Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(13):5802-5814. doi:10.7150/thno.44772 This issue Cite
Research Paper
89Zr-PET imaging of DNA double-strand breaks for the early monitoring of response following α- and β-particle radioimmunotherapy in a mouse model of pancreatic ductal adenocarcinoma
Sophie Poty1 ![]() , Komal Mandleywala1, Edward O'Neill2, James C. Knight2,3, Bart Cornelissen2, Jason S. Lewis1,4,5,6
, Komal Mandleywala1, Edward O'Neill2, James C. Knight2,3, Bart Cornelissen2, Jason S. Lewis1,4,5,6 ![]()
1. Department of Radiology, Memorial Sloan Kettering Cancer Center, NY, USA
2. CRUK/MRC Oxford Institute of Radiation Oncology, Department of Oncology, University of Oxford, Oxford, UK
3. School of Natural and Environmental Sciences, Newcastle University, Newcastle upon Tyne, UK
4. Radiochemistry and Molecular Imaging Probes Core, Memorial Sloan Kettering Cancer Center, New York, NY, USA
5. Weill Cornell Medical College, New York, NY, USA
6. Molecular Pharmacology Program, Memorial Sloan Kettering Cancer Center, New York, NY, USA
Received 2020-2-10; Accepted 2020-4-13; Published 2020-4-27
Abstract

Rationale: The evaluation of early treatment response is critical for patient prognosis and treatment planning. When the current methods rely on invasive protocols that evaluate the expression of DNA damage markers on patient biopsy samples, we aim to evaluate a non-invasive PET imaging approach to monitor the early expression of the phosphorylated histone γH2AX in the context of pancreatic cancer targeted radionuclide therapy. Pancreatic ductal adenocarcinoma has a poor patient prognosis due to the absence of curative treatment for patients with advanced disease. There is therefore a critical need for the fast clinical translation of new therapeutic options. In line with these observations, our group has been focusing on the development of radiotheranostic agents based on a fully human monoclonal antibody (5B1) with exceptional affinity for CA19.9, an antigen overexpressed in PDAC. Two on-going clinical trials resulted from these efforts, one with 89Zr (diagnosis) and one with 177Lu (β-particle therapy). More recently, we successfully developed and evaluated in PDAC mouse models a targeted α-therapy strategy with high clinical translation potential. We aim to expedite the clinical translation of the developed radioimmunotherapy approaches by investigating the early therapeutic response and effect of radiation therapy in a PDAC mouse model via PET imaging.
Methods: Mice bearing BxPC3 tumor xenografts were treated with α- and β-particle pretargeted radioimmunotherapy (PRIT), external beam radiotherapy (EBRT), or sham-treated (vehicle). The phosphorylated histone γH2AX produced as a response to DNA double strand breaks was quantified with the PET radiotracer, [89Zr]Zr-DFO-anti-γH2AX-TAT.
Results: PET imaging studies in BxPC3 PDAC mouse models demonstrated increased uptake of [89Zr]Zr-DFO-anti-γH2AX-TAT (6.29 ± 0.15 %IA/g) following β-PRIT in BxPC3 PDAC xenografts as compared to the saline control group (4.58 ± 0.76 %IA/g) and EBRT control group (5.93 ± 0.76 %IA/g). Similarly, significantly higher uptake of [89Zr]Zr-DFO-anti-γH2AX-TAT was observed in tumors of the 225Ac-PRIT and EBRT (10 Gy) cohorts (7.37 ± 1.23 and 6.80 ± 1.24 %IA/g, respectively) compared to the negative control cohort (5.08 ± 0.95 %IA/g). Ex vivo γH2AX immunohistochemistry and immunofluorescence analysis correlated with in vivo 89Zr-anti-γH2AX PET/CT imaging with increased γH2AX positive cell and γH2AX foci per cell in the treated cohorts. When α-PRIT resulted in prolonged overall survival of treated animals (107.5 days) as compared to β-PRIT (73.0 days), no evidence of difference in [89Zr]Zr-DFO-anti-γH2AX-TAT uptake at the tumor site was observed, highlighting that DNA damage is not the sole radiobiology paradigm and that off-targeted (bystander) effects should be considered.
Conclusions: PET imaging studies with [89Zr]Zr-DFO-anti-γH2AX-TAT following α- and β-particle PRIT in a BxPC3 PDAC subcutaneous xenograft mouse model allowed the monitoring of tumor radiobiological response to treatment.
Keywords: Radioimmunotherapy, DNA damage, γH2AX, PET, pancreatic cancer
Introduction
With a 5-year survival rate in the US of 9%, pancreatic ductal adenocarcinoma (PDAC) is a uniformly lethal disease [1,2]. This dire prognosis is partly due to a lack of screening and diagnostic tools, but also limited therapeutic options [1]. To solve these issues, our group has been developing clinically translatable diagnostic and therapeutic approaches based on antibody radioconjugates for the positron emission tomography (PET) imaging and radioimmunotherapy (RIT) of PDAC [3-6].
The Sialyl-Lewisa (sLea), also known as CA19.9 antigen, is one of the most widely studied pancreatic cancer biomarkers as patients' serum levels correlate with cancer stage, prognosis or treatment response [7-9]. The fully human 5B1 antibody, generated from blood lymphocytes from a patient immunized with a sLea - KLH vaccine, demonstrates exceptional affinity and high specificity for an extracellular epitope of CA19.9 [10,11]. In 2016, the clinical potential of CA19.9 as a therapeutic target was investigated in patients with pancreatic cancers or CA19.9-positive malignancies (NCT02672917) via the weekly intravenous administration of 5B1 (MVT-5873). This antibody was also evaluated as a platform for the specific delivery of radioisotopes to PDAC tumor sites. Two on-going clinical trials with 5B1-based radioimmunoconjugates resulted from these studies: one in which the antibody is labeled with 89Zr for PET imaging (NCT02687230) and a second in which the antibody is labeled with 177Lu for β-RIT (NCT03118349). More recently, our group evaluated the potential of 225Ac labeled 5B1-conjugates for α-RIT in PDAC mouse models [6]. During these preclinical investigations, a pretargeted administration method relying on the inverse electron demand Diels-Alder reaction [4,5,12-15] was compared to conventional RIT. Pretargeted RIT (PRIT) allowed the delivery of α-radioactive payloads to PDAC tumor sites while reducing the mean absorbed dose to healthy tissues, enabling prolonged survival and reduced haematotoxicity in subcutaneous and orthotopic models of PDAC as compared to conventional RIT [6]. The clinical translation of α-based PRIT for PDAC would complement the two on-going 5B1-based clinical trials for the diagnosis (89Zr) and β-therapy (177Lu) of PDAC and has the potential to extend the population of patients eligible for CA19.9-targeted radiotherapy to include those with residual metastatic lesions as well as patients that develop resistance to β-therapy.
To expedite the clinical translation of new targeted radiotherapies (for small molecule, peptide and antibody-based systems), we aim to validate a PET imaging platform for the evaluation of early treatment response. Conventional methods to assess therapeutic response rely on the monitoring of tumor dimensions through anatomic imaging techniques such as X-ray, CT or MRI [16,17]. The visualization and quantification of cellular or biochemical responses to treatment through molecular imaging techniques offer the incredible opportunity for early treatment response monitoring when anatomical changes cannot be measured yet [18]. The monitoring of therapeutic response after radiotherapy can be gauged by the quantification of DNA damage [19]. DNA double-strand breaks (DSBs) are the most severe forms of DNA damage and one of the main mechanisms through which radiotherapy leads to apoptosis [20]. As DNA DSBs are not present in large numbers, they do not represent an ideal target for molecular imaging. An alternative strategy lays in the targeting of DNA damage response proteins such as phosphorylated γH2AX which is present exclusively in the cell nucleus as foci containing thousands of copies around individual DNA DSBs [21-23]. In a clinical setting, quantification of γH2AX foci is performed by the immunohistochemistry evaluation of biopsied samples [24]. However, this method is invasive, associated with risks (e.g. bleeding, infection), not adequate for a disseminated disease and can be biased by tumor heterogeneity. At the preclinical stage, non-invasive SPECT (111In) and PET (89Zr) imaging techniques were developed to quantify γH2AX expression levels in vivo. Cornelissen et al. functionalized an anti-γH2AX antibody that binds both human and mouse γH2AX with a cell penetrating peptide (TAT) that contains a nuclear localization sequence and a chelator (DFO or DTPA) for further radiolabeling with 89Zr or 111In [25-27]. The uptake of the SPECT imaging agent, [111In]In-anti-γH2AX-TAT, and the PET imaging agent, [89Zr]Zr-DFO-anti-γH2AX-TAT, was shown to be linearly dependent of the number of γH2AX foci per cell as well as the absorbed dose of radiation deposited at the tumor site by external beam radiation [25,27]. The ability of [89Zr]Zr-DFO-anti-γH2AX-TAT to monitor early therapeutic response following chemotherapy was investigated in a PDAC mouse model [28]. It was demonstrated that [89Zr]Zr-DFO-anti-γH2AX-TAT allows early indication of therapy response when the clinical standard, [18F]F-FDG, did not provide any indication after initiation nor completion of the treatment [28]. Recently, [111In]In-anti-γH2AX-TAT allowed the non-invasive visualization of DNA damage following molecular targeted radionuclide therapy with [177Lu]Lu-DOTA-TATE in a preclinical mouse model of pancreatic neuroendocrine cancer [29]. A dual isotope SPECT imaging strategy, allowing the simultaneous imaging of 177Lu and 111In, highlighted the correlation of the 177Lu dose heterogeneity to the uptake of [111In]In-anti-γH2AX-TAT. This study opened the path toward the evaluation of the DNA damage imaging approach for a larger range of targeted radionuclide therapy approaches including α- and β-RIT.
In this study, we investigated the ability of [89Zr]Zr-DFO-anti-γH2AX-TAT to monitor the activation of DNA damage response following α- and β-PRIT in a subcutaneous PDAC mouse model. We hypothesized that the in vivo quantification of γH2AX via PET imaging will provide an early readout of α-/β-PRIT efficacy. Such approach to early radiotherapy response is a critical tool for the clinical translation of new radiotherapy approaches, as it would ultimately expedite the translation evaluation time. Furthermore, this technology will be applicable to numerous radiotherapeutic delivery platforms, including small molecules, peptides, antibodies and nanoparticles, and will help streamline their development and translation.
Materials and Methods
Radiochemistry
The synthesis of DOTA-PEG7-Tz was performed using the previously published synthetic pathway [5]. The conjugation of TCO-NHS to 5B1 was performed according to previously published methods [5,6]. 177Lu was obtained from either ITG (Germany) or the University of Missouri Research Reactor through the United States Department of Energy Office of Science. 225Ac was supplied by the United States Department of Energy Office of Science by the Isotope Program in the Office of Nuclear Physics. 177Lu- and 225Ac-radiolabeling was performed according to protocols previously published by our group [5,30]. 225Ac-radiopharmaceuticals for in vitro and in vivo evaluation were prepared and used at least 4 h after the purification of the radiotracers to allow actinium to reach a pseudo-equilibrium state and have an accurate reading of the activity used/injected. 225Ac samples from in vitro and in vivo assays were measured on a gamma counter once secular equilibrium was reached (> 24 hours).
The anti-γH2AX immunoconjugate for molecular imaging of DNA damage response was prepared following the published protocol [27]. Succinctly, the free lysine residues of a mouse monoclonal anti-γH2AX antibody (Merck, clone JBW-301) were activated with an N-hydroxysuccinimidyl ester. The activated antibody was then conjugated to a TAT (GRKKRRQRRRPPQGYG) peptide, a cell penetrating peptide with a non-canonical nuclear localization sequence [25]. The resulting immunoconjugate was then reacted with pSCN-Bn-deferoxamine (DFO) for further radiolabeling with 89Zr. 89Zr was produced through proton-beam bombardment of yttrium foil and isolated in high purity as 89Zr-oxalate at Memorial Sloan Kettering Cancer Center according to a previously published procedure [31]. 89Zr-oxalate (10 MBq) was neutralized to pH 6.9-7.2 with 1 M Na2CO3. The DFO-anti-γH2AX-TAT in PBS buffer (pH 7.4) was added (100 μg), and the reaction was incubated at room temperature for 1 h with a gentle shaking (400 rpm).
Purity and radiolabeling efficacies were quantified through instant thin-later chromatography (iTLC) with a 50 mM ethylenediaminetetraacetic acid (pH 5, 177Lu and 225Ac), or a 0.1 M sodium citrate (pH 5.0, 89Zr) mobile phase. Radiochemical purity was routinely >99%.
Cell lines and xenograft models
CA19.9-positive BxPC3 cells were grown in RPMI medium modified to contain 4.5 g/L sodium bicarbonate and supplemented with 10 % (vol/vol) heat-inactivated FCS, 100 IU penicillin, 100 μg/mL streptomycin, 10 mM HEPES, and 10 cc/L nonessential amino acids. All animals were treated according to the guidelines approved by the Research Animal Resource Center and Institutional Animal Care and Use Committee at Memorial Sloan Kettering Cancer Center. Female athymic homozygous nude mice (strain: Crl:NU(NCr)-Foxn1nu, Charles River Laboratories, Wilmington, MA, aged 6-8 weeks) were xenografted subcutaneously with 5×106 BxPC3 cells suspended in 150 μL of a 1:1 Matrigel (Becton Dickson, Bedford, MA) and cell culture medium mixture.
Cell viability
Cell viability 48 hours after [177Lu]Lu-DOTA-5B1 and [225Ac]Ac-DOTA-5B1 treatment was evaluated. BxPC3 cells (2x104 cells) were seeded in 96 well plates and incubated for 4 h. [177Lu]Lu-DOTA-5B1 (220 MBq/mL to 0.55 kBq/mL) and [225Ac]Ac-DOTA-5B1 (2.97 MBq/mL to 7.4 Bq/mL) were added to the cells for 48h. Each concentration was run as quadruplicates. After 48 hours of treatment, a tetrazolium salt MTT (10 μL, 50 μg/mL) was added and incubated at 37°C for 1 hour. After that, a 0.04 M HCl solution in 10% Triton-x isopropanol (100 μL) was added. Absorption at 570 nm was detected on a SpectraMax M5 (Molecular Devices) plate reader. This experiment was performed twice; ED50 values are presented as the mean value of these two replicates.
Clonogenic survival
BxPC3 cells (1,000-3,000) were plated in 6-well plates in growth media and incubated for 4 h at 37°C in 5% CO2. Cells were then treated with [177Lu]Lu-DOTA-5B1 (0.22 to 1.51 MBq/mL), [225Ac]Ac-DOTA-5B1 (0.08 to 5.01 kBq/mL), or sham treated (cell media + radiopharmaceutical vehicle). After 48 h, the treatment-containing supernatant media was removed and cells were washed with PBS. Cells were then incubated with 2.5 mL of growth media for 12 days at 37°C in 5% CO2. After 12 days, the media was removed and cells were gently washed with PBS. Cells were then fixed and stained with 3 mL of a mixture of 6% glutaraldehyde and 0.5% crystal violet. Colonies of at least 50 cells (~ 0.5 mm) were counted manually. The plating efficiency, defined as the ratio of the number of colonies formed by the number of cells seeded, was determined using the sham treated cells. The surviving fraction is defined as the number of colonies formed after treatment divided by the number of cells seeded multiplied by the platting efficiency.
CA19.9 pretargeted radioimmunotherapy
Tumor volumes were monitored with a Peira TM900 imaging device (Peira, Belgium). Four weeks after PDAC cells implantation, mice were randomized into cohorts (n = 8) according to their tumor volume. The mean tumor volume of each cohort was approximately equal to 150 mm3. The study was then blinded for the technical person responsible of tumor measurements so that the volume acquired were not biased. One day after randomization, mice receiving PRIT were injected with 5B1-TCO (200 μg, 1 nmol; in 200 μL of PBS). Seventy-two hours later, mice were injected with the appropriate radioactive payload: either [177Lu]Lu-DOTA-PEG7-Tz (18.5 MBq, 0.4 nmol) or [225Ac]Ac-DOTA-PEG7-Tz (37 kBq, 0.4 nmol) for PRIT. The control cohorts were injected with either vehicle (0.9 % sterile saline) or 5B1-TCO alone). Tumor volume and body weight were measured twice a week until 120 days or until mice reached the set volume endpoint (> 2000 mm3). Mice were monitored for outward signs of toxicity, including lethargy, loss of appetite, or disseminated intravascular coagulation.
In vivo γH2AX PET imaging
Four weeks post-subcutaneous PDAC cells implantation, mice with an average tumor volume of about 120 mm3 were randomized into cohorts (n=5/cohort) and used for PET/CT imaging studies. Three days post-administration of 177Lu-/225Ac-PRIT (18.5 MBq/0.037 MBq, 0.4 nmol), [89Zr]Zr-DFO-anti-γH2AX-TAT (0.5 MBq, 5.0 μg) was administered intravenously. As a positive control, γH2AX was induced in mice by X-ray irradiation of the tumor (X-RAD 320). One hour before the injection of the PET imaging probe, positive control mice were irradiated (10 Gy) at a dose rate of 1.2 Gy/min. As negative control, mice were mock-treated (0 Gy). PET/CT imaging was performed on an Inveon PET/CT scanner (Siemens) up to 4 days post-injection of [89Zr]Zr-DFO-anti-γH2AX-TAT. Images were processed using the Inveon Research Workplace software package. Volume of interests (VOIs) analysis was performed using 3D Slicer. VOIs were drawn around the major organs of interest and the tumor. CT scans were used for volume measurements (cm3) when PET scans were used for quantification of the activity in the organ of interest. VOIs are presented as percent injected activity per cm3 (%IA/cm3). After imaging, mice were euthanized; organs were collected, weighted and counted on an automatic gamma counter. Results are presented as the injected activity per gram (%IA/g).
Ex vivo γH2AX staining
Separate mice cohorts (n=3/cohort) were treated as previously described. However, three days post-injection of the radionuclide therapy, mice were euthanized and their tumors were collected for γH2AX staining. Immunohistochemistry and immunofluorescence were carried out on formalin-fixed, paraffin-embedded 10-μm-thick tumor sections. Sections were submitted to MSKCC Molecular Cytology Core Facility for γH2AX as well as hematoxylin and eosin staining. Slides were scanned using a Pannoramic Flash 250 slide scanner from 3DHistech (Budapest, Hungary) - Zeiss 20x/0.8NA objective. Immunohistochemistry was used for the determination of γH2AX positive cells when immunofluorescence was used for the detection of γH2AX foci per cell. Immunohistochemistry cell positive analysis was performed in random tumor areas (n = 8/sample) using the open source software QuPath [32]. The total number of cells was determined using the hematoxylin staining whereas the DAB staining was used to quantify the percentage of positive cells. Immunofluorescence analysis was performed using the open source image processing package Fiji [33]. The mean FITC fluorescence intensity was normalized to the DAPI fluorescent signal and the number of foci/cell were counted in random tumor areas (n =6/sample).
Statistical analyses
All data are represented as mean value ± standard deviation (n = 5-8, unless otherwise noted). The sample sizes were selected taking into account both statistical consideration and the exigencies of funding. For the PET imaging and biodistribution studies, a minimum of 4 animals were required to obtain a statistical power > 0.8 (bilateral test, δ = 1.3, σ = 0.6, α = 0.05). For the therapy studies, a minimum of 8 animals were required to obtain a statistical power > 0.8 (bilateral test, δ = 25, σ = 17, α = 0.05). The significance analyses were performed using Prism 8.3, employing one-way ANOVA followed by a Dunnett's multiple comparisons test and log rank Mantel-Cox test as detailed in figure legends. A P value of <0.05 was considered significant.
Results
Exposure to α- and β-RIT results in differential viability in BxPC3 PDAC cell line
Viability of the BxPC3 PDAC cell line after treatment with [225Ac]Ac-DOTA-5B1 and [177Lu]Lu-DOTA-5B1 was evaluated through an MTT assay. Reduced viability of the BxPC3 cell line was observed after a 48-hour incubation with [225Ac]Ac-DOTA-5B1 at activity concentrations greater than 0.002 MBq/mL (Figure 1A). The mean ED50 value of two replicate experiments was calculated to 0.26 ± 0.17 MBq/mL. After a 48-hour incubation with [177Lu]Lu-DOTA-5B1, a reduced cell viability was observed as well. However, much higher activity concentrations (> 8 MBq/mL) were required (Figure 1A, Figure S1). The ED50 value after [177Lu]Lu-DOTA-5B1 treatment was calculated to 122 ± 74 MBq/mL. The ratio of cytotoxic effects between the α- and β-RIT treatments was calculated to be 483 ± 39.
In vitro and in vivo evidence of the higher cytotoxic potential of α-RIT compared to β-RIT A. Viability of BxPC3 PDAC cells after a 48 hours incubation with various activity concentration of [177Lu]Lu-DOTA-5B1 and [225Ac]Ac-DOTA-5B1 (n=4 per concentration). B. Clonogenic survival of BxPC3 cells exposed to [177Lu]Lu-DOTA-5B1 (n=6 per concentration). C. Clonogenic survival of BxPC3 cells exposed to [225Ac]Ac-DOTA-5B1 (n=6 per concentration). D. Schematic representation of the PRIT dosing schedule. E. Percent survival as function of time after PRIT injection. Mice were sacrificed when tumor >2000 mm3. Survival data reflect the progression of primary subcutaneous tumors (n=8 per cohort). Values are represented as means, and error bars represent standard deviations.
