Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Immunoregulatory Cell Types...
3. Recent Advances in...
4. Conclusions and Future...
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(7):3099-3117. doi:10.7150/thno.42998 This issue Cite
Review
Modulation of tumor microenvironment for immunotherapy: focus on nanomaterial-based strategies
Yun Liu1, Jianfeng Guo1,2 ![]() , Leaf Huang1
, Leaf Huang1 ![]()
1. Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States
2. School of Pharmaceutical Sciences, Jilin University, Changchun 130021, China
Received 2019-12-12; Accepted 2020-1-19; Published 2020-2-10
Abstract

Recent advances in the field of immunotherapy have profoundly opened up the potential for improved cancer therapy and reduced side effects. However, the tumor microenvironment (TME) is highly immunosuppressive, therefore, clinical outcomes of currently available cancer immunotherapy are still poor. Recently, nanomaterial-based strategies have been developed to modulate the TME for robust immunotherapeutic responses. In this review, the immunoregulatory cell types (cells relating to the regulation of immune responses) inside the TME in terms of stimulatory and suppressive roles are described, and the technologies used to identify and quantify these cells are provided. In addition, recent examples of nanomaterial-based cancer immunotherapy are discussed, with particular emphasis on those designed to overcome barriers caused by the complexity and diversity of TME.
Keywords: tumor immunology, characterization and quantification of immunoregulatory cells, nanoparticles, drug delivery, combination therapy
1. Introduction
Recent knowledge of the crosstalk between cancer cells and the host immune system (termed the cancer-immunity cycle) has opened up the potential for cancer immunotherapy [1]. The clinical promise of different strategies such as monoclonal antibodies (mAbs, conjugated with and without drugs) [2], cancer vaccines [3], adoptive T cell therapy [4] and immune checkpoint inhibitors (mostly antibodies) [5], has underscored the status of immunotherapy as a pillar of cancer treatment. However, the neoplastic foci (unlike hematologic malignancies) is normally surrounded by immune cells, fibroblasts, soluble signaling molecules, blood vessels, and the extracellular matrix (ECM) (see more details in [6]). These cells/components in the tumor microenvironment (TME) cause serious resistance to currently available immune-based therapies [6]. For example, the adoptive transfer of genetically engineered T cells expressed with chimeric antigen receptors (CARs) has achieved promising results in the treatment of acute lymphocytic leukemia (one of blood cancers) with up to 90% of five-year overall survival, but this treatment has been significantly limited in solid tumors [7]. Therapeutic efficacy of antibody-drug conjugates and cancer vaccines is also largely attenuated by immunosuppression caused by the TME [8]. In addition, the blockade of immune checkpoint molecules (e.g. programmed cell death protein 1, PD-1; cytotoxic T lymphocyte-associated protein 4, CTLA-4) using antibodies has demonstrated great promise for sculpting tumor immunogenicity in certain solid tumors (e.g. melanoma and non-small cell lung cancer) [8]; however, response rates to immune checkpoint inhibitors tremendously vary in different tumor types, which is mainly attributed to the complex nature of TME [9].
Recently, increasing research in nanomaterials has offered great potential for the improvement of cancer immunotherapy [10], but the immunosuppressive TME still limits the efficacy. Therefore, it is really of critical importance to understand the complexity and diversity of TME. Recent advances in technologies such as high-solution imaging, flow cytometry and next-generation sequencing are anticipated to provide a comprehensive view of TME constituents [6], which will inspire the development of novel nanoformulations to advance cancer immunotherapy. In this review, the immunoregulatory cells (cells relating to the regulation of immune responses) in terms of stimulatory and suppressive roles in the TME are described, and the techniques used to characterize and quantify them are provided. This review will also discuss different nanoparticle (NP) strategies under investigation for cancer immunotherapy, with specific emphasis on those designed for circumventing barrages caused by the TME.
2. Immunoregulatory Cell Types in TME
Tumorigenesis as a complex and dynamic process is generally comprised of three phases namely initiation, development and metastasis. The interactions between malignant/non-malignant cells and cellular/non-cellular components form a microenvironment surrounding the neoplastic foci [6]. Inside there, the ECM (a complex network of proteins, proteoglycans and enzymes) provides the physical and biochemical support for surrounding cells, and the crosstalk between tumor cells, immune cells and stromal cells via the secretion of cytokines and chemokines causes the escape of immunosurveillance for tumor progression [11]. The details of tumor/non-tumor cell communications and cell-ECM interactions have been substantially studied [12], which assist in understanding the structural and physiological obstacles associated with the TME and consequently improving cancer therapies. Generally, cells inside the TME [13] include: immune cells (e.g. dendritic cells, lymphocytes, macrophages and myeloid-derived suppressor cells (MDSCs)), cells of mesenchymal origin (e.g. fibroblasts, myofibroblasts, mesenchymal stromal cells), and vascular cells (e.g. endothelial cells and pericytes).
In this section, we will discuss the cell types within the TME in terms of immunostimulatory and immunosuppressive roles (Table 1) and describe technologies for characterization and quantification of these cells, hoping to understand the mechanisms of immunotherapy resistance, identify potential therapeutic targets, and advance antitumor immunity for long-term effects and/or eradication of cancer.
The commonly used phenotypic markers for immune cells within the TME in terms of stimulatory/suppressive roles.
| Cell Subtypes | Markers | Ref. |
|---|---|---|
| Immunostimulatory | ||
| DCs | CD11b+ MHCII+ | [14] |
| Cytotoxic T cells | CD3+ CD8+ | [15] |
| Helper T cells | CD3+ CD4+ | [16] |
| Memory T cells | CD44+ CD62L+ CD3+ | [17] |
| Follicular B cells | IgD+CD21+CD22+ CD23+ | [18] |
| Plasma cells | CD138+CD38+ | [19] |
| Memory B cells | CD20+CD27+CD40+CD80+ | [20] |
| NK cells | CD16+ CD56+ CD57+ NK1.1+/NK1.2+ | [21] [22] |
| M1 cells | F4/80+ CD86+ CD80+ | [23] |
| Immunosuppressive | ||
| MDSCs | CD11b+ Gr-1+ | [24] |
| M2 cells | F4/80+ CD206+ CD163+ | [23] |
| Tregs | CD3+ CD4+ CD25+ Foxp3+ TIM-3+ | [25] [26] |
| Bregs | CD19+ IL-10+ | [27] |
2.1. Immunostimulatory cells
Dendritic cells (DCs): It is known that a large number of genetic mutations and the failure of normal cellular regulatory processes are evident in cancers. These abnormalities cause the presence of neoantigens, differentiation antigens, or cancer/testis antigens (together termed tumor-associated antigens (TAAs)), which result in the presentation of peptides bound to the major histocompatibility class I (MHC-I), distinguishing cancer cells from the normal cell types [28]. At the beginning of cancer-immunity cycle depicted by Chen and Mellman (step 1 of Figure 1) [1], TAAs are released from dying tumor cells and captured by antigen-presenting cells (APCs). The professional APCs mainly include DCs, macrophages and B cells, and among these, DCs play critical roles in starting and regulating the anticancer immunity [29]. Subsequently, TAAs are bound to MHC-I/MHC-II of mature DCs (step 2 of Figure 1). When mature DCs migrate into tumor-draining lymph nodes, they present TAAs to T cells, which lead to the priming and activation of effector T cells against TAAs (step 3 of Figure 1). The function of DCs can be regulated by a complex network involving cytokines, chemokines and damage-associated molecular patterns (DAMPs) (Figure 1; also see review in [29]).
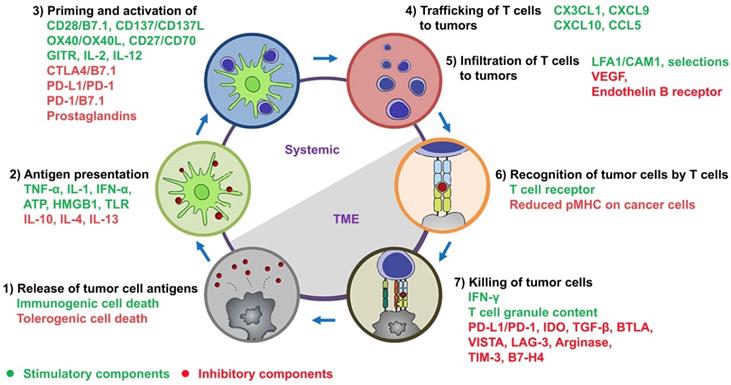
The cancer-immunity cycle in tandem with a summary of stimulatory and inhibitory components. As depicted by Chen and Mellman, this cycle is comprised of 1) release of tumor cell antigens by dying cancer cells, 2) antigen presentation by DCs, 3) priming and activation of T cells, 4) trafficking and 5) infiltration of activated T cells to tumors, 6) recognition of tumor cells by activated T cells, and 7) killing of tumor cells. The stimulatory and inhibitory factors together form an immune regulatory network for the modulation of cancer-immunity cycle. This figure has been modified from [1] and [10].
The phenotypic maturation of DCs is associated with the upregulation of surface markers such as CD80, CD83 and CD86 along with the MHC molecules, whereas the expression of these markers is negative or low in immature or semi-mature DCs [30]. When DCs become mature, they secrete medium/high levels of pro-inflammatory or immunostimulatory cytokines (e.g. IL-12, IL-23 and IL-1β) and low level of immunosuppressive cytokines (e.g. IL-10 and TGF-β) [30]. In contrast to mature DCs, immature or semi-mature counterparts are devoid of the capacity to prime and activate T cells against tumors, or may even cause T cell anergy and induce tolerance therefore compromising antitumor immunity [29]. Recently, the stimulatory and inhibitory factors associated with DC maturation have been used as therapeutic means or targets for developing novel nanomaterial-based strategies.
Cytotoxic T lymphocytes (CTLs): As shown in step 3 of Figure 1, naive CD8 T cells become CTLs when TAAs bound on MHC-I of DCs are interacted with the T-cell receptor (TCR, a disulfide-linked membrane-anchored heterodimeric protein complex composed of CD3 and highly variable alpha and beta chains [31]). CTLs are capable of trafficking through tissues (e.g. blood and lymphatic vessels) (step 4 of Figure 1) and infiltrating into tumors (step 5 of Figure 1). It is known that the trafficking and infiltration of CTLs are tightly upregulated by a complex interactions between T cells and endothelial cells, mainly including [31] 1) the expression of homing molecules (e.g. PSGL-1 and CD44) on CTLs that can facilitate them to migrate into tumors; 2) a temporary attachment of CTLs onto the endothelium by binding P- and E-selectins via homing molecules; 3) the expression of chemokine receptors (e.g. CXCR3) on CTLs that can bind chemokines (e.g. CXCL9 and CXCL10) released from the TME; 4) the activation of integrins (e.g. LFA-1 and VLA-4) on CTLs that can bind integrin ligands (e.g. ICAM-1 and VCAM-1), which form a firm adhesion between CTLs and the endothelium, leading to extravasation of CTLs into the tumor bed. Subsequently, CTLs release the cytotoxic mediators such as IFN-γ, granzymes or perforin to kill cancer cells in a TCR-dependent manner (steps 6 and 7 of Figure 1).
However, when CTLs enter the TME, they encounter an immunosuppressive milieu, in which inhibitory components derived from tumor cells and stromal cells can affect the phenotype and function of CTLs and finally turn them into “exhausted” state (e.g. decreased proliferation and reduced production of cytotoxic mediators). For example, the activity of CTLs is significantly dampened by immunosuppressive mediators produced by tumor cells such as indoleamine 2,3-dioxygenase 1 (IDO-1), programmed death-ligand 1 (PD-L1), cyclooxygenase type 2 (COX‐2), and signal transducer and activator of transcription 3 (STAT3) [32]. In addition, a number of cytokines released from tumor-associated fibroblasts (TAFs), myeloid-derived suppressor cells (MDSCs), macrophage type 2 (M2) cells and regulatory T cells (Tregs) can negatively regulate CTL-mediated cancer killing (step 7 of Figure 1; see below discussion). Therefore, the nanomaterial-based strategies targeting these aforemetioned inhibitory mediators may potentially relieve the exhaustion of CTLs and rescue their cytotoxic function for antitumor immunity.
T helper (Th) cells: The cell-mediated antitumor immunity (an immune response that is not involved with antibodies) has been largely attributed to CD8+ CTLs, however, emerging evidence indicates that CD4+ Th cells also play significant roles in the initiation and maintenance of antitumor effects. When antigens bound onto MHC-II of APCs interact with the TCR, naive CD4+ T cells are generally differentiated into Th1, Th2, Th17 and Th9 subtypes [33].
