Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Abnormal microenvironment and...
The Effects of Lipid Metabolism...
Important signalling pathways...
Targeting lipid metabolism for...
Summary and Perspectives
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(16):7053-7069. doi:10.7150/thno.41388 This issue Cite
Review
Lipid metabolism alteration contributes to and maintains the properties of cancer stem cells
Huangcan Li1, Zhuoying Feng1, Ming-Liang He1,2 ![]()
1. Department of Biomedical Sciences, City University of Hong Kong, Hong Kong, China.
2. CityU Shenzhen Research Institute, Nanshan, Shenzhen, China.
Received 2019-10-22; Accepted 2020-4-28; Published 2020-5-30
Abstract

Lipids, the basic components of the cell membrane, execute fundamental roles in almost all the cell activities including cell-cell recognition, signalling transduction and energy supplies. Lipid metabolism is elementary for life sustentation that balances activity between synthesis and degradation. An accumulating amount of data has indicated abnormal lipid metabolism in cancer stem cells (CSCs), and that the alteration of lipid metabolism exerts a great impact on CSCs' properties such as the capability of self-renewal, differentiation, invasion, metastasis, and drug sensitivity and resistance. CSCs' formation and maintenance cannot do without the regulation of fatty acids and cholesterol. In normal cells and embryonic development, fatty acids and cholesterol metabolism are regulated by some important signalling pathways (such as Hedgehog, Notch, Wnt signalling pathways); these signalling pathways also play crucial roles in initiating and/or maintaining CSCs' properties, and such signalling is shown to be commonly modulated by the abnormal lipid metabolism in CSCs; on the other hand, the altered lipid metabolism in turn modifies the cell signalling and generates additional impacts on CSCs. Metabolic rewiring is considered as an ideal hallmark of CSCs, and metabolic alterations would be promising therapeutic targets of CSCs for aggressive tumors. In this review, we summarize the most updated findings of lipid metabolic abnormalities in CSCs and prospect the potential applications of targeting lipid metabolism for anticancer treatment.
Keywords: Cancer stem cells, lipid metabolism, signalling pathways, self-renewal
Introduction
Cancer stem cell (CSC) is a proportion of abnormal cell lineages involved in tumor initiation, progression and metastasis during tumorigenesis (Figure 1), are believed the major cause of drug resistance and recurrence after a period of anticancer chemotherapies. CSCs are similar to or even enhanced self-renewal of the normal pluripotent and multipotent stem cells but lose a certain degree of differentiation capacity [1,2]. Two potential origins of CSCs are suggested--either derived from normal stem/progenitor cells through transformation/reprogramming or be transformed from fully differentiated cells caused by genetic instability and epigenetic abnormality during neoplasia pathology [3]. Evidence shows that adenomatous polyposis coli (APC) deleted crypt stem cells could induce intestinal microadenomas by activating the Wnt signalling pathway [4]. Besides, CSC surface markers, such as CD133+, CD44+, CD34+ CD166+, CD24+ and ALDH1+, also are well known stem cell markers. The study on cell-type plasticity shows that the Wnt-activation in intestinal epithelial cells (IECs) induces non-stem cancer cells' dedifferentiation and crypt stem cell expansion [5]. Results from methylation-specific polymerase chain reaction (PCR) experiments reveal that the activated IL-6/JAK2/STAT3 signalling promotes the proliferation of lung cancer stem cells by suppressing p53/p21 expression via DNA hypermethylation [6]. During epithelial-mesenchymal transition (EMT), a complicated, multistage process for epithelial cells to enhance motility, weaken cell polarity and degrade the extracellular matrix (ECM) [7], both normal and cancer cells acquire stem-cell like properties to allow migration and invasion to foreign tissues [8].
The hallmarks of cancer stem cells (CSCs). CSCs may originate from either normal stem cells (including progenitor cells) or transformation of differentiated cells through reprogramming by genetic instability and epigenetic abnormality under long-term stress conditions (e.g., microenvironment factors, hypoxia, virus invasion, etc.). CSCs display a close association with tumor microenvironment. The self-renewal capability of CSCs is significantly enhanced to certain degrees similar to or even stronger than that of the normal pluripotent and multipotent stem cells, but the differentiation capacity is somewhat lost. During the epithelial-mesenchymal transition (EMT) process, cancer cells acquire stem-cell like properties to allow migration and invasion to foreign tissues. CSCs are hierarchical plasticity subpopulations, which are the main causes for tumorigenesis, immune evasion, EMT, tumor progression, metastasis, and drug resistance. These characters of CSCs are regulated by several signalling pathways, such as Notch, Wnt, Hippo cascades, Hedgehog, hypoxia-inducible factor 1 (HIF), etc.
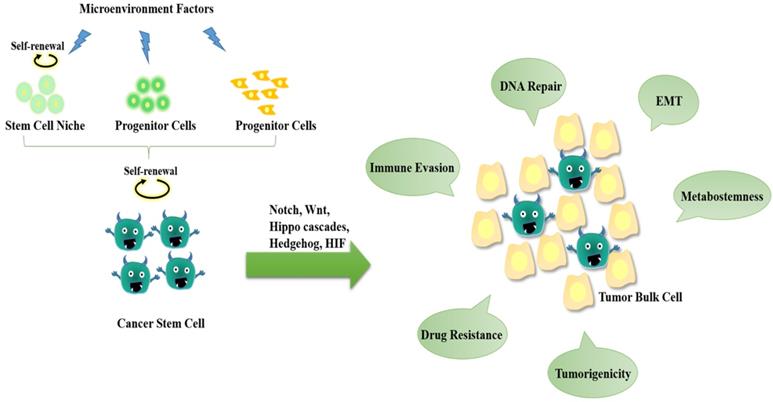
CSCs have been detected in different types of tumors, including lung cancer [6], breast cancer [9], ovarian cancer [10], pancreatic cancer [11], human acute myeloid leukemia (AML) [12], and glioblastoma (GBM) [13]. Regardless of the origin, CSCs are hierarchical plasticity subpopulations driving tumor progression and chemotherapy resistance, thereby hindering tumor prognosis and promoting tumor recurrence. Besides the abnormalities of signalling activations, increasing data have shown that the abnormalities of lipid metabolism exhibit great impacts on CSC properties.
Abnormal microenvironment and metabolic pattern in tumors and CSCs
CSCs are a group of subpopulation cells in carcinoma. Numerous studies demonstrate that CSCs are responsible for driving tumor growth, epithelial-mesenchymal transition (EMT), metastases and drug resistance. Alternated nutrient consumption between tumor bulk cells and CSCs in tumor microenvironment (TME) is associated with tumor immune evasion and progression. Induced by oncogenes, CSCs facilitate adaptive metabolic changes to sustain increasing energy need for growth and anabolic functions. Similar to stem cells, CSCs exhibit high plasticity in response to the metabolic changes in maintaining self-renewal, proliferation, and survival [14]. The metabolic phenotype of CSCs may be heavily decided by microenvironmental conditions. The metabolisms of CSCs are specifically varied, dependent on tumor types and the site of metastasis. Metabolic alternation of CSCs has been proposed as a functional marker and promising therapeutic target.
Warburg effect
The suffering of cancer cells from abnormal limitations in nutrient supply (such as glucose and oxygen) is referred to as the “Warburg effect” [15]. Warburg effect describes a metabolic shift from oxidative phosphorylation (OXPHOS) to glycolysis in pentose phosphate shunt and an accumulation of lactate in exchange for sustained ATP production in TME [16]. Emerging evidence has suggested that the glycolytic metabolism of Warburg effect plays a role in stemness and the EMT process [17]. R406, a Syk inhibitor for immune thrombocytopenia (ITP), inhibits neurosphere formation and triggers apoptosis in GBM through inducing a metabolic shift from glycolysis to OXPHOS and subsequently producing excessive reactive oxygen species (ROS) in glioma stem cells (GSCs) [18]. In the basal-like breast cancer (BLBC) EMT process, Snail-mediated promoter methylation of fructose-1,6-biphosphatase (FBP1) gives rise to enhanced CSC-like properties and tumorigenicity through increasing glucose uptake and macromolecules biosynthesis, as well as inhibiting oxygen consumption through suppressing mitochondrial complex I activity [19]. Internal tandem duplication (ITD) mutation in Fms-like tyrosine kinase 3 gene (FLT3/ITD) accounts for approximately 30% of acute myeloid leukemia (AML) cases. FLT3/ITD is extensively present in leukemia stem cells (LSCs) and is proposed to be a primary event in leukemogenesis in possessing CD123 (IL-3RA) stage, and is the main cause of poor prognosis in patients [20]. Results from both in vitro and in vivo studies demonstrate that FLT3/ITD upregulates aerobic glycolysis through activating mitochondrial hexokinase (HK2) in an AKT-dependent manner. Glycolytic inhibitors cause severe ATP depletion and massive cell death in FLT3/ITD positive leukemia cells [21]. Recent findings suggest that Warburg effect persist stem cell metabolism in tumors, as a failure of differentiation [13,22]. Clinical studies reveal that lower-level uptake of 18F-fluorodeoxyglucose occurs in well-differentiated tumors while higher level uptake happens in the poorly differentiated group. In GBMs, CSCs under nutrient deprivation shift toward the use of pentose phosphate shunt, which promotes CSCs' self-renewal, proliferation and survival [15].
Oxidative phosphorylation (OXPHOS)
As opposed to differentiated bulk tumor cells that suffer from the “Warburg” effect, CSCs exhibit a distinct metabolic phenotype--being highly glycolytic or OXPHOS dependent. Cancers can be clustered along the differentiation pathways into two groups, utilizing either glycolysis or oxidative phosphorylation. Each group is decided by tumor subtypes, specific phenotype of CSCs, and tumor microenvironment [23]. In an inducible pancreatic cancer mouse model, a subpopulation of dormant tumor cells is found to rely on oxidative phosphorylation (OXPHOS) for survival [24]. OXPHOS happens in the mitochondria, with the generation of ROS. In gliomaspheres, CSC expansion also depends on OXPHOS in the mitochondrial respiratory chain to produce energy for survival [25]. AML employs higher mitochondria oxidative phosphorylation as compared to non-malignant CD34+ hematopoietic progenitor cells [26,27]. In an AML xenograft model, the bone marrow stromal cell is deprived of mitochondria through deriving tunnelling nanotubes in the stimulation of superoxide by NOX2. Inhibition of NOX2 interrupts mitochondrial transfer, increases AML apoptosis, and improves AML mouse survival [28]. On the contrary, lung CSCs derived from A549 cells display a low quantity of mtDNA, high mitochondrial membrane potential, low oxygen and glucose consumption and a low intracellular concentration of ATP and ROS [29]. Similarly, mitophagy, a selective cleansing of mitochondria through autophagy, facilitates the generation and proliferation of liver CSCs by inhibiting p53 expression [30].
Lipid metabolism
Lipids are typically classified as lipoids (phospholipid, cholesterol and cholesterol ester, etc.) and fats (triglycerides, TG). Lipoids are essential for a variety of cellular functions, including membrane construction, signalling transduction and other biological activities. TG is the main source of cellular energy. Lipid metabolism is elementary for life sustentation that balances synthesis and degradation. As a prerequisite to maintain cell survival, lipid homeostasis is coordinated by integrated systems to quickly respond to metabolic changes. In an energy-deficient or a nutrient exhausted condition, the cell demand for metabolic intermediates for nutrient synthesis and energy production is substantial. Hence, the role of TGs and cholesterol is especially indispensable in cancer and related diseases. Accordingly, disorder or alternation of lipid metabolisms has been linked significantly with pathogenic infection (bacteria, fungi, and virus), lipid-related diseases (hyperlipidemia, lipid storage disease, obesity, etc.) and pathological cancers. Currently, lipid metabolism has been heralded as a novel and significant target for cancer therapy. Emerging evidence has revealed cancer cell alternations in several aspects including membranes formation, lipids synthesis and degradation, and cellular signalling driven by lipids. In the following sections, we focus on the importance and latest findings of fatty acid and cholesterol metabolisms in CSCs, as well as relevant and promising therapeutic targets for cancer therapy.
The Effects of Lipid Metabolism Alterations in CSCs
Accumulating evidence has shed light on alterations in lipid metabolism and related pathways. Recently, it has been shown that lipids and lipoproteins, either exogenous (or dietary) uptake or endogenous synthesis, have been shown to have a great impact on maintaining CSCs' properties in tumorigenesis. For example, the fatty acid synthase (FASN), a rate-limiting enzyme for de novo lipid synthesis, is consistently found to facilitate in multiple types of CSCs. Furthermore, lipids and cholesterol are increasingly uptaken or generated through hyper-activating the metabolic routes in tumor stem cells. Single-probe mass spectrometry (MS) study has revealed a remarkable metabolic pattern of live CSCs at the single cell level [31]. As compared to non-stem counterpart, CSCs generate more active tricarboxylic acid (TCA) and more abundant unsaturated lipids. Previous studies showed that CSCs require more monounsaturated fatty acids (MUFAs) than their non-stem counterparts in ovarian tumors and glioblastoma, suggesting that lipid desaturation may be an ideal biomarker for CSCs [32-35]. Furthermore, a comparison of lipidomic profiles between CSCs and non-stem cancer cells suggests that MUFAs affect the formation and stemness of CSCs [32]. As the structural components of cellular membranes, the membrane fluidity is highly dependent on the degree of lipid unsaturation. Low membrane fluidity inhibits metastasis and stemness in breast cancers [36]. Of note, treating with saturated fatty acids (SFAs) in proportion with glycerophospholipids suppresses hepatocellular carcinogenesis [37]. The high proportion of saturated fatty acids attenuates membrane tension and inhibits symmetric division or pluripotent deficiency, indicating the importance of MUFAs in maintaining CSCs [38]. The unsaturated lipids regulated by stearoyl-CoA desaturase-1 (SCD1), nuclear factor κB (NF-κB) and aldehyde dehydrogenases 1 A1 (ALDH1A1) significantly promotes the stemness of colorectal CSCs [31]. To further clarify this assumption, a study on a series of 577 breast carcinomas shows that the highly elevated ALDH1 level is correlated with poor prognosis [39]. Results obtained from both in vitro and in vivo studies have highlighted the importance of ALDH activity in CSCs' self-renewal and tumorigenicity. Additionally, elevated fatty acid oxidation (FAO) ensures the energy supplies for the extreme environment alternation in CSCs. Hence, carnitine palmitoyl transferase 1 (CPT1), the critical accelerator of FAO, promotes breast cancer stemness and chemoresistance [40]. Besides the glycolysis or oxidative phosphorylation, lipids from adipocytes residing in the microenvironment are also used as an energy source in ovarian cancer and prostate cancer [41,42]. CSCs require an accelerated FAO to obtain sufficient metabolic intermediates, such as acetyl-CoA and NADH, to satisfy the needs of ATP generation for self-maintenance and proliferation. In hepatocellular carcinoma (HCC) cells and leukemia-initiating cells, FAO is linked to stem-like properties with de novo fatty acid synthesis [43]. Leukemia-initiating cells co-opt the adipose tissue niche to create a supportive microenvironment for leukemic growth and chemoresistance [44]. Cholesterol is one of the key components in the cell membrane and lipid raft for signalling transduction in pro-oncogenic and anti-apoptotic pathways. Interfering cholesterol biosynthesis may bring large, additional impacts on the cholesterol content in lipid rafts and the signalling transduction for CSCs' proliferation [45,46]. Lipid droplets (LDs) are cytoplasmic organelles originating from the endoplasmic reticulum and/or the Golgi apparatus for fatty acids and cholesteryl ester storage. Studies from Groupwise comparisons show that the accumulation of LDs has a close relationship with tumor proliferation and aggression potential [47]. In colorectal CSCs, as revealed by Raman spectroscopy imaging, a high level of LDs is a distinctive marker of CSCs. LDs' level also fluctuates with other well-accepted CSC markers such as CD133, activated Wnt pathway, etc. [48]. Furthermore, a statistical analysis of the overall lipid droplets from cancer cells has been considered as an ideal marker of tumor aggressiveness [49].
Fatty Acids (FAs) homeostasis
Fatty acid (FA) metabolism is the core of lipid status harmonization, which maintains the energy and supplies for the living of organisms. FA synthesis produces the fundamental component of various lipids for cell membrane construction, signal transduction, energy storage, and biological functions. FA catabolic pathway generates energy via FA degradation conducted by FA oxidation (FAO), or commonly known as β-oxidation.
In the last years, the importance of lipid metabolism in cancer cells has been repeatedly emphasized, and a series of significant advances have been made to provide useful reference indicators and directions for cancer therapy [50,51]. Tumor cells proliferate rapidly while angiogenesis becomes abnormal, thus cancer cells are under hypoxic, hyper-oxidative, acidic and malnutrition conditions. CSCs alter their basic metabolisms to encounter those unfavorable microenvironments. Lipid metabolism presents a massive and complex network of flexible pathways, feedback loops and cross talks that maintains the metabolic requirement for cancer cells. FA homeostasis and balance of FA synthesis, storage, and degradation control the core node of the framework. FA synthesis generates various metabolic intermediates that are fed to anabolic metabolisms for cellular membrane maintenance or signal transduction in inducing oncogenic cascades, resulting in malignancy, chemoresistance and cancer stemness. As a supplement, FAO retrieves acetyl CoA to initiate FA synthesis, indicating that FA synthesis and FAO are mutually complemented. More distinctive than glycolysis contribution in CSCs, lipid metabolism may contribute to CSCs in varied aspects. A recent study shows that fatty acids metabolisms, both FA synthesis and FAO, contribute to the pluripotency and reprogramming of embryonic and somatic stem cells [38,52,53]. Compared to the non-stem counterparts, CSCs reduce the utilization of glycolysis but maintain a sufficient amount of ATP generation, indicating the significance of alteration in lipid metabolism [54]. Distinctive lipidomics changes have been demonstrated to vary in CSCs and bulk cancer cells in glioblastoma multiform [32]. Moreover, the intermediates generated by glycolysis are fed to FA synthesis for CSCs self-renewal [55]. Indeed, fatty acid homeostasis, or the balance of catabolic/anabolic state, keeps the grip of pluripotency, self-renewal, proliferation and formation of the CSCs (Figure 2).
The abnormal lipid metabolisms and current therapeutic targets in tumors and CSCs. Cancer cells, especially CSCs, increase lipids catabolic/anabolic activities, such as FA synthesis (Yellow arrows), FA oxidation (Green arrows), and cholesterol synthesis (Blue arrows). The ectopic lipid metabolisms facilitate the pluripotency, self-renewal, proliferation and formation of CSCs. Currently, key enzymes (Red letters) dominating lipid metabolisms have been considered as ideal therapeutic targets or prognosis for cancers. Abbreviation: ACC, acetyl-CoA carboxylase; ACLY, ATP-citrate lyase; ACSS2, acyl-CoA synthetase short-chain family member 2; CPT1, carnitine palmitoyl-transferase 1; FA, fatty acid; FAO, fatty acid oxidation; FASN, fatty acid synthase; SCD1, stearoyl-CoA desaturase1; TCA cycle, tricarboxylic acid cycle; MUFA, monounsaturated FA; SFA, saturated FA; PUFA, polyunsaturated FA. The figure was produced using Servier Medical Art (http://www.servier.com).
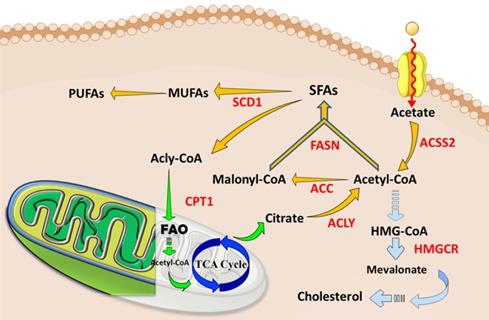
FA synthesis promotes CSCs
Limited to the absorption and consumption of dietary lipids, cancer cells urgently require FA synthesis to meet energy needs and structural construction. Overall, key players in FA synthesis, such as ATP-citrate lyase (ACLY), acetyl-CoA carboxylase (ACC) and fatty acid synthase (FASN), are elevated in cancer cells. These enzymes are emerging as the hallmark of cancer and even ideal markers for cancer stemness [51,56]. Unlike their non-stem counterpart, CSCs may absorb glycolytic metabolic intermediates for lipid biosynthesis to improve self-renewability under the Warburg effect [57]. By measuring the 14C-glucose and 14C-acetate incorporation as the carbon source for de novo lipogenesis, studies show GSC requires more lipogenesis than bulk cancer cells in glioblastoma [58]. Emerging evidence has emphasized the impact of fatty acid synthesis deficiency in multiple carcinogenesis and cancer stemness, recognizing the inevitable role of de novo fatty acid synthesis in CSC self-renewal and survival [59]. Here, we mainly compare each key player for its role in constituting fatty acid synthesis and further discuss the potential therapeutic strategies in eliminating CSCs via the anti-lipogenesis method.
ATP citrate lyase (ACLY)
ACLY catalyzes the conversion of citrate into acetyl CoA in the cytoplasm, which is the significant building block of fatty acid and cholesterol synthesis. Elevated expression level and activation of ACLY have been broadly reported in multiple tumors. Elevated ACLY activity positively enhances malignant phenotypes and poorer prognosis [38,60,61]. On the contrary, inhibition of ACLY suppresses tumor growth and EMT [62,63]. ACLY is also indicated as a fundamental factor of cancer stemness. Inhibition of ACLY by siRNAs or chemical inhibitors significantly impairs the growth of CSCs derived from human non-small cell lung carcinoma or breast cancer [64-66]. ACLY inhibition decreases the proliferation of lung CSCs activated by Ras or by epidermal growth factor receptor (EGFR) mutations. ACLY knockdown significantly reduces the CSC population in breast cancer cells [62,66]. Snail, a crucial transcription factor for EMT induction, is a potential target of ACLY in the process of Ras-induced cancer stemness. The phosphorylation of ACLY at serine 454 by phosphatidylinositol 3‑kinase (PI3K)/protein kinase B (AKT) pathway is up-regulated with the stage, the tumor differentiation grade, and poor prognosis in non-small cell lung cancer [67]. Phosphorylated forms are suggested to profoundly increase the effect of ACLY in CSCs.
Acetyl-CoA carboxylase (ACC)
ACC, which carboxylates acetyl-CoA into malonyl-CoA, exhibits up-regulation in the breast, gastric, and lung cancers [68-70]. Furthermore, the distinctive elevation of ACC and FASN in iPSC emphasizes the importance of lipogenesis in stemness and beacons potential therapeutic utilization in CSCs. Cytosolic ACC inhibition mediated by phosphorylation at serine 80 has been considered as a necessary feature for metastasis and invading behaviour in breast and lung cancers, and this concept may be universal in other types of cancers [70]. A study on ACC function in breast cancer indicates an unexpected enzymatic feature, in that the regulation of ACC in metastasis and tumor recurrence depends on the accumulation of acetyl-CoA and protein acetylation instead of its native duty in fatty acids synthesis [62]. Wnt/β-catenin signalling also participates in the regulation of ACC in CSCs, because silence of β-catenin induces ACC expression [71].
Fatty acid synthase (FASN)
Fatty acid synthase (FASN), the crucial enzyme for the de novo lipogenesis, also plays an important role in maintaining cancer stemness. Elevated FASN levels have been reported in a variety of cancers, including liver, prostate, breast, ovarian, endometrial and pancreatic cancers [59]. The increase of FASN is highly correlated with poor prognosis and disease recurrence [72]. Notably, FASN is overexpressed in iPSCs, while FASN deficiency impairs the reprogramming ability of iPSCs [73]. Similarly, adult neural stem and progenitor cells (NSPCs) show a strong dependence on FASN. Inhibition of FASN-mediated lipogenesis decreases NSPC proliferation [74]. FASN expression level is regulated by β-catenin [75], and associated with the level of stemness markers (SOX2, CD133 and Nestin) in GSCs [58]. In certain cases, FASN levels seems to be positively correlated with ACC expression in CSCs. FASN is suggested to be a more vulnerable target in CSCs than in the bulk cancer cells.
Stearoyl-CoA desaturase (SCD)
Several studies also revealed how signalling pathways to promote CSCs via regulation of unsaturated fatty acids. Importantly, NF-κB, the main regulator of tumors and CSCs, directly regulates the expression and activation of lipid desaturases, whereas the abrogation of lipogenesis through desaturases inhibition inactivates AKT/ERK-mediated NF-κB signalling [35,76]. Meanwhile, the level of SCD-dependent MUFAs also directly regulates CSCs through Wnt/β-catenin pathway, one of the most significant signalling both in stem cells and in CSCs [77,78]. Hippo pathway regulated by YES-associated protein (YAP) and tafazzin (TAZ) promotes embryonic and somatic stem cell renewal and differentiation [79]. Interestingly, the activation of SCD1 positively regulates the stabilization and nuclear localization of YAP/TAZ, indicating a significant impact on cancer stemness and the chemotherapy resistance in lung cancer stem cells [80].
In humans, SCDs have two isoforms, SCD1 and SCD5. SCD1 is the major enzyme catalysing desaturation in all tissues while SCD5 mainly expresses in the pancreas and brain [81]. Consistent with the performance of MUFAs in CSCs, the increased expression level of SCD1 in the lung, ovarian, breast, and glioblastoma cancer stem cells further emphasizes the importance of MUFAs, speculating a significant role of SCD1 for lipid component regulation in CSCs [80,82-84]. Additionally, SCD1 expression level also increases and corresponds with the maintenance of some stem cells, such as bone marrow mesenchymal stem cells, pluripotent stem cells and hair stem cells [85-87]. It is found that SCD1 also regulates Wnt signalling in CSCs [75,88]. Correspondingly, SCD1 inhibition preferentially diminishes CSCs population in the lung, brain, ovarian, lymphatic and colon cancers; while the functional failure is rescued by the MUFAs, such as oleic acid [32,35,80,82,89,90]. However, SCD5 expression and activity may not be prominent in most cancer cells. Intriguingly, the SCD5 expression level is reduced in melanoma; while restoration of SCD5 suppresses the formation of malignant melanoma through a reversed EMT-like process and induction of cancer cell differentiation [91].