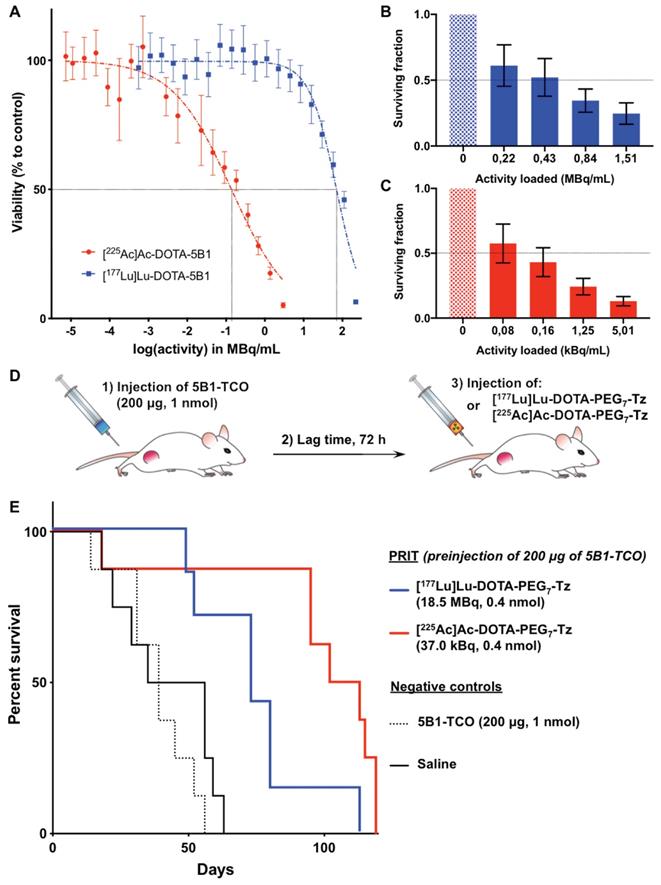
As the method of choice to determine cell reproductive death after ionizing radiation, clonogenic assays were performed and confirmed the higher potency of [225Ac]Ac-DOTA-5B1 as compared to [177Lu]Lu-DOTA-5B1 (Figure 1B-C). Only 0.2 kBq/mL of [225Ac]Ac-DOTA-5B1 were required to achieve a surviving fraction of 0.5 (SF0.5), when 560 kBq/mL were needed to result in similar effects with [177Lu]Lu-DOTA-5B1. The ratio of cytotoxic effects between the α- and β-RIT treatments was here calculated to 2,800.
Based on these two cytotoxic experiments and the different physical properties of the radionuclides used, the relative biological effectiveness between our α- and β-RIT was determined (see SI for calculation details). In the conditions of our MTT assay, [225Ac]Ac-DOTA-5B1 was 3.7 ± 0.3 times more cytotoxic than [177Lu]Lu-DOTA-5B1. Comparatively, a 21-fold ratio was found between [225Ac]Ac-DOTA-5B1 and [177Lu]Lu-DOTA-5B1 in the settings of our clonogenic experiment (SF0.5). The variability of the relative biological effectiveness between α- and β-RIT calculated for these two experiments highlights the importance of the selected survival endpoint.
α-PRIT results in prolonged survival as compared to β-PRIT without sign of toxicity
For α- and β-RIT therapy studies, the pretargeting administration method was chosen due to its demonstrated potency and its ability to reduce absorbed dose to healthy tissues and thereof toxicities (Figure 1D). Based on the aforementioned in vitro assays and our previously published PRIT studies with 177Lu and 225Ac, the injected activities for [177Lu]Lu-DOTA-PEG7-Tz and [225Ac]Ac-DOTA-PEG7-Tz were set to 18.5 MBq and 0.037 MBq, respectively [5,6]. With these injected activities, a factor of 500, in the same range as the ratio observed for equitoxic effects in our MTT assay, applies between the 177Lu- and 225Ac-PRIT. For both PRIT approaches, the injected mass of 5B1-TCO and Tz-radioligand were equal. Our group previously reported similar pharmacokinetics and tumoral uptake between the α- and β-PRIT strategies [5,6]. The absorbed dose delivered to the tumor was estimated using mouse dosimetry extrapolated from biodistribution studies previously performed with 177Lu-/225Ac-PRIT [5,6]. The tumor absorbed dose was evaluated to 102 Gy for 177Lu-PRIT and 75 Gy for 225Ac-PRIT. The accepted threshold for a meaningful response with EBRT in solid tumors being 100 Gy, a significant therapeutic effect was expected for both PRIT treatment group. Control groups were injected with either saline (vehicle) or 5B1-TCO alone.
Survival analysis suggested therapeutic effects in both PRIT cohorts compared with control groups (P values <0.01) (Figure 1E). Median survival for the group treated with 177Lu-PRIT (73.0 days) and 225Ac-PRIT (107.5 days) highlighted the superior therapeutic potential of α-PRIT in our setting (P value < 0.05). Mean tumor volume are provided in supporting information and correlated with the survival results (Figure S2). Even though, the tumor absorbed dose was 25 % less in the α-PRIT cohort, the higher therapeutic efficacy of this treatment as compared to β-PRIT suggests that the relative biological effectiveness of 5 recommended by the US Department of Energy for effective dose calculations with α-particle emitters might be under-estimated. The body weight progression for 225Ac-PRIT showed a transient deviation from control groups and the 177Lu-PRIT cohort from day 3 to day 21 after the administration of the systemic radiotherapy (Figure S2). Nevertheless, no weight loss exceeded the 20% threshold that would have required the early euthanasia of the animal. No other sign of gross acute toxicity was observed. All the mice were sacrificed when their tumor reached the set endpoint of > 2000 mm3 or at 120 days.
Exposure to β-PRIT in BxPC3 PDAC xenografts induces increased uptake of [89Zr]Zr-DFO-anti-γH2AX-TAT as compared to controls
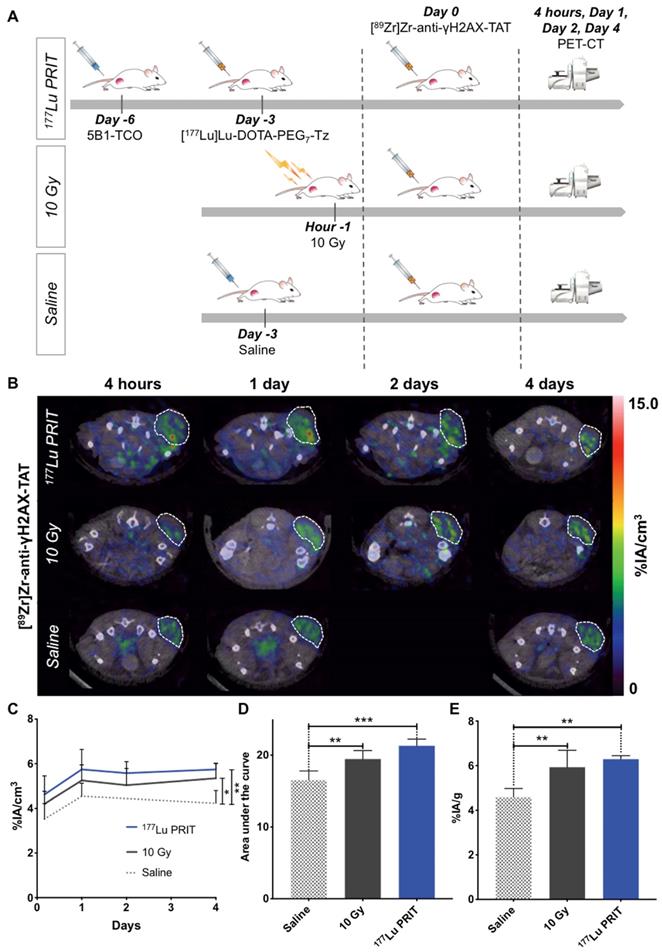
In an attempt to measure in vivo the response to 177Lu-PRIT, [89Zr]Zr-DFO-anti-γH2AX-TAT was used to visualize DNA damage via PET imaging. Mice were treated with 177Lu-PRIT (18.5 MBq) 72 hours prior the injection of the PET anti-γH2AX tracer to allow sufficient dose deposition and induction of γH2AX at the tumor site (Figure 2A) [34]. Positive and negative control cohorts were treated according to established protocols (Figure 2A). PET images were recorded at multiple time points after the injection of the PET tracer to define the best time point for the visualization of DNA damage after PRIT. Representative transverse images are shown in Figure 2B, coronal sections and maximum intensity projections are shown in the supporting information (Figure S3). VOI analyses of tumors were performed at the different time points (Figure 2C, Figure S4A). At the 4 days imaging time point, a statistically significant (P value < 0.01) increased uptake of [89Zr]Zr-DFO-anti-γH2AX-TAT was observed in the 177Lu-PRIT cohort as compared to the negative control. An increasing uptake trend was also observed with the positive control (10 Gy) cohort (P < 0.05). Analysis of the area under the curve revealed a statistically significant increased accumulation of [89Zr]Zr-DFO-anti-γH2AX-TAT over the 4 day imaging period in both 10 Gy and 177Lu-PRIT cohorts (Figure 2D). In every cohort, the uptake of [89Zr]Zr-DFO-anti-γH2AX-TAT in the tumor increases up to 24 hours post-injection of the 89Zr-PET radiotracer before reaching a plateau. Consequently, the 24 hours imaging time point will as of now be used in this study for the in vivo evaluation of DNA damage with [89Zr]Zr-DFO-anti-γH2AX-TAT. After the final image acquisition, mice were sacrificed and organs of interest were counted on a γ-counter to confirm the PET imaging results. The biodistribution confirmed the significantly higher accumulation of [89Zr]Zr-DFO-anti-γH2AX-TAT in irradiated tumors (Figure 2E). Cohorts irradiated with 10 Gy of external beam radiation or 177Lu-PRIT showed increased retention of the 89Zr-PET anti-γH2AX tracer with uptake of 5.93 ± 0.76 and 6.29 ± 0.15 %IA/g, respectively. The uptake in our negative control cohort was significantly lower with 4.58 ± 0.76 %IA/g. The full biodistribution profile of [89Zr]Zr-DFO-anti-γH2AX-TAT (Figure S4C) matches the expectations for a full-length IgG radioimmunoconjugates with an hepatobiliary clearance and prolonged half-life in the blood and vascularized organs. We observed a significantly higher accumulation of [89Zr]Zr-DFO-anti-γH2AX-TAT in the blood, well-perfused organs (e.g. heart, lungs) and in the spleen of irradiated animals (Figure S4B-C). In the case of the 177Lu-PRIT cohort, the uptake of [177Lu]Lu-DOTA-PEG7-Tz in the blood 72 hours post-injection is 1.51 ± 0.21 %IA/g and the mean absorbed dose was calculated to be 48.8 cGy/MBq, therefore approximately 9.0 Gy was delivered to the blood [5]. Even though no hematotoxicity was reported in our previous studies with 177Lu-PRIT, such a dose could result in the generation of off-target toxicities and therefore elevated γH2AX level in the blood. In the case of the 10 Gy external beam radiation cohort, the mice were positioned on the outside X-ray field and only their lower right side were exposed to the radiation. Therefore, according to the dose delivered and their position we didn't expected any significant toxicity to the blood. The radiation exposure is consequently not expected to be the source of the increased uptake of 89Zr-PET anti-γH2AX tracer.
In vivo imaging of DNA damage with [89Zr]Zr-DFO-anti-γH2AX-TAT following β-PRIT in a PDAC mouse model. A. Schematic representation of the β-PRIT and 89Zr-PET imaging schedule in subcutaneous BxPC3 xenografted athymic nude mice. B. Representative transverse PET/CT images up to 4 days post-intravenous injection of [89Zr]Zr-DFO-anti-γH2AX-TAT (n=4 per cohort). Tumors are delineated with white dashed lines. C. Tumor VOI analysis of the PET/CT images up to 4 days p.i. (n=4 per cohort). D. Area under the curve of the tumor VOI analysis between 4 hours and 4 days p.i. (n=4 per cohort). E. Tumor uptake as determined through ex vivo gamma-counting post-animal sacrifice (n=5 per cohort). Values are represented as means, and error bars represent standard deviations. One-Way ANOVA followed by a Dunnett's multiple comparisons test was applied. Adjusted P values: *** P ≤ 0.001, ** P≤ 0.01, * P≤ 0.05, n.s. = non significant.
α-PRIT results in an increased [89Zr]Zr-DFO-anti-γH2AX-TAT tumoral uptake that correlates with the prolonged survival of the animals as compared to β-PRIT
Following the protocol established with 177Lu-PRIT, PET imaging of DNA damage following 225Ac-PRIT (37 kBq) was undertaken. Representative transverse images of [89Zr]Zr-DFO-anti-γH2AX-TAT uptake at the tumor site 24 hours post-intravenous injection of the PET radiotracer are reported in Figure 3A. Coronal sections and maximum intensity projections are provided in supporting information (Figure S5A). Quantitative VOI analysis of the 89Zr-PET signal revealed a statistically significant (P < 0.05) increase of the tumor [89Zr]Zr-DFO-anti-γH2AX-TAT uptake in the 225Ac-PRIT cohort (Figure 3B). VOI analysis was compared to the results determined through ex vivo gamma-counting post-animal sacrifice (Figure S6). The uptake of the 89Zr-PET DNA damage radiotracer in the 225Ac-PRIT and positive control (10 Gy) cohorts was 7.37 ± 1.23 and 6.80 ± 1.24 %IA/g, respectively, when the negative control cohort uptake was 5.08 ± 0.95 %IA/g (Figure 3C, Figure S5B). These results were in perfect agreement with the PET VOI values.
In vivo imaging of DNA damage with [89Zr]Zr-DFO-anti-γH2AX-TAT following α-PRIT in a PDAC mouse model. Comparison with β-PRIT A. Representative transverse PET/CT images 24 hours post-intravenous injection of [89Zr]Zr-DFO-anti-γH2AX-TAT (n=4 per cohort). Tumors are delineated with white dashed lines. B. Tumor VOI analysis of the PET/CT images 24 hours p.i. (n=4 per cohort). C. Tumor uptake as determined through ex vivo gamma-counting post-animal sacrifice (n=5 per cohort). D. Comparison of the β-PRIT and α-PRIT DNA damage imaging cohorts in terms of [89Zr]Zr-DFO-anti-γH2AX-TAT tumor and muscle uptake as determined through ex vivo gamma-counting post-animal sacrifice (4 days and 24 hours post-administration of the PET radiotracer, respectively). Values are represented as means, and error bars represent standard deviations. One-Way ANOVA followed by a Dunnett's multiple comparisons test was applied. Adjusted P values: ** P≤ 0.01, * P≤ 0.05, n.s. = non significant.
