crest progenitor state to initiate melanoma [23].
Histone modifications “Writers”
Several studies have highlighted a role for “chromatin writers” in melanoma progression (Figure 1). Using metastatic melanomas from patient-derived tumors, Bossi et al., performed the first in vivo genetic screen targeting chromatin players with specific shRNA libraries [24]. Their study identified an unprecedented number of genes essential for tumor growth (e.g BAZ1B, SMARCA4, CHD4, KMT2D) and a certain interpatient heterogeneity. Importantly, these genes were not mutated in the same patients suggesting that the signaling pathways in these tumors are activated in a patient-specific manner. The authors focused on KMT2D, the major methyltransferase for H3K4me1 enhancer in mammals, implicated therefore in gene expression program [25]. KMT2D-silencing leads to the inactivation of a subset of KMT2D-bound enhancers with a decrease of H3K4me1 and H3K27ac along with a down-regulation of genes which are critical for cell migration (e.g. MFGE8 and RPL39L). Of note, the most proximal genes to these enhancers were KMT2D target genes suggesting that KMT2D deregulates enhancer activity to promote tumorigenesis [24]. However, an important heterogeneity was observed within the patients analyzed, suggesting distinct signaling pathways involved, most likely reflecting tumor-specific environment or genetic context. Usually, best candidates for development of targeted therapies are genes harboring biologically relevant mutations. However, taking into consideration the patient heterogeneity and that most genes identified in that study critical for tumor maintenance are not somatically mutated, the clinical impact of this study is demonstrated by their increased number of potential druggable genes for each patient [24].
Role of “Writers” in melanoma. Epigenetic mechanisms driven by the histone lysine methyltransferases ((A) KMT2D, (B) SETDB1 and (C) EZH2) in melanoma progression. Few epigenetic players can be targeted by small molecules to reverse the chromatin state and decrease tumorigenicity.
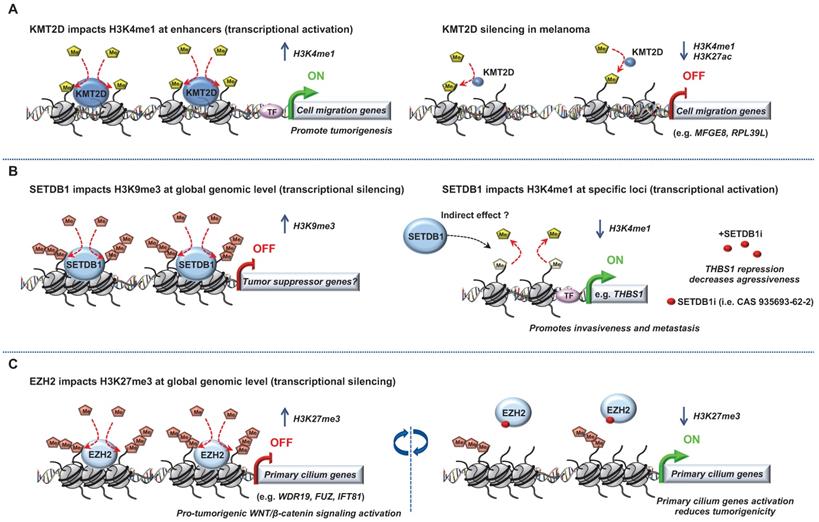
Another “chromatin writers” implicated in melanoma is the SET domain bifurcated 1 (SETDB1), a member of the SUV39 family of histone lysine methyltransferases, catalyzing methylation of lysine 9 on the histone 3 which leads to epigenetically mediated gene expression silencing [26]. Interestingly, the deposition of H3K9me3 on histones by SETDB1 occurs upon its recruitment to methylated CpG islands via a methyl-CpG-binding domain [27]. Linking DNA methylation with heterochromatin formation at specific loci suggest a precise transcriptional repression control for a more accurate gene expression program. Strikingly, SETDB1 is amplified in human melanoma compared to nevus or normal skin and accelerates melanoma development in the same zebrafish BRAFV600E model system described above [28]. Recently, the study from Orouji et al., unraveled a SETDB1-mediated epigenetic mechanism in melanoma progression. They showed that the activation of thombospondin-1 (THBS1), known to promote invasiveness and metastasis formation in melanoma, is induced by SETDB1. In this case, in addition to H3K9me3, SETDB1 alters the methylation patterns related to H3K4. Indeed, they identified enrichment for H3K4me1 upstream of the THBS1 gene which was reversely influenced by SETDB1 expression suggesting that SETDB1 may act not only on regulating H3K9me3 distribution but also on additional epigenetic marks to impact gene activation or repression. Finally, treatment with a small molecule inhibitor for H3K9me-specific histone methyltransferase to block the SETDB1 protein significantly decreased melanoma cell viability. Of note, to temper the impact of other H3K9 histone methyltransferases, the authors focused on melanoma cell lines with high levels of endogenous SETDB1 only. Interestingly, melanoma cells with low levels of SETDB1 were not affected suggesting SETDB1 as a promising new therapeutic target in melanoma [29].
Another histone methyltransferase involved in melanoma is enhancer of zeste homolog 2 (EZH2), the catalytic subunit of the polycomb repressive complex 2 (PRC2) catalyzing trimethylation of lysine 27 on histone 3 subsequently repressing transcription. EZH2 expression is elevated and associated with poor survival in melanoma. Its conditional ablation inhibits tumor growth and metastases in a NRASQ61K melanoma mouse model [30]. Conversely, the most common human EZH2Y646N gain of function somatic mutation (Y641F in mouse) through H3K27me3 accumulation and gene repression, favors melanoma progression [31-33]. EZH2 has been shown to exert its effect through stimulation of the noncanonical NF-kB pathway leading to senescence bypass [34] and epigenetic silencing of primary cilium genes that results in activation of the pro-tumorigenic WNT/β-catenin signaling [31].
A specific cooperation between Ezh2Y641F and B-RafV600E but not N-RasQ61R in inducing melanoma in mice was also reported [33]. Of note, the role of EZH2 and its associated change in histone trimethylation seems more complex than expected. Indeed, Souroullas et al., showed that although Ezh2Y641F triggers H3K27me3 accumulation, it also caused a vast reorganization of chromatin structure, including a loss of H3K27me3 that was associated with increased transcription at many loci [33]. Together, the abovementioned studies have demonstrated that EZH2 function can be effectively inhibited by a number of small molecules reducing melanoma cell growth and metastases. The translation of EZH2 inhibitors into clinical trials have shown preliminary evidence of clinical response (CR) in cancer [35]. Notably, a recent study identified a molecular mechanism linking MAPK signaling activation mediated by BRAFV600E mutation and downstream H3K27me3 remodeling for maintenance of gene expression silencing in tumorigenesis. The authors demonstrated that c-Myc regulates transcription of the PRC2 complex components (i.e. Ezh2, Suz12 and Jarid2) as well as their post-transcriptional levels. This regulation mediated by c-Myc is essential for BRAFV600E-induced H3K27me3 deposition and gene silencing in tumorigenesis [36].
Histone modifications “Readers”
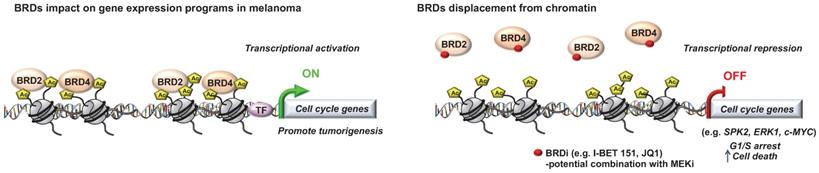
Critical role for a number of “chromatin readers” in melanoma has now been confirmed by several groups (Figure 2). These “reader” proteins are able to recognize a specific chromatin modification, subsequently initiating downstream regulatory processes. We will focus here on the bromodomain and extra-terminal domain (BET) proteins (BRD2, BRD3, BRD4 and BRDT), which bind to acetylated lysine residues of histone. Briefly, it has been shown that these proteins render nucleosomes marked by acetylation permissive to the passage of elongating RNA polymerase II, and therefore couple histone acetylation to gene expression regulation [37]. Among this family, it has been shown that BRD2 and BRD4 are overexpressed in melanoma tissues and are required for tumor maintenance by controlling the expression of key cell cycle and survival genes. In particular, using the bromodomain (BrD) containing proteins inhibitor I-BET151, Gallagher et al., observed a selective inhibition of the NF-κB signaling pathway with genes involved in induction of inflammation (e.g. VEGF, CCL-20), cell cycle regulation (e. g. CDK6) and a downregulation of cytokines production such as IL-6 and IL-8 mainly via BRD2 displacement [38]. The same group has also shown that I-BET151 inhibits melanoma growth in vivo and induces apoptosis which is caspase-dependent and associated with G1 cell cycle arrest in melanoma cells [39]. Interestingly, by using the BrDi MS436 or MS417, another BrDi previously reported to have higher binding affinity and specificity for BET family members, similar observations (cytostatic effect along with G1 arrest) were reported [40]. The authors showed that BET displacement downregulates the key cell cycle genes SKP2, ERK1 and c-MYC along with the accumulation of cyclin-dependent kinase inhibitors (e.g. p21 and p27) [40]. These studies suggest that specific inhibition of BET family members similarly impairs melanoma cells growth in vitro and in vivo than general BrDi. Moreover, transcriptomic analysis of melanocytes and melanoma cells exposed to the BET inhibitor JQ1 identified the transmembrane protein, AMIGO2, as a BET target gene essential for melanoma cell survival [41]. The authors showed that AMIGO2 is regulated by a melanoma-specific BRD2/4-bound promoter and super-enhancer configuration [41]. Importantly, these studies reported that BETi efficacy was not influenced by BRAF or NRAS mutational status, supporting the inhibition of BrD proteins, especially BET family members, to modify epigenetic mechanisms of gene expression to correct disease states in patients for whom no effective targeted therapy is offered. On that note, an effective combination treatment for NRAS-mutant melanoma remains a therapeutic challenge in this field. A recent study used this model to test compound combinations to avert resistance. In that study, using a combination of BET and MEK inhibitors, they identified a critical role for the transcription factor TCF19 in cell cycle checkpoints regulation. TCF19 downregulation triggers a substantial transcriptional perturbation and activates pro-apoptotic signaling leading to cell death [42]. Underlying the clinical aspect of these studies, the co-targeting of BET and MEK has been proposed in NRAS mutant melanoma cell lines or for melanomas with no other therapeutic options to offset resistance to targeted and/or immunotherapies [42].
Role of “Readers” in melanoma. Key roles of the bromodomain and extraterminal domain (BET) proteins in melanoma progression that can be targeted to decrease tumorigenicity.
Histone modifications “Erasers”
The most studied of these modifying enzymes are the HDACs removing acetyl groups on the histone tails (Figure 3). Typically, HDACs belong to either the histone deacetylase family or the Sir2 regulator family. In humans, HDACs are divided into four classes. The class I proteins (HDAC1, HDAC2, HDAC3, and HDAC8) mostly nuclear, the class II proteins (HDAC4, HDAC5, HDAC6, HDAC7, HDAC9, and HDAC10) that shuttled between the nucleus and cytoplasm, the class III proteins (SIRT1, SIRT2, SIRT3, SIRT4, SIRT5, SIRT6, and SIRT7) that are the NAD dependent sirtuins and the class IV protein (HDAC11) [43]. Studies of epigenomic alterations using non-tumorigenic melanocytes from nevi and tumorigenic melanocytes from melanomas revealed a loss of histone acetylation and H3K4me2/3 on regulatory regions proximal to specific cancer-regulatory genes involved in important signaling pathway-driving melanoma [44]. Treatment with HDAC inhibitors (HDACi) prevented excessive proliferation in human melanoma cells, suggesting functional roles of observed chromatin state transitions in driving hyper-proliferative phenotype [44].