FAO enhances CSCs
FAO mediated energy generation is particularly critical to cancer cell survival and metastasis, especially to non-glycolytic prostate adenocarcinoma and diffuse large B-cell lymphoma [92,93]. Elevated FAO helps cancer cell survival in nutrient deficiency and anoxic microenvironments [94]. Ectopic activation of FAO maintains CSCs under conditions of glycolytic deficiency [95,96]. FAO is also found to dominate the stemness in mesenchymal stem cells (MSCs) isolated from the advanced stage of gastric cancer (GC), indicating an effective target for reducing chemoresistance [97]. Consistently, inhibition of FAO by perhexiline impairs cancer stem cell self-renewal and increases the sensitivity of breast CSCs to chemotherapy [98]. Increased FAO has also been speculated to interact with Src oncoprotein and participate in the generation of triple-negative breast cancer stem cells [99]. Emerging evidence has revealed the mechanisms behind mitochondrial FAO's contribution to CSCs. ROS reduction caused by FAO impairs stem cells [95], explaining the efficient therapeutic effect through redox defence blockage in CSCs [100]. Additionally, mitochondrial FAO contributes to cellular activity and pluripotency in haematopoietic stem cells [101] and adult neural stem cells [102]. Therefore, inhibition of FAO exacerbates the symmetric differentiation of adult neural stem cells at the expense of self-renewal abilities [103]. On the other hand, elevated peroxisome FAO benefits Tie2+ hematopoietic stem cell proliferation by activation of mitochondrial clearance [101]. Though no direct evidence pinpoints the influence of FAO on Notch signalling, Notch1 coordinates FAO for the regulation of lipid accumulation in the liver [104] and redox homeostasis in quiescent endothelial cells [105]. FAO and its functions in lipid accumulation provide the grounds for CSCs' for survival under nutrition, environment and energy stress.
Cholesterol homeostasis
Cholesterol homeostasis mainly relies on two mechanisms [106]. On one hand, Cholesterol levels can be upregulated by synthesizing de novo from acetyl CoA provided by glycolysis, glutamine metabolism, TCA cycle or exogenous uptake by low density lipoprotein receptors (LDLRs), as well as the so-called reverse cholesterol transport (RCT), allowing peripheral cholesterol to be returned to the liver in low density lipoprotein (LDL) [107]. The process of cholesterol synthesis is mediated by the mevalonate (MVA) pathway [108]. On the other hand, cholesterol levels can be negatively regulated through the inhibition of the MVA pathway or the activation of liver X receptors (LXRs). The MVA pathway can be reduced through proteolytic processing or nuclear import of sterol regulatory element binding proteins (SREBP2), while LXRs can be activated through cholesterol conversion to oxysterols [109]. The activation of LXRs/PPAR pathway, in turn, activates the transcription of the E3 ubiquitin ligase IDOL, which ubiquitinates LDLR and upregulates the cholesterol efflux pump ABCA1 and ABCG1 [110]. Retrospective and experiment data show that both circulating LDL-cholesterol and elevated dietary cholesterol are associated with a poor progression free survival time (PFS), while statins show protective effects in ovarian cancer [111,112], non-small cell lung cancer [113], breast cancer [114], pancreatic tumour [115], colorectal cancer [116], and so on. SREBP2 is found to promote stem cell-like properties and metastasis by transcriptional activation of c-Myc in prostate cancer [117].
Cholesterol is the major sterol in mammals and is especially critical for cell growth and function. Besides acting as a precursor for sterol hormones, bile acids, vitamin D and oxysterols, as well as major components for membrane reinforcement, cholesterol regulates cell signalling via lipid rafts. Various of proteins have been discovered in lipid rafts, such as caveolins [118], src family kinases [119], MAP kinase (MAPK), protein kinase C, EGFRs [120], flotillins, low molecular weight heterotrimeric G proteins [121,122], platelet-derived growth factor (PDGF) receptors [123], endothelin receptors and so on. Proteins are selectively included or excluded from the membrane microdomains, which serve as rafts for the transportation of specific domains or relay stations for transducing intracellular signalling [124]. In most cases, lipid rafts act as signalling platforms that combine the necessary components, manage their interactions and transduce pathway signalling [125]. Different lipid rafts could also be united as complementary components of a signalling pathway. In turn, pathway signalling could be regulated by lipid rafts compartmentalization through locational and physical separation of proteins [121,126,127]. For example, CD133+ pancreatic tumor initiating cells (TIC) shows high-level expression of MAPK with high cholesterol content. Furthermore, the study also shows that CD133 is localized in the lipid rafts. Disruption of lipid rafts decreases metastatic potential and chemoresistance in CD133+ cells but does not affect the CD133- cells, resulting in deregulation of focal adhesion kinase (FAK)-signalling [128].
Cholesterol facilitates CSCs tumorigenesis
Cancer was first linked to nutrition by epidemiological studies, demonstrating that environmental factors such as diet and nutrition are important in carcinogenesis [129]. Caloric intake, types and amount of fats, proteins, amino acids, vitamins, minerals, fibers, and other dietary constituents have been studied regarding their influence on tumorigenesis. In particular, increased cholesterogenesis is associated with tumorigenesis through activation of tissue growth and loss in feedback control. Early laboratory studies elicited the role of cholesterol in cancer development and progression [45]. Lipoproteins are capable of stimulating growth and metastasis of cancer cells in vivo and in vitro [49,130,131]. Accelerated evidence also shows that cholesterol and FA metabolism are the hallmarks of cancer, contributing to malignant transformation due to the obligatory requirement of cholesterol for cell membrane functions. In mammosphere models generated from breast patient-derived xenograft (PDX) tumors, GO enrichment analysis identifies cholesterol biosynthesis to be the most significantly activated process [132]. Furthermore, the high levels of cholesterol biosynthesis-related proteins are associated with short relapse-free survival in basal-like breast cancer patients [33]. In a cohort of 615 basal-like breast cancer patients, except for DHCR7 or LSS, all cholesterol synthesis-associated proteins show a significant correlation between higher level of gene expression and shorter relapse-free survival [9]. In another analysis, enzymes of the MVA metabolic pathway are overexpressed in breast cancer stem cell tumorspheres as compared to cognate adherent cells. A small-molecule inhibitor of the geranylgeranyl transferase (GGTI) reduces the breast CSC population both in vitro and in vivo [133]. Phospholipid remodeling enzyme lysophosphatidylcholine acetyltransferase 3 (LPCAT3), which incorporates polyunsaturated fatty acids into phospholipids, is a crucial determinant of membrane lipid composition. Lack of LPCAT3 in intestinal stem cells leads to an excess of cholesterol production in response to changes in phospholipid composition, resulting in intestinal stem cell hyperproliferation [46]. In multiple brain tumor-initiating cells, several genes in MVA pathway, including HMGCR, PMVK, MVK, MVD, IDI1 and FDPS, are highly expressed [134].
Bile acids and oxysterol are two chemical by-products in the MVA pathway. They act as ligands for a family of nuclear receptors (including FXR, VDR, LXR and PXR) and G-protein-coupled receptors [135] [136]. As reported in colorectal cancer, the high-fat diet and dysregulated WNT signalling pathway alter bile acids profiles, activate FXR, and drive malignant transformations in Lgr5+ subpopulation CSCs [137]. Oxysterols are a group of Janus molecules result from enzymatic oxidation of cholesterol's side chain, can induce both the early inflammatory reaction against cancer expansion or apoptosis and sustain a complex survival signalling pathway in favor of the neoplastic process [138].
Abnormal cholesterol metabolism in CSCs
Cancer cells adapt to maintain high intracellular cholesterol similar to the normal homeostasis including accelerated endogenous production of cholesterol and fatty acids regulated by the SREBPs, or by reducing cholesterol efflux trough ABC class A transporters such as ABCA1, or by increasing the uptake of LDL.
Loss of phospholipid-remodelling enzyme Lpcat3 or activation of SREBP-2 in APC-defect mice markedly promotes intestinal tumor formation by modulating intestinal stem cell homeostasis and tumorigenesis [139]. HMG-CoAR is the rate-limiting enzyme in the MVA pathway and the popular cholesterol synthesis lowering agents [131]. Statins, the inhibitors for HMG-CoAR, reduces tumor-like sphere formation and exhibits high therapeutic indices [140]. This study indicates that HMGCR may be a predictive marker for statin therapy [141]. Overexpression of ABCA1 contributes to drug resistant in subpopulations of CSCs (EpCAM+ CD45+ CD133+ and CD117+ CD44+) in epithelial ovarian carcinoma patients [142]. The scavenger receptor, class B type 1 (SRB1), is a multiligand membrane receptor protein that functions as high-density lipoprotein (HDL) influx receptor of HDL-derived cholesteryl esters into cells and tissues [136]. SRB1 also facilitates the efflux of cholesterol from peripheral tissues back to the liver [143]. SRB1 may be responsible for an increased cholesterol uptake by the tumor and indirectly regulate tumor development. In the western diet mice models, SRB1 is highly expressed in the transformed prostatic epithelial cells and is responsible for an increased cholesterol uptake sustaining tumor development [144]. The higher affinity of LDL in tumor cells is detected, the increased activity of HMG-CoAR is observed [145,146]. LDL macromolecule has been developed as a specific delivery for cytotoxic drugs or radio nucleotides [147], specifically in CML patients where the poor prognosis is linked to low plasma lipid concentrations [148].
Signalling modulated by cholesterol in CSCs
MVA pathway is highly conserved in all the eukaryotes. Other intermediate products, such as farnesyl pyrophosphate, squalene, isoprenoids, lanosterol, bile acids, steroid hormones and vitamin D, participate in cellular physiological functions. Deregulation of cholesterol homeostasis has been proven a powerful way to suppress oncogenic receptors' signalling for inhibiting tumor growth. Cholesterol mainly promotes cancer signalling in two ways. Cholesterol itself as well as chemical by-products from the mevalonate cascade exerts a tumor progression effect. On the other hand, as a critically important component in lipid rafts, cholesterol can affect receptor affinity and the activity of proteins via sterol-sensing domains, including EGFR family, G-protein coupled receptors (GPCRs), PI3K and AKT [146].
In the Hh signalling pathway, lipid modification is crucial for its biological function. Cholesterol covalently bound to the cleaved C-terminal end Gly257 of the N-peptide in Shh translation is an essential step for Shh maturation. The genetic defects in cholesterol biosynthesis can cause a subset of genetically determined anatomical defects, termed holoprosencephaly (HPE), which results from Shh signalling blockage in embryonic development [149]. Besides, the GPCR-like protein Smoothened (SMO), as an orphan GPCR that transmits the Hh signal, can be directly activated by cholesterol. Cholesterol together with some other oxysterols, such as 20(S)-hydroxylcholesterol, 22(S)-hydroxylcholesterol and 7-keto-25-hydroxylcholesterol, is identified as potent activators of SMO through its extracellular cysteine-rich domain (CRD) [150,151]. Sterol depletion reduces SMO accumulation on the primary cilium. However, neither the Notch signalling nor the Hippo cascades could directly interact with cholesterol. Instead, Notch signalling can be modulated by the lipid composition of the cell membrane, in addition to the O-glycosylation of the receptor [152]. A high-content with high-throughput screening on FDA -approved drug library shows the strongest YAP/TAZ inhibitory effect in all of the five statins present in the library [152]. MVA pathway activity, mainly the geranylgeranyl pyrophosphate (GGPP), is required to sustain the YAP/TAZ gene expression program. Only the geranylgeranyl transferase inhibitor GGTI-298 is shown to rescue the effect of statins on YAP/TAZ localization, while the squalene synthase inhibitor (YM-53601) or farnesyl transferase inhibitor (FTI-227) fails to converse the effect. GGPP, crucial for the enzymatic activity of Rho small GTPases located in the plasma membrane, reduces the inhibitory phosphorylation of YAP/TAZ and sustains YAP/TAZ nuclear accumulation [153]. Inhibitor of the geranylgeranyl transferase effectively reduces the growth of breast CSCs both in vitro and in vivo [133]. CSCs' sphere-forming is suppressed through inhibiting RhoA and increasing P27kip1 accumulation that finally leads to inhibition of RB phosphorylation and cell cycle arrest in cancer cells. These findings are considered as promising perspectives for target-based cancer therapy [131,133].
Cholesterol metabolism has also been explored its important roles in immunity. Current studies indicate a significant connection between cholesterol metabolism and immunotherapy resistance. The plasticity and ability of CSCs enable them to interact with TME, to modulate and shape immune responses, thus resulting in immune impairment and tumor recurrence [154,155]. Among the numerous immune cells, T cells (especially CD8+ T cells) have exerted an important function in IFN-γ and TNF (tumor necrosis factor) signalling [156]. However, the activity of CD8+ T cells is suppressed in tumor microenvironments. Nevertheless, the increased cholesterol level decreases the T cell antigen receptor (TCR) activity and nanoclusters, which are critical for antigens recognition and binding to major histocompatibility complex molecules (MHC) on other cells [157].