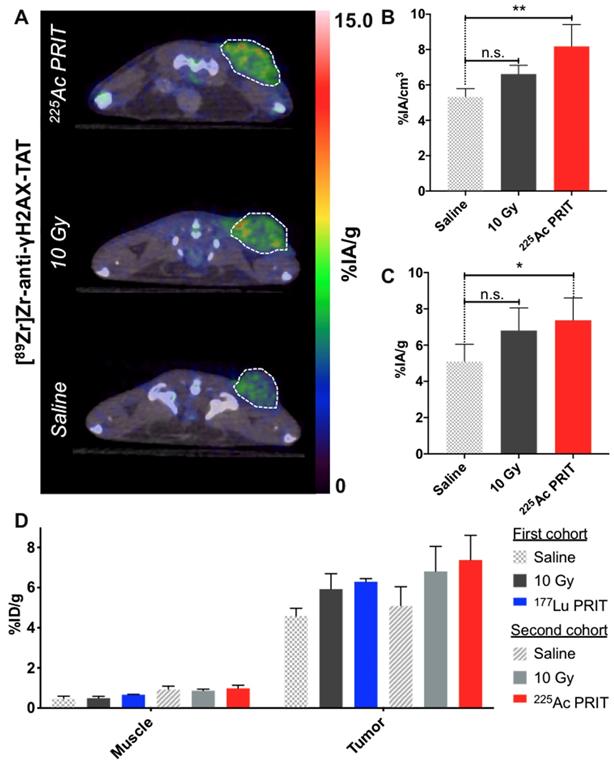
Comparing the 225Ac-PRIT and 177Lu-PRIT imaging cohorts is rendered difficult due to confounding factors that include different animal cohorts, PET radiotracer synthesis, or PET imaging endpoint (4 days post-radiotracer administration for 177Lu-PRIT and 24 hours for 225Ac-PRIT). However, there is no evidence of difference between the two different negative and positive control cohorts. No statistically significant differences can be observed between the 225Ac-PRIT and 177Lu-PRIT cohorts with our DNA damage PET imaging tracer (Figure 3D), even though 225Ac-PRIT resulted in prolonged survival of the treated animal as compared to 177Lu-PRIT. This study seems to indicate that DNA damage is not the only apoptotic pathway involved in the therapeutic efficacy of our PRIT approaches.
Ex vivo γH2AX analysis correlates with in vivo 89Zr-anti-γH2AX PET/CT imaging
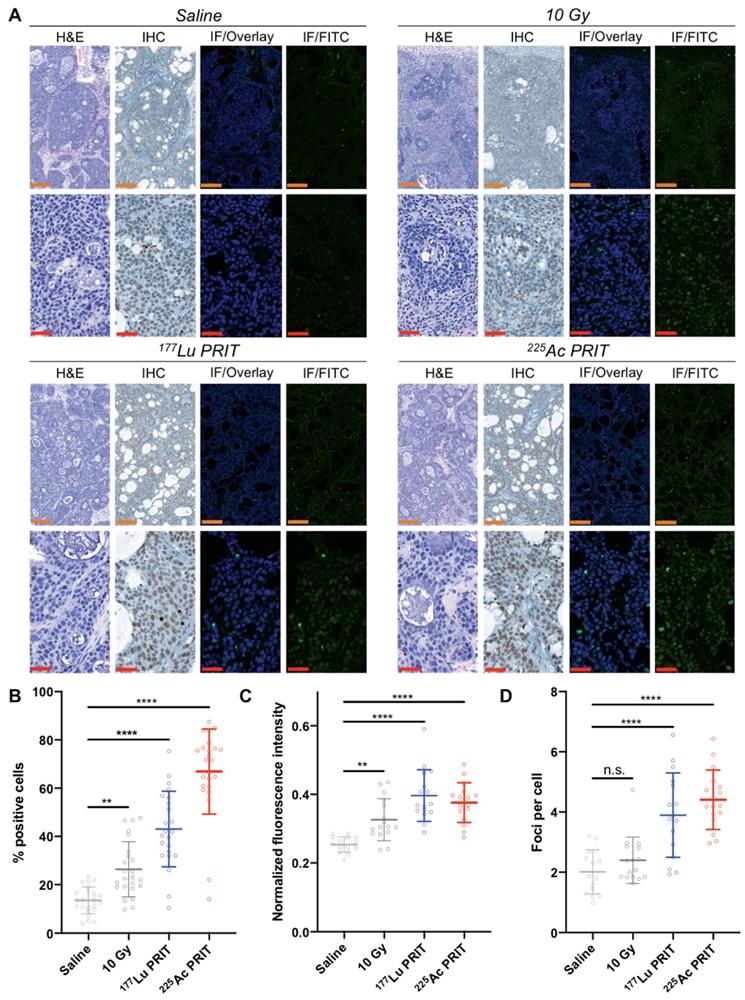
PET/CT results were confirmed via ex vivo immunohistochemistry and fluorescence staining of γH2AX foci in tumor sections following treatment with α-PRIT (37 kBq) and β-PRIT (18.5 MBq) (Figure 4A, Figure S7). With immunohistochemistry, the percentage of cells staining positive for γH2AX expression was significantly higher (P < 0.0001) in the cohort treated with α- and β-PRIT as well as in the 10 Gy positive control cohort (P < 0.01) when compared to the saline negative control cohort (Figure 4B). 225Ac-PRIT treated tumors showed the highest percentage of γH2AX positive cells (66.9 ± 17.6 %) followed by the 177Lu-PRIT treated tumors (43.1 ± 15.7 %), the 10 Gy cohort (26.4 ± 11.4 %), and finally the negative control (13.5 ± 5.51 %). Similarly, immunofluorescence γH2AX staining revealed increased fluorescence intensity and number of γH2AX foci per cell in the α-/β-PRIT treated cohorts (P < 0.0001). Notably, the detected number of γH2AX foci was twice as high in α-/β-PRIT cohorts (4.7 ± 1.0 and 4.2 ± 1.4 foci/cell, respectively) as compared to the saline treated cohort (1.9 ± 0.9 foci/cell).
Ex vivo γH2AX immunostaining for the monitoring of α-/β-PRIT in tumor sections of PDAC mouse model 72 hours post-irradiation (n=3 per cohort). A. Representative microscopy images (orange scale = 200 μm, red scale = 50 μm). Zoomed-in area are provided in Supporting information (Figure S8) B. Percentage of γH2AX positive cells after immunohistochemistry analysis of tumor sections following α-/β-PRIT as compared to negative and positive controls (n=8 per slide). Quantification of: C. γH2AX fluorescent signal normalized to DAPI signal (n=8 per slide), and D. γH2AX foci per cell (n=6 per slide), after immunofluorescence analysis of tumor sections following α-/β-PRIT as compared to negative and positive controls. Values are represented as means, and error bars represent standard deviations. One-Way ANOVA followed by a Dunnett's multiple comparisons test was applied. Adjusted P values: **** P≤ 0.0001, ** P≤ 0.01, n.s. = non significant.
Discussion
There is presently no established non-invasive clinical approach for the local and systemic evaluation of radiation-induced biomarkers following targeted radionuclide therapy. Instead, targeted radionuclide therapies are not personalized to individual radiation response but rather administered at a set activity/kg based on observed general population response. When the field of targeted radionuclide therapy should transition to a personalized approach based on dosimetry, it is critical to develop in parallel non-invasive approaches for the evaluation of radiation-induced biomarker for early assessment and prediction of therapeutic response. Herein, we have demonstrated that 89Zr-PET imaging of DNA damage response is an effective non-invasive tool for the monitoring of the early therapeutic response following α- and β-particle molecular radiotherapy in a preclinical mouse model of PDAC.