The differentiation of Th1 requires IL-2, IL-12 and IFN-γ, and Th1 cells release IFN-γ, IL-2 and TNF-α for the assistance of CTL differentiation, activation of macrophage type 1 (M1) cells, and mediation of cell-mediated immunity [34]. The differentiation of Th2 requires IL-4, IL-6 and IL-10, and Th2 cells secrete IL-4, IL-5 and IL-13 for coordinating humoral immunity (an immune response that is involved with antibodies) (see below discussion). Although Th1 and Th2 subsets are both known to induce antitumor immunity, IFN-γ-secreting Th1 cells have demonstrated better efficacy in this role [33]. However, the level of Th2 cytokines within the TME is significantly higher than that of Th1 cytokines, which prevent the production of Th1 cells and activation of CTLs [34]. In addition, Th17 cells as an independent CD4+ lineage from either Th1 or Th2 have demonstrated a paradox of its function in tumor immunity [35]. Although Th17 mediates antitumor immune responses by means of stimulating effector CTLs, they may increase tumor progression through promoting angiogenesis and immunosuppressive events [35]. Recently, it has been reported that a subset of CD4+ Th cells namely Th9 possess less-exhausted cytolytic function as strong as Th1 cells and demonstrate hyperproliferative feature to persist as long as Th17 cells [36]. Th9-mediated anticancer efficacy is highly relied on IL-9 and upregulated expression of Eomes (Eomesodermin; the effector master regulator that controls granzyme expression) and Traf6 (tumor necrosis factor receptor (TNFR)-associated factor 6; one of NF-κB upstream signaling proteins). As a result, tumor-specific Th9 cells eliminated the advanced late-stage melanoma and protected surviving animals against the tumor rechallenge [36], indicating the significant role of Th9 cells in adoptive cancer therapy. As Th subsets are generally supposed as a double-edged sword in tumor immunology, nanomaterial-based therapeutic approaches that balance these Th cells hold the promise for cancer immunotherapy.
B cells: The critical contributions of T cells in antitumor immunity have been substantially investigated and well established. In contrast, the immunologic roles of B cells in response to tumors are less well studied. B cells are comprised of functionally distinct subpopulations, and the balance among these has a significant impact on tumoricidal activity [37]. When B cells are activated under the stimulation of B cell receptor (BCR) pathway, microRNA pathway, and Toll-like receptor (TLR) pathway [38], they exert antitumor immunity by means of producing antibodies, cytokines and chemokines [39], acting as local APCs [40], and forming tertiary lymphoid structures (TLS, ectopic lymphoid-like structures for long-term antitumor immunity) [41]. A subpopulation of B cells termed plasma cells can produce antibodies for antitumor responses, which mainly include antibody-depedent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). In addition, mature follicular B cells (FOB, a subset of B cells) can differentiate into Be-1 and Be-2 cells, which produce cytokines such as IFN-γ, TNF-α, IL-2 and IL-12 for enhancing the antitumor immunity of T and NK cells [39]. When stimulated by the CD40-CD40 ligand (CD40L) signaling pathway, B cells become local APCs in tumors, which maintain the survival and proliferation of tumor infiltrating T cells for durable antitumor responses [40]. However, B cells are significantly shaped inside the TME, which impair the activity of immunostimulatory B cells but result in differentiation of B cells into an immunosuppressive subtype termed regulatory B cells (Bregs, see below review).
Natural Killer (NK) cells: They have long been known as a subclass of cytotoxic lymphocytes that are critical for innate immunity against virus-infected and malignant cells [42]. Recently, emerging evidence has displayed that abnormal cells can be distinguished from healthy cells through a group of functional receptors (e.g. inhibitory and activating receptors) on the surface of NK cells [42]. The acquisition of corresponding ligands in combination with reduced expression of MHC-I molecules on aberrant cells will exert the cytotoxicity of NK cells against assaults (e.g. viruses and cancers) while ensuring self-tolerance [43]. The cytotoxic activity of NK cells is relied on cytokines such as IL-2, IL-12, IL-15 and IFN-α/β [43]. The NK cells release IFN-γ to promote the expression levels of MHC-I on cancer cells and MHC-II on APCs, facilitating the connection of innate and adaptive immunities [44]. The NK cells are also able to govern the growth and differentiation of DCs and T cells; for example, IFN-γ secreted by NK cells can activate DCs for priming subsequent T-cell responses [43, 44]. However, the development of therapeutic means based on NK cells remains a challenge, as the inhibitory factors produced by the TME can significantly cause the dysfunction of NK cells [45]. Recent strategies (e.g. checkpoint inhibitors and therapeutic antibodies) have demonstrated the potential to reverse NK cell dysfunction therefore boosting antitumor immunity [46].
Macrophage type 1 (M1) cells: Tumor-associated macrophages (TAMs) are a population of immune cells within the TME of solid tumors [47]. TAMs are recruited into tumors by chemokines (e.g. CCL2), cytokines (e.g. VEGF, PDGF and M-CSF), and other factors (e.g. fibronectin, fibrinogen, cleavage products of ECM proteins). As two key subtypes of TAMs, the classically and alternatively activated macrophages (termed M1 and M2 respectively) play distinct roles in the processes of immunosurveillance and angiogenesis underlying tumor formation, development, and metastasis [47]. When TAMs are under the stimulation of bacterial products (e.g. lipopolysaccharide, LPS) and Th1 cytokines (e.g. IFN-γ and TNF-α), they are driven towards M1. The M1 subtype is normally characterized by immunostimulatory activity and antitumor function [48]. For example, M1 cells release Th1 cytokines (e.g. IFN-γ, IL-2 and TNF-α) and chemokines (e.g. CXCL9 and CXCL10) for directly killing tumor cells as well as for indirectly augmenting the cytotoxic activity of T cells [48]. In addition, M1 cells are capable of normalizing the tortuous vasculature [49], which can remodel the TME and overcome resistance to cancer therapy. In contrast, M2 cells have a significant impact on tumor progression by promoting genetic instability, supporting tumor growth and metastasis, and orchestrating tumor immunity (see below discussion).
2.2. Immunosuppresive cells
Myeloid-derived suppressor cells (MDSCs): It is known that mononuclear cells (monocytes, they are terminally differentiated into macrophages and DCs) and granulocytes (for example, neutrophils as the most abundant representative) originate from hematopoietic stem cells via common myeloid progenitors within the bone marrow (BM) [50]. The activity of these myeloid cells is tightly governed by a network of signals from pathogens in the form of TLR ligands, DAMPs and/or pathogen-associated molecular patterns (PAMPs) [50]. These signals are often strong but end in a short duration. The response to the signals leads to a rapid mobilization of monocytes and neutrophils from the BM, the significantly enhanced phagocytosis, a generation of pro-inflammatory cytokines, and the upregulation of MHC class II and costimulatory molecules [51]. In contrast, the signals generated by chronic conditions (e.g. cancers) are relatively weak but sustain for a long while [52]. When the nature of myeloid cells is deformed under cancerous condition, monocytes/neutrophils demonstrate immature phenotype and morphology, ineffective phagocytic activity, and high expression of anti-inflammatory cytokines [53]. Consequently, these immature myeloid cells are proliferated and converted to MDSCs. MDSCs always coexist with normal monocytes and neutrophils in cancer patients, but the number of MDSCs is increased during tumor progression and becomes dominant, which suppress the adaptive immunity and facilitate tumor progression and metastasis [53].
MDSCs consist of two main subpopulations namely monocytic (M-) [54] and polymorphonuclear (PMN-) [55] MDSCs. Increasing evidence indicates that M-MDSCs are phenotypically and morphologically similar to monocytes, and PMN-MDSCs are similar to neutrophils [56]. M-MDSCs (CD11b+Gr1low phenotype) rapidly differentiate into TAMs within tumors, in which these terminally differentiated myeloid cells (most likely macrophage type 2, see below discussion) inhibit immune responses and promote tumor development [56]. On the other hand, PMN-MDSCs (often referred as immunosuppressive neutrophils, with a CD11b+Gr1high phenotype) are propagated inside tumors in which they become the dominant subpopulation of neutrophils [57]. As MDSCs and monocytes/neutrophils share a common number of markers and are identical in morphology, there is still a debate associated with the relationship between these cells. Therefore, future studies are needed to solve the controversy and confusion surrounding the true nature of MDSCs (see review in [58]).
The migration of MDSCs to tumors is achieved by chemokines, and among these, CCL2 and CCL5 are considered the main chemokines underlying the MDSC migration [59, 60]. The other chemokines such as CCL15, CXCL5, CXCL6, CXCL8 and CXCL12 have also been reported to induce the recruitment of MDSCs into the TME [59]. As one of the major cellular components of the TME, MDSCs exert immunosuppressive activities mainly by the upregulation of inhibitory PD-L1 on the surface, release of immunosuppressive cytokines such as transforming growth factor (TGF-β) and IL-10, and production of chemokines (e.g. CCL4 and CCL5) for Tregs into tumors [60]. As MDSCs are phenotypically and morphologically similar to monocytes and neutrophils, therapeutic strategies that can specifically target MDSCs may provide better therapeutic benefits (see review in [59, 60]).
Macrophage type 2 (M2) cells: In contrast to M1 cells that act preferentially in pro-inflammatory responses and antitumor cytotoxic function, M2 counterparts exert anti-inflammatory and tissue remodeling/regenerative roles [61]. MDSCs may drive TAMs towards the M2 phenotype by increasing the secretion of IL-10 and alleviating the production of IL-12 [48]. In addition, when TAMs are infiltrated into tumors, they are preferentially differentiated into M2 cells under the stimulation of cytokines (e.g. IL-4, IL-13, IL-21 and IL-33) and chemokines (e.g. CCL2 and CXCL4) [48]. The M2 subclass is functionally characterized by the immunosuppression and the promotion of tissue remodeling (e.g. angiogenesis). For example, M2 macrophages express different chemokines such as CCL17, CCL22 and CCL24, and these chemokine receptors are present on Th2 and Treg cells [48, 61]. As such, the release of M2 chemokines can lead to the recruitment of immunosuppressive cells into tumors. The activation of M2 cells exerts inhibitory activity against DCs and T cells by releasing suppressive cytokines (e.g. IL-10 and TGF-β) [62], produces inhibitory metabolites for T cell suppression/anergy/death by triggering IDO-1 mediated pathway [63], and induces immune tolerance by expressing checkpoint molecules (e.g. PD-L1 and CD47) [64]. M2 macrophages also facilitate neovascularization by the release of pro-angiogenic mediators such as IL-8, vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), and epidermal growth factor (EGF) [65]. Recently, the inhibition of TAM recruitment and depletion of M2 macrophages have provided therapeutic opportunities to restrain tumor growth and metastasis [66]. In addition, due to the controversial (supportive and inhibitory) role of TAMs, strategies reprogramming the phenotype from M2 to M1 to rescue antitumor immunity have presented significant antitumor potential [61].
Regulatory T cells (Tregs): As a subtype of T cells, Tregs play important roles in the maintenance of immunological tolerance in the periphery (e.g. autoimmune diseases) by suppressing the host immunity against self- and nonself-antigens [67]. The most physiologically relevant Tregs are characterized by the expression of surface markers CD4/CD25 and transcription factor Forkhead box protein 3 (FoxP3) [68]. Accumulating evidence indicates that an elevated number of CD4+CD25+FoxP3+ Tregs are infiltrated into tumors, and their abundant presence is considered a major hurdle to effective immunotherapy [69]. It has been reported that Tregs in patients with tumors, as compared to those in healthy populations, are often evident with high expression of chemokine receptors such as CCR4, CCR5 and CXCR4, and the corresponding chemokines derived from the TME can facilitate the infiltration of Tregs into tumors [70]. Treg-mediated suppressive mechanisms mainly include: 1) they scarcely produce IL-2 but express the high-affinity IL-2 receptor α chain (CD25) to deprive this cytokine from the neigbour, which may limit the activation and proliferation of effector T cells [71]; 2) CTLA-4 expressed on Tregs has higher affinity for CD80 and CD86 (co-stimulatory molecules) on DCs than CD28 expressed on T cells does (the interaction between CD28 and CD80/CD86 provides co-stimulatory signals required for the activation and survival of T cells), thus hindering co-stimulation of T cells [72]. In addition, the binding of CTLA-4 with CD80/CD86 may downregulate the expression of these co-stimulatory molecules, further causing the inactivation of T cells [73]; 3) Tregs can produce immunosuppressive cytokines (e.g. IL-10 and TGF-β) to skew the function of DCs and T cells, and may even cause direct killing of these immunostimulatory cells by secreting granzymes and perforin [74]. Recent progress in tumor immunotherapy targeting Treg-mediated immunosuppressive mechanisms holds great promise for cancer patients [75].
Regulatory B cells (Bregs): As discussed above, B cells are subveted toward Bregs inside the TME, which is accomplished by pathways of TLR, CD40/CD40L, B-cell activating factor (BAFF), BCR, and CD80/CD86 [76]. Bregs negatively regulate antitumor immunity through different mechanisms: 1) they produce immunosuppressive mediators such as cytokines (e.g. IL-10, TGF-β and IL-35) and IDO-1, which can suppress the proliferation and activation of T and NK cells [40]; 2) Bregs inactivate these immunostimulatory cells by expressing immune checkpoints (e.g. PD-L1) [77]; 3) when Bregs express the death-inducing molecule Fas ligand (FASL), they will induce the apoptosis of effector T cells [78]; 4) Bregs promote tumor progression by secreting TGF-β for epithelial-mesenchymal transition (EMT) [79]. In addition, the expression of suppressive markers (e.g. FoxP3 and CTLA-4) on Tregs can be promoted by Bregs by cell-to-cell contact [80]. Therefore, strategies used to target or reshape Bregs may provide therapeutic potential for rescuing antitumor immunotherapy.
Stromal cells: As important cell types inside the TME, stromal cells (e.g. fibroblasts, vascular endothelial cells and pericytes) usually facilitate the development and maintenance of tumors by supporting tumor cells, remodeling ECM, and promoting angiogenesis [81]. Recently, accumulating evidence has indicated that stromal cells also play immunosuppressive roles within the TME [81]. As one of the prominent stromal cells inside the TME, tumor-associated fibroblasts (TAFs) are composed of heterogeneous subtypes that are derived from different cellular origins (e.g. local fibroblasts and mesenchymal stem cells) [82]. The fibroblasts are usually quiescent in healthy tissues and early-stage cancers, however, they become activated and are turned into TAFs following a serial of physiological and biochemical changes during tumor progression (see review in [83]). TAFs are involved in ECM remodeling, tumor immunity, angiogenesis, and cancer cell proliferation and metastasis, which have previously been reviewed [84, 85]. In addition, the highly heterogeneous tumor vasculature is also a key component associated with the TME in many solid tumors, which result in abnormal blood flow into under-perfused tumor areas [86]. Due to the lack of functional intratumor lymphatic vessels, the elevated interstitial fluid pressure disrupts the transport of therapeutic agents to the TME [86]. The tumor blood and lymphatic vascular networks can hinder immunosurveillance mechanisms and suppress antitumor immunity, which have been discussed elsewhere [87]. Therefore, novel therapeutic strategies used to remodel these stromal cells also hold great promise for overcoming immunotherapy resistance [87].
Recently, the reprogramming of immunoregulatory cells has been achieved using nanomaterial-based approaches, which can profoundly improve immune therapy against cancers. The methods of engineering nanomaterial-based approaches for targeted modulation of immunoregulatory cells have been extensively reviewed by Shi et al. [88] and Yu et al. [89], demonstrating the significant promise of NPs for enhancing the efficacy of current immunotherapies (see reviews for more details). It is known that the reprogramming of one single cell type is normally not sufficient to achieve antitumor efficacy, whereas the modulation of different cell populations simultaneously may lead to satisfactory therapeutic outcome. Notably, the concepts of immunoregulatory cells still remain debatable due to controversial issues such as the origin and nature of these cells and their distinctive biological roles at different stages of cancer (so called the double-edged sword) [90]. Therefore, technologies that precisely discriminate these cells are urgently required to solve these controversies, obtain a deeper insight into the definition of distinctive cell types, and confirm therapeutic targets for nanomaterial-based immunotherapeutics (nanoimmunotherapeutics).
2.3. Technologies for characterization and quantification of immunoregulatory cells
As described above, the TME is composed of a heterogeneous population of tumor cells and distinct resident/infiltrating non-tumor cells such as immune cells, fibroblasts, endothelial cells, pericytes [91], and adipocytes [92]. Tumorigenesis is profoundly affected by reciprocal interactions between these cells through cell-to-cell contact, secreted factors, and ECM proteins/peptides [6]. Recent studies have suggested the impact of resident/tumor-infiltrating host cells on cancer prognosis and clinical outcome of immune-based therapies [9], indicating the importance of immunoregulatory cells in the TME. In addition, a deeper analysis of complexity and diversity of immunoregulatory cells may facilitate a better understanding of how these cells affect the TME, which will enable the prediction of therapeutic responsiveness and reveal new therapeutic targets. The commonly used technologies to identify and quantify immunoregulatory cells in terms of phenotypic and functional analyses are selectively discussed in here.
Analysis of immunological phenotypes: As shown in Table 1, immunoregulatory cells represent a heterogeneous population, which differ in their cell surface antigens and intracellular markers in a spatiotemporal manner (e.g. early stage v.s. late stage and tumor-infiltrating v.s. blood circultating). These molecules can be characterized and quantified using antibody-based imaging and cellular phenotypic techniques, such as immunohistochemical (IHC) staining assay [93], immunofluorescent (IF) microscopy [94], and flow cytometry [95]. The IHC- and IF-based analyses can be used to study the expression and location of antigens of interest from in vitro, in vivo and clinical samples. These techniques are also useful to investigate the trafficking, internalization, and recycling of surface antigens/receptors. In addition, the co-localization of cells with cells may also be assessed using these technologies. However, it is worth noting that IHC- and IF-based analyses are often associated with practical pitfalls [96] and subjective interpretation [93], therefore, experienced researchers and qualified pathologists are required to perform experimental procedures and data analyses. Also, it is difficult to track different antigens inside individual cells from the same slice of a sample using IHC- and IF-based analyses. In contrast to these techniques, flow cytometry may provide greater sensitivity and specificity for single cells [95], and therefore has long been considered a preferred analysis method in the field of immunology. Recently, the incorporation of imaging, spectrometric and cytometric technologies including the mass spectrometry IHC (MSIHC) [97], quantitative immunofluorescence (QIF) [98], imaging flow cytometry (IFC) [99] and mass cytometry (flow cytometry coupled with mass spectroscopy) [100], may provide more reliable and reproducible antibody-based technologies for characterization and quantification of immunoregulatory cells. In addition, clinical imaging modalities such as positron emission tomography (PET) and magnetic resonance imaging (MRI) have also been used for the detection of tumor-associated immune cells (e.g. macrophages) in animal models and patients [101].
It is worth noting that although the imaging and cellular phenotypic technologies are widely applied, they can only provide partial information about the “immune fingerprint” due to their limited ability for characterizing a tremendous number of immune subpopulations in tumors. In recent years, bioinformatics, which is defined as a subject that combines biology, computer science, information engineering and mathematics/statistics, has become one of fastest growing technologies in the fields of biology and medicine [102]. Bioinformatics has earned its place as a high-throughput computational tool to analyze large collections of biological data (e.g. DNA/RNA sequences, protein samples and cell populations) in a whole genome pattern [103]. This technique can be used for discovering novel candidate genes/proteins underlying disease progression as well as for identifying new therapeutic targets [104]. Computational genomic tools, which are categorized into two methods namely gene set enrichment analysis (GSEA) and deconvolution, can be used to comprehensively analyze immunophenotype in the TME [105]. Both methods are relied on a matrix of expression profiles (e.g. gene expression profiles, DNA methylation profiles or IHC profiles) for individual cell populations, and the detail has been substantially reviewed [105, 106]. Among these single-cell analyses, single-cell RNA sequencing (scRNA-seq) has received increasing attention due to its ability to uncover complex and rare cell populations, reveal relationships between genes, and delineate distinct cell lineages during early development [107]. By means of isolating individual cells, obtaining the transcripts, and establishing sequencing libraries (the transcripts are mapped to single cells) [108], scRNA-seq also allows researchers to assess highly diverse immune cell populations in healthy and malignant sites/states [109]. For example, Szabo et al. utilized scRNA-seq to define the heterogeneity of T cells isolated from the blood, bone marrow, lungs and lymph nodes from healthy donors [110]. By analysis of over 50,000 resting and activated T cells throughout these tissues, authors described T cell signatures (e.g. distinct effector states for CD8+ T cells and an interferon-response state for CD4+ T cells) and generated a healthy baseline dataset [110]. Subsequently, the comparison between the scRNA-seq profiles of tumor-associated T cells published by others and the reference map of healthy dataset generated by authors revealed the predominant activities of T cells at different tumor sites, providing insights of how to define the origin, composition and function of immune cells in malignant diseases [110]. Therefore, it is expected that the heterogeneity and dynamics of immune cell infiltrates in tumors can also be characterized using scRNA-seq in response to NP-based immunotherapy.
In addition to characterization and quantification between immunoregulatory cells, a variety of computational methods and software tools (see guidelines in [105, 106]) may be used to unravel tumor-immune cell interactions for better understanding of tumor immunology, predict neoantigens for therapeutic cancer vaccination, and determine mechanistic principles for combination treatment with synergistic effects [111].
Analysis of immunological functions: As shown in Figure 1, immunoregulatory cells produce a variety of stimulatory and suppressive cytokines and chemokines to manipulate the crosstalk between cancer cells and the host immune system. In order to accurately detect and quantitate the immune responses within the TME, a number of techniques such as real-time quantitative polymerase chain reaction (qPCR), enzyme-linked immunosorbent assay (ELISA), enzyme-linked immunospot (ELISPOT) and flow cytometry, can be carried out to evaluate the in vitro and in vivo expression of cytokines and chemokines. The level of cytokine mRNA transcripts from in vitro and in vivo models can be measured using qPCR. The in vitro and in vivo release of cytokines by immune cells may be assessed by either quantifying bulk cytokine production using ELISA [112] or measuring individual cytokine-producing cells using ELISPOT [113]. Detection of intracellular cytokines from tumor tissues, lymph nodes and peripheral blood may also be carried out using flow cytometry [114]; for example, CD8 and IFN-γ double-positive T cells are considered effector CTLs [115]. In addition, immunostimulatory cells will proliferate in response to successful immune-based therapies, whereas immunosuppressive counterparts will decline. The proliferative states of T cells may be evaluated by flow cytometry according to the level of proliferation markers (e.g. Ki67) and the intensity of proliferation tracking fluorescent dyes (e.g. carboxyfluorescein succinimidyl ester (CFSE)) [116].
It is worth noting that these phenotypic and functional analysis technologies have certain limitations (see discussion in [93, 96, 106]), therefore, it is critical to understand their ability and availability, in order to assist in the selection of appropriate and accurate ones. In fact, a combination of these techniques is preferred to provide high-accuracy for characterization and quantification of immunoregulatory cells.
3. Recent Advances in Nanomaterial-Based Strategies for Cancer Immunotherapy via Modulation of TME
The TME, which contains immunosuppressive cells and soluble signaling molecules, disorganized blood vessels and the dense ECM, is highly resistant to currently available immune-based therapies. Recent advances in the fields of nanotechnology and biomedical engineering provide great potential for the delivery of immunoregulatory agents to modulate the TME systemically (lymph nodes) and locally (tumors) [117], in order to restore the cancer-immunity cycle (Figure 1). Nanomaterial-based delivery strategies designed for immunotherapy, when applied alone or in combination with chemotherapy, gene therapy, phototherapy and radiotherapy, have profoundly revolutionized cancer therapy [118]. In vivo studies using a variety of immunotherapeutics are summarized in Table 2 according to the material type, nanoformulation strategy, and immunologic modulation. In this section, selected recent examples will be discussed based on the “fuel the engine, release the brake” rules of cancer immunotherapy.