Role of “Erasers” in melanoma. Epigenetic mechanisms driven by (A) histone lysine deacetylases (HDACs) or (B) histone lysine demethylases (KDM5B, KDM1A, KDM4C and KDM6B) in melanoma progression. Few epigenetic players can be targeted by small molecules to reverse the chromatin state and decrease tumorigenicity.
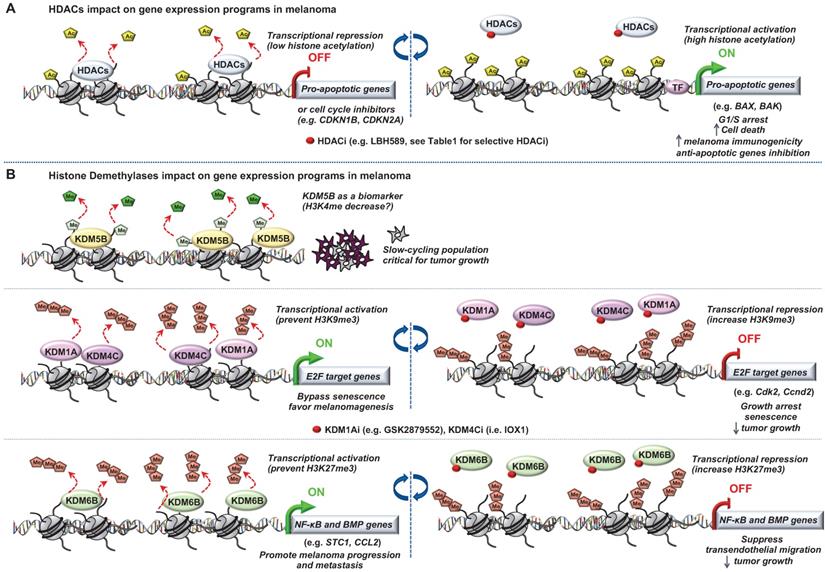
In this context, the locus INK4a-ARF, which plays key role in melanomagenesis [45], is subjected to histone acetylation modifications leading to the deregulation of the cognate tumor suppressor genes expression p14ARF and p16INK4A [46,47]. With the emergence of a role for HDACs in melanoma pathogenesis and the increasing availability of HDACi being developed, epigenetic-targeted therapies have gained some attention. Several groups have previously reported the effect of pan‐HDACi on tumor cells in a variety of cancers [48,49]. In melanoma, it has been shown that the pan‐HDACi panobinostat (LBH589) exerts a dual effect on melanoma cells by affecting growth/survival through increased apoptosis along with a G1 cell cycle arrest and by increasing melanoma immunogenicity [50]. Another study showed that the combination of panobinostat with BRAFi have synergistic effects on BRAFi-resistant melanoma by decreasing the PI3K pathway activity and by changing the balance between pro- and anti-apoptotic proteins [51]. Vorinostat, another HDACi, is known to induce ROS [52]. Since elevated levels of ROS are found in drug-resistant cells, vorinostat was used to further increase these ROS levels to trigger apoptotic death selectively in the drug-resistant tumor cells [53]. However, one of the most challenging issues with the use of HDACi is to attribute the effect to a single HDAC or to a particular sub‐group of HDACs and determine the HDAC(s) responsible for these anti‐tumor effects. To address this question, Waon et al., evaluated the effect of various pan‐ and selective-HDACi on a broad panel of human melanoma cell lines. They assessed effects of pan-HDACi (LBH589 and TSA), class I and IV inhibitor (MGCD0103), or the HDAC6 inhibitors (Tubastatin A [54] and Nexturastat A [55]). Interestingly, selectively inhibiting HDAC6 recapitulated the anti-proliferative effects of pan-HDACi [56]. Further, the same group showed that HDAC6 is an important regulator of the JAK/STAT3 pathway via production of IL6 in response to LPS [57] and in turn PD-L1 expression rending its selective inhibition as a potential immuno‐modulatory option in current therapies [57].
In melanoma, T-box 2 (Tbx2) downregulates expression of the cell cycle inhibitor CDKN1A (p21) by targeting HDAC1 to its promoter. This leads to senescence bypass and melanoma progression [58].
Wilmott et al., have shown that increased percentage of nuclear HDAC3 and cytoplasmic HDAC8 is associated with better prognosis from the time of diagnosis of primary melanoma [59]. Collectively, this suggests that nuclear expression of some HDAC, such as HDAC3, are good prognostic factors and that some HDAC such as HDAC8 could exert good prognosis when having cytoplasmic functions beyond their classical role. However, a recent study showed that multiple stress exposure on melanomas such as BRAFi and MEKi combination, increased HDAC8 expression and lead to a drug-resistant phenotype [60]. The authors showed that HDAC8 is implicated in MAPK and AP-1 signaling regulation by deacetylation of c-Jun increasing its transcriptional activity. Importantly, xenograft studies supported a critical role for HDAC8 in therapeutic response upon non-selective (panobinostat) or HDAC8 specific inhibitor (PCI-34051) treatment by increasing targeted therapy durability [60]. Moreover, additional studies described HDACs not only as histone modifiers but also to have the capacity to modify other proteins unrelated to the chromatin environment [61,62].
Noteworthy, most of the US Food and Drug Administration (FDA)-approved HDACi showed significant CR for the treatment of lymphomas, Cutaneous T-cell lymphoma (CTCL) and Peripheral T-cell lymphoma (PTCL). Unfortunately, HDACi monotherapy has not demonstrated similar success in solid tumors. The poor efficacy of HDACi in solid tumors compared to hematological malignancies is still poorly understood. One possibility could be that HDACi reach their therapeutic concentrations more efficiently in hematological malignancies so that the short-life may not affect their activities as it could be the case in solid tumors. Moreover, in phase II clinical trials with HDACi as monotherapies against solid tumors, only a small subset of patients presented CR or partial response along with severe adverse effects [63]. A Phase II clinical trial with entinostat (inhibitor of class I HDAC enzymes) for patients with metastatic melanoma pretreated with systemic therapies (at least one and no more than two) reported adverse events (e.g. nausea, hypophosphatemia, pain in extremeties, diarrhea) [64]. Recently, Maertens et al., highlighted the use of HDACi to potentiate MAPKi effects in melanoma [65]. They have shown that genetic or chemical suppression of HDAC3 using entinostat potently cooperates with the combination dabrafenib/trametinib in BRAF-mutant melanoma and in difficult-to-treat NRAS- and NF1-mutant tumors. Moreover, they found that MGMT expression serves as a biomarker for this triple BRAF/MEK/HDAC inhibitor combination efficacy. Mechanistically, this combination triggers severe DNA repair defects by suppressing the expression of ELK which regulates key genes involved in this process (e.g. BRIP1, PARP3, XRCC5) ultimately leading to enhanced melanoma cells death [65].
Of note, deregulation of histone demethylases resulting in aberrant histone methylation patterns have been linked to melanoma pathogenesis (Figure 3). Using the H3K4me3 demethylase JARID1B (i.e KDM5B) as a biomarker, Roesch et al., characterized the existence of a slow cycling subpopulation of cells, within the rapidly proliferating main population [66]. The slow-cycling JARID1B-positive subpopulation shows increased in vitro self-renewal and knockdown of JARID1B caused exhaustion of melanoma cells [66]. Collectively, JARID1B-positive cells are critical for continuous melanoma tumor growth.
This subpopulation was found to be highly dynamic underlying the variable nature of the epigenetic landscape of melanoma [66]. Importantly, the discovery of this slow-cycling subpopulation was of critical clinical importance considering its role in melanoma maintenance since the majority of current therapies target proliferating cells. Further, characterization of the slow-cycling JARID1B (high) phenotype revealed a high expression of mitochondrial bioenergetic enzymes and blocking the mitochondrial respiratory chain overcomes intrinsic multidrug resistance in melanoma [67].
Moreover, it has been shown that two different types of histone H3 lysine 9 demethylases, Lysine-Specific Histone Demethylase 1A (LSD1 i.e KDM1A) and Jumonji Domain-Containing Protein 2D (JMJD2C i.e KDM4C), promote the bypass of oncogenic HRasG12V- or BrafV600E-induced senescence by preventing H3K9 Trimethylation at E2F target gene promoters, thereby favoring melanomagenesis. Inhibition of these H3K9 demethylases restored senescence and growth arrest [68]. Interestingly, a recent study described an effective dual pharmacological inhibitor of the CoREST complex containing HDAC1 along with LSD1 in slowing tumor growth [69]. However, the regulation of methylation is complex since histone hypermethylation induced by low glutamine in tumor core regions or in patient-derived BRAFV600E melanoma cells resulted in cancer cell de-differentiation or resistance to targeted therapy which will be discussed later. Importantly, knockdown of the H3K27-specific demethylase KDM6B (i.e jumonji domain-containing 3, JMJD3) reproduced the low-glutamine effects in vitro and in vivo, whereas EZH2 knockdown (described in the “writers”) attenuates them [70]. Another study also involved KDM6B in melanoma pathogenesis. Indeed, the authors identified a novel epigenetic mechanism by which KDM6B transcriptionally upregulates several targets of NF-κB and BMP (Bone Morphogenic Protein) signaling to promote melanoma progression and metastasis [71].
Importantly, some of these studies suggested their respective histone demethylases or deacetylases as a potential target for melanoma treatment supporting the reversible aspect to explore previously mentioned (Table 1 for selective inhibitors).