First, we validated that exposure to CA19.9 targeted α- and β-particle molecular radiotherapy resulted in reduced viability and clonogenic survival in BxPC3 PDAC cells. Using the pretargeting administration method [5,6], we confirmed in vivo the higher therapeutic potential of the CA19.9 pretargeted α-particle radiotherapy as compared to β-particle radiotherapy, when administered in activity ratios that resulted in equitoxic effects in vitro. Such results are not surprising due to the higher RBE of α-particle and correlates with results previously reported with 225Ac- and 177Lu-somatostatin receptor targeted radiotherapy [35]. However, they highlight the need for an in vivo predictor of radiation-induced effects, independent of the radiation type.
PET imaging studies performed following β-particle PRIT in a BxPC3 PDAC subcutaneous xenograft mouse model allowed the establishment of an imaging protocol that enabled the monitoring of the tumor radiobiological response through γH2AX measurement. This non-invasive molecular imaging approach was successfully translated to α-particle PRIT. This study further highlights the versatile potential of the [89Zr]Zr-DFO-anti-γH2AX-TAT radiotracer for the imaging of response following the induction of DNA double-strand break damage from multiple sources (EBRT, α/β-particle) [25,29]. No evidence of a difference in [89Zr]Zr-DFO-anti-γH2AX-TAT uptake at the tumor site after α-PRIT and β-PRIT could be observed when α-PRIT resulted in prolonged survival compared to β-PRIT. This lack of evidence could be due to inter-experiment confounding factors but more importantly highlights that DNA damage is not the sole radiobiology paradigm and that off-targeted (bystander) effects should be considered when trying to compare different targeted radionuclide therapy approaches [36]. Our in vivo results were confirmed later with ex vivo γH2AX immunohistochemistry and immunofluorescence staining. Non-invasive therapeutic response assessment is critical at the preclinical stage to validate the potential of novel molecular radiotherapy approaches and expedite the clinical translation phase. In addition, such approach offers great clinical prospects as an in vivo biodosimeter for treatment response assessment at the molecular level to better tailor therapeutic regimens to patient response and elucidate the heterogeneous response observed between patients but also between different tumor sites or within the same tumor. The first clinical evaluation of this DNA damage radiotracer should investigate the correlation of early [89Zr]Zr-DFO-anti-γH2AX-TAT PET signal following targeted radionuclide therapy or EBRT to patient response evaluated at later time point according to the conventional RECIST/PERCIST criteria. Such evaluation should allow us to determine a γH2AX PET signal cut-off between responders and non-responders.
A major limitation for the clinical translation and use of molecular radiotherapy approaches lies in the irradiation of healthy tissues following target engagement (on-target toxicity) or the non-specific accumulation of the radiopharmaceutical (off-target toxicity) [37,38]. γH2AX stands as a potential biomarker for radiation-induced toxicities and allowed previously the detection of radiotherapy-induced DNA damage in healthy human tissues, such as peripheral blood lymphocytes, hair follicles or kidneys [39-41]. Our DNA damage PET imaging should therefore allow the monitoring of on-/off-target toxicities. Our study did not allow such observation. Increased accumulation of our PET tracer in the blood and vascularized tissues was observed in the 177Lu-PRIT and first EBRT cohort, however this phenomenon was unlikely radiation-induced as it was not observed with the second EBRT cohort or with 225Ac-PRIT. Further preclinical studies should evaluate the potential of DNA damage PET imaging after targeted radionuclides therapies that demonstrate known toxicities to healthy tissues.
In conclusion, we have demonstrated in a preclinical PDAC mouse model the potential of [89Zr]Zr-anti-γH2AX-TAT as an in vivo biodosimeter for α- and β-particle targeted radionuclide therapy response assessment.
Supplementary Material
Supplementary methods and figures.
Acknowledgements
The authors gratefully acknowledge the Radiochemistry and Molecular Imaging Probes core facility, which was supported in part by NIH grant P30 CA08748. They gratefully acknowledge the William H. and Alice Goodwin and the Commonwealth Foundation for Cancer Research, the Center for Experimental Therapeutics of Memorial Sloan Kettering Cancer Center (JSL), an MSK IMRAS grant (JSL), the US National Institutes of Health grants NCI R35 CA232130 (JSL) and U01 CA221046 (JSL), and the François Wallace Monahan Fellowship from the JLM Benevolent Fund (SP). BC, EON and JK were supported by the CRUK/MRC Oxford Institute for Radiation Oncology. BC and EON were further supported by MRC (MR/P018661/1).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kleeff J, Korc M, Apte M, La Vecchia C, Johnson CD, Biankin A V. et al. Pancreatic cancer. Nat Rev Dis Prim. 2016;2:16022
2. Howlader N, Noone AM, Krapcho M, Miller D, Brest A, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ CK. Cancer Statistics Review, 1975-2016 - SEER Statistics [Internet]. National Cancer Institute. Bethesda, MD,. Available from: https://seer.cancer.gov/csr/1975_2016/.
3. Viola-Villegas NT, Rice SL, Carlin S, Wu X, Evans MJ, Sevak KK. et al. Applying PET to broaden the diagnostic utility of the clinically validated CA19.9 serum biomarker for oncology. J Nucl Med. 2013;54:1876-82
4. Houghton JL, Zeglis BM, Abdel-Atti D, Sawada R, Scholz WW, Lewis JS. Pretargeted Immuno-PET of Pancreatic Cancer: Overcoming Circulating Antigen and Internalized Antibody to Reduce Radiation Doses. J Nucl Med. 2016;57:453-9
5. Houghton JL, Membreno R, Abdel-Atti D, Cunanan KM, Carlin S, Scholz WW. et al. Establishment of the In Vivo Efficacy of Pretargeted Radioimmunotherapy Utilizing Inverse Electron Demand Diels-Alder Click Chemistry. Mol Cancer Ther. 2017;16(1):124-33
6. Poty S, Carter LM, Mandleywala K, Membreno R, Abdel-Atti D, Ragupathi A. et al. Leveraging bioorthogonal click chemistry to improve 225Ac-radioimmunotherapy of pancreatic ductal adenocarcinoma. Clin Cancer Res. 2019;25(2):868-80
7. Ballehaninna UK, Chamberlain RS. The clinical utility of serum CA 19-9 in the diagnosis, prognosis and management of pancreatic adenocarcinoma: An evidence based appraisal. J Gastrointest Oncol. 2012;3:105-19
8. Humphris JL, Chang DK, Johns AL, Scarlett CJ, Pajic M, Jones MD. et al. The prognostic and predictive value of serum CA19.9 in pancreatic cancer. Ann Oncol. 2012;23:1713-22
9. Poruk KE, Gay DZ, Brown K, Mulvihill JD, Boucher KM, Scaife CL. et al. The clinical utility of CA 19-9 in pancreatic adenocarcinoma: diagnostic and prognostic updates. Curr Mol Med. 2013;13:340-51
