Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Brief history of YAP/TAZ -...
YAP/TAZ inhibiting drugs -...
Group I drugs
Group II modalities
Group III drugs
Major challenges
Conclusions and future...
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(8):3622-3635. doi:10.7150/thno.40889 This issue Cite
Review
A combat with the YAP/TAZ-TEAD oncoproteins for cancer therapy
Ajaybabu V. Pobbati ![]() , Wanjin Hong
, Wanjin Hong ![]()
Institute of Molecular and Cell Biology, 61 Biopolis Drive, 138673, Singapore
Received 2019-10-4; Accepted 2019-12-20; Published 2020-2-18
Abstract

The transcriptional co-regulators YAP and TAZ pair primarily with the TEAD family of transcription factors to elicit a gene expression signature that plays a prominent role in cancer development, progression and metastasis. YAP and TAZ endow cells with various oncogenic traits such that they sustain proliferation, inhibit apoptosis, maintain stemness, respond to mechanical stimuli, engineer metabolism, promote angiogenesis, suppress immune response and develop resistance to therapies. Therefore, inhibiting YAP/TAZ- TEAD is an attractive and viable option for novel cancer therapy. It is exciting to know that many drugs already in the clinic restrict YAP/TAZ activities and several novel YAP/TAZ inhibitors are currently under development. We have classified YAP/TAZ-inhibiting drugs into three groups. Group I drugs act on the upstream regulators that are stimulators of YAP/TAZ activities. Many of the Group I drugs have the potential to be repurposed as YAP/TAZ indirect inhibitors to treat various solid cancers. Group II modalities act directly on YAP/TAZ or TEADs and disrupt their interaction; targeting TEADs has emerged as a novel option to inhibit YAP/TAZ, as TEADs are major mediators of their oncogenic programs. TEADs can also be leveraged on using small molecules to activate YAP/TAZ-dependent gene expression for use in regenerative medicine. Group III drugs focus on targeting one of the oncogenic downstream YAP/TAZ transcriptional target genes. With the right strategy and impetus, it is not far-fetched to expect a repurposed group I drug or a novel group II drug to combat YAP and TAZ in cancers in the near future.
Keywords: TEAD, YAP, TAZ, Hippo, cancer therapy
Brief history of YAP/TAZ - mediated oncogenesis
The transcriptional co-regulators YAP (Yes-associated protein) and TAZ (transcriptional co-activator with PDZ-binding motif) are key players that mediate various oncogenic processes and targeting their activities has emerged as an attractive option for potential cancer therapy. YAP, as the name suggests, was initially identified as a protein that associates with Yes, a src family kinase (SFK) [1]. The exact function of YAP remained elusive until it was demonstrated to be a potent transcriptional activator [2]. YAP's paralog TAZ, identified from a screen for 14-3-3 interacting proteins, is also a transcriptional co-activator [3] (Figure 1).
The oncogenic milestones of the transcriptional co-regulators YAP and TAZ. Discovery of YAP/TAZ and TEAD functions predate the discovery of the Hippo pathway. Role of YAP/TAZ in the Hippo pathway and the discovery of their oncogenic abilities in cell and animal models are considered significant. The initial studies from the groups that linked YAP/TAZ to oncogenic signaling pathway, stemness, actin cytoskeleton, fusion genes, drug resistance, metabolism, angiogenesis and immune suppression are also listed.
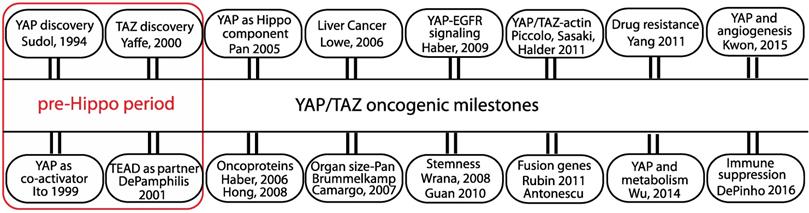
YAP and TAZ do not have a DNA-binding domain and they need to associate with a transcription factor in order to access DNA. It has now emerged that YAP/TAZ use predominantly the TEAD (TEA domain) family of transcription factors [4] to elicit most of their biologically relevant gene expression programs. ChIP-Seq data unraveled a significant overlap in YAP/TAZ and TEAD peaks throughout the genome, and also showed that some YAP/TAZ-responsive genes are also synergistically regulated by AP-1 transcription factors [5, 6]. In addition to its interaction with TEADs, YAP/TAZ also communicates with the mediator complex and chromatin modeling enzymes like the methyltransferase and SWI/SNF complex to elicit changes in gene expression [7-9]. YAP/TAZ also suppress gene expression and should be regarded as co-regulators rather than co-activators [10].
YAP/TAZ are now considered as effectors of a physiologically and pathologically important signaling pathway - popularly called the Hippo pathway [11]. The Hippo pathway was initially identified in a genetic mosaic screen in Drosophila but the pathway components are evolutionarily conserved. It is now known that the primary function of the Hippo pathway is to suppress the activity of Yorkie - the Drosophila homolog of YAP [12]. The Hippo pathway in mammals also inhibits YAP/TAZ through phosphorylation by the large tumor suppressor (LATS) family of Hippo core kinases [13], which leads to cytoplasmic sequestration via interaction with 14-3-3 proteins and/or degradation via ubiquitin proteasome pathway [14, 15].
YAP and TAZ were first shown to transform mammary epithelial cells [16, 17]. The oncogenic role of YAP became apparent when it was shown to be a driver gene in a mouse model of liver cancer [18] (Figure 1). In a conditional transgenic mouse model, YAP overexpression dramatically increases liver size and the mouse eventually develops hepatocellular carcinoma [19, 20]. In addition to causing primary tumor growth, YAP also helps in the metastatic dissemination of tumor cells [21].
Over a decade of research has revealed that YAP/TAZ integrates the inputs of various oncogenic signaling pathways, such as EGFR, TGFβ, Wnt, PI3K, GPCR and KRAS. Through expression of the ligand AREG, YAP was first shown to communicate with the EGFR pathway [22] (Figure 1). The genes regulated by YAP/TAZ collectively coordinate various oncogenic processes, such as stemness, mechanotransduction, drug resistance, metabolic reprogramming, angiogenesis and immune suppression (Figure 1), many of which are considered to be cancer hallmarks [23].
YAP and TAZ regulate the expression of crucial transcription factors like Sox2, Nanog and Oct4 and are able to maintain pluripotency or stemness in human embryonic stem cells (ESCs) and in induced pluripotent stem (iPS) cells [24, 25] (Figure 1). More specifically, TAZ has been shown to confer self- renewal and tumorigenic capabilities to cancer stem cells [26]. Within the microenvironmental landscape of tissues, YAP/TAZ are increasingly recognized as mechanosensors that respond to extrinsic and cell-intrinsic mechanical cues. To this end, mechanical signals related to extracellular matrix (ECM) stiffness, cell morphology and cytoskeletal tension rely on YAP/TAZ for a mechano-activated transcriptional program [27-29]. YAP/TAZ target genes, CTGF and CYR61, cause resistance to chemotherapy drugs like Taxol [30] and YAP/TAZ has emerged as a widely used alternate survival pathway that is adopted by drug-resistant cancer cells [31]. YAP/TAZ activity is regulated by glucose metabolism and is connected to the activity of the central metabolic sensor AMP-activated protein kinase (AMPK) [32-35]. YAP/TAZ reprograms glucose, nucleotide and amino acid metabolism in order to increase the supply of energy and nutrients to fuel cancer cells [36]. Through expression of proangiogenic factors like VEGF and angiopoetin-2 [37, 38], YAP is able to stimulate blood vessel growth to support tumor angiogenesis [39]. YAP is also shown to recruit myeloid-derived suppressor cells in prostate cancers in order to maintain an immune suppressive environment [40]. Active YAP also recruits M2 macrophages to evade immune clearance [41].
A TAZ fusion gene (TAZ-CAMTA1) alone, in the absence of any other chromosomal alteration or mutation, is sufficient to drive epithelioid hemangioendothelioma (EHE), a vascular sarcoma [42, 43]. Furthermore, comprehensive analysis of human tumors across multiple cancer types from the TCGA database unraveled that YAP and TAZ are frequently amplified in squamous cell cancers in a mutually exclusive manner [44]. In human cancers, there is also a good correlation between YAP/TAZ target gene signature and poor prognosis. To date, a proportion of every solid tumor type has been shown to possess aberrant YAP/TAZ activity. Further, many of the upstream Hippo components that negatively regulate YAP/TAZ are found inactivated across many cancer types [45]. Thus, all of this paint a clear picture of the prominent role played by YAP and TAZ at the roots of cancers [46, 47].
YAP/TAZ inhibiting drugs - combat strategies
There are more than fifty drugs that have been shown to inhibit YAP/TAZ activity [48], however, with the exception of verteporfin; none act directly on YAP/TAZ. The unstructured nature of YAP and TAZ renders them difficult to target using small molecules. Therefore, YAP/TAZ inhibition is achieved indirectly through targeting their stimulators or partners. In this review, we focus on small molecules, antibody and peptide-based drugs, as the majority of the drugs in the clinic belong to this class. Less attention is given to nucleotide-based molecules and to small molecule YAP/TAZ inhibitors whose targets are unknown. We classify the YAP/TAZ-inhibiting drugs into three groups with each group having its own combating strategy to counter YAP/TAZ activity (Figure 2). Group I drugs target the upstream YAP/TAZ stimulators and enhance the LATS-dependent inhibitory phosphorylation of YAP/TAZ in order to restrain their transcriptional output. Group II drugs/candidates act directly on YAP/TAZ or TEAD and may either interfere with the formation of the YAP/TAZ-TEAD complex or inhibit TEADs directly and hence affect YAP/TAZ-TEAD transcriptional outcomes. Group III drugs' combat strategy is to target the oncogenic proteins that are transcriptionally upregulated by YAP and TAZ.
Classification ofYAP/TAZ-TEAD inhibiting drugs into three groups. Group I drugs (red font) act upstream and prevent the nuclear entry of YAP and TAZ, group I drug targets for potential pharmacological exploitation in order to generate repurposed YAP/TAZ-inhibiting drugs are circled. Group II drugs (green font) disrupt the formation of the YAP/TAZ-TEAD complex and they primarily bind to the TEAD family of transcription factors. Group Ill drugs (blue font) act on the downstream transcriptional targets in order to prevent YAP/TAZ-mediated oncogenicity.
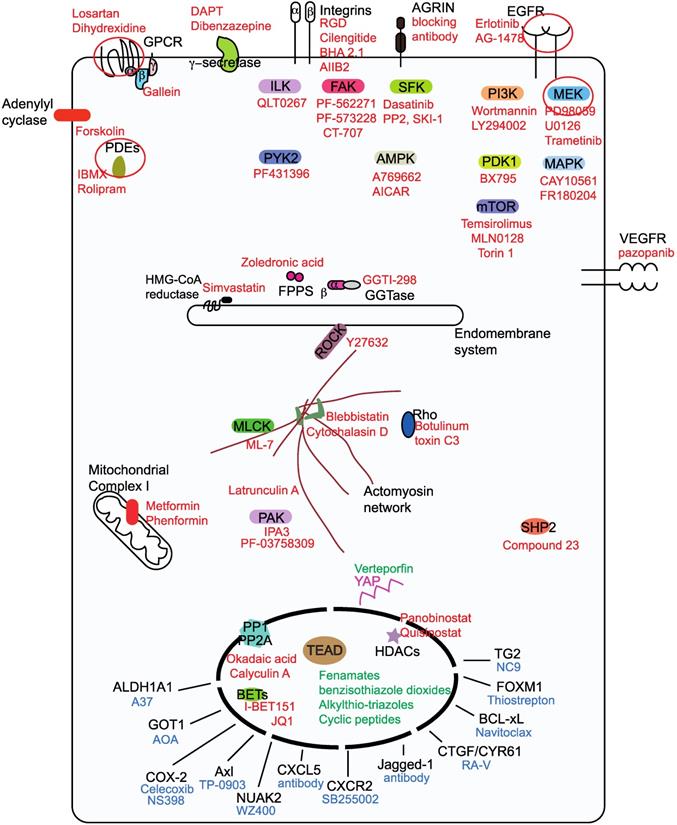
Group I drugs
Group I drugs target the upstream proteins (Figure 2), inhibition of which culminates in the enhancement of the LATS-dependent inhibitory phosphorylation of YAP/TAZ [49, 50]. However, some group I targets like SFKs [51-53], AMPK [33, 34] and phosphatases [54-56] act directly on YAP and TAZ and activate them. Majority of group I drugs are kinase inhibitors, in addition to restricting YAP/TAZ nuclear entry; they intriguingly promote TAZ, but not YAP degradation. A possible explanation for this is the presence of two phosphodegrons that render TAZ more prone to degradation [15]. Some group I drugs, such as MEK/MAPK inhibitors [57, 58] and γ-secretase inhibitors (GSIs) [59] have the ability to actively reduce both YAP and TAZ levels. HDAC inhibitors however, reduce YAP, but not TAZ levels [60]. Here, we have classified the group I drugs based on the nature of the drug target.
Cell surface receptors
Drugs targeting the EGFR, GPCR, Integrin, VEGFR and adenylyl cyclase families as well as those targeting receptors like the γ-secretase complex and Agrin are shown to inhibit YAP/TAZ activity [51, 61-64].
YAP/TAZ exploits the transformative abilities of the ErbB receptors (EGFR family) to drive cell proliferation. By transcribing ErbB ligands, such as AREG [22, 65], TGF-α [66], NRG1 [67] as well as the ErbB receptors EGFR and ErbB3 [67], YAP is able to activate ErbB signaling and promote tumorigenesis. Sustained EGFR signaling also disassembles the Hippo core complexes leading to an increased active pool of YAP/TAZ [68] that is ready to transcribe more ErbB ligands/receptors. Under these conditions, EGFR inhibitors like Erlotinib [22] and AG-1478 [66] (Figure 2) are able to act as YAP/TAZ inhibitors and may be used for EGFR-driven cancers requiring YAP/TAZ transcription.
Signaling from G-protein coupled receptors (GPCRs), transduced by the associated Gα subunit or by the Gβγ subunits, modulates YAP/TAZ activities [69]. Inhibiting Gαq/11 sub-type signaling, using losartan [70], or stimulating Gαs sub-type, using dihydrexidine, has been shown to stimulate YAP inhibitory phosphorylation [69]. Agonism of Gαs has been recently exploited to facilitate YAP/TAZ inhibition that reverses fibrosis in mice [71]. Gβγ inhibition using gallein has also been shown to restrict YAP/TAZ [72]. Activating mutations in the Gαq/11 types of GPCRs present in approximately 80% of the uveal melanoma patients generate an active pool of YAP [73, 74] but the signal transduction occurs via Trio-Rho/Rac signaling and not through the canonical Hippo pathway [74].
Integrin signaling negatively regulates the Hippo pathway complexes to drive YAP/TAZ activity [75, 76]. Although blocking integrin activity using RGD peptides [63], cilengitide (cyclic RGD peptide) [77], function-blocking antibodies - BHA 2.1 [76] and clone AIIB2 [78] has been shown to increase YAP/ TAZ's inhibitory phosphorylation, disappointingly, the efficacy of integrin- blocking drugs against cancers has not been clinically proven [79]. Interestingly, a function-blocking antibody against Agrin, an extrinsic stimulator of integrin signaling, abrogates YAP-dependent proliferation in mouse models [63, 80].
Among the kinase inhibitors tested in a biosensor screen for LATS activity, the VEGFR inhibitors are shown to potently activate LATS and thereby inhibit YAP and TAZ activity [81]. Further, VEGFR2 signaling is also shown to induce actin cytoskeletal changes and promote YAP/TAZ activation [82]. Therefore, VEGFR inhibitors like SU4312, Apatinib, Axitinib and pazopanib are able to inhibit the expression of YAP/TAZ-responsive genes in endothelial cells. But whether these drugs work as YAP/TAZ inhibitors in cancer cells remains to be seen.
Enhancing cyclic AMP (cAMP) levels using the adenylyl cyclase activator forskolin activates the LATS kinases through Protein kinase A (PKA) and Rho [69], therefore forskolin is also a YAP/TAZ inhibitor. cAMP is degraded by the cyclic nucleotide phosphodiesterases (PDE), the use of PDE inhibitors like theophylline, IBMX, ibudilast and rolipram also promotes YAP/TAZ-inhibitory phosphorylation [83, 84].
Notch and YAP/TAZ signaling are also closely linked, inhibiting notch activity by targeting the γ-secretase complex, either using DAPT or dibenzazepine has been shown to decrease YAP/TAZ expression levels in mouse livers and also reduce YAP activation and YAP-induced dysplasia in the intestine [20, 51, 59].
Intracellular kinases
Integrin signaling activates focal adhesion kinase (FAK), SFK and integrin- linked kinase (ILK). Growth factor and GPCR signaling occurs through mitogen-activated protein kinase (MAPK) and phosphoinositide 3-OH kinase (PI3K) signaling. There is also significant crosstalk in the signaling from these membrane receptors. Given the availability of potent small molecule drugs targeting the downstream kinases, they are leveraged on to inhibit YAP or TAZ activities.
Members of downstream integrin signaling pathway including FAK, its counterpart PYK2, and ILK have emerged as negative regulators of the core Hippo pathway and thus activate YAP/TAZ. Membrane receptors, such as ErbB and GPCRs are unable to activate YAP upon genetic deletion of ILK. Therefore, pharmacological inhibition of ILK using a specific ILK inhibitor, QLT0267 potently inhibits YAP-dependent tumor growth in xenograft models [85]. The FAK inhibitors PF-562271 and PF-573228 have also been shown to enhance the LATS-mediated inhibitory phosphorylation of YAP [63, 75]. A multi-kinase inhibitor CT-707 that predominantly inhibits FAK, anaplastic lymphoma kinase (ALK) and PYK2 is able to render cancer cells vulnerable to hypoxia through YAP inhibition [86]. Inhibiting PYK2 activity using the dual PYK2/FAK inhibitor PF431396 destabilizes TAZ and also inhibits YAP/TAZ activity in triple negative breast cancer cells [87].
The SFK member Src prevents the activation of LATS [75, 88], thereby relieves YAP/TAZ inhibition by LATS. Interestingly, SFKs, Src and YES are also shown to activate YAP through direct tyrosine phosphorylation [51-53]. Treating cells with SFK inhibitors, such as Dasatinib, PP2, SU6656, AZD0530 and SKI-1 inactivates YAP [51-53, 75, 88]. In β-catenin-driven cancers, YES facilitates the formation of a tripartite complex comprising β-catenin, YAP and TBX5 that drives cell survival and tumor growth [53, 89]. The SFK inhibitor dasatinib also serves as YAP inhibitor in these cancers [53]. Dasatinib, in addition to inhibiting SFKs may also potently inhibit PDGFR and Ephrin receptors, both of which are known to activate YAP/TAZ [90, 91]. However, FAK and SFK inhibitors have shown very limited efficacy against solid tumors in clinical trials therefore their utility in YAP-driven cancers remains to be seen.
MEK (MAP kinase kinase) and YAP interact with each other and maintain transformed phenotypes in liver cancer cells [57]. MEK inhibitors PD98059, U0126 and trametinib or MAPK inhibitors CAY10561 and FR180204 are able to trigger degradation of YAP in a Hippo-independent manner [57, 58]. The finding that MEK inhibition causes YAP degradation is, however, difficult to reconcile if YAP and TAZ are shown to mediate resistance to the MEK inhibitor trametinib [92]. The efficacy of trametinib is also being evaluated in EHE, a cancer that is caused by the TAZ-CAMTA1 fusion gene (NCT03148275).