Epigenetic players and their impact on melanoma
| Categories | Players | Target / Site | Impact on melanoma | Selective inhibitors in melanoma | References |
|---|---|---|---|---|---|
| Writers: | SETDB1 | methylation of H3K9 | favors melanoma development | CAS 935693-62-2 [29] | [28, 29] |
| EZH2 | methylation of H3K27 | favors melanoma progression -associated with poor survival -senescence bypass | GSK503 [31] EZH2i [35] | [30, 31, 32, 33] [34] | |
| Readers: | BRD2, BRD4 | acetylated histones | essential for tumor maintenance and melanoma cell survival | JQ1 I-BET151 | [38, 39, 40, 41, 42] |
| Erasers: | HDAC6 | regulates JAK/STAT3 and PD-L1 expression | role in immunosurveillance | Tubastatin A Nexturastat A | [54, 55, 56, 57] |
| HDAC1 | p21 promoter | senescence bypass | Corin [69] | [58] | |
| KDM1A, KDM4C | demethylation of H3K9 | favors melanomagenesis by senescence bypass | GSK2879552, IOX1 [68] Corin [69] | [68] | |
| HDAC3 | High nuclear staining | associated with improved survival of patients with stage IV metastatic melanoma | Entinostat [64] | [59] | |
| HDAC8 | High cytoplasmic staining | -associated with improved survival of patients with stage IV metastatic melanoma -regulates MAPK and AP-1 signaling | PCI-34051 [59] | [59] [60] | |
| KDM5B | demethylation of H3K4 | critical for melanoma tumor growth | NA | [66] | |
| KDM6B | demethylation of H3K27 | upregulates several targets of NF-κB and BMP to promote melanoma progression and metastasis | GSK-J4 | [71] | |
| Chromatin remodeling complexes: | ARID2, ARID1A ARID1B, SMARCA4 | chromatin remodeling | tumor suppressor? | NA | [72, 73] |
| SMARCA4, SMARCA2 | chromatin remodeling | required for melanoma tumorigenicity | NA | [74] | |
| SMARCA4 | -Recruited by MITF and SOX10 to a subset of MITF-associated regulatory elements (MAREs) at active enhancers -Regulates MITF dynamics genomic occupancy | -essential for transcription regulation in melanocyte and melanoma cell physiology -progression of oncogenic Braf-driven mouse melanoma | NA | [75, 76] | |
| BPTF | chromatin remodeling essential for the melanocyte gene expression program | -regulates proliferation, migration and morphology of murine melanoblasts in vivo -essential for differentiation of adult melanocyte stem cells -progression of oncogenic Braf-driven mouse melanoma | NA | [76, 77] | |
| ATRX | chromatin remodeling | decreased ATRX expression correlates with melanoma progression | NA | [83] | |
| Histone variants: | macroH2A | replace canonical H2A associated with transcription repression | -macroH2A suppresses melanoma progression via transcriptional repression of CDK8 -macroH2A loss promotes tumor growth and metastatic potential | NA | [85] |
| H3.3 | replace canonical H3 | overexpression triggers senescence via E2F target genes repression | NA | [86] | |
| H2A.Z.2 | replace canonical H2A, binds and stabilizes BRD2 | H2A.Z.2 correlates with poor patient survival and promotes cell cycle progression via E2F target genes transcription control | NA | [87] |
Chromatin remodeling complexes
Importantly, SWI/SNF member's alterations have been linked to melanoma. Especially, loss-of-function mutation in components of this complex such as ARID2, ARID1A, ARID1B or SMARCA4 are found in 13% of melanomas, suggesting a tumor suppressor role and highlighting the importance of chromatin remodeling in melanomagenesis [72,73]. Interestingly, at least one of the ATPase subunits BRG1 or BRM is required for melanoma tumorigenicity and most likely promote expression of distinct target genes [74]. It has been shown that BRG1 takes part in a novel form of the PBAF chromatin remodeling complex along with CHD7, and interacts with the Microphthalmia-associated transcription factor (MITF) [75]. The authors showed that MITF and SOX10 actively recruit BRG1 to a set of MITF-associated regulatory elements (MAREs) at active enhancers and that BRG1 also regulates the dynamics of MITF genomic occupancy. This interplay along with additional transcription factors is essential for transcription regulation and many aspects of melanocyte and melanoma cell physiology [75]. In line with this study, using a mouse melanoma model conditionally expressing BRAFV600E along with Pten inactivation that rapidly develop melanoma, it has been shown that somatic inactivation of Brg1 and Bptf (the defining subunit of the NURF complex) delay tumor formation and deregulate a substantial and common gene expression programs critical for normal tumor cell growth. These two subunits also coregulate with Mitf and Sox10 many genes supporting a cooperation between transcription factors and chromatin remodeling complexes to dictate fundamental gene expression programs in melanoma [76]. The same group also reported that the NURF complex interacts with MITF and uncovers a role for the defining subunit of this complex, BPTF. The study shows that Bptf regulates proliferation, migration and morphology of murine melanoblasts in vivo and is essential for differentiation of adult melanocyte stem cells [77]. These studies are of critical importance since MITF is a key driver of plasticity [78,79], allowing the transition of melanoma cells between a differentiated-proliferative phenotype and a stem cell like slow cycling-motile phenotype [80-82]. Finally, it has been shown that ATRX loss (another SWI/SNF chromatin remodeler) correlates with melanoma progression [83].
Of note, several subunits in the SWI/SNF complex, including SMARCA4, SMARCA2, BRD9, and PBRM1 contain druggable bromodomains. Synthetic lethal interactions involving several of these subunits have opened the possibility of new therapeutic strategies [84].
Histone variants
With different sequences and properties, histone variants replace the canonical histones into defined regions of the genome driven by histone chaperones and therefore modify chromatin structure and gene expression [12]. We will discuss here few of the first discoveries including variants of H2A and H3 in melanoma pathogenesis. MacroH2A is generally considered to be transcriptionally repressive. It suppresses melanoma progression via transcriptional repression of CDK8, which is required for proliferation of melanoma cells [85]. On the other hand, macroH2A loss directly contributes to melanoma progression by promoting tumor growth and metastatic potential [85]. Overexpression of H3.3, a variant of H3, represses E2F target genes and triggers senescence [86]. More recently, a role for a specific isoform of the H2A.Z variant, H2A.Z.2, has been described in melanomagenesis [87]. H2A.Z.2 is highly expressed in melanoma and high levels correlate with poor patient survival. That study demonstrated that H2A.Z.2 binds and stabilizes BRD2 to promote cell cycle progression by controlling the transcriptional output of E2F target genes. Finally, H2A.Z.2 deficiency increased sensitivity of melanoma cells to chemo- and targeted therapies (MEKi) [87]. The essential role of these variants in melanoma or the chaperones upstream depositing them into defined region of the genome, should be taken in consideration to explore potential therapeutic strategies to alter sensitivity of melanoma cells to current therapies.
Epigenetic impact on targeted therapy efficiency
MAPK pathway and targeted therapies
Hyperactivation of the MAPK signaling pathway via mutations in BRAF, NRAS, or NF1, drives CM progression, underlining the fundamental role of controlled MAPK signaling for melanocyte homeostasis [88-90]. As such, efforts have been directed towards this pathway for targeted cancer therapies. BRAFV600E is the most common mutation in CM (>50%), which leads to constitutive activation of MEK/ERK signaling independently of upstream Receptor Tyrosine Kinases (RTK) or RAS activation, resulting in recurrent positive regulation of genes involved in cell proliferation and survival [90], that is, uncontrolled cell proliferation.
BRAF inhibitors (BRAFi: vemurafenib, dabrafenib, encorazfenib) and more recently, the combination of a BRAFi and MEKi (cobimetinib, trametinib, binimetinib) have shown remarkable clinical activity in advanced metastatic CM in patients with mutant BRAFV600E/K [92-95]. However, their use is conditioned by the presence of the activating mutation and therefore can only benefit up to 50% of patients. Moreover, ~60% of patients with this mutation respond well to ERK signaling inhibitors, however patients almost invariably develop resistance and relapse within a 6-9 month period [88].
Mechanisms underlying acquired resistance to ERK signaling inhibitors include alterations of BRAFV600E (overexpression, amplification and aberrant splicing) [96,97], upregulation of kinases (e.g. Tpl2/Cot) [98] and RTK [99] or RAS mutations [100], amongst others - all of which reactivate ERK signaling. However, these mechanisms account for a fraction of acquired resistance. Collectively, novel targets and therapies are still critically needed.
Epigenetic mechanisms of resistance to targeted therapies
The place that epigenetics is taking in melanoma pathogenesis is undeniable over the last few years. Not only epigenetic alterations were proved to be involved in melanoma development but also in mechanisms underlying acquired resistance to targeted- and immunotherapies that we will discuss here after (Table 2).
Epigenetic players and their impact on melanoma resistance to therapies
| Players | Therapies | Mechanisms of resistance | Alternative treatments proposed in the study | References | |
|---|---|---|---|---|---|
| Targeted therapies | KDM5A demethylation of H3K4 | Pan-RAF inhibitor (AZ628) | Elevated expression in drug-tolerant cells and IGF-1R signaling activation | combined with IG1-1Ri (AEW541) | [110] |
| KDM5B demethylation of H3K4 | Vemurafenib | Elevated expression in slow cycling cells with increase in oxidative phosphorylation | combined with mitochondrial oxidative-ATP-synthesis (e.g. oligomycin, Bz-423) | [67] | |
| KDM1B, KDM5A, KDM5B demethylation of H3K4 KDM6A, KDM6B demethylation of H3K27 | Vemurafenib or Trametinib | Elevated expression in induced drug-tolerant cells (IDTC) and undifferentiated state transition which increases aggressiveness | combined with HDACi, IGF-1Ri, PI3/AKTi to eliminate parental cells prior transition to IDTC | [111] | |
| KDM5B demethylation of H3K4 | Vemurafenib or Vemurafenib+Trametinib | Elevated expression to shift into a drug-tolerant state (e.g. decrease in Gdf15, Ldlr epxression) | combined with pan-KDM5i (e.g. KDM5-C70, CPI-48) | [112] | |
| KDM6B demethylation of H3K27 EZH2 methylation of H3K27 | Vemurafenib | Involved in glutamin-induced histone methylation impacting vemurafenib response | combined with EZH2i? | [70] | |
| TADA2B, TADA1 acetylation of histones H3 and H4 | Vemurafenib | Loss promotes resistance; Mechanisms unknown Impacting histone acetylation and gene expression? | combined with HDACi | [114] | |
| SIRT1 | Vemurafenib | Elevated expression in vemurafenib resistant cells | combined with SIRT1i (e.g. sirtinol, EX-527) | [116] | |
| SIRT2 | Vemurafenib or Selumetinib | SIRT2 knockdown increases ERK signaling | NA | [117] | |
| SIRT6 | Dabrafenib or Dabrafenib+Trametinib | SIRT6 haploinsufficiency activates the IGF1-R and downstream AKT signaling | combined with IGF-1Ri (i.e. linsitinib) | [119] | |
| Immunotherapies | EZH2 methylation of H3K27 | anti-CTLA4 and IL-2 | Increased EZH2 activity dependent on T cells and TNF-α promoting dedifferentiation, loss of immunogenicity and PD-1/PD-L1 axis upregulation | combined with EZH2i (i.e. GSK503) | [134] |
| ARID2, PBRM1, BRD7 | anti-PD1/CTLA4 T-cell mediating killing | e.g. regulates mTORC signaling pathway | NA | [135] | |
| LSD1 | anti-PD1 | Represses endogenous retroviral element and interferon response with inhibition of tumor responses to host immunity | combined with GSK-LSD1 | [136] |
In a panel of tumor biopsies that have acquired resistance to MAPK inhibitors (MAPKi), for approximately 40% of them, no validated mutational mechanism was identified [7]. Recent reports have implicated DNA methylation, transcriptional changes, microRNA alterations, as well as microenvironmental stressors in promoting melanoma drug resistance to MAPKi in BRAFV600-mutant melanoma [7,101-104]. These highly recurrent non-genetic mechanisms clearly support the necessity to dig into transcriptomic and epigenetic alterations in addition to genetic events to better grasp the burden of melanoma resistance and to develop new combinations of therapeutic strategies.
Importantly, it suggests that epigenetic alterations may play a key role in rewiring the chromatin landscape of melanoma cells to allow adaptation to current therapies. Owing to the fact that chromatin-mediated changes are reversible processes, the most clinically relevant observations implicating such regulations in drug resistance would be the “drug holiday” concept. It consists of a treatment “break” or intermittent treatment, which delays resistance whereas a genetic regulated drug resistance would not be affected. Indeed, re-challenging two patients with BRAFi after a treatment free period and disease progression upon BRAFi or BRAFi+MEKi administration resulted in a significant CR indicating that resistance to BRAF-selective inhibitors can be reversible following treatment interruption [105]. Supporting that, a recent retrospective study for patients retreated with BRAF-targeted therapy after disease progression and treatment “break” showed 43% of clinically significant response [106]. The emergence of clinical evidences of a reversible aspect for drug resistance highlights previous findings almost a decade ago where slow cycling subpopulations of cancer cells, including those of melanomas, have been implicated in reversible drug tolerance. However, additional mechanisms are not to be excluded such as induction of cancer cell drug addiction, matrix remodeling and secretome adaptation promoting temporary resistance to BRAFi [107-109]. Using anti-cancer agents in several tumor cell lines, Sharma et al., consistently identified a small fraction of cells surviving treatment with drug concentration 100-fold higher than the IC50 [110]. This study was one of the first shedding light on such a drug-tolerant subpopulation. Importantly, a “drug holiday” period re-sensitized this subpopulation to the initial treatment. Briefly, this drug-tolerant cells displayed elevated expression of the histone demethylase KDM5A (JARID1A) and consequent reduced level of its target, the histone modifications H3K4me3/2, therefore altering their chromatin state. Finally, RNAi-mediated knockdown of KDM5A confirmed that this histone demethylase was important for the establishment of the reversible drug-tolerant state [110]. Cells displaying elevated expression of another H3K4 demethylase, KDM5B (JARID1B), were also found to be enriched upon BRAFi treatment that has been linked to this drug-tolerant state phenotype as well. Importantly, inhibition of mitochondrial respiration blocks the emergence of the KDM5B(high) subpopulation and sensitized melanoma cells to therapy [67]. Pushing this concept further, another study showed that chronic exposure to external stressors such as hypoxia, nutrient starvation and drug treatment give rise to an induced drug-tolerant cells (IDTCs) rather than a selection of a pre-existing subpopulation [110]. In this study, microarray analyses from IDTCs cells revealed elevated expression levels for the H3K4 demethylases KDM1B, KDM5A, KDM5B and for the H3K27 specific demethylases KDM6A, KDM6B. A concomitant chromatin modification state was observed with a decrease of the histone marks targeted by these enzymes, respectively H3K4me3 and H3K27me3, highlighting epigenetic remodeling. On that note, loss of differentiation markers such as melan-A and tyrosinase which are MITF target genes, was also observed in IDTCs suggesting the transition into an undifferentiated state in accordance with increased aggressiveness [111]. Recently, another study highlighted tumor heterogeneity as a major challenge for cancer treatment. In mouse melanomas, CD34+ and CD34- tumor subpopulations have been characterized as melanoma-propagating cells exhibiting some key properties from stem cell or progenitor cell. Moreover, differences in tumorigenic properties, heterogeneity recapitulation and resistance have been observed in these two subpopulations [112]. The authors demonstrate that CD34+ and CD34- subpopulations harboring the BRAFV600E mutation have differential response to targeted BRAFi. Linking epigenetics to tumor heterogeneity, upon exposure to targeted therapies, elevated KDM5B expression shifts melanoma cells to a more drug-tolerant CD34- state while KDM5B loss shifts melanoma cells to a more sensitive CD34+ state. Together, these studies support a critical role for KDM5B in epigenetic regulation to orchestrate the transition of subpopulations with distinct drug sensitivity.
