Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Material and Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(14):6467-6482. doi:10.7150/thno.43865 This issue Cite
Research Paper
MGAT3-mediated glycosylation of tetraspanin CD82 at asparagine 157 suppresses ovarian cancer metastasis by inhibiting the integrin signaling pathway
Jun Li1, Jiawen Xu1, Luhan Li1, Alessandro Ianni2, Poonam Kumari2, Shuo Liu1, Peiqing Sun3, Thomas Braun2, Xiaoyue Tan1, Rong Xiang1 ![]() , Shijing Yue1
, Shijing Yue1 ![]()
1. State Key Laboratory of Medicinal Chemical Biology, School of Medicine, Nankai University, Tianjin, China
2. Department of Cardiac Development and Remodeling, Max-Planck-Institute for Heart and Lung Research, Bad Nauheim, Germany
3. Department of Cancer Biology, Wake Forest University School of Medicine, Winston-Salem, NC, USA
Received 2020-1-12; Accepted 2020-5-4; Published 2020-5-16
Abstract

Background: Tetraspanins constitute a family of transmembrane spanning proteins that function mainly by organizing the plasma membrane into micro-domains. CD82, a member of tetraspanins, is a potent inhibitor of cancer metastasis in numerous malignancies. CD82 is a highly glycosylated protein, however, it is still unknown whether and how this post-translational modification affects CD82 function and cancer metastasis.
Methods: The glycosylation of CD82 profiles are checked in the paired human ovarian primary and metastatic cancer tissues. The functional studies on the various glycosylation sites of CD82 are performed in vitro and in vivo.
Results: We demonstrate that CD82 glycosylation at Asn157 is necessary for CD82-mediated inhibition of ovarian cancer cells migration and metastasis in vitro and in vivo. Mechanistically, we discover that CD82 glycosylation is pivotal to disrupt integrin α5β1-mediated cellular adhesion to the abundant extracellular matrix protein fibronectin. Thereby the glycosylated CD82 inhibits the integrin signaling pathway responsible for the induction of the cytoskeleton rearrangements required for cellular migration. Furthermore, we reveal that the glycosyltransferase MGAT3 is responsible for CD82 glycosylation in ovarian cancer cells. Metastatic ovarian cancers express reduced levels of MGAT3 which in turn may result in impaired CD82 glycosylation.
Conclusions: Our work implicates a pathway for ovarian cancers metastasis regulation via MGAT3 mediated glycosylation of tetraspanin CD82 at asparagine 157.
Keywords: Ovarian cancer, Metastasis, Glycosylation, CD82, Exosomes
Introduction
Ovarian cancer is a frequent gynecological malignancy characterized by a high rate of metastasis and lethality. Differently from other kind of tumors, ovarian cancer only rarely metastasizes through the vasculature, but it rather invades the adjacent tissues or spreads to the distal organs through passive peritoneal dissemination and it is often associated with ascites formation [1-4]. The members of integrin family of transmembrane cellular receptors mediate cellular adhesion to the extracellular matrix (ECM) and play crucial roles in the transmission of intracellular signals required for cellular migration.
Cell migration is orchestrated by a highly elaborate complex of molecular pathways, which promotes rapid turnover of the focal adhesion proteins and complex dynamic changes in the cytoskeleton structure, such as the formation of stress fibers and assembly of other specialized structures. Binding of integrins to ECM induces integrin clustering, which in turn leads to the recruitment of cytosolic associated proteins such as the focal adhesion kinase (FAK). Upon recruitment and subsequent autophosphorylation, FAK promotes the phosphorylation of downstream targets, which control the signaling cascade involved in cell migration [5-7]. Therefore, integrin-mediated signaling plays an important role in the initiation and establishment of metastasis.
Within the last decade, tetraspanins have been recognized to interfere with cancer metastasis without affecting the growth of the primary tumor. Different studies demonstrated that tetraspanins can directly or indirectly bind to integrins and modulate the activation of the molecular pathway downstream to these receptors [8, 9]. CD82, also known as KAI1, is a member of the tetraspanin superfamily and has been identified as a potent inhibitor of cancer metastasis. Consistently, downregulation of CD82 in different metastatic cancers as compared to primary tumors was reported [10, 11]. Proteomics-based studies demonstrated that CD82 is glycosylated at three specific residues: Asn129, Asn157 and Asn198, which are located in the extracellular loop (ECL2) domain [12]. However, the role of CD82 post-translational modification and its impact on ovarian cancer metastasis remain largely uncharacterized.
In this study, we demonstrate that glycosylation of CD82 at Asn157 is fundamental to ensure CD82-mediated inhibition of ovarian cancer cell migration and metastasis both in vitro and in vivo. Mechanistically, we show that CD82 glycosylation at Asn157 is required for efficient binding to integrin α5β1 and subsequent disruption of the integrin-based cell adhesion to fibronectin. We further uncovered a previously undescribed mechanism that MGAT3 is a critical glycosyltransferase, which promotes CD82 glycosylation in ovarian cancer cells. Interestingly, MGAT3 is significantly downregulated in ovarian cancer metastasis that contributes at least in part to provoke low glycosylation of CD82.
Material and Methods
Cell lines and culture
Human ovarian cancer cell lines ES2 and SKOV3 were purchased from ATCC and cultivated in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. HEK-293T cells were purchased from ATCC and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS and 1% penicillin/streptomycin.
Patient derived samples
Ovarian cancer clinical samples were obtained from The Central Maternity Hospital of Tianjin. This study was approved by the ethics committees of The Central Maternity Hospital of Tianjin. All patients signed an informed consent. Patients' information is reported in Table S1.
Plasmids and transfections
The human CD82 cDNA was fused with Flag-peptide at the C-terminal and cloned into the lentiviral expression vector pLV-EF1α-MCS-IRES-Bsd/puro (Biosettia, San Diego, CA, USA). The CD82-Flag NQ mutants (N129Q, N157Q, N198Q, N157/198Q, 3NQ) were generated by site-directed mutagenesis using the pLV-CD82-Flag expression vector as a template. All constructs were verified by DNA sequencing. The primers used for the generation of the CD82 point mutants are listed in Table S2.
Quantitative reverse transcription PCR (RT-qPCR)
Total RNA was extracted using TRIZOL reagent (Invitrogen) following manufacturer's instructions. cDNA was then synthesized using the Trans Script First-Strand cDNA Synthesis Super Mix Kit (Trans Gen Biotech, Beijing, China). qPCR was performed in a CFXTM real-time thermal cycler (Bio-Rad, Hercules, CA, USA) using a Trans Start Top Green qPCR Supper Mix kit (Trans Gen Biotech). Data analysis was performed with the comparative ΔCt method using GAPDH as internal control [13]. The sequences of the primers used in this study are listed in Table S3.
Generation of stable cell lines by lentiviral delivery system
Stable ovarian cancer cells expressing wild type CD82-Flag, the glycosylation deficient mutants CD82-Flag NQ or control cells were established using a lentiviral-based delivery system. For the generation of lentiviral particles, 4x106 HEK293T cells were seeded in a 10 cm culture dish and cultivated overnight. Cells were co-transfected with 9 µg of empty or CD82-containing pLV-EF1α-MCS-IRES-Bsd/puro plasmids together with packaging and envelope vectors (4.5 µg pMDLg/pRRE, 1.8 µg pRSV-REV, 2.7 µg pCMV-VSV-G purchased from Addgene) using Lipofectamine 2000 (Invitrogen). Culture medium containing lentiviral particles was collected 48 h post-transfection and filtered through a 0.45 µm membrane. Lentiviral particles were diluted 1:2 in fresh medium and supplemented with 8 μg/mL polybrene. For lentiviral infection, target cells were incubated with lentiviral particles for 6-8 h. After incubation, cells were supplemented with fresh medium and incubated for additional 48 h. Stable cells were selected using puromycin (1 µg/mL).
Animal experiments
BALB/c nude (nu/nu) mice were purchased from Vital River Laboratory Animal Technology Co. Ltd (Beijing, China) and maintained in pathogen-free facility of the Nankai University. All of the animal experiments were approved by the Nankai University Animal Care and Use Committee and handled according to the Nankai University Animal Welfare Guidelines.
For the mice xenograft model, 1×106 ES2 cells stably expressing wild type (WT) CD82, the glycosylation deficient mutant N157Q or a control vector (MCS), were subcutaneously or intraperitoneally injected into 6-week-old female nude mice (n= 6 per group). Tumor volume was measured starting from day 10 post-injection every 5 days and calculated using the standard equation: V = 1/2 × L × W2, where V is the tumor volume, L the tumor length and W the tumor width. The number of metastatic foci were counted in three randomly selected fields and weighted.
Western blot analysis and immunoprecipitation
Western blot analysis was performed as described previously [14]. Briefly, adherent cells were washed twice with PBS and collected by scraping. Cellular pellets were re-suspended in RIPA lysis buffer (25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitor cocktail (Sigma‐Aldrich, St. Louis, MO, USA) and incubated on ice for 30 min. Protein lysates were cleared by centrifugation (16200 g for 15 min at 4 °C) and the protein concentration was assessed by BCA Protein Assay (Bio-Rad). Proteins were separated onto polyacrylamide gels and transferred to PVDF membranes. The protein samples were heated into 100 ℃ for 5 min and the blots were carried out in reducing condition. Membranes were blocked for 1h in 5% milk in TBS-T (w/v) and incubated overnight at 4 °C with primary antibodies on a shaking platform. On the next day, membranes were washed 5 times in TBS-T and incubated with appropriate HRP-conjugated secondary antibodies for 1 h. After incubation, membranes were extensively washed in TBS-T prior to signal detection using the Tanon Chemiluminescent Imaging System (Tanon, Shanghai, China). In these studies the following primary antibodies were used: Anti-Flag tag (Sigma-Aldrich; F1804), CD82 (CST; D7G6H), integrin β1 (SCBT; sc-8978), integrin α5 (CST; 4705S), p-FAK (CST; 3283S), FAK (CST; 3285S), p-SRC (CST; 2105S), SRC (CST; 2108S), paxillin (BD; 610620) integrin β4 (SCBT; sc-9090), Talin (SCBT; SC-15336), MGAT3 (proteintech; 17869-1-AP), Fibronectin (BD; 610078), Caveolin (SCBT; sc-984), Alix (SCBT; SC-99010), TSG101 (SCBT; SC-22774), p62 (SCBT; sc-166870), GM130 (SCBT; sc-55591).
For immunoprecipitation, cell lysates were collected in IP buffer (50 mM Tris-HCl; pH 7.4, 150 mM NaCl, 0.1% NP-40, 5 mM EDTA) supplemented with protease inhibitor cocktail (Sigma-Aldrich) and cleared by centrifugation. Protein lysates were incubated with specific antibodies as indicated in the figure legends or with non-immune immunoglobulin (IgG; negative control) and with G-agarose beads overnight on a rotating wheel. Next day, the beads were washed in ice-cold IP buffer, resuspended in 2x western blot loading buffer (65 mM Tris-HCl pH 6.8, 25% glycerol, 2% SDS, 0.01 % bromophenol blue, 50 mM DTT) and boiled at 95 °C for 5 min prior to western blot analysis.
Transwell assay
Transwell assay was performed as already described with minor modifications [14]. Briefly 2x105 cells were resuspended in RMPI-1640 medium supplemented with 1% FBS and seeded in the upper chamber of a 24-well transwell plate with 8 μm pore size. The lower chamber was filled with RMPI-1640 supplemented with 10% FBS. 6 hours after seeding, the transmigrated cells were fixed with 4% paraformaldehyde and stained with crystal violet. Pictures were acquired using an Olympus BX51 microscope and the number of cells was counted using imaging J software.
Cell adhesion and RGD blocking assay
96-well plates were coated with 10 mg/mL Fibronectin (FN, Millipore, Darmstadt, Germany) overnight at 4 °C. Ovarian cancer cell lines were stained with the fluorescent dye Calcein AM (abcam, MA, USA). For treatment with RGD-motif containing peptides (RGDs), Calcein stained cells were incubated with 1μg/mL RGDs (GL Biochem; Shangai) or BSA for 1 h at 37 ℃. After incubation, 5x103 cells were seeded on fibronectin pre-coated plates for 1 h and unattached cells were removed by washing twice in PBS. Adherent cells were photographed using Olympus BX51 microscope and quantified by green fluorescence measurement using the Glomax 96 microplate luminometer (Promega, WI, USA).
Live-cell imaging
Live-cell imaging for cell migration analysis was performed using AS MDW live-cell imaging system (Leica Microsystems) controlled by the Image Pro software (version 6.3; Media Cybernetics). Cells were cultured at 37 °C in a humidified 5% CO2 atmosphere. Bright-field images were acquired every 5 min over 4.5 h. Cell migration was tracked manually using the ImageJ plugin MTrackJ and plotted with the origin of migration superimposed at 0.0.
High-content assay
High content assay was performed on 24-well plates pre-coated with 10 μg/mL fibronectin overnight at 4 °C. Cells were serum deprived for 8 h, washed twice in PBS, trypsinized with 0.25% trypsin and then re-suspended in RPM1-1640 medium supplemented with 1% FBS. 1x105 cells were seeded in each well and the migration was monitored by incubation for 4 h at 37 °C by Operetta High-Content Imaging System (PerkinElmer, US) and analyzed by Harmony Analysis System (PerkinElmer, US).
Immunofluorescence
Cells were seeded onto fibronectin pre-coated glass slides, fixed with 4% formaldehyde in PBS for 15 min and washed twice in ice-cold PBS. Cells were blocked for 1 h in 5% goat serum in PBS at room temperature for 1 h and incubated overnight at 4 °C in primary antibody. In this study, the following antibodies were used: Anti-Flag tag (Sigma-Aldrich; F1804), FAK (CST; 3285S), paxillin (BD; 610620). Next day, cells were washed 3 times in PBS and incubated with Alexa Fluor 488 or Alexa Fluor 594 conjugated antibodies (ZSGB-BIO, Beijing, China) for 1 h at room temperature. Cells were counterstained with DAPI. Pictures were captured using a confocal microscope (Olympus, Tokyo, Japan).
Immunohistochemistry (IHC)
IHC staining was performed as described previously [14]. Briefly, mice or human tissue sections were incubated with antibodies against CD82 or MGAT3 overnight at 4 °C. Next day, samples were incubated with a biotin-conjugated secondary antibody for 2 h at room temperature and then incubated with an avidin-biotin-peroxidase complex. The antibody detection was performed using 3-amino-9-ethylcarbazole chromogen.
Hematoxylin and eosin (HE)
HE staining was performed using standard procedures. Briefly, the sections were deparaffinized by 2 times xylene, 5 min, and following 100% EtOH, 90% EtOH, 80% EtOH, 70% EtOH, 5 min, and 2 times H2O, 2 min each. Then, the sections were stained with hematoxylin-eosin kit (C0105, Beyotime, Shanghai, China). The sections were stained with hematoxylin solution for 1-2 min, and following the rinse with running tap water for 10 min. The sections were stained with eosin solution for 30 s to 2 min. Thereafter, the sections were dehydrated with 70% EtOH, 80% EtOH, 90% EtOH, 100% EtOH for 10 s, and 2 times xylene 5 min each. Finally, the sections were mounted with resinous mounting medium.
Exosomes isolation, characterization and labelling
Exosomes were purified from cells cultured in serum-free medium for 36 h using already described procedures [15]. Purified exosomes were suspended in physiological saline and characterized using limited trypsin digestion, immunoelectron microscopy (IEM), and western blotting. Limited trypsin digestion was performed as already described [16]. Briefly, exosomes were incubated with 0.05% trypsin for 5 min at room temperature to digest surface proteins prior to western blot analysis.
IEM was performed using standard procedures. Purified exosomes were re-suspended in 2% paraformaldehyde and adsorbed into carbon-coated formvar EM grids (Electron Microscopy Sciences) for 20 min. The grids were then washed in physiological saline and transferred to 50 mM glycine/PBS for 3 min 3 times. The grids were washed in blocking buffer (5% BSA in PBS) for 10 min and incubated with 20 µl of anti-Flag antibody diluted in blocking buffer (1:200) for 30 min. The grids were transferred into 1% glutaraldehyde for 5 min and incubated with 20 µl of goat anti-mouse IgG/Gold antibody (Bioss, Beijing) for 30 min. Finally, the grids were embedded with 30μl of uranyl-oxalate solution for 90 s and air dried. Pictures were captured using FEI Talos™ F200C transmission electron microscope.
Statistical analysis
Data are expressed as the average ± standard deviation of at least 3 independent experiments. Statistical significance was assessed using the Student´s t-test and GraphPad Prism 5 software (Inc., La Jolla, CA).
Results
CD82 is glycosylated in ovarian cancer cells
Numerous studies demonstrated that CD82 plays a pivotal role as inhibitor of tumor metastasis in various cancers [10, 17, 18]. However, the role of CD82 in ovarian cancer metastasis has not been investigated. We explored CD82 expression using The Cancer Genome Atlas database (TCGA). No significant changes of CD82 expression were found at different stages of ovarian cancer (Figure 1A). Moreover, no association between CD82 expression and lymphovascular invasion was observed (Figure 1B). The expression of CD82 in human biopsies from primary and metastatic ovarian cancers was analyzed using IHC. We did not observe any significant change in CD82 expression in ovarian metastatic and paired primary tumors (Figure 1C, S1A). Interestingly, western blotting data indicate that CD82 mainly detected at around 55 kDa in primary tumors, while a reduction of 55 kDa and a significant increase of smaller between 35 and 55 kDa detected in metastatic tissues (Figure 1D, S1B). The mRNA levels of CD82 were no different in metastatic and primary tumors by real time PCR detection (Figure S1C).
CD82 is glycosylated in ovarian cancer. A. Mass-spectrometry based proteomics data for CD82 expression in ovarian cancers at different progression stages was obtained from the Cancer Genome Atlas database (TCGA) and indicate no significant difference. Cancer staging was performed according to the American Joint Committee on Cancer (AJCC). NS: not significant. B. Mass spectrometry-based proteomics data indicate no significant difference in CD82 expression between ovarian cancers with or without lymphovascular invasion. C. Immunohistochemistry analysis of CD82 expression in clinical samples from primary and metastatic ovarian cancers. Note that CD82 expression is not significant change between primary tumors and metastasis. D. Western blot analysis of CD82 expression in primary and metastatic ovarian cancers. Note the reduction in the expression of the 55 kDa fraction of CD82 in metastasis and the concomitant increase in lower molecular weight fractions. E. Western blot analysis of CD82 expression in lung, pancreatic and ovarian cancer cell lines. F. ES2 and SKOV3 ovarian cancer cell lines lysates were either left untreated or incubated with recombinant glycosidase (peptide-N-glycosidase F; PNGase F) as indicated, prior to western blot analysis. A representative western blot of three independent experiments is shown. G. Schematic representation of the CD82 glycosylation deficient mutants used in this study. The crosses indicate substitutions of asparagine (N) with glutamine (Q). H. Cellular lysates derived from ES2 ovarian cancer cells stably expressing Flag-tagged CD82 and NQ point mutants were either untreated or incubated with PNGase F and analyzed by western blotting. Note the reduction in the molecular size of CD82 glycosylation deficient mutants as compared with CD82.