Important signalling pathways involved in lipid metabolism of CSCs
In stem cells, several important signalling pathways involved in lipid metabolism participate in controlling self-renewal, embryonic development and lineage specification. Since CSCs can be derived from stem cells through genetic mutations and epigenetic alteration, it is highly likely that these pathways are hijacked to maintain the unrestrained proliferation, invasion and drug resistance [158]. In CSCs, a series of pathways involved in lipid metabolism maintains the undifferentiating state, guide the lineage progeny and sustain their survival as well as proliferation (Figure 3), including Notch signalling [159-161], Hippo cascades, Hedgehog (Hh) signalling, and Wnt signalling [9,153,154].
A diagram of cholesterol homeostasis. Cholesterol uptake is mediated by LDLR through EGFR dependent pathway. Cholesterol synthesis goes through MVA pathway. LXR plays a crucial role in both the negative control of cholesterol uptake and regulation of cholesterol efflux. Abbreviation: EGFR, epidermal growth factor receptor; LDLR, low density lipoprotein receptors; ABCA1, ATP-binding cassette transporter 1; PI3K/Akt, phosphatidylinositol 3‑kinase/protein kinase B; SREBP-1, sterol regulatory element binding proteins; MVA, mevalonate; IDOL, inducible degrader of the low-density lipoprotein receptor; PPAR/LXR, lipid-activated transcription factors /liver X receptors.
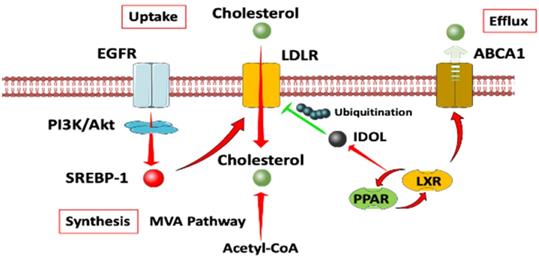
Notch signalling pathway
Notch signalling pathway is one of the most conserved signalling pathways activated in embryonic vasculature development [160]. In Drosophila, Notch signalling is sensitive to environmental sterol levels. The expression level of Notch signalling is modulated by dietary cholesterol, resulting in intestinal cell differentiation from stemness status [162]. In cancer cells, Notch pathway plays a critical role in angiogenesis, EMT and CSCs proliferation [159,163]. The low-sterol diet restricts the growth of enteroendocrine tumors by decrease of Notch responses [162]. Interestingly, Notch1 controls FAO to achieve intermediate lipid homeostasis and redox homeostasis in CSCs [104,105]. Exogenous lipids are demonstrated to positively regulate Notch signalling. In human beings, Notch signalling can be modulated by the lipid composition of the cell membrane [153].
Hippo signalling
Emerging evidence shows the intimated association and the importance of hippo-YAP/TAZ signalling in lipid metabolisms in regulating cancer stemness [80,164]. Hippo signalling mainly functions through phosphorylating the plasma membrane receptor MST1/2, followed by phosphorylation and activation of MOB1A/B and LATS1/2, causing sequester and proteasomal degradation of YAP and TAZ by the 14-3-3 phosphopeptide binding proteins. In CSCs, the activation of YAP or TAZ sustains self-renewal and tumor-initiation capacities [165], promotes cell pluripotency [166] and drug resistance [167], and is highly related to EMT process [167-171]. The main MUFAs regulator SCD1 contributes to cancer stemness through the regulating YAP/TAZ in both expression and nuclear localization [80]. As an intermediate controlled by the MVA pathway, GGPP is a sufficient factor for the stabilization of YAP/TAZ [153].
Hedgehog (Hh) signalling
When the three Hh ligands, Sonic (Shh), Indian (Ihh), and Desert (Dhh), binding and inhibiting the Patched 1 (Ptch1) and/or Patched 2 (Ptch2) receptors, the repression of Smoothened (SMO) is relieved, followed by activation of the GLI transcription factors (GLI-TFs) GLI1, GLI2, and GLI3 [172]. The Hh signalling cascade leads to transcription of Hh-targeted genes, such as Cyclin D1 and c-Myc [173]. Hh ligands are found to activate in colon cancer, as well as other solid tumors. The enhanced Hh signalling accelerates the progression of advanced neoplasms [174]. Hh signalling also plays a crucial role in CSCs. Hh signalling modulates the postnatal mammary stem cells (MASCs) proliferation and generates the complex ductal structure of the adult mammary gland [175]. In breast cancer EMT programs, primary ciliogenesis activates the Hh signalling that enables the stemness and the tumor-forming capacity of stem cell-like tumor-initiating cells [176]. Lipid metabolism is also known to regulate hedgehog signalling and its ligand properties [177]. Cholesterol is crucial for Shh maturation and can directly activate the SMO receptor in Hh signalling [149,150]. The genetic defects in cholesterol biosynthesis causes a subset of anatomical defect holoprosencephaly (HPE), resulting from Shh signalling blockage in embryonic development [149]. Recently, SMO inhibitors and GLI inhibitors are used to target the Hh signalling pathway in clinical trials [178,179].
The Wnt signalling pathway
In the canonical Wnt pathway, Wnt ligands bind to the transmembrane receptor Frizzled (Fzd) family, leading to activating Dishevelled (Dvl) and then triggering the stabilization and accumulation of nuclear β-catenin transcriptional activity, in cooperation with T-cell factor (TCF)/lymphoid enhancer factor (LEF) family. Other co-receptors, such as low-density lipoprotein-related protein (LRP5/6) or tyrosine kinase receptors (PTK7, ROR, RYK), may also act as cofactors in the canonical Wnt pathway. The non-canonical signalling (β-catenin-independent pathway) consists of the Wnt/Ca2+ pathway and the planar cell polarity (PCP) pathway. The Wnt signalling pathway plays a highly evolutionarily conserved role in embryonic proliferative tissue development (such as hematopoietic system, skin and intestine) for body axis patterning, cell fate specification, cell proliferation and migration [180]. In tumorigenesis, the Wnt signalling promotes tumor migration and invasion by upregulating genes involved in cell adhesion, including Eph/Ephrins, E-cadherin and MMPs [181]. However, in the hypoxic GBM patient-derived cell lines, TCF1 and HIF-1α together inhibit the expression of stemness markers Nestin and CD133 through activation of Wnt signalling that reduces the GBM stem cell frequency and strongly increases acquisition of neuronal traits [182,183]. In squamous cell carcinoma, depletion of β-catenin halts tumor progression, suggesting its roles in the maintenance of cutaneous CSCs-like properties [181]. The Wnt signalling also cooperates with lipogenesis in cancer cells [75]. The Wnt/β-catenin signalling significantly modulates de novo lipogenesis, which is characterized by a significantly increased expression of ACC, FASN, and sterol regulatory element binding protein-1c (SREBP1-c) in breast cancer cells [71]. FAO has been identified as an enhancer for β-catenin expression in HCC [184]. Importantly, SCDs have been considered as the key factor in regulating Wnt signalling in CSCs [75,88]. In colorectal cancer, a high-fat diet and dysregulated WNT signalling pathway alter bile acids profiles, activate FXR, and drive malignant transformation in Lgr5+ subpopulation CSCs, which promote an adenoma-to- adenocarcinoma progression [137].
Other signalling pathways involved in lipid metabolism in CSCs
Other pathways, such as EGFR signalling [185,186], signal transducer and activator of transcription (STAT) signalling, PI3K/PTEN/Akt/mTORC1/GSK-3 pathway [186], ephrins and bone morphogenetic proteins (BMPs) signalling [187], and NF-κB signalling, have been studied in CSCs for many years. Pharmacological agonists/inhibitors targeting such pathways are in clinical trials [188]. For example, the member of STAT family ultimately regulates tumor stem cell self-renewal, differentiation, and apoptosis [189]. Activation of JAK/STAT3 signalling promotes CPT1 expression, resulting in the reinforcement of cancer stemness and chemoresistance in breast cancer [98].
Targeting lipid metabolism for anticancer treatment
The importance of lipid metabolism in CSCs has been continuously studied and emphasized that the inhibitors targeting each participant in FAS, FAO and cholesterol metabolisms are widely tested in cancer treatment and chemotherapy assistance.
Targets on FAS
Promisingly, therapeutic targets on ACC and FASN achieve reliable results in elimination of CSCs or cancer therapy. ACC inhibitor, such as Soraphen A, has been considered as a treatment option by targeting lipogenesis in breast CSCs [190]. Additionally, chemical compounds with the same binding site as Soraphen A can inhibit the growth and proliferation in non-small cell lung cancer (NSCLC) and hepatocellular carcinoma cells [191,192], indicating the significance and potential of ACC in both CSCs inhibition and cancer therapy. Similarly, FASN plays an essential part in CSCs' survival and proliferation. Both pharmacological inhibitor and RNA silencing of FASN diminished a variety of CSCs through different disruptive activities, including the destruction of mammosphere formation, weakened invasion, inhibition of proliferation and apoptosis [58,193-195]. Currently, FASN inhibitor, such as TVB-2640, is in the phase II trials as the assistant drug for chemotherapy (Paclitaxel, and Trastuzumab) in patients with human epidermal growth factor receptor 2 (HER2) positive advanced breast cancer. Meanwhile, a phase I trial of TVB-2640 is also in patients with colon or other cancers (National Cancer Institute, NCI).
However, therapy by targeting the ACLY seems to be tangled in a whack-a-mole effect. Currently, the progress of ACLY inhibition in CSCs has still been stuck in vitro since the year 2013 [64]. The most controversial issue haunting in the progress is the compensation effect after ACLY inhibition. For example, inhibition or knockdown of ACLY undoubtedly inhibits the growth of certain cancers, but other key players in the fatty acid and cholesterol synthesis pathways, such as FASN and HMGCR, are stimulated in accordingly to reimburse for the effects of ACLY deficiency [55]. Furthermore, the duty of ACLY in lipogenesis that converses acetate into acetyl CoA can also be substituted by acetyl-CoA synthetase short-chain family member 2 (ACSS2) in mammals [196]. ACSS2 is particularly prominent in the absence of ACLY [60]. Therefore, previous studies have speculated that ACSS2 supplements the acetyl CoA required by cells to restore the effects of ACYL inactivation [197, 198]. Additionally, ACSS2 also maintains cancer growth under lipid deficiency, and ACSS2 knockdown inhibits tumor xenografts in vivo [199]. Of note, phosphorylation of ACLY can be conducted by other kinases such as nucleoside diphosphate kinase [200] and cyclic AMP-dependent protein kinase [201]. Therapeutic strategies focusing on ACLY phosphorylation also encounters an obstacle, because dephosphorylation and inactivation of ACLY with PI3K inhibitors have no significant effect on lung cancer cell therapy. Though the compensatory effect may not completely rescue the consequences under the absence of ACLY in vitro [64, 202], there is still a gap in the effect of ACLY deficiency in cancer research.
Targets on FAs desaturation
A strong relevance between SCD1 and CSCs suggests a promising therapeutic target for identification and elimination of CSCs. Previous studies aiming at the importance of unsaturated lipids in CSCs also show that SCD1 inhibition by chemical compounds such as CAY10566, A939572, effectively interferes with cancer stemness, tumor formation and proliferation [35, 90]. However, it remains unclear what a consequence on blockage of systemic metabolism would be in normal cells. Ben-David et al showed that an SCD1 inhibitor PluriSIn-1 effectively eliminates hPSCs while it reserves a sufficient amount of progenitor and differentiated cells [86]. Another SCD1 inhibitor CVT-11127 induces programmed cell death in lung cancer without impairing the proliferation of normal human fibroblasts [89]. The current progress of SCD1 inhibitor for cancer therapy mainly stays at the animal test. Comparatively, a liver-specific SCD1inhibitor MK-8245 is proven to treat diabetes and dyslipidemia without liver toxicity at Phase II clinical trials [203]. Noticeably, cocktail inhibitors targeting both the Wnt and Hippo-YAP signallings effectively suppress triple-negative breast cancer in both mesenchymal and epithelial states [204]. This finding shows that alteration of lipid metabolism may be a synergy from both the Wnt and YAP pathways in CSCs, indicating an ideal therapeutic strategy. Cocktail inhibitors may be a better option for treating CSCs. No further reports show that SCD1 inhibitor can selectively affect CSCs by sparing normal somatic cells. ALDH family, which is related to the lipid desaturation, is considered as an ideal marker and target for in clinic application. ALDH inhibitors, such as disulfiram and its derivative, achieve periodic results in the promotion of chemosensitization of lung cancer [205].
Targets on FAO
The therapeutic targets on FAO mainly focus on inhibitors of the rate-limited enzyme CPT1, which locates on the outer mitochondrial membrane. Typically, CPT1 is associated with the increasing level of FAO in glioblastoma and breast, prostate, ovarian, and lung cancer [206]. CPT1 inhibitors, such as etomoxir, ranolazine, reduce chemoresistance and pluripotency of cancer cells [40,97,105,207]. Because JAK/STAT3 interferes with cancer stemness through the regulation of CPT1, a first-in-class STAT3 inhibitor displays strong anti-CSC effects in numerous cancers [179]. Napabucasin (BBI608) is in phase III clinical trials for metastatic colorectal carcinoma and pancreatic cancer [189]. Therefore, the potency of FAO inhibition may also create an effective combination for eliminating cancer stemness.
Targets on cholesterol metabolisms
In melanoma mice models, inhibiting cholesterol esterification by cholesterol acyltransferase (ACAT) inhibitor avasimibe leads to enhanced effector function and proliferation of CD8+ instead of CD4+ T cells [208]. Furthermore, ACAT1-deficient CD8+ T cells show better control in melanoma growth and metastasis in mice models. In preclinical research, the combination of ACAT inhibitor and an anti-PD-1 antibody exhibits more promising anti-tumor efficiency than monotherapies.
Besides, the activation of LXRs in cancer cells can be induced by disrupting cholesterol metabolism, which regulate inflammation and innate and acquired immunity. In NK cells, activated LXRs leads to overexpression of major histocompatibility complex class I chain-related molecule A and B (MICA and MICB), ligands in melanoma cells rendering the tumor cells more sensitive to recognition, degranulation, and killing by NK cells [209]. Cholestane-3β, 5α, 6β-triol (abbreviated as triol) is one of the most abundant and active oxysterols. Triol exhibits anti-cancer activity against human prostate cancer cells. Triol treatment results in reduced expression of Akt1, phospho-Akt Ser473, phospho-Akt Thr308, PDK1, c-Myc [134], and Skp2 as well as accumulation of the cell cycle inhibitor p27kip[210]. In a phase II study, atorvastatin, a lipophilic statin, is found to prolong survival in HMGCR-positive breast cancer. These observations suggest a novel immune-mediated mechanism involving modulation of intracellular cholesterol levels in cancer cells.
Cancer stemness related signalling pathways involved in the lipid metabolisms in CSCs. Notch, Hippo, Hh, and Wnt signalling participate in lipid metabolism to maintain the properties of cancer stem cells. Abbreviation: GGPP, geranylgeranyl pyrophosphate; YAP/TAZ, yes-associated protein (YAP)/ tafazzin (TAZ); JAK/STAT3, Janus kinase/signal transducers and activators of transcription 3; SMO, Smoothened; Hh, Hedgehog. The figure was produced using Servier Medical Art (http://www.servier.com).
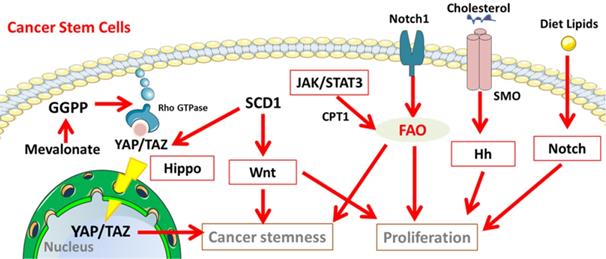
Summary and Perspectives
Due to the excessive demand for energy and structural component than 'normal' cancer cells, cancer stem cells urgently rely on lipid metabolism to maintain cell survival and proliferation. Dramatically, the known cancer stemness associated signalling pathways, such as Notch, Hippo, Wnt, and Hh, have a close relationship with lipid metabolisms. Therefore, the alternation of 'alternated' lipid metabolisms has been indicated as promising therapeutic targets for CSCs suppression and cancer therapy. Specifically, because of the relatively simple and maneuverability for those key regulators in the pathway, therapeutic targets on fatty acid and cholesterol metabolism contribute to several impressive progress on the inhibition of CSCs and reduction of chemoresistance both in vivo and in vitro.
However, the application also encounters several challenges when commencing in the clinical trials. For example, fatty acid synthesis is drastically upregulated under pathogen infection, a wide range of diseases including cardiovascular disease, insulin resistance of type 2 diabetes and cancers mentioned above [211,212]. Hence, the co-occurring of FASN and cancer stemness markers, such as SOX2, CD133 and Nestin, would be hard to hallmark cancer stem cells within multiple pathogenic or metabolic disorder circumstances, especially in tissue with excessive metabolic activities. Secondly, the compensation conversed by other metabolic pathways or uptake from extracellular environment spare cancer cells from the shortage of energy and intermediates for metabolisms. The last but not least, the dilemma crushing on the cancer therapy remains to be the same problem for current treatment; i.e., lipid metabolism-associated inhibitors may also affect surrounding healthy cells, resulting in inevitable side-effects.
Notably, there are several interesting related progresses for therapeutic targets on lipid metabolisms, solving those concerned or problems haunting in current cancer therapy. For instance, the combination of lipid metabolism-associated inhibitors and chemotherapy agents, or immunotherapy (such as PD-1 antibodies), does significantly promote anticancer efficiency. Interestingly, the current study shows another novel strategy by utilizing engineered adipocytes or lipids to deliver the anticancer drug [213,214]. By intratumoral or postsurgical injection, this drug design and deliver strategy enable those 'greedy' cancer cells to suffer their consequences. As the global profiles of lipid metabolisms have been well unveiled in cancer or CSCs, we may continuously exploit the combination of exciting therapeutic strategy or a novel treatment, and lipid-associated drugs to ameliorate chemoresistance and even the cure for cancers.
Acknowledgements
We thank Jason He for reading and editing this manuscript. The work was partially supported by grants from The Science Technology and Innovation Committee of Shenzhen Municipality [JCYJ2018050718162705, JCYJ20170818100531426], grants from National Science Foundation of China [81671995], and strategic match fund from City University of Hong Kong [9680149].
Competing Interests
The authors have declared that no competing interest exists.
References
1. Fiala S. The cancer cell as a stem cell unable to differentiate. A theory of carcinogenesis. Neoplasma. 1968;15(6):607-22
2. da Silva-Diz V, Lorenzo-Sanz L, Bernat-Peguera A, Lopez-Cerda M, Munoz P. Cancer cell plasticity: Impact on tumor progression and therapy response. Semin Cancer Biol. 2018;53:48-58
3. Feinberg AP, Koldobskiy MA, Gondor A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet. 2016;17:284-99
4. Barker RAR. et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457(7229):608-611
5. Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Goktuna SI, Ziegler PK. et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell. 2013;152:25-38
6. Liu CC, Lin JH, Hsu TW, Su K, Li AF, Hsu HS. et al. IL-6 enriched lung cancer stem-like cell population by inhibition of cell cycle regulators via DNMT1 upregulation. Int J Cancer. 2015;136:547-59
7. Liu X, Fan D. The epithelial-mesenchymal transition and cancer stem cells: functional and mechanistic links. Curr Pharm Des. 2015;21:1279-91
8. Luo M, Brooks M, Wicha MS. Epithelial-mesenchymal plasticity of breast cancer stem cells: implications for metastasis and therapeutic resistance. Curr Pharm Des. 2015;21:1301-10
9. Ehmsen S, Pedersen MH, Wang GS, Terp MG, Arslanagic A, Hood BL. et al. Increased Cholesterol Biosynthesis Is a Key Characteristic of Breast Cancer Stem Cells Influencing Patient Outcome. Cell Rep. 2019;27:3927-29
10. Ahmed N, Escalona R, Leung D, Chan E, Kannourakis G. Tumour microenvironment and metabolic plasticity in cancer and cancer stem cells: Perspectives on metabolic and immune regulatory signatures in chemoresistant ovarian cancer stem cells. Semin Cancer Biol. 2018;53:265-81
11. Perusina Lanfranca M, Thompson JK, Bednar F, Halbrook C, Lyssiotis C, Levi B. et al. Metabolism and epigenetics of pancreatic cancer stem cells. Semin Cancer Biol. 2018;57:19-26
12. Thomas D, Majeti R. Biology and relevance of human acute myeloid leukemia stem cells. Blood. 2017;129:1577-85
13. Justin D Lathia SCM, Erin E Mulkearns-Hubert, Claudia LL Valentim, Jeremy N Rich. Cancer stem cells in glioblastoma. Genes Dev. 2015;29(12):1203-1217
14. Polyak K, Haviv I, Campbell IG. Co-evolution of tumor cells and their microenvironment. Trends In Genetics. 2009;25:30-8
15. Kathagen A SA, Balcke G, Phillips HS, Martens T, Matschke J, Günther HS, Soriano R, Modrusan Z, Sandmann T, Kuhl C, Tissier A, Holz M, Krawinkel LA, Glatzel M, Westphal M, Lamszus K. Hypoxia and oxygenation induce a metabolic switch between pentose phosphate pathway and glycolysis in glioma stem-like cells. Acta Neuropathol. 2013;126(5):763-80
16. Koren E, Fuchs Y. The bad seed: Cancer stem cells in tumor development and resistance. Drug Resist Updat. 2016;28:1-12
17. Aguilar E, Marin de Mas I, Zodda E, Marin S, Morrish F, Selivanov V. et al. Metabolic Reprogramming and Dependencies Associated with Epithelial Cancer Stem Cells Independent of the Epithelial-Mesenchymal Transition Program. Stem Cells. 2016;34:1163-76
18. Sun S, Xue D, Chen Z, Ou-Yang Y, Zhang J, Mai J. et al. R406 elicits anti-Warburg effect via Syk-dependent and -independent mechanisms to trigger apoptosis in glioma stem cells. Cell Death Dis. 2019;10:358
19. Dong C, Yuan T, Wu Y, Wang Y, Fan TW, Miriyala S. et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell. 2013;23:316-31
20. Al-Mawali A GD, Lewis I. Immunoprofiling of leukemic stem cells CD34+/CD38-/CD123+ delineate FLT3/ITD-positive clones. J Hematol Oncol. 2016;9(1):61
21. Ju HQ, Zhan G, Huang A, Sun Y, Wen S, Yang J. et al. ITD mutation in FLT3 tyrosine kinase promotes Warburg effect and renders therapeutic sensitivity to glycolytic inhibition. Leukemia. 2017;31:2143-50
22. Riester M, Xu Q, Moreira A, Zheng J, Michor F, Downey RJ. The Warburg effect: persistence of stem-Cell Metab in cancers as a failure of differentiation. Annals Of Oncology. 2018;29:264-70
23. Zhao Y, Dong QZ, Li JH, Zhang KL, Qin J, Zhao JG. et al. Targeting cancer stem cells and their niche: perspectives for future therapeutic targets and strategies. Semin Cancer Biol. 2018;53:139-55
24. Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M. et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514:628-32
25. Janiszewska M, Suva ML, Riggi N, Houtkooper RH, Auwerx J, Clement-Schatlo V. et al. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012;26:1926-44
26. Suganuma K, Miwa H, Imai N, Shikami M, Gotou M, Goto M. et al. Energy metabolism of leukemia cells: glycolysis versus oxidative phosphorylation. Leuk Lymphoma. 2010;51:2112-9
27. Skrtic M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z. et al. Inhibition of Mitochondrial Translation as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell. 2011;20:674-88
28. Marlein CR, Zaitseva L, Piddock RE, Robinson SD, Edwards DR, Shafat MS. et al. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood. 2017;130:1649-60
29. Ye XQ, Li Q, Wang GH, Sun FF, Huang GJ, Bian XW. et al. Mitochondrial and energy metabolism-related properties as novel indicators of lung cancer stem cells. Int J Cancer. 2011;129:820-31
30. Liu K, Lee J, Kim JY, Wang L, Tian Y, Chan ST. et al. Mitophagy Controls the Activities of Tumor Suppressor p53 to Regulate Hepatic Cancer Stem Cells. Mol Cell. 2017;68:281-92
31. Sun M, Yang ZB. Metabolomic Studies of Live Single Cancer Stem Cells Using Mass Spectrometry. Anal Chem. 2019;91:2384-91
32. Song M, Lee H, Nam M-H, Jeong E, Kim S, Hong Y. et al. Loss-of-function screens of druggable targetome against cancer stem-like cells. FASEB J. 2017;31:625-35
33. Kim WY. Therapeutic targeting of lipid synthesis metabolism for selective elimination of cancer stem cells. Arch Pharm Res. 2019;42:25-39
34. Mukherjee A, Kenny HA, Lengyel E. Unsaturated Fatty Acids Maintain Cancer Cell Stemness. Cell Stem Cell. 2017;20(3):291-292