Furthermore, cancer cell dedifferentiation and histone methylation were attributed to resistance following BRAF inhibitor treatment [70]. In that case however, the authors described a role for histone hypermethylation (H3K27me3) in tumor core regions specifically, that resulted in cancer cell dedifferentiation and resistance to BRAF inhibitor treatment [70]. The variability of these studies regarding the role of H3K27me3 in resistance to BRAFi could be explained by the difference in the models used (e.g. melanoma cell lines and tumors). Nevertheless, these studies involved chromatin remodeling such as loss or gain of histones post-translational modifications impacting melanoma cells response to external stressors (hypoxia, drug exposure, low nutrients) and highlight the importance of the microenvironment impact onto epigenetic alterations, intra-tumoral heterogeneity and the therapeutic response.
The emergence of new technologies over the last years (e.g. single cell analysis or genome editing using the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-associated protein 9 (Cas9) system) have become attractive and powerful tools for biological discoveries and the identification of novel drug targets. Analyses at the single cell level of human melanoma cells allowed Shaffer et al., to identify a transcriptional variability in which the cells with the capability to resist drug treatment can be predicted [113]. Expression of few resistance markers are found at high levels in a small number of cells and the addition of drug triggered an epigenetic reprogramming to switch from a transient transcriptional state into a stably resistant state. Especially, this transition is mediated first by a dedifferentiation state followed by activation of new signaling pathways mediated respectively by loss of SOX10 (regulates neural crest development in melanocytes) and activation of TEAD (regulates invasion in melanoma) among others [113].
Taking advantage of the simplicity of programming the CRISPR-Cas9 to modify specific genomic loci, Shalem et al., interrogated on a genome-wide scale, gene function in melanoma resistance by screening for genes whose loss is involved in resistance to vemurafenib [114]. This study identified members of the STAGA HAT complex (e.g. TADA2B, TADA1), consistent with a critical role for histone acetylation in melanoma drug resistance [114]. Taking all this in consideration, there is no doubt today that shedding light onto the transcriptomic and epigenetic alterations underlying acquired MAPKi resistance in melanoma is of critical importance to improve patients' clinical outcome.
Moreover, several studies highlighted a role in melanoma resistance for sirtuin proteins constituting the class III HDACs. Of note, SIRT1, 2, and 6 are considered the nuclear SIRTUINs and are chromatin-bound [115]. Previous report on sirtuins in melanoma showed that SIRT1 inhibition decreases melanoma cell growth and rescues the sensitivity to PLX4032 of PLX4032-resistant BRAFV600E-mutated melanoma cells [116]. On the other hand, an shRNA screen identified that SIRT2 depletion conferred resistance to MAPKi in BRAFV600E melanoma cells through ERK reactivation [117]. Interestingly, the role of SIRT6 in the pathogenesis of several cancers has been controversial. Indeed, in the last years, SIRT6 has been described either as a tumor suppressor or an oncogene in tumorigenesis regulation through diverse biological pathways [118]. This pleiotropism, that can be extended to the Sirtuin family, adds a layer of difficulty in studying the cellular mechanisms by which sirtuins impact cancer or biological processes but makes it extremely exciting.
We favored the hypothesis that epigenetic mechanisms altering gene expression programs contribute to ERK signaling inhibitor resistance. Using a CRISPR-Cas9 screen approach focused on chromatin factors to identify epigenetic players in melanoma drug resistance, we identified the chromatin associated histone deacetylase SIRT6 as a regulator of resistance to the clinically relevant BRAFi (dabrafenib) and BRAFi+MEKi (dabrafenib+trametinib) combination [119]. Interestingly, we have also identified the histone acetyltransferases (HATs) KAT1 (HAT1) and KAT2B (PCAF), again supporting the importance of the reversible aspect in chromatin mediated processes and the balance of opposing factors in orchestrating the chromatin state in diseases. However, the role of KAT1 and KAT2B remain to be explored in melanoma drug resistance.
In our study, we uncovered a new role for the NAD-dependent chromatin-associated deacetylase SIRT6 in melanoma drug resistance [119]. Using, a combination of transcriptomic, epigenomic and proteomic analyses we demonstrated that haploinsufficiency of SIRT6 in BRAF-mutant melanoma cells decreases sensitivity to MAPKi independently of the ERK signaling pathway. This allows cells to survive by increasing their IGFBP2 expression which in turn activates their IGF-1R receptor and downstream AKT survival signaling in presence of these inhibitors [119]. Consistent with our results, a link between the insulin/IGF and sirtuin pathways has been reported previously in the development of cardiac hypertrophy and heart failure [120]. On the other hand, previous studies have also suggested that increased activation of the AKT signaling pathway plays a role in MAPKi resistance [99,121,122]. Importantly, a recent study identified IGFBP2 as part of a gene signature in response to MAPKi “drug-tolerant persisters” [102] and there is little evidence for its use as a biomarker. Strikingly, we observed that IGFBP2 protein levels correlated with resistance to MAPKi in several BRAF-mutant melanoma cell lines and are associated with poor prognosis in primary melanomas. Importantly, we showed that co-targeting the MAPK and IGF-1R pathways can prevent/delay resistance to targeted MAPKi therapies, particularly for patients with high levels of IGFBP2, highlighting the importance of early detection as previously mentioned.
Intriguingly, we also observed that melanoma cells devoid of SIRT6 undergo chromatin reorganization reflected by increased open chromatin and H3K56ac at these sites [119]. Such potential for genomic instability is consistent with increased DNA damage and impaired tumor growth such as previously reported in acute myeloid leukemia (AML) [123]. Together, these data suggest additional functions for SIRT6 to explore in melanoma biology, in which we could consider SIRT6 “complete” depletion as a novel strategy in melanoma pathogenesis to enhance their sensitivity to current targeted MAPK therapies. Our study and others, highlight here the importance of the epigenetic balance and how the levels of chromatin factors can be critical in disease biology, in our case, with two opposite outcome observed for SIRT6 haploinsufficiency versus SIRT6 deficiency [119].
While we can refer to these cells as “IDTCs; drug tolerant persisters or slow cycling drug tolerant cells”, we now know that the burden of acquired melanoma resistance not only arise from genetic alterations but also from the emergence of these subpopulations. Considering that acquired drug resistance may involve multiple distinct molecular mechanisms taking place independently within the same patient, strategies to overcome such resistance with a single targeted agent remain extremely challenging. Therefore, early treatment to short-circuit this drug tolerant state to prevent or delay drug resistance is particularly attractive (Table 2).
Epigenetic impact on immunotherapy efficiency
Immune regulation and checkpoint inhibitors therapies
The second therapeutic revolution in melanoma came from a deeper understanding of the tumor microenvironment and immunophenotype of tumors. Melanomas are not isolated entities but are dependent on their surrounding cells whom they are in constant dialogue. These include fibroblasts, immune cells, and endothelial cells that constitute the tumor stroma and that allow tumor cells to adapt to their changing microenvironment and therefore survive and replicate [124]. Stroma composition is heterogeneous and dynamic. The immune system is also very heterogeneous in itself and can lead to a state of immunosurveillance allowing efficient tumor elimination or a state of immunotolerance promoting tumor escape and therefore tumor survival. Thus, the challenge of effective immunotherapy is to not only directly promote tumor death but importantly, to modulate its microenvironment in order to obtain a state of immunosurveillance [125].
In this regard, one of the great breakthroughs in medicine over the last 10-15 years have been the discovery of how the immune system fights against cancer thanks to the work by Allison and Honio whose Nobel-winning research led to the development of immune checkpoint inhibitors (ICI). ICI is a form of cancer immunotherapy which targets immune checkpoints, that is, immune cells which dampen the immune response following an immunologic stimulus. The rationale behind this strategy is to stimulate patient's own immune system to recognize and destroy their cancer cells more effectively. This is achieved by blocking checkpoint proteins such as programmed cell-death protein 1 (PD-1) and cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) which are inhibitory receptors expressed normally on activated effector T lymphocytes which act as 'brakes' to turn off T-cell responses and prevent T cells attacking their own cells. By avoiding detection, malignant cells are able to spread uninhibited. The ligand for PD-1 (PD-L1) is strongly expressed by melanoma cells. Immunotherapy aims to block the interaction between PD-1 on T cells and its ligand PD-L1 on tumor cell or alternatively, blocking the interaction between CTLA-4 on T cells and its co-stimulatory receptors CD80, CD88 or B7 on antigen presenting cells. The use of anti-CTLA-4 antibody with Ipilimumab (now also with Tremelimumab) was the first immune checkpoint inhibitor drug that prolonged overall survival in patients with advanced cutaneous melanoma [126]. Later, anti-PD-1 (Nivolumab, Pembrolizumab and most recently Cemiplimab) and anti-PD-L1 (Atezolizumab and Avelubam) antibodies have similarly shown improved survival and progression-free survival in these patients [126]. The IFN-γ, JAK/STAT pathway appears critical for the response to immune checkpoint blockers [127].
Although ICIs have revolutionized the treatment of patients with advanced melanoma, approximately one third of patients benefit from anti-PD1 treatment but responses are often associated with immune-related adverse events (irAE) which can be serious and life-threatening [128]. Albeit such progress, the alarming trend is that the global incidence of cutaneous melanoma has been steadily increasing over the last 60 years with two thirds of all patients with advanced forms of the disease who still fail to get a long-term benefit of the treatments. This area of research clearly warrants urgent attention to identify potential new therapeutic targets.
One of the pathways which clearly needs fine-tuning is the more specific recognition of neoplastic cells to reduce the incidence of irAE. In this regard, the innate immune system which relies on the potent and critical anti-tumor function of natural killer (NK) cells acting in concert with surrounding dendritic cells and macrophages to destroy the tumor is likely to be an attractive target. The prompt response of NKs is due to their release of pre-formed cytotoxic mediators and expression of surface ligands that are able to trigger death receptors on target cells (innate response), as well as their ability to produce a wide variety of chemokines and cytokines to recruit and instruct other immune cells for subsequent priming (adaptive response). In this regard, the latest paradigm shift which demonstrates innate immune cells to have memory properties may offer a lot of promise [129]. The mechanisms behind this trained immunity include changes in intracellular metabolism and epigenetic regulation at the level of histone modification [130,131]. Epigenetic remodeling is a common feature of human melanoma and other tumor types and plays a key role in the immune escape of neoplastic cells from antigen specific T cell recognition. Epigenetic drugs have been demonstrated to improve recognition of cancer cells, to have strong immunomodulatory activity and to be able to reverse epigenetically driven immune alterations suggesting a combination of agents that target both epigenetic and immunotherapy approaches may improve the efficacy and specificities of treatment against tumor [132].
Epigenetic mechanisms of resistance to immunotherapies
Unfortunately, as for any therapies in melanoma, resistance ultimately develops. Despite recent studies pointing out epigenetic regulations contributing to tumor immune escape, antigen expression or presentation, regulating tumor cell killing or T-cell response (and we refer the reader this review for more details [133]) our knowledge in epigenetic mechanisms involved in resistance to immunotherapies is still in its infancy. Indeed, to the best of our knowledge, only a few studies to date reported epigenetic players in melanoma immunotherapy resistance (Table 2). For instance, Zingg et al., described a role for EZH2 in acquired resistance to cancer immunotherapy [134]. In particular, they showed that EZH2 is upregulated upon anti-CTLA-4 or IL-2 immunotherapies in cancer cells, leading to a loss of tumor control. Mechanistically, activation of EZH2 promotes H3K27 trimethylation and consequent suppression of essential immune-related genes [134]. Importantly, GSK503 an inhibitor of the methyltransferase activity of EZH2, restored tumor immunogenicity and T-cell infiltration and suppressed melanoma growth upon immunotherapy. In that study, Zingg et al., provided an insight into the effects of EZH2 activation which resulted in adaptive cancer resistance to immunotherapy and they provided a rationale for combinatorial epigenetic immunotherapy approach. Using a genome-scale CRISPR-Cas9 screen, a recent study identified key chromatin regulators of tumor immune resistance [135]. Briefly, inactivation of either ARID2, PBRM1 and BRD7 of the PBAF complex sensitized melanoma cells to cytotoxic T-cells via an enhanced response to IFN-γ [135]. Of note, PBAF-deficient tumor cell lines produced higher amounts of chemokines, therefore allowing more efficient T-cell infiltration into the tumor site. Another study implicated the histone H3K4 demethylase LSD1 in resistance to anti-PD1 therapy [136]. The authors showed that double-stranded RNA (dsRNA) stress, resulting from LSD1 loss, led to potent anti-tumor T cell immunity. Importantly, LSD1 depletion renders refractory mouse tumors responsive to anti-PD-1 therapy [136]. These studies highlight the role of chromatin regulators whereby their inhibition reverses certain features of adaptive resistance of tumors to immunotherapy, supporting again the importance of the reversible aspect of epigenetic processes (Table 2). Moreover, this provides a strong rationale for implementing epigenetically-based immunotherapies in cancer patients.
Therapies targeting anti-tumor T-cell responses were successful in a variety of diseases, unfortunately, most patients still do not respond, underlying a critical need for approaches which improve immunotherapeutic efficacy. For instance, combination of HDACi with immunotherapy (anti-Pmel T-cell transfer plus Pmel peptide-pulsed DC vaccine) decreased tumor volume by 70% compared to untreated mice, where reductions in tumor volumes of 49% and 21%, were achieved with immunotherapy or HDACi alone, respectively [137]. The efficacy of combining epigenetic modulators such as the DNA hypomethylating agent 5-aza-2'-deoxycytidine (5-AZA-CdR) and immunotherapy caused a 77-81% reduction in tumor volume and was much more effective than the effect mediated by single agents [138]. On the same note, a recent study from Laino et al., highlighted a role for HDAC6 on immune function of melanoma patient T-cells [139]. Using the HDAC6-specific inhibitors, ACY-1215 and ACY-241, on T-cells from metastatic melanoma patients, the authors observed decreased cytokine production (IL-4, IL-5 and IL-6), decreased FOXP3 expression (a master regulator of regulatory T cells) and higher T-cell infiltration in melanoma upon treatment [139]. This study demonstrated that the use of HDAC6-specific inhibitors decreases immunosuppression and enhances immune function of melanoma patient T-cell giving a rationale for a potential translation into clinic. Taking all this in consideration, efforts taken in studying the different mechanisms of intrinsic and extrinsic resistance post immunotherapy need to be intensified to improve combinatorial epigenetic immunotherapy approaches. A summary of epigenetic drugs in combination with immunotherapy in ongoing clinical trials for the treatment of melanomas is discussed in Table 3.
Epigenetic drugs in combination with immunotherapy agents in melanoma
| HDACi | Combination | Phase clinical trial | Identifier |
|---|---|---|---|
| Panobinostat (LBH589) | Ipilimumab | I | NCT02032810 |
| Entinostat (MS275-SNDX-275) | Pembrolizumab | II | NCT02697630 |
| HBI-8000 | Nivolumab | I/II | NCT02718066 |
| 4SC202 | Pembrolizumab | I/II | NCT03278665 |
| CPI-1205 | Ipilimumab | I/II | NCT03525795 |
NIH clinical trial database: www.clinicaltrials.gov
Epigenetics in uveal melanomas (UM)
While most melanomas do form on the skin, it can also arise in the eye, known as ocular melanoma. Large majority of ocular melanomas originate from uvea (95%), involving the posterior uvea (choroid 90% and the ciliary body 5%) and anterior uvea (iris 5%). Uveal melanoma (UM) is the most common primary cancer of the eye in adults. In U.S. and Europe, UM has an incidence of 5 cases per million people per year. Despite successful treatment of the primary lesion, liver metastases develop in half of these patients [140]. The etiopathogenesis and biological behaviors of UM are very different from cutaneous melanoma [141]. They display distinct landscapes of genetic alterations with different metastatic routes and tropisms. Hence, therapeutic improvements achieved in the last few years for the treatment of CM have failed to improve the clinical outcomes of these patients.
Genetic alterations most often observed in UM are somatic activating mutations in the G-protein coupled receptor GNAQ signaling cascade [142-145] associated with mutations prognostically significant of the metastatic risk in BAP1, SF3B1, and EIF1AX (BSE mutations) [146]. In addition, copy-number variations can also be detected in the context of the BSE mutational status and specific gene expression signature. Collectively, these different alterations predict UM subtypes. Currently, there are no approved systemic treatments for UM once it has spread [147]. 90% of patients will die within 6 months after diagnosis of metastases (review [141]). Thus, this is really an area of urgent need for research to find more efficient treatments for UM.
Changes in the epigenetic landscape, including DNA methylation, histone modification and small non-coding RNA have also been reported in UM. Given the reversible nature of some epigenetic regulations, inhibition of the epigenetic enzymes in cancer cells might switch these modifications back to a “normal-like” chromatin landscape. As for CM described above, only histone modifications will be addressed in detail.
As mentioned above, one of the most prominent alterations found in UM is the loss of the tumor suppressor BRCA-1 associated protein-1 (BAP1) gene. BAP1 has one copy that is often lost via monosomy of chromosome 3 and the second copy by mutation. BAP1 is the catalytic subunit of the PR-DUB complex that deubiquitinates histone H2A [148]. Consistently, knockdown of BAP1 in UM cells induced a marked increase in H2A ubiquitination [149]. Ubiquitinated H2A is the most prevalent ubiquitin conjugate in cells which is linked to the Polycomb protein complex 1 (PRC1) ubiquitin ligase activity [150]. In the nucleosomes, ubiquitinated H2A is situated close to linker histone H1. Deubiquitination of H2A initiates transcriptional activation via linker histone H1 dissociation [151] and via trans-histone cross-talk with H3K4 di- and trimethylation [152].
Depletion of BAP1 in cultured cells induces a switch in transcriptional programs from differentiated poorly aggressive Class 1 to dedifferentiated highly aggressive Class 2 gene expression profile and re-programmation of UM cells towards a stem-like phenotype [149,153]. The stem-like phenotype is associated with quiescence and motile ability, thereby suggesting that the loss of BAP1 may be mechanistically linked to the metastatic ability. A recent study from Field et al., went further on characterizing the impact of BAP1 loss on DNA methylation in UM [154]. Here, the authors analyzed global DNA methylation in 47 Class 1 and 45 Class 2 primary UMs and in engineered UM cells where BAP1 was inducibly depleted. Moreover, they analyzed RNA-seq data from 80 UM samples and engineered UM cells. They observed hypermethylation on chromosome 3 coupled with decreased gene expression at several loci among which, BAP1 is located. The deregulated genes identified are involved in axon guidance and melanogenesis with many located on chromosome 3 (e.g. MITF, SATB1, ROBO1 or SEMA3B). Interestingly, BAP1 itself might be epigenetically regulated since a hypermethylated site was identified in the BAP1 locus for all the class 2 tumors. By inducibly knocking down BAP1 expression, a methylomic repatterning was observed and enriched for genes similar to UM tumors. This study supports previous work and suggests a chronological order for UM divergence from Class 1 to Class 2 with loss of one chromosome 3 copy, a BAP1 mutation on the other copy leading to a methylomic redistribution characteristic of Class 2 UMs, thereby a more aggressive state [154].
Meantime, HDAC inhibitors are reported to decrease histone H2A ubiquitination through transcriptional repression of the PRC1 component BMI1 [155]. Hence, HDACi emerged as promising drugs in the treatment of UM to fight the H2A hyperubiquitination phenotype caused by BAP1-deficiency. HDAC inhibitors such as the pan-HDACi suberoylanilide hydroxamic acid (SAHA, Vorinostat) and class I-selective HDACi Romidepsin (FK-228) have been FDA-approved to treat patients with cutaneous T cell lymphoma and are well tolerated [156,157].
In the context of UM, HDACi including valproic acid (VPA), trichostatin A (TSA), LBH-589, and SAHA have been assessed. They reverse the H2A hyperubiquitination caused by BAP1 loss and convert highly aggressive UM cells to a low-grade, differentiated state [149]. A phase 1 clinical trial is assessing the ability of vorinostat to induce the switch of class 2 UM cells into a cell phenotype that resembles normal melanocytes (NCT03022565).
VPA, LBH-589, TSA and SAHA have been described to inhibit proliferation in vitro, yet they did not induce much cell death. VPA has been also reported to inhibit UM tumor growth in vivo [149]. Of note, BAP1-deficient UM cells seem more sensitive to HDACi than BAP1-proficient cells [149]. A phase 2 clinical trial is testing the effect of VPA on tumor growth in Class 2 metastatic UM patients (NCT01587352).
Other HDACi including JSL-1 [158], quisinostat [159] and the Sirtuin 1-2 inhibitor Tenovin-6 [160] have been shown to induce apoptosis in vitro. However, it appears that HDACi possess poor anti-cancer clinical activity against solid tumors when used as a monotherapy.
Moreover, the clinical use of MEKi in UM is limited since acquisition of resistance has been observed along with adverse effects [161,162]. Thus, combining epigenetic drugs and chemotherapy, immunotherapy or targeted therapy may prove to have clinical value especially in the case of UM where loss of BAP1 and epigenetic alterations are critically involved in the pathogenesis. Several lines of evidence indicate that the combined therapy could be promising. Using multiomics approaches and drug screens, a recent study identified the pan-HDACi panobinostat to restrain MEKi resistance [163]. The authors identified several potential pathways to target that were upregulated upon MEKi including the PI3K/AKT, ROR1/2 and IGF-1R signaling pathways. They also observed increased GPCR expression leading to therapeutic escape through YAP signaling. Finally, their screen compounds identified panobinostat as a potential inhibitor to suppress YAP and AKT signaling activation upon MEKi that was validated in vivo with a long-term decrease of tumor growth [163]. This study provides a rationale for the use of HDACi in combination with MEKi in patients with advanced UM. Combination of quisinostat and pan-CDK inhibitor flavopiridol [159] as well as the combination of Tenovin-6 and vinblastine [160] are synergistic in inducing apoptosis of several UM cell lines. Interestingly, Tenovin-6 purges cancer stem cells [160].