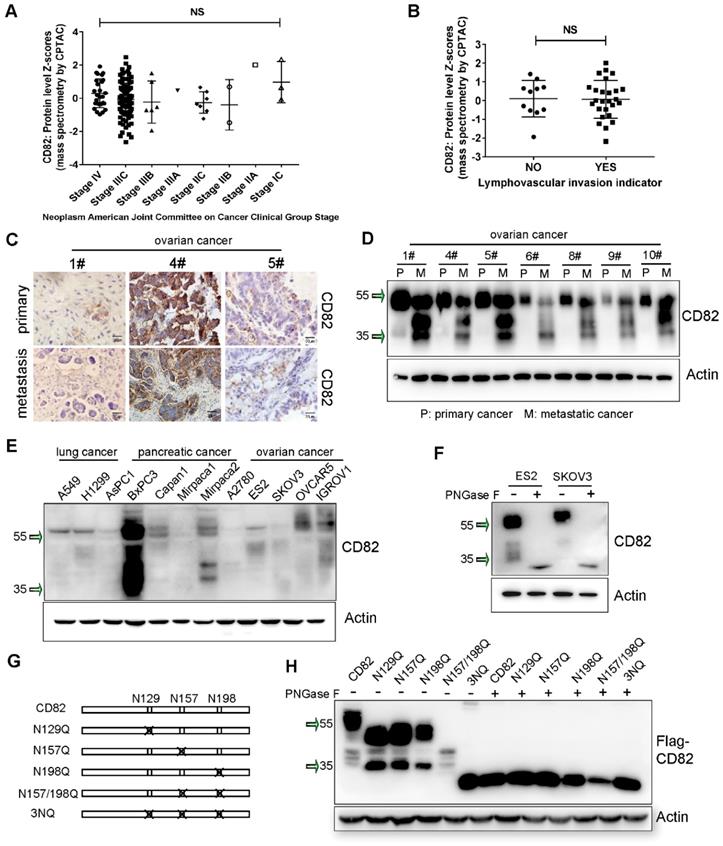
Furthermore, expression pattern of CD82 in lung, pancreatic and ovarian cancer cell lines revealed a molecular weight ranging between 35 and 55 kDa (Figure 1E). It was previously demonstrated that tetraspanins are highly glycosylated proteins [9]. We subjected protein lysates derived from ES2 and SKVO3 ovarian cancer cell lines to treatment with recombinant glycosidase (peptide-N-glycosidase F; PNGase F) to remove the presumed N-glycan structures. CD82 protein was detected at around 30 kDa in PNGase F treatment, while was detected at 55 kDa in untreated cells (Figure 1F).
Former proteomics based-analysis identified three major residues in the EC2 domain of CD82, which are N-glycosylated: Asn129, Asn157 and Asn198 (Figure 1G) [12]. To prove whether the glycosylation is caused the separation pattern of CD82 in western blotting, we generated CD82 glycosylation deficient point mutants. Each asparagine (N) was substituted with glutamine (Q) to generate the single mutants N129Q, N157Q, N198Q, the double mutant N157/198Q and the triple mutant N129/N157/N198Q (3NQ) (Figure 1G). The five glycosylation deficient mutants and the CD82 were stably overexpressed in ES2 cells using a lentiviral-based system. The subsequent western blot analysis revealed that the abolishment of single or combined glycosylation residues generated protein bands at the lower molecular weight (Figure 1H). The triple mutant 3NQ could be mainly detected at around 30 kDa. This molecular weight was also obtained with all glycosylation mutants upon treatment with PNGase F (Figure 1H). These results were confirmed by analysis of the glycosylation deficient mutants overexpressed in SKOV3 cells (Figure S2A). All these data corroborate that CD82 is highly glycosylated in ovarian cancer cells and indicate that the reduction of 55 kDa CD82 and the concomitant increase in smaller molecular weight fractions in metastatic ovarian cancer can be attributed to impaired CD82 glycosylation.
Glycosylation of CD82 at Asn157 impairs ovarian cancer cells migration in vitro
We next investigated whether the glycosylation of CD82 has any impact on ovarian cancer cells migration. A transwell migration assay demonstrated that CD82, N129Q and N198Q mutants significantly reduced migration of ES2 cells. In contrast, the N157Q mutant, the double mutant N157/198Q and the triple mutant 3NQ did not exert any significant effect on migration of ES2 cells (Figure 2A). Similar results were observed in scratch assay (Figure 2B). Consistent results were also obtained in SKOV3 stable cell lines (Figure S2D-E). Taken together these data strongly imply that the glycosylation of CD82 at N157 is sufficient for CD82-mediated inhibition of ovarian cancer cells migration. To provide a more exact assessment of the migratory potential, we analyzed the migration distance of control, CD82 and N157Q mutant cells using time-lapse assay. As demonstrated in Figure 2C CD82 significantly reduced the migration of ES2 cells whereas no effect was caused by N157Q mutant. The cell migration videos and the relative quantification of the migration velocity (Videos S1A-C, Figure 2D) further confirmed these data.
Glycosylation at Asn 157 is required for CD82-mediated inhibition of ovarian cancer cell migration. A. Transwell migration assay of stable control (MCS), CD82- and NQ mutants overexpressing ES2 cells. The transmigrated cells were stained with crystal violet (left panel). Scale bar 100 µm. A quantification of the number of migrated cells per field ± SD is shown in the histogram on the right. **p<0.01, *p<0.05, NS: not significant. B. Stable ES2 cells as in A were subjected to scratch assay. Representative pictures at time 0 and 12 h after scratching are shown. Red lines highlight the scratch margin. The relative distance of wound edges ± SD is given in the graph on the right (n=3). **p<0.01, *p<0.05, NS: not significant. C. Superimposed migration trajectories of stable control, CD82 and N157Q overexpressing ES2 cells as determined by live cell imaging. 6 cells for each group were analyzed (represented with different color lines). D. Normalized migration velocity of stable ES2 cells as in C. E. Western blot analysis of whole cell and exosomes protein lysates from stable ES2 cells. Western blot membranes where probed with antibodies against the exosomes markers Alix and TSG101 or against p62 and GM130 as negative control. CD82 overexpression was verified with anti-Flag antibody. F. Transmigration assay of ES2 cells incubated with control BSA or with exosomes derived from control, CD82 and N157Q overexpressing cells. A representative picture out of three independent experiments is shown on the left. A quantification of the average number of transmigrated cells per field ± SD of all performed experiments is shown in the histogram on the right. **p<0.01, *p<0.05.
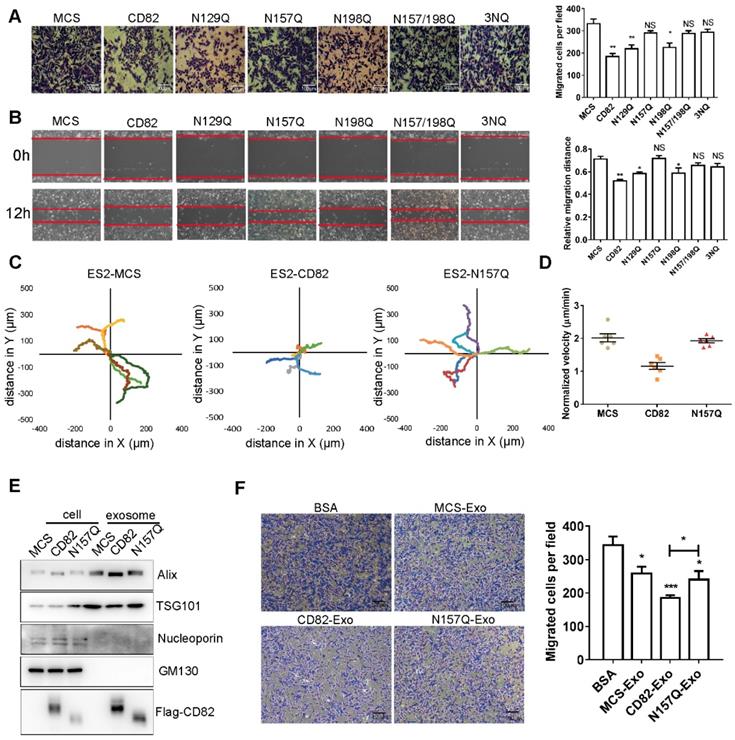
Exosomes were isolated from control, CD82 and N157Q mutant cells. The characteristics were assessed using Alix and TSG101 as exosome markers and nucleoporin and GM130 as negative controls (Figure 2E, S2B). In addition, we confirmed the localization of CD82 in exosomes using immunoelectron microscopy (IEM) and limited trypsin digestion assay (Figure S2C, S2G). Next, we treated ES2 cells with exosomes, which were pre-labeled with Dir dye and confirmed the internalization of exogenous exosomal CD82 by immunofluorescence using anti-Flag antibody (Figure S2H). CD82-enriched exosomes significantly impaired ovarian cancer cell migration in contrast to exosomes derived from control and N157Q mutant cells (Figure 2F, S2F). Altogether these data demonstrate that CD82 glycosylation at N157 is a determinant post-translational modification responsible for CD82-mediated inhibition of ovarian cancer cell migration in vitro.
CD82 glycosylation at Asn157 impairs fibronectin-integrin pathway
We were further interested to get more insight into the molecular mechanism by which CD82 glycosylation at Asn157 inhibits ovarian cancer cells migration. Different studies demonstrated that tetraspanins function as scaffolding proteins for membrane receptors such as integrins, modulating thereby the activation of downstream signaling pathways [9]. Integrins including α5β1, α4β1 and αvβ3 play pivotal roles in the anchorage of cells to the ECM especially through the binding to the extracellular matrix protein fibronectin [19-21]. The binding of integrins to fibronectin is in turn essential for the transmission of intracellular signals, which promote cellular migration and motility [21]. Since previous studies suggested that CD82 suppresses cell migration by association with integrins and disruption of the fibronectin-integrin axis [22], we reasoned that the glycosylation of CD82 at N157 might impact its interaction with integrins. Consistently with previous reports, we could show in co-immunoprecipitation experiments that Flag-tagged CD82 stably expressed in ES2 cells interacts with endogenous integrin α5β1 but does not interact with the β4 subunit (Figure 3A). Noticeably, we could further demonstrate that the glycosylation deficient mutant N157Q, but not the N198Q mutant, displayed an impaired capacity of binding to integrin α5β1, indicating that the glycosylation of CD82 at Asn157 is required for the efficient interaction of CD82 with integrins (Figure 3A).
CD82 glycosylation at N157 promotes CD82 binding to integrin α5β1 and disrupts the integrin-fibronectin signaling pathway. A. Coupled immunoprecipitation (anti-Flag antibody; IP: Flag) and western blot analysis (anti-α5, β1 and β4 antibodies) of stable ES2 cells overexpressing Flag-tagged CD82, N157Q and N198Q mutants. B. Cell adhesion assay on fibronectin pre-coated plates that the stable control, CD82 or N157Q overexpressing cells were plated for 30 min. Adherent cells were visualized by Calcein fluorescence. A representative picture of three independent experiments is shown on the left. Scale bar 100 µm. Quantification of the relative fluorescence ± SD of three independent experiments is shown in the histogram on the right. ***p<0.005. NS: Not significant. C. Coupled immunoprecipitation (anti-β1 integrin antibody; IP: β1) and western blot analysis (anti-fibronectin; FN, α5, β1, FAK and Talin antibodies) of stable control (MCS) and CD82 overexpressing cells grown in presence of fibronectin. IgG was used as a negative control. A representative western blot out of three independent experiments is shown. D. Coupled immunoprecipitation (anti-α5 integrin antibody; IP: α5) and western blot analysis (β1, Paxillin, Talin, and anti-Flag antibodies) of cellular lysates derived from stable ES2 cells expressing CD82, N157Q and N198Q. A representative picture of three independent experiments is shown. E. Western blot analysis of phosphorylated and total FAK and Src in stable control and CD82 overexpressing cells at different time points after incubation with Fibronectin. A representative western blot of three independent experiments is shown. Note the reduction in the phosphorylation of FAK (p-FAK) and Src (p-Src) in CD82 as compared with MCS and N157Q expressing cells. F. Representative western blot (n=3) of phosphorylated and total FAK and phosphorylated paxillin (p-Paxillin) in stable ES2 cells upon treatment with fibronectin. Actin was used as a loading control. G. Representative immunofluorescence pictures of the subcellular localization of exogenous CD82 (Flag) and FAK in MCS, CD82 and N157Q overexpressing ES2 cells. Cellular nuclei were counterstained with DAPI. Scale bar 10 µm. H. Immunofluorescence analysis of Paxillin subcellular localization in MCS, CD82 and N157Q overexpressing cells (green). Exogenous CD82 was stained using anti-Flag antibody (red). Cell nuclei were counterstained with DAPI. Scale bar 10 µm. A representative picture of three independent experiments is shown.
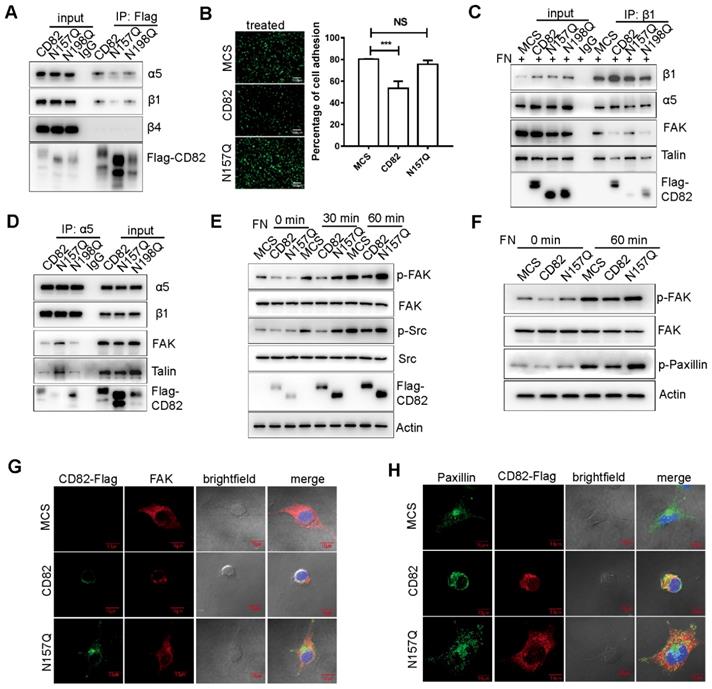
Since integrin α5β1 contributes to cellular adhesion to fibronectin, we wondered whether the N157 glycosylated CD82 might inhibit the adhesion of ovarian cancer cells by disrupting the integrin-fibronectin interaction. Indeed, ES2 cells overexpressing CD82 possess an impaired capacity to adhere to fibronectin coated plates in contrast to control and N157Q overexpressing cells (Figure 3B, S3A). We could further confirm the inhibitory function of N157 glycosylation on cell adhesion using exosomes. Exosomes enriched in CD82 significantly inhibited ovarian cancer cells adhesion in contrast to control and N157Q enriched exosomes (Figure S3B). To univocally demonstrate that CD82 glycosylation at Asn157 is responsible for the disruption of the integrin-fibronectin interaction, we assessed this interaction in presence of CD82 and the N157Q and N198Q mutants. Since the interaction between integrins and fibronectin is pivotal for the recruitment of cytosolic integrin-binding adaptor proteins such as FAK and Talin [23], we also estimated the amount of FAK and Talin co-immunoprecipitated with integrin β1. A significantly reduced interaction of these molecules with integrin β1 in ES2 cells overexpressing CD82 and N198Q mutant but not in control and N157Q lent an additional support for the proposed inhibitory role of Asn157 glycosylation (Figure 3C). Consistently, co-immunoprecipitation experiments using anti-integrin α5 antibody demonstrated a dramatic increase in the association of integrin α5 with FAK and Talin in ES2 cells overexpressing the glycosylation deficient N157Q mutant as compared with CD82 and N198Q mutant overexpressing cells (Figure 3D).
Binding of integrins to fibronectin induces the recruitment and also phosphorylation of FAK, which in turn activates the integrin downstream pathway leading to the phosphorylation of adaptor proteins such as Src and Paxillin [23]. To further confirm that the glycosylation of CD82 contributes to diminished activation of the integrin pathway, we analyzed the phosphorylation levels of FAK and Src in stably transfected ES2 cells upon treatment with fibronectin. As expected, cells overexpressing CD82 displayed a significant reduction in the phosphorylation of FAK and Src. While FAK and Src phosphorylation was clearly induced in response to fibronectin in control and N157Q mutant cells (Figure 3E). Consistently, a reduced phosphorylation of another down-stream integrin target, Paxillin, was observed in CD82 but not N157Q mutant overexpressing cells (Figure 3F). Since the interaction of integrin with fibronectin increases the recruitment of FAK and Paxillin to the focal adhesions, the subcellular localization of these molecules was estimated. In agreement with our expectations, FAK and Paxillin were mainly distributed at the cellular periphery in control and N157Q mutant expressing cells, while in CD82 overexpressing cells they were constricted rather to the perinuclear space (Figure 3G-H, S3C).
Moreover, since the generation of driving forces required for cellular migration results in cellular spreading, we evaluated the impact of CD82 glycosylation on the spreading capacity of ovarian cancer cell lines. In agreement with our assumption, CD82 overexpressing cells possess less spreading capacity as compared with control ES cells or N157Q mutant cells (Figure 4A). Because activation of integrin pathway has been connected to the formation of stress fibers, we further analyzed the F-actin organization. F-actin organization mainly produce and transmit mechanical tension required for cellular migration [24, 25]. In fact, overexpression of CD82 but not N157Q mutant strongly inhibited stress fiber formation in ovarian cancer cells (Figure 4B, S3D).
RGD-loop containing peptides (RGDs) abolished the inhibition of CD82 on integrin signaling pathway. A. Representative bright field image of cell spreading assay performed in stable ES2 cells. Note the inhibition of cell spreading in CD82 overexpressing cells as compared with MCS and N157Q. B. Representative fluorescent picture of stress fiber formation in stable ES2 cells as visualized by F actin staining. Cell nuclei were counterstained with DAPI. Scale bar 10 µm. C. Calcein stained ES2 cells were either pre-incubated with BSA or with RGDs and subjected to adhesion assay on fibronectin coated plates. A representative picture of three independent experiments is shown. A relative quantification of the number of adherent cells ± SD of three independent experiments is shown in the histogram on the right. **p<0.01, NS: not significant; scale bar 100 µm. D. Representative western blot analysis (n=3) of phosphorylated and total FAK and Src expression in stable ES2 cells upon treatment with fibronectin (FN) and RGDs. E. Representative pictures of transmigration assay performed in stable ES2 cells pre-incubated with BSA or RGDs. Quantification of the number of transmigrated cells ± SD is reported in the graph on the right. **p<0.01, NS: not significant. F. Representative graph of the relative migration velocity of stable ES2 cells in presence or absence of RGDs as established by high content analysis. G. Immunofluorescence analysis of FAK subcellular localization and the focal adhesion spots formation in stable ES2 cells overexpressing CD82 or N157Q after treatment with BSA or RGDs. Scale bar 10µm. Quantification of the number of focal adhesion spots per cell for 10 random fields is reported in the graph on the right. ***p<0.005, NS: not significant.
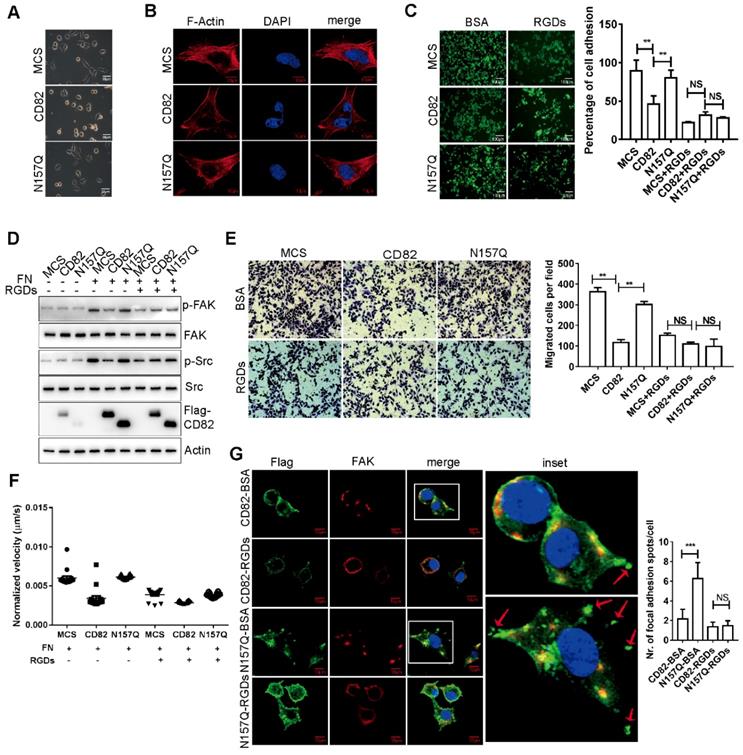
Since it is known that fibronectin-derived Arg-Gly-Asp (RGD) motif-containing peptides can abolish the association of integrin α5β1 to fibronectin by competitive inhibition and therefore inhibit down-stream cellular events [26, 27], we analyzed the adhesion capacity of control, CD82 and N157Q mutant overexpressing cells on fibronectin coated plates upon treatment with RGD motif-containing peptides (RGDs) or in presence of BSA as a negative control. Noticeably, RGDs treatment strongly impaired cell adhesion in all cells tested (Figure 4C, S3E). However, while a reduced adhesion capacity was apparent in ES2 cells overexpressing CD82 upon treatment with BSA no further statistically significant differences were detected between the different cell lines after incubation with RGDs (Figure 4C, S3E). This finding strongly suggests that CD82 interrupts integrin-fibronectin contact efficiently enough to exclude the further interference by RGDs. Next, we estimated the extent of FAK and Src phosphorylation in stably transfected ES2 cells upon incubation with RGDs. In agreement with the previous data, cells overexpressing CD82 displayed a significant reduction in the activation of the integrin signaling pathway in absence of RGDs as compared to N157Q mutant and control cells. Treatment with RGDs peptides dramatically reduced the phosphorylation of FAK and Src in control and N157Q mutant cells but, again, no further reduction was detectable in CD82 overexpressing cells (Figure 4D). RGDs treatment and N157 glycosylation leads also to a slower rate of cell migration besides inhibiting cell adhesion. Indeed, using transwell and high-content analysis we could demonstrate that the RGD-mediated inhibition of the integrin-fibronectin binding in control and N157Q mutant overexpressing cells reduced the transmigration capacity and cellular migration velocity to the levels of untreated CD82 overexpressing cells and did not exert any additional inhibitory influence in these latter cells (Figure 4E-F, S3F and Videos 2A-F). Furthermore, the focal adhesion spots formation is a key step of cell adhesion or migration. We revealed that CD82 overexpressing cells significantly reduced the focal adhesion spots but not in N157Q mutant overexpressing cells (Figure 4G). The treatment with RGDs reduced the focal adhesion spots formation in both CD82 and N157Q mutant overexpressing cells (Figure 4G). All together these data strongly demonstrate that the glycosylation of CD82 at Asn157 inhibits cell motility and migration by disrupting the integrin-mediated anchorage to fibronectin leading thereby to a blunted activation of the signaling pathway downstream of integrin receptors.
CD82 glycosylation at Asn157 inhibits ovarian cancer metastasis in vivo
The results reported till now clearly demonstrate a pivotal role of CD82 glycosylation at N157 in the inhibition of ovarian cancer cell motility in vitro. We subcutaneously injected control, CD82 and N157Q mutant cells using a xenograft cancer model for 6 mice per group (N=6) in nude mice [28, 29] and analyzed the tumor growth (Figure 5A). According to the accepted role of CD82 as a metastasis suppressor, no impact on the growth rate of the primary tumors was discernible (Figure 5B-C). In agreement with the postulated CD82 function, mice injected with CD82 cells displayed a reduced incidence of lung and liver metastasis. Notably, mice injected with control or N157Q mutant cells showed higher metastasis formation (Figure 5D-E, S4A and Table S4). This finding provides evidence for the critical role of N157 glycosylation in metastasis inhibition in vivo.
Glycosylation of CD82 at N157 is fundamental for CD82-mediated inhibition of ovarian cancer metastasis in vivo. A. Schematic representation of the subcutaneous mouse xenograft model. 1x106 stable ES2 cells were injected subcutaneously in 4-6 weeks old female BALB/c nude mice. The tumor volume was measured every five days starting from day 10 post-injection. B. Graph representing the measurement of the tumor volume at the indicated time for the experiment described in a. C. Macroscopic images of excised tumors of the experiment described in a and b, 30 days after injection. D-E. Hematoxylin-eosin staining of lung and liver sections from the mouse xenograft model described in a. Note the impaired incidence of lung and liver metastasis in mice injected with CD82 overexpressing cells as compared with MCS or N157Q N=5. Quantification of the percentage of the metastasis area is reported in the graph on the right N=5. ***p<0.005, NS: not significant. F. Schematic representation of the intraperitoneal mouse xenograft model. 1x106 stable ES2 cells were injected intraperitoneally in 4-6 weeks old female BALB/c nude mice. Mice were sacrificed 4 weeks post injection N=4. G. Macroscopic analysis of total tumor weight per mouse and mesenteric metastasis in the mouse xenograft model described in f. Quantification of the tumor weight per mouse is represented in the graph on the right. **p<0.01; N=4. H. Representative macroscopic pictures of ascites from mice xenografts described in f-g. Quantification of the total volume of ascites per mouse is shown in the graph on the right. N=3; ***p<0.005.
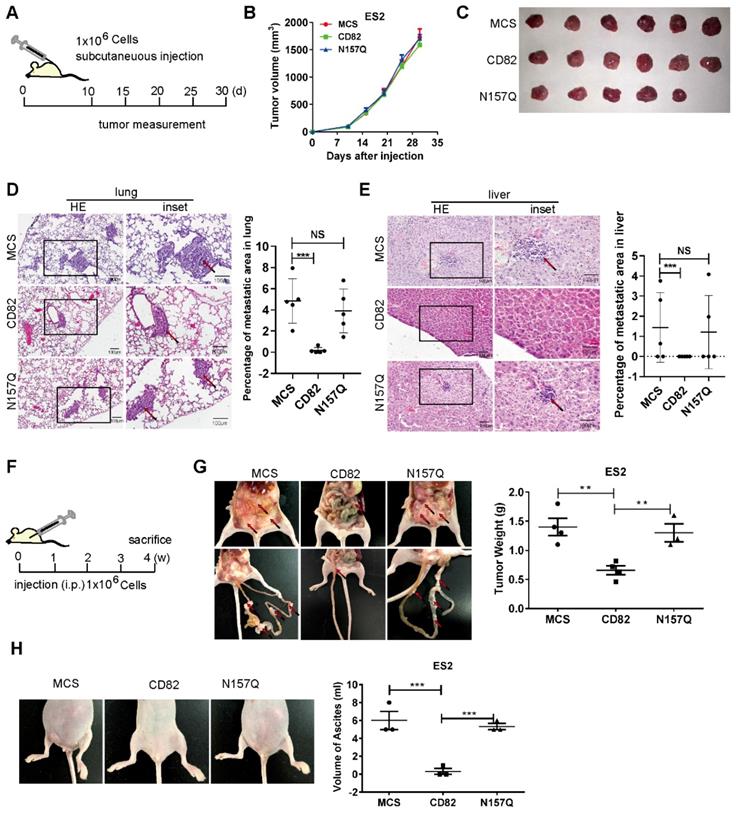
To investigate the impact of glycosylated CD82 on the peritoneal dissemination of ovarian cancer, ES2 cells were injected intraperitoneally for 4 mice per group (N=4) and analyzed for tumor growth four weeks post-injection (Figure 5F). Mice injected with CD82 cells exhibited significantly reduced the tumor cell dissemination as compared with control or N157Q cells by the total tumor weight per mouse (Figure 5G, S4B). In addition, we demonstrate a clear reduction in the total size and volume of ascites per mouse injected with CD82 cells as compared with control or N157Q cells (Figure 5H). Our findings provide compelling evidence for a pivotal role of the glycosylation at N157 in CD82-mediated inhibition of ovarian cancer invasion and metastasis.
MGAT3 mediates glycosylation of CD82 in metastatic ovarian cancer
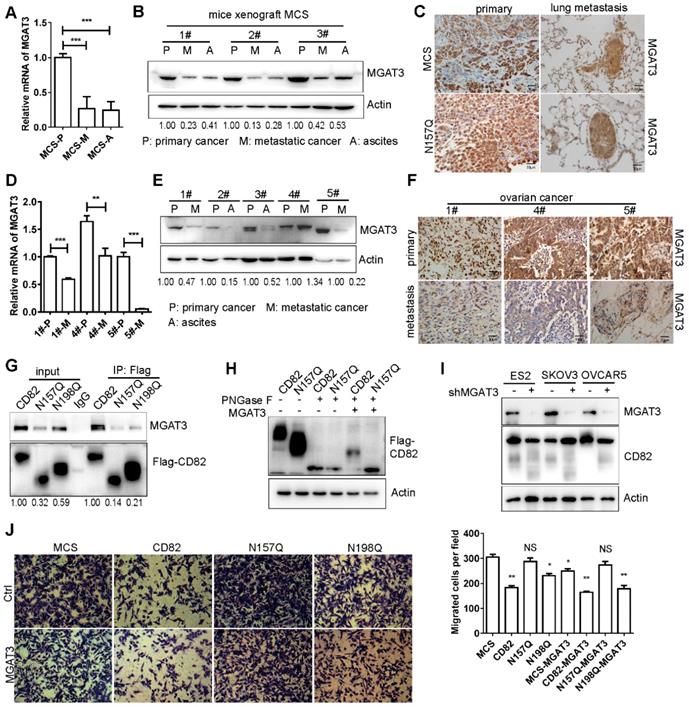
CD82 appears to be hypo-glycosylated in human metastatic ovarian tumors (Figure 1D). Thereafter we uncovered the possible cause responsible for CD82 hypo-glycosylation. Wang and collaborators have demonstrated that CD82 glycosylation mainly consists of N-acetylglucosamine, (α-2, 6) N-acetylneuraminic acid, and a core fucose [12]. Noticeably, the modification is catalyzed by the enzyme MGAT3 [30]. Interestingly, studies reported an association between reduced expression of MGAT3 and cancer metastasis [31-36]. A significant reduction of the mRNA and protein levels of MGAT3 was detected in metastatic tumors as well as in the ascites compared to primary tumors (Figure 6A-B, S4C). A lower MGAT3 expression was observed in lung metastasis compared with primary tumors using IHC (Figure 6C). In agreement with the results obtained in mice, a dramatic reduction of MGAT3 expression was detected in human metastatic ovarian tumors at the mRNA and protein levels (Figure 6D-F). A molecular complex formation between CD82 and MGAT3 would be required for CD82 glycosylation. Co-immunoprecipitation shows that endogenous MGAT3 efficiently co-precipitates with CD82 (Figure 6G, S4D-E). To unequivocally prove that MGAT3 is the enzyme responsible for CD82 glycosylation in ovarian cancer cells, we treated protein lysates from CD82 and N157Q mutant cells with recombinant PNGase F to remove the glycosylation moiety and subsequently subjected these lysates to treatment with recombinant MGAT3. In line with our expectation, treatment with recombinant MGAT3 after PNGase F addition increased the molecular weight of CD82 but not N157Q (Figure 6H). These data strongly indicate that MGAT3 specifically promotes CD82 glycosylation at N157. To verify the function of MGAT3 mediated CD82 glycosylation, we performed the MGAT3 knockdown and overexpression assays. Knockdown MGAT3 in the ovarian cancer cell lines using shRNA impaired the glycosylation of CD82 (Figure 6I). The overexpression of MGAT3 significantly reduced the transmigration capacity of control, CD82, and N198Q mutant but not of N157Q mutant cells (Figure 6J). The efficient overexpression of MGAT3 was confirmed by western blotting (Figure S4F). These data clearly demonstrate that MGAT3 is responsible for CD82 glycosylation and suggest that MGAT3 downregulation in metastatic ovarian cancer may promote metastasis.
MGAT3 impaired CD82 glycosylation and downregulation in metastatic ovarian cancers. A. RT-qPCR analysis of MGAT3 expression in primary and metastatic tumors as well as in ascites derived from injection of MCS cells in the intraperitoneal xenograft model described in Figure. 4F-H. The relative expression of MGAT3 was calculated using the ΔCt method and GAPDH as normalization control. The graph represents the average of MGAT3 relative expression ± SD of 3 different mice. ***p<0.005. B. Western blot analysis of MGAT3 expression in primary (P) and metastatic (M) tumors as well as in ascites (A) of three different mice as in a. Actin was used as loading control. C. Immunohistochemistry analysis of MGAT3 expression in primary tumors and lung metastasis from MCS and N157Q mice in the intraperitoneal xenograft model described in Figure. 4F-H. D. RT-qPCR analysis of MGAT3 expression in clinical samples of primary (P) and metastatic (M) ovarian cancers. Expression of MGAT3 in three different patients (1#, 4# and 5#) is shown. The histogram represents the average of MGAT3 relative expression ± SD of three technical replicates for each patient. **p<0.01, ***p<0.005. E. Western blot analysis of MAGAT3 expression in primary and metastatic ovarian cancers as well as in ascites derived from different patients. F. Immunohistochemistry analysis of MGAT3 expression in primary ovarian cancers and lung metastasis of three different patients (1#, 4# and 5#). G. Coupled immunoprecipitation (anti-Flag antibody) and western blot analysis (anti-MGAT3 antibody) of cellular lysates derived from stable CD82, N157Q and N198Q overexpressing ES2 cells. The picture is representative of three independent experiments. H. Cellular lysates derived from stable ES2 cells overexpressing CD82 or N157Q were treated with recombinant MGAT3 or PNGase F as indicated prior to western blot analysis. I. MGAT3 knockdown was performed in three ovarian cancer cell lines: ES2, SKOV3, OVCAR5. The glycosylation of CD82 is impaired by the MGAT3 knockdown by western blot analysis. J. Transwell migration assay of MCS, CD82, N157Q and N198Q overexpressing ES2 cells transfected with an empty vector or with MGAT3 as indicated. The transmigrated cells were stained with crystal violet. A quantification of the number of migrated cells per field ± SD is reported in the histogram on the right. **p<0.01, *p<0.05, NS: not significant.
Discussion
CD82 is a ubiquitously expressed transmembrane protein which has been implicated in a vast number of biological functions such as immune-response, differentiation, cancer growth and metastasis [9]. Numerous reports demonstrated that CD82 is a potent inhibitor of cancer metastasis and downregulated in metastatic cancers [10, 11, 37]. The mechanisms that lead to CD82 downregulation in cancer still remain poorly characterized. Promoter hypermethylation, gene mutations and loss of heterozygosity only infrequently provoke CD82 downregulation in cancers, although reduced CD82 promoter activity due to aberrant availability of specific transcription factors might account for this process [38-42]. In addition, impaired CD82 protein stability might contribute to CD82 downregulation in some metastatic tumors [43].
CD82 is a highly post-translational modified protein which is mainly N-glycosylated and palmitoylated [10]. The essential role of glycosylation for CD82-mediated functions was proposed by Ono and collaborators. The authors demonstrate that the effect of CD82 in the inhibition of cellular motility requires the restoration of efficient N-glycosylation [44]. Marjon et al. recently demonstrated that N-glycosylation and palmitoylation of CD82 are critical modifications that control CD82-dependent regulation of bone marrow homing of acute myeloid leukemia (AML). The authors demonstrated that glycosylation and palmitoylation exert opposite functions in the trafficking of AML cells in the bone marrow through differential regulation of membrane clustering of the adhesion molecule N-cadherin [45]. Zhou et al. demonstrated that the palmitoylation of CD82 is required for its inhibitory role on migration and invasion of prostate cancer cell lines [46]. The subcellular distribution of CD82 and its association with other tetraspanins are dependent on the palmitoylation. Palmitoylation regulates the specific tetraspanin-enriched microdomains formation or disrupts other tetraspanin/tetraspanin interactions [47]. CD82 inhibits cancer cells dissemination from primary tumors by promoting cell-cell adhesion [48]. Numerous studies demonstrated that CD82 plays a pivotal role as inhibitor of cancer cells motility by interfering integrin-mediated cellular adhesion and migration [49-53]. CD82 promotes integrin α6 internalization resulting thereby in reduced adhesion of cancer cells to laminin, and subsequent reduction of cell migration [54]. In addition, CD82 inhibits cell surface expression of β1 integrin by reducing its glycosylation and maturation in prostate cancer cell lines [55, 56]. Moreover, studies demonstrated that CD82 impacts the expression and activation of adaptor proteins downstream of the integrin pathway [56, 57]. It is also known that CD82 regulates the localization in microdomains and the ubiquitylation of EGFR. EGFR also impact CD82 regulated integrin signaling to impair the cell adhesive/invasive ability [58].
In this study, we dissected a previously undescribed mechanism controlling ovarian cancer metastasis. We demonstrated that glycosylation of CD82 at Asn 157 is a fundamental post-translational modification which is required for CD82-mediated inhibition of ovarian cancer metastasis in vitro and in vivo. These findings are in line with previous data obtained in ldlD cells that the extensive N-glycosylation impairs CD82 interaction with α3 and α5 integrins [59]. Numerous glycoproteins are recognized as possible markers for cancer diagnosis and prognosis [60-63]. Studies reported that the glycosyltransferase MGAT3 controls cancer proliferation, migration and metastasis although these effects may be beneficial or detrimental in a cell type-dependent manner [64-70]. Another glycosyltransferase MGAT5 has also been reported to catalyze β1-6 GlcNAc-branched N-glycans, which is increased in highly metastatic tumor cell lines [71, 72]. MGAT3 transfers GlcNAc to a β1-4 mannose in N-glycans to form a bisecting GlcNAc, which may suppress β1-6 GlcNAc branching formation catalyzed by MGAT5. We find that the glycosylation of CD82 is mainly catalyzed by the glycosyltransferase MGAT3. Interestingly, we additionally demonstrated that treatment of ovarian cells with CD82-enriched exosomes efficiently inhibits ovarian cancer cells adhesion and migration in vitro that may represent novel strategies for ovarian cancer metastasis therapies.