PI3K inhibitors Wortmannin/LY294002 as well as the drug BX795, an inhibitor of its effector 3'-phosphoinositide-dependent kinase-1 (PDK1) prevents nuclear entry of YAP [68]. PI3K is closely linked to the mammalian target of rapamycin (mTOR) pathway. mTOR inhibitors temsirolimus and MLN0128 have been shown to inhibit YAP activity in patients with idiopathic pulmonary fibrosis and in a mouse model of cholangiocarcinoma, respectively [93, 94]. YAP levels in TSC1 mutant mouse could also be reduced by blocking mTOR using torin1 treatment that induces the autophagy-lysosomal pathway [95].
YAP/TAZ inhibition is an additional unexpected activity possessed by the few kinase inhibitors mentioned above. However, apart from YAP/TAZ inhibition, all other signaling events initiated by the target kinase are also shut down due to inhibitor treatment. If these signaling events are critical for cellular homeostasis, then, toxic side effects will outweigh clinical benefits and this cannot be uncoupled from YAP/TAZ inhibition. Therefore, kinase inhibitors that failed in the trials due to unacceptable toxcity or poor pharmacokinetics may not be repurposed as YAP/TAZ inhibitors in the clinic. Focus should be on the kinase inhibitors that are already in the clinic like EGFR, VEGFR, MEK, PI3K or mTOR inhibitors but efficacy needs to be proven in order to repurpose them as YAP/TAZ inhibitors. The kinase targeted by the inhibitor must activate YAP/TAZ in tumors, for the treatment to be efficacious and this restricts the use of kinase inhibitors to selective tumor types. Intriguingly, YAP/TAZ activation has emerged as a prominent survival strategy adapted by cancers that cause drug resistance to EGFR and its downstream MEK/MAPK inhibitors [31]. In such scenarios, coupling a group II YAP/TAZ inhibitor with a EGFR pathway inhibitor might offer the intended treatment benefits.
Mevalonate pathway inhibitors
The mevalonate pathway is essential for the biosynthesis of isoprenoids, cholesterol and steroid hormones. Statins as well as other mevalonate pathway inhibitors like zoledronic acid and GGTI-298 that target farnesyl pyrophosphate synthase and geranylgeranyltransferase, respectively are identified as drugs that restrict the nuclear entry of YAP and TAZ [96, 97]. Studies have also shown that combining statins like simvastatin with the EGFR inhibitor gefitinib provides stronger anti-neoplastic effects [98]. Atorvastatin and zoledronic acid have entered Phase II clinical trials in triple negative breast cancer to test if they improve the pathological complete response rates (NCT03358017).
Actin modulators
Actin polymerization promotes YAP/TAZ nuclear localization and therefore, polymerization inhibitors like latrunculin A [27] and cytochalasin D [28, 29] inhibit YAP/TAZ. Myosin or myosin light-chain kinase inhibitors like blebbistatin and ML-7, respectively have a similar effect [27, 29]. Interfering with the actomyosin cytoskeleton through other means, such as Rho inhibition (toxin C3 treatment), or by using Rho kinase inhibitors like Y27632 has also been shown to have an inhibitory effect on YAP/TAZ [27, 29]. p21 activated kinase (PAK) family kinases are cytoskeletal regulators as well as Hippo inhibitors. The PAK allosteric inhibitor IPA3 prevents YAP's nuclear entry [63, 99], further, the PAK4 inhibitor PF-03758309 is also shown to reduce YAP levels [77].
Phosphatase inhibitors
YAP/TAZ inhibitory phosphorylation is dynamic and the protein phosphatases PP1 and PP2A are shown to associate with YAP/TAZ and aid in their dephosphorylation and activation. Inhibiting these phosphatases using okadaic acid or calyculin A increases YAP/TAZ phosphorylation and shifts YAP/TAZ to the cytoplasm [54-56]. Some of the oncogenic functions of YAP/TAZ are also mediated by the protein-tyrosine phosphatase SHP2 [100], therefore SHP2 inhibitors have also been shown to attenuate YAP/TAZ activity [101].
Cellular energy stress modulators
Cellular energy stress is closely linked with attenuation of YAP/TAZ activities [32]. Drugs that enhance energy stress like the mitochondrial complex I inhibitors metformin and phenformin, enhance YAP/TAZ inhibitory phosphorylation, cytoplasmic localization and suppression of YAP/TAZ- mediated transcription [32]. The energy stress induced by these drugs activates AMPK, which is shown to phosphorylate and stabilize AMOTL1 - a YAP/TAZ negative regulator [32]. AMPK is also shown to directly phosphorylate and inactivate YAP by disrupting its interaction with TEADs [33, 34]. Therefore, AMPK activators A769662 and AICAR (an AMP-mimetic) are YAP inhibitors [32-34].
Epigenetic modulators
Histone deacetylases (HDACs) are uniquely positioned to alter the transcription of target genes. Interestingly, HDAC inhibitors panobinostat, quisinostat, dacinostat, vorinostat and Trichostatin A transcriptionally repress the expression of YAP but not TAZ, and thereby reduce YAP-addicted tumorigenicity [60]. Treatment of cholangiocarinoma cells with the HDAC inhibitor CG200745 is also shown to decrease YAP levels [102]. Although HDAC inhibitors are used to treat hematological malignancies their efficacy in solid cancers is questionable, however, combining HDAC inhibitor panobinostat with BET (bromodomain and extra-terminal) inhibitor I-BET151 achieves more effective YAP inhibition [103]. There is also a clinical trial designed to evaluate the efficacy of HDAC/BET inhibitor combination in solid tumors and determination of YAP expression level after drug treatment is used as one of the objectives (NCT03925428). The BET family protein BRD4 is a part of the YAP/TAZ-TEAD transcriptional complex and inhibiting BRD4 using BET inhibitor JQ1 inhibits YAP upregulation and YAP-mediated transcription in KRAS mutant cells [104].
Many group I drugs can potentially be repurposed to treat YAP/TAZ- driven cancers [105]. Among the group I drugs, only statins, trametinib and HDAC/BET inhibitors are being evaluated in clinical trials to test if they act against YAP/TAZ. Our prediction is that group I drugs that facilitate YAP/ TAZ inhibitory phosphorylation as well as degradation will have greater success in combating YAP/TAZ in cancers as YAP/TAZ degradation prevents their reactivation through other mechanisms. Importantly, the repurposing of group I drugs would also allow YAP/TAZ and its target gene(s) expression-based stratification amongst cancer patients.
Group II modalities
Modalities that target either the TEAD family of transcription factors or YAP/TAZ are classified under this group (Figure 3). The majority of the modalities, with the exception of verteporfin [106], target TEADs and are therefore predicted to act in the nucleus. By pairing with the TEAD family of transcription factors, YAP and TAZ upregulate the expression of many oncoproteins. The C-terminus of all TEADs possesses the YAP/TAZ-binding domain. The partnership between YAP/TAZ and TEAD is essential for the initiation of transcriptional program to drive oncogenesis. YAP is no longer oncogenic when sequestered by a dominant negative TEAD that lacks the DNA-binding domain [106]. Similarly, a naturally occurring DNA-binding deficient TEAD isoform is also able to inhibit YAP/TAZ-mediated oncogenicity [107]. Therefore, directly inhibiting TEAD or preventing YAP/TAZ-TEAD interaction is a promising and most direct strategy that warrants special attention [108].
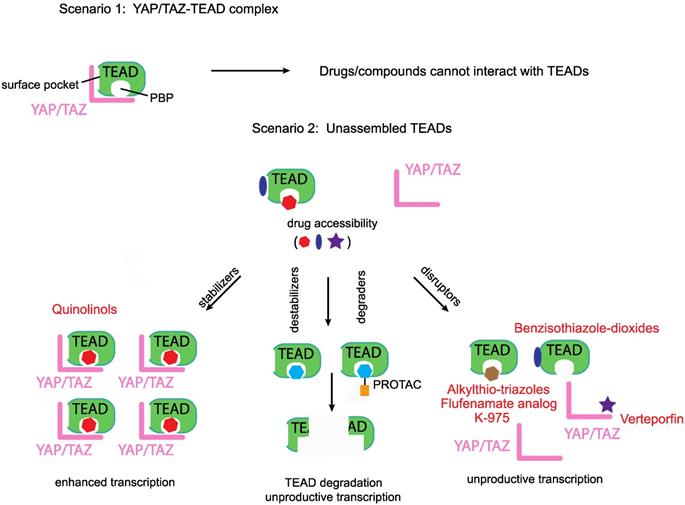
Disruptors, stabilizers and destabilizers/degraders. A preformed YAP/TAZ-TEAD complex prevents access to drugs that occupy either the TEADs' surface or the palmitate-binding pocket (PBP), however, unassembled TEADs are accessible to drugs. Majority of the known YAP/TEAD-binding compounds are disruptors as they prevent the formation of the YAP/TAZ-TEAD complex. Two other classes of TEAD-binding compounds are stabilizers and destabilizers/degraders. Stabilizers either stabilize TEAD expression levels or enhance the formation of the YAP/TAZ-TEAD complex. Destabilizers bind to TEADs' surface or PBP and reduce TEAD expression levels through in situ denaturation, degraders on the other hand direct TEADs for proteasomal degradation.
Group I drugs target the upstream YAP/TAZ-activating proteins like the EGFR, GPCR, Src, or Integrins. As there are so many upstream YAP/TAZ activators, group I drugs are vulnerable to oncogenic bypass where inhibition of one group I YAP/TAZ activator leads to selection of cancer cells that activate YAP/TAZ via another group I activator. Strategically, Group II drugs may address this issue by directly targeting YAP/TAZ or TEAD, the converging points for various pathways and also the effectors for oncogenic transcription. However, Group II targeting modalities are still at the exploratory stage and it remains to be seen whether it is feasible to develop a Group II modality that works in clinic. We also need to be mindful of the possible associated toxicities due to YAP/TAZ-TEAD inhibition [109].
Most of the reported Group II modalities are disruptors; they target YAP/TAZ or TEAD and prevent their binary interaction. However, in addition to disruptors, in the future, we predict the emergence two other classes of group II compounds that would act as TEAD stabilizers and destabilizers/degraders (Figure 3).
Disruptors targeting YAP
A small molecule benzoporphyrin drug named Verteporfin (VP) was shown to have the ability to bind to YAP and disrupt the YAP-TEAD interaction [106]. VP is also able to inhibit YAP-induced excessive cell proliferation in YAP- inducible transgenic mice and in NF2 (upstream Hippo pathway component) liver-specific knockout mouse models [106]. Although we do not understand the molecular details of VP binding to YAP, it is still undoubtedly the most popular YAP inhibitor within the scientific community. However, we need to be cautious as some of the tumor-inhibitory effects of VP are reported to be YAP- independent [110, 111]. VP is photosensitive and proteotoxic and there is a need for better derivatives. A VP derivative, a symmetric divinyldipyrrine was shown to inhibit YAP/TAZ-dependent transcription but it is not clear if the compound specifically binds to YAP [112].
Disruptors targeting the TEADs' surface
YAP and TAZ bind on the TEADs' surface; Inventiva Pharma has identified several compounds with benzisothiazole-dioxide scaffold that bind to the TEADs' surface and disrupt the YAP/TAZ-TEAD interaction. These compounds are currently in the lead optimization stage and have the potential to treat malignant pleural mesothelioma as well as lung and breast cancers that are driven by YAP/TAZ [113].
YAP cyclic peptide (peptide 17) and cystine-dense peptide (TB1G1) are also disruptors of YAP/TAZ-TEAD interaction in vitro but they have poor cell-penetrating abilities [114, 115]. Interestingly, a peptide derived from the co-regulator Vgll4 appears to have remarkable cell-penetrating abilities and inhibits YAP-mediated tumorigenesis in animal models [116]. Vgll proteins, named Vgll1-4 in mammals, belong to another class of co-regulators that pair with TEADs in a structurally similar, and therefore, in a mutually exclusive manner with YAP and TAZ [117, 118].
Disruptors targeting TEADs' palmitate-binding pocket (PBP)
We identified a novel druggable pocket in the center of the TEADs' YAP/TAZ- binding domain [119] that could be occupied by fenamate drugs. Palmitate was subsequently shown to occupy this pocket, hereafter referred to as the palmitate-binding pocket (PBP). TEAD palmitoylation is shown to be important for stability and for the interaction with YAP [120, 121]. Although the fenamate drug flufenamic acid competes with palmitate for binding to TEAD, higher concentrations are needed for it to be effective and it is not a disruptor of the interaction between YAP/TAZ with TEADs [122]. However, covalently linking the fenamate to TEAD, using a chloromethyl ketone substitution, enables it to disrupt the YAP-TEAD interaction [123]. The non-fused tricyclic compounds identified by Vivace Therapeutics could also be considered as fenamate analogs but it remains to be seen if they function as disruptors [124]. Through structure-based virtual screen, vinylsulfonamide derivatives were identified as compounds that bind to PBP [125]. Optimization of these derivatives yielded DC-TEADin02 a covalent TEAD autopalmitoylation inhibitor with an IC50 value of 200 nM. Interestingly, DC-TEADin02 is able to inhibit TEAD activity without disrupting the YAP-TEAD interaction.
Palmitate, by occupying the PBP, allosterically modulates YAP's interaction with TEAD [121], therefore it is conceivable that there might be small molecules that occupy the PBP and allosterically disrupt YAP/TAZ's interaction with TEADs. To this end, Xu Wu's group has identified and patented several potent compounds with alkylthio-triazole scaffold as PBP- occupying compounds that prevent YAP-TEAD interaction in cells [126]. Another potent TEAD inhibitor that occupies the PBP is the small molecule K-975 [127]. K-975 also disrupts the YAP-TAZ-TEAD interaction and displayed anti-tumorigenic properties in malignant pleural mesothelioma cell lines much akin to the loss of YAP. Although palmitate is covalently attached to TEAD, it is a reversible modification and addition of PBP-occupying small molecules reduce the cellular palmitoylation status of TEADs [126]. Moreover, the palmitoyl group is also removed from TEADs by depalmitoylases [128].
Being predominantly unstructured, YAP and TAZ are difficult to target directly. However, TEADs offer two attractive ways for targeting, one is to directly block the YAP/TAZ-binding pocket on the TEADs' surface with small molecules or peptides, whilst the other is to leverage on the PBP and allosterically disrupt YAP/TAZ interaction or inhibit TEADs (Figure 3). However, the molecular determinants that confer YAP/TAZ disrupting ability to PBP-occupying small molecules are not clear. We do not know why flufenamate and DC-TEADin02 are unable to disrupt YAP/TAZ-TEAD interaction, like chloromethyl fenamate, K-975 and compounds with alkylthio-triazole scaffold.
Stabilizers and destabilizers/degraders
The PBP could also be leveraged to allosterically enhance YAP/TAZ-TEAD stability or interaction. This prediction is subject to the identification of small molecules that functionally mimic the ligand palmitate (Figure 3). Compounds with such an ability will enhance TEAD-dependent transcription and may have therapeutic value for regenerative medicine where enhancement of YAP/TAZ- TEAD activity is needed to repair damaged tissues [129]. We recently identified that quinolinols occupy the PBP, stabilize YAP/TAZ levels and upregulate TEAD-dependent transcription [130]. Enhanced YAP/TAZ levels increase the pool of assembled YAP/TAZ complex and therefore quinolinols could be considered as stabilizers (Figure 3).
We identified a few chemical scaffolds that have the ability to occupy the PBP and destabilize TEAD (unpublished results). Addition of these compounds unfolds the TEADs' YAP/TAZ-binding domain and we call these compounds destabilizers (Figure 3). Degraders could be generated when potent and selective TEAD surface or PBP-occupying compounds are coupled to proteolysis targeting chimera (PROTAC) [131] to direct TEAD proteasomal degradation. Therefore, destabilizers aim to reduce the cellular concentration of TEADs through in situ unfolding and degraders reduce TEAD levels through proteasomal degradation. Reducing the levels of their interacting partners deprives YAP/TAZ of their ability to activate transcription.
Any TEAD-binding compounds (disruptors, stabilizers or destabilizers/degraders) can only access unbound TEADs, as binding of YAP and TAZ blocks both the surface and the palmitate-binding pockets (Figure 3). After accessing unbound TEADs, the disruptors and destabilizers/degraders reduce, whereas the stabilizers enhance, the formation of the YAP/TAZ-TEAD complex.
Group III drugs
YAP/TAZ-mediated tumor development is due to the collective action of the repertoire of proteins that are expressed under their influence. However, some proteins are able to drive oncogenesis much better than others and they vary depending on the solid tumor and context. Therefore, drugs against these downstream YAP/TAZ targets including metabolic enzymes, kinases, ligands and proteins, such as BCL-xL, FOXM1 and TG2 are also used to combat YAP/TAZ-mediated oncogenicity (Figure 2).
Metabolic enzymes
TAZ-dependent expression of ALDH1A1 (aldehyde dehydrogenase) is shown to impart stemness and tumorigenic ability; inhibition of ALDH1A1 using A37 reverses this transformation [132]. GOT1 - the aspartate transaminase induced by YAP/TAZ, confers glutamine dependency to breast cancer cells and targeting this metabolic vulnerability using aminooxyacetate (AOA) represses breast cancer cell proliferation [133]. Targeting the YAP/TAZ transcriptional target cyclooxygenase 2 (COX-2) using celecoxib inhibits cell proliferation and tumorigenesis in NF2 mutant cells [134]. Interestingly, a positive feedback is seen in hepatocellular carcinoma cell lines where COX-2 is also shown to increase the expression of YAP [135]. Inhibiting COX-2 using NS398 stimulates LATS-dependent phosphorylation of TAZ [136].
Kinases
In hepatocellular carcinoma, Axl kinase has been shown to be crucial for mediating several YAP-driven oncogenic functions like cell proliferation and invasion [137]. Similarly, YAP-driven Axl expression has been implicated in the development of resistance against EGFR inhibitors in lung cancer and sensitivity could be restored through Axl inhibition using TP-0903 [138]. YAP is shown to upregulate the expression of the kinase NUAK2 [139] that, in turn activates YAP/TAZ by inhibiting LATS. Specific pharmacological inhibition of NUAK2 using WZ400 shifts YAP/TAZ to the cytoplasm and reduces cancer cell proliferation [140].
Ligands
In a mouse model of prostate adenocarcinoma, the YAP-TEAD complex promotes the expression of the chemokine ligand CXCL5 that facilitates myeloid-derived suppressor cells (MDSC) infiltration and adenocarcinoma progression. Administering CXCL5 neutralizing antibody, or blocking CXCL5 receptor using the inhibitor SB255002, inhibits MDSC migration and tumor burden [40]. The notch ligand Jagged-1 that is upregulated by YAP/TAZ is crucial for liver tumorigenesis [59, 141]. Treating liver tumor cells with Jagged-1 neutralizing antibody greatly reduces oncogenic traits. The levels of integrin ligands CTGF and CYR61 that are also YAP target genes, could be reduced using the cyclopeptide RA-V (deoxybouvardin) leading to a reduction of YAP- mediated tumorigenesis in mst1/2 (Hippo homolog) knockout mouse model [142]. Although neutralizing CTGF (FG-3019/pamrevlumab) and CYR61 (093G9) antibodies are available, they have not been effectively used against YAP/TAZ-driven cancers.
BCL-xL
YAP mediates drug resistance to RAF- and MEK-targeted therapies in BRAF V600E cells, in part through the expression of the anti-apoptotic protein BCL- xL. BCL-xL inhibition using navitoclax sensitizes these cells to targeted therapies [92].
FOXM1
YAP-mediated proliferation through its target gene FOXM1 could be prevented in sarcoma cell lines and mouse models through the administration of thiostrepton that reduces FOXM1 levels [143].
Transglutaminase 2
Transglutaminase 2 (TG2) - the multifunctional transamidase is a YAP/TAZ target gene that is important for cancer stem cell survival and for maintaining integrin expression. TG2 inhibition using NC9 dramatically reduces tumorigenicity [144, 145].
We are aware that many of these target proteins also act upstream and stimulate YAP/TAZ by forming a positive feedback but we would nevertheless consider them in this group and not as group I as their expression is influenced by the TEAD-binding motif and YAP/TAZ.
Major challenges
Although attractive, toxicity issues and the identification of responsive patient population could be challenges in the successful implementation of the YAP/TAZ inhibitors in the clinic. YAP/TAZ inhibition might elicit toxicity [146]; homozygous disruption of YAP in mice causes embryonic lethality, whereas TAZ knockouts are viable [147-150]. Tissue-specific deletions of YAP in the heart [151], lung [152] or kidney [153] cause hypoplasia, whereas YAP/TAZ deletion in the liver cause hepatomegaly and liver injury [154]. Surprisingly, YAP/TAZ knockouts in the intestine are well tolerated with no apparent tissue defects [155]. All of these suggest that YAP and TAZ are crucial for development. However, they appear to be dispensable for adult tissue homeostasis. In most adult tissues, under normal homeostasis, YAP/TAZ are found restricted to the cytoplasm and are activated primarily in response to injury to initiate tissue regeneration. Therefore, it is predictable that administration of a YAP/TAZ inhibitor may not elicit severe toxicity. However, given the dynamic shuttling of YAP/TAZ/Yorkie between nucleus and cytoplasm [156-158], it is feasible that they still have a role in normal tissue homeostasis. Fittingly, YAP has been identified to be important for podocyte homeostasis and its functional inactivation compromises the glomerular filtration barrier and cause renal disease [109]. Along similar lines, renal toxicity was observed in mice administered with K-975 - a YAP/TAZ-TEAD inhibitor [127]. Renal toxicity in targeted therapy is very common and is seen in most of the kinase inhibitors used in oncology [159]. Yet these kinase inhibitors are in the clinic as there is a therapeutic window, where the drug could be dosed to improve patient survival without causing much toxicity. The same could be envisaged for YAP/TAZ-inhibiting drugs.
Several drugs that act as YAP/TAZ inhibitors target multiple signaling pathways. Targeting multiple pathways could be a boon or a bane. Drug resistance is minimized in a multi-targeted approach as potential bypass mechanisms are also targeted. However, toxicity becomes an issue when the drug targets multiple important signaling pathways. For instance, raising cAMP through the use of PDE inhibitors activates a multitude of proteins like PKA, EPACs, ion channels and small GTPases. Similarly, GPCR modulators influence multiple pathways through signaling via G proteins, arrestins or GPCR kinases. To reduce toxic side effects, there are options available like selective targeting or biased signaling. Instead of hitting all the PDEs, the PDE enzyme that is the most potent activator of YAP/TAZ should be selectively targeted. Nonspecific PDE inhibitors cause more severe side effects than sub-type selective PDE inhibitors [160]. Similarly, through stabilizing a particular GPCR conformation, certain small molecule GPCR modulators are able to effect signaling bias where one GPCR effector is preferentially activated over others, say G proteins over β-arrestins, this way only a subset of signaling pathways get activated [161].
Another major challenge is the identification of patients responding to a YAP/TAZ inhibitor. YAP/TAZ expression is low in normal tissues and their levels are significantly elevated in cancers. Is YAP or TAZ positivity in tumors sufficient criteria to administer a YAP/TAZ inhibitor? YAP and TAZ might not be transcriptionally active or drivers in all tumors. Further, they could be expressing target genes that negatively regulate their activity [162, 163]. There are also tumor types where YAP/TAZ or TEAD levels have no prognostic significance [46]. These YAP/TAZ positive tumors are unlikely to respond to a YAP/TAZ inhibitor. Barring a few such scenarios, in many solid tumors, YAP or TAZ expression levels correlate well with higher-grade cancers or poor prognosis. Tumors with nuclear YAP or TAZ that are also positive for the downstream oncogenic YAP/TAZ target genes are likely to respond to a YAP/TAZ inhibitor and this should be used as criteria for patient stratification. As many of the YAP/TAZ-TEAD target genes are secreted proteins, the expression levels of these in the serum could also be estimated in addition to assessing their levels through immunohistochemistry.
Conclusions and future perspectives
As YAP and TAZ contribute to the acquisition of many hallmarks of cancer traits, targeting them is predicted to be more relevant for the management of several cancer types. It is still early to expect a newly developed drug against YAP/TAZ but it is nevertheless disconcerting to see that there are hardly any clinical trials that evaluate if known drugs could be repurposed as YAP/TAZ- inhibitors. Group I drugs are well suited to repurpose [105] but only statins (NCT03358017); trametinib (NCT03148275) and epigenetic modulators (NCT03925428) are being evaluated in clinical trials, assessment of the expression levels of YAP/TAZ after drug treatment is used as one of the clinical trial objectives. It is essential that we bolster our pharmacological arsenal so that we are prepared to combat YAP and TAZ. Group I drugs that failed in oncology trials are not expected to fare any better against YAP/TAZ. However, drugs that are already in the clinic like the kinase inhibitors targeting the EGFR or MEK, PDE inhibitors as well as GPCR modulators could be repurposed to combat YAP/TAZ. The cancer types need to be carefully stratified to ensure they are driven by YAP/TAZ through the upstream stimulator targeted by the drug. To overcome potential bypass mechanism or drug resistance, combinatory use of group I and II drugs could also serve as an avenue for cancer treatment. For the group III drugs, the situation may not be as promising, as they target only one of the many possible oncogenic proteins regulated by YAP/TAZ. Again, combinatory inhibition of few downstream target genes could be considered if they are collectively essential for oncogenic manifestation of YAP/TAZ-driven transcription. As they are new and untested, there is much excitement and progress in the development of novel group II compounds as drugs against YAP/TAZ. We are at an exciting juncture in the Hippo field where we could potentially see a novel group II drug or a repurposed group I drug to combat YAP/TAZ in the near future.
Acknowledgements
A.V. Pobbati and W. Hong are supported by the Agency for Science, Technology, and Research (A*STAR), Singapore. We thank Sayan Chakraborty, Gandhi T.K.B. and John Hellicar for critical reading of this review. We apologize to all authors whose work was not cited due to space constraints.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sudol M. Yes-associated protein (YAP65) is a proline-rich phosphoprotein that binds to the SH3 domain of the Yes proto-oncogene product. Oncogene. 1994;9:2145-52
2. Yagi R, Chen LF, Shigesada K, Murakami Y, Ito Y. A WW domain-containing yes-associated protein (YAP) is a novel transcriptional co-activator. EMBO J. 1999;18:2551-62
3. Kanai F, Marignani PA, Sarbassova D, Yagi R, Hall RA, Donowitz M. et al. TAZ: a novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J. 2000;19:6778-91
4. Vassilev A, Kaneko KJ, Shu H, Zhao Y, DePamphilis ML. TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes Dev. 2001;15:1229-41
5. Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B. et al. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol. 2015;17:1218-27
6. Stein C, Bardet AF, Roma G, Bergling S, Clay I, Ruchti A. et al. YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers. PLoS Genet. 2015;11:e1005465
7. Galli GG, Carrara M, Yuan WC, Valdes-Quezada C, Gurung B, Pepe-Mooney B. et al. YAP Drives Growth by Controlling Transcriptional Pause Release from Dynamic Enhancers. Mol Cell. 2015;60:328-37
8. Oh H, Slattery M, Ma L, White KP, Mann RS, Irvine KD. Yorkie promotes transcription by recruiting a histone methyltransferase complex. Cell Rep. 8:449-59.
9. Skibinski A, Breindel JL, Prat A, Galvan P, Smith E, Rolfs A. et al. The Hippo transducer TAZ interacts with the SWI/SNF complex to regulate breast epithelial lineage commitment. Cell Rep. 2014;6:1059-72
10. Kim M, Kim T, Johnson RL, Lim DS. Transcriptional co-repressor function of the hippo pathway transducers YAP and TAZ. Cell Rep. 2015;11:270-82
11. Zheng Y, Pan D. The Hippo Signaling Pathway in Development and Disease. Dev Cell. 2019;50:264-82
12. Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell. 2005;122:421-34
13. Bae SJ, Luo X. Activation mechanisms of the Hippo kinase signaling cascade. Biosci Rep. 2018:38
14. Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta- TRCP). Genes Dev. 2010;24:72-85
15. Liu CY, Zha ZY, Zhou X, Zhang H, Huang W, Zhao D. et al. The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCF{beta}-TrCP E3 ligase. J Biol Chem. 2010;285:37159-69
16. Overholtzer M, Zhang J, Smolen GA, Muir B, Li W, Sgroi DC. et al. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci U S A. 2006;103:12405-10
17. Chan SW, Lim CJ, Guo K, Ng CP, Lee I, Hunziker W. et al. A role for TAZ in migration, invasion, and tumorigenesis of breast cancer cells. Cancer Res. 2008;68:2592-8
18. Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J. et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell. 2006;125:1253-67
19. Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA. et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130:1120-33
20. Camargo FD, Gokhale S, Johnnidis JB, Fu D, Bell GW, Jaenisch R. et al. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol. 2007;17:2054-60
21. Lamar JM, Stern P, Liu H, Schindler JW, Jiang ZG, Hynes RO. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc Natl Acad Sci U S A. 2012;109:E2441-50
22. Zhang J, Ji JY, Yu M, Overholtzer M, Smolen GA, Wang R. et al. YAP- dependent induction of amphiregulin identifies a non-cell-autonomous component of the Hippo pathway. Nat Cell Biol. 2009;11:1444-50
23. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
24. Varelas X, Sakuma R, Samavarchi-Tehrani P, Peerani R, Rao BM, Dembowy J. et al. TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat Cell Biol. 2008;10:837-48
25. Lian I, Kim J, Okazawa H, Zhao J, Zhao B, Yu J. et al. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev. 2010;24:1106-18
26. Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C. et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell. 2011;147:759-72
27. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M. et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179-83
28. Sansores-Garcia L, Bossuyt W, Wada K, Yonemura S, Tao C, Sasaki H. et al. Modulating F-actin organization induces organ growth by affecting the Hippo pathway. EMBO J. 2011;30:2325-35
29. Wada K, Itoga K, Okano T, Yonemura S, Sasaki H. Hippo pathway regulation by cell morphology and stress fibers. Development. 2011;138:3907-14
30. Lai D, Ho KC, Hao Y, Yang X. Taxol resistance in breast cancer cells is mediated by the hippo pathway component TAZ and its downstream transcriptional targets Cyr61 and CTGF. Cancer Res. 2011;71:2728-38
31. Nguyen CDK, Yi C. YAP/TAZ Signaling and Resistance to Cancer Therapy. Trends Cancer. 2019;5:283-96
32. DeRan M, Yang J, Shen CH, Peters EC, Fitamant J, Chan P. et al. Energy stress regulates hippo-YAP signaling involving AMPK-mediated regulation of angiomotin-like 1 protein. Cell Rep. 2014;9:495-503
33. Wang W, Xiao ZD, Li X, Aziz KE, Gan B, Johnson RL. et al. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat Cell Biol. 2015;17:490-9
34. Mo JS, Meng Z, Kim YC, Park HW, Hansen CG, Kim S. et al. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nature cell biology. 2015;17:500-10
35. Kuo CC, Ling HH, Chiang MC, Chung CH, Lee WY, Chu CY. et al. Metastatic Colorectal Cancer Rewrites Metabolic Program Through a Glut3-YAP-dependent Signaling Circuit. Theranostics. 2019;9:2526-40
36. Koo JH, Guan KL. Interplay between YAP/TAZ and Metabolism. Cell Metab. 2018;28:196-206
37. Choi HJ, Zhang H, Park H, Choi KS, Lee HW, Agrawal V. et al. Yes- associated protein regulates endothelial cell contact-mediated expression of angiopoietin-2. Nat Commun. 2015;6:6943
38. Ma B, Chen Y, Chen L, Cheng H, Mu C, Li J. et al. Hypoxia regulates Hippo signalling through the SIAH2 ubiquitin E3 ligase. Nat Cell Biol. 2015;17:95-103
39. Boopathy GTK, Hong W. Role of Hippo Pathway-YAP/TAZ Signaling in Angiogenesis. Front Cell Dev Biol. 2019;7:49
40. Wang G, Lu X, Dey P, Deng P, Wu CC, Jiang S. et al. Targeting YAP- Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov. 2016;6:80-95
41. Guo X, Zhao Y, Yan H, Yang Y, Shen S, Dai X. et al. Single tumor-initiating cells evade immune clearance by recruiting type II macrophages. Genes Dev. 2017;31:247-59
42. Tanas MR, Sboner A, Oliveira AM, Erickson-Johnson MR, Hespelt J, Hanwright PJ. et al. Identification of a disease-defining gene fusion in epithelioid hemangioendothelioma. Sci Transl Med. 2011;3:98ra82
43. Errani C, Zhang L, Sung YS, Hajdu M, Singer S, Maki RG. et al. A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosomes Cancer. 2011;50:644-53
44. Wang Y, Xu X, Maglic D, Dill MT, Mojumdar K, Ng PK. et al. Comprehensive Molecular Characterization of the Hippo Signaling Pathway in Cancer. Cell Rep. 2018;25:1304-17
45. Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC. et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell. 2018;173:321-37
46. Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the Roots of Cancer. Cancer Cell. 2016;29:783-803
47. Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer. 2013;13:246-57
48. Juan WC, Hong W. Targeting the Hippo Signaling Pathway for Tissue Regeneration and Cancer Therapy. Genes (Basel). 2016:7
49. Park HW, Guan KL. Regulation of the Hippo pathway and implications for anticancer drug development. Trends in pharmacological sciences. 2013;34:581-9
50. Calses PC, Crawford JJ, Lill JR, Dey A. Hippo Pathway in Cancer: Aberrant Regulation and Therapeutic Opportunities. Trends Cancer. 2019;5:297-307
51. Taniguchi K, Wu LW, Grivennikov SI, de Jong PR, Lian I, Yu FX. et al. A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature. 2015;519:57-62
52. Li P, Silvis MR, Honaker Y, Lien WH, Arron ST, Vasioukhin V. alphaE-catenin inhibits a Src-YAP1 oncogenic module that couples tyrosine kinases and the effector of Hippo signaling pathway. Genes Dev. 2016;30:798-811
53. Rosenbluh J, Nijhawan D, Cox AG, Li X, Neal JT, Schafer EJ. et al. beta-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell. 2012;151:1457-73
54. Schlegelmilch K, Mohseni M, Kirak O, Pruszak J, Rodriguez JR, Zhou D. et al. Yap1 acts downstream of alpha-catenin to control epidermal proliferation. Cell. 144: 782-95.
55. Liu CY, Lv X, Li T, Xu Y, Zhou X, Zhao S. et al. PP1 cooperates with ASPP2 to dephosphorylate and activate TAZ. J Biol Chem. 2011;286:5558-66
56. Wang P, Bai Y, Song B, Wang Y, Liu D, Lai Y. et al. PP1A-mediated dephosphorylation positively regulates YAP2 activity. PLoS One. 6: e24288.
57. Li L, Wang J, Zhang Y, Zhang Y, Ma L, Weng W. et al. MEK1 promotes YAP and their interaction is critical for tumorigenesis in liver cancer. FEBS Lett. 2013;587:3921-7
58. You B, Yang YL, Xu Z, Dai Y, Liu S, Mao JH. et al. Inhibition of ERK1/2 down-regulates the Hippo/YAP signaling pathway in human NSCLC cells. Oncotarget. 2015;6:4357-68
59. Kim W, Khan SK, Gvozdenovic-Jeremic J, Kim Y, Dahlman J, Kim H. et al. Hippo signaling interactions with Wnt/beta-catenin and Notch signaling repress liver tumorigenesis. J Clin Invest. 2017;127:137-52
60. Han H, Yang B, Nakaoka HJ, Yang J, Zhao Y, Le Nguyen K. et al. Hippo signaling dysfunction induces cancer cell addiction to YAP. Oncogene. 2018
61. Yu FX, Guan KL. The Hippo pathway: regulators and regulations. Genes Dev. 2013;27:355-71
62. Zhang Y, Xia H, Ge X, Chen Q, Yuan D, Chen Q. et al. CD44 acts through RhoA to regulate YAP signaling. Cell Signal. 2014;26:2504-13
63. Chakraborty S, Njah K, Pobbati AV, Lim YB, Raju A, Lakshmanan M. et al. Agrin as a Mechanotransduction Signal Regulating YAP through the Hippo Pathway. Cell Rep. 2017;18:2464-79
64. Oku Y, Nishiya N, Shito T, Yamamoto R, Yamamoto Y, Oyama C. et al. Small molecules inhibiting the nuclear localization of YAP/TAZ for chemotherapeutics and chemosensitizers against breast cancers. FEBS Open Bio. 2015;5:542-9
65. Yang N, Morrison CD, Liu P, Miecznikowski J, Bshara W, Han S. et al. TAZ induces growth factor-independent proliferation through activation of EGFR ligand amphiregulin. Cell Cycle. 2012;11:2922-30
66. He C, Mao D, Hua G, Lv X, Chen X, Angeletti PC. et al. The Hippo/YAP pathway interacts with EGFR signaling and HPV oncoproteins to regulate cervical cancer progression. EMBO Mol Med. 2015;7:1426-49
67. He C, Lv X, Hua G, Lele SM, Remmenga S, Dong J. et al. YAP forms autocrine loops with the ERBB pathway to regulate ovarian cancer initiation and progression. Oncogene. 2015;34:6040-54
68. Fan R, Kim NG, Gumbiner BM. Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:2569-74
69. Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH. et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell. 2012;150:780-91
70. Wennmann DO, Vollenbroker B, Eckart AK, Bonse J, Erdmann F, Wolters DA. et al. The Hippo pathway is controlled by Angiotensin II signaling and its reactivation induces apoptosis in podocytes. Cell Death Dis. 2014;5:e1519
71. Haak AJ, Kostallari E, Sicard D, Ligresti G, Choi KM, Caporarello N. et al. Selective YAP/TAZ inhibition in fibroblasts via dopamine receptor D1 agonism reverses fibrosis. Sci Transl Med. 2019:11
72. Mi W, Lin Q, Childress C, Sudol M, Robishaw J, Berlot CH. et al. Geranylgeranylation signals to the Hippo pathway for breast cancer cell proliferation and migration. Oncogene. 2015;34:3095-106
73. Yu FX, Luo J, Mo JS, Liu G, Kim YC, Meng Z. et al. Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell. 2014;25:822-30
74. Feng X, Degese MS, Iglesias-Bartolome R, Vaque JP, Molinolo AA, Rodrigues M. et al. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell. 2014;25:831-45
75. Kim NG, Gumbiner BM. Adhesion to fibronectin regulates Hippo signaling via the FAK-Src-PI3K pathway. J Cell Biol. 2015;210:503-15
76. Wong KF, Liu AM, Hong W, Xu Z, Luk JM. Integrin alpha2beta1 inhibits MST1 kinase phosphorylation and activates Yes-associated protein oncogenic signaling in hepatocellular carcinoma. Oncotarget. 2016;7:77683-95
77. Cosset E, Ilmjarv S, Dutoit V, Elliott K, von Schalscha T, Camargo MF. et al. Glut3 Addiction Is a Druggable Vulnerability for a Molecularly Defined Subpopulation of Glioblastoma. Cancer Cell. 2017;32:856-68
78. Wei SC, Fattet L, Tsai JH, Guo Y, Pai VH, Majeski HE. et al. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat Cell Biol. 2015;17:678-88
79. Ley K, Rivera-Nieves J, Sandborn WJ, Shattil S. Integrin-based therapeutics: biological basis, clinical use and new drugs. Nat Rev Drug Discov. 2016;15:173-83
80. Chakraborty S, Lakshmanan M, Swa HL, Chen J, Zhang X, Ong YS. et al. An oncogenic role of Agrin in regulating focal adhesion integrity in hepatocellular carcinoma. Nat Commun. 2015;6:6184
81. Azad T, Janse van Rensburg HJ, Lightbody ED, Neveu B, Champagne A, Ghaffari A. et al. A LATS biosensor screen identifies VEGFR as a regulator of the Hippo pathway in angiogenesis. Nat Commun. 2018;9:1061
82. Wang X, Freire Valls A, Schermann G, Shen Y, Moya IM, Castro L. et al. YAP/TAZ Orchestrate VEGF Signaling during Developmental Angiogenesis. Dev Cell. 2017;42:462-78
83. Yu FX, Zhang Y, Park HW, Jewell JL, Chen Q, Deng Y. et al. Protein kinase A activates the Hippo pathway to modulate cell proliferation and differentiation. Genes & development. 2013;27:1223-32
84. Kim M, Kim M, Lee S, Kuninaka S, Saya H, Lee H. et al. cAMP/PKA signalling reinforces the LATS-YAP pathway to fully suppress YAP in response to actin cytoskeletal changes. EMBO J. 2013;32:1543-55
85. Serrano I, McDonald PC, Lock F, Muller WJ, Dedhar S. Inactivation of the Hippo tumour suppressor pathway by integrin-linked kinase. Nature communications. 2013;4:2976
86. Zhu H, Wang DD, Yuan T, Yan FJ, Zeng CM, Dai XY. et al. Multi-kinase inhibitor CT-707 targets liver cancer by interrupting the hypoxia-activated IGF- 1R-YAP axis. Cancer Res. 2018
87. Kedan A, Verma N, Saroha A, Shreberk-Shaked M, Muller AK, Nair NU. et al. PYK2 negatively regulates the Hippo pathway in TNBC by stabilizing TAZ protein. Cell Death Dis. 2018;9:985
88. Si Y, Ji X, Cao X, Dai X, Xu L, Zhao H. et al. Src Inhibits the Hippo Tumor Suppressor Pathway through Tyrosine Phosphorylation of Lats1. Cancer Res. 2017;77:4868-80
89. Rosenbluh J, Nijhawan D, Cox AG, Li X, Neal JT, Schafer EJ. et al. beta-Catenin-Driven Cancers Require a YAP1 Transcriptional Complex for Survival and Tumorigenesis. Cell. 151: 1457-73.
90. Smoot RL, Werneburg NW, Sugihara T, Hernandez MC, Yang L, Mehner C. et al. Platelet-derived growth factor regulates YAP transcriptional activity via Src family kinase dependent tyrosine phosphorylation. J Cell Biochem. 2018;119:824-36
91. Edwards DN, Ngwa VM, Wang S, Shiuan E, Brantley-Sieders DM, Kim LC. et al. The receptor tyrosine kinase EphA2 promotes glutamine metabolism in tumors by activating the transcriptional coactivators YAP and TAZ. Sci Signal. 2017:10
92. Lin L, Sabnis AJ, Chan E, Olivas V, Cade L, Pazarentzos E. et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat Genet. 2015:250-56
93. Gokey JJ, Sridharan A, Xu Y, Green J, Carraro G, Stripp BR. et al. Active epithelial Hippo signaling in idiopathic pulmonary fibrosis. JCI Insight. 2018:3
94. Zhang S, Song X, Cao D, Xu Z, Fan B, Che L. et al. Pan-mTOR inhibitor MLN0128 is effective against intrahepatic cholangiocarcinoma in mice. J Hepatol. 2017;67:1194-203
95. Liang N, Zhang C, Dill P, Panasyuk G, Pion D, Koka V. et al. Regulation of YAP by mTOR and autophagy reveals a therapeutic target of tuberous sclerosis complex. J Exp Med. 2014;211:2249-63
96. Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S. et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nature cell biology. 2014;16:357-66
97. Wang Z, Wu Y, Wang H, Zhang Y, Mei L, Fang X. et al. Interplay of mevalonate and Hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc Natl Acad Sci U S A. 2014;111:E89-98
98. Xia H, Dai X, Yu H, Zhou S, Fan Z, Wei G. et al. EGFR-PI3K-PDK1 pathway regulates YAP signaling in hepatocellular carcinoma: the mechanism and its implications in targeted therapy. Cell Death Dis. 2018;9:269
99. Sabra H, Brunner M, Mandati V, Wehrle-Haller B, Lallemand D, Ribba AS. et al. beta1 integrin-dependent Rac/group I PAK signaling mediates YAP activation of Yes-associated protein 1 (YAP1) via NF2/merlin. J Biol Chem. 2017;292:19179-97
100. Tsutsumi R, Masoudi M, Takahashi A, Fujii Y, Hayashi T, Kikuchi I. et al. YAP and TAZ, Hippo signaling targets, act as a rheostat for nuclear SHP2 function. Dev Cell. 2013;26:658-65
101. Xie J, Si X, Gu S, Wang M, Shen J, Li H. et al. Allosteric Inhibitors of SHP2 with Therapeutic Potential for Cancer Treatment. J Med Chem. 2017;60:10205-19
102. Jung DE, Park SB, Kim K, Kim C, Song SY. CG200745, an HDAC inhibitor, induces anti-tumour effects in cholangiocarcinoma cell lines via miRNAs targeting the Hippo pathway. Sci Rep. 2017;7:10921
103. Heinemann A, Cullinane C, De Paoli-Iseppi R, Wilmott JS, Gunatilake D, Madore J. et al. Combining BET and HDAC inhibitors synergistically induces apoptosis of melanoma and suppresses AKT and YAP signaling. Oncotarget. 2015;6:21507-21
104. Zanconato F, Battilana G, Forcato M, Filippi L, Azzolin L, Manfrin A. et al. Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat Med. 2018;24:1599-610
105. Elisi GM, Santucci M, D'Arca D, Lauriola A, Marverti G, Losi L. et al. Repurposing of Drugs Targeting YAP-TEAD Functions. Cancers (Basel). 2018:10
106. Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA. et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012:1300-5
107. Qi Y, Yu J, Han W, Fan X, Qian H, Wei H. et al. A splicing isoform of TEAD4 attenuates the Hippo-YAP signalling to inhibit tumour proliferation. Nat Commun. 2016;7:ncomms11840
108. Holden JK, Cunningham CN. Targeting the Hippo Pathway and Cancer through the TEAD Family of Transcription Factors. Cancers (Basel). 2018:10
109. Schwartzman M, Reginensi A, Wong JS, Basgen JM, Meliambro K, Nicholas SB. et al. Podocyte-Specific Deletion of Yes-Associated Protein Causes FSGS and Progressive Renal Failure. J Am Soc Nephrol. 2016;27:216-26
110. Zhang H, Ramakrishnan SK, Triner D, Centofanti B, Maitra D, Gyorffy B. et al. Tumor-selective proteotoxicity of verteporfin inhibits colon cancer progression independently of YAP1. Sci Signal. 2015;8:ra98
111. Dasari VR, Mazack V, Feng W, Nash J, Carey DJ, Gogoi R. Verteporfin exhibits YAP-independent anti-proliferative and cytotoxic effects in endometrial cancer cells. Oncotarget. 2017;8:28628-40
112. Gibault F, Bailly F, Corvaisier M, Coevoet M, Huet G, Melnyk P. et al. Molecular Features of the YAP Inhibitor Verteporfin: Synthesis of Hexasubstituted Dipyrrins as Potential Inhibitors of YAP/TAZ, the Downstream Effectors of the Hippo Pathway. ChemMedChem. 2017;12:954-61
113. Barth MC S, Montalbetti C, Spitzer L. Preparation of new 4-(1,1-Dioxo- 1,2-benzothiazol-3-yl)hydrazono]methyl]-2-methoxyphenols as Inhibitors of the YAP/TAZ-TEAD Interaction and their use in the treatment of Malignant Mesothelioma. WO 2017064277 A1 20170420: Inventiva. 2017
114. Zhang Z, Lin Z, Zhou Z, Shen HC, Yan SF, Mayweg AV. et al. Structure- Based Design and Synthesis of Potent Cyclic Peptides Inhibiting the YAP-TEAD Protein-Protein Interaction. ACS medicinal chemistry letters. 2014;5:993-8
115. Crook ZR, Sevilla GP, Friend D, Brusniak MY, Bandaranayake AD, Clarke M. et al. Mammalian display screening of diverse cystine-dense peptides for difficult to drug targets. Nat Commun. 2017;8:2244
116. Jiao S, Wang H, Shi Z, Dong A, Zhang W, Song X. et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell. 2014;25:166-80
117. Pobbati AV, Chan SW, Lee I, Song H, Hong W. Structural and functional similarity between the Vgll1-TEAD and the YAP-TEAD complexes. Structure. 2012;20:1135-40
118. Pobbati AV, Hong W. Emerging roles of TEAD transcription factors and its coactivators in cancers. Cancer Biol Ther. 2013;14:390-8
119. Pobbati AV, Han X, Hung AW, Weiguang S, Huda N, Chen GY. et al. Targeting the Central Pocket in Human Transcription Factor TEAD as a Potential Cancer Therapeutic Strategy. Structure. 2015;23:2076-86
120. Noland CL, Gierke S, Schnier PD, Murray J, Sandoval WN, Sagolla M. et al. Palmitoylation of TEAD Transcription Factors Is Required for Their Stability and Function in Hippo Pathway Signaling. Structure. 2016;24:179-86
121. Chan P, Han X, Zheng B, DeRan M, Yu J, Jarugumilli GK. et al. Autopalmitoylation of TEAD proteins regulates transcriptional output of the Hippo pathway. Nat Chem Biol. 2016;12:282-9
122. Li Y, Liu S, Ng EY, Li R, Poulsen A, Hill J. et al. Structural and ligand-binding analysis of the YAP-binding domain of transcription factor TEAD4. Biochem J. 2018;475:2043-55
123. Bum-Erdene K, Zhou D, Gonzalez-Gutierrez G, Ghozayel MK, Si Y, Xu D. et al. Small-Molecule Covalent Modification of Conserved Cysteine Leads to Allosteric Inhibition of the TEADYap Protein-Protein Interaction. Cell Chem Biol. 2018 P378-89
124. Konradi AaL T. Non-fused tricyclic compounds. WO2018204532: Vivace Therapeutics. 2018
125. Lu W, Wang J, Li Y, Tao H, Xiong H, Lian F. et al. Discovery and biological evaluation of vinylsulfonamide derivatives as highly potent, covalent TEAD autopalmitoylation inhibitors. Eur J Med Chem. 2019;184:111767
126. Wu X. Tead transcription factor autopalmitoylation inhibitors. WO2017053706 A1. 2016
127. Kaneda Aea. Discovery of a first-in-class TEAD inhibitor which directly inhibits YAP/TAZ-TEAD protein-protein interaction and shows a potent anti- tumor effect in malignant pleural mesothelioma. Abstract 3086 AACR Annual meting. 2019
128. Kim NG, Gumbiner BM. Cell contact and Nf2/Merlin-dependent regulationof TEAD palmitoylation and activity. Proc Natl Acad Sci U S A. 2019;116:9877-82
129. Moya IM, Halder G. Hippo-YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat Rev Mol Cell Biol. 2019;20:211-26
130. Pobbati AV, Mejuch T, Chakraborty S, Karatas H, Bharath SR, Gueret SM. et al. Identification of Quinolinols as Activators of TEAD-Dependent Transcription. ACS Chem Biol. 2019;14:2909-21
131. Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2001;98:8554-9
132. Yu J, Alharbi A, Shan H, Hao Y, Snetsinger B, Rauh MJ. et al. TAZ induces lung cancer stem cell properties and tumorigenesis by up-regulating ALDH1A1. Oncotarget. 2017;8:38426-43
133. Yang CS, Stampouloglou E, Kingston NM, Zhang L, Monti S, Varelas X. Glutamine-utilizing transaminases are a metabolic vulnerability of TAZ/YAP- activated cancer cells. EMBO Rep. 2018:19
134. Guerrant W, Kota S, Troutman S, Mandati V, Fallahi M, Stemmer-Rachamimov A. et al. YAP Mediates Tumorigenesis in Neurofibromatosis Type 2 by Promoting Cell Survival and Proliferation through a COX-2-EGFR Signaling Axis. Cancer Res. 2016;76:3507-19
135. Xu G, Wang Y, Li W, Cao Y, Xu J, Hu Z. et al. COX-2 Forms Regulatory Loop with YAP to Promote Proliferation and Tumorigenesis of Hepatocellular Carcinoma Cells. Neoplasia. 2018;20:324-34
136. Bendinelli P, Maroni P, Matteucci E, Luzzati A, Perrucchini G, Desiderio MA. Hypoxia inducible factor-1 is activated by transcriptional co-activator with PDZ-binding motif (TAZ) versus WWdomain-containing oxidoreductase (WWOX) in hypoxic microenvironment of bone metastasis from breast cancer. Eur J Cancer. 2013;49:2608-18
137. Xu MZ, Chan SW, Liu AM, Wong KF, Fan ST, Chen J. et al. AXL receptor kinase is a mediator of YAP-dependent oncogenic functions in hepatocellular carcinoma. Oncogene. 2010:1229-40
138. Ghiso E, Migliore C, Ciciriello V, Morando E, Petrelli A, Corso S. et al. YAP- Dependent AXL Overexpression Mediates Resistance to EGFR Inhibitors in NSCLC. Neoplasia. 2017;19:1012-21
139. Yuan WC, Pepe-Mooney B, Galli GG, Dill MT, Huang HT, Hao M. et al. NUAK2 is a critical YAP target in liver cancer. Nat Commun. 2018;9:4834
140. Gill MK, Christova T, Zhang YY, Gregorieff A, Zhang L, Narimatsu M. et al. A feed forward loop enforces YAP/TAZ signaling during tumorigenesis. Nat Commun. 2018;9:3510
141. Tschaharganeh DF, Chen X, Latzko P, Malz M, Gaida MM, Felix K. et al. Yes- associated protein up-regulates Jagged-1 and activates the Notch pathway in human hepatocellular carcinoma. Gastroenterology. 2013;144:1530-42
142. Ji X, Song L, Sheng L, Gao A, Zhao Y, Han S. et al. Cyclopeptide RA-V Inhibits Organ Enlargement and Tumorigenesis Induced by YAP Activation. Cancers (Basel). 2018:10
143. Eisinger-Mathason TS, Mucaj V, Biju KM, Nakazawa MS, Gohil M, Cash TP. et al. Deregulation of the Hippo pathway in soft-tissue sarcoma promotes FOXM1 expression and tumorigenesis. Proc Natl Acad Sci U S A. 2015;112:E3402-11
144. Fisher ML, Kerr C, Adhikary G, Grun D, Xu W, Keillor JW. et al. Transglutaminase Interaction with alpha6/beta4-Integrin Stimulates YAP1- Dependent DeltaNp63alpha Stabilization and Leads to Enhanced Cancer Stem Cell Survival and Tumor Formation. Cancer Res. 2016;76:7265-76
145. Liu CY, Pobbati AV, Huang Z, Cui L, Hong W. Transglutaminase 2 Is a Direct Target Gene of YAP/TAZ-Letter. Cancer Res. 2017;77:4734-5
146. Kakiuchi-Kiyota S, Schutten MM, Zhong Y, Crawford JJ, Dey A. Safety Considerations in the Development of Hippo Pathway Inhibitors in Cancers. Front Cell Dev Biol. 2019;7:156
147. Morin-Kensicki EM, Boone BN, Howell M, Stonebraker JR, Teed J, Alb JG. et al. Defects in yolk sac vasculogenesis, chorioallantoic fusion, and embryonic axis elongation in mice with targeted disruption of Yap65. Mol Cell Biol. 2006;26:77-87
148. Hossain Z, Ali SM, Ko HL, Xu J, Ng CP, Guo K. et al. Glomerulocystic kidney disease in mice with a targeted inactivation of Wwtr1. Proc Natl Acad Sci U S A. 2007;104:1631-6
149. Tian Y, Kolb R, Hong JH, Carroll J, Li D, You J. et al. TAZ promotes PC2 degradation through a SCFbeta-Trcp E3 ligase complex. Mol Cell Biol. 2007;27:6383-95
150. Makita R, Uchijima Y, Nishiyama K, Amano T, Chen Q, Takeuchi T. et al. Multiple renal cysts, urinary concentration defects, and pulmonary emphysematous changes in mice lacking TAZ. Am J Physiol Renal Physiol. 2008;294:F542-53
151. Xin M, Kim Y, Sutherland LB, Qi X, McAnally J, Schwartz RJ. et al. Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci Signal. 2011;4:ra70
152. Mahoney JE, Mori M, Szymaniak AD, Varelas X, Cardoso WV. The hippo pathway effector Yap controls patterning and differentiation of airway epithelial progenitors. Dev Cell. 2014;30:137-50
153. Reginensi A, Scott RP, Gregorieff A, Bagherie-Lachidan M, Chung C, Lim DS. et al. Yap- and Cdc42-dependent nephrogenesis and morphogenesis during mouse kidney development. PLoS Genet. 2013;9:e1003380
154. Lu L, Finegold MJ, Johnson RL. Hippo pathway coactivators Yap and Taz are required to coordinate mammalian liver regeneration. Exp Mol Med. 2018;50:e43
155. Azzolin L, Panciera T, Soligo S, Enzo E, Bicciato S, Dupont S. et al. YAP/TAZ incorporation in the beta-catenin destruction complex orchestrates the Wnt response. Cell. 2014;158:157-70
156. Chan SW, Lim CJ, Loo LS, Chong YF, Huang C, Hong W. TEADs mediate nuclear retention of TAZ to promote oncogenic transformation. J Biol Chem. 2009;284:14347-58
157. Ege N, Dowbaj AM, Jiang M, Howell M, Hooper S, Foster C. et al. Quantitative Analysis Reveals that Actin and Src-Family Kinases Regulate Nuclear YAP1 and Its Export. Cell Syst. 2018;6:692-708
158. Manning SA, Dent LG, Kondo S, Zhao ZW, Plachta N, Harvey KF. Dynamic Fluctuations in Subcellular Localization of the Hippo Pathway Effector Yorkie In Vivo. Curr Biol. 2018;28:1651-60
159. Jhaveri KD, Wanchoo R, Sakhiya V, Ross DW, Fishbane S. Adverse Renal Effects of Novel Molecular Oncologic Targeted Therapies: A Narrative Review. Kidney Int Rep. 2017;2:108-23
160. Knott EP, Assi M, Rao SN, Ghosh M, Pearse DD. Phosphodiesterase Inhibitors as a Therapeutic Approach to Neuroprotection and Repair. Int J Mol Sci. 2017:18
161. Smith JS, Lefkowitz RJ, Rajagopal S. Biased signalling: from simple switches to allosteric microprocessors. Nat Rev Drug Discov. 2018;17:243-60
162. Moroishi T, Park HW, Qin B, Chen Q, Meng Z, Plouffe SW. et al. A YAP/TAZ-induced feedback mechanism regulates Hippo pathway homeostasis. Genes & development. 2015;29:1271-84
163. Chen Q, Zhang N, Xie R, Wang W, Cai J, Choi KS. et al. Homeostatic control of Hippo signaling activity revealed by an endogenous activating mutation in YAP. Genes Dev. 2015;29:1285-97
Author contact
![]() Corresponding author: ajaybabuvpa-star.edu.sg
Corresponding author: ajaybabuvpa-star.edu.sg