35. Li J, Condello S, Thomes-Pepin J, Ma X, Xia Y, Hurley TD. et al. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell. 2017;20:303-14
36. Zhao W, Prijic S, Urban BC, Tisza MJ, Zuo Y, Li L. et al. Candidate Antimetastasis Drugs Suppress the Metastatic Capacity of Breast Cancer Cells by Reducing Membrane Fluidity. Cancer Res. 2016;76:2037-49
37. Lin L, Ding Y, Wang Y, Wang Z, Yin X, Yan G. et al. Functional lipidomics: Palmitic acid impairs hepatocellular carcinoma development by modulating membrane fluidity and glucose metabolism. Hepatology. 2017;66:432-48
38. Wang L, Zhang T, Wang L, Cai Y, Zhong X, He X. et al. Fatty acid synthesis is critical for stem cell pluripotency via promoting mitochondrial fission. EMBO J. 2017;36:1330-47
39. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M. et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555-67
40. Tan Z, Xiao L, Tang M, Bai F, Li J, Li L. et al. Targeting CPT1A-mediated fatty acid oxidation sensitizes nasopharyngeal carcinoma to radiation therapy. Theranostics. 2018;8:2329
41. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR. et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011;17:1498-503
42. Gazi E, Gardner P, Lockyer NP, Hart CA, Brown MD, Clarke NW. Direct evidence of lipid translocation between adipocytes and prostate cancer cells with imaging FTIR microspectroscopy. J Lipid Res. 2007;48:1846-56
43. Chen C-L, Kumar DBU, Punj V, Xu J, Sher L, Tahara SM. et al. NANOG metabolically reprograms tumor-initiating stemlike cells through tumorigenic changes in oxidative phosphorylation and fatty acid metabolism. Cell Metab. 2016;23(1):206-19
44. Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M. et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell. 2016;19:23-37
45. Danilo C, Frank PG. Cholesterol and breast cancer development. Curr Opin Pharmacol. 2012;12:677-682
46. Strzyz P. Stem cells: A balancing act of lipids. Nat Rev Mol Cell Biol. 2018;19:141
47. de Gonzalo-Calvo D, Lopez-Vilaro L, Nasarre L, Perez-Olabarria M, Vazquez T, Escuin D. et al. Intratumor cholesteryl ester accumulation is associated with human breast cancer proliferation and aggressive potential: a molecular and clinicopathological study. Bmc Cancer. 2015 15
48. Tirinato L, Liberale C, Di Franco S, Candeloro P, Benfante A, La Rocca R. et al. Lipid droplets: a new player in colorectal cancer stem cells unveiled by spectroscopic imaging. Stem Cells. 2015;33:35-44
49. Tirinato L, Pagliari F, Limongi T, Marini M, Falqui A, Seco J. et al. An Overview of Lipid Droplets in Cancer and Cancer Stem Cells. Stem Cells Int. 2017;2017:1656053
50. Luo X, Cheng C, Tan Z, Li N, Tang M, Yang L. et al. Emerging roles of lipid metabolism in cancer metastasis. Mol Cancer. 2017;16(1):76
51. Yi M, Li J, Chen S. et al. Emerging role of lipid metabolism alterations in Cancer stem cells [published correction appears in J Exp Clin Cancer Res. 2018 Jul 16;37(1):155]. J Exp Clin Cancer Res. 2018;37(1):118
52. Horikoshi Y, Yan Y, Terashvili M. et al. Fatty Acid-Treated Induced Pluripotent Stem Cell-Derived Human Cardiomyocytes Exhibit Adult Cardiomyocyte-Like Energy Metabolism Phenotypes. Cells. 2019;8(9):1095
53. Lin Z, Liu F, Shi P, Song A, Huang Z, Zou D. et al. Fatty acid oxidation promotes reprogramming by enhancing oxidative phosphorylation and inhibiting protein kinase C. Stem Cell Res Ther. 2018;9:47
54. Vlashi E, Lagadec C, Vergnes L, Matsutani T, Masui K, Poulou M. et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc Natl Acad Sci. 2011;108:16062-7
55. Corominas-Faja B, Cuyàs E, Gumuzio J, Bosch-Barrera J, Leis O, Martin ÁG. et al. Chemical inhibition of acetyl-CoA carboxylase suppresses self-renewal growth of cancer stem cells. Oncotarget. 2014;5:8306
56. Santos CR, Schulze A. Lipid metabolism in cancer. FEBS J. 2012;279(15):2610-2623
57. Menendez JA, Joven J, Cufí S, Corominas-Faja B, Oliveras-Ferraros C, Cuyàs E. et al. The Warburg effect version 2.0. Cell Cycle. 2013;12:1166-79
58. Yasumoto Y, Miyazaki H, Vaidyan LK, Kagawa Y, Ebrahimi M, Yamamoto Y. et al. Inhibition of Fatty Acid Synthase Decreases Expression of Stemness Markers in Glioma Stem Cells. PLOS ONE. 2016;11:e0147717
59. Kuo CY, Ann DK. When fats commit crimes: Fatty acid metabolism, cancer stemness and therapeutic resistance. Cancer Commun (Lond). 2018;38(1):47
60. Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D. et al. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. 2005;8:311-21
61. Qian X, Hu J, Zhao J, Chen H. ATP citrate lyase expression is associated with advanced stage and prognosis in gastric adenocarcinoma. Int J Clin Exp Med. 2015;8:7855
62. Rios Garcia M, Steinbauer B, Srivastava K, Singhal M, Mattijssen F, Maida A. et al. Acetyl-CoA Carboxylase 1-Dependent Protein Acetylation Controls Breast Cancer Metastasis and Recurrence. Cell Metab. 2017;26:842-55
63. Zaidi N, Swinnen JV, Smans K. ATP-Citrate Lyase: A Key Player in Cancer Metabolism. Cancer Res. 2012;72:3709-14
64. Hanai J-i, Doro N, Seth P, Sukhatme VP. ATP citrate lyase knockdown impacts cancer stem cells in vitro. Cell Death Dis. 2013;4:e696
65. Liu H, Huang X, Ye T. MiR-22 down-regulates the proto-oncogene ATP citrate lyase to inhibit the growth and metastasis of breast cancer. Am J Transl Res. 2018;10:659-69
66. Lucenay KS, Doostan I, Karakas C, Bui T, Ding Z, Mills GB. et al. Cyclin E Associates with the Lipogenic Enzyme ATP-Citrate Lyase to Enable Malignant Growth of Breast Cancer Cells. Cancer Res. 2016;76:2406-18
67. Migita T, Narita T, Nomura K, Miyagi E, Inazuka F, Matsuura M. et al. ATP citrate lyase: Activation and therapeutic implications in non-small cell lung cancer. Cancer Res. 2008;68:8547-54
68. Moncur JT, Park JP, Memoli VA, Mohandas TK, Kinlaw WB. The "Spot 14" gene resides on the telomeric end of the 11q13 amplicon and is expressed in lipogenic breast cancers: Implications for control of tumor metabolism. Proc Natl Acad Sci U S A. 1998;95:6989-94
69. Svensson RU, Parker SJ, Eichner LJ, Kolar MJ, Wallace M, Brun SN. et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat Med. 2016;22:1108-19
70. Fang W, Cui H, Yu D, Chen Y, Wang J, Yu G. Increased expression of phospho-acetyl-CoA carboxylase protein is an independent prognostic factor for human gastric cancer without lymph node metastasis. Med Oncol. 2014;31:15
71. Vergara D, Stanca E, Guerra F, Priore P, Gaballo A, Franck J. et al. β-Catenin Knockdown Affects Mitochondrial Biogenesis and Lipid Metabolism in Breast Cancer Cells. Front Physiol. 2017;8:544
72. Cai Y, Wang J, Zhang L, Wu D, Yu D, Tian X. et al. Expressions of fatty acid synthase and HER2 are correlated with poor prognosis of ovarian cancer. Med Oncol. 2015;32:391
73. Vazquez-Martin A, Corominas-Faja B, Cufi S, Vellon L, Oliveras-Ferraros C, Menendez OJ. et al. The mitochondrial H(+) -ATP synthase and the lipogenic switch. Cell Cycle. 2013;12:207-18
74. Knobloch M, Braun SMG, Zurkirchen L, von Schoultz C, Zamboni N, Araúzo-Bravo MJ. et al. Metabolic control of adult neural stem cell activity by Fasn-dependent lipogenesis. Nature. 2013;493:226-30
75. El-Sahli S, Xie Y, Wang L, Liu S. Wnt Signaling in Cancer Metabolism and Immunity. Cancers. 2019;11(7):904
76. Fritz V, Benfodda Z, Rodier G, Henriquet C, Iborra F, Avances C. et al. Abrogation of De novo Lipogenesis by Stearoyl-CoA Desaturase 1 Inhibition Interferes with Oncogenic Signaling and Blocks Prostate Cancer Progression in Mice. Mol Cancer Ther. 2010;9:1740-54
77. Lai KKY, Kweon S-M, Chi F, Hwang E, Kabe Y, Higashiyama R. et al. Stearoyl-CoA Desaturase Promotes Liver Fibrosis and Tumor Development in Mice via a Wnt Positive-Signaling Loop by Stabilization of Low-Density Lipoprotein-Receptor-Related Proteins 5 and 6. Gastroenterology. 2017;152:1477-91
78. Zheng B, Jarugumilli GK, Chen B, Wu X. Chemical Probes to Directly Profile Palmitoleoylation of Proteins. Chem BioChem. 2016;17:2022-7
79. Hiemer SE, Varelas X. Stem cell regulation by the Hippo pathway. Biochim Biophys Acta. 2013;1830(2):2323-2334
80. Noto A, De Vitis C, Pisanu ME, Roscilli G, Ricci G, Catizone A. et al. Stearoyl-CoA-desaturase 1 regulates lung cancer stemness via stabilization and nuclear localization of YAP/TAZ. Oncogene. 2017;36:4573-84
81. Wang J, Yu L, Schmidt RE, Su C, Huang X, Gould K. et al. Characterization of HSCD5, a novel human stearoyl-CoA desaturase unique to primates. Biochem Biophys Res Commun. 2005;332:735-42
82. Li L, Wang C, Calvisi DF, Evert M, Pilo MG, Jiang L. et al. SCD1 Expression is dispensable for hepatocarcinogenesis induced by AKT and Ras oncogenes in mice. PloS one. 2013;8(9):e75104
83. Lobello N, Biamonte F, Pisanu ME, Faniello MC, Jakopin Ž, Chiarella E. et al. Ferritin heavy chain is a negative regulator of ovarian cancer stem cell expansion and epithelial to mesenchymal transition. Oncotarget. 2016;7:62019-33
84. Colacino JA, McDermott SP, Sartor MA, Wicha MS, Rozek LS. Transcriptomic profiling of curcumin-treated human breast stem cells identifies a role for stearoyl-coa desaturase in breast cancer prevention. Breast Cancer Res Treat. 2016;158:29-41
85. Stoffel W, Schmidt-Soltau I, Jenke B, Binczek E, Hammels I. Hair Growth Cycle Is Arrested in SCD1 Deficiency by Impaired Wnt3a-Palmitoleoylation and Retrieved by the Artificial Lipid Barrier. J Investig Dermatol. 2017;137:1424-33
86. Ben-David U, Gan Q-F, Golan-Lev T, Arora P, Yanuka O, Oren Yifat S. et al. Selective Elimination of Human Pluripotent Stem Cells by an Oleate Synthesis Inhibitor Discovered in a High-Throughput Screen. Cell Stem Cell. 2013;12:167-79
87. Lu Y, Zhou Z, Tao J, Dou B, Gao M, Liu Y. Overexpression of stearoyl-CoA desaturase 1 in bone marrow mesenchymal stem cells enhance the expression of induced endothelial cells. Lipids Health Dis. 2014;13:53
88. Choi S, Yoo YJ, Kim H, Lee H, Chung H, Nam MH. et al. Clinical and biochemical relevance of monounsaturated fatty acid metabolism targeting strategy for cancer stem cell elimination in colon cancer. Biochem Biophys Res Commun. 2019;519(1):100-105
89. Hess D, Chisholm JW, Igal RA. Inhibition of StearoylCoA Desaturase Activity Blocks Cell Cycle Progression and Induces Programmed Cell Death in Lung Cancer Cells. PLoS ONE. 2010;5:e11394
90. Tracz-Gaszewska Z, Dobrzyn P. Stearoyl-CoA Desaturase 1 as a Therapeutic Target for the Treatment of Cancer. Cancers. 2019;11:948
91. Puglisi R, Bellenghi M, Pontecorvi G, Gulino A, Petrini M, Felicetti F. et al. SCD5 restored expression favors differentiation and epithelial-mesenchymal reversion in advanced melanoma. Oncotarget. 2018;9:7567-81
92. Caro P, Kishan AU, Norberg E, Stanley IA, Chapuy B, Ficarro SB. et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer cell. 2012;22:547-60
93. Li J, Cheng J-X. Direct Visualization of De novo Lipogenesis in Single Living Cells. Sci Rep. 2015;4:6807
94. Kamphorst JJ, Cross JR, Fan J, de Stanchina E, Mathew R, White EP. et al. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci. 2013;110:8882-7
95. Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer. 2013;13:227-32
96. Raulien N, Friedrich K, Strobel S, Rubner S, Baumann S, von Bergen M. et al. Fatty Acid Oxidation Compensates for Lipopolysaccharide-Induced Warburg Effect in Glucose-Deprived Monocytes. Front Immunol. 2017;8:609
97. He W, Liang B, Wang C, Li S, Zhao Y, Huang Q. et al. MSC-regulated lncRNA MACC1-AS1 promotes stemness and chemoresistance through fatty acid oxidation in gastric cancer. Oncogene. 2019;38:4637-54
98. Wang T, Fahrmann JF, Lee H, Li Y-J, Tripathi SC, Yue C. et al. JAK/STAT3-Regulated Fatty Acid β-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab. 2018;27:136
99. Park Jun H, Vithayathil S, Kumar S, Sung P-L, Dobrolecki Lacey E, Putluri V. et al. Fatty Acid Oxidation-Driven Src Links Mitochondrial Energy Reprogramming and Oncogenic Properties in Triple-Negative Breast Cancer. Cell Rep. 2016;14:2154-65
100. Yoshida GJ, Saya H. Therapeutic strategies targeting cancer stem cells. Cancer Sci. 2016;107:5
101. Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE. et al. A PML-PPARδ pathway for fatty acid oxidation regulates haematopoietic stem cell maintenance. Nat Med. 2012;18:1350
102. Knobloch M, Pilz G-A, Ghesquière B, Kovacs WJ, Wegleiter T, Moore DL. et al. A Fatty Acid Oxidation-Dependent Metabolic Shift Regulates Adult Neural Stem Cell Activity. Cell Rep. 2017;20:2144
103. Xie Z, Jones A, Deeney JT, Hur SK, Bankaitis VA. Inborn Errors of Long Chain Fatty Acid β-Oxidation Link Neural Stem Cell Self-Renewal to Autism. Cell Rep. 2016;14:991
104. Song N-J, Yun UJ, Yang S, Wu C, Seo C-R, Gwon A-R. et al. Notch1 deficiency decreases hepatic lipid accumulation by induction of fatty acid oxidation. Sci Rep. 2016;6:19377
105. Kalucka J, Bierhansl L, Conchinha NV, Missiaen R, Elia I, Brüning U. et al. Quiescent Endothelial Cells Upregulate Fatty Acid β-Oxidation for Vasculoprotection via Redox Homeostasis. Cell Metab. 2018;28:881-94
106. Hafiane A, Gasbarrino K, Daskalopoulou SS. The role of adiponectin in cholesterol efflux and HDL biogenesis and metabolism. Metabolism. 2019;100:153953
107. Munir R, Lisec J, Swinnen JV, Zaidi N. Lipid metabolism in cancer cells under metabolic stress. Br J Cancer. 2019;120:1090-8
108. Hong C, Marshall SM, McDaniel AL, Graham M, Layne JD, Cai L. et al. The LXR-Idol axis differentially regulates plasma LDL levels in primates and mice. Cell Metab. 2014;20:910-8
109. Ahmad F, Sun Q, Patel D, Stommel JM. Cholesterol Metabolism: A Potential Therapeutic Target in Glioblastoma. Cancers (Basel). 2019;11(2):146
110. Spite M. Resolving lipids: lipoxins regulate reverse cholesterol transport. Cell Metab. 2014;20:935-7
111. He S, Ma L, Baek AE. et al. Host CYP27A1 expression is essential for ovarian cancer progression. Endocr Relat Cancer. 2019;26(7):659-675
112. Akinwunmi B, Vitonis AF, Titus L, Terry KL, Cramer DW. Statin therapy and association with ovarian cancer risk in the New England Case Control (NEC) study. Int J Cancer. 2019;144:991-1000
113. Otahal A, Aydemir D, Tomasich E, Minichsdorfer C. Delineation of cell death mechanisms induced by synergistic effects of statins and erlotinib in non-small cell lung cancer cell (NSCLC) lines. Sci Rep. 2020;10:959
114. Bjarnadottir O, Feldt M, Inasu M, Bendahl P-O, Elebro K, Kimbung S. et al. Statin use, HMGCR expression, and breast cancer survival - The Malmö Diet and Cancer Study. Sci Rep. 2020;10:558
115. Cordes T, Metallo CM. Statins Limit Coenzyme Q Synthesis and Metabolically Synergize with MEK Inhibition in Pancreatic Tumors. Cancer Res. 2020;80(2):151-152
116. Wei T-T, Lin Y-T, Chen W-S, Luo P, Lin Y-C, Shun C-T. et al. Dual Targeting of 3-Hydroxy-3-methylglutaryl Coenzyme A Reductase and Histone Deacetylase as a Therapy for Colorectal Cancer. EBioMedicine. 2016;10:124-36
117. Li X, Wu JB, Li Q, Shigemura K, Chung LW, Huang WC. SREBP-2 promotes stem cell-like properties and metastasis by transcriptional activation of c-Myc in prostate cancer. Oncotarget. 2016;7(11):12869-12884
118. Smart EJ, Graf GA, McNiven MA, Sessa WC, Engelman JA, Scherer PE. et al. Caveolins, liquid-ordered domains, and signal transduction. Mol Cell Biol. 1999;19:7289-304
119. Shenoyscaria AM, Dietzen DJ, Kwong J, Link DC, Lublin DM. Cysteine(3) Of Src Family Protein-Tyrosine Kinases Determines Palmitoylation And Localization In Caveolae. J Cell Biol. 1994;126:353-63
120. Mineo C, James GL, Smart EJ, Anderson RG. Localization of epidermal growth factor-stimulated Ras/Raf-1 interaction to caveolae membrane. J Biol Chem. 1996;271:11930-5
121. Incardona JP ES. Cholesterol in signal transduction. Curr Opin Cell Biol. 2000;12(2):193-203
122. Chun M, Liyanage UK, Lisanti MP, Lodish HF. Signal transduction of a G protein-coupled receptor in caveolae: colocalization of endothelin and its receptor with caveolin. Proc Natl Acad Sci U S A. 1994;91:11728-32
123. Liu P, Ying Y, Ko YG, Anderson RG. Localization of platelet-derived growth factor-stimulated phosphorylation cascade to caveolae. J Biol Chem. 1996;271:10299-303
124. Pralle A, Keller P, Florin EL, Simons K, Horber JKH. Sphingolipid-cholesterol rafts diffuse as small entities in the plasma membrane of mammalian cells. J Cell Biol. 2000;148:997-1007
125. Brown DA, London E. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J Biol Chem. 2000;275:17221-4
126. Chhuon C PI, Borot F, Tondelier D, Lipecka J, Fritsch J, Chanson M, Edelman A, Ollero M, Guerrera IC. Changes in lipid raft proteome upon TNF-α stimulation of cystic fibrosis cells. J Proteomics. 2016;145:246-253
127. Gilbert RJC, Serra MD, Froelich CJ, Wallace MI, Anderluh G. Membrane pore formation at protein-lipid interfaces. Trends Biochem Sci. 2014;39:510-6
128. Gupta VK, Sharma NS, Kesh K, Dauer P, Nomura A, Giri B. et al. Metastasis and chemoresistance in CD133 expressing pancreatic cancer cells are dependent on their lipid raft integrity. Cancer Lett. 2018;439:101-112
129. Alcantara EN SE. Diet, nutrition, and cancer. Am J Clin Nutr. 1976;9:1035-47
130. Marzagalli M, Raimondi M, Fontana F, Montagnani Marelli M, Moretti RM, Limonta P. Cellular and molecular biology of cancer stem cells in melanoma: Possible therapeutic implications. Semin Cancer Biol. 2019;59:221-235
131. Mancini R, Noto A, Pisanu ME, De Vitis C, Maugeri-Sacca M, Ciliberto G. Metabolic features of cancer stem cells: the emerging role of lipid metabolism. Oncogene. 2018;37:2367-78
132. So-Yeon Park J-HC, Jeong-Seok Nam. Targeting Cancer Stem Cells in Triple-Negative Breast Cancer. Cancers (Basel). 2019;11(7):965
133. Ginestier C, Monville F, Wicinski J, Cabaud O, Cervera N, Josselin E. et al. Mevalonate Metabolism Regulates Basal Breast Cancer Stem Cells and Is a Potential Therapeutic Target. Stem Cells. 2012;30:1327-37
134. Wang XX, Huang Z, Wu QL, Prager BC, Mack SC, Yang KL. et al. MYC-Regulated Mevalonate Metabolism Maintains Brain Tumor-Initiating Cells. Cancer Res. 2017;77:4947-60
135. Joyce SA, Gahan CGM. Bile Acid Modifications at the Microbe-Host Interface: Potential for Nutraceutical and Pharmaceutical Interventions in Host Health. Annu Rev Food Sci Technol, Vol 7. 2016;7:313-33
136. Hafiane A, Gasbarrino K, Daskalopoulou SS. The role of adiponectin in cholesterol efflux and HDL biogenesis and metabolism. Metabolism. 2019;100:153953
137. Fu T, Coulter S, Yoshihara E, Oh TG, Fang S, Cayabyab F. et al. FXR Regulates Intestinal Cancer Stem Cell Proliferation. Cell. 2019;176:1098-112
138. Poli G, Biasi F. Potential modulation of cancer progression by oxysterols. Mol Aspects Med. 2016;49:47-8
139. Wang B, Rong X, Palladino END, Wang J, Fogelman AM, Martin MG. et al. Phospholipid Remodeling and Cholesterol Availability Regulate Intestinal Stemness and Tumorigenesis. Cell Stem Cell. 2018;22:206-20
140. Wang X HZ, Wu Q, Prager BC, Mack SC, Yang K, Kim LJY, Gimple RC, Shi Y, Lai S, Xie Q, Miller TE, Hubert CG, Song A, Dong Z, Zhou W, Fang X, Zhu Z, Mahadev V, Bao S, Rich JN. MYC-regulated mevalonate metabolism maintains brain tumorinitiating cells. Cancer Res. 2017;77(18):4947-4960
141. Bjarnadottir O, Romero Q, Bendahl PO, Jirstrom K, Ryden L, Loman N. et al. Targeting HMG-CoA reductase with statins in a window-of-opportunity breast cancer trial. Breast Cancer Res Treat. 2013;138:499-508
142. Akhter MZ, Sharawat SK, Kumar V, Kochat V, Equbal Z, Ramakrishnan M. et al. Aggressive serous epithelial ovarian cancer is potentially propagated by EpCAM(+)CD45(+) phenotype. Oncogene. 2018;37:2089-103
143. Shen WJ, Azhar S, Kraemer FB. SR-B1: A Unique Multifunctional Receptor for Cholesterol Influx and Efflux. Annu Rev Physiol. 2018;80:95-116
144. Llaverias G, Danilo C, Wang Y, Witkiewicz AK, Daumer K, Lisanti MP. et al. A Western-type diet accelerates tumor progression in an autochthonous mouse model of prostate cancer. Am J Pathol. 2010;177:3180-91
145. Gal D, Ohashi M, Macdonald PC, Buchsbaum HJ, Simpson ER. Low-Density Lipoprotein as a Potential Vehicle for Chemotherapeutic-Agents And Radionucleotides In the Management Of Gynecologic Neoplasms. American Journal of Obstetrics And Gynecology. 1981;139:877-85
146. Gabitova L, Gorin A, Astsaturov I. Molecular pathways: sterols and receptor signaling in cancer. Clin Cancer Res. 2014;20:28-34
147. Ghalaut VS, Pahwa MB. et al. Alteration in lipid profile in patients of chronic myelold leukemia before and after chemotherapy. Clinica Chimica Acta. 2006;366:239-42
148. Zhou PX, Hatziieremia S, Elliott MA, Scobie L, Crossan C, Michie AM. et al. Uptake of synthetic Low Density Lipoprotein by leukemic stem cells - a potential stem cell targeted drug delivery strategy. J Control Release. 2010;148:380-7
149. Hu A, Song BL. The interplay of Patched, Smoothened and cholesterol in Hedgehog signaling. Curr Opin Cell Biol. 2019;61:31-8
150. Luchetti G, Sircar R, Kong JH, Nachtergaele S, Sagner A, Byrne EF. et al. Cholesterol activates the G-protein coupled receptor Smoothened to promote Hedgehog signaling. Elife. 2016;5:e20304
151. Wu F, Zhang Y, Sun B, McMahon AP, Wang Y. Hedgehog Signaling: From Basic Biology to Cancer Therapy. Cell Chem Biol. 2017;24:252-80
152. Suckling RJ, Korona B, Whiteman P, Chillakuri C, Holt L, Handford PA. et al. Structural and functional dissection of the interplay between lipid and Notch binding by human Notch ligands. EMBO J. 2017;36:2204-15
153. Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S. et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat Cell Biol. 2014;16:357-66
154. Maccalli C, Rasul KI, Elawad M, Ferrone S. The role of cancer stem cells in the modulation of anti-tumor immune responses. Semin Cancer Biol. 2018;53:189-200
155. Steinbichler TB DJ, Skvortsov S, Ganswindt U, ´Riechelmann H, Skvortsova I-Ida. Therapy resistance mediated by cancer stem cells. Semin Cancer Biol. 2018;53:156-167
156. Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol. 2009;9:271-85
157. Wang F, Beck-Garcia K, Zorzin C, Schamel WW, Davis MM. Inhibition of T cell receptor signaling by cholesterol sulfate, a naturally occurring derivative of membrane cholesterol. Nat Immunol. 2016;17:844-50
158. Reya T MS, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105-11
159. He Y, Zou L. Notch-1 inhibition reduces proliferation and promotes osteogenic differentiation of bone marrow mesenchymal stem cells. Exp Ther Med. 2019;18:1884-90
160. Patel NS LJ, Generali D, Poulsom R, Cranston DW, Harris AL. Up-regulation of delta-like 4 ligand in human tumor vasculature and the role of basal expression in endothelial cell function. Cancer Res. 2005;65(19):8690-7
161. Tsoumas D, Nikou S, Giannopoulou E. et al. ILK Expression in Colorectal Cancer Is Associated with EMT, Cancer Stem Cell Markers and Chemoresistance. Cancer Genomics Proteomics. 2018;15(2):127-141
162. Obniski R, Sieber M, Spradling AC. Dietary Lipids Modulate Notch Signaling and Influence Adult Intestinal Development and Metabolism in Drosophila. Dev Cell. 2018;47:98-111
163. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983-8
164. Shu Z, Gao Y, Zhang G, Zhou Y, Cao J, Wan D. et al. A functional interaction between Hippo-YAP signalling and SREBPs mediates hepatic steatosis in diabetic mice. J Cell Mol Med. 2019;23:3616-28
165. Bhat KP, Salazar KL, Balasubramaniyan V, Wani K, Heathcock L, Hollingsworth F. et al. The transcriptional coactivator TAZ regulates mesenchymal differentiation in malignant glioma. Genes Dev. 2011;25:2594-609
166. Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C. et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell. 2011;147:759-72
167. Huo X ZQ, Liu AM, Tang C, Gong Y, Bian J, Luk JM, Xu Z, Chen J. Overexpression of Yes-associated protein confers doxorubicin resistance in hepatocellullar carcinoma. Oncol Rep. 2013Feb;29(2):840-6
168. Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the Roots of Cancer. Cancer Cell. 2016;29:783-803
169. Okazaki T KT, Kuwayama K, Kitazato KT, Mure H, Hara K, Morigaki R, Mizobuchi Y, Matsuzaki K, Nagahiro S. Up-regulation of endogenous PML induced by a combination of interferon-beta and temozolomide enhances p73/YAP-mediated apoptosis in glioblastoma. Cancer Lett. 2012;323(2):199-207
170. Kulkarni M TT, Syed Sulaiman NB, Lamar JM, Bansal P, Cui J, Qiao Y, Ito Y. RUNX1 and RUNX3 protect against YAP-mediated EMT, stem-ness and shorter survival outcomes in breast cancer. Oncotarget. 2018;9(18):14175-14192
171. Sundqvist A MM, Ren J, Vasilaki E, Kawasaki N, Kobayashi M, Koinuma D, Aburatani H, Miyazono K, Heldin CH, van Dam H, Ten Dijke P. JUNB governs a feed-forward network of TGFβ signaling that aggravates breast cancer invasion. Nucleic Acids Res. 2018;46(3):1180-1195
172. Briscoe J, Therond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol. 2013;14:416-29
173. Lim Y, Gondek L, Li L. et al. Integration of Hedgehog and mutant FLT3 signaling in myeloid leukemia [published correction appears in Sci Transl Med. 2015 Jul 8;7(295):295er6. Ma, Haley [corrected to Ma, Hayley]]. Sci Transl Med. 2015;7(291):291ra96
174. Gerling M BN, Kirn LM, Joost S, Frings O, Englert B, Bergström Å, Kuiper RV, Blaas L, Wielenga MC, Almer S, Kühl AA, Fredlund E, van den Brink GR, Toftgård R. Stromal Hedgehog signalling is downregulated in colon cancer and its restoration restrains tumour growth. Nat Commun. 2016;7:12321
175. Miyazaki Y, Matsubara S, Ding Q, Tsukasa K, Yoshimitsu M, Kosai K. et al. Efficient elimination of pancreatic cancer stem cells by hedgehog/GLI inhibitor GANT61 in combination with mTOR inhibition. Mol Cancer. 2016;15:49
176. Guen VJ CT, Kröger C, Ye X, Weinberg RA, Lees JA. EMT programs promote basal mammary stem cell and tumor-initiating cell stemness by inducing primary ciliogenesis and Hedgehog signaling. Proc Natl Acad Sci USA. 2017;114(49 E):10532-10539
177. Blassberg R, Jacob J. Lipid metabolism fattens up hedgehog signaling. BMC Biol. 2017;15(1):95
178. Long B, Wang LX, Zheng FM, Lai SP, Xu DR, Hu Y. et al. Targeting GLI1 Suppresses Cell Growth and Enhances Chemosensitivity in CD34+ Enriched Acute Myeloid Leukemia Progenitor Cells. Cell Physiol Biochem. 2016;38:1288-302
179. Zhang Y, Jin Z, Zhou H, Ou X, Xu Y, Li H. et al. Suppression of prostate cancer progression by cancer cell stemness inhibitor napabucasin. Cancer Med. 2016;5:1251-8
180. Teo R, Möhrlen F, Plickert G, Müller WA, Frank U. An evolutionary conserved role of Wnt signaling in stem cell fate decision. Dev Biol. 2006;289(1):91-99
181. Lang CMR, Chan CK, Veltri A, Lien WH. Wnt Signaling Pathways in Keratinocyte Carcinomas. Cancers (Basel). 2019;11(9):1216
182. Liu XS, Lin XK, Mei Y, Ahmad S, Yan CX, Jin HL. et al. Regulatory T Cells Promote Overexpression of Lgr5 on Gastric Cancer Cells via TGF-beta1 and Confer Poor Prognosis in Gastric Cancer. Front Immunol. 2019;10:1741
183. Boso D RE, Zanon C, Bresolin S, Maule F, Porcù E, Cani A, Della Puppa A, Trentin L, Basso G, Persano L. HIF-1α/Wnt signaling-dependent control of gene transcription regulates neuronal differentiation of glioblastoma stem cells. Theranostics. 2019;9(17):4860-4877
184. Senni N, Savall M, Cabrerizo Granados D, Alves-Guerra MC, Sartor C, Lagoutte I. et al. beta-catenin-activated hepatocellular carcinomas are addicted to fatty acids. Gut. 2019;68(2):322-334
185. Sulaiman A MS, Han X, Liu S, Wang L CSCs in Breast Cancer-One Size Does Not Fit All. Therapeutic Advances in Targeting Heterogeneous Epithelial and Mesenchymal CSCs. Cancers (Basel). 2019;11(8):1128
186. Timothy L. Fitzgerald KL, Lucio Cocco, Alberto M. Martelli, Massimo, Libra SC, Giuseppe Montalto, Melchiorre Cervello, Linda Steelman, Stephen L. Abrams JAM, Ph.D. Roles of EGFR and KRAS and their Downstream Signaling Pathways in Pancreatic Cancer and Pancreatic Cancer Stem Cells. Adv Biol Regul. 2015;59:65-81
187. Yan K, Wu QL, Yan DH, Lee CH, Rahim N, Tritschler I. et al. Glioma cancer stem cells secrete Gremlin1 to promote their maintenance within the tumor hierarchy. Genes Dev. 2014;28:1085-100
188. Du FY, Zhou QF, Sun WJ, Chen GL. Targeting cancer stem cells in drug discovery: Current state and future perspectives. World J Stem Cells. 2019;11:398-420
189. Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14:736-46
190. Corominas-Faja B, Cuyàs E, Gumuzio J, Bosch-Barrera J, Leis O, Martin ÁG. et al. Chemical inhibition of acetyl-CoA carboxylase suppresses self-renewal growth of cancer stem cells. Oncotarget. 2014;5:8306-16
191. Svensson R, Harriman G, Greenwood J, Bhat S, Harwood HJ, Kapeller R. et al. Abstract 2679: Acetyl-CoA carboxylase inhibition by ND646 reduces fatty acid synthesis and inhibits cell proliferation in human non-small cell lung cancer cells. Cancer Res. 2014;74:2679
192. DePeralta DK, Wei L, Harriman G, Greenwood J, Bhat S, Westlin W. et al. Abstract 1427: Liver selective Acetyl-CoA Carboxylase inhibition by ND-654 decreases hepatocellular carcinoma development in cirrhotic rats. Cancer Res. 2014;74:1427
193. Pandey PR, Okuda H, Watabe M, Pai SK, Liu W, Kobayashi A. et al. Resveratrol suppresses growth of cancer stem-like cells by inhibiting fatty acid synthase. Breast Cancer Res Treat. 2011;130:387-98
194. Wahdan-Alaswad RS, Cochrane DR, Spoelstra NS, Howe EN, Edgerton SM, Anderson SM. et al. Metformin-Induced Killing of Triple-Negative Breast Cancer Cells Is Mediated by Reduction in Fatty Acid Synthase via miRNA-193b. Horm Cancer. 2014;5:374-89
195. Brandi J, Dando I, Pozza ED, Biondani G, Jenkins R, Elliott V. et al. Proteomic analysis of pancreatic cancer stem cells: Functional role of fatty acid synthesis and mevalonate pathways. J Proteom. 2017;150:310-22
196. Yoshii Y, Furukawa T, Yoshii H, Mori T, Kiyono Y, Waki A. et al. Cytosolic acetyl-CoA synthetase affected tumor cell survival under hypoxia: the possible function in tumor acetyl-CoA/acetate metabolism. Cancer Sci. 2009;100:821-7
197. Zaidi N, Royaux I, Swinnen JV, Smans K. ATP Citrate Lyase Knockdown Induces Growth Arrest and Apoptosis through Different Cell- and Environment-Dependent Mechanisms. Mol Cancer Ther. 2012;11:1925-35
198. Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-Citrate Lyase Links Cellular Metabolism to Histone Acetylation. Science. 2009;324:1076-80
199. Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS. et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell. 2015;27:57-71
200. Wagner PD, Vu ND. Phosphorylation of ATP-citrate lyase by nucleoside diphosphate kinase. J Biol Chem. 1995;270:21758-64
201. Pierce MW, Palmer JL, Keutmann HT, Avruch J. ATP-citrate lyase. Structure of a tryptic peptide containing the phosphorylation site directed by glucagon and the cAMP-dependent protein kinase. J Biol Chem. 1981;256:8867-70
202. Zhao S, Torres A, Henry RA, Trefely S, Wallace M, Lee JV. et al. ATP-Citrate Lyase Controls a Glucose-to-Acetate Metabolic Switch. Cell Rep. 2016;17:1037-52
203. Oballa RM, Belair L, Black WC, Bleasby K, Chan CC, Desroches C. et al. Development of a Liver-Targeted Stearoyl-CoA Desaturase (SCD) Inhibitor (MK-8245) to Establish a Therapeutic Window for the Treatment of Diabetes and Dyslipidemia. J Med Chem. 2011;54:5082-96
204. Sulaiman A, McGarry S, Li L, Jia D, Ooi S, Addison C. et al. Dual inhibition of Wnt and Yes-associated protein signaling retards the growth of triple-negative breast cancer in both mesenchymal and epithelial states. Mol Oncol. 2018;12:423-40
205. Cole AJ, Fayomi AP, Anyaeche VI, Bai S, Buckanovich RJ. An evolving paradigm of cancer stem cell hierarchies: therapeutic implications. Theranostics. 2020;10(7):3083-3098
206. Qu Q, Zeng F, Liu X, Wang QJ, Deng F. Fatty acid oxidation and carnitine palmitoyltransferase I: emerging therapeutic targets in cancer. Cell Death Dis. 2016;7:e2226
207. Iwamoto H, Abe M, Yang Y, Cui D, Seki T, Nakamura M. et al. Cancer Lipid Metabolism Confers Antiangiogenic Drug Resistance. Cell Metab. 2018;28:104-17
208. Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X. et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature. 2016;531:651-5
209. Bilotta MT AM, Molfetta R, Scarno G, Fionda C, Zingoni A, Soriani A, Garofalo T, Petrucci MT, Ricciardi MR, Paolini R, Santoni A, Cippitelli M. Activation of liver X receptor up-regulates the expression of the NKG2D ligands MICA and MICB in multiple myeloma through different molecular mechanisms. FASEB J. 2019;33(8):9489-9504
210. Lin CY HC, Kuo LK, Hiipakka RA, Jones RB, Lin HP, Hung Y, Su LC, Tseng JC, Kuo YY, Wang YL, Fukui Y. Cholestane-3β, 5α, 6β-triol suppresses proliferation, migration, and invasion of human prostate cancer cells. PLoS One. 2013;8(6 e):65734
211. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7:763-77
212. Boodhoo N, Kamble N, Kaufer BB, Behboudi S. Targeted induction of de novo Fatty acid synthesis enhances MDV replication in a COX-2/PGE2α dependent mechanism through EP2 and EP4 receptors engagement. bioRxiv. 2018:323840.
213. Wen D, Wang J, Van Den Driessche G, Chen Q, Zhang Y, Chen G. et al. Adipocytes as Anticancer Drug Delivery Depot. Matter. 2019;1(5):1203-1214
214. Shen H, Shi S, Zhang Z, Gong T, Sun X. Coating Solid Lipid Nanoparticles with Hyaluronic Acid Enhances Antitumor Activity against Melanoma Stem-like Cells. Theranostics. 2015;5(7):755-771
Author contact
![]() Corresponding author: Dr. Ming-Liang He, E-mail: mlhe7788com.
Corresponding author: Dr. Ming-Liang He, E-mail: mlhe7788com.