Further, the class I-specific HDACi MS-275 (Entinostat) can synergize with the pro-apoptotic ligand of the TNF family TRAIL to promote apoptosis of UM cells. MS-275 increases in a variable manner expression of the TRAIL receptors DR4, DR5, and procaspase 8 as well as recurrently inhibits expression of the anti-apoptotic effector cFLIP expression [164,165].
A phase 2 clinical trial, evaluating the efficacy of concomitant use of pembrolizumab and entinostat in UM (NCT02697630) has been launched.
Bap1 has also been shown to alter other histone marks. In a model of hematopoietic transformation in mice, Bap1 loss is associated with decreased H4K20me1 at the EZH2 locus, allowing its expression and in turn catalyzes H3K27me3 [166]. Regulation of H4K20me1 is mediated through SETD8 the only known methyltransferase that places H4K20me1 on chromatin [167].
Further, the myeloproliferation syndrome associated with Bap1-KO mice is reduced by treatment with the small-molecule EZH2 inhibitor EPZ011989, suggesting that EZH2 might represent a therapeutic target in BAP1-deficient malignancies, including UM cells.
However, targeting EZH2 might not have clinical value in UM for several reasons. Indeed, the expression of EZH2 appears similar in BAP1-deficient or proficient-UM cells regardless of the BAP1 mutational status or protein expression levels. Moreover, UM cell line are not sensitive to the EZH2i EPZ-6438 [168]. Thus, the effect of EZH2 might be context dependent. Considering the lack of treatments in UM there is an urgent need for research and for efficient therapeutics to defeat metastatic UM and improve patient survival.
Epigenetic modifications and therapeutics
The significant role that is taking epigenetic dysregulation in melanoma inspires scientists to orientate their studies into compounds that target epigenetic regulators. As described in this review, there are three main categories: “writers, readers and erasers”. Few drugs inhibiting epigenetic writers and erasers have been FDA-approved for the treatment of cancer. To the best of our knowledge, the only compounds targeting histone function approved in the clinic for melanomas patients are HDACi. However, the studies discussed in this review raise several points and highlight important aspect in using HDACi as potential combined anti-melanoma options. First, would it not be better to focus our effort in developing selective HDACi and to better understand their biological functions to reduce unwanted side effect observed with pan‐HDACi in clinical trials [49]? On that note, although reversing gene expression repression remains attractive, a major issue to date in the use of HDACi lies on their capacity to alter epigenetic process for a specific subset of genes or cell type. For instance, the increase in acetylated histones upon such treatments could impact previously silenced tumor suppressor genes as much as oncogenes. We also need to take into consideration how this epigenetic landscape rewiring impacts normal cells? A possibility could be a malignant transformation leading to carcinogenesis due to genomic instability upon treatment. Moreover, HDACs have a variety of functions and display different expression profiles in normal cells, the effects of HDACi in cancer cells will most likely be tissue-dependent [169], underlining the importance of validating targets in a tissue-specific manner prior to treatment. Another question is whether their inhibition would benefit patients with melanoma? This is highly relevant since some HDAC can exert good prognosis when functioning in cell cytoplasm.
As previously discussed, another issue for the use of HDACi in melanoma patients is their poor efficacy in solid tumors. Further studies are essential to determine the additional functions of HDACs unrelated to the chromatin state modeling. Therefore, while preliminary results for a clinical use of HDACi are encouraging, it still has to be taken with a “grain of salt” for the reasons mentioned above. However, taking into consideration all the studies discussed in this review, it is undeniable that targeting general epigenetic players and/or transcriptional regulators would significantly potentiate anti-tumor effects although low toxicity level for normal tissues remains challenging.
Conclusion
Several lines of evidence demonstrate that epigenetic modifications play an important role in melanoma initiation, progression, and metastasis. Along with the accumulation of knowledge on the multiplicity of epigenetic alterations that impact melanoma due to their reversible nature, here we highlight the latest research which is still in its infancy with the new but promising molecules in pipeline for future possible treatment of the disease (Table 1-3). Targeting epigenetic modifications is of intense interest in the treatment against cancer and epigenetic drug discovery is a rapidly advancing field. To date, these drugs have few limitations including substrate specificity, therefore many challenges remain to be resolved.
Our next challenge is to better understand the mechanisms at the origin of aberrant epigenetics that would help to identify new relevant therapeutic targets. So far, the only approved epigenetic drugs are HDACi and DNA methyl transferase (DNMT) inhibitors. Unfortunately, HDACi monotherapy has been shown to have limited efficacy against solid tumors. However, they can function synergistically with a variety of compounds or immune therapies, some of which are currently in clinical trials. Over the last few years, HMTs have been particularly attractive (e.g. SETDB1, EZH2) in the light of the structural and mechanistic data suggesting a potential modulation by small compounds. A success in one of these clinical trials (Table 3) would offer new hope for melanoma patients for whom other treatments have previously failed.
In the interim, histone modification signatures may guide prognosis by predicting treatment outcome and thus may support clinical decision-making in treatment of melanoma patients. We focused in this review on histone modifications among various other mechanisms, but we have to keep in mind that they are working in concert with other epigenetic aberrations such as DNA methylation or miRNA dysregulations and genetic alterations. In particular, aberrant DNA methylation associated with transcriptional repression is a major phenomenon observed in cancers. Although the exact sequence of events between histone modifications and DNA methylation remains unclear to date, these events likely drive epigenetic mechanisms together leading to malignant transformation. The classical view is that histone modifications might be the first step of epigenetic silencing, which orchestrates the recruitment of DNA methylation machinery. In that case, aberrant DNA methylation potentiate the transcriptional silencing already existing such as a lock-off mechanism. Another view is that aberrant DNA methylation has a key role in the reversion of the epigenetic state at specific genomic loci and activates silent genes. Regardless the sequence of these events, a dual inhibition using HDACi with DNMTi remains an attractive area of interest in the clinic.
It is undeniable that personalized treatments based onto the genetic and epigenetic characteristics have to be considered. Combinations of epigenetic drugs with other anti-cancer agents such as targeted therapy or immunomodulatory drugs is a promising avenue for improving the effectiveness of treatments in both cutaneous and uveal melanoma.
Acknowledgements
We thank Dr. MK. Tulic (C3M, Nice, France) for critical review of the manuscript and for helpful discussions. This work was supported by Institut National du Cancer (INCa_10573) and La Fondation ARC (20171206312) to C.B. The team is “Equipe Labellisée Fondation ARC” to R.B. The first author also acknowledges the financial support provided by La Fondation pour la Recherche Médicale (FRM ARF201809006989).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wolchok JD, Saenger YM. Current topics in melanoma. Curr Opin Oncol. 2007;19:116-20
2. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell. 2017;168:707-23
3. Long G V, Stroyakovskiy D, Gogas H. et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877-88
4. Long G V, Stroyakovskiy D, Gogas H. et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet. 2015;386:444-51
5. Robert C, Karaszewska B, Schachter J. et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-9
6. Long G V, Hauschild A, Santinami M. et al. Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N Engl J Med. 2017;377:1813-23
7. Hugo W, Shi H, Sun L. et al. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell. 2015;162:1271-85
8. Jones PA. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484-92
9. Lee JJ, Murphy GF, Lian CG. Melanoma epigenetics: novel mechanisms, markers, and medicines. Lab Invest. 2014;94:822-38
10. Goldberg AD, Allis CD, Bernstein E. Epigenetics: A Landscape Takes Shape. Cell. 2007;128:635-8
11. Bernstein E, Hake SB. The nucleosome : a little variation goes a long way. Biochem Cell Biol. 2006;84:505-7
12. Vardabasso C, Hasson D, Ratnakumar K, Chung CY, Duarte LF, Bernstein E. Histone variants: Emerging players in cancer biology. Cell Mol Life Sci. 2014;71:379-404
13. Lawrence M, Daujat S, Schneider R. Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends Genet. 2016;32:42-56
14. Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17:487-500
15. Zentner GE, Henikoff S. Regulation of nucleosome dynamics by histone modifications. Nat Struct Mol Biol. 2013:259-66
16. Heintzman ND, Stuart RK, Hon G. et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311-8
17. HniszBrian D, Hnisz D, Abraham BJ. et al. Super-Enhancers in the Control of Cell Identity and Disease. Cell. 2013;155:934-47
18. Lu C, Allis CD. SWI/SNF complex in cancer. Nat Genet. 2017:178-9
19. Marmorstein R, Zhou MM. Writers and readers of histone acetylation: Structure, mechanism, and inhibition. Cold Spring Harb Perspect Biol. 2014;6:a018762
20. Bernstein BE, Mikkelsen TS, Xie X. et al. A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells. Cell. 2006;125:315-26
21. Mikkelsen TS, Ku M, Jaffe DB. et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553-60
22. Patton EE, Widlund HR, Kutok JL. et al. BRAF mutations are sufficient to promote nevi formation and cooperate with p53 in the genesis of melanoma. Curr Biol. 2005;15:249-54
23. Kaufman CK, Mosimann C, Fan ZP. et al. A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation. Science (80- ). 2016;351:aad2197
24. Bossi D, Cicalese A, Dellino GI. et al. In Vivo genetic screens of patient-derived tumors revealed unexpected frailty of the transformed phenotype. Cancer Discov. 2016;6:650-63
25. Guo C, Chen LH, Huang Y. et al. KMT2D maintains neoplastic cell proliferation and global histone H3 lysine 4 monomethylation. Oncotarget. 2013;4:2144-53
26. Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ. SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002;16:919-32
27. Sarraf SA, Stancheva I. Methyl-CpG binding protein MBD1 couples histone H3 methylation at lysine 9 by SETDB1 to DNA replication and chromatin assembly. Mol Cell. 2004;15:595-605
28. Ceol CJ, Houvras Y, Jane-Valbuena J. et al. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature. 2011;471:513-7
29. Orouji E, Federico A, Larribère L. et al. Histone methyltransferase SETDB1 contributes to melanoma tumorigenesis and serves as a new potential therapeutic target. Int J Cancer. 2019;145:3462-77
30. Zingg D, Debbache J, Schaefer SM. et al. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat Commun. 2015;6:6051
31. Zingg D, Debbache J, Peña-Hernández R. et al. EZH2-Mediated Primary Cilium Deconstruction Drives Metastatic Melanoma Formation. Cancer Cell. 2018;34:69-84
32. Barsotti AM, Ryskin M, Zhong W. et al. Epigenetic reprogramming by tumor-derived EZH2 gain-of-function mutations promotes aggressive 3D cell morphologies and enhances melanoma tumor growth. Oncotarget. 2015;6:2928-38
33. Souroullas GP, Jeck WR, Parker JS. et al. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat Med. 2016;22:632-40
34. De Donatis GM, Le Pape E, Pierron A. et al. NF-kB2 induces senescence bypass in melanoma via a direct transcriptional activation of EZH2. Oncogene. 2016;35:2735-45
35. Kim KH, Roberts CWM. Targeting EZH2 in cancer Kimberly. Nat Med. 2016;22:128-34
36. Qu Y, Yang Q, Liu J. et al. C-Myc is required for BRAFV600E-induced epigenetic silencing by H3K27me3 in tumorigenesis. Theranostics. 2017;7:2092-107
37. LeRoy G, Rickards B, Flint SJ. The Double Bromodomain Proteins Brd2 and Brd3 Couple Histone Acetylation to Transcription. Mol Cell. 2008;30:51-60
38. Gallagher SJ, Mijatov B, Gunatilake D. et al. Control of NF-kB activity in human melanoma by bromodomain and extra-terminal protein inhibitor I-BET151. Pigment Cell Melanoma Res. 2014;27:1126-37
39. Gallagher SJ, Mijatov B, Gunatilake D. et al. The epigenetic regulator I-BET151 induces BIM-dependent apoptosis and cell cycle arrest of human melanoma cells. J Invest Dermatol. 2014;134:2795-2805
40. Segura MF, Fontanals-Cirera B, Gaziel-Sovran A. et al. BRD4 sustains melanoma proliferation and represents a new target for epigenetic therapy. Cancer Res. 2013;73:6264-6276
41. Fontanals-Cirera B, Hasson D, Vardabasso C. et al. Harnessing BET Inhibitor Sensitivity Reveals AMIGO2 as a Melanoma Survival Gene. Mol Cell. 2017;68:731-44
42. Echevarría-Vargas IM, Reyes-Uribe PI, Guterres AN. et al. Co-targeting BET and MEK as salvage therapy for MAPK and checkpoint inhibitor-resistant melanoma. EMBO Mol Med. 2018;10:e8446
43. Seto E, Yoshida M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb Perspect Biol. 2014;6:a018713
44. Fiziev P, Akdemir KC, Miller JP. et al. Systematic Epigenomic Analysis Reveals Chromatin States Associated with Melanoma Progression. Cell Rep. 2017;19:875-89
45. Sharpless NE, Chin L. The INK4α/ARF locus and melanoma. Oncogene. 2003;22:3092-8
46. Venza M, Visalli M, Biondo C. et al. Epigenetic regulation of p14ARF and p16INK4A expression in cutaneous and uveal melanoma. Biochim Biophys Acta - Gene Regul Mech. 2015;1849:247-56
47. Valentini A, Gravina P, Federici G, Bernardini S. Valproic acid induces apoptosis, p16INK4Aupregulation and sensitization to chemotherapy in human melanoma cells. Cancer Biol Ther. 2007;6:185-91
48. Ceccacci E, Minucci S. Inhibition of histone deacetylases in cancer therapy: Lessons from leukaemia. Br J Cancer. 2016;114:605-11
49. Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone deacetylase inhibitors as anticancer drugs. Int J Mol Sci. 2017;18:1414
50. Woods DM, Woan K, Cheng F. et al. The antimelanoma activity of the histone deacetylase inhibitor panobinostat (LBH589) is mediated by direct tumor cytotoxicity and increased tumor immunogenicity. Melanoma Res. 2013;23:341-8
51. Gallagher SJ, Gunatilake D, Beaumont KA. et al. HDAC inhibitors restore BRAF-inhibitor sensitivity by altering PI3K and survival signalling in a subset of melanoma. Int J Cancer. 2018;142:1926-37
52. Petruccelli LA, Dupéré-Richer D, Pettersson F, Retrouvey H, Skoulikas S, Miller WH. Vorinostat induces reactive oxygen species and dna damage in acute myeloid leukemia cells. PLoS One. 2011;6:e20987
53. Wang L, Leite de Oliveira R, Huijberts S. et al. An Acquired Vulnerability of Drug-Resistant Melanoma with Therapeutic Potential. Cell. 2018;173:1413-25
54. Kozikowski AP, Tapadar S, Luchini DN, Ki HK, Billadeau DD. Use of the Nitrile Oxide Cycloaddition (NOC) reaction for molecular probe generation: A new class of enzyme selective histone deacetylase inhibitors (HDACIs) showing picomolar activity at HDAC6. J Med Chem. 2008;51:4370-3
55. Bergman JA, Woan K, Perez-Villarroel P, Villagra A, Sotomayor EM, Kozikowski AP. Selective histone deacetylase 6 inhibitors bearing substituted urea linkers inhibit melanoma cell growth. J Med Chem. 2012;55:9891-9
56. Woan K V, Lienlaf M, Perez-Villaroel P. et al. Targeting histone deacetylase 6 mediates a dual anti-melanoma effect: Enhanced antitumor immunity and impaired cell proliferation. Mol Oncol. 2015;9:1447-57
57. Lienlaf M, Perez-Villarroel P, Knox T. et al. Essential role of HDAC6 in the regulation of PD-L1 in melanoma. Mol Oncol. 2016;10:735-50
58. Vance KW, Carreira S, Brosch G, Goding CM. Tbx2 is overexpressed and plays an important role in maintaining proliferation and suppression of senescence in melanomas. Cancer Res. 2005;65:2260-8
59. Wilmott JS, Colebatch AJ, Kakavand H. et al. Expression of the class 1 histone deacetylases HDAC8 and 3 are associated with improved survival of patients with metastatic melanoma. Mod Pathol. 2015;28:884-94
60. Emmons MF, Faião-Flores F, Sharma R. et al. HDAC8 regulates a stress response pathway in melanoma to mediate escape from BRAF inhibitor therapy. Cancer Res. 2019;79:2947-61
61. Woan K V, Sahakian E, Sotomayor EM, Seto E, Villagra A. Modulation of antigen-presenting cells by HDAC inhibitors: Implications in autoimmunity and cancer. Immunol Cell Biol. 2012;90:55-65
62. Villagra A, Sotomayor EM, Seto E. Histone deacetylases and the immunological network: Implications in cancer and inflammation. Oncogene. 2010;29:157-73
63. McClure JJ, Li X, Chou CJ. Advances and Challenges of HDAC Inhibitors in Cancer Therapeutics. Adv Cancer Res. 2018;138:183-211
64. Hauschild A, Trefzer U, Garbe C. et al. Multicenter phase II trial of the histone deacetylase inhibitor pyridylmethyl-N-oyl]-benzyl}-carbamate in pretreated metastatic melanoma. Melanoma Res. 2008;18:274-8
65. Maertens O, Kuzmickas R, Manchester HE. et al. MAPK pathway suppression unmasks latent dna repair defects and confers a chemical synthetic vulnerability in BRAF-, NRAS-, and NF1 -mutant melanomas. Cancer Discov. 2019;9:526-45
66. Roesch A, Fukunaga-Kalabis M, Schmidt EC. et al. A Temporarily Distinct Subpopulation of Slow-Cycling Melanoma Cells Is Required for Continuous Tumor Growth. Cell. 2010;141:583-94
67. Roesch A, Vultur A, Bogeski I. et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell. 2013;23:811-25
68. Yu Y, Schleich K, Yue B. et al. Targeting the Senescence-Overriding Cooperative Activity of Structurally Unrelated H3K9 Demethylases in Melanoma. Cancer Cell. 2018;33:322-36
69. Kalin JH, Wu M, Gomez A V. et al. Targeting the CoREST complex with dual histone deacetylase and demethylase inhibitors. Nat Commun. 2018;9:53
70. Pan M, Reid MA, Lowman XH. et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat Cell Biol. 2016;18:1090-101
71. Park WY, Hong BJ, Lee J, Choi C, Kim MY. H3K27 demethylase JMJD3 employs the NF-κB and BMP signaling pathways to modulate the tumor microenvironment and promote melanoma progression and metastasis. Cancer Res. 2016;76:161-70
72. Hodis E, Watson IR, Kryukov G V. et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251-63
73. Mehrotra A, Mehta G, Aras S, Trivedi A, de la Serna IL. SWI/SNF chromatin remodeling enzymes in melanocyte differentiation and melanoma. Crit Rev Eukaryot Gene Expr. 2014;24:151-61
74. Keenen B, Qi H, Saladi S V, Yeung M, De La Serna IL. Heterogeneous SWI/SNF chromatin remodeling complexes promote expression of microphthalmia-associated transcription factor target genes in melanoma. Oncogene. 2010;29:81-92
75. Laurette P, Strub T, Koludrovic D. et al. Transcription factor MITF and remodeller BRG1 define chromatin organisation at regulatory elements in melanoma cells. Elife. 2015;4:e06857
76. Laurette P, Coassolo S, Davidson G. et al. Chromatin remodellers Brg1 and Bptf are required for normal gene expression and progression of oncogenic Braf-driven mouse melanoma. Cell Death Differ. 2019 doi:10.1038/s41418-019-0333-6
77. Koludrovic D, Laurette P, Strub T. et al. Chromatin-Remodelling Complex NURF Is Essential for Differentiation of Adult Melanocyte Stem Cells. PLoS Genet. 2015;11:e1005555
78. Cheli Y, Guiliano S, Botton T. et al. Mitf is the key molecular switch between mouse or human melanoma initiating cells and their differentiated progeny. Oncogene. 2011;30:2307-18
79. Ohanna M, Giuliano S, Bonet C. et al. Senescent cells develop a parp-1 and nuclear factor-κB-associated secretome (PNAS). Genes Dev. 2011;25:1245-61
80. Cheli Y, Bonnazi VF, Jacquel A. et al. CD271 is an imperfect marker for melanoma initiating cells. Oncotarget. 2014;5:5272-83
81. Cheli Y, Giuliano S, Fenouille N. et al. Hypoxia and MITF control metastatic behaviour in mouse and human melanoma cells. Oncogene. 2012;31:2461-70
82. Ohanna M, Cerezo M, Nottet N. et al. Pivotal role of NAMPT in the switch of melanoma cells toward an invasive and drug-resistant phenotype. Genes Dev. 2018;32:448-61
83. Qadeer ZA, Harcharik S, Valle-Garcia D. et al. Decreased expression of the chromatin remodeler ATRX associates with melanoma progression. J Invest Dermatol. 2014;134:1768-72
84. Schiaffino-Ortega S, Balinas C, Cuadros M, Medina PP. SWI/SNF proteins as targets in cancer therapy. J Hematol Oncol. 2014;7:81
85. Kapoor A, Goldberg MS, Cumberland LK. et al. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature. 2010;468:1105-9
86. Duarte LF, Young ARJ, Wang Z. et al. Histone H3.3 and its proteolytically processed form drive a cellular senescence programme. Nat Commun. 2014;5:5210
87. Vardabasso C, Gaspar-Maia A, Hasson D. et al. Histone Variant H2A.Z.2 Mediates Proliferation and Drug Sensitivity of Malignant Melanoma. Mol Cell. 2015;59:75-88
88. Nissan MH, Solit DB. The “SWOT” of BRAF inhibition in melanoma: RAF inhibitors, MEK inhibitors or both? Curr Oncol Rep. 2011;13:479-87
89. Flaherty KT, Robert C, Hersey P. et al. Improved Survival with MEK Inhibition in BRAF-Mutated Melanoma. N Engl J Med. 2012;367:107-14
90. Bollag G, Hirth P, Tsai J. et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596-9
91. Davies H, Bignell GR, Cox C. et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949-54
92. Flaherty KT, Infante JR, Daud A. et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694-703
93. Chapman PB, Hauschild A, Robert C. et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N Engl J Med. 2011;364:2507-16
94. Long G V, Weber JS, Infante JR. et al. Overall survival and durable responses in patients with BRAF V600-mutant metastatic melanoma receiving dabrafenib combined with trametinib. J Clin Oncol. 2016;34:871-8
95. Luke JJ, Flaherty KT, Ribas A, Long G V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat Rev Clin Oncol. 2017;14:463-82
96. Shi H, Moriceau G, Kong X. et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012;3:724
97. Poulikakos PI, Persaud Y, Janakiraman M. et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 2011;480:387-90
98. Johannessen CM, Boehm JS, Kim SY. et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968-72
99. Villanueva J, Vultur A, Lee JT. et al. Acquired Resistance to BRAF Inhibitors Mediated by a RAF Kinase Switch in Melanoma Can Be Overcome by Cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683-95
100. Nazarian R, Shi H, Wang Q. et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973-7
101. Koetz-Ploch L, Hanniford D, Dolgalev I. et al. MicroRNA-125a promotes resistance to BRAF inhibitors through suppression of the intrinsic apoptotic pathway. Pigment Cell Melanoma Res. 2017;30:328-38
102. Song C, Piva M, Sun L. et al. Recurrent tumor cell-intrinsic and -extrinsic alterations during mapki-induced melanoma regression and early adaptation. Cancer Discov. 2017;7:1248-65
103. Shen C-H, Kim SH, Trousil S. et al. Loss of cohesin complex components STAG2 or STAG3 confers resistance to BRAF inhibition in melanoma. Nat Med. 2016;22:1056-61
104. Somasundaram R, Zhang G, Fukunaga-Kalabis M. et al. Tumor-associated B-cells induce tumor heterogeneity and therapy resistance. Nat Commun. 2017;8:607
105. Seghers AC, Wilgenhof S, Lebbé C, Neyns B. Successful rechallenge in two patients with BRAF-V600-mutant melanoma who experienced previous progression during treatment with a selective BRAF inhibitor. Melanoma Res. 2012;22:466-72
106. Valpione S, Carlino MS, Mangana J. et al. Rechallenge with BRAF-directed treatment in metastatic melanoma: A multi-institutional retrospective study. Eur J Cancer. 2018;91:116-24
107. Das Thakur M, Salangsang F, Landman AS. et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494:251-5
108. Hirata E, Girotti MR, Viros A. et al. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin β1/FAK Signaling. Cancer Cell. 2015;27:574-88
109. Obenauf AC, Zou Y, Ji AL. et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature. 2015;520:368-72
110. Sharma S V, Lee DY, Li B. et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69-80
111. Menon DR, Das S, Krepler C. et al. A stress-induced early innate response causes multidrug tolerance in melanoma. Oncogene. 2015;34:4545-4545
112. Liu X, Zhang SM, McGeary MK. et al. KDM5B promotes drug resistance by regulating melanoma-propagating cell subpopulations. Mol Cancer Ther. 2019;18:706-17
113. Shaffer SM, Dunagin MC, Torborg SR. et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature. 2017;546:431-5
114. Shalem O, Sanjana NE, Hartenian E. et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84-7
115. Rajendran R, Garva R, Krstic-Demonacos M, Demonacos C. Sirtuins: Molecular traffic lights in the crossroad of oxidative stress, chromatin remodeling, and transcription. J Biomed Biotechnol. 2011;2011:368276
116. Ohanna M, Bonet C, Bille K. et al. SIRT1 promotes proliferation and inhibits the senescence-like phenotype in human melanoma cells. Oncotarget. 2014;5:2085-95
117. Bajpe PK, Prahallad A, Horlings H, Nagtegaal I, Beijersbergen R, Bernards R. A chromatin modifier genetic screen identifies SIRT2 as a modulator of response to targeted therapies through the regulation of MEK kinase activity. Oncogene. 2014;34:531-6
118. Desantis V, Lamanuzzi A, Vacca A. The role of SIRT6 in tumors. Haematologica. 2018:103
119. Strub T, Ghiraldini FG, Carcamo S. et al. SIRT6 haploinsufficiency induces BRAFV600E melanoma cell resistance to MAPK inhibitors via IGF signalling. Nat Commun. 2018;9:3440
120. Sundaresan NR, Vasudevan P, Zhong L. et al. The sirtuin SIRT6 blocks IGF-Akt signaling and development of cardiac hypertrophy by targeting c-Jun. Nat Med. 2012;18:1643-50
121. Jiang CC, Lai F, Thorne RF. et al. MEK-independent survival of B-RAFV600Emelanoma cells selected for resistance to apoptosis induced by the RAF inhibitor PLX4720. Clin Cancer Res. 2011;17:721-30
122. Shao Y, Aplin AE. Akt3-mediated resistance to apoptosis in B-RAF-targeted melanoma cells. Cancer Res. 2010;70:6670-81
123. Cagnetta A, Soncini D, Orecchioni S. et al. Depletion of SIRT6 enzymatic activity increases acute myeloid leukemia cells' vulnerability to DNA-damaging agents. Haematologica. 2018;103:80-90
124. Villanueva J, Herlyn M. Melanoma and the tumor microenvironment. Curr Oncol Rep. 2008;10:439-46
125. Patel SJ, Sanjana NE, Kishton RJ. et al. Identification of essential genes for cancer immunotherapy. Nature. 2017;548:537-42
126. Chae YK, Arya A, Iams W. et al. Current landscape and future of dual anti-CTLA4 and PD-1/PD-L1 blockade immunotherapy in cancer; lessons learned from clinical trials with melanoma and non-small cell lung cancer (NSCLC). J Immunother Cancer. 2018;6:39
127. Ayers M, Lunceford J, Nebozhyn M. et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127:2930-40
128. Puzanov I, Diab A, Abdallah K. et al. Managing toxicities associated with immune checkpoint inhibitors: Consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J Immunother Cancer. 2017;5:95
129. Netea MG, Joosten LAB, Latz E. et al. Trained immunity: A program of innate immune memory in health and disease. Science (80- ). 2016;352:aaf1098
130. Arts RJW, Carvalho A, La Rocca C. et al. Immunometabolic Pathways in BCG-Induced Trained Immunity. Cell Rep. 2016;17:2562-71
131. Saeed S, Quintin J, Kerstens HHD. et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science (80- ). 2014;345:1251086
132. Dunn J, Rao S. Epigenetics and immunotherapy: The current state of play. Mol Immunol. 2017;87:227-39
133. Arenas-Ramirez N, Sahin D, Boyman O. Epigenetic mechanisms of tumor resistance to immunotherapy. Cell Mol Life Sci. 2018;75:4163-76
134. Zingg D, Arenas-Ramirez N, Sahin D. et al. The Histone Methyltransferase Ezh2 Controls Mechanisms of Adaptive Resistance to Tumor Immunotherapy. Cell Rep. 2017;20:854-67
135. Pan D, Kobayashi A, Jiang P. et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science (80- ). 2018;359:770-5
136. Sheng W, LaFleur MW, Nguyen TH. et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell. 2018;174:549-63
137. Vo DD, Prins RM, Begley JL. et al. Enhanced antitumor activity induced by adoptive T-cell transfer and adjunctive use of the histone deacetylase inhibitor LAQ824. Cancer Res. 2009;69:8693-9
138. Covre A, Coral S, Nicolay H. et al. Antitumor activity of epigenetic immunomodulation combined with CTLA-4 blockade in syngeneic mouse models. Oncoimmunology. 2015;4:e1019978
139. Laino AS, Betts BC, Veerapathran A. et al. HDAC6 selective inhibition of melanoma patient T-cells augments anti-tumor characteristics. J Immunother Cancer. 2019;7:33
140. Rietschel P, Panageas KS, Hanlon C, Patel A, Abramson DH, Chapman PB. Variates of survival in metastatic uveal melanoma. J Clin Oncol. 2005;23:8076-80
141. Pandiani C, Béranger GE, Leclerc J, Ballotti R, Bertolotto C. Focus on cutaneous and uveal melanoma specificities. Genes Dev. 2017;31:724-43
142. Johansson P, Aoude LG, Wadt K. et al. Deep sequencing of uveal melanoma identifies a recurrent mutation in PLCB4. Oncotarget. 2016;7:4624-31
143. Moore AR, Ceraudo E, Sher JJ. et al. Recurrent activating mutations of G-protein-coupled receptor CYSLTR2 in uveal melanoma. Nat Genet. 2016;48:675-80
144. Van Raamsdonk CD, Bezrookove V, Green G. et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457:599-602
145. Van Raamsdonk CD, Griewank KG, Crosby MB. et al. Mutations in GNA11 in Uveal Melanoma. N Engl J Med. 2010;363:2191-9
146. Field MG, Durante MA, Anbunathan H. et al. Punctuated evolution of canonical genomic aberrations in uveal melanoma. Nat Commun. 2018;9:116
147. Carvajal RD, Schwartz GK, Tezel T, Marr B, Francis JH, Nathan PD. Metastatic disease from uveal melanoma: Treatment options and future prospects. Br J Ophthalmol. 2017;101:38-44
148. Scheuermann JC, de Ayala Alonso AG, Oktaba K. et al. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature. 2010;465:243-7
149. Landreville S, Agapova OA, Matatall KA. et al. Histone deacetylase inhibitors induce growth arrest and differentiation in uveal melanoma. Clin Cancer Res. 2012;18:408-16
150. Wang H, Wang L, Erdjument-Bromage H. et al. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 2004;431:873-8
151. Zhu P, Zhou W, Wang J. et al. A Histone H2A Deubiquitinase Complex Coordinating Histone Acetylation and H1 Dissociation in Transcriptional Regulation. Mol Cell. 2007;27:609-21
152. Nakagawa T, Kajitani T, Togo S. et al. Deubiquitylation of histone H2A activates transcriptional initiation via trans-histone cross-talk with H3K4 di- and trimethylation. Genes Dev. 2008;22:37-49
153. Matatall KA, Agapova OA, Onken MD, Worley LA, Bowcock AM, Harbour JW. BAP1 deficiency causes loss of melanocytic cell identity in uveal melanoma. BMC Cancer. 2013;13:371
154. Field MG, Kuznetsov JN, Bussies PL. et al. BAP1 Loss Is Associated with DNA Methylomic Repatterning in Highly Aggressive Class 2 Uveal Melanomas. Clin Cancer Res. 2019;25:5663-73
155. Bommi P V, Dimri M, Sahasrabuddhe AA, Khandekar JD, Dimri GP. The polycomb group protein BMI1 is a transcriptional target of HDAC inhibitors. Cell Cycle. 2010;9:2663-73
156. Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA Approval Summary: Vorinostat for Treatment of Advanced Primary Cutaneous T-Cell Lymphoma. Oncologist. 2007;12:1247-52
157. Robertson FM, Woodward WA, Pickei R. et al. Suberoylanilide hydroxamic acid blocks self-renewal and homotypic aggregation of inflammatory breast cancer spheroids. Cancer. 2010;116:2760-7
158. Wang Y, Liu M, Jin Y, Jiang S, Pan J. In vitro and in vivo anti-uveal melanoma activity of JSL-1, a novel HDAC inhibitor. Cancer Lett. 2017;400:47-60
159. Heijkants R, Willekens K, Schoonderwoerd M. et al. Combined inhibition of CDK and HDAC as a promising therapeutic strategy for both cutaneous and uveal metastatic melanoma. Oncotarget. 2018;9:6174-87
160. Dai W, Zhou J, Jin B, Pan J. Class III-specific HDAC inhibitor Tenovin-6 induces apoptosis, suppresses migration and eliminates cancer stem cells in uveal melanoma. Sci Rep. 2016;6:22622
161. Carvajal RD, Sosman JA, Quevedo JF. et al. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: A randomized clinical trial. JAMA - J Am Med Assoc. 2014;311:2397-405
162. Carvajal RD, Piperno-Neumann S, Kapiteijn E. et al. Selumetinib in combination with dacarbazine in patients with metastatic uveal melanoma: A Phase III, Multicenter, Randomized Trial (SUMIT). J Clin Oncol. 2018;36:1232-9
163. Faião-Flores F, Emmons MF, Durante MA. et al. HDAC Inhibition Enhances the In Vivo Efficacy of MEK Inhibitor Therapy in Uveal Melanoma. Clin Cancer Res. 2019;25:5686-701
164. Safa AR. c-FLIP, a master anti-apoptotic regulator. Exp Oncol. 2012;34:176-84
165. Venza I, Visalli M, Oteri R, Teti D, Venza M. Class I-specific histone deacetylase inhibitor MS-275 overrides TRAIL-resistance in melanoma cells by downregulating c-FLIP. Int Immunopharmacol. 2014;21:439-46
166. Lafave LM, Béguelin W, Koche R. et al. Loss of BAP1 function leads to EZH2-dependent transformation. Nat Med. 2015;21:1344-9
167. Nishioka K, Rice JC, Sarma K. et al. PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent Chromatin. Mol Cell. 2002;9:1201-13
168. Schoumacher M, Le Corre S, Houy A. et al. Uveal melanoma cells are resistant to EZH2 inhibition regardless of BAP1 status. Nat Med. 2016;22:577-8
169. Schrump DS. Cytotoxicity mediated by histone deacetylase inhibitors in cancer cells: Mechanisms and potential clinical implications. Clin Cancer Res. 2009;15:3947-57
Author contact
![]() Corresponding author: Thomas Strub, Thomas.Strubfr
Corresponding author: Thomas Strub, Thomas.Strubfr