Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(16):7111-7130. doi:10.7150/thno.45286 This issue Cite
Research Paper
Fgfr3 mutation disrupts chondrogenesis and bone ossification in zebrafish model mimicking CATSHL syndrome partially via enhanced Wnt/β-catenin signaling
Xianding Sun1#, Ruobin Zhang1#, Hangang Chen1#, Xiaolan Du1, Shuai Chen1, Junlan Huang1, Mi Liu1, Meng Xu1, Fengtao Luo1, Min Jin1, Nan Su1, Huabing Qi1, Jing Yang1, Qiaoyan Tan1, Dali Zhang1, Zhenhong Ni1, Sen Liang1, Bin Zhang1, Di Chen2, Xin Zhang3, Lingfei Luo4 ![]() , Lin Chen1
, Lin Chen1 ![]() , Yangli Xie1
, Yangli Xie1 ![]()
1. Department of Wound Repair and Rehabilitation Medicine, State Key Laboratory of Trauma, Burns and Combined Injury, Daping Hospital, Army Medical University, Chongqing 400042, China.
2. Research Center for Human Tissues and Organs Degeneration, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen 518055, China.
3. Departments of Ophthalmology, Pathology and Cell Biology, Columbia University, New York, NY 10032, USA.
4. Key Laboratory of Freshwater Fish Reproduction and Development, Ministry of Education, Laboratory of Molecular Developmental Biology, School of Life Sciences, Southwest University, Beibei, Chongqing 400715, China.
#These authors contributed equally to this work.
Received 2020-2-24; Accepted 2020-5-21; Published 2020-5-30
Abstract

CATSHL syndrome, characterized by camptodactyly, tall stature and hearing loss, is caused by loss-of-function mutations of fibroblast growth factor receptors 3 (FGFR3) gene. Most manifestations of patients with CATSHL syndrome start to develop in the embryonic stage, such as skeletal overgrowth, craniofacial abnormalities, however, the pathogenesis of these phenotypes especially the early maldevelopment remains incompletely understood. Furthermore, there are no effective therapeutic targets for this skeleton dysplasia.
Methods: We generated fgfr3 knockout zebrafish by CRISPR/Cas9 technology to study the developmental mechanisms and therapeutic targets of CATSHL syndrome. Several zebrafish transgenic lines labeling osteoblasts and chondrocytes, and live Alizarin red staining were used to analyze the dynamical skeleton development in fgfr3 mutants. Western blotting, whole mount in situ hybridization, Edu labeling based cell proliferation assay and Wnt/β-catenin signaling antagonist were used to explore the potential mechanisms and therapeutic targets.
Results: We found that fgfr3 mutant zebrafish, staring from early development stage, showed craniofacial bone malformation with microcephaly and delayed closure of cranial sutures, chondroma-like lesion and abnormal development of auditory sensory organs, partially resembling the clinical manifestations of patients with CATSHL syndrome. Further studies showed that fgfr3 regulates the patterning and shaping of pharyngeal arches and the timely ossification of craniofacial skeleton. The abnormal development of pharyngeal arch cartilage is related to the augmented hypertrophy and disordered arrangement of chondrocytes, while decreased proliferation, differentiation and mineralization of osteoblasts may be involved in the delayed maturation of skull bones. Furthermore, we revealed that deficiency of fgfr3 leads to enhanced IHH signaling and up-regulated canonical Wnt/β-catenin signaling, and pharmacological inhibition of Wnt/β-catenin could partially alleviate the phenotypes of fgfr3 mutants.
Conclusions: Our study further reveals some novel phenotypes and underlying developmental mechanism of CATSHL syndrome, which deepens our understanding of the pathogenesis of CATSHL and the role of fgfr3 in skeleton development. Our findings provide evidence that modulation of Wnt/β-catenin activity could be a potential therapy for CATSHL syndrome and related skeleton diseases.
Keywords: FGFR3, CATSHL syndrome, Zebrafish, Skeletal development, Wnt/β-catenin
Introduction
CATSHL (camptodactyly, tall stature, and hearing loss) syndrome (OMIM 610474) is a genetic disorder first named and described by Toydemir et al. in 1996 [1]. Toydemir et al. evaluated a multigenerational family with 27 living affected members, all of whom possessed an autosomal dominant syndrome of camptodactyly, tall stature, and hearing loss [1]. Then, Makrythanasis et al. reported an autosomal recessive case and Escobar et al. reported an autosomal dominant family with CATSHL Syndrome [2,3]. Other clinical features of CATSHL syndrome include microcephaly, wormian skull bones, a high palate, pectus excavatum, scoliosis/kyphoscoliosis, tall vertebral bodies with irregular borders, broad femoral metaphysis, osteochondromas of the femur, tibia, or phalanx, and intellectual disabilities [1-3]. Hearing loss in CATSHL syndrome appears to be sensorineural, congenital, and progressive in affected individuals [1].
CATSHL syndrome is caused by mutations in the fibroblast growth factor receptors 3 (FGFR3) gene on chromosome 4p16 [1-3]. In humans, FGFR3 is one of the four membrane-spanning receptor tyrosine kinases that serve as high affinity receptors for multiple fibroblast growth factors [4]. It plays an essential role in the development of a wide range of tissues, especially the skeleton [5]. Patients with activating FGFR3 mutations exhibit skeletal dysplasia characterized by short-limbed dwarfism including achondroplasia (ACH), hypochondroplasia, thanatophoric dysplasia I/II, Muenke syndrome, Crouzon syndrome with acanthosis nigricans, severe ACH with developmental delay and acanthosis nigricans (SADDAN) and lacrimo-auriculo-dental-digital (LADD) syndrome [4,6-8]. Some patients with Muenke syndrome or Crouzon syndrome with acanthosis nigricans have premature closure of cranial sutures. Activated FGFR3 results in disordered endochondral bone growth and skeletal dysplasia through impaired proliferation and differentiation of growth plate chondrocytes [9,10]. Similar to the phenotype of patients with CATSHL syndrome, Fgfr3 deficient mice also have skeletal overgrowth due to enhanced proliferation of growth plate chondrocytes, and sensorineural deafness [11,12].
Physiological and pathological development of skeleton are gradually finished, starting from early embryonic stage. Although there are well designed studies about the skeleton phenotypes and underlying mechanisms of CATSHL patients or mice with FGFR3 deficiency, majority of them studied the skeleton phenotypes at perinatal or postnatal stages. The reason for few studies about the early skeleton phenotypes of Fgfr3 mutants is that it is very difficult to analyze the early phenotype development process dynamically in current animal models such as mice. Furthermore, the Fgfr3 deficient mice have no apparent phenotype in craniofacial skeleton [12,13], while CATSHL syndrome patients exhibit craniofacial skeleton phenotypes such as microcephaly, high palate and wormian bones [1-3]. The reason for the discrepant in the craniofacial phenotypes between mouse model and patients remains to be studied. In addition, the molecular mechanism of the skeleton phenotypes especially the early maldevelopment of CATSHL syndrome is not well understood and therefore there are no effective therapies to alleviate the skeletal phenotypes.
Zebrafish (Danio rerio) has become an immensely useful and popular model for genetic and developmental studies. Due to its short developing time, external development, transparency and strong breeding ability, zebrafish are suitable for various genetic manipulation, living imaging observation and drug screens [14-17]. Especially, the superior imaging technology available in zebrafish has provided unprecedented insights into the dynamics of skeletal development [18,19]. In addition, the craniofacial bones of zebrafish are formed through both endochondral ossification and intramembranous ossification like humans. Especially the ceratohyal bone in zebrafish is composed of epiphysis and epiphysial growth plates and formed through endochondral ossification similar to that of mammalian [20]. Previous studies have shown that mutation of rmrp and nans genes, which cause chondrodysplasia in humans, can also lead to maldevelopment of craniofacial bone such as ceratohyal cartilage in zebrafish [19,21]. Furthermore, the fundamental signaling pathways and cellular events that sculpt the nascent craniofacial skeleton in the embryo have been proven to be highly conserved from fish to human [22,23]. Therefore, zebrafish is a suitable model for studying the role and mechanism of FGFs/FGFRs in long bone and craniofacial skeleton development.
The roles of Fgfrs in zebrafish have been examined using morpholinos and dominant-negative approaches. Fgfr1a, Fgfr2 and Fgfrl1 were found to control the cranial cartilage development. Fgfr3c is required for the early embryogenesis and the anterior-posterior patterning of zebrafish [24-26]. However, the function of Fgfr3 in zebrafish development especially the skeleton development has not been illuminated. The expression pattern of fgfr3 in zebrafish has been found to be similar to those in higher vertebrates. Fgfr3 is expressed in chondrocytes of the head cartilages, osteoblasts, ventricular zone of the brain, undifferentiated mesenchymal cells of the skin, and eye lens epithelia [27].
In this study, we generated fgfr3 knockout zebrafish to study the function of fgfr3 in zebrafish skeleton development and gain novel insight into the mechanisms underlying the maldevelopment of skeleton, especially the early skeleton development of CATSHL syndrome. We found that fgfr3 mutants, staring from 10 days post fertilization (dpf), gradually showed craniofacial bone malformation with smaller cranial skull bones and delayed closure of cranial sutures, as well as dysregulated development of pharyngeal arch cartilage with abnormal hypertrophy and disordered arrangement of chondrocytes. Further studies showed that fgfr3 regulates the proliferation and differentiation of chondrocytes, as well as the osteogenic differentiation and mineralization of zebrafish craniofacial bone, and up-regulated IHH and Wnt/β-catenin pathway is involved in the maldevelopment of skeleton especially the cartilage in mutants.
Materials and Methods
Zebrafish strains and generation of transgenic lines
Zebrafish (Danio rerio) of the AB genetic background were used. The Tg(col2a1a:EGFP) was described before [28]. The Tg(osterix:EGFP) is a gift from Chung-Der Hsiao (Chung Yuan Christian University, Chung-Li, Taiwan). All zebrafish were housed in semi-closed recirculation housing systems (ESEN, Beijing, China) and were kept at a constant temperature (27-28°C) on a 14:10 hour light: dark photoperiod. All zebrafish lines were raised and maintained according to standard protocols as previously described [29]. All in vivo experiments and protocols were approved by Institutional Animal Care and Use Committee of the Research Institute of Surgery, Daping Hospital IACUC protocol SYXK-(Army) 2017-0057.
Multiple sequence alignment of FGFR3
ClustalW software was used for multiple sequence alignment. Amino acids of GenBank Accession numbers: human FGFR3: NM_000142, mouse FGFR3: NM_001163215 and zebrafish Fgfr3: NM_131606 were used for alignment. Consensus elements were highlighted with BOXSHADE.
In situ hybridizations
The template for fgfr3 antisense probe synthesis was amplified with the following primer, forward primer: 5'-GGTGACCTTGGAAAGGATTACTG-3', reverse primer: 5'-CCTGCCTCGTCCTCATCTTCATC-3'. Primers for other probes are available from the corresponding authors upon request. PCR products were cloned into the pGEMT-easy vector (Promega). Digoxigenin-labelled probes were generated by in vitro transcription (DIG RNA Labeling Kit, Roche) and in situ hybridization was carried out as previously described [30]. The WISH images were captured using a SteREO Discovery 20 microscope (Carl Zeiss).
Generation of fgfr3 mutants using CRISPR/Cas9 system
Fgfr3 mutants were generated by targeting the 11th exon of fgfr3 with CRISPR/Cas9 technology. The process was performed as previously described [31]. The fgfr3 target sequence was 5'-GGGAGAGGGCTGCTTTGGGC-3', and the target region was amplified by PCR using the following primers: forward primer: 5'-GCCTAACGTGTCTGAACTTGAAC-3', reverse primer: 5'-GATGGCATTTGAATATACACTCAC-3'. Founder fish (F0) carrying deletion mutations and corresponding F1 embryos were raised up. F1 fish with heterozygote fgfr3 mutation were crossed to identify mutants with phenotypes.
X -ray and micro-computed tomography
3-month adult zebrafish (equal to 26.0 mm SL (standard length)) were harvested, stored in 75% ethanol at 4°C, and were subjected to high-resolution X-rays examination using Faxitron MX20. The zebrafish were scanned with micro-CT (viva CT-40, Scanco Medical AG, Switzerland). Image acquisition was performed with the condition of 45 kV and 177 μA in high-resolution scans (10.5µm voxel resolution). Two-dimensional images were used to generate three-dimensional reconstructions. Every measurement used the same filtering and segmentation values. The images were analyzed by micro-CT Evolution Program V6.5 software.
Edu cell proliferation assay
The Edu cell proliferation assay was applied for S-phase labeling according to the manufacturer's instructions (Click-iT Edu Imaging Kits, Invitrogen, C10340). Wild-type (WT) and fgfr3 mutants were injected with 10 nL 0.2 mM Edu (Life Technology, E10187) at 1 month (SL 10.0 mm) and incubated at 28.5°C incubator for 2 hours. After being fixed with 4% formaldehyde at 4°C overnight, the samples were subjected to frozen section and EdU assay.
Western blot analysis
Zebrafish head skeleton (n = 20) at 20 dpf (SL 7.5 mm) and 40 dpf (SL 13.0 mm) were dissected and lysed using RIPA lysis buffer containing protease inhibitors (Roche). Equal amount of protein samples was resolved on a 12% SDS-PAGE gel and transferred onto a PVDF membrane (Millipore). Then samples were probed with primary antibody specific for non-phospho (active) β-catenin (1:1000; Cell Signaling Technology, 8814S), phospho-β-catenin (1:1000; Cell Signaling Technology, 9561S), IHH (1:1000; Abcam, ab52919), followed by secondary antibodies. The signal was detected using the chemiluminescent signal (Pierce, NCI4106) according to the manufacturer's instructions. Western blot analysis was performed for three times. Quantitative analysis for western blotting was conducted using ImageJ software.
Skeletal analysis by whole skeleton staining, histology and imaging
Zebrafish in vivo skeletal staining was incubated with 0.05% Alizarin red (Sigma, A5533) for 1 hour and then washed with system water three times. Alizarin red and Alcian blue whole skeleton staining was performed as described [12]. For histological analysis, samples were fixed in 4% paraformaldehyde, decalcified in 15% EDTA and embedded in paraffin as described [32]. Sections (5-μm thick) were stained with Safranin O/Fast Green and hematoxylin and eosin (H & E). Picric-sirius red staining was used to detect collagen fiber density and organization as described [33]. Zebrafish embryos were imaged with a SteREO Discovery 20 microscope (Carl Zeiss) or LSM880NLO confocal microscope with a 20× water immersion objective (Carl Zeiss). The ceratohyal length measurements, the relative area of osterix labeled osteoblasts measurements and the mineralization intensity measurement were conducted using Image J.
Drug treatment
XAV939 was purchased from Selleck Chemicals and was dissolved with DMSO to get a stock solution of 10 mM. The drug treatment was performed for three times. In each experiment, the self-crossing offspring of fgfr3 heterozygous mutants were randomly assigned to be treated with 2.5 µM XAV939 or 0.1% DMSO control from 10 dpf (SL 5.0 mm) to 20 dpf (SL 7.5 mm) to investigate the effect of Wnt signaling antagonist on the development of pharyngeal arches cartilage. In each experiment, more than 60 WT and 20 fgfr3 mutants were treated with DMSO or XAV939, respectively.
Statistical analysis
All numeric data are presented as mean ± SD. Error bars indicate SD. Differences between two groups were evaluated using Unpaired Student's t test, and ANOVA was used for comparisons of multiple groups. When significant levels (P<0.05) were achieved, Tukey's Post Hoc test was performed. All statistical analyses were performed using GraphPad PRISM 7.0 software, and P-values were considered significant at * p < 0.05, ** p < 0.01, *** p < 0.001.
Results
Zebrafish fgfr3 is highly conserved across multiple species
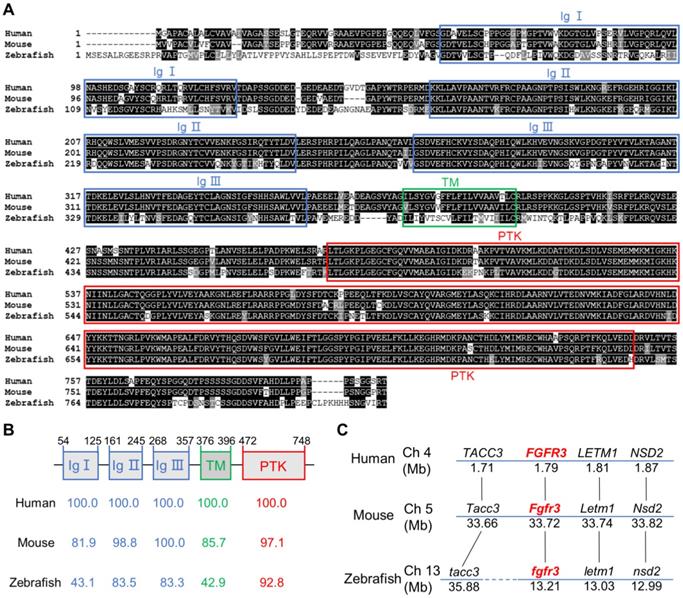
To determine the sequence conservation of zebrafish fgfr3, we did multiple sequence alignments for coding sequence and amino acids of FGFR3 in human, mouse and zebrafish. We found that the amino acid identity of FGFR3 was 76.9% between zebrafish and human, and 74.6% between zebrafish and mouse (Figure 1A). The homology of multiple domains of FGFR3 is variable with the amino acid identity of tyrosine-protein kinase catalytic domain as high as 92.8% between zebrafish and human, while that of three immunoglobulin (Ig)-like domain (Ig I-III) was 43.1%, 83.5%, 83.3% respectively. In contrast, the transmembrane domain shared only 42.9% identity (Figure 1A, B). We further compared the conservation of coding sequence of fgfr3 and found a 59.41% sequence identity between zebrafish and human. We also performed a synteny analysis for fgfr3 gene in zebrafish, human and mouse (Figure 1C). It showed that the surrounding genes (LETM1, NSD2, TACC3) of FGFR3 in human and mouse have the similar positions as the surrounding genes (letm1, nsd2, tacc3) linked to fgfr3 in zebrafish. These data suggest that zebrafish fgfr3 gene is syntenic to human FGFR3 and mouse Fgfr3 gene. Together, these data suggest that zebrafish fgfr3 is highly conserved especially in important catalytic domain and may play a conservative role across these species.
Zebrafish fgfr3 is highly conserved across multiple species. (A) Multiple alignment of amino acids of FGFR3 for human (806 aa), mouse (800 aa) and zebrafish (819 aa). Identical amino acids are shaded. The three Ig-like domains (Ig I-III), transmembrane domain (TM), and tyrosine-protein kinase domain (PTK) are marked with blue, green and red boxes, respectively. (B) The identity of multiple domains of each FGFR3 protein for human, mouse and zebrafish, as referred to human FGFR3. (C) Conserved synteny analysis for FGFR3 gene in zebrafish, human and mouse. Numbers next to the gene names represent megabase pair (Mbp) of gene location on the chromosome. Chromosome segments are represented with blue lines and dashed blue lines represent discontinuous segments. Orthologs are connected with black lines.
Expression pattern of fgfr3 during early development of zebrafish
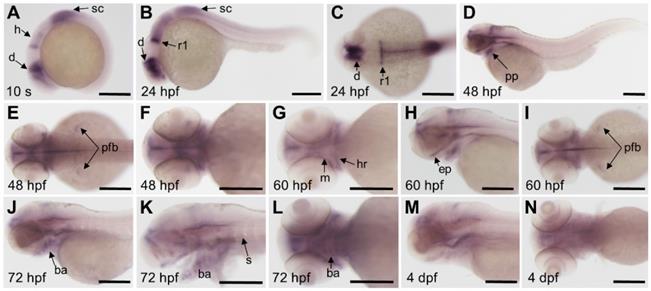
We detected the expression pattern of fgfr3 gene by whole mount in situ hybridization during early development stage. Results showed that fgfr3 transcripts were first detected weakly at tailbud stage in the prospective diencephalon and the anterior hindbrain (Figure 2A). From segmentation stage to 24 hours post fertilization (hpf), fgfr3 showed expression in diencephalon, anterior hindbrain, anterior spinal cord, especially was intensely expressed in the posterior rhombomere 1 (Figure 2B, C). Fgfr3 transcripts were detected in pharyngeal pouches and the pectoral fin bud at 48 hpf (Figure 2D-F), and then in mandibular and hyoid arches cartilage at 60 hpf. It was also detected extensively in ethmoid plate at 60 hpf (Figure 2G-I). By 72 hpf, fgfr3 was expressed in chondrocytes of cartilage of branchial arch 1-5 (Figure 2J-L), and gradually diminished at 4 dpf (Figure 2M, N). Besides, fgfr3 was weakly expressed in the somites. These observations suggest a potential role for fgfr3 in the development of various tissues, including cartilage.
Expression patterns of fgfr3 detected by whole mount in situ hybridization in wild-type zebrafish. (A) Lateral view of 10-somite stage; (B-C) show 24 hpf embryo in lateral view (B) and head region in dorsal view (C). (D-F) show 48 hpf embryo in lateral view (D) and head region in dorsal view (E) and in ventral view (F). (G-I) show head region of 60 hpf embryo in ventral view (G), in lateral view (H) and in dorsal view (I). (J-L) show head region of 72 hpf embryo in lateral view with yolk (J) and in lateral view without yolk (K), 72 hpf embryo in ventral view (L); (M-N) show head region of 4 dpf embryo in lateral view (M) and in ventral view (N). n = 30 embryos for A-N. Abbreviations: ba, branchial arch; d, diencephalon; ep, ethmoid plate; h, hindbrain; ha, hyoid arch; m, mandibular arch; pfb, pectoral fin bud; pp, pharyngeal pouches; r1, rhombomere 1; s, somites; sc, spinal cord. Scale bars: 200 µm in A-N.
Fgfr3 deletion in zebrafish disrupts craniofacial skeleton development
In order to generate the zebrafish model of CATSHL and study the role of fgfr3 in development, we used CRISPR/Cas9 genome editing approach to knockout fgfr3 by targeting the tyrosine-protein kinase domain of Fgfr3 protein. We identified two stable homozygous mutant lines in F2 generation, which have 2-bp and 17-bp deletion, respectively (Figure 3A). Both two mutations caused a frameshift, predictably resulting in premature stop codon with a truncated Fgfr3 protein lacking the tyrosine kinase domain (Figure 3A). We performed real-time quantitative PCR (RT-qPCR) and found that the expression level of fgfr3 was significantly decreased in both two fgfr3 mutant lines (Figure 3B). We analyzed the gross phenotypes of fgfr3 knockout zebrafish in F3 generation. Since we found the same phenotype in these two mutant lines, we chose the 17-bp deletion line as fgfr3 mutant in our following studies. Fgfr3 heterozygous mutants were normal as wild-type (WT) siblings. Homozygous mutant embryos showed no obvious morphological defects until at 1 month (SL 10.0 mm) when the jawbone and skull bone were gradually formed. The fgfr3 mutants were readily identified by their mandibular deformity with hyoid arch drooping toward the ventral side and mild domed-shaped skulls at 1 month (SL 10.0 mm) (Figure 3C), which resemble the microcephaly and high palate in the skulls of CATSHL patients [3], but the body length of WT and fgfr3 mutant had no significant change at 1 month (SL 10.0 mm). Some fgfr3 mutants can't grow to adult stage due to severe cranial deformity, while mutants survived to adult stage had smaller body size than that of WT siblings (Figure 3D). From gross appearance and X-rays, we can find that fgfr3 mutants exhibited varying degrees of mandibular deformity with remarkable domed-shaped skulls and microcephaly. Especially in fgfr3 mutants with severe phenotypes, their mouth failed to close due to severe jaw deformity with resultant eating disorder (Figure 3D, E).
Fgfr3 knockout in zebrafish disrupts craniofacial development. (A) The WT and two fgfr3 mutant lines in F2 generation generated by CRISPR/Cas9 technology. Left panel show the target sequence (red), PAM sequence (bold dashed line) and 2 bp or 17 bp deletion of the mutant lines. Right panel diagram domains of WT and predicted mutants of Fgfr3 protein. (B) RT-qPCR analysis of the expression level of fgfr3 in 2 bp deletion mutant line and 17 bp deletion mutant line at 20 dpf (SL 7.5 mm). n = 3 independent experiments. ***p < 0.001. (C, D) Bright-field images showing the WT and fgfr3 mutant at 1 month (SL 10.0 mm) (C) and the adult stage about 3 months (SL 26.0 mm) with different degrees of skull deformity (D). The quantification of body length for (C) and (D) is show in the right panel. n = 10, ***p < 0.001, no significance (ns). (E) X-rays of the corresponding zebrafish of (D) in lateral view (left) and dorsal view (right). (F, G) The micro-CT reconstruction images of WT and fgfr3 mutant, the (F) are magnified in the (G). Note that the domed-shaped skull (arrowheads), drooped hyoid arch (arrow), malformed jaw bone and delayed closure of cranial sutures in fgfr3 mutants. Scale bars: 1 mm in C, F-G.
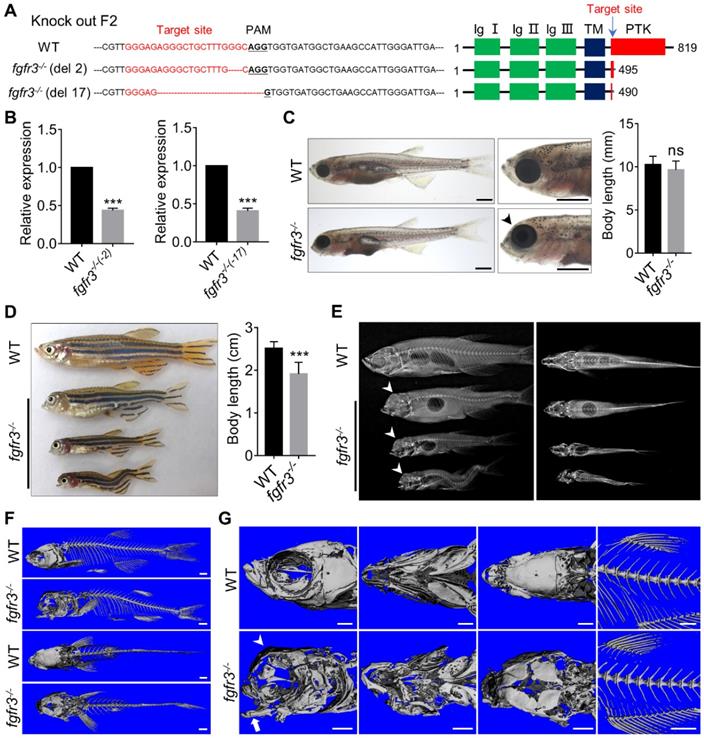
We performed high resolution micro-CT and three-dimensional skeletal reconstructions of adult zebrafish and found that the ossification of the craniofacial bone including mandibular bone and skull bone was severely decreased in fgfr3 mutants (Figure 3F, G). The fgfr3 mutants exhibited grossly deformed pharyngeal arches, domed skulls, microcephaly, midface hypoplasia, and delayed closure of cranial sutures (Figure 3 F, G). The spine in most mild and moderate fgfr3 mutants was morphologically normal, but exhibited kyphosis and mild scoliosis in severe fgfr3 mutants (Figure 3E). The vertebral body, neural arches and haemal arches had no significant abnormality in fgfr3 mutants compared to the WT (Figure 3F, G).
Alizarin red and Alcian blue whole skeleton staining were performed to further examine the changes of skeleton in fgfr3 mutants (Figure S1A, B). We found the consistent morphological changes of craniofacial skeleton as micro-CT revealed. We then dissected the craniofacial bone and interestingly found some small bone islands at the margin of the cranial bone in fgfr3 mutants (Figure S1A), which resemble wormian bones in the skull of CATSHL patients [2]. Cranial base will affect the shape of skulls [34]; we found delayed synchondrosis closure and irregular morphology of the cranial base in fgfr3 mutants (Figure S1A). Both the mandibular arch and hyoid arch had symmetrical and regular shape in WT, but they exhibited asymmetrical and irregular morphology in fgfr3 mutants (Figure 3G and Figure S1A). The morphology of precaudal vertebrae and caudal vertebrae showed no marked abnormality in fgfr3 mutants in contrast to WT. But the joint between the precaudal vertebrae and the ribs had abnormal morphology and more regions positively stained by Alcian blue. Besides, the caudal fin vertebrae were slightly bent with abnormal morphology (Figure S1B).
Fgfr3 regulates timely ossification of craniofacial skeleton
It has been reported that both CATSHL patients and Fgfr3 knockout mice have abnormal bone ossification [1,13]. To study the effect of fgfr3 mutation on bone formation in zebrafish at early embryonic development, we used live Alizarin red staining and transgenic line Tg(osterix:EGFP) that labels osteoblasts to examine the ossification in fgfr3 mutants from 5 dpf (SL 4.0 mm) to 20 dpf (SL 7.5 mm). At 5 dpf (SL 4.0 mm), confocal microscopy revealed no significant difference in the bone ossification between WT and fgfr3 mutants (Figure S2A, B). At 10 dpf (SL 5.0 mm), however, the fgfr3 mutants showed delayed ossification of ceratohyal perichondrium and slightly delayed formation of branchiostegal rays (Figure 4A). Similar results were also observed using Alizarin red staining after fixation, which showed that the perichondral ossification of occipital arch in fgfr3 mutants was delayed at 10 dpf (SL 5.0 mm) (Figure S3A). Whole mount in situ hybridization revealed that the expression of col10a1, which is related to osteoblast differentiation in zebrafish [35], was down-regulated in fgfr3 mutants at 8 dpf (Figure S3B). We also found that the expressions of col1a2, osteopontin (spp1), osteonectin (osn), which were the later markers of osteoblast differentiation, were decreased in fgfr3 mutants (Figure S3C). From 15 dpf (SL 6.0 mm) to 20 dpf (SL 7.5 mm), although the morphology of skulls still showed no gross change, confocal microscopy revealed that the ossification of intramembranous bone especially the opercle and branchiostegal rays was strongly inhibited (Figure 4A and Figure S4A). Besides, there were less osteoblasts at the mineralized bone collars of ceratohyal bone in fgfr3 mutants (Figure 4A). These data indicate that the formation of both intramembranous bone and endochondral bone was delayed in fgfr3 mutants in contrast to WT.
Fgfr3 is required for the timely bone ossification. (A) Confocal imaging of WT and fgfr3 mutants in Tg(osterix:EGFP) background live stained with Alizarin red at 10 dpf (SL 5.0 mm) (left), 15 dpf (SL 6.0 mm) (middle) and 20 dpf (SL 7.5 mm) (right). White arrowheads indicate less osteoblasts at the mineralized bone collars of ceratohyal perichondrium. (B-D) Stereo fluorescence microscope imaging of WT and fgfr3 mutants at 30 dpf (SL 10.0 mm) (B), 45 dpf (SL 14.0 mm) (C) and 60 dpf (SL 18.0 mm) (D) in lateral view (left) and ventral view (right) after live stained with Alizarin red in Tg(osterix:EGFP). White arrows indicate some small bone islands at the margin of the opercle bone. (E) Bright field (left), Tg(osterix:EGFP) images (middle) and Alizarin red staining images (right) showing the development of parietal (p) and frontal (f) bones of WT and fgfr3 mutants in dorsal view at 30 dpf (SL 10.0 mm) (left), 45 dpf (SL 14.0 mm) (middle) and 60 dpf (SL 18.0 mm) (right). White arrows indicate several small separated bones in fgfr3 mutants. Abbreviations: Bsr, branchiostegal rays; Ch, ceratohyal bone; Op, opercle. Scale bars: 100 µm in A, 400 µm in B-E.
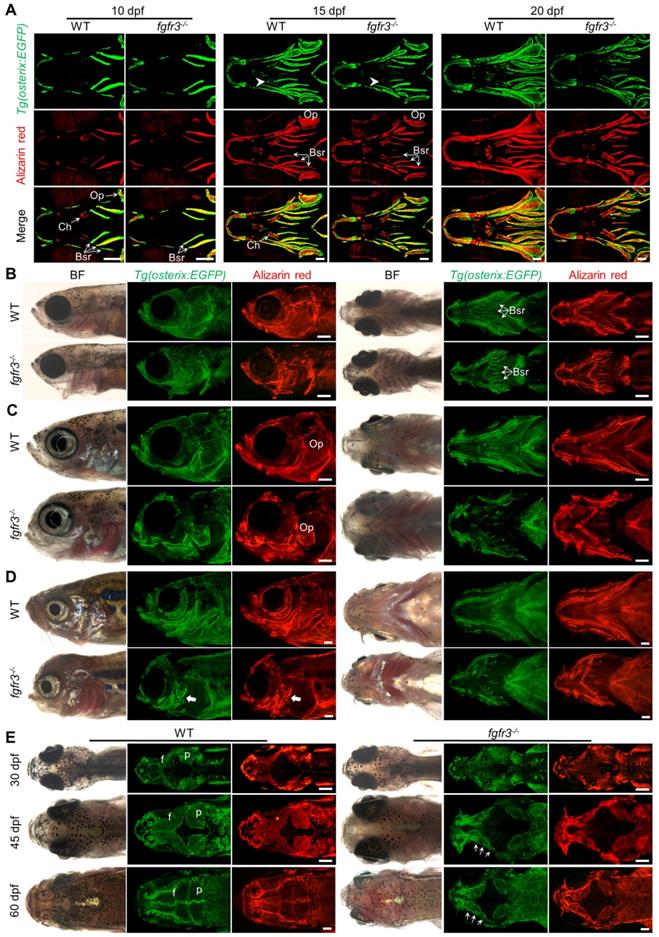
From 1 month (SL 10.0 mm), 1.5 month (SL 14.0 mm) to 2 months (SL 18.0 mm), both intramembranous bone and endochondral bone were gradually developed into a matured skeleton in WT skulls, however, the ossification of most intramembranous bones especially the opercle and branchiostegal rays were severely delayed in fgfr3 mutants (Figure 4B-D and Figure S4B). In the margins of these immature intramembranous bones, there were small scattered bone islands that cannot develop into fully matured bones, indicating that the osteoblastic differentiation and mineralization were severely affected in fgfr3 mutants. Besides, the symmetrical and regular shapes of mandibular arch and hyoid arch were gradually lost and became asymmetrical from 1 month (SL 10.0 mm) to 2 months (SL 18.0 mm) in fgfr3 mutants (Figure 4B-D). Moreover, the gills were still exposed without being covered by the opercle and branchiostegal rays in mutants at 2 months (SL 18.0 mm) due to severe mineralization defect (Figure 4D).
As gain-of-function mutations of FGFR3 lead to craniosynostoses in both ACH patients and mouse model [7,36,37], Alizarin red staining and transgenic line Tg(osterix:EGFP) that labels osteoblasts were employed to image the cranium development in vivo dynamically from 1 month (SL 10.0 mm), 1.5 month (SL 14.0 mm) to 2 months (SL 18.0 mm). The cranial vault development is normally coordinated with that of brain. Parietal and frontal bones are formed through intramembranous ossification within a layer of mesenchyme positioned between the dermal mesenchyme and meninges surrounding the brain in zebrafish [38]. In contrast to the WT, the intramembranous ossification of parietal and frontal bones was severely delayed in fgfr3 mutants, leading to smaller bone that is unable to cover the brain at 2 months (SL 18.0 mm) (Figure 4E and Figure S4C). Both the frontal suture and coronal suture were not closed even at adult stage in fgfr3 mutants. Besides, compared to the single whole bone in WT, the parietal and frontal bones in the mutants were consisted of several small and separately located bones that were unable to develop into fully matured single bone (Figure 3G, 4E, Figure S1A and Figure S4C), indicating that the osteoblastic differentiation and mineralization of cranial vault was severely blocked in fgfr3 mutants.
Fgfr3 knockout inhibits scale formation
Zebrafish scales are part of the exoskeleton; their formation is needed for proper bone mineralization. Fgf signaling has been implicated in zebrafish scale development: fgfr1a and fgf20a mutations affect scale number and size in adult zebrafish [39-41]. In addition to study the effect of fgfr3 deficiency on craniofacial skeleton bone ossification, we also investigated the scale formation in fgfr3 mutants. Using Tg(osterix:EGFP) transgenic line, we found that there were four rows of scales formed in WT at 35 dpf (SL 12.0 mm), but fgfr3 mutants with similar body length only had one row of scales that just began to be formed (Figure S5A). At 50 dpf (SL 16.0 mm), the scales in WT exhibited shapes with regular smooth arc, but the mutant scales were smaller with irregular shapes. Besides, using live Alizarin red staining, we found that the mineralization of scales was reduced in fgfr3 mutants (Figure S5B, C). These data indicate that fgfr3 regulates the mineralization of scales, which is necessary for the scale formation.
Fgfr3 deficiency disrupts the patterning and shaping of pharyngeal arches
It has been reported that both CATSHL syndrome patients and Fgfr3 deficient mice exhibit skeletal overgrowth due to enhanced proliferation of growth plate chondrocytes [1-3, 11, 12]. We used transgenic line Tg(col2a1a:EGFP) that labels chondrocytes to examine the early cartilage development in fgfr3 mutants from 5 dpf (SL 4.0 mm) to 20 dpf (SL 7.5 mm). Confocal microscopy revealed that the morphology and arrangement of mandibular chondrocytes were similar to that in the WT at 5 dpf (SL 4.0 mm) (Figure S2C) and 10 dpf (SL 5.0 mm) (Figure 5A), but the perichondral ossification of ceratohyal cartilage was delayed in fgfr3 mutants at 10 dpf (SL 5.0 mm) (Figure 5A). At 20 dpf (SL 7.5 mm), the gross features of pharyngeal arches still had no significant change, but the arrangement of ceratohyal and basihyal chondrocytes was gradually disordered in fgfr3 mutants, with variable cellular sizes and polarity in contrast to the regular size and arrangement of chondrocytes in WT (Figure 5B). In addition, the perichondral ossification of ceratohyal cartilage was delayed severely (Figure 5B). At 30 dpf (SL 10.0 mm), the fgfr3 mutants can be grossly distinguishable by their abnormally drooped hyoid arches (Figure 5C). Confocal microscopy revealed that the endochondral ossification of ceratohyal and palatoquadrate cartilage was delayed with disorganized chondrocyte arrangement especially in the ceratohyal cartilage of fgfr3 mutants (Figure 7C, D). In addition, the morphology of mutant pharyngeal arches including the mandibular joint was abnormal with loss of symmetry (Figure 5C). At 3 months (SL 26 mm), the endochondral bones were developed into a mature skeleton in WT, however, in fgfr3 mutants there were still some unossified cartilage such as ceratohyal and basihyal cartilage (Figure 5D). These data indicate that fgfr3 is required for the patterning and shaping of pharyngeal arches.
Fgfr3 regulates the patterning and shaping of pharyngeal arche. (A-B) Confocal imaging of WT and fgfr3 mutants in Tg(col2a1a:EGFP) background live stained with Alizarin red at 10 dpf (SL 5.0 mm) (A) and 20 dpf (SL 7.5 mm) (B). From left to right are 3D view of Tg(col2a1a:EGFP) image, Alizarin red staining image, merged 3D view image and merged single layer image. The right panel are the quantification of relative mineralization intensity of ceratohyal perichondrium for WT and fgfr3 mutants. White arrowheads indicate that perichondral ossification of ceratohyal cartilage was delayed in fgfr3 mutants. White arrows indicate disarrangement of chondrocytes of ceratohyal cartilage and basihyal cartilage in fgfr3 mutants in contrast to WT. n = 10, **p < 0.01. (C) Stereo fluorescence microscope imaging of WT and fgfr3 mutants at 30 dpf (SL 10.0 mm) in lateral view (left) and ventral view (right) after live stained with Alizarin red in Tg(col2a1a:EGFP). (D) Stereo fluorescence microscope imaging of WT and fgfr3 mutants at about 3 months (SL 26.0 mm) in lateral view (left) and ventral view (right) in Tg(col2a1a:EGFP). Ch: ceratohyal cartilage; Bh: basihyal cartilage. Scale bars: 50 µm in A and B, 400 µm in C and D.
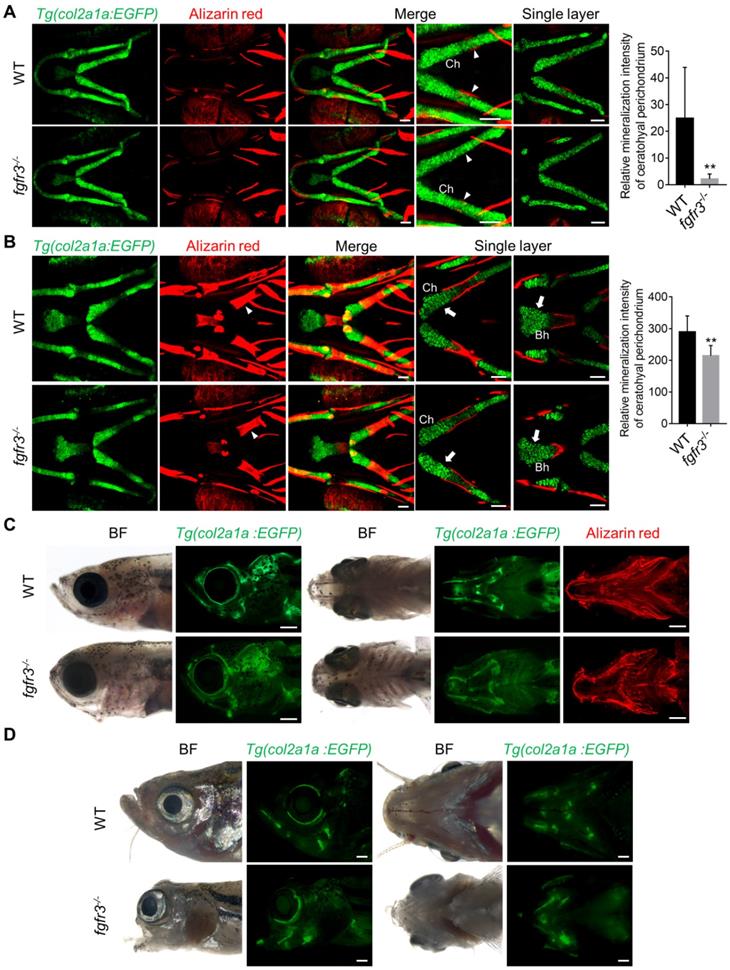
Fgfr3 mutation leads to abnormal chondrocyte hypertrophy and arrangement. (A) Images show the dissected Meckel's cartilage of WT and fgfr3 mutants at 2 months (SL 18.0 mm) in Tg(col2a1a:EGFP) background. The top panel is the stereo fluorescence microscope image, the middle and bottom panel are the confocal images. Red arrows indicate the uniform arrangement of chondrocytes in WT and the white arrows indicate abnormal hypertrophy and disorganized chondrocyte orientation in fgfr3 mutants. (B) Images show the dissected basihyal bone of fgfr3 mutants at 2 months (SL 18.0 mm) in Tg(col2a1a:EGFP) background. The left panel shows the stereo fluorescence microscope image, the right panel shows the confocal images. White arrows indicate the abnormally enlarged chondrocytes and the nuclei in basihyal cartilage of fgfr3 mutants. (C-D) Confocal imaging of WT and fgfr3 mutants in Tg(col2a1a:EGFP) background live stained with Alizarin red at 30 dpf (SL 10.0 mm) in lateral view (C) and ventral view (D). Boxed regions in the top bright field images are magnified in the bottom 3D confocal image. The right panel show the single layer image. White arrows indicate abnormal hypertrophy and disarrangement of ceratohyal and palatoquadrate chondrocytes in fgfr3 mutants. (E) Alcian blue staining (left) and Safranin O-Fast Green staining (right) of epiphysial growth plate of ceratohyal cartilage in WT and fgfr3 mutants at 1.5 month (SL 14.0 mm). The right panel are the quantification of the proportion of various sized chondrocytes in epiphysial growth plates of ceratohyal cartilage. n = 5, ***p<0.001, no significance (ns). (F) Alizarin red and Alcian blue whole skeleton staining of dissected ceratohyal bone of WT and fgfr3 mutants at 1.5 months (SL 14.0 mm). Boxed regions are magnified in the right panel. Brackets indicate that the chondrocytes in growth plate were ordered in WT and disorganized in fgfr3 mutants. (G) Picric-sirius red staining for epiphysial growth plate of WT and fgfr3 mutants, the left panel show the image from ordinary light microscope and the right panel show the image from polarized light microscopy. Scale bars: 100 µm in A, C and D, 20 µm in B, 50 µm in E-G.
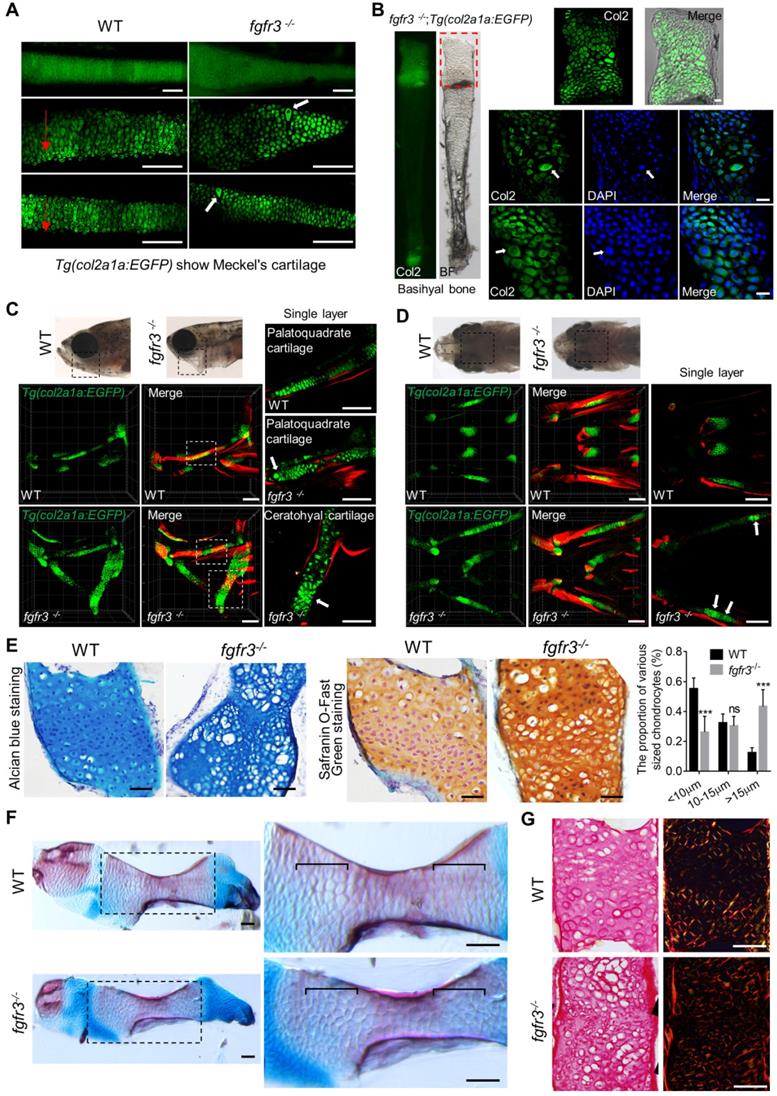
We also used Alizarin red and Alcian blue whole skeleton staining to further examine the development of craniofacial and axial skeleton in fgfr3 mutants at 20 dpf (SL 7.5 mm) and 30 dpf (SL 10.0 mm) (Figure 6A, C). We found the similar skeleton changes in mutants as those observed using in vivo imaging. These data revealed that both the intramembranous ossification and endochondral ossification were inhibited in fgfr3 mutants. Among them, the ossification of intramembranous bone in opercle, branchiostegal rays and dentary were strongly inhibited, while the endochondral ossification of ceratohyal, mentomeckelian, retroarticular, basihyal, hypobranchial, palatoquadrate and ceratobranchial bone were all delayed with abnormal morphology of mandibular arch and hyoid arch (Figure 6A, C). Interestingly, we found that the length of ceratohyal cartilage was increased in fgfr3 mutants in contrast to WT at 20 dpf (SL 7.5 mm) (Figure 6B, D). The increased length of ceratohyal cartilage in mutant zebrafish is consistent with the increased long bone length in CATSHL syndrome patients and Fgfr3-deficient mice.
The phenotype of fgfr3 mutant zebrafish detected by Alizarin red and Alcian blue whole skeleton staining. (A, C) Alizarin red and Alcian blue whole skeleton staining of the WT and fgfr3 mutant at 20 dpf (SL 7.5 mm) (A) and 30 dpf (SL 10.0 mm) (C). Left two panels show the lateral view and dorsal view of the craniofacial bone. Right two panels show the dissected pharyngeal arches and the magnified ceratohyal bone. (B, D) Quantification of ceratohyal cartilage length for WT and fgfr3 mutants at 20 dpf (SL 7.5 mm) (B) and 30 dpf (SL 10.0 mm) (D). n = 6, *p<0.05, no significance (ns). (E) Lateral view (left) and ventral view (right) of the Weberian apparatus of the WT and fgfr3 mutant at 30 dpf (SL 10.0 mm). Ch: ceratohyal cartilage. Scale bar, 200 µm in A, C and E.
Fgfr3 mutation leads to abnormal hypertrophy and disarrangement of chondrocytes
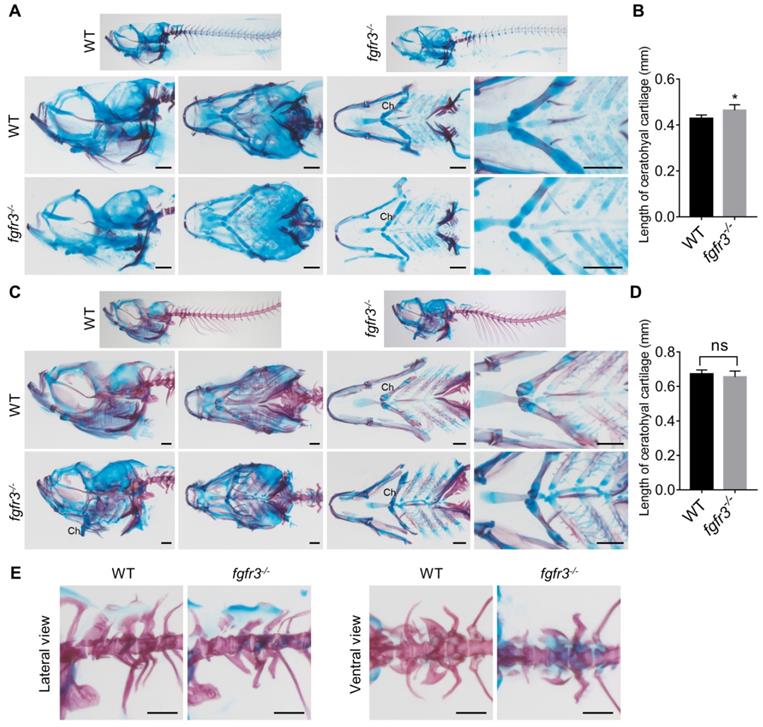
To clarify the underlying mechanisms for the morphological abnormality of craniofacial skeleton in fgfr3 mutants, we dissected and examined multiple craniofacial bones. We found that there was irregular chondrocyte orientation with resultant disorganized chondrocyte column in Meckel's and basihyal cartilage in fgfr3 mutants, and some chondrocytes in these cartilage were abnormally hypertrophied (Figure 7A, B). In particular, the chondrocytes and their nuclei had variable sizes and shapes, some nuclei were abnormally enlarged in basihyal cartilage of fgfr3 mutants (Figure 7B). Abnormal enlarged hypertrophic chondrocytes were also found in the palatoquadrate cartilage of 1-month mutants (SL 10.0 mm) (Figure 7C, D). Alcian blue and Safranin O-Fast Green staining revealed orderly arranged reserve, proliferation and hypertrophic chondrocyte zone in WT ceratohyal cartilage, which was completely disrupted in mutant cartilage with variably sized and shaped chondrocytes (Figure 7E). We also found that the proportion of chondrocytes with abnormally enlarged volume and hypertrophy was increased significantly in fgfr3 mutants (Figure 7E). The disorganized epiphysial growth plates and chondrocytes were also confirmed by Alizarin red and Alcian blue whole skeleton staining in mutant zebrafish (Figure 7F).
To determine whether the organization of collagen fibers in cartilage was related with the disrupted chondrocyte arrangement in fgfr3 mutant cartilage, we did Picric-sirius red staining for epiphysial growth plate and utilized polarized light microscopy, a common method to study collagen networks in tissues [42]. We found that the chondrocytes in WT were well defined with symmetric and well-organized sharp fibers, however, the collagen fibers lost its symmetry with disrupted organization in fgfr3 mutant chondrocytes (Figure 7G). The results suggest that fgfr3 mutation affected the organization of collagen fibers in the cartilage matrix.
Toydemir et al. reported that there are osteochondroma in the long bones of several members of a family with CATSHL syndrome, and our previous study has found that loss of Fgfr3 function leads to the formation of multiple chondroma-like lesions in mice [43]. We performed histological analyses on the epiphysial growth plate of ceratohyal cartilage at adult stage. Consistently, chondroma-like lesions such as cartilaginous lobules with irregular architecture and cellular pleomorphism composed of many small chondrocytes and large hypertrophic chondrocytes were also found in some fgfr3 mutant zebrafish (Figure S6A). We did histological analyses of the joint between the precaudal vertebrae and the ribs, which consist of cartilaginous element and are formed through endochondral ossification. In contrast to the WT, some fgfr3 mutants also showed chondroma-like lesions in the rib attached to the precaudal vertebrae (Figure S6B). Briefly, our results suggest that Fgfr3 similarly regulates chondrocyte differentiation and growth plate development in zebrafish as it does in mice and humans.
Fgfr3 knockout leads to abnormal development of swim bladder and auditory sensory organs
Swim bladder occupies the same position as the lungs of higher vertebrates and is regarded as homologous to the lungs of mammal [44]. It has been reported that FGFR3 and FGFR4 function cooperatively to promote the formation of alveoli during lung development in murine [45,46]. Interestingly, we found that the dysregulated development of swim bladder in fgfr3 mutants. The swim bladder in the WT consist of an anterior and a posterior sac-like chambers, but in fgfr3 mutants, there was only single round chamber, or even no chamber at all (Figure 3C, E). These data suggest that fgfr3 deficiency may affect the formation of swim bladder in zebrafish.
A hallmark phenotype of CATSHL syndrome patients is progressive sensorineural hearing loss [1-3]. Fgfr3 knockout mice also have sensorineural deafness [11]. We detected whether fgfr3 mutant zebrafish also have abnormalities in auditory sensory organs. The auditory sensory organ of zebrafish consists of the inner ear, Weberian apparatus and lateral lines [47]. The gross morphology and size of otoliths in the inner ear of fgfr3 mutants were normal as the WT at 5 dpf (SL 4.0 mm) and 10 dpf (SL 5.0 mm) (Figure S2A and S6A). Using transgenic line Tg(col2a1a:EGFP) that labels the otic vesicle [28], we found that the early development of the otic vesicle including its size and morphology appeared to be normal in fgfr3 mutants at 10 dpf (SL 5.0 mm) (Figure S7A-B). However, the Weberian apparatus, which include scaphium, claustrum, intercalarium, tripus bones and other elements, were maldeveloped in fgfr3 mutants. The ossification of these bones was delayed with abnormal morphology from 20 dpf (SL 7.5 mm) to 30 dpf (SL 10.0 mm) (Figure 6E). Besides, at adult stage, the Weberian apparatus still had abnormal morphology in contrast to the WT as evaluated by micro-CT, Alizarin red and Alcian blue whole skeleton staining (Figure 3G, Figure S1A). The lengths of both tripus and suspensorium bone were reduced in fgfr3 mutants. Furthermore, the anterior chamber of swim bladder was tightly attached to the Weberian apparatus in the WT, but not in the fgfr3 mutants (Figure 3E). The Weberian apparatus transfers acoustic signals from the swim bladders to the inner ear that will increase the auditory sensitivity at high frequencies [47]. The maldevelopment of Weberian apparatus and swim bladder may be involved in the reduced hearing ability. Above results suggest that the deafness in CATSHL syndrome patients may be related to the abnormal development of auditory sensory organs.
Fgfr3 mutation inhibits osteoblast proliferation, promotes chondrocyte proliferation and enhances IHH signaling
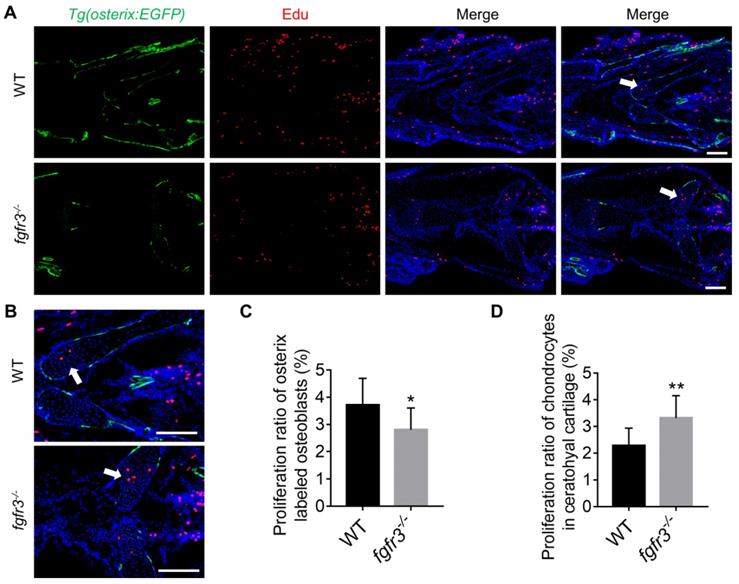
Since intramembranous ossification and endochondral ossification were both inhibited in fgfr3 mutants, we investigated the effects of fgfr3 mutation on cell proliferation using Edu incorporation assay in fgfr3 mutants of Tg(osterix:EGFP) transgenic background (Figure 8A). We found that the Edu labeled Osterix positive cells in the head region was decreased in 1-month-old (SL 10.0 mm) fgfr3 mutants (Figure 8C), while the number of Edu labeled chondrocytes in the growth plate of ceratohyal cartilage was increased in fgfr3 mutants in contrast to WT (Figure 8B, D). These data suggest that fgfr3 mutation in zebrafish promotes chondrocyte proliferation but inhibits osteoblast proliferation.
Fgfr3 mutation promotes chondrocyte proliferation and inhibits osteoblast proliferation. (A) Edu incorporation assay in WT and fgfr3 mutants of Tg(osterix:EGFP) background at 1 month (SL 10.0 mm), (B) is the higher magnification of ceratohyal cartilage. White arrows indicate the increased proliferation of chondrocytes in ceratohyal cartilage of fgfr3 mutants. (C) Quantification of the proliferation ratio of osterix labeled osteoblasts in (A), n = 10, *p < 0.05. (D) Quantification of the proliferation ratio of chondrocytes in ceratohyal cartilage in (A), n = 10, **p < 0.01. Scale bars: 100 µm in A.
IHH and PTHrP signaling are important regulators of the cartilage development [48]. We and others have shown that constitutively activated FGFR3 results in downregulation of Ihh mRNA in growth plate chondrocytes [10, 49, 50]. Moreover, our previous study in mice showed that FGFR3 deficiency causes multiple chondroma-like lesions by upregulating hedgehog signaling [43]. Therefore, we examined whether IHH signaling is perturbed after fgfr3 mutation in zebrafish. Western blot using the extracts from the head skeleton at 20 dpf (SL 7.5 mm) and 40 dpf (SL 13.0 mm) showed that Ihh protein was significantly increased in fgfr3 mutants (Figure 9A). These results suggest that the enhanced Ihh signaling may be associated with the abnormal development of pharyngeal cartilage in fgfr3 mutants.
Fgfr3 mutation upregulates canonical Wnt/β-catenin signaling and Wnt inhibition partially alleviates the phenotype of fgfr3 mutants. (A) Western blot detecting the protein levels of phosphorylated-β-catenin, non-phosphorylated β-catenin (activated β-catenin) and IHH in WT and mutant zebrafish at 20 dpf (SL 7.5 mm) and 40 dpf (SL 13.0 mm), β-actin was used as loading control. Quantitative analyses of the relative expressions of IHH and non-phosphorylated β-catenin are show in the right panel. n = 3 independent experiments, **p < 0.01, *p < 0.05. (B) Expression level of axin2 examined by in situ hybridization at 7 dpf. Black arrows indicate pharyngeal cartilage. (C) RT-qPCR analysis of the expression level of axin2 at 20 dpf (SL 7.5 mm). n = 3 independent experiments, *p < 0.05. (D-F) WT and fgfr3 mutants treated with 2.5 µM XAV939 (right) or DMSO (left) from 10 dpf (SL 5.0 mm) to 20 dpf (SL 7.5 mm). (D) show the lateral view of head region detected by light microscopy at 30 dpf (SL 10.0 mm) (top) and 40 dpf (SL 13.0 mm) (bottom). Arrows indicate that the mandibular deformity with hyoid arch drooping toward the ventral side was partially rescued in XAV939-treated mutants. (E) show the lateral view (top) and ventral view (bottom) of craniofacial bone at 40 dpf (SL 13.0 mm) with living Alizarin red staining. (F) is the confocal images showing the ceratohyal cartilage (Ch) at 30 dpf (SL 10.0 mm) of WT and fgfr3 mutants in Tg (col2a1a:EGFP) transgenic background. Boxed regions in the 3D confocal image are showed the single layer in the bottom. Scale bar, 100 µm in C, 400 µm in D and E, 50 µm in F.
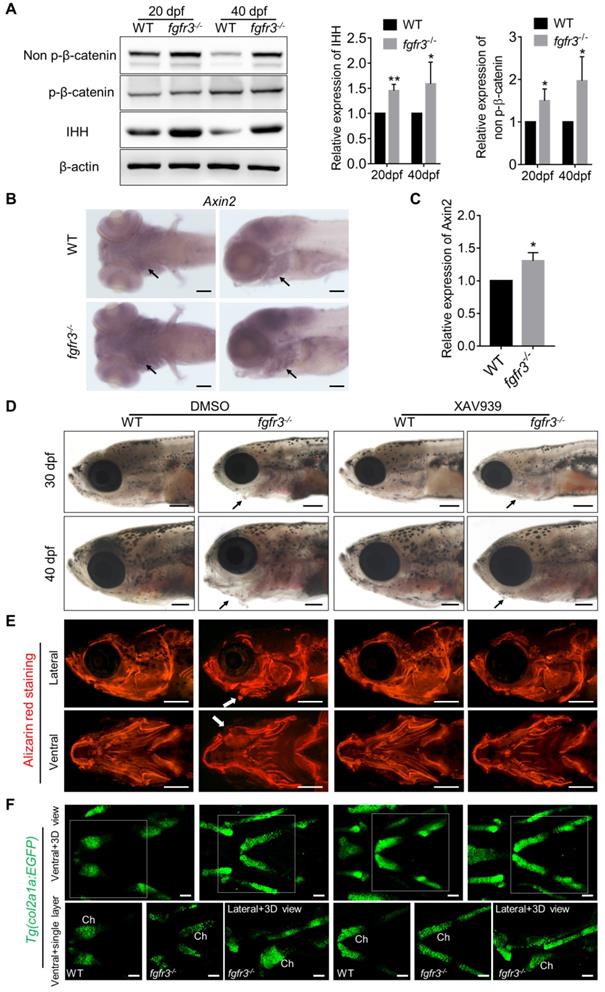
Fgfr3 mutation upregulates canonical Wnt/β-catenin signaling and Wnt inhibition partially alleviates the phenotypes of fgfr3 mutants
We next explored for the possible signaling pathway involved in skeletal maldevelopment of fgfr3 mutants. Recent study has showed the functional link between Hh signaling and Wnt/β-Catenin signaling in skeletal growth and cartilage/bone tumor formation [51]. Shung et al. found that gain-of-function mutations of FGFR3K650E led to down-regulated of β-catenin level and activity in growth plate chondrocytes [52]. Thus, we examined the levels of accumulated β-catenin protein in mutant and WT cartilage. Western blot using the extracts from the head skeleton at 20 dpf (SL 7.5 mm) and 40 dpf (SL 13.0 mm) showed that the level of non-phosphorylated β-catenin (activated-β-catenin) was remarkably up-regulated in fgfr3 mutants, while phosphorylated-β-catenin had no significant change (Figure 9A). Furthermore, in situ hybridization showed that the level of Wnt/β‐catenin target gene axin2 was increased in the pharyngeal cartilage of fgfr3 mutants (Figure 9B). Consistently, RT‐qPCR analysis showed that the expression level of axin2 was increased in head skeleton of fgfr3 mutants at 20 dpf (SL 7.5 mm) (Figure 9C). These results suggest that Wnt/β-catenin signaling was up-regulated in fgfr3 mutants.
We then asked whether pharmacological inhibition of Wnt/β-catenin signaling could rescue the craniofacial bone malformation caused by fgfr3 mutation. We used a β-catenin inhibitor XAV939, which stimulates β-catenin degradation by stabilizing axin, one suppressor of β-catenin destruction complex [53,54]. 2.5 µM XAV939 or DMSO control were used from 10 dpf (SL 5.0 mm) to 20 dpf (SL 7.5 mm). We analyzed their effect at 30 dpf (SL 10.0 mm) and 40 dpf (SL 13.0 mm), and found that the abnormal drooping of hyoid arch in fgfr3 mutants was partially alleviated by XAV939 (Figure 9D and Figure S8A), the domed-shaped skulls and microcephaly also were partially alleviated (Figure 9D). Alizarin red staining showed that the asymmetrical morphology of pharyngeal arches in fgfr3 mutants was partially corrected after XAV939 treatment (Figure 9E), but the delayed intramembranous ossification of opercle and branchiostegal rays was not significantly changed in XAV939-treated mutants (Figure 9E). We detected the distance between Meckel's and ceratohyal bone (M-Ch distance) and the angle between vertical and horizontal axis (V-H angle) of skulls after XAV939 treatment, and showed that XAV939 treatment partially rescued the mandibular asymmetry in fgfr3 mutants (Figure S8B, C). We then used transgenic line Tg(col2a1a:EGFP) to detect the cartilage development, and revealed that XAV939-treated mutants had their chondrogenic dysplasia especially the dysplasia of ceratohyal cartilage partially alleviated. The abnormal arrangement and hypertrophy of chondrocytes in ceratohyal cartilage were both alleviated in XAV939-treated mutants (Figure 9F). These results suggest that skeletal dysplasia especially the chondrodysplasia in fgfr3 mutants is associated with up-regulated Wnt/β-catenin signaling, which could be partially alleviated by inhibiting Wnt/β-catenin signaling.
Discussion
The critical roles of FGFs/FGFR3 in human skeleton development and diseases have been well documented [4]. It's established that FGFR3 negatively regulates the development of skeleton formed through endochondral ossification [6, 8]. However, the underlying mechanism of the negative regulatory role of FGFR3 in endochondral bone development is not fully clarified, for example there is still dispute about the effect of FGFR3 on chondrocyte hypertrophy. Moreover, it is still highly debatable about the roles of FGFR3 in the development of skeleton formed through intramembranous ossification, i.e., skull vaults. For example, both mice with either FGFR3 deficiency or gain-of-function mutations have decreased bone mass and mineralization [10,13,55]. Activated mutations of FGFR3 lead to craniosynostoses with macrocephaly and midfacial hypoplasia in patients and mouse models due to premature closure of cranial sutures and cranial base synchondrosis [36,37,56,57]. While CATSHL syndrome patients are also reported to have microcephaly and wormian skull bones [2,3]. In contrast, the Fgfr3 deficient mice have no apparent phenotype in craniofacial skeleton [12,13]. One important reason for these seemingly controversial phenotypes is that the mouse models and patients are not suitable for dynamically studying the pathological process especially the early maldevelopment of skeleton.
Because of its in vitro and fast development, and transparency, zebrafish is very good for dynamically studying the early development of skeleton [14,16,18]. Moreover, the development of craniofacial skeleton has proven to be highly conserved from zebrafish to humans [22,23]. These facts prompted us to utilize zebrafish to answer those yet to be clarified questions mentioned above.
In this study, we generated a zebrafish model mimicking human CATSHL syndrome by deleting fgfr3 in zebrafish, to study the pathogenesis and potential therapeutic targets for CATSHL syndrome. We found that the fgfr3 mutant zebrafish have severe craniofacial bone malformation, as evidenced by microcephaly with smaller cranial skull bones and delayed closure of cranial sutures, drooped hyoid arch and midface hypoplasia, which to some extent resembles the clinical manifestations of CATSHL syndrome patients [3]. These changes suggest that, compared with mouse CATSHL model, our fgfr3 knockout zebrafish seems to better mimic the craniofacial skeleton phenotypes of CATSHL syndrome.
Using this zebrafish model, we analyzed the early development process and clarified the underlying mechanisms of craniofacial bone malformation in fgfr3 mutants. We first dynamically examined bone mineralization and osteoblast differentiation using transgenic fish labeling osteoblasts and live Alizarin red staining from 5 dpf (SL 4.0 mm) to 2 months (SL 18.0 mm). We found that the osteoblastic differentiation and mineralization is severely affected in fgfr3 mutants. The intramembranous bones including opercle, branchiostegal rays, parietal and frontal bones were consisted of several small and separately located extra bones, which were unable to develop into fully matured single bone in fgfr3 mutants. The proliferation of osteoblasts and expressions of genes related to osteoblast differentiation were down-regulated, which may be responsible for the severely delayed ossification of intramembranous bones in fgfr3 mutants. In particular, the skull vault in fgfr3 mutants consisted of multiple, irregular cranial bones, which is very similar to that of osterix/sp7 mutants with extra sutures and irregular skull bones [58], suggesting that the skull malformations in fgfr3 mutants may be related to the delayed osteoblast differentiation and bone maturation due to down-regulated osterix expression. Besides, the delayed scale formation in fgfr3 mutants also confirms that fgfr3 mutation inhibits osteoblast differentiation and bone mineralization in zebrafish.
In addition to the role of osteogenesis, the abnormal chondrogenesis of jaw bone and skull base cartilage is also involved in the craniofacial malformation [34]. We then dynamically examined the early development of cartilage from 5 dpf (SL 4.0 mm) to 1 month (SL 10.0 mm) using Tg(col2a1a:EGFP) that label chondrocytes. We found that the ordered arrangement of chondrocytes was gradually disrupted with some abnormally hypertrophied chondrocytes in fgfr3 mutants. The chondrocyte zone of ceratohyal cartilage growth plate was completely disrupted with chondrocytes of variable sizes and shapes. We suspect that the gradually maldeveloped cartilage structure may lead to mechanical changes of jaw cartilage, resulting in drooped hyoid arch and malformed jaw bone, which may also be related to the high arched palate in CATSHL syndrome patients [3]. Besides, we also found delayed synchondrosis closure and irregular morphology in the cranial base of fgfr3 mutants. In brief, we revealed disrupted chondrogenesis in the mutants, which, together with the dysregulated bone ossification of skull bones, results in the craniofacial skeleton malformation in fgfr3 mutant zebrafish (Figure 10).
Schematic diagram of the mechanisms underlying the role of fgfr3 in zebrafish skeleton development. Deletion of Fgfr3 in zebrafish results in enhanced IHH signaling and up-regulated canonical Wnt/β-catenin signaling that may lead to increased chondrocyte proliferation, abnormal hypertrophy and disordered arrangement of chondrocytes in growth plates. Fgfr3 mutation leads to decreased proliferation and differentiation of osteoblasts and decreased mineralization in skull bone. A combination of above mechanisms may lead to disrupted chondrogenesis and bone ossification resulting in craniofacial skeleton malformation in fgfr3 mutant zebrafish.
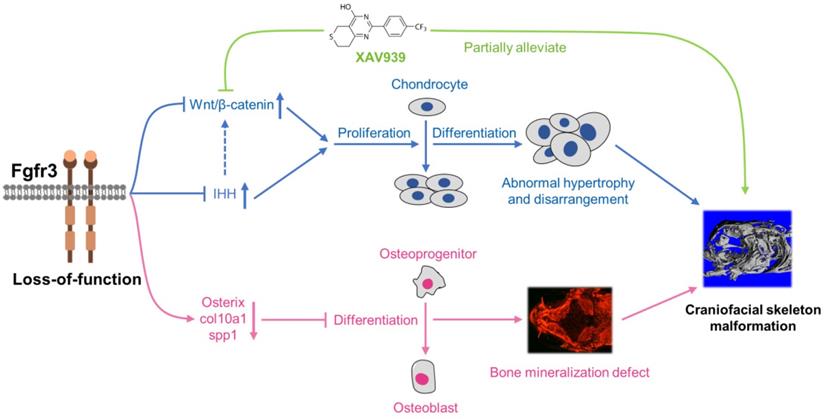
We explored the possible signaling pathway involved in the skeletal maldevelopment of fgfr3 mutants. FGFR3 has close crosstalk with several signaling pathways that play critical roles in regulating skeleton development, including IHH, PTHLH/PTHrP, BMP and IGF1 [4,59,60]. Firstly, we found that IHH signaling was increased in mutant zebrafish. As recent study showed that Wnt/β-catenin signaling acts downstream of Hh signaling in skeletal growth and tumor formation [51], we thus speculated that fgfr3 deletion may also exert its effect on skeleton development through modulating Wnt/β-catenin signaling. Indeed, we found that canonical Wnt/β‐catenin signaling was up-regulated in fgfr3 mutants, and pharmacological inhibition of Wnt/β‐catenin could partially correct the phenotype of craniofacial malformation in fgfr3 mutants including the abnormal arrangement of chondrocytes, although the delayed intramembranous bone ossification was not significantly rescued. These data indicate that up-regulated Wnt/β-catenin signaling in fgfr3 mutants may be mainly involved in the chondrodysplasia phenotype, while the disrupted bone ossification may be result from other mechanism. We found that XAV939 treatment also partially rescue the domed-shaped skulls and microcephaly of fgfr3 mutants, we speculated that this is via improving the chondrogenic phenotype such as in jaw cartilage and cranial base cartilage, which may lead to improvement of cranial skeletal asymmetry and abnormal mechanics in fgfr3 mutants. In addition, the rescue of drooped hyoid arch could improve the malformed mouth, which may also improve the overall skull phenotype by improving food intake in fgfr3 mutants.
So far, the crosstalk between FGF/FGFR3 pathways and Wnt/β-catenin signaling in chondrogenesis is not fully understood. Shung et al. found that gain-of-function mutation of FGFR3K650E led to down-regulation of β-catenin levels and activity in growth plate chondrocytes, as well as decreased β-catenin levels and transcriptional activity in cultured mesenchymal cells [52]. In contrast, some studies in cultured chondrocytes showed that FGF signaling activates Wnt/β-catenin signaling through MAP kinase-mediated phosphorylation of LRP6 [61, 62]. Since there are no studies dissecting the role of Wnt signaling in the skeleton maldevelopment resulting from FGFR3 deletion, our results for the first time reveal that fgfr3 deficiency upregulates the Wnt/β-catenin pathway, which may be involved in the pathogenesis of the abnormal skeleton phenotype in CATSHL syndrome. However, the mechanism through which fgfr3 mutation upregulates Wnt/β‐catenin signaling remains elusive, whether Fgfr3 negatively modulates Wnt pathway in human and mouse models requires additional studies.
The dwarfism phenotypes in ACH patients and mouse models, as well as the skeletal overgrowth with tall stature in CATSHL syndrome patients and Fgfr3 deficient mice strongly demonstrate that FGFR3 is a negative regulator of development of endochondral bone [4,8]. However, the body size of fgfr3 mutant fish was smaller at adult stage, and some fgfr3 mutants even died before adult stage. These may be due to the eating disorders resulting from craniofacial bone maldevelopment especially the jaw bone malformation. In addition, FGFR3 mainly regulates growth plate development of the long bones and vertebral bodies, while the zebrafish have no limbs and long bones, and the formation of their vertebral bodies is not through endochondral ossification as zebrafish vertebral body has no growth plate. All these differences between zebrafish and mammals may be responsible for the absence and presence of longer body length in fgfr3 mutant zebrafish and mice/humans. However, it is important that some craniofacial bones in zebrafish such as the ceratohyal bones have growth plates and are formed through endochondral ossification like humans [20]. Fgfr3 expression was also detected in the proliferative and hypertrophic chondrocytes of growth plates of these zebrafish bones [27]. Consistently, we found that the length of ceratohyal bone in fgfr3 mutants was significantly increased with enhanced proliferation of its growth plate chondrocytes in contrast to WT at 20 dpf (SL 7.5 mm). Our data suggest that the development of cartilage is conserved between zebrafish and mammals, and our fgfr3 deficient zebrafish model can be used to study the role of FGFR3 in chondrogenesis and screen therapeutic molecules for FGFR3 related genetic diseases including CATSHL and ACH by taking the advantages of zebrafish.
Similar to the variation in the phenotype severity in CATSHL syndrome patients [3], fgfr3 mutant zebrafish also exhibited varying degrees of craniofacial bone malformations. The reasons for the variable phenotypes in mutant zebrafish are not clear presently. We think one important reason is the secondary effects of malformed mouth, which will strongly affect the food intaking. Indeed, we noticed that the body length of WT and fgfr3 mutant had no significant change at 1 month (SL 10.0 mm), hereafter, the cranial deformity and phenotypic differences gradually appeared apparently.
The zebrafish is recognized as an excellent model system for the study of the development of the vertebrate inner ear and its functions of balance and hearing. Many of the molecular mechanisms of inner ear development, endolymph generation, and hair cell development and function in zebrafish are conserved with those in the mammals [47]. As hearing loss is a hallmark features of CATSHL syndrome patients, although we found that the gross morphology and size of otoliths and otic vesicle appeared to be normal in fgfr3 mutants, the Weberian apparatus and swim bladder were abnormally maldeveloped. These malformations may lead to reduced hearing function in fgfr3 mutant zebrafish. Besides, the larval zebrafish jaw is evolutionarily homologous to the mammalian middle ear ossicles [63]. The disrupted patterning and shaping of pharyngeal arches in fgfr3 mutants indicate that FGFR3 may regulate the development of mammalian middle ear ossicles. In Fgfr3 knockout mice the inner ear defects include failure of pillar cell differentiation and tunnel of Corti formation that result in profound deafness [11]. However, whether the hair cell have developmental defect in the inner ear of fgfr3 mutant zebrafish needs more studies.
In conclusion, we generated a zebrafish model for CATSHL syndrome that partially resembles the clinical manifestations especially the craniofacial malformation of patients with CATSHL syndrome. Using this zebrafish model, we found that fgfr3 deficiency upregulates canonical Wnt/β‐catenin signaling, and inhibition of Wnt/β‐catenin signaling could partially alleviate the phenotypes of fgfr3 mutants. Our zebrafish model for CATSHL syndrome will help to better understand the underlying pathogenetic mechanisms of FGFR3 related human genetic syndromes including CATSHL and ACH, and explore their possible therapies.
Abbreviations
ACH: achondroplasia; CATSHL syndrome: camptodactyly, tall stature and hearing loss syndrome; dpf: days post fertilization; FGFR3: fibroblast growth factor receptors 3; hpf: hours post fertilization; SL: standard length; WT: wild-type.
Supplementary Material
Supplementary figures.
Acknowledgements
We thank Chung-Der Hsiao for the fish lines Tg(osterix:EGFP). We thank all the members of Drs. Chen's and Luo's laboratory for technical support and fruitful discussions. This work is supported by research funding from the National Key Research and Development Program of China (2018YFA0800802), the National Natural Science Foundation of China (No. 81530071, 81830075, 81772306, 81721001), the Key Program of Innovation Project of Military Medical Science (16CXZ016) and Innovative Research Team in University (IRT1216).
Authors' Contributions
Authors' roles: Study design: XS, YX, LC and LL. Study conduct: XS, RZ, HC, SC, JH, ML, MX, FL, DZ, SL and BZ. Data collection: XS, RZ and HC. Technical and material support: LL, XD and LC. Data analysis and interpretation: XS, YX, LC, LL, MJ, NS, HQ, JY, ZN and DC. Drafting and revising manuscript: XS, YX, LC, LL, DC and XZ. All authors approved the final version of the manuscript. YX, LC and LL take responsibility for the integrity of the data analysis.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Toydemir RM, Brassington AE, Bayrak-Toydemir P, Krakowiak PA, Jorde LB, Whitby FG. et al. A novel mutation in FGFR3 causes camptodactyly, tall stature, and hearing loss (CATSHL) syndrome. Am J Hum Genet. 2006;79:935-41
2. Makrythanasis P, Temtamy S, Aglan MS, Otaify GA, Hamamy H, Antonarakis SE. A novel homozygous mutation in FGFR3 causes tall stature, severe lateral tibial deviation, scoliosis, hearing impairment, camptodactyly, and arachnodactyly. Hum Mutat. 2014;35:959-63
3. Escobar LF, Tucker M, Bamshad M. A second family with CATSHL syndrome: Confirmatory report of another unique FGFR3 syndrome. Am J Med Genet A. 2016;170:1908-11
4. Ornitz DM, Marie PJ. Fibroblast growth factors in skeletal development. Curr Top Dev Biol. 2019;133:195-234
5. Du X, Xie Y, Xian CJ, Chen L. Role of FGFs/FGFRs in skeletal development and bone regeneration. J Cell Physiol. 2012;227:3731-43
6. Foldynova-Trantirkova S, Wilcox WR, Krejci P. Sixteen years and counting: the current understanding of fibroblast growth factor receptor 3 (FGFR3) signaling in skeletal dysplasias. Hum Mutat. 2012;33:29-41
7. Muenke M, Gripp KW, McDonald-McGinn DM, Gaudenz K, Whitaker LA, Bartlett SP. et al. A unique point mutation in the fibroblast growth factor receptor 3 gene (FGFR3) defines a new craniosynostosis syndrome. Am J Hum Genet. 1997;60:555-64
8. Narayana J, Horton WA. FGFR3 biology and skeletal disease. Connect Tissue Res. 2015;56:427-33
9. Iwata T, Chen L, Li C-l, Ovchinnikov DA, Behringer RR, Francomano CA. et al. A neonatal lethal mutation in FGFR3 uncouples proliferation and differentiation of growth plate chondrocytes in embryos. Hum Mol Genet. 2000;9:1603-13
10. Chen L, Adar R, Yang X, Monsonego EO, Li C, Hauschka PV. et al. Gly369Cys mutation in mouse FGFR3 causes achondroplasia by affecting both chondrogenesis and osteogenesis. J Clin Invest. 1999;104:1517-25
11. Colvin JS, Bohne BA, Harding GW, McEwen DG, Ornitz DM. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat Genet. 1996;12:390-7
12. Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911-21
13. Valverde-Franco G, Liu H, Davidson D, Chai S, Valderrama-Carvajal H, Goltzman D. et al. Defective bone mineralization and osteopenia in young adult FGFR3-/- mice. Hum Mol Genet. 2004;13:271-84
14. Luderman LN, Unlu G, Knapik EW. Zebrafish Developmental Models of Skeletal Diseases. Curr Top Dev Biol. 2017;124:81-124
15. Bergen DJM, Kague E, Hammond CL. Zebrafish as an Emerging Model for Osteoporosis: A Primary Testing Platform for Screening New Osteo-Active Compounds. Front Endocrinol (Lausanne). 2019;10:6
16. Busse B, Galloway JL, Gray RS, Harris MP, Kwon RY. Zebrafish: An Emerging Model for Orthopedic Research. J Orthop Res. 2019
17. van Opbergen CJM, Koopman CD, Kok BJM, Knopfel T, Renninger SL, Orger MB. et al. Optogenetic sensors in the zebrafish heart: a novel in vivo electrophysiological tool to study cardiac arrhythmogenesis. Theranostics. 2018;8:4750-64
18. Hammond CL, Moro E. Using transgenic reporters to visualize bone and cartilage signaling during development in vivo. Front Endocrinol (Lausanne). 2012;3:91
19. Sun X, Zhang R, Liu M, Chen H, Chen L, Luo F. et al. Rmrp mutation disrupts chondrogenesis and bone ossification in zebrafish model of cartilage-hair hypoplasia via enhanced Wnt/beta-catenin signaling. J Bone Miner Res. 2019
20. Weigele J, Franz-Odendaal TA. Functional bone histology of zebrafish reveals two types of endochondral ossification, different types of osteoblast clusters and a new bone type. J Anat. 2016;229:92-103
21. van Karnebeek CD, Bonafe L, Wen XY, Tarailo-Graovac M, Balzano S, Royer-Bertrand B. et al. NANS-mediated synthesis of sialic acid is required for brain and skeletal development. Nat Genet. 2016;48:777-84
22. Mork L, Crump G. Zebrafish Craniofacial Development: A Window into Early Patterning. Curr Top Dev Biol. 2015;115:235-69
23. Y Z, D J, L L, S Y, H Z, X D. ClC-7 Regulates the Pattern and Early Development of Craniofacial Bone and Tooth. Theranostics. 2019;9:1387-400
24. Hall C, Flores MV, Murison G, Crosier K, Crosier P. An essential role for zebrafish Fgfrl1 during gill cartilage development. Mech Dev. 2006;123:925-40
25. Larbuisson A, Dalcq J, Martial JA, Muller M. Fgf receptors Fgfr1a and Fgfr2 control the function of pharyngeal endoderm in late cranial cartilage development. Differentiation. 2013;86:192-206
26. Ota S, Tonou-Fujimori N, Yamasu K. The roles of the FGF signal in zebrafish embryos analyzed using constitutive activation and dominant-negative suppression of different FGF receptors. Mech Dev. 2009;126:1-17
27. Ledwon JK, Turin SY, Gosain AK, Topczewska JM. The expression of fgfr3 in the zebrafish head. Gene Expr Patterns. 2018;29:32-8
28. Dale RM, Topczewski J. Identification of an evolutionarily conserved regulatory element of the zebrafish col2a1a gene. Dev Biol. 2011;357:518-31
29. Westerfield M. The zebrafish book: a guide for the laboratory use of zebrafish (Danio rerio): M. Westerfield. 2007
30. Sun X, Zhou Y, Zhang R, Wang Z, Xu M, Zhang D. et al. Dstyk mutation leads to congenital scoliosis-like vertebral malformations in zebrafish via dysregulated mTORC1/TFEB pathway. Nat Commun. 2020;11:479
31. Chang N, Sun C, Gao L, Zhu D, Xu X, Zhu X. et al. Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res. 2013;23:465-72
32. Su N, Li X, Tang Y, Yang J, Wen X, Guo J. et al. Deletion of FGFR3 in Osteoclast Lineage Cells Results in Increased Bone Mass in Mice by Inhibiting Osteoclastic Bone Resorption. J Bone Miner Res. 2016;31:1676-87
33. Killion CH, Mitchell EH, Duke CG, Serra R. Mechanical loading regulates organization of the actin cytoskeleton and column formation in postnatal growth plate. Mol Biol Cell. 2017;28:1862-70
34. Luo F, Xie Y, Xu W, Huang J, Zhou S, Wang Z. et al. Deformed Skull Morphology Is Caused by the Combined Effects of the Maldevelopment of Calvarias, Cranial Base and Brain in FGFR2-P253R Mice Mimicking Human Apert Syndrome. Int J Biol Sci. 2017;13:32-45
35. Renn J, Buttner A, To TT, Chan SJ, Winkler C. A col10a1:nlGFP transgenic line displays putative osteoblast precursors at the medaka notochordal sheath prior to mineralization. Dev Biol. 2013;381:134-43
36. Di Rocco F, Biosse Duplan M, Heuze Y, Kaci N, Komla-Ebri D, Munnich A. et al. FGFR3 mutation causes abnormal membranous ossification in achondroplasia. Hum Mol Genet. 2014;23:2914-25
37. Nah HD, Koyama E, Agochukwu NB, Bartlett SP, Muenke M. Phenotype profile of a genetic mouse model for Muenke syndrome. Childs Nerv Syst. 2012;28:1483-93
38. Topczewska JM, Shoela RA, Tomaszewski JP, Mirmira RB, Gosain AK. The Morphogenesis of Cranial Sutures in Zebrafish. PLoS One. 2016;11:e0165775
39. Aman AJ, Fulbright AN, Parichy DM. Wnt/beta-catenin regulates an ancient signaling network during zebrafish scale development. Elife. 2018 7
40. Rohner N, Bercsenyi M, Orban L, Kolanczyk ME, Linke D, Brand M. et al. Duplication of fgfr1 permits Fgf signaling to serve as a target for selection during domestication. Curr Biol. 2009;19:1642-7
41. Daane JM, Rohner N, Konstantinidis P, Djuranovic S, Harris MP. Parallelism and Epistasis in Skeletal Evolution Identified through Use of Phylogenomic Mapping Strategies. Mol Biol Evol. 2016;33:162-73
42. Junqueira LC, Bignolas G, Brentani RR. Picrosirius staining plus polarization microscopy, a specific method for collagen detection in tissue sections. Histochem J. 1979;11:447-55
43. Zhou S, Xie Y, Tang J, Huang J, Huang Q, Xu W. et al. FGFR3 Deficiency Causes Multiple Chondroma-like Lesions by Upregulating Hedgehog Signaling. PLoS Genet. 2015;11:e1005214
44. Zhang Y, Liu H, Yao J, Huang Y, Qin S, Sun Z. et al. Manipulating the air-filled zebrafish swim bladder as a neutrophilic inflammation model for acute lung injury. Cell Death Dis. 2016;7:e2470
45. Weinstein M, Xu X, Ohyama K, Deng CX. FGFR-3 and FGFR-4 function cooperatively to direct alveogenesis in the murine lung. Development. 1998;125:3615-23
46. Li R, Herriges JC, Chen L, Mecham RP, Sun X. FGF receptors control alveolar elastogenesis. Development. 2017;144:4563-72
47. Abbas L, Whitfield TT. The zebrafish inner ear. In: Perry SF, Ekker M, Farrell AP, Brauner CJ, editors. Fish Physiology. London: Academic Press. 2010 p. 123-71
48. Long F, Ornitz DM. Development of the endochondral skeleton. Cold Spring Harb Perspect Biol. 2013;5:a008334
49. Chen L, Li C, Qiao W, Xu X, Deng C. A Ser(365)->Cys mutation of fibroblast growth factor receptor 3 in mouse downregulates Ihh/PTHrP signals and causes severe achondroplasia. Hum Mol Genet. 2001;10:457-65
50. Li C, Chen L, Iwata T, Kitagawa M, Fu XY, Deng CX. A Lys644Glu substitution in fibroblast growth factor receptor 3 (FGFR3) causes dwarfism in mice by activation of STATs and ink4 cell cycle inhibitors. Hum Mol Genet. 1999;8:35-44
51. Deng Q, Li P, Che M, Liu J, Biswas S, Ma G. et al. Activation of hedgehog signaling in mesenchymal stem cells induces cartilage and bone tumor formation via Wnt/beta-Catenin. Elife. 2019 8
52. Shung CY, Ota S, Zhou ZQ, Keene DR, Hurlin PJ. Disruption of a Sox9-beta-catenin circuit by mutant Fgfr3 in thanatophoric dysplasia type II. Hum Mol Genet. 2012;21:4628-44
53. Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA. et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461:614-20
54. Hamad S, Derichsweiler D, Papadopoulos S, Nguemo F, Saric T, Sachinidis A. et al. Generation of human induced pluripotent stem cell-derived cardiomyocytes in 2D monolayer and scalable 3D suspension bioreactor cultures with reduced batch-to-batch variations. Theranostics. 2019;9:7222-38
55. Su N, Sun Q, Li C, Lu X, Qi H, Chen S. et al. Gain-of-function mutation in FGFR3 in mice leads to decreased bone mass by affecting both osteoblastogenesis and osteoclastogenesis. Hum Mol Genet. 2010;19:1199-210
56. Matsushita T, Wilcox WR, Chan YY, Kawanami A, Bukulmez H, Balmes G. et al. FGFR3 promotes synchondrosis closure and fusion of ossification centers through the MAPK pathway. Hum Mol Genet. 2009;18:227-40
57. Agochukwu NB, Solomon BD, Doherty ES, Muenke M. Palatal and oral manifestations of Muenke syndrome (FGFR3-related craniosynostosis). J Craniofac Surg. 2012;23:664-8
58. Kague E, Roy P, Asselin G, Hu G, Simonet J, Stanley A. et al. Osterix/Sp7 limits cranial bone initiation sites and is required for formation of sutures. Dev Biol. 2016;413:160-72
59. Wang X, Qi H, Wang Q, Zhu Y, Wang X, Jin M. et al. FGFR3/fibroblast growth factor receptor 3 inhibits autophagy through decreasing the ATG12-ATG5 conjugate, leading to the delay of cartilage development in achondroplasia. Autophagy. 2015;11:1998-2013
60. Jin M, Du X, Chen L. Cross-talk between FGF and other cytokine signalling pathways during endochondral bone development. Cell Biol Int. 2012;36:691-6
61. Krejci P, Aklian A, Kaucka M, Sevcikova E, Prochazkova J, Masek JK. et al. Receptor tyrosine kinases activate canonical WNT/beta-catenin signaling via MAP kinase/LRP6 pathway and direct beta-catenin phosphorylation. PLoS One. 2012;7:e35826
62. Buchtova M, Oralova V, Aklian A, Masek J, Vesela I, Ouyang Z. et al. Fibroblast growth factor and canonical WNT/beta-catenin signaling cooperate in suppression of chondrocyte differentiation in experimental models of FGFR signaling in cartilage. Biochim Biophys Acta. 2015;1852:839-50
63. Anthwal N, Joshi L, Tucker AS. Evolution of the mammalian middle ear and jaw: adaptations and novel structures. J Anat. 2013;222:147-60
Author contact
![]() Corresponding authors: Lingfei Luo. School of Life Sciences, Southwest University, 2 Tian-sheng Road, Beibei, Chongqing 400715, China. Tel: +86-23-68367957, E-mail address: lluoedu.cn; Lin Chen. Department of Wound Repair and Rehabilitation Medicine, Daping Hospital, Army Medical University, Chongqing 400042 China. Tel: +86-23-68757041, E-mail address: linchen70com; Yangli Xie: Department of Wound Repair and Rehabilitation Medicine, Daping Hospital, Army Medical University, Chongqing 400042 China. Tel: +86-23-68757040, E-mail address: xieyangli841015com.
Corresponding authors: Lingfei Luo. School of Life Sciences, Southwest University, 2 Tian-sheng Road, Beibei, Chongqing 400715, China. Tel: +86-23-68367957, E-mail address: lluoedu.cn; Lin Chen. Department of Wound Repair and Rehabilitation Medicine, Daping Hospital, Army Medical University, Chongqing 400042 China. Tel: +86-23-68757041, E-mail address: linchen70com; Yangli Xie: Department of Wound Repair and Rehabilitation Medicine, Daping Hospital, Army Medical University, Chongqing 400042 China. Tel: +86-23-68757040, E-mail address: xieyangli841015com.