10. Ragupathi G, Damani P, Srivastava G, Srivastava O, Sucheck SJ, Ichikawa Y. et al. Synthesis of sialyl Lewis a (sLe a, CA19-9) and construction of an immunogenic sLe a vaccine. Cancer Immunol Immunother. 2009;58(9):1397-405
11. Sawada R, Sun SM, Wu X, Hong F, Ragupathi G, Livingston PO. et al. Human monoclonal antibodies to sialyl-Lewis a (CA19.9) with potent CDC, ADCC, and antitumor activity. Clin Cancer Res. 2011;17(5):1024-32
12. Meyer JP, Houghton JL, Kozlowski P, Abdel-Atti D, Reiner T, Pillarsetty NVK. et al. 18F-Based Pretargeted PET Imaging Based on Bioorthogonal Diels-Alder Click Chemistry. Bioconjug Chem. 2016Feb17;27(2):298-301
13. Meyer JP, Kozlowski P, Jackson J, Cunanan KM, Adumeau P, Dilling TR. et al. Exploring Structural Parameters for Pretargeting Radioligand Optimization. J Med Chem. 2017Oct12;60(19):8201-17
14. Rondon A, Schmitt S, Briat A, Ty N, Maigne L, Quintana M. et al. Pretargeted radioimmunotherapy and SPECT imaging of peritoneal carcinomatosis using bioorthogonal click chemistry: probe selection and first proof-of-concept. Theranostics. 2019;9(22):6706-18
15. Cheal SM, Xu H, Guo H fen, Patel M, Punzalan B, Fung EK. et al. Theranostic pretargeted radioimmunotherapy of internalizing solid tumor antigens in human tumor xenografts in mice: Curative treatment of HER2-positive breast carcinoma. Theranostics. 2018;8(18):5106-25
16. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R. et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228-47
17. Wahl RL, Jacene H, Kasamon Y, Lodge MA. From RECIST to PERCIST: Evolving Considerations for PET response criteria in solid tumors. J Nucl Med. 2009May;50(Suppl 1):122S-50S
18. Zhu A, Lee D, Shim H. Metabolic positron emission tomography imaging in cancer detection and therapy response. Semin Oncol. 2011Feb;38(1):55-69
19. Goldstein M, Kastan MB. The DNA damage response: implications for tumor responses to radiation and chemotherapy. Annu Rev Med. 2015;66:129-43
20. Eriksson D, Stigbrand T. Radiation-induced cell death mechanisms. Tumour Biol. 2010Aug;31(4):363-72
21. Kuo LJ, Yang L-X. γH2AX - A novel biomarker for DNA double-strand breaks. In Vivo (Brooklyn). 2008;22(3):305-9
22. Sharma A, Singh K, Almasan A. Histone H2AX Phosphorylation: A Marker for DNA Damage BT - DNA Repair Protocols. In: Methods in molecular biology. 2012
23. Nakamura AJ, Rao VA, Pommier Y, Bonner WM. The complexity of phosphorylated H2AX foci formation and DNA repair assembly at DNA double-strand breaks. Cell cycle. 2010Jan15;9(2):389-97
24. Qvarnström OF, Simonsson M, Johansson K-A, Nyman J, Turesson I. DNA double strand break quantification in skin biopsies. Radiother Oncol. 2004Sep;72(3):311-7
25. Cornelissen B, Kersemans V, Darbar S, Thompson J, Shah K, Sleeth K. et al. Imaging DNA damage in vivo using γH2AX-targeted immunoconjugates. Cancer Res. 2011;71(13):4539-49
26. Cornelissen B, Able S, Kartsonaki C, Kersemans V, Allen PD, Cavallo F. et al. Imaging DNA damage allows detection of preneoplasia in the BALB-neuT model of breast cancer. J Nucl Med. 2014Dec;55(12):2026-31
27. Knight JC, Topping C, Mosley M, Kersemans V, Falzone N, Fernández-Varea JM. et al. PET imaging of DNA damage using 89Zr-labelled anti-γH2AX-TAT immunoconjugates. Eur J Nucl Med Mol Imaging. 2015;42(11):1707-17
28. Knight JC, Mosley MJ, Bravo LC, Kersemans V, Allen PD, Mukherjee S. et al. 89Zr-anti-γH2AX-TAT but not 18F-FDG Allows Early Monitoring of Response to Chemotherapy in a Mouse Model of Pancreatic Ductal Adenocarcinoma. Clin Cancer Res. 2017Nov1;23(21):6498-504
29. O'Neill E, Kersemans V, Allen PD, Terry SYA, Torres JB, Mosley M. et al. Imaging DNA Damage Repair in vivo Following 177Lu-DOTATATE Therapy. J Nucl Med. 2019 Nov 22;jnumed.119.232934
30. Poty S, Membreno R, Glaser JM, Ragupathi A, Scholz WW, Zeglis BM. et al. The inverse electron-demand Diels-Alder reaction as a new methodology for the synthesis of 225Ac-labelled radioimmunoconjugates. Chem Commun. 2018;54(21):2599-602
31. Holland JP, Sheh Y, Lewis JS. Standardized methods for the production of high specific-activity zirconium-89. Nucl Med Biol. 2009;36(7):729-39
32. Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD. et al. QuPath: Open source software for digital pathology image analysis. Sci Rep. 2017 Dec 1;7(1)
33. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T. et al. Fiji: An open-source platform for biological-image analysis. Vol. 9, Nature Methods. 2012 p. 676-82
34. Bañuelos CA, Banáth JP, Kim JY, Aquino-Parsons C, Olive PL. γH2AX expression in tumors exposed to cisplatin and fractionated irradiation. Clin Cancer Res. 2009;15(10):3344-53
35. Graf F, Fahrer J, Maus S, Morgenstern A, Bruchertseifer F, Venkatachalam S. et al. DNA double strand breaks as predictor of efficacy of the alpha-particle emitter Ac-225 and the electron emitter Lu-177 for somatostatin receptor targeted radiotherapy. PLoS One. 2014 Feb 7;9(2)
36. Pouget JP, Georgakilas AG, Ravanat JL. Targeted and Off-Target (Bystander and Abscopal) Effects of Radiation Therapy: Redox Mechanisms and Risk/Benefit Analysis. Antioxidants Redox Signal. 2018;29(15):1447-87
37. Kratochwil C, Bruchertseifer F, Rathke H, Bronzel M, Apostolidis C, Weichert W. et al. Targeted a-therapy of metastatic castration-resistant prostate cancer with 225Ac-PSMA-617: Dosimetry estimate and empiric dose finding. J Nucl Med. 2017;58(10):1624-31
38. Bergsma H, Konijnenberg MW, van der Zwan WA, Kam BLR, Teunissen JJM, Kooij PP. et al. Nephrotoxicity after PRRT with 177Lu-DOTA-octreotate. Eur J Nucl Med Mol Imaging. 2016;43(10):1802-11
39. Siva S, Lobachevsky P, MacManus MP, Kron T, Möller A, Lobb RJ. et al. Radiotherapy for non-small cell lung cancer induces DNA damage response in both irradiated and out-of-field normal tissues. Clin Cancer Res. 2016;22(19):4817-26
40. Ivashkevich A, Redon CE, Nakamura AJ, Martin RF, Martin OA. Use of the γ-H2AX assay to monitor DNA damage and repair in translational cancer research. Cancer Lett. 2012;327(1-2):123-33
41. Pellegrini G, Siwowska K, Haller S, Antoine DJ, Schibli R, Kipar A. et al. A short-term biological indicator for long-term kidney damage after radionuclide therapy in mice. Pharmaceuticals. 2017 Jun 21;10(2)
Author contact
![]() Corresponding authors: Jason S. Lewis, Department of Radiology, 1275 York Avenue, Memorial Sloan Kettering Cancer Center, NY, 10065, USA; Email: lewisj2org. Sophie Poty, Department of Radiology, 1275 York Avenue, Memorial Sloan Kettering Cancer Center, NY, 10065, USA; Email: sophie.potyfr
Corresponding authors: Jason S. Lewis, Department of Radiology, 1275 York Avenue, Memorial Sloan Kettering Cancer Center, NY, 10065, USA; Email: lewisj2org. Sophie Poty, Department of Radiology, 1275 York Avenue, Memorial Sloan Kettering Cancer Center, NY, 10065, USA; Email: sophie.potyfr