A brief summary of in vivo studies on delivery of immunoregulatory agents using nanoparticles and natural carriers, including material types, nanoformulation strategy, and immunologic modulation. (↑ = upregulation, ↓ = downregulation)
| Material type | Nanoformulation strategy | Immunologic modulation | Ref. |
|---|---|---|---|
| Lipids & Liposomes | LPD with PD-L1 trap for colorectal cancer | DC, CD8+, CD4+ and Memory T ↑ Th17 ↓ | [119] |
| LPD with pLPS trap for colorectal cancer | DC, CD8+ and CD4+ T, M1/M2 ↑ Treg, MDSC ↓ | [120] | |
| LPD with IL-10 and CXCL12 traps for pancreatic cancer | DC, CD8+ T, NK ↑ M2, MDSC ↓ | [121] | |
| LCP with pRLN for liver cancer | DC, CD8+ and CD4+ T, M1/M2 ↑ Treg, TAF, MDSC ↓ | [122] | |
| LCP with CXCL12 trap for liver metastasis | CD8+ T ↑ Treg, MDSC, TAF ↓ | [123] | |
| LCP with BRAF peptide for melanoma | DC, CD8+ T, M1/M2 ↑ Treg ↓ | [124] | |
| Liposome with HDZ to increase NP tumor penetration in desmoplastic melanoma | DC, CD8+ and CD4+ T, NK, M1/M2 ↑ MDSC, TAF ↓ | [125] | |
| Lipid NP with OxP and DHA for colorectal cancer | DC, CD8+ and Memory T, M1↑ | [126] | |
| Polymers | PMP/OVA/siRNA nanovaccine for melanom | DC, CD8+ and CD4+ T ↑ Treg, MDSC ↓ | [127] |
| AC-NP for melanoma | DC, CD8+ T, CD8+ T/Treg, CD4+ T/Treg ↑ | [128] | |
| PLGA-R847@Cat NP enhanced radiotherapy for colon cancer | DC, CD8+ and CD4+ T ↑ Treg, M2 ↓ | [129] | |
| NanoNO to normalize tumor vasculature for liver cancer | CD8+ and CD4+ T, M1 ↑ TAF, M2 ↓ | [130] | |
| TPGS-based nanoemulsion with quercetin and alantolactone for colorectal cancer | DC, NK, CD8+ and CD4+ T ↑ Treg, MDSC ↓ | [131] | |
| DINP with aPD1 and aOX40 for melanoma | CD8+ and memory T ↑ | [132] | |
| BCPN with oxaliplatin prodrug and NLG919 for colorectal and breast cancers | DC, CD8+ T ↑ Treg ↓ | [133] | |
| H1-NB NP with OVA for melanoma | DC, CD8+ T ↑ | [134] | |
| Cellax NP with DTX for metastatic pancreatic cancer | TAF ↓ | [135] | |
| Inorganic materials | CaCO3 NP gel with aPD-1 and zebularine for melanoma | DC, CD8+ and CD4+ T ↑ MDSC ↓ | [136] |
| CaCO3 NP gel with aCD47 for melanoma | CD8+ T, M1 ↑ Treg, M2, MDSC ↓ | [137] | |
| H-MnO2 NP for TME modulation for triple negative breast cancer | CD8+ T, M1 ↑ Treg, M2 ↓ | [138] | |
| Fe3O4-ZnO nanovaccines for colorectal cancer | DC, CD8+ and CD4+ T ↑ | [139] | |
| Hollow mesoporous silica nanosphere as cancer immunoadjuvant for lung cancer | CD8+ and CD4+ T ↑ | [140] | |
| AuNP-DNA photothermal immunotherapy for tumor | DC, HSP70 ↑ | [141] | |
| MoS2-PEG-CpG for photothermal cancer immunotherapy | DC ↑ | [142] | |
| Cell membrane coated system | Erythrocyte membrane coated NP as cancer vaccine for melanoma | DC, CD8+ T ↑ | [143] |
| Cancer cell membrane-coated NP as cancer vaccine for melanoma | DC, CD8+ T ↑ | [144] | |
| Cancer cell membrane-coated NP for anticancer vaccine for melanoma | DC, CD8+ T ↑ | [145] | |
| NP coated bacterial as oral DNA vaccines for melanoma | CD8+ and CD4+ T ↑ | [146] | |
| Natural carrier mimics | Lipoprotein NP for antigen delivery for colorectal cancer and melanoma | CD8+ , CD4+ and memory T ↑ | [147] |
| Lipoprotein NP with DOX for colorectal cancer | DC, CD8+ T ↑ | [148] | |
| T cells conjugated with IL-15 and IL-21 loaded NP for melanoma | CD8+, CD4+ and memory T ↑ | [149] | |
| T cells with amphiphilic ligands for melanoma and glioma | CD8+ and CD4+ T ↑ | [150] | |
| T cells conjugated with NSC-87877 loaded NP for prostate cancer | CD8+ T ↑ | [151] | |
| Platelets loaded aPD-L1 for melanoma and triple negative breast cancer | CD8+ and CD4+ T ↑ Treg ↓ | [152] | |
| Photothermal therapy for tumor infiltration and antitumor activity of CAR T Cells in melanoma | CD8+ and CD4+ T ↑ | [153] | |
3.1. Promoting immunostimulatory effects to “fuel the engine”
Methods for the initiation of antitumor immunity including the antigen release, presentation and T cell priming/activation (step 1 to step 3, Figure 1) have been substantially studied (Figure 2). Several nanovaccines are currently investigated in clinical trials for certain solid tumors [88, 89]. Recently, biomimetic nanovaccines have been developed for overcoming the barriers involving traditional platforms, by means of improving the stability of antigens, targeted delivery, and long-term release [154-156]. The modification of NPs with peptides, proteins and antibodies has also been achieved to produce biomimetic nanovaccines with the enhanced potency, which may allow better reprogramming of immune responses [154-156]. The approaches of engineering biomimetic nanovaccines and their application in remodeling the TME for cancer immunotherapy have been extensively reviewed (see more details in [154-157]).
Development of nanovaccines for promoting immunostimulatory effects to “fuel the engine” A) LCP-based delivery of mRNA vaccine for an enhanced immune response against melanoma. Adapted with permission from [160], copyright 2017 Elsevier. B) Albumin-mediated enhanced CAR-T cell activity for solid tumors. Adapted with permission from [150], copyright 2019 American Association for the Advancement of Science C) Cancer cell membrane-coated adjuvant NPs with mannose modification for anticancer vaccination. Adapted with permission from [144], copyright 2018 American Chemical Society. D) Erythrocyte membrane-coated NPs as vaccine for antitumor immunity against melanoma. Adapted with permission from [143], copyright 2015 American Chemical Society.
Nanomaterials alone or when formulated with antigens in a form as DNA, RNA or peptides can be designed for delivery into APCs in the lymph nodes, which boost T cell priming and activation for antitumor immunity [158, 159]. Recently, Wang et al. have developed a mannose-targeted PEGylated lipid-coated calcium phosphate (LCP) NP for co-delivery of mRNA (encoding tyrosinase-related protein 2 (TRP2), a melanoma-associated antigen) and siRNA (targeting PD-L1 mRNA) to DCs in the lymph nodes [160]. The LCP-mediated expression of TRP2 in DCs elicited a robust antigen-specific CTL response as well as the production of serum immunoglobulin G against the full-length TRP2 protein in mice with melanoma [160]. In addition, the PD-L1 expression in DCs was significantly downregulated by LCP-mediated siRNA, resulting in enhancement of T cell activation and proliferation. Consequently, this LCP nanovaccine remarkably inhibited tumor growth and metastasis [160].
It has been recently reported that stimulator of interferon genes (STING, a signaling molecule) plays a significant role in the regulation of intracellular DNA-mediated IFN-dependent innate immunity [161], demonstrating the potential of STING-mediated cancer immunotherapy. The details of molecular pathways associated with STING, STING agonists/inhibitors, and how to activate STING using nanomaterial-based strategies for cancer immunotherapy have been substantially summarized in [162, 163]. Recently, Luo et al. demonstrated a nanovaccine by physical mixture of an antigen and a synthetic polymeric NP (termed PC7A NP) [164]. In this study, the delivery of tumor antigens to APCs in the draining lymph nodes was achieved using PC7A NP, resulting in the surface presentation while simultaneously activating STING-dependent type I interferon-stimulated genes [164]. As a result, this nanovaccine significantly inhibited the tumor growth in mice with melanoma, colon cancer, and human papilloma virus-E6/E7 cancer [164]. In addition, cyclic dinucleotide (CDN) agonists of STING have demonstrated a promising role in the activation of tumor immunogenicity [165]. However, the therapeutic efficacy of CDNs, due to the hydrophilicity, negative charge and sensitivity to enzymatic degradation, is limited by in vivo delivery barriers. Therefore, Shae and co-workers developed a polymeric NP (polymersome) for enhanced intracellular delivery of 2'3' cyclic guanosine monophosphate-adenosine monophosphate (cGAMP, the endogenous ligand for STING) [166]. The resultant formulation (termed STING-NPs) significantly increased the cytosolic activity of cGAMP, promoted the STING signaling in the TME and sentinel lymph nodes, and turned immunosuppressive tumors into immunogenic [166]. Consequently, the therapeutic outcomes including the suppression of tumor growth, long-term survival, and induction of immunological memory were successfully achieved by STING-NPs in mice with melanoma [166].
In addition to design of nanovaccines for delivery into APCs in lymph nodes, NPs containing certain therapeutic agents may convert cancer cells into their own vaccine. When tumor cells undergo immunogenic cell death (ICD, also known as immunogenic apoptosis), the DAMPs released by dying tumor cells, which mainly include the exposure of calreticulin (CRT) onto cell surface, secretion of adenosine triphosphate (ATP), and release of high mobility group protein B1 (HMGB1), will activate DCs [167]. Consequently, ICD makes the dying cancer cells operate as a vaccine that can trigger a tumor-specific immune response [167]. ICD can be induced by certain chemotherapeutic drugs (e.g. anthracyclines, mitoxantrone, oxaliplatin, and bortezomib) [168], physical treatments (e.g. UV irradiation and photodynamic therapy) [169], and oncolytic viruses [170]. In addition, the details of ICD-associated signaling pathways, the ICD inducers, and NP-based ICD-mediated cancer immune therapy have been extensively described in [168, 171, 172]. Recently, Liu and co-workers have developed an amino ethylanisamide (AEAA, targeting Sigma-1 receptors overexpressed on cancers [173])-targeted PEGylated polymeric NP for co-delivery of mitoxantrone (the ICD inducer) and celastrol (a pentacyclic triterpene extracted from Tripterygium wilfordii) in mice with desmoplastic melanoma. Consequently, the resultant formulation containing two agents at the optimal ratio significantly induced ICD-mediated immunotherapeutic effects, reprogram the fibrotic and immunosuppressive TME, and promote the progression-free survival and sustained immunosurveillance in diseased mice [174].
3.2. Overcoming immunosuppressive barriers to “release the brake”
The efficacy of antitumor immunity including the trafficking/infiltration of T cells, recognition of tumor cells by T cells and killing of tumor cells (step 4 to step 7, Figure 1) is significantly dampened by the immunosuppressive TME. Therefore, approaches used to overcome such immune tolerance have been extensively investigated (Figure 3). Recent advances in nanoengineered strategies for delivery of checkpoint inhibitors have been reviewed [175]. These NP-based approaches enable the selective delivery of checkpoint inhibitors into tumors, which can reduce immune-related toxic issues. Consequently, they significantly reprogram immunosuppressive cells and improve the activity and persistence of effectors T cells. In addition, a number of NP-based delivery approaches have been recently developed for delivery of therapeutic components (e.g. chemotherapeutics, antibody and siRNA) to target immunosuppressive soluble mediators such as TGF‐β, IDO, COX‐2 and epidermal growth factor receptor (EGFR), which can significantly remodel the suppressive TME and restore the antitumor effects with reduced systemic toxicity (see review in [175]).
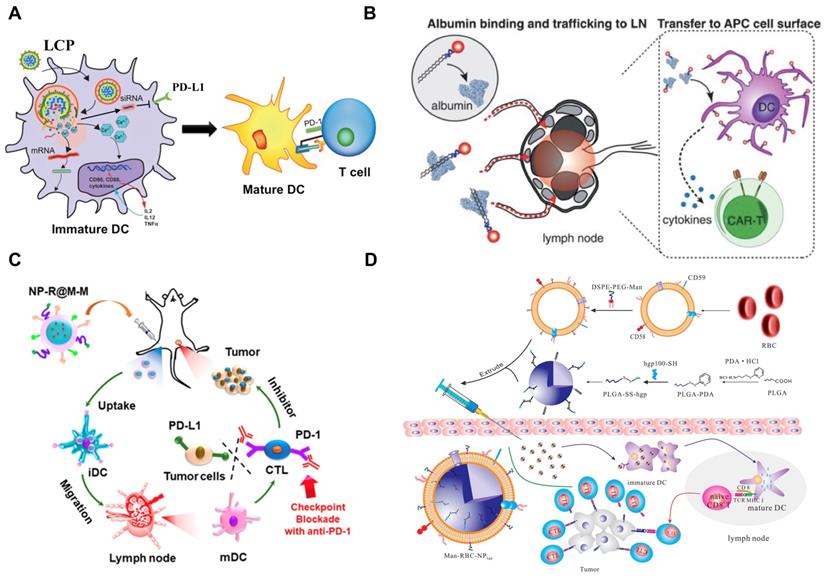
Development of nanoimmunotherapeutics for overcoming immunosuppressive barriers to “releasing the brake”. A) Local blockade of IL-10 and CXCL 12 using LPD for antitumor response for pancreatic cancer. Adapted with permission from [121], copyright 2018 American Chemical Society. B) Inhibiting PI3 kinase-γ using AEAA-targeted PLGA in both myeloid and plasma cells to remodel the suppressive TME in pancreatic cancer. Adapted with permission from [173], copyright 2019 Elsevier. C) Liposome-mediated delivery of vasodilator hydralazine for nanoparticle penetration in advanced desmoplastic melanoma. Adapted with permission from [125], copyright 2019 American Chemical Society. D) Immunotherapeutic strategy for melanoma via dual-targeting NPs delivering siRNA to TAMs. Adapted with permission from [176], copyright 2017 American Chemical Society.
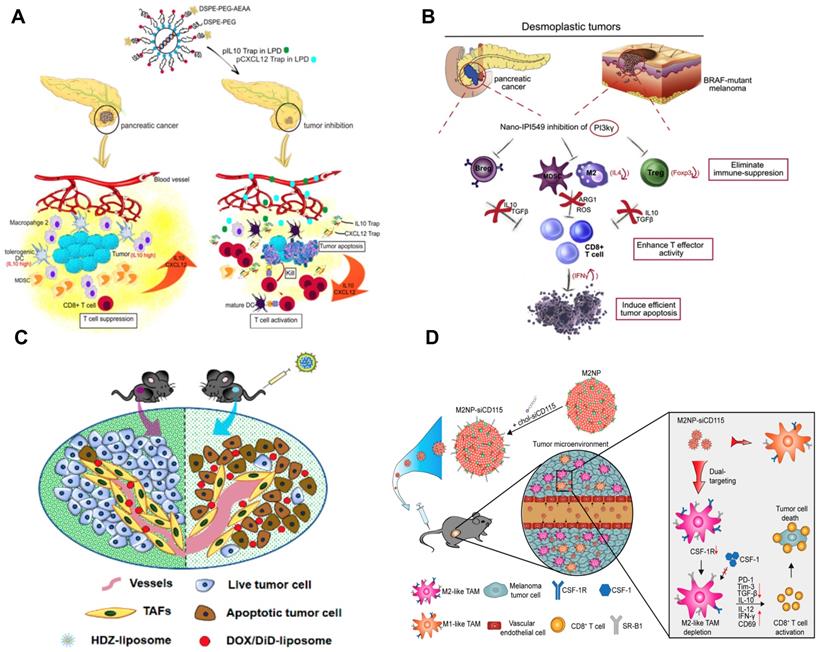
TAMs have recently become a promising therapeutic target; however, it is still challenging to deliver therapeutic agents to them. Recently, a liposomal NP has been developed with the modification of α-peptide (a scavenger receptor B type 1 (SR-B1) targeting peptide) and M2pep (an M2 macrophage binding peptide) [176]. Following intravenous (i.v.) injection this dual-targeted NP demonstrated higher binding affinity to M2-like TAMs than to tissue-resident macrophages in healthy tissues. As a result, the inhibition of survival signals in M2-like macrophages as well as the depletion of this cell type from melanoma were achieved using this dual-targeted NP containing siRNA against colony stimulating factor-1 receptor (CSF-1R), which was observed along with the increase of immunogenic cytokines (IL-12 and IFN-γ) and reduction of immunosuppressive cytokines (IL-10 and TGF-β) [176]. In addition, Rodell et al. developed a β-cyclodextrin NP (CDNP) for delivery of R848 (an agonist of TLR7 and TLR8) in a range of tumor models in mice [177]. As a result, CDNP-R848 significantly altered the TAMs toward the M1 phenotype, which slowed down tumor growth and protected mice against tumor rechallenge [177]. More importantly, improved antitumor immune responses were achieved by CDNP-R848 when applied in combination with anti-PD-1 therapy, confirming the potential of NP-based strategies to effectively remodel TAMs for cancer immunotherapy [177].
Stromal cells as one of the key cellular components in the TME usually facilitate the development and maintenance of tumors by supporting tumor cells, remodeling ECM, and promoting angiogenesis [81]. It has been reported that the development of liver metastasis is often associated with activated hepatic stellate cell (aHSC)-mediated liver fibrosis, and the relaxin (RLN, an anti-fibrotic peptide) can deactivate aHSCs and therefore resolve liver fibrosis [122]. Therefore, an AEAA-targeted PEGylated LCP NP containing the RLN plasmid was developed by Hu and co-workers to target cancer cells and aHSCs within the metastatic lesion and use them as an in situ factory for the production of RLN protein. Consequently, the stromal microenvironment in liver metastases was effectively reversed by LCP-mediated expression of RLN protein, which significantly inhibited metastatic progression and prolonged the survival of animals, accompanied with the upregulation of immunogenic cells/cytokines and downregulation of immunosuppressive counterparts [122].
Although NPs may take advantage of the enhanced permeability and retention (EPR) effect for tumor accumulation [178], the elevated interstitial fluid pressure, high density of ECM and disorganized blood vessels (particularly in desmoplastic tumors) cause significant hurdles for particle penetration. To address these issues, Chen and colleagues developed a hydralazine (HDZ, a routine medication used to treat high blood pressure and heart failure)-containing liposomal NP to reshape tumor blood vasculature in advanced desmoplastic melanoma [125]. The i.v. injection of HDZ-liposome favorably modulated the vascular dilation, tumor hypoxia, and tumor permeability, which were accompanied with the TME modulation (Figure 3). As a result, the HDZ-liposome significantly improved the therapeutic efficacy of liposomal doxorubicin as the second-wave treatment in mice with tumor size over 400 mm3 [125]. In addition, it has been reported that high concentration of perivascular nitric oxide (NO) can facilitate tumor vascular normalization and further the chemotherapy efficacy [130]. Despite the promising anticancer effect, the clinical application of NO is limited by the short half-life, low bioavailability, and poor tumor targeting behavior [130]. Recently, Sung et al. have developed a poly(lactic-co-glycolic acid) (PLGA)-based delivery system (NanoNO) containing dinitrosyl iron complex (DNIC, the NO donor) [130]. In murine hepatocellular carcinoma model, NanoNO was able to provide sustained NO release into tumors, which resulted in effective normalization of tumor vasculature and improve the delivery of follow-up chemotherapy for the suppression of primary tumors and metastases [130]. Immunological analyses revealed that NanoNO at a lower dose could reprogram the immunosuppressive TME therefore improving the anticancer efficacy [130].
3.3. The combination therapy
Schemes that simultaneously target stimulatory and inhibitory mechanisms potentially provide synergistic antitumor immunotherapeutic effectiveness (Figure 4). It has been reported that the blockage of immune checkpoint molecules using systemically administrated mAbs may reverse the immune tolerance, but autoimmune-like side effects are unavoidable for healthy tissues or organs [179]. Alternatively, local delivery of immune checkpoint inhibitors in the TME may alleviate the immune-related adverse effects (irAEs). Therefore, Song and colleagues developed an AEAA-targeted lipid-protamine-DNA (LPD) NP for delivery of plasmid encoded with PD-L1 trap (a small antibody-like fusion protein targeting PD-L1) in mice with colorectal cancer. Consequently, the expression of PD-L1 trap by LPD in tumors led to a synergistic chemo-immunotherapeutic outcome in combination with oxaliplatin (OxP)-mediated ICD effects, resulting in longer animal survival time and lower level of irAEs, in comparison with free PD-L1 mAb and OxP [119].
Development of nanoimmunotherapeutics for combination therapy. A) NP-mediated co-delivery of mitoxantrone (MIT) and celastrol (CEL) to induce chemo-immunotherapy for cancer inhibition and tumor dormancy in desmoplastic melanoma. Adapted with permission from [174], copyright 2018 American Chemical Society. B) NP-based co-delivery of Quercetin (Q) and Alantolactone (A) for antitumor responses through synergistic ICD. Adapted with permission from [131], copyright 2019 American Chemical Society. C) Synergistic and low adverse effect cancer immunotherapy by LPD-mediated immunogenic chemotherapy and locally expressed PD-L1 trap in combination with oxaliplatin for colorectal cancer. Adapted with permission from [119], copyright 2018 Nature Publishing Group. D) LCP-mediated relaxin gene delivery for synergistic effect with checkpoint inhibition in liver metastasis. Adapted with permission from [122], copyright 2019 Nature Publishing Group.
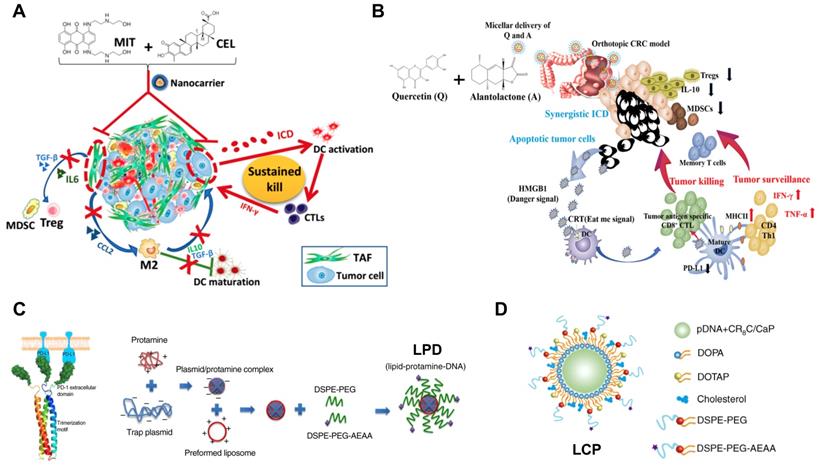
It is known that IDO‐1 is one of tryptophan catabolic enzymes that can facilitate the conversion of tryptophan (Trp) to kynurenine (Kyn) [180]. The downregulation of Trp can suppress the proliferation and activity of CTLs and NKs, and the upregulation of Kyn can activate Tregs and MDSCs [181]. Therefore, approaches against IDO-1 hold great promises for tumor immunotherapy. Indeed, the combination immunotherapy has been achieved using the co-delivery of IDO-1 inhibitor and immune checkpoint inhibitor [182]. In addition, it has been reported that IFN-γ released by ICD-mediated CTLs can positively regulate tumor immunogenicity, but may also cause the production of IDO-1, which dampen the immunotherapeutic efficacy [183]. To address such paradox, Feng et al. developed an amphiphilic polymeric NP for co-delivery of OxP prodrug and NLG919 (an IDO-1 inhibitor) to induce OxP-mediated ICD effects and reverse IDO-1 mediated immunosuppression, respectively [133]. Consequently, the resultant nanoformulation (BCPN) could achieve significantly better tumor inhibition at primary and metastatic sites than the combination of free OxP and NLG919 [133].
In addition, a light-sensitive in situ gelation system was reported by Meng et al. for the combination of photodynamic therapy and immunotherapy [184]. In this study, the photosensitizer (Chlorin e6, Ce6) modified-catalase (CAT, an enzyme triggers the rapid decomposition of H2O2) was conjugated with poly(ethylene glycol) double acrylate (PEGDA) to form Ce6-CAT-PEGDA. Subsequently, the Ce6-CAT-PEGDA was mixed with imiquimod (R837)-loaded PLGA NPs (RPNPs, the immune adjuvant), forming a polymeric matrix (Ce6-CAT-PEGDA-RPNPs) [184]. When locally applied to tumors and exposed under 660 nm red light, Ce6-CAT-PEGDA-RPNPs significantly reversed the immunosuppressive TME by the production of O2 that can relieve the tumor hypoxia [184]. Consequently, the photodynamic therapy-mediated ICD together with immune adjuvant could mediate a significantly stronger “abscopal effect” for tumor inhibition at primary and distant sites [184].
4. Conclusions and Future Perspectives
In recent years, an improved understanding of cancer biology [185] and the discovery of cellular and molecular mechanisms for innate and adaptive immunologic responses [186] have significantly revolutionized the fields of cancer immunology and immunotherapy. These have remarkably encouraged researchers to investigate the possibility of restoring the cancer-immunity cycle using nanomaterial-based immunotherapeutics (nanoimmunotherapeutics) [187-189]. Several studies of NP-based cancer immunotherapy are currently undertaken in clinical trials (see the summaries in [88, 89]). Despite the potential of nanoimmunotherapeutics for solid tumors [89], none of them have reached the clinic for patients. One major reason for the lack of clinical translation is the presence of the immunosuppressive TME. As shown in Table 2, substantial studies have been undertaken for investigating the capacity and availability of NP-based delivery of immunoregulatory agents to systemically and locally modulate the suppressive milieu within the TME. These works provide proof of concept for NP-based TME-modulating methods and illustrate the potential of nanoimmunotherapeutics to advance the “fuel the engine, release the brake” rules (see reviews in [175, 190-192]).
In addition, one of the major remaining challenges associated with clinical translation of nanoimmunotherapeutics (nanomedicine as well) is still the lack of efficient, safe and widely applied delivery strategies to facilitate the transport of therapeutic agents to tumor sites following systemic administration [193]. Although NPs may accumulate into tumors following the EPR effect, the delivery efficacy is extremely low [194]. In addition, a large number of intratumoral NPs may be either isolated by the ECM or taken up by non-specific cells. The high density of ECM and tortuous blood vessels (particularly in desmoplastic tumors) may be overcome by NPs containing a variety of TME modulators [122, 125, 130, 195, 196], which relieve the harsh niches associated with the failure of drug delivery and enhance the follow-up treatment of targeted nanoimmunotherapeutics that act specifically in cells of interest.
In addition, it should be borne in mind that complicated modifications of nanomaterials, which is hoped to achieve multifunctional delivery formulations with stabilizing groups, targeting ligands and bioresponsive linkers, may complicate the large-scale and reproducible production. In addition, such extensive modifications may also cause unexpected toxicity. Therefore, further investigation must be performed to keep balance between the therapeutic benefit, the complexity of formulation preparation/scale-up and the risk of toxicity before nanoimmunotherapeutics can be satisfactorily applied for cancer patients.
Abbreviations
ADCC: antibody-dependent cell-mediated cytotoxicity; AEAA: amino ethylanisamide; aHSC: Activated hepatic stellate cell; APC: antigen-presenting cell; ATP: adenosine triphosphate; BAFF: B-cell activating factor; BCR: B-cell receptor; bFGF: basic fibroblast growth factor; Breg: regulatory B cell; CAR: chimetic antigen receptors; CCL2: C-C motif chemokine ligand 2; CDC: complement-dependent cytotoxicity; CDN: cyclic dinucleotide; CD40L: CD40 ligand; PD-1: programmed cell death protein 1; Ce6: chlorin e6; CFSE: carboxyfluorescein succinimidyl ester; cGAMP: 2'3' cyclic guanosine monophosphate-adenosine monophosphate; COX-2: cyclooxygenase type 2; CRT: calreticulin; CSF-1R: colony stimulating factor-1 receptor; CTL: cytotoxic T lymphocyte; CTLA-4: cytotoxic T lymphocyte-associated protein 4; CXCL9: C-X-C motif chemokine ligand 9; CXCL10: C-X-C motif chemokine ligand 10; DAMP: damage-associated molecular pattern; DC: dendritic cell; DNIC: dinitrosyl iron complex; ECM: extracellular matrix; EGF: epidermal growth factor; ELISA: enzyme-linked immunosorbent assay; ELISPOT: enzyme-linked immune absorbent spot; EMT: epithelial-mesenchymal transition; EPR: enhanced permeability and retention; FASL: Fas ligand; FOB: follicular B; Foxp3: forkhead box protein 3; GSEA: gene set enrichment analysis; HDZ: hydralazine; HMGB1: high mobility group protein B1; ICAM-1: Intercellular adhesion molecule 1; ICD: immunogenic cell death; IDO-1: indoleamine 2,3-dioxygenase 1; IF: immunofluorescent; IFC: imaging flow cytometry; IFN-γ: interferon gamma; IHC: immunohistochemical; IL: interleukin; irAE: immune-related adverse effect; Kyn: kynurenine; LCP: lipid-coated calcium phosphate; LFA-1: lymphocyte function associated antigen 1; LPD: lipid-protamine-DNA; LPS: lipopolysaccharide; M-CSF: macrophage colony stimulating factor; MDSC: macrophages and myeloid-derived suppressor cell; MHC-I: major histocompatibility class I; MHC-II: major histocompatibility class II; M-MDSC: monocytic-myeloid derived suppressor cell; MRI: magnetic resonance imaging; MSIHC: mass spectrometry IHC; M1: macrophage type 1; M2: macrophage type 2; NK: natural killer; NO: nitric oxide; NP: nanoparticle; OxP: oxaliplatin; PDGF: platelet-derived growth factor; PD-L1: programmed death-ligand 1; PET: positron emission tomography; PLGA: poly (lactic-co-glycolic acid); PMN-MDSC: polymorphonuclear-myeloid derived suppressor cell; PSGL-1: p-selectin glycoprotein ligand 1; QIF: quantitative immunofluorescence; qPCR: quantitative polymerase chain reaction; RLN: relaxin; scRNA-sq: single-cell RNA sequencing; SR-B1: scavenger receptor B type 1; STAT3: signal transducer and activator of transcription 3; STING: stimulator of interferon gene; TAA: tumor associated antigen; TAF: tumor-associated fibroblast; TAM: tumor associated macrophages; TCR: T-cell receptor; Th: T helper; TGF-β: transforming growth factor beta; TME: tumor microenvironment; TNF-α: tumor necrosis factor alpha; TLR: Toll-like receptor; TLS: tertiary lymphoid structure; Treg: regulatory T; TRP2: tyrosinase-related protein 2; Trp: tryptophan; VCAM-1: vascular cell adhesion molecule 1; VEGF: vascular endothelial growth factor; VLA-4: very late antigen 4.
Acknowledgements
We acknowledge the Carolina Center for Cancer Nanotechnology Excellence (NIH grant CA198999) for the support in funding our research. This work is also supported by “Talents Cultivation Program” of Jilin University.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1-10
2. Weiner LM, Surana R, Wang SZ. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 2010;10:317-27
3. Sahin U, Tureci O. Personalized vaccines for cancer immunotherapy. Science. 2018;359:1355-60
4. Xu X, Li T, Shen SY, Wang JQ, Abdou P, Gu Z. et al. Advances in Engineering Cells for Cancer Immunotherapy. Theranostics. 2019;9:7889-905
5. Curran MA, Glisson BS. New Hope for Therapeutic Cancer Vaccines in the Era of Immune Checkpoint Modulation. Annu Rev Med. 2019;70:409-24
6. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M. et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24:541-50
7. D'Aloia MM, Zizzari IG, Sacchetti B, Pierelli L, Alimandi M. CAR-T cells: the long and winding road to solid tumors. Cell Death Dis. 2018;9:282
8. O'Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. 2019;16:151-67
9. Fridman WH, Zitvogel L, Sautes-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. 2017;14:717-34
10. Song W, Musetti SN, Huang L. Nanomaterials for cancer immunotherapy. Biomaterials. 2017;148:16-30
11. Belli C, Trapani D, Viale G, D'Amico P, Duso BA, Della Vigna P. et al. Targeting the microenvironment in solid tumors. Cancer Treat Rev. 2018;65:22-32
12. Hinshaw DC, Shevde LA. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019;79:4557-66
13. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309-22
14. Mellman I. Dendritic cells: master regulators of the immune response. Cancer Immunol Res. 2013;1:145-9
15. Zhang N, Bevan MJ. CD8(+) T cells: foot soldiers of the immune system. Immunity. 2011;35:161-8
16. Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood. 2008;112:1557-69
17. Drobek A, Moudra A, Mueller D, Huranova M, Horkova V, Pribikova M. et al. Strong homeostatic TCR signals induce formation of self-tolerant virtual memory CD8 T cells. EMBO J. 2018;37:e98518
18. Cariappa A, Boboila C, Moran ST, Liu H, Shi HN, Pillai S. The recirculating B cell pool contains two functionally distinct, long-lived, posttransitional, follicular B cell populations. J Immunol. 2007;179:2270-81
19. Robillard N, Wuilleme S, Moreau P, Bene MC. Immunophenotype of normal and myelomatous plasma-cell subsets. Front Immunol. 2014;5:137
20. Kaminski DA, Wei C, Qian Y, Rosenberg AF, Sanz I. Advances in human B cell phenotypic profiling. Front Immunol. 2012;3:302
21. Veluchamy JP, Delso-Vallejo M, Kok N, Bohme F, Seggewiss-Bernhardt R, van der Vliet HJ. et al. Standardized and flexible eight colour flow cytometry panels harmonized between different laboratories to study human NK cell phenotype and function. Sci Rep. 2017;7:43873
22. Paul S, Lal G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front Immunol. 2017;8:1124
23. Wang YC, He F, Feng F, Liu XW, Dong GY, Qin HY. et al. Notch signaling determines the M1 versus M2 polarization of macrophages in antitumor immune responses. Cancer Res. 2010;70:4840-9
24. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. 2018;19:108-19
25. Kondelkova K, Vokurkova D, Krejsek J, Borska L, Fiala Z, Ctirad A. Regulatory T cells (TREG) and their roles in immune system with respect to immunopathological disorders. Acta Medica (Hradec Kralove). 2010;53:73-7
26. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG. et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501
27. LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood. 2008;112:1570-80
28. Schumacher TN, Scheper W, Kvistborg P. Cancer Neoantigens. Annu Rev Immunol. 2019;37:173-200
29. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20:7-24
30. Dudek AM, Martin S, Garg AD, Agostinis P. Immature, Semi-Mature, and Fully Mature Dendritic Cells: Toward a DC-Cancer Cells Interface That Augments Anticancer Immunity. Front Immunol. 2013;4:438
31. Slaney CY, Kershaw MH, Darcy PK. Trafficking of T cells into tumors. Cancer Res. 2014;74:7168-74
32. Farhood B, Najafi M, Mortezaee K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: A review. J Cell Physiol. 2019;234:8509-21
33. Kim HJ, Cantor H. CD4 T-cell subsets and tumor immunity: the helpful and the not-so-helpful. Cancer Immunol Res. 2014;2:91-8
34. Kennedy R, Celis E. Multiple roles for CD4(+) T cells in anti-tumor immune responses. Immunol Rev. 2008;222:129-44
35. Asadzadeh Z, Mohammadi H, Safarzadeh E, Hemmatzadeh M, Mahdian-Shakib A, Jadidi-Niaragh F. et al. The paradox of Th17 cell functions in tumor immunity. Cell Immunol. 2017;322:15-25
36. Lu Y, Wang Q, Xue G, Bi E, Ma X, Wang A. et al. Th9 Cells Represent a Unique Subset of CD4(+) T Cells Endowed with the Ability to Eradicate Advanced Tumors. Cancer Cell. 2018;33:1048-60 e7
37. Tsou P, Katayama H, Ostrin EJ, Hanash SM. The Emerging Role of B Cells in Tumor Immunity. Cancer Res. 2016;76:5597-601
38. Guo FF, Cui JW. The Role of Tumor-Infiltrating B Cells in Tumor Immunity. J Oncol. 2019;2019:2592419
39. Nelson BH. CD20+ B cells: the other tumor-infiltrating lymphocytes. J Immunol. 2010;185:4977-82
40. Sarvaria A, Madrigal JA, Saudemont A. B cell regulation in cancer and anti-tumor immunity. Cell Mol Immunol. 2017;14:662-74
41. Sautes-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer. 2019;19:307-25
42. Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol. 2016;17:1025-36
43. Habif G, Crinier A, Andre P, Vivier E, Narni-Mancinelli E. Targeting natural killer cells in solid tumors. Cell Mol Immunol. 2019;16:415-22
44. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9:503-10
45. Bi J, Tian Z. NK Cell Dysfunction and Checkpoint Immunotherapy. Front Immunol. 2019;10:1999
46. Pahl J, Cerwenka A. Tricking the balance: NK cells in anti-cancer immunity. Immunobiology. 2017;222:11-20
47. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14:399-416
48. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889-96
49. Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J. et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci U S A. 2012;109:17561-6
50. Gabrilovich DI. Myeloid-Derived Suppressor Cells. Cancer Immunol Res. 2017;5:3-8
51. Kruger P, Saffarzadeh M, Weber AN, Rieber N, Radsak M, von Bernuth H. et al. Neutrophils: Between host defence, immune modulation, and tissue injury. PLoS Pathog. 2015;11:e1004651
52. Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res. 2014;2014:149185
53. Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011;32:19-25
54. Mandruzzato S, Brandau S, Britten CM, Bronte V, Damuzzo V, Gouttefangeas C. et al. Toward harmonized phenotyping of human myeloid-derived suppressor cells by flow cytometry: results from an interim study. Cancer Immunol Immunother. 2016;65:161-9
55. Condamine T, Dominguez GA, Youn JI, Kossenkov AV, Mony S, Alicea-Torres K. et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol. 2016;1:aaf8943
56. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253-68
57. Granot Z. Neutrophils as a Therapeutic Target in Cancer. Front Immunol. 2019;10:1710
58. Pawelec G, Verschoor CP, Ostrand-Rosenberg S. Myeloid-Derived Suppressor Cells: Not Only in Tumor Immunity. Front Immunol. 2019;10:1099
59. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016;37:208-20
60. Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest. 2015;125:3356-64
61. Ugel S, De Sanctis F, Mandruzzato S, Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest. 2015;125:3365-76
62. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49-61
63. Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013;34:137-43
64. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S. et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14-20
65. Riabov V, Gudima A, Wang N, Mickley A, Orekhov A, Kzhyshkowska J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front Physiol. 2014;5:75
66. Mantovani A, Allavena P. The interaction of anticancer therapies with tumor-associated macrophages. J Exp Med. 2015;212:435-45
67. Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295-307
68. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4(+)CD25(+) regulatory T cells. Nat Immunol. 2003;4:330-6
69. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27:109-18
70. Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer. 2010;127:759-67
71. Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y. et al. An essential role for the IL-2 receptor in Treg cell function. Nat Immunol. 2016;17:1322-33
72. Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N. et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192:303-10
73. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM. et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332:600-3
74. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR. et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity. 2007;27:635-46
75. Han S, Toker A, Liu ZQ, Ohashi PS. Turning the Tide Against Regulatory T Cells. Front Oncol. 2019;9:279
76. Rosser EC, Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity. 2015;42:607-12
77. Khan AR, Hams E, Floudas A, Sparwasser T, Weaver CT, Fallon PG. PD-L1hi B cells are critical regulators of humoral immunity. Nat Commun. 2015;6:5997
78. Peng B, Ming Y, Yang C. Regulatory B cells: the cutting edge of immune tolerance in kidney transplantation. Cell Death Dis. 2018;9:109
79. Jensen-Jarolim E, Fazekas J, Singer J, Hofstetter G, Oida K, Matsuda H. et al. Crosstalk of carcinoembryonic antigen and transforming growth factor-beta via their receptors: comparing human and canine cancer. Cancer Immunol Immunother. 2015;64:531-7
80. Kessel A, Haj T, Peri R, Snir A, Melamed D, Sabo E. et al. Human CD19(+)CD25(high) B regulatory cells suppress proliferation of CD4(+) T cells and enhance Foxp3 and CTLA-4 expression in T-regulatory cells. Autoimmun Rev. 2012;11:670-7
81. Turley SJ, Cremasco V, Astarita JL. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat Rev Immunol. 2015;15:669-82
82. Kobayashi H, Enomoto A, Woods SL, Burt AD, Takahashi M, Worthley DL. Cancer-associated fibroblasts in gastrointestinal cancer. Nat Rev Gastro Hepat. 2019;16:282-95
83. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16:582-98
84. Ishii G, Ochiai A, Neri S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv Drug Deliver Rev. 2016;99:186-96
85. Ziani L, Chouaib S, Thiery J. Alteration of the Antitumor Immune Response by Cancer-Associated Fibroblasts. Front Immunol. 2018;9:414
86. De Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer. 2017;17:457-74
87. Schaaf MB, Garg AD, Agostinis P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis. 2018;9:115
88. Gao S, Yang D, Fang Y, Lin X, Jin X, Wang Q. et al. Engineering Nanoparticles for Targeted Remodeling of the Tumor Microenvironment to Improve Cancer Immunotherapy. Theranostics. 2019;9:126-51
89. Saeed M, Gao J, Shi Y, Lammers T, Yu H. Engineering Nanoparticles to Reprogram the Tumor Immune Microenvironment for Improved Cancer Immunotherapy. Theranostics. 2019;9:7981-8000
90. Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235-71
91. Paiva AE, Lousado L, Guerra DAP, Azevedo PO, Sena IFG, Andreotti JP. et al. Pericytes in the Premetastatic Niche. Cancer Res. 2018;78:2779-86
92. Quail DF, Dannenberg AJ. The obese adipose tissue microenvironment in cancer development and progression. Nat Rev Endocrinol. 2019;15:139-54
93. Matos LL, Trufelli DC, de Matos MG, da Silva Pinhal MA. Immunohistochemistry as an important tool in biomarkers detection and clinical practice. Biomark Insights. 2010;5:9-20
94. Lin JR, Izar B, Wang S, Yapp C, Mei S, Shah PM. et al. Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes. Elife. 2018;7:e31657
95. Adan A, Alizada G, Kiraz Y, Baran Y, Nalbant A. Flow cytometry: basic principles and applications. Crit Rev Biotechnol. 2017;37:163-76
96. Kim SW, Roh J, Park CS. Immunohistochemistry for Pathologists: Protocols, Pitfalls, and Tips. J Pathol Transl Med. 2016;50:411-8
97. Rimm DL. Next-gen immunohistochemistry. Nat Methods. 2014;11:381-3
98. Gerdes MJ, Sevinsky CJ, Sood A, Adak S, Bello MO, Bordwell A. et al. Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc Natl Acad Sci U S A. 2013;110:11982-7
99. Han Y, Gu Y, Zhang AC, Lo YH. Review: imaging technologies for flow cytometry. Lab Chip. 2016;16:4639-47
100. Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM. et al. Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell. 2017;168:487-502
101. Mukherjee S, Sonanini D, Maurer A, Daldrup-Link HE. The yin and yang of imaging tumor associated macrophages with PET and MRI. Theranostics. 2019;9:7730-48
102. Hagen JB. The origins of bioinformatics. Nat Rev Genet. 2000;1:231-6
103. Ding L, Wendl MC, McMichael JF, Raphael BJ. Expanding the computational toolbox for mining cancer genomes. Nat Rev Genet. 2014;15:556-70
104. Rodriguez R, Miller KM. Unravelling the genomic targets of small molecules using high-throughput sequencing. Nat Rev Genet. 2014;15:783-96
105. Hackl H, Charoentong P, Finotello F, Trajanoski Z. Computational genomics tools for dissecting tumour-immune cell interactions. Nat Rev Genet. 2016;17:441-58
106. Finotello F, Rieder D, Hackl H, Trajanoski Z. Next-generation computational tools for interrogating cancer immunity. Nat Rev Genet. 2019;20:724-46
107. Hwang B, Lee JH, Bang D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp Mol Med. 2018;50:96
108. AlJanahi AA, Danielsen M, Dunbar CE. An Introduction to the Analysis of Single-Cell RNA-Sequencing Data. Mol Ther Methods Clin Dev. 2018;10:189-96
109. Shalek AK, Satija R, Adiconis X, Gertner RS, Gaublomme JT, Raychowdhury R. et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature. 2013;498:236-40
110. Szabo PA, Levitin HM, Miron M, Snyder ME, Senda T, Yuan J. et al. Single-cell transcriptomics of human T cells reveals tissue and activation signatures in health and disease. Nat Commun. 2019;10:4706
111. Chen YP, Zhang Y, Lv JW, Li YQ, Wang YQ, He QM. et al. Genomic Analysis of Tumor Microenvironment Immune Types across 14 Solid Cancer Types: Immunotherapeutic Implications. Theranostics. 2017;7:3585-94
112. Chiswick EL, Duffy E, Japp B, Remick D. Detection and quantification of cytokines and other biomarkers. Methods Mol Biol. 2012;844:15-30
113. Janetzki S, Price L, Schroeder H, Britten CM, Welters MJ, Hoos A. Guidelines for the automated evaluation of Elispot assays. Nat Protoc. 2015;10:1098-115
114. Maino VC, Picker LJ. Identification of functional subsets by flow cytometry: intracellular detection of cytokine expression. Cytometry. 1998;34:207-15
115. Verma V, Shrimali RK, Ahmad S, Dai W, Wang H, Lu S. et al. PD-1 blockade in subprimed CD8 cells induces dysfunctional PD-1(+)CD38(hi) cells and anti-PD-1 resistance. Nat Immunol. 2019;20:1231-43
116. Tang L, Zheng Y, Melo MB, Mabardi L, Castano AP, Xie YQ. et al. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat Biotechnol. 2018;36:707-16
117. Goldberg MS. Improving cancer immunotherapy through nanotechnology. Nat Rev Cancer. 2019;19:587-602
118. Sang W, Zhang Z, Dai YL, Chen XY. Recent advances in nanomaterial-based synergistic combination cancer immunotherapy. Chem Soc Rev. 2019;48:3771-810
119. Song W, Shen L, Wang Y, Liu Q, Goodwin TJ, Li J. et al. Synergistic and low adverse effect cancer immunotherapy by immunogenic chemotherapy and locally expressed PD-L1 trap. Nat Commun. 2018;9:2237
120. Song W, Tiruthani K, Wang Y, Shen L, Hu M, Dorosheva O. et al. Trapping of Lipopolysaccharide to Promote Immunotherapy against Colorectal Cancer and Attenuate Liver Metastasis. Adv Mater. 2018;30:e1805007
121. Shen L, Li J, Liu Q, Song W, Zhang X, Tiruthani K. et al. Local Blockade of Interleukin 10 and C-X-C Motif Chemokine Ligand 12 with Nano-Delivery Promotes Antitumor Response in Murine Cancers. ACS Nano. 2018;12:9830-41
122. Hu M, Wang Y, Xu L, An S, Tang Y, Zhou X. et al. Relaxin gene delivery mitigates liver metastasis and synergizes with check point therapy. Nat Commun. 2019;10:2993
123. Goodwin TJ, Zhou Y, Musetti SN, Liu R, Huang L. Local and transient gene expression primes the liver to resist cancer metastasis. Sci Transl Med. 2016;8:364ra153
124. Liu Q, Zhu H, Liu Y, Musetti S, Huang L. BRAF peptide vaccine facilitates therapy of murine BRAF-mutant melanoma. Cancer Immunol Immunother. 2018;67:299-310
125. Chen Y, Song W, Shen L, Qiu N, Hu M, Liu Y. et al. Vasodilator Hydralazine Promotes Nanoparticle Penetration in Advanced Desmoplastic Tumors. ACS Nano. 2019;13:1751-63
126. Duan X, Chan C, Han W, Guo N, Weichselbaum RR, Lin W. Immunostimulatory nanomedicines synergize with checkpoint blockade immunotherapy to eradicate colorectal tumors. Nat Commun. 2019;10:1899
127. Luo Z, Wang C, Yi H, Li P, Pan H, Liu L. et al. Nanovaccine loaded with poly I:C and STAT3 siRNA robustly elicits anti-tumor immune responses through modulating tumor-associated dendritic cells in vivo. Biomaterials. 2015;38:50-60
128. Min Y, Roche KC, Tian S, Eblan MJ, McKinnon KP, Caster JM. et al. Antigen-capturing nanoparticles improve the abscopal effect and cancer immunotherapy. Nat Nanotechnol. 2017;12:877-82
129. Chen Q, Chen J, Yang Z, Xu J, Xu L, Liang C. et al. Nanoparticle-Enhanced Radiotherapy to Trigger Robust Cancer Immunotherapy. Adv Mater. 2019;31:e1802228
130. Sung YC, Jin PR, Chu LA, Hsu FF, Wang MR, Chang CC. et al. Delivery of nitric oxide with a nanocarrier promotes tumour vessel normalization and potentiates anti-cancer therapies. Nat Nanotechnol. 2019;14:1160-9
131. Zhang J, Shen L, Li X, Song W, Liu Y, Huang L. Nanoformulated Codelivery of Quercetin and Alantolactone Promotes an Antitumor Response through Synergistic Immunogenic Cell Death for Microsatellite-Stable Colorectal Cancer. ACS Nano. 2019;13:12511-24
132. Mi Y, Smith CC, Yang F, Qi Y, Roche KC, Serody JS. et al. A Dual Immunotherapy Nanoparticle Improves T-Cell Activation and Cancer Immunotherapy. Adv Mater. 2018;30:e1706098
133. Feng B, Zhou F, Hou B, Wang D, Wang T, Fu Y. et al. Binary Cooperative Prodrug Nanoparticles Improve Immunotherapy by Synergistically Modulating Immune Tumor Microenvironment. Adv Mater. 2018;30:e1803001
134. Lee C, Jose L, Shim K, An SSA, Jang S, Song JK. et al. Influenza mimetic protein-polymer nanoparticles as antigen delivery vehicles to dendritic cells for cancer immunotherapy. Nanoscale. 2019;11:13878-84
135. Ernsting MJ, Hoang B, Lohse I, Undzys E, Cao P, Do T. et al. Targeting of metastasis-promoting tumor-associated fibroblasts and modulation of pancreatic tumor-associated stroma with a carboxymethylcellulose-docetaxel nanoparticle. J Control Release. 2015;206:122-30
136. Ruan H, Hu Q, Wen D, Chen Q, Chen G, Lu Y. et al. A Dual-Bioresponsive Drug-Delivery Depot for Combination of Epigenetic Modulation and Immune Checkpoint Blockade. Adv Mater. 2019;31:e1806957
137. Chen Q, Wang C, Zhang X, Chen G, Hu Q, Li H. et al. In situ sprayed bioresponsive immunotherapeutic gel for post-surgical cancer treatment. Nat Nanotechnol. 2019;14:89-97
138. Yang G, Xu L, Chao Y, Xu J, Sun X, Wu Y. et al. Hollow MnO2 as a tumor-microenvironment-responsive biodegradable nano-platform for combination therapy favoring antitumor immune responses. Nat Commun. 2017;8:902
139. Cho NH, Cheong TC, Min JH, Wu JH, Lee SJ, Kim D. et al. A multifunctional core-shell nanoparticle for dendritic cell-based cancer immunotherapy. Nat Nanotechnol. 2011;6:675-82
140. Wang X, Li X, Ito A, Watanabe Y, Sogo Y, Tsuji NM. et al. Stimulation of In Vivo Antitumor Immunity with Hollow Mesoporous Silica Nanospheres. Angew Chem Int Ed Engl. 2016;55:1899-903
141. Yata T, Takahashi Y, Tan M, Nakatsuji H, Ohtsuki S, Murakami T. et al. DNA nanotechnology-based composite-type gold nanoparticle-immunostimulatory DNA hydrogel for tumor photothermal immunotherapy. Biomaterials. 2017;146:136-45
142. Han Q, Wang X, Jia X, Cai S, Liang W, Qin Y. et al. CpG loaded MoS2 nanosheets as multifunctional agents for photothermal enhanced cancer immunotherapy. Nanoscale. 2017;9:5927-34
143. Guo Y, Wang D, Song Q, Wu T, Zhuang X, Bao Y. et al. Erythrocyte Membrane-Enveloped Polymeric Nanoparticles as Nanovaccine for Induction of Antitumor Immunity against Melanoma. ACS Nano. 2015;9:6918-33
144. Yang R, Xu J, Xu L, Sun X, Chen Q, Zhao Y. et al. Cancer Cell Membrane-Coated Adjuvant Nanoparticles with Mannose Modification for Effective Anticancer Vaccination. ACS Nano. 2018;12:5121-9
145. Kroll AV, Fang RH, Jiang Y, Zhou J, Wei X, Yu CL. et al. Nanoparticulate Delivery of Cancer Cell Membrane Elicits Multiantigenic Antitumor Immunity. Adv Mater. 2017;29:1703969
146. Hu Q, Wu M, Fang C, Cheng C, Zhao M, Fang W. et al. Engineering nanoparticle-coated bacteria as oral DNA vaccines for cancer immunotherapy. Nano Lett. 2015;15:2732-9
147. Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A, Moon JJ. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat Mater. 2017;16:489-96
148. Kuai R, Yuan W, Son S, Nam J, Xu Y, Fan Y. et al. Elimination of established tumors with nanodisc-based combination chemoimmunotherapy. Sci Adv. 2018;4:eaao1736
149. Stephan MT, Moon JJ, Um SH, Bershteyn A, Irvine DJ. Therapeutic cell engineering with surface-conjugated synthetic nanoparticles. Nat Med. 2010;16:1035-41
150. Ma L, Dichwalkar T, Chang JYH, Cossette B, Garafola D, Zhang AQ. et al. Enhanced CAR-T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science. 2019;365:162-8
151. Stephan MT, Stephan SB, Bak P, Chen J, Irvine DJ. Synapse-directed delivery of immunomodulators using T-cell-conjugated nanoparticles. Biomaterials. 2012;33:5776-87
152. Wang C, Sun WJ, Ye YQ, Hu QY, Bomba HN, Gu Z. In situ activation of platelets with checkpoint inhibitors for post-surgical cancer immunotherapy. Nat Biomed Eng. 2017;1:0011
153. Chen Q, Hu Q, Dukhovlinova E, Chen G, Ahn S, Wang C. et al. Photothermal Therapy Promotes Tumor Infiltration and Antitumor Activity of CAR T Cells. Adv Mater. 2019;31:e1900192
154. Vijayan V, Mohapatra A, Uthaman S, Park IK. Recent Advances in Nanovaccines Using Biomimetic Immunomodulatory Materials. Pharmaceutics. 2019;11:E534
155. Kroll AV, Jiang Y, Zhou J, Holay M, Fang RH, Zhang L. Biomimetic Nanoparticle Vaccines for Cancer Therapy. Adv Biosyst. 2019;3:1800219
156. Zhang Y, Lin S, Wang XY, Zhu G. Nanovaccines for cancer immunotherapy. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2019;11:e1559
157. Zhou J, Kroll AV, Holay M, Fang RH, Zhang L. Biomimetic Nanotechnology toward Personalized Vaccines. Adv Mater. 2019 e1901255
158. Luo M, Samandi LZ, Wang Z, Chen ZJ, Gao J. Synthetic nanovaccines for immunotherapy. J Control Release. 2017;263:200-10
159. Zhu G, Zhang F, Ni Q, Niu G, Chen X. Efficient Nanovaccine Delivery in Cancer Immunotherapy. ACS Nano. 2017;11:2387-92
160. Wang Y, Zhang L, Xu Z, Miao L, Huang L. mRNA Vaccine with Antigen-Specific Checkpoint Blockade Induces an Enhanced Immune Response against Established Melanoma. Mol Ther. 2018;26:420-34
161. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788-92
162. Su T, Zhang Y, Valerie K, Wang XY, Lin S, Zhu G. STING activation in cancer immunotherapy. Theranostics. 2019;9:7759-71
163. Flood BA, Higgs EF, Li S, Luke JJ, Gajewski TF. STING pathway agonism as a cancer therapeutic. Immunol Rev. 2019;290:24-38
164. Luo M, Wang H, Wang Z, Cai H, Lu Z, Li Y. et al. A STING-activating nanovaccine for cancer immunotherapy. Nat Nanotechnol. 2017;12:648-54
165. Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE. et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015;11:1018-30
166. Shae D, Becker KW, Christov P, Yun DS, Lytton-Jean AKR, Sevimli S. et al. Endosomolytic polymersomes increase the activity of cyclic dinucleotide STING agonists to enhance cancer immunotherapy. Nat Nanotechnol. 2019;14:269-78
167. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12:860-75
168. Dudek AM, Garg AD, Krysko DV, De Ruysscher D, Agostinis P. Inducers of immunogenic cancer cell death. Cytokine Growth Factor Rev. 2013;24:319-33
169. Adkins I, Fucikova J, Garg AD, Agostinis P, Spisek R. Physical modalities inducing immunogenic tumor cell death for cancer immunotherapy. Oncoimmunology. 2014;3:e968434
170. Workenhe ST, Mossman KL. Oncolytic virotherapy and immunogenic cancer cell death: sharpening the sword for improved cancer treatment strategies. Mol Ther. 2014;22:251-6
171. Lim S, Park J, Shim MK, Um W, Yoon HY, Ryu JH. et al. Recent advances and challenges of repurposing nanoparticle-based drug delivery systems to enhance cancer immunotherapy. Theranostics. 2019;9:7906-23
172. Duan X, Chan C, Lin W. Nanoparticle-Mediated Immunogenic Cell Death Enables and Potentiates Cancer Immunotherapy. Angew Chem Int Ed Engl. 2019;58:670-80
173. Zhang X, Shen L, Liu Q, Hou L, Huang L. Inhibiting PI3 kinase-gamma in both myeloid and plasma cells remodels the suppressive tumor microenvironment in desmoplastic tumors. J Control Release. 2019;309:173-80
174. Liu Q, Chen F, Hou L, Shen L, Zhang X, Wang D. et al. Nanocarrier-Mediated Chemo-Immunotherapy Arrested Cancer Progression and Induced Tumor Dormancy in Desmoplastic Melanoma. ACS Nano. 2018;12:7812-25
175. Phuengkham H, Ren L, Shin IW, Lim YT. Nanoengineered Immune Niches for Reprogramming the Immunosuppressive Tumor Microenvironment and Enhancing Cancer Immunotherapy. Adv Mater. 2019;31:e1803322
176. Qian Y, Qiao S, Dai Y, Xu G, Dai B, Lu L. et al. Molecular-Targeted Immunotherapeutic Strategy for Melanoma via Dual-Targeting Nanoparticles Delivering Small Interfering RNA to Tumor-Associated Macrophages. ACS Nano. 2017;11:9536-49
177. Rodell CB, Arlauckas SP, Cuccarese MF, Garris CS, Li R, Ahmed MS. et al. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat Biomed Eng. 2018;2:578-88
178. Park J, Choi Y, Chang H, Um W, Ryu JH, Kwon IC. Alliance with EPR Effect: Combined Strategies to Improve the EPR Effect in the Tumor Microenvironment. Theranostics. 2019;9:8073-90
179. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L. et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015;372:2521-32
180. Liu X, Shin N, Koblish HK, Yang G, Wang Q, Wang K. et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood. 2010;115:3520-30
181. Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N. et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269-74
182. Cheng K, Ding Y, Zhao Y, Ye S, Zhao X, Zhang Y. et al. Sequentially Responsive Therapeutic Peptide Assembling Nanoparticles for Dual-Targeted Cancer Immunotherapy. Nano Lett. 2018;18:3250-8
183. Mellor AL, Munn DH. Creating immune privilege: active local suppression that benefits friends, but protects foes. Nat Rev Immunol. 2008;8:74-80
184. Meng Z, Zhou X, Xu J, Han X, Dong Z, Wang H. et al. Light-Triggered In Situ Gelation to Enable Robust Photodynamic-Immunotherapy by Repeated Stimulations. Adv Mater. 2019;31:e1900927
185. Li X, Wenes M, Romero P, Huang SC, Fendt SM, Ho PC. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat Rev Clin Oncol. 2019;16:425-41
186. Sanmamed MF, Chen L. A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization. Cell. 2018;175:313-26
187. Xiong Y, Wang Y, Tiruthani K. Tumor immune microenvironment and nano-immunotherapeutics in colorectal cancer. Nanomedicine. 2019;21:102034
188. Chen Q, Chen M, Liu Z. Local biomaterials-assisted cancer immunotherapy to trigger systemic antitumor responses. Chem Soc Rev. 2019;48:5506-26
189. Zhang P, Zhai Y, Cai Y, Zhao Y, Li Y. Nanomedicine-Based Immunotherapy for the Treatment of Cancer Metastasis. Adv Mater. 2019;31:e1904156
190. Zhang B, Hu Y, Pang Z. Modulating the Tumor Microenvironment to Enhance Tumor Nanomedicine Delivery. Front Pharmacol. 2017;8:952
191. Le QV, Suh J, Oh YK. Nanomaterial-Based Modulation of Tumor Microenvironments for Enhancing Chemo/Immunotherapy. AAPS J. 2019;21:64
192. Han S, Huang K, Gu Z, Wu J. Tumor immune microenvironment modulation-based drug delivery strategies for cancer immunotherapy. Nanoscale. 2020;12:413-36
193. Yang VC. Personal perspectives and concerns over the so-called nanomedicine. J Control Release. 2019;311-312:322-3
194. Dai Q, Wilhelm S, Ding D, Syed AM, Sindhwani S, Zhang Y. et al. Quantifying the Ligand-Coated Nanoparticle Delivery to Cancer Cells in Solid Tumors. ACS Nano. 2018;12:8423-35
195. Han X, Li Y, Xu Y, Zhao X, Zhang Y, Yang X. et al. Reversal of pancreatic desmoplasia by re-educating stellate cells with a tumour microenvironment-activated nanosystem. Nat Commun. 2018;9:3390
196. Min H, Wang J, Qi Y, Zhang Y, Han X, Xu Y. et al. Biomimetic Metal-Organic Framework Nanoparticles for Cooperative Combination of Antiangiogenesis and Photodynamic Therapy for Enhanced Efficacy. Adv Mater. 2019;31:e1808200
Author contact
![]() Corresponding authors: Leaf Huang and Jianfeng Guo. leafhunc.edu; jguoedu.cn
Corresponding authors: Leaf Huang and Jianfeng Guo. leafhunc.edu; jguoedu.cn