Abbreviations
ECM: Extracellular matrix; EC1/2: Extracellular loop 1/2; FAK: Focal adhesion kinase; FBS: Fetal bovine serum; DMEM: Dulbecco's modified Eagle's medium; WT: Wild type; IP: Immunoprecipitation; IF: Immunofluorescence; IHC: Immunohistochemistry; H&E: Hematoxylin and eosin; IEM: Immunoelectron microscopy; TCGA: The Cancer Genome Atlas database; RGD: Arg-Gly-Asp.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Nr. 81773120, 81972882), the Fundamental Research Funds for the Central Universities, Nankai University (Nr. 63191214), the State Commission of Science Technology of China Projects (2018YFE0114300).
Author Contributions
J.L. designed and performed the experiments, analyzed data and wrote the manuscript; J.X., L.L. and S.L. performed experiments and analyzed data; A.I., P.K., P.S., X.T., T.B. and R.X. provided scientific input and wrote the manuscript; S.Y supervised the entire project, designed the experiments, analyzed data and wrote the manuscript. All authors read and approved the final manuscript.
Availability of data and materials
The detailed procedures of methods, figures, tables and videos are attached.
Ethics approval and consent to participate
For all of the ovarian cancer clinical samples were obtained from The Central Maternity Hospital of Tianjin. This study was approved by the ethics committees of The Central Maternity Hospital of Tianjin. All patients signed an informed consent. All of the animal experiments were approved by the Nankai University Animal Care and Use Committee and handled according to the Nankai University Animal Welfare Guidelines.
Consent for publication
The authors confirmed that they have obtained written consent from the patient to publish the manuscript.
Supplementary Material
Supplementary figures and tables.
Supplementary video 1a.
Supplementary video 1b.
Supplementary video 1c.
Supplementary video 2a.
Supplementary video 2b.
Supplementary video 2c.
Supplementary video 2d.
Supplementary video 2e.
Supplementary video 2f.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bast RC Jr, Hennessy B, Mills GB. The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer. 2009;9:415-28
2. Cho KR, Shih IM. Ovarian Cancer. Annu Rev Pathol-Mech. 2009;4:287-313
3. Moffitt L, Karimnia N, Stephens A, Bilandzic M. Therapeutic Targeting of Collective Invasion in Ovarian Cancer. Int J Mol Sci. 2019;20:1466
4. Yeung TL, Leung CS, Yip KP, Au Yeung CL, Wong ST, Mok SC. Cellular and molecular processes in ovarian cancer metastasis. A Review in the Theme: Cell and Molecular Processes in Cancer Metastasis. Am J Physiol Cell Physiol. 2015;309:C444-56
5. Seguin L, Desgrosellier JS, Weis SM, Cheresh DA. Integrins and cancer: regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol. 2015;25:234-40
6. Jin H, He Y, Zhao P, Hu Y, Tao J, Chen J. et al. Targeting lipid metabolism to overcome EMT-associated drug resistance via integrin β3/FAK pathway and tumor-associated macrophage repolarization using legumain-activatable delivery. Theranostics. 2019;9:265-78
7. Millard M, Odde S, Neamati N. Integrin targeted therapeutics. Theranostics. 2011;1:154-88
8. Hemler ME. Tetraspanin proteins promote multiple cancer stages. Nat Rev Cancer. 2014;14:49-60
9. Termini CM, Gillette JM. Tetraspanins Function as Regulators of Cellular Signaling. Front Cell Dev Biol. 2017;5:34
10. Tonoli H, Barrett JC. CD82 metastasis suppressor gene: a potential target for new therapeutics? Trends Mol Med. 2005;11:563-70
11. Feng J, Huang C, Wren JD, Wang DW, Yan J, Zhang J. et al. Tetraspanin CD82: a suppressor of solid tumors and a modulator of membrane heterogeneity. Cancer Metastasis Rev. 2015;34:619-33
12. Wang H, Zhang W, Zhao J, Zhang L, Liu M, Yan G. et al. N-Glycosylation pattern of recombinant human CD82 (KAI1), a tumor-associated membrane protein. J Proteomics. 2012;75:1375-85
13. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402-8
14. Li J, Yin J, Shen W, Gao R, Liu Y, Chen Y. et al. TLR4 Promotes Breast Cancer Metastasis via Akt/GSK3beta/beta-Catenin Pathway upon LPS Stimulation. Anat Rec (Hoboken). 2017;300:1219-29
15. Yue S, Mu W, Erb U, Zoller M. The tetraspanins CD151 and Tspan8 are essential exosome components for the crosstalk between cancer initiating cells and their surrounding. Oncotarget. 2015;6:2366-84
16. Plebanek MP, Angeloni NL, Vinokour E, Li J, Henkin A, Martinez-Marin D. et al. Pre-metastatic cancer exosomes induce immune surveillance by patrolling monocytes at the metastatic niche. Nat Commun. 2017;8:1319
17. Wang GM, Zhang L, Zhou Y, Sun Q, Xu HB, Cai F. et al. KAI1/CD82 Genetically Engineered Endothelial Progenitor Cells Inhibit Metastasis of Human Nasopharyngeal Carcinoma in a Mouse Model. Med Sci Monitor. 2018;24:3146-52
18. Neumann E, Schwarz MC, Hasseli R, Hulser ML, Classen S, Sauerbier M. et al. Tetraspanin CD82 affects migration, attachment and invasion of rheumatoid arthritis synovial fibroblasts. Ann Rheum Dis. 2018;77:1619-26
19. Danen EH, Sonneveld P, Brakebusch C, Fassler R, Sonnenberg A. The fibronectin-binding integrins alpha5beta1 and alphavbeta3 differentially modulate RhoA-GTP loading, organization of cell matrix adhesions, and fibronectin fibrillogenesis. J Cell Biol. 2002;159:1071-86
20. Benito-Jardon M, Klapproth S, Gimeno LI, Petzold T, Bharadwaj M, Muller DJ. et al. The fibronectin synergy site re-enforces cell adhesion and mediates a crosstalk between integrin classes. Elife. 2017 6
21. Huttenlocher A, Horwitz AR. Integrins in cell migration. Cold Spring Harb Perspect Biol. 2011;3:a005074
22. Lee J, Byun HJ, Lee MS, Jin YJ, Jeoung D, Kim YM. et al. The metastasis suppressor CD82/KAI1 inhibits fibronectin adhesion-induced epithelial-to-mesenchymal transition in prostate cancer cells by repressing the associated integrin signaling. Oncotarget. 2017;8:1641-54
23. Huveneers S, Danen EH. Adhesion signaling - crosstalk between integrins, Src and Rho. J Cell Sci. 2009;122:1059-69
24. Horton ER, Humphries JD, Stutchbury B, Jacquemet G, Ballestrem C, Barry ST. et al. Modulation of FAK and Src adhesion signaling occurs independently of adhesion complex composition. J Cell Biol. 2016;212:349-64
25. Kechagia JZ, Ivaska J, Roca-Cusachs P. Integrins as biomechanical sensors of the microenvironment. Nat Rev Mol Cell Biol. 2019
26. Kong F, Garcia AJ, Mould AP, Humphries MJ, Zhu C. Demonstration of catch bonds between an integrin and its ligand. J Cell Biol. 2009;185:1275-84
27. Takagi J, Strokovich K, Springer TA, Walz T. Structure of integrin alpha5beta1 in complex with fibronectin. EMBO J. 2003;22:4607-15
28. Agrawal P, Fontanals-Cirera B, Sokolova E, Jacob S, Vaiana CA, Argibay D. et al. A Systems Biology Approach Identifies FUT8 as a Driver of Melanoma Metastasis. Cancer cell. 2017;31:804-19 e7
29. Le Magnen C, Dutta A, Abate-Shen C. Optimizing mouse models for precision cancer prevention. Nat Rev Cancer. 2016;16:187-96
30. Bhaumik M, Seldin MF, Stanley P. Cloning and chromosomal mapping of the mouse Mgat3 gene encoding N-acetylglucosaminyltransferase III. Gene. 1995;164:295-300
31. Song Y, Aglipay JA, Bernstein JD, Goswami S, Stanley P. The bisecting GlcNAc on N-glycans inhibits growth factor signaling and retards mammary tumor progression. Cancer Res. 2010;70:3361-71
32. Pinho SS, Reis CA, Paredes J, Magalhaes AM, Ferreira AC, Figueiredo J. et al. The role of N-acetylglucosaminyltransferase III and V in the post-transcriptional modifications of E-cadherin. Hum Mol Genet. 2009;18:2599-608
33. Pinho SS, Oliveira P, Cabral J, Carvalho S, Huntsman D, Gartner F. et al. Loss and recovery of Mgat3 and GnT-III Mediated E-cadherin N-glycosylation is a mechanism involved in epithelial-mesenchymal-epithelial transitions. PloS One. 2012;7:e33191
34. Miwa HE, Song Y, Alvarez R, Cummings RD, Stanley P. The bisecting GlcNAc in cell growth control and tumor progression. Glycoconj J. 2012;29:609-18
35. Huang H, Liu Y, Yu P, Qu J, Guo Y, Li W. et al. MiR-23a transcriptional activated by Runx2 increases metastatic potential of mouse hepatoma cell via directly targeting Mgat3. Sci Rep. 2018;8:7366
36. Allam H, Aoki K, Benigno BB, McDonald JF, Mackintosh SG, Tiemeyer M. et al. Glycomic analysis of membrane glycoproteins with bisecting glycosylation from ovarian cancer tissues reveals novel structures and functions. J Proteome Res. 2015;14:434-46
37. Tsai YC, Weissman AM. Dissecting the diverse functions of the metastasis suppressor CD82/KAI1. FEBS Lett. 2011;585:3166-73
38. Tagawa K, Arihiro K, Takeshima Y, Hiyama E, Yamasaki M, Inai K. Down-regulation of KAI1 messenger RNA expression is not associated with loss of heterozygosity of the KAI1 gene region in lung adenocarcinoma. Jpn J Cancer Res. 1999;90:970-6
39. Dong JT, Suzuki H, Pin SS, Bova GS, Schalken JA, Isaacs WB. et al. Down-regulation of the KAI1 metastasis suppressor gene during the progression of human prostatic cancer infrequently involves gene mutation or allelic loss. Cancer Res. 1996;56:4387-90
40. Liu FS, Dong JT, Chen JT, Hsieh YT, Ho ES, Hung MJ. Frequent down-regulation and lack of mutation of the KAI1 metastasis suppressor gene in epithelial ovarian carcinoma. Gynecol Oncol. 2000;78:10-5
41. Jackson P, Millar D, Kingsley E, Yardley G, Ow K, Clark S. et al. Methylation of a CpG island within the promoter region of the KAI1 metastasis suppressor gene is not responsible for down-regulation of KAI1 expression in invasive cancers or cancer cell lines. Cancer Lett. 2000;157:169-76
42. Marreiros A, Dudgeon K, Dao V, Grimm MO, Czolij R, Crossley M. et al. KAI1 promoter activity is dependent on p53, junB and AP2: evidence for a possible mechanism underlying loss of KAI1 expression in cancer cells. Oncogene. 2005;24:637-49
43. Tsai YC, Mendoza A, Mariano JM, Zhou M, Kostova Z, Chen B. et al. The ubiquitin ligase gp78 promotes sarcoma metastasis by targeting KAI1 for degradation. Nat Med. 2007;13:1504-9
44. Ono M, Handa K, Withers DA, Hakomori S. Motility inhibition and apoptosis are induced by metastasis-suppressing gene product CD82 and its analogue CD9, with concurrent glycosylation. Cancer Res. 1999;59:2335-9
45. Marjon KD, Termini CM, Karlen KL, Saito-Reis C, Soria CE, Lidke KA. et al. Tetraspanin CD82 regulates bone marrow homing of acute myeloid leukemia by modulating the molecular organization of N-cadherin. Oncogene. 2016;35:4132-40
46. Zhou B, Liu L, Reddivari M, Zhang XA. The palmitoylation of metastasis suppressor KAI1/CD82 is important for its motility- and invasiveness-inhibitory activity. Cancer Res. 2004;64:7455-63
47. Charrin S, Manie S, Oualid M, Billard M, Boucheix C, Rubinstein E. Differential stability of tetraspanin/tetraspanin interactions: role of palmitoylation. FEBS Lett. 2002;516:139-44
48. Abe M, Sugiura T, Takahashi M, Ishii K, Shimoda M, Shirasuna K. A novel function of CD82/KAI-1 on E-cadherin-mediated homophilic cellular adhesion of cancer cells. Cancer Lett. 2008;266:163-70
49. Mannion BA, Berditchevski F, Kraeft SK, Chen LB, Hemler ME. Transmembrane-4 superfamily proteins CD81 (TAPA-1), CD82, CD63, and CD53 specifically associated with integrin alpha 4 beta 1 (CD49d/CD29). J Immunol. 1996;157:2039-47
50. Iwata S, Kobayashi H, Miyake-Nishijima R, Sasaki T, Souta-Kuribara A, Nori M. et al. Distinctive signaling pathways through CD82 and beta1 integrins in human T cells. Eur J Immunol. 2002;32:1328-37
51. Berditchevski F, Odintsova E. Characterization of integrin-tetraspanin adhesion complexes: role of tetraspanins in integrin signaling. J Cell Biol. 1999;146:477-92
52. Lee JH, Seo YW, Park SR, Kim YJ, Kim KK. Expression of a splice variant of KAI1, a tumor metastasis suppressor gene, influences tumor invasion and progression. Cancer Res. 2003;63:7247-55
53. Ruseva Z, Geiger PX, Hutzler P, Kotzsch M, Luber B, Schmitt M. et al. Tumor suppressor KAI1 affects integrin alphavbeta3-mediated ovarian cancer cell adhesion, motility, and proliferation. Exp Cell Res. 2009;315:1759-71
54. He B, Liu L, Cook GA, Grgurevich S, Jennings LK, Zhang XA. Tetraspanin CD82 attenuates cellular morphogenesis through down-regulating integrin alpha6-mediated cell adhesion. J Biol Chem. 2005;280:3346-54
55. Jee BK, Lee JY, Lim Y, Lee KH, Jo YH. Effect of KAI1/CD82 on the beta1 integrin maturation in highly migratory carcinoma cells. Biochem Biophys Res Commun. 2007;359:703-8
56. Lee HA, Park I, Byun HJ, Jeoung D, Kim YM, Lee H. Metastasis suppressor KAI1/CD82 attenuates the matrix adhesion of human prostate cancer cells by suppressing fibronectin expression and beta1 integrin activation. Cell Physiol Biochem. 2011;27:575-86
57. Delaguillaumie A, Lagaudriere-Gesbert C, Popoff MR, Conjeaud H. Rho GTPases link cytoskeletal rearrangements and activation processes induced via the tetraspanin CD82 in T lymphocytes. J Cell Sci. 2002;115:433-43
58. Odintsova E, van Niel G, Conjeaud H, Raposo G, Iwamoto R, Mekada E. et al. Metastasis Suppressor Tetraspanin CD82/KAI1 Regulates Ubiquitylation of Epidermal Growth Factor Receptor. J Biol Chem. 2013;288:26323-34
59. Ono M, Handa K, Withers DA, Hakomori S. Glycosylation effect on membrane domain (GEM) involved in cell adhesion and motility: a preliminary note on functional alpha3, alpha5-CD82 glycosylation complex in ldlD 14 cells. Biochem Biophys Res Commun. 2000;279:744-50
60. Peixoto A, Relvas-Santos M, Azevedo R, Santos LL, Ferreira JA. Protein Glycosylation and Tumor Microenvironment Alterations Driving Cancer Hallmarks. Front Oncol. 2019;9:380
61. Munkley J, Elliott DJ. Hallmarks of glycosylation in cancer. Oncotarget. 2016;7:35478-89
62. Padler-Karavani V. Aiming at the sweet side of cancer: Aberrant glycosylation as possible target for personalized-medicine. Cancer Lett. 2014;352:102-12
63. Reily C, Stewart TJ, Renfrow MB, Novak J. Glycosylation in health and disease. Nat Rev Nephrol. 2019;15:346-66
64. Yoshimura M, Ihara Y, Nishiura T, Okajima Y, Ogawa M, Yoshida H. et al. Bisecting GlcNAc structure is implicated in suppression of stroma-dependent haemopoiesis in transgenic mice expressing N-acetylglucosaminyltransferase III. Biochem J. 1998;331:733-42
65. Yoshimura M, Ihara Y, Ohnishi A, Ijuhin N, Nishiura T, Kanakura Y. et al. Bisecting N-acetylglucosamine on K562 cells suppresses natural killer cytotoxicity and promotes spleen colonization. Cancer Res. 1996;56:412-8
66. Akama R, Sato Y, Kariya Y, Isaji T, Fukuda T, Lu L. et al. N-acetylglucosaminyltransferase III expression is regulated by cell-cell adhesion via the E-cadherin-catenin-actin complex. Proteomics. 2008;8:3221-8
67. Tan Z, Wang C, Li X, Guan F. Bisecting N-Acetylglucosamine Structures Inhibit Hypoxia-Induced Epithelial-Mesenchymal Transition in Breast Cancer Cells. Front Physiol. 2018;9:210
68. Miwa HE, Koba WR, Fine EJ, Giricz O, Kenny PA, Stanley P. Bisected, complex N-glycans and galectins in mouse mammary tumor progression and human breast cancer. Glycobiology. 2013;23:1477-90
69. Allam H, Johnson BP, Zhang M, Lu Z, Cannon MJ, Abbott KL. The glycosyltransferase GnT-III activates Notch signaling and drives stem cell expansion to promote the growth and invasion of ovarian cancer. J Biol Chem. 2017;292:16351-9
70. Zhang X, Wang Y, Qian Y, Wu X, Zhang Z, Liu X. et al. Discovery of specific metastasis-related N-glycan alterations in epithelial ovarian cancer based on quantitative glycomics. PLoS One. 2014;9:e87978
71. Nagae M, Kizuka Y, Mihara E, Kitago Y, Hanashima S, Ito Y. et al. Structure and mechanism of cancer-associated N-acetylglucosaminyltransferase-V. Nat Commun. 2018;9:3380
72. Taniguchi N, Kizuka Y. Glycans and cancer: role of N-glycans in cancer biomarker, progression and metastasis, and therapeutics. Adv Cancer Res. 2015;126:11-51
Author contact
![]() Corresponding authors: R. Xiang, School of Medicine, Nankai University, 94 Weijin Road, Tianjin, 300071, China, Tel: 86 2223503994, e-mail: rxiangedu.cn. S. Yue, School of Medicine, Nankai University, 94 Weijin Road, Tianjin, 300071, China, Tel: 86 2223503994, e-mail: shijingyueedu.cn
Corresponding authors: R. Xiang, School of Medicine, Nankai University, 94 Weijin Road, Tianjin, 300071, China, Tel: 86 2223503994, e-mail: rxiangedu.cn. S. Yue, School of Medicine, Nankai University, 94 Weijin Road, Tianjin, 300071, China, Tel: 86 2223503994, e-mail: shijingyueedu.cn