Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(18):8098-8110. doi:10.7150/thno.45363 This issue Cite
Research Paper
Mcl-1 inhibition overcomes intrinsic and acquired regorafenib resistance in colorectal cancer
Xiangping Song1,2,3, Lin Shen1,3,4, Jingshan Tong1,3, Chaoyuan Kuang1, Shan Zeng4, Robert E. Schoen1,5, Jian Yu1,6, Haiping Pei2 ![]() , Lin Zhang1,3
, Lin Zhang1,3 ![]()
1. UPMC Hillman Cancer Center, Pittsburgh, PA 15213, USA.
2. Department of Gastrointestinal Surgery, Xiangya Hospital, Central South University, Changsha, Hunan, 410008, P.R. China.
3. Department of Pharmacology and Chemical Biology, University of Pittsburgh School of Medicine, Pittsburgh, PA 15213, USA.
4. Department of Oncology, Xiangya Hospital, Central South University, Changsha, Hunan, 410008, P.R. China.
5. Department of Medicine, University of Pittsburgh School of Medicine, Pittsburgh, PA 15213. USA.
6. Department of Pathology, University of Pittsburgh School of Medicine, Pittsburgh, PA 15213. USA.
Received 2020-2-26; Accepted 2020-6-19; Published 2020-7-9
Abstract

Intrinsic and acquired resistance to targeted therapies is a significant clinical problem in cancer. We previously showed that resistance to regorafenib, a multi-kinase inhibitor for treating colorectal cancer (CRC) patients, can be caused by mutations in the tumor suppressor FBW7, which block degradation of the pro-survival Bcl-2 family protein Mcl-1. We tested if Mcl-1 inhibition can be used to develop a precision combination therapy for overcoming regorafenib resistance.
Methods: Small-molecule Mcl-1 inhibitors were tested on CRC cells with knock-in (KI) of a non-degradable Mcl-1. Effects of Mcl-1 inhibitors on regorafenib sensitivity were determined in FBW7-mutant and -wild-type (WT) CRC cells and tumors, and in those with acquired regorafenib resistance due to enriched FBW7 mutations. Furthermore, translational potential was explored by establishing and analyzing FBW7-mutant and -WT patient-derived organoid (PDO) and xenograft (PDX) tumor models.
Results: We found that highly potent and specific Mcl-1 inhibitors such as S63845 overcame regorafenib resistance by restoring apoptosis in multiple regorafenib-resistant CRC models. Mcl-1 inhibition re-sensitized CRC tumors with intrinsic and acquired regorafenib resistance in vitro and in vivo, including those with FBW7 mutations. Importantly, Mcl-1 inhibition also sensitized FBW7-mutant PDO and PDX models to regorafenib. In contrast, Mcl-1 inhibition had no effect in FBW7-WT CRCs.
Conclusions: Our results demonstrate that Mcl-1 inhibitors can overcome intrinsic and acquired regorafenib resistance in CRCs by restoring apoptotic response. FBW7 mutations might be a potential biomarker predicting for response to the regorafenib/Mcl-1 inhibitor combination.
Keywords: Mcl-1, FBW7, regorafenib, colorectal cancer, apoptosis
Introduction
Colorectal cancer (CRC) is the second leading cause of cancer-related deaths in the US [1]. Metastatic, chemotherapy-refractory CRC patients are treated with targeted drugs such as the approved multi-kinase inhibitor regorafenib [2]. However, most CRCs are not responsive to regorafenib treatment [2], and CRCs treated with targeted therapies almost invariably develop resistance shortly after initial therapy [3, 4]. Understanding the mechanisms of intrinsic and acquired resistance and developing rational combination therapies for overcoming such resistance is pivotal for improving efficacy of targeted therapies.
Regorafenib inhibits the RAS/RAF/MEK/ERK axis, which is aberrantly activated in most CRCs due to prevalent RAS and RAF mutations [5]. We recently showed that the therapeutic activity of regorafenib in CRC cells is associated with apoptosis induction and proteasomal degradation of myeloid cell leukemia 1 (Mcl-1), a pro-survival Bcl-2 family protein [6]. CRC cells that contain mutations in F-box and WD repeat domain-containing 7 (FBW7) are intrinsically resistant to regorafenib. FBW7 is a tumor suppressor and E3 ubiquitin ligase frequently mutated in CRCs [7, 8]. In response to regorafenib treatment, FBW7 binds to phosphorylated Mcl-1 and promotes Mcl-1 ubiquitination and subsequent degradation [9]. Blocking Mcl-1 phosphorylation by a knock-in approach abrogates Mcl-1 binding to FBW7. This subsequently leads to Mcl-1 stabilization, which suppresses regorafenib-induced killing of CRC cells [9]. Importantly, CRC cells with acquired regorafenib resistance were found to have blocked Mcl-1 degradation and enriched FBW7 hot-spot mutations at R505, R465 and R479 [6]. The critical role of Mcl-1 stabilization and FBW7 mutations in intrinsic and acquired resistance to regorafenib suggests that Mcl-1 is an attractive target for developing a precision combination therapy for overcoming regorafenib resistance in FBW7-mutant CRCs.
Targeting pro-survival Bcl-2 family proteins is a promising therapeutic strategy and has led to the approval of the Bcl-2-selective inhibitor Venetoclax [10]. A number of small-molecule Mcl-1 inhibitors with distinct chemical structures have been described, among which the most promising ones include S63845, AMG176, AZD5991, and VU661013 [11-14]. However, most of the preclinical and clinical studies on the Mcl-1 inhibitors have been carried out on hematopoietic malignancies in which Mcl-1 by itself is required for maintenance of cell survival [11-14]. The rationale and efficacy for targeting Mcl-1 in solid tumors such as CRC have yet to be demonstrated, in part due to lack of biomarkers for predicting response to Mcl-1 inhibition.
In this study, we evaluated the effects of Mcl-1 inhibitors on regorafenib sensitivity by using isogenic cell line, xenograft, patient-derived organoid (PDO) and patient-derived xenograft (PDX) models. Our results revealed a striking efficacy of Mcl-1 inhibitors in sensitizing regorafenib-resistant CRCs to regorafenib. We demonstrate the critical role of FBW7 mutations in promoting Mcl-1-dependent resistance to regorafenib, as well as how this resistance can be reversed by Mcl-1 inhibition. Our study provides a compelling rationale for developing a precision combination therapy on CRCs that are refractory to regorafenib treatment using Mcl-1 inhibitors.
Materials and Methods
Cell culture
Human CRC cell lines, including HCT116, DLD1, Lim1215, Lim2405, RKO, SW837, SW48, LoVo, SW1463, HCT-8 and LS411N, and the mouse CRC cell line CT26 were obtained from ATCC. HCT116 cells with knock-in of the Mcl-1 phosphorylation site mutant S121A/E125A/S159A/T163A (Mcl-1-KI) were generated by homologous recombination as described previously [9]. Regorafenib-resistant HCT116 (HCT116-R) and Lim1215 (Lim1215-R) were generated by 4 cycles of regorafenib selection as previously described [6]. Isogenic FBW7-KO HCT116 cells was obtained from Horizon Discovery [7]. All cell lines were authenticated by genotyping and analysis of protein expression by western blotting throughout the study, and also checked for mycoplasma contamination by PCR. Cell lines were maintained at 37°C and 5% CO2 in McCoy's 5A modified media (Invitrogen) supplemented with 10% defined FBS (HyClone), 100 U/mL penicillin, and 100 mg/mL streptomycin (Invitrogen). For drug treatment, cells were plated in 12-well plates at 20-30% density 24 hours before treatment. Anticancer agents including regorafenib, sorafenib, S63845, AZD5991, and AMG176 were dissolved in dimethylsulfoxide (DMSO) and diluted to appropriate concentrations with cell culture medium.
Analysis of cell viability
Cells seeded in 96-well plates at a density of 1×104 cells/well were treated with regorafenib at different concentrations +/- Mcl-1 inhibitor for 72 hours. Cell viability was analyzed by MTS (3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxy-phenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assay using the MTS Assay Kit (Promega) as described [6]. Cells plated in 12-well plates at 20-30% confluency were treated with regorafenib +/- Mcl-1 inhibitor at different concentrations for 48 hours, followed by staining of viable cells by crystal violet as described [15]. MTS and crystal violet staining results were quantified using a WallacVictor1420 Multilabel Counter (PerkinElmer). Each assay was conducted in triplicate and repeated three times. Combination index (CI) was calculated using the CompuSyn program (ComboSyn Inc).
Western blotting
Western blotting was performed using antibodies listed in Table S1 as previously described [16].
Cellular thermal shift assay (CETSA)
Binding of Mcl-1 inhibitors to endogenous Mcl-1 was analyzed by CETSA based on a published protocol [17]. Briefly, cells were treated with Mcl-1 inhibitors or control 0.1% DMSO for 2 hours in T-75 flasks. After treatment, cells were harvested, washed once with 1× PBS, resuspend in 750 μL HBSS, and lysed by 4 cycles of freezing (dry ice/ethanol for 5 min) and thawing (37 °C for 5 min). Samples were then distributed equally into 0.2-mL PCR tubes and heated at different temperatures for 3 min on a thermal cycler (Hybaid), followed by centrifugation at 13,200× rpm for 5 min and analysis of supernatants by western blotting.
Immunoprecipitation (IP)
Cell lysis, preparation of cell lysates, and IP using 1-2 µg of IP antibodies (Table S1) was performed as previously described [18]. The precipitates suspended in 2× Laemmli sample buffer were analyzed by SDS-PAGE and western blotting.
Analysis of apoptosis
Adherent and floating cells were harvested after treatment. Apoptosis was measured by counting condensed and fragmented nuclei after nuclear staining with Hoechst 33258 (Invitrogen) as previously described [19]. At least 300 cells were analyzed for each sample. Annexin V/propidium iodide (PI) staining was performed using Annexin-Alexa Fluor 488 (Invitrogen) and PI as described [6]. Colony formation assays were performed by plating treated cells in 6-well plates at appropriate dilutions, followed by crystal violet staining 14 days later as described [6]. Each experiment was performed in triplicate and repeated at least twice.
Genomic PCR and sequencing
To verify Mcl-1-KI cell lines and detect FBW7 hotspot mutations in regorafenib-resistant CRC cells and patient-derived samples, genomic DNA was isolated by using ZR-96 Quick-gDNA Kit (ZYMO Research) according to the manufacturer's instructions. One μL out of 50 μL genomic DNA preparation was amplified by PCR using previously described cycle conditions [9] and primer pairs for: Mcl-1 KI: 5'-GGGTCTTCCCCAGTTTTCTC-3'/5'-AATGAACCCCCTTACCTTGG-3'; FBW7 R465: 5'-CCCAACTTCCCATTCCCTTA-3'/5'-ATTAGTATGCCCCTGCAACG-3'; and FBW7 R479/R505: 5'-GGTGGAGTATGGTCATCACAAA-3'/5'-CAAAACGCTATGGCTTTCCT-3'.
Analysis of patient-derived CRC organoids
Patient-derived CRC organoids were established using surgically resected CRC tissues from the Pitt Biospecimen Core (PBC) at University of Pittsburgh as described [20]. Tissues were acquired with informed consent and approval by the University of Pittsburgh Ethics Committee. CRC organoids were cultured in Matrigel (Corning) incubated with advanced DMEM/F12 (Invitrogen) medium with supplements (Table S1), including 50% (v/v) L-WRN-conditioned medium containing Wnt3a, R spondin , and Noggin prepared as described [20], 1× penicillin/streptomycin (Invitrogen), 10 mM HEPES (Invitrogen), 2 mM GlutaMAX (Invitrogen), 1× B27 (Invitrogen), 1× N2 (Invitrogen), 1 mM N-Acetylcysteine (Sigma), 10 nM [leu-15]-Gastrin (Sigma), 10 mM nicotinamide (Sigma), 10 μM SB202190 (Sigma), 50 ng/mL recombinant murine EGF (Peprotech), and 0.5 μM A83-01 (Tocris Bioscience).
Before treatment, organoids were digested into small clumps and seeded into 24-well or 96-well plates at appropriate density and cultured for 2 days. After treatment, organoid cell viability was analyzed by using the CellTiter-Glo® 3D Cell Viability Assay Kit (Promega) according to the manufacture's protocol. Active caspase 3 in organoids was analyzed by immunostaining as described [21]. Quantitation of active caspase 3 was analyzed by using SensoLyte ® Homogeneous AMC Caspase-3/7 Assay Kit (AnaSpec). Results were obtained from at least three independent experiments with triplicate wells in each experiment.
Animal experiments
All animal experiments were approved by the University of Pittsburgh Institutional Animal Care and Use Committee. Mice were housed in a sterile environment with micro isolator cages and allowed access to water and chow ad libitum. Cell line xenografts were established by subcutaneously injecting 4×106 HCT116, Mcl-1-KI, or HCT116-R cells into both flanks of 5-6-week-old female Nu/Nu mice (Charles River). Syngeneic tumors were established by injecting 5×105 CT26 cells in to into both flanks of 5-6-week-old BABL/cJ mice (Jackson Laboratory). PDX tumors were established and propagated in 5-6-week-old female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (Jackson Laboratory) as described [22]. PDX1 was established from a FBW7-mutant and microsatellite stable (MSS) tumor (T4N0M1) in the sigmoid colon of a 77-year-old male. PDX2 was established from a FBW7-WT and MSS tumor (T2N0) in the right colon of a 69-year-old male. Xenograft tumors reached ~60 mm3 in size before treatment.
Tumor-bearing mice were randomized into different groups and treated with regorafenib (oral gavage; 20 mg/kg) with or without a combination with S63845 (i.p.; 20 mg/kg). S63845 was dissolved in 25 mM HCl, 20% hydroxypropyl-β-cyclodextrin (Sigma). Regorafenib was dissolved in Cremephor EL/95% ethanol (50:50) as a 4× stock solution and diluted to the final concentration with sterile water before use. Six mice were included in each group. Tumor growth was monitored by calipers, and tumor volumes were calculated according to the formula 1/2×length×width2. Ethical endpoint was defined as a time point when a tumor reached 1.5 cm or more in any dimension.
Tumor tissues were dissected and fixed in 10% formalin and embedded in paraffin. Terminal deoxynucleotidyl transferase mediated dUTP Nick End Labeling (TUNEL; EMD Millipore) and active caspase 3 (Cell Signaling Technology) immunostaining was performed on 5 μm paraffin-embedded tumor sections as described [9]. Signals were detected by using AlexaFluor 488-conjugated secondary antibody (Invitrogen) with nuclear counter staining by 4'6-Diamidino-2-phenylindole (DAPI).
Statistical Analysis
Statistical analyses were performed using GraphPad Prism Ⅵ software. P values were calculated by the Student t test between two groups or one-way ANOVA in three or more groups and considered significant if P< 0.05. The mean ± s.d. was displayed in the figures.
Results
Mcl-1 inhibitors restore regorafenib sensitivity in CRC cells expressing non-degradable Mcl-1
Our previous study showed that knock-in (KI) of an Mcl-1 phosphorylation site mutant (S121A/E125A/S159A/T163A) in regorafenib-sensitive HCT116 CRC cells (Figure S1A) blocks regorafenib-induced Mcl-1 degradation and subsequent apoptotic response [9]. We tested whether small-molecule Mcl-1 inhibitors can restore regorafenib sensitivity in an Mcl-1-specific manner in isogenic Mcl-1-KI cells relative to the parental HCT116 cells. Treating Mcl-1-KI cells with the Mcl-1 inhibitor S63845, AZD5991, or AMG176 completely restored regorafenib sensitivity in Mcl-1-KI cells, indicated by regorafenib inhibitory concentration 50 (IC50) (Figure 1A), crystal violet staining of viable cells (Figure 1B), and long-term cell survival assayed by colony formation (Figure 1C). A marked synergism between regorafenib and S63845, as indicated by CI (combination index) < 0.8, was observed in Mcl-1-KI cells, but not in WT HCT116 cells (Figure 1D and Figure S1B-C). Mcl-1 inhibitors also fully restored regorafenib-induced apoptosis, as determined by nuclear fragmentation (Figure 1E), annexin V staining (Figure S1D), and activation of caspases 3, 8 and 9 (Figure 1F). Analysis of different Bcl-2 family proteins showed that PUMA induction and Mcl-1 degradation are critical events in regorafenib-induced apoptosis in CRC cells (Figure S2A) [9, 23]. These results demonstrate that regorafenib resistance is mediated by Mcl-1 in the Mcl-1-KI model, and that pharmacologic inhibition of Mcl-1 can restore sensitivity to regorafenib.
Mcl-1 inhibitors restores apoptosis response and sensitivity to regorafenib in Mcl-1-KI HCT116 cells. (A) MTS analysis of wild-type (WT) and Mcl-1-KI HCT116 cells treated with regorafenib at indicated concentrations with or without a combination with an indicated Mcl-1 inhibitor (5 μM) for 72 hours. (B) Crystal Violet staining of WT and Mcl-1-KI HCT116 cells treated with regorafenib (40 μM), an indicated Mcl-1 inhibitor (5 μM), or their combination for 48 hours. (C) Colony formation of WT and Mcl-1-KI HCT116 cells treated with regorafenib (40 μM), S63845 (5 μM), or their combination for 48 hours. Left panel: representative pictures of colonies visualized by crystal violet staining 2 weeks after treatment; right panel: enumeration of colony numbers. (D) Combination index (CI) and fraction affected of regorafenib and S63845 combining at different concentrations in WT and Mcl-1-KI HCT116 cells treated for 48 hours (shown in Figure S1B-C) were analyzed by the CompuSyn program (ComboSyn). (E) Apoptosis in cells treated as in B was analyzed by counting condensed and fragment nuclei after nuclear staining with Hoechst 33258. (F) Western blotting of Mcl-1 and cleaved (C) caspases 3, 8 and 9 in cells treated as in C. (G) Cellular thermal shift assay (CETSA) for the binding of indicated inhibitors to endogenous Mcl-1 in HCT116 cells. Cells were treated with the control DMSO or an indicated Mcl-1 inhibitor (5 μM) for 2 hours, followed by heating of cell lysates at indicated temperatures for 3 minutes and probing of supernatants by Mcl-1 western blotting. (H) Immunoprecipitation (IP) analysis of the binding between endogenous Mcl-1 and PUMA in cells treated as in C for 24 hours. In A, C and E, results were expressed as means ± s.d. of three independent experiments. **, P <0.01; ***, P <0.001.
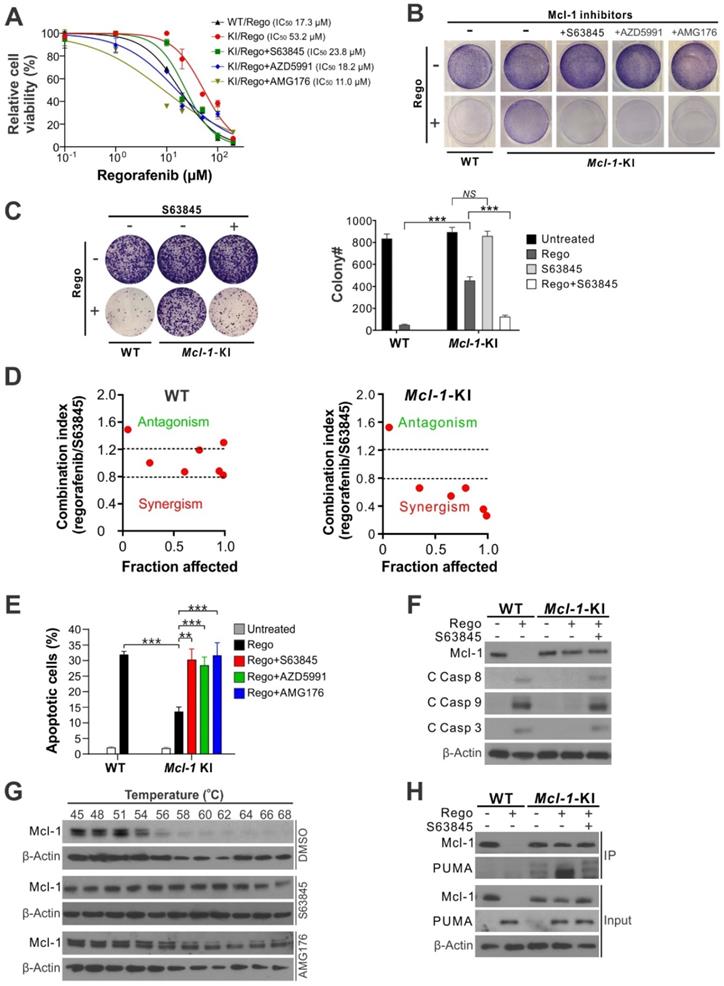
To further investigate the biochemical activity of the Mcl-1 inhibitors S63845 and AMG176, we utilized a cellular thermal shift assay (CETSA) on the parental HCT116 cells. This analysis showed that treatment with S63845 or AMG176 markedly protected endogenous Mcl-1 from heat-induced denaturation, indicating strong binding of these inhibitors to Mcl-1 (Figure 1G). Immunoprecipitation of Mcl-1 protein demonstrated strong binding of Mcl-1 to PUMA in regorafenib-treated Mcl-1-KI cells. However, this binding was disrupted by treatment with S63845 (Figure 1H). PUMA is a proapoptotic BH3-only Bcl-2 family protein required for regorafenib-induced apoptosis [23]. Consistent with the results from HCT116 cells (Figure 1), Mcl-1 inhibitors alone do not affect viability or induce apoptosis in different CRC cell lines (Figure S2B) [9]. These results demonstrate that the Mcl-1 inhibitors can overcome Mcl-1-mediated regorafenib resistance by relieving its inhibition on proapoptotic proteins such as PUMA.
Mcl-1 inhibitors re-sensitize FBW7-mutant CRC cells to regorafenib by restoring apoptotic response
FBW7, a tumor suppressor and E3 ubiquitin ligase of Mcl-1, is frequently mutated in CRCs, which underlies intrinsic regorafenib resistance in CRC cells [6]. Compared to FBW7-WT cells, FBW7-mutant CRC cells were much more refractory to regorafenib (Figure 2A and Figure S3A), and deficient in regorafenib-induced Mcl-1 degradation and apoptosis (Figure 2B and Figure S3B) [6]. Combining regorafenib with any Mcl-1 inhibitor markedly lowered regorafenib IC50 and restored regorafenib-induced loss of cell viability, apoptosis, and caspase activation in FBW7-mutant CRC cell lines [6], including SW837, SW48, LoVo, SW1463, HCT-8, and LS411N (Figure 2C-G and Figure S3C-D). In contrast, Mcl-1 inhibition had virtually no effect on regorafenib sensitivity and apoptosis in WT cell lines [6], including RKO, HCT116, DLD1, Lim1215, and Lim2405 (Figure 2C and Figure S3C-D). Similar observations were made in FBW7-WT and -mutant cells treated with sorafenib (Figure S3E-F), an analog of regorafenib approved for treating liver cancer and other gastrointestinal malignancies [24].
Mcl-1 inhibitors re-sensitize FBW7-mutant CRC cells to regorafenib. (A) MTS analysis of indicated WT (black) and FBW7-mutant (red) CRC cell lines treated with regorafenib at indicated concentrations for 72 hours. (B) Western blotting of Mcl-1 in indicated WT and -mutant CRC cell lines treated with regorafenib (40 μM) for 8 hours. (C) Comparison of regorafenib IC50 in indicated WT and FBW7-mutant CRC cell lines treated with regorafenib alone or in combination with S63845 (5 μM) for 72 hours. (D) Crystal violet staining of FBW7-mutant LoVo, HCT-8, and SW48 cells treated with regorafenib (40 μM) alone or in combination with S63845 (5 μM) for 48 hours. (E) MTS analysis of FBW7-mutant LoVo, HCT-8, and SW48 cells treated with regorafenib at indicated concentrations alone or in combination with an indicated Mcl-1 inhibitor (5 μM) for 72 hours. (F) Apoptosis in cells treated as in D and E for 48 hours was analyzed by counting condensed and fragment nuclei after nuclear staining. (G) Western blotting of cleaved (C) caspases 3, 8 and 9 in cells treated as in D. In A, C, E, and F, results were expressed as means ± s.d. of three independent experiments. *, P < 0.05; **, P <0.01; ***, P <0.001.
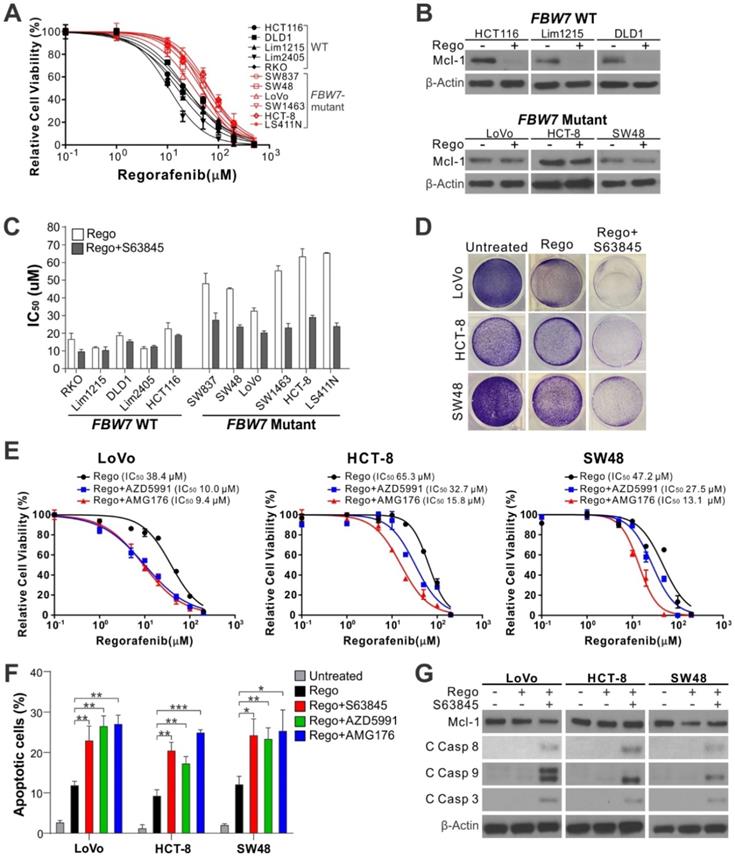
To verify if FBW7 status is a key factor in determining the response to Mcl-1 inhibition in CRC cells, we analyzed isogenic FBW7-knockout (KO) HCT116 cells generated by homologous recombination [7]. Similar to FBW7-mutant cells, FBW7-KO cells were much less sensitive to regorafenib and deficient in regorafenib-induced apoptosis and caspase activation (Figure S4A-E). Mcl-1 inhibition by S63845, AZD5991, or AMG176 restored regorafenib sensitivity, apoptosis, and caspase activation in FBW7-KO cells (Figure S4A-E). Similar observations were also made on FBW7-KO cells treated with sorafenib (Figure S4F). Therefore, the results from both FBW7-mutant and FBW7-KO CRC cells demonstrate that Mcl-1 inhibitors can overcome regorafenib resistance in FBW7-deficient CRC cells.
Mcl-1 inhibitors overcome acquired resistance to regorafenib in CRC cells by restoring apoptotic response
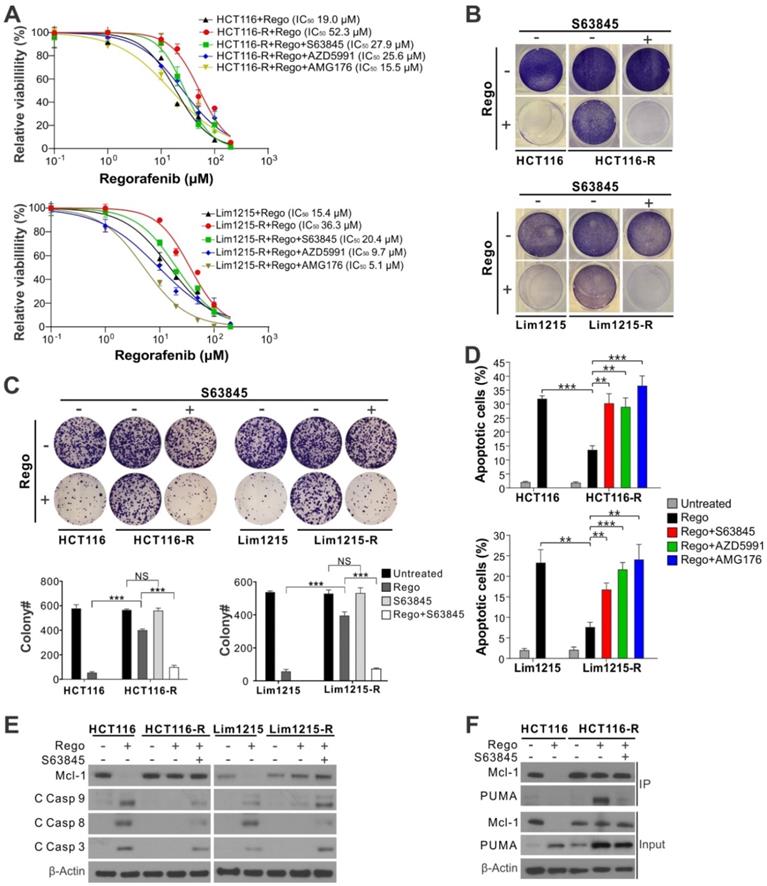
Our previous study showed that regorafenib-resistant CRC cells established by multiple rounds of drug selection are deficient in Mcl-1 degradation and often enriched in FBW7 hotspot mutations [6]. We tested if Mcl-1 inhibitors can be used to restore regorafenib sensitivity in regorafenib-resistant HCT116 (HCT116-R) and Lim1215 (Lim1215-R) cells, which contain enriched FBW7 R505C and R465C hotspot mutations, respectively [6]. Treating HCT116-R and Lim1215-R cells with regorafenib combined with S63845, AZD5991 or AMG176 completely restored regorafenib sensitivity relative to the parental HCT116 and Lim1215 cells, as shown by decreased IC50, loss of cell viability, and suppression of colony formation (Figure 3A-C). Induction of apoptosis and caspase activation were also restored (Figure 3D-E and Figure S4G), as well as the dissociation of Mcl-1 and PUMA (Figure 3F). These data indicate that Mcl-1 inhibition can overcome acquired regorafenib resistance by liberating PUMA from Mcl-1 and subsequently restoring apoptosis.
Mcl-1 inhibitors re-sensitize CRC cells with acquired resistance to regorafenib. (A) MTS analysis of parental and regorafenib-resistant HCT116 (HCT116-R) and Lim1215 (Lim1215-R) cells treated with regorafenib at indicated concentrations alone or in combination with an indicated Mcl-1 inhibitor (5 μM) for 72 hours. (B) Crystal violet staining of parental and regorafenib-resistant HCT116 and Lim1215 cells treated with regorafenib (40 μM) alone or in combination with S63845 (5 μM) for 48 hours. (C) Colony formation of cells treated as in (B). Upper panel: representative pictures of colonies visualized by crystal violet staining 2 weeks after treatment; lower panel: enumeration of colony numbers. (D) Apoptosis in cells treated with regorafenib (40 μM) alone or in combination with indicated Mcl-1 inhibitor (5 μM) for 48 hours was analyzed by counting condensed and fragment nuclei. (E) Western blotting of Mcl-1 and cleaved (C) caspases 3, 8, and 9 in indicated cells treated as in C. (F) IP analysis of the binding between endogenous Mcl-1 and PUMA in parental HCT116 and HCT116-R cells treated as in C for 24 hours. In A, C and D, results were expressed as means ± s.d. of three independent experiments. NS, not significant, P >0.05; **, P <0.01; ***, P <0.001.
Mcl-1 inhibition overcomes regorafenib resistance in xenograft tumors
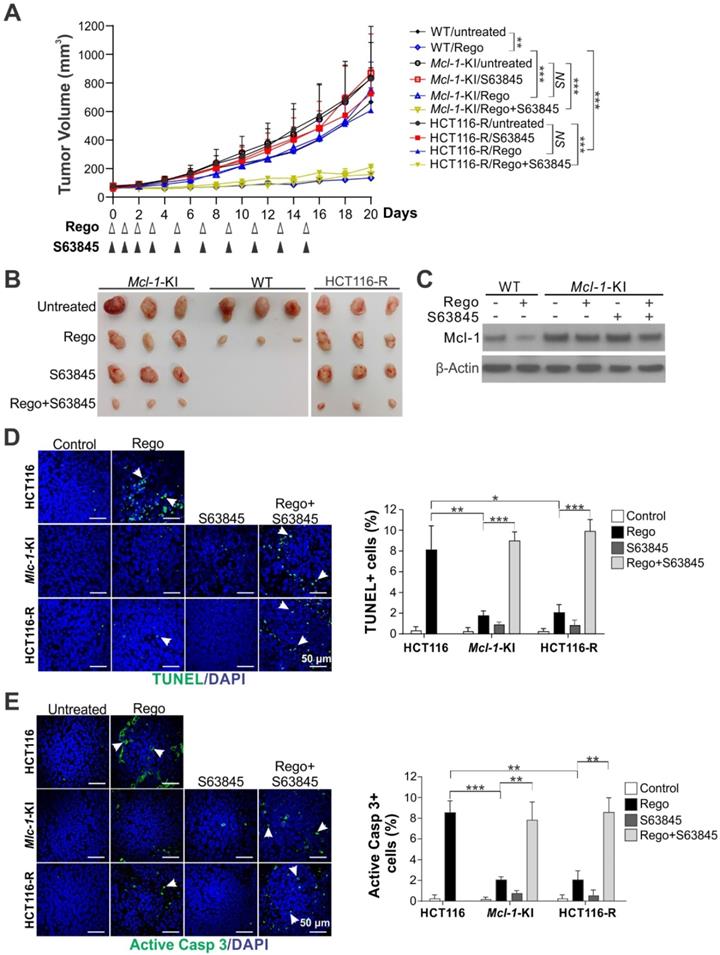
To determine if Mcl-1 inhibition can reverse intrinsic and acquired resistance to regorafenib in vivo, we compared the effects of regorafenib alone or in combination with S63845 on parental HCT116, Mcl-1-KI, and HCT116-R xenograft tumors established in nude mice. Regorafenib (oral gavage; 20 mg/kg) alone significantly suppressed the growth of the parental HCT116 tumors and induced Mcl-1 degradation; but had little effect on Mcl-1-KI and HCT116-R tumors (Figure 4A-C). S63845 treatment (IP; 20 mg/kg), though having no effect by itself, completely reversed regorafenib resistance in Mcl-1-KI and HCT116-R tumors when used in combination with regorafenib (Figure 4A-B). TUNEL and active caspase 3 immunostaining showed that S63845 fully restored regorafenib-induced apoptosis in Mcl-1-KI and HCT116-R tumors relative to the parental HCT116 tumors (Figure 4D-E). S63845 with or without regorafenib was well tolerated and did not significantly decrease animal weight (Figure S5A), or cause cell loss or histological changes in normal tissues from heart, liver, spleen, lung and kidney (Figure S5B). Furthermore, S63845 had no effect on FBW7-WT CT26 syngeneic tumors in BABL/cJ mice with or without regorafenib (Figure S6) [25]. These results indicate that Mcl-1 inhibition can overcome in vivo resistance to regorafenib caused by Mcl-1 degradation deficiency.
Mcl-1 inhibition overcomes intrinsic and acquired resistance to regorafenib in xenograft tumors. (A) Nude mice were injected s.c. with 4 × 106 parental HCT116, Mcl-1-KI or HCT116-R cells. After tumor volume reached ~60 mm3, mice were treated with regorafenib (oral gavage; 20 mg/kg) alone or in combination with S63845 (i.p.; 20 mg/kg) as indicated. Tumor volume at indicated time points after treatment was calculated and plotted (n=6 in each group). (B) Representative pictures of tumors at the end of the experiment in A. (C) Xenograft tumors established and treated as in A for 4 consecutive days were randomly selected and analyzed for Mcl-1 by Western blotting. (D) and (E), Paraffin-embedded sections of tumor tissues from C were analyzed by (D) TUNEL and (E) active caspase 3 staining. Left, representative staining pictures with nuclear counterstaining by DAPI; right, quantification of positive cells. In D and E, arrows indicate example cells with positive staining. Results were expressed as means ± s.d. of three independent experiments. *, P < 0.05; **, P <0.01; ***, P <0.001.
Mcl-1 inhibition sensitizes FBW7-mutant but not WT PDOs to regorafenib
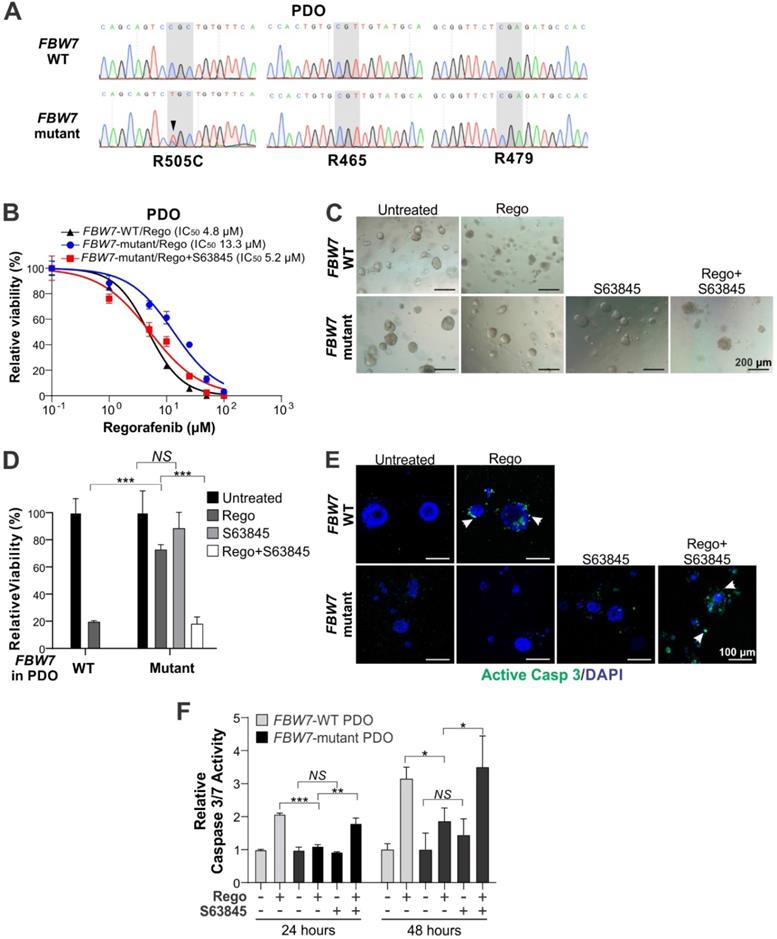
To explore the translational potential of Mcl-1 inhibitors in CRC, we tested if the regorafenib/S63845 combination is efficacious against patient-derived CRC samples containing FBW7 mutations. We established a panel of patient-derived organoids (PDOs) using surgical specimens from 12 CRC patients. Upon targeted genomic sequencing for FBW7 mutational hotspots including R505, R465 and R479, we identified a PDO with a heterozygous dominant R505C mutation (Figure 5A). This mutation was previously shown to mediate intrinsic and acquired regorafenib resistance in LoVo and HCT116-R cells, respectively, by blocking Mcl-1 degradation [6]. Interestingly, this FBW7-mutant PDO was from a T4N0M1 tumor with KRAS G13D and NRAS G12D and MSS, a profile associated with lack of clinical response to EGFR-targeted therapy and anti-PD-1 immunotherapy [26, 27]. Compared to the control FBW7-WT PDO from a different patient, this FBW7-mutant PDO was substantially less sensitive to regorafenib (IC50 13.3 vs. 4.8 µM) (Figure 5B), and showed significantly less growth inhibition, loss of cell viability, and activation of caspase 3/7 (Figure 5C-F). Importantly, S63845 fully restored regorafenib sensitivity as well as the induction of cell death and caspase 3/7 activation in this FBW7-mutant PDO (Figure 5B-F). In contrast, S63845 had little effect on the FBW7-WT PDO with or without regorafenib (Figure S7A-B). These results suggest that PDOs may be useful for predicting the response of Mcl-1 inhibitor and regorafenib combination therapy in FBW7 mutated CRCs.
Mcl-1 inhibition re-sensitizes FBW7-mutant PDO to regorafenib. (A) DNA sequencing of the targeted genomic regions in CRC patient-derived organoids (PDOs) highlighting WT and corresponding FBW7 mutant sequences. (B) Indicated CRC PDOs were treated with regorafenib at different concentrations alone or in combination with S63845 (1 μM) for 72 hours. Cell viability was assessed by 3D Cell Viability Assay Kit. (C) and (D), Representative images (C) and quantification (D) of CRC PDOs treated with regorafenib (20 μM) alone or in combination with S63845 (1 μM) for 48 hours. (E) CRC PDOs treated as in (C) were analyzed by active caspase 3 staining images. Representative images are shown with DAPI for nuclear counter staining. (F) CRC PDOs treated as in C for 24 or 48 hours were analyzed by Caspase 3/7 Activity Assay. Results in B, D and F were expressed as means ± s.d. of three independent experiments. NS, P > 0.05; *, P < 0.05; **, P <0.01; ***, P <0.001.
Mcl-1 inhibition re-sensitizes FBW7-mutant PDX to regorafenib
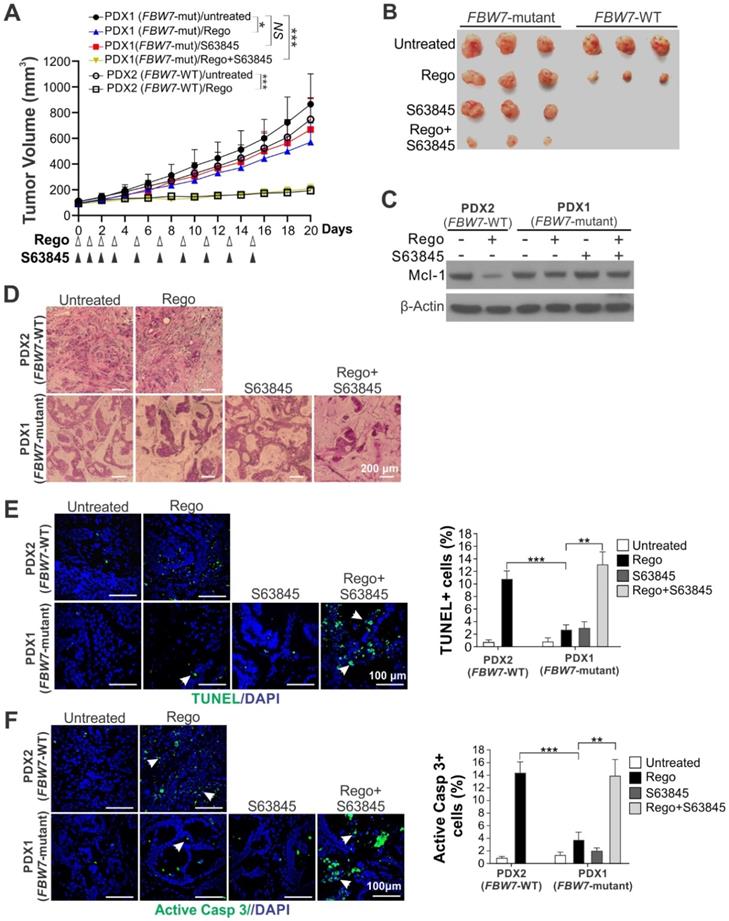
Compared with other in vivo models, PDX models better recapitulate heterogeneity, histology, and molecular alterations of patient tumors [28]. We further explored the efficacy of the regorafenib/S63845 combination in a CRC PDX model with WT or mutant (R505C) FBW7 (Figure S7C). Regorafenib was able to potently suppress the growth of the WT PDX, but caused significantly less growth suppression of the FBW7-mutant PDX (Figure 6A-B). Regorafenib was also able to induce Mcl-1 degradation in the WT PDX, but unable to induce a similar Mcl-1 degradation in the FBW7-mutant PDX (Figure 6C). S63845 completely reversed regorafenib resistance in the FBW7-mutant PDX (Figure 6A-B), and increased tumor cell loss as analyzed by H&E staining (Figure 6D), cell death by TUNEL staining (Figure 6E), and activation of caspase 3 (Figure 6F). No obvious toxicity or weight loss was observed in the NOD/SCID hosts with the treatment (Figure S7D). Collectively, our results demonstrate that Mcl-1 inhibitors can be used to develop a precision combination therapy for overcoming regorafenib resistance in CRCs.
Mcl-1 inhibition re-sensitizes FBW7-mutant PDX to regorafenib. (A) NSG mice were subcutaneously implanted with PDX1 (FBW7-mutant) and PDX2 (FBW7-WT) tumors. After tumor volume reached ~60 mm3, mice were treated with regorafenib (oral gavage; 20 mg/kg) alone or in combination with S63845 (i.p.; 20 mg/kg) as indicated. Tumor volume at indicated time points after treatment was calculated and plotted (n=6 in each group). (B) Representative pictures of tumors at the end of the experiment in A. (C) PDX1 and PDX2 tumors established and treated as in A for 4 consecutive days were randomly selected and analyzed for Mcl-1 by Western blotting. (D)-(F), Paraffin-embedded sections of tumor tissues from C were analyzed by (D) H&E, (E) TUNEL, and (F) active caspase 3 staining. In D and E, arrows indicate example cells with positive staining. Left, representative staining pictures with nuclear counterstaining by DAPI; right, quantification of positive cells. Results were expressed as means ± s.d. of three independent experiments. NS, P > 0.05; *, P < 0.05; **, P <0.01; ***, P <0.001.
Discussion
Metastatic CRC is one of the most deadly cancers characterized by poor prognosis and low five-year survival rate of just 11% [29]. Metastatic CRC patients are typically treated with conventional cytotoxic chemotherapy, targeted therapy, and more recently with anti-PD-1 immunotherapy [26]. However, most CRCs are either inherently insensitive to therapeutic treatment or acquire resistance upon relapse [3]. There is a critical need for developing novel and more effective CRC therapies, especially for those with intrinsic or acquired resistance to existing treatments [30].
Our results from isogenic cell line, xenograft, and patient-derived models demonstrate that inhibiting Mcl-1 is an effective approach for re-sensitizing CRCs with intrinsic and acquired resistance to regorafenib. Regorafenib is a multi-kinase inhibitor approved for treating CRC and other gastrointestinal malignancies [2, 31]. The antitumor activity of regorafenib relies on degradation of the antiapoptotic protein Mcl-1 [6]. Aberrant Mcl-1 expression is frequently found in CRCs and significantly correlated with advanced tumor stages, lymph node metastasis, resistance to chemotherapy, and poor patient survival [32-35]. Blocking Mcl-1 degradation abolished the response of CRC cells to a variety of anticancer agents, such as inhibitors of different kinases, heat shock proteins, and histone deacetylases [9, 18, 36]. Targeting pro-survival Bcl-2 family proteins has led to recent approval of the Bcl-2-selective inhibitor Venetoclax [10]. However, lack of Mcl-1 binding has limited the applications of Venetoclax and other Bcl-2 inhibitors, and Mcl-1 accumulation can cause resistance to these inhibitors [37, 38]. Together, these studies provide compelling evidence that Mcl-1 is an attractive therapeutic target against CRC and other solid tumors, especially those that are refractory to other therapies.
The observed differences between WT and FBW7-mutant CRCs suggest that FBW7 status is critical in determining the response to the regorafenib/Mcl-1 inhibitor combination. FBW7 is frequently mutated in human cancers including 10-15% of CRCs [7, 8]. FBW7 mutations have a broad functional role in determining therapeutic responses of cancer cells [8], and can modulate responses to γ secretase inhibitors in leukemia cells [39], to histone deacetylase inhibitors in squamous tumor cells [40], and to antimitotic drugs in CRC cells [41]. FBW7 encodes an F-box protein that functions as a substrate receptor for SCF-type of ubiquitin ligase complexes [42]. In addition to Mcl-1, FBW7 also mediates ubiquitination and degradation of other substrates, including Jun, Myc, cyclin E, and Notch [8]. Due to the multi-functional nature of FBW7, the same mutation could produce highly variable outcomes depending on the tumor type and treatment type. Therefore, it is essential to understand the exact functional role of FBW7 mutations to explore their use as a biomarker of therapeutic response.
The current study highlights multiple areas of translation. First, our results suggest that the presence of FBW7 hotspot mutations can predict for poorer response to regorafenib. FBW7 mutations may appear during the course of regorafenib treatment and indicate an acquired resistance mechanism, or they may be present in regorafenib-naïve patients and signal intrinsic resistance to regorafenib. Anticancer therapies including targeted therapies often generate highly heterogeneous patient responses and are associated with substantial toxicities. Hepatotoxicity and other side effects have been observed in regorafenib-treated patients [43]. Using biomarkers to identify potential responders and stratify patients can prevent unnecessary treatments and avoid therapy-associated adverse effects. For example, KRAS and BRAF mutations have been routinely used to exclude anti-EGFR therapy in CRC patients [44], while MSI (microsatellite instable) predicts responsiveness of CRCs to anti-PD-1 immunotherapy [45]. Second, the results from this study suggest that establishing and analyzing patient-derived tumor models, such as PDO and PDX models, can be used to screen tumors for identifying potential responders, and for designing personalized treatment. Finally, we have developed highly sensitive PCR assays that may be useful for monitoring responses and FBW7 mutations in liquid biopsies [6].
In conclusion, we demonstrate that Mcl-1 inhibitors can be used to overcome mutant-FBW7-driven regorafenib resistance and help to develop a precision therapy for improving CRC treatment. The potential use of this strategy needs to be further assessed in clinical studies to determine the safety and efficacy of this combination, as well as the utility of FBW7 mutations and Mcl‐1 level to predict therapeutic response.
Abbreviations
CETSA: cellular thermal shift assay; CI: combination index; CRC: colorectal cancer; DAPI: 4' 6-Diamidino-2-phenylindole; DMSO: dimethylsulfoxide; FBW7: F-box and WD repeat domain-containing 7; IP: immunoprecipitation; KI: knock-in; KO: knockout; Mcl-1: myeloid cell leukemia 1; MSI: microsatellite instable; MSS: microsatellite stable; MTS: 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium; NSG: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ; PDO: patient-derived organoid; PDX: patient-derived xenograft; PI: propidium iodide; RT-PCR: reverse transcriptase PCR; TUNEL: terminal deoxynucleotidyl transferase mediated dUTP nick end labeling; WT: wild-type.
Supplementary Material
Supplementary figures and table.
Acknowledgements
We thank our lab members for critical reading and discussion and the China Scholarship Council for supporting the visit of X. Song in our lab. This work is supported by U.S. National Institutes of Health grants (R01CA213028 to L. Zhang and U01CA152753 to R. E. Schoen). This project used the Hillman Cancer Center Animal Facility, Cytometry Facility, and Tissue and Research Pathology Services, which are supported in part by award P30CA047904.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7-34
2. Grothey A, Van Cutsem E, Sobrero A, Siena S, Falcone A, Ychou M. et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:303-12
3. Zhang L, Yu J. Role of apoptosis in colon cancer biology, therapy, and prevention. Curr Colorectal Cancer Rep. 2013;9:331-40
4. Park SH, Jo MJ, Kim BR, Jeong YA, Na YJ, Kim JL. et al. Sonic hedgehog pathway activation is associated with cetuximab resistance and EPHB3 receptor induction in colorectal cancer. Theranostics. 2019;9:2235-51
5. Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ. et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108-13
6. Tong J, Tan S, Zou F, Yu J, Zhang L. FBW7 mutations mediate resistance of colorectal cancer to targeted therapies by blocking Mcl-1 degradation. Oncogene. 2017;36:787-96
7. Rajagopalan H, Jallepalli PV, Rago C, Velculescu VE, Kinzler KW, Vogelstein B. et al. Inactivation of hCDC4 can cause chromosomal instability. Nature. 2004;428:77-81
8. Davis RJ, Welcker M, Clurman BE. Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer Cell. 2014;26:455-64
9. Tong J, Wang P, Tan S, Chen D, Nikolovska-Coleska Z, Zou F. et al. Mcl-1 Degradation Is Required for Targeted Therapeutics to Eradicate Colon Cancer Cells. Cancer Res. 2017;77:2512-21
10. Del Poeta G, Postorino M, Pupo L, Del Principe MI, Dal Bo M, Bittolo T. et al. Venetoclax: Bcl-2 inhibition for the treatment of chronic lymphocytic leukemia. Drugs Today (Barc). 2016;52:249-60
11. Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G. et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538:477-82
12. Caenepeel S, Brown SP, Belmontes B, Moody G, Keegan KS, Chui D. et al. AMG 176, a Selective MCL1 Inhibitor, Is Effective in Hematologic Cancer Models Alone and in Combination with Established Therapies. Cancer Discov. 2018;8:1582-97
13. Tron AE, Belmonte MA, Adam A, Aquila BM, Boise LH, Chiarparin E. et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat Commun. 2018;9:5341
14. Ramsey HE, Fischer MA, Lee T, Gorska AE, Arrate MP, Fuller L. et al. A Novel MCL1 Inhibitor Combined with Venetoclax Rescues Venetoclax-Resistant Acute Myelogenous Leukemia. Cancer Discov. 2018;8:1566-81
15. Knickelbein K, Tong J, Chen D, Wang YJ, Misale S, Bardelli A. et al. Restoring PUMA induction overcomes KRAS-mediated resistance to anti-EGFR antibodies in colorectal cancer. Oncogene. 2018;37:4599-610
16. Chen D, Tong J, Yang L, Wei L, Stolz DB, Yu J. et al. PUMA amplifies necroptosis signaling by activating cytosolic DNA sensors. Proc Natl Acad Sci U S A. 2018;115:3930-5
17. Jafari R, Almqvist H, Axelsson H, Ignatushchenko M, Lundback T, Nordlund P. et al. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc. 2014;9:2100-22
18. Tong J, Zheng X, Tan X, Fletcher R, Nikolovska-Coleska Z, Yu J. et al. Mcl-1 Phosphorylation without Degradation Mediates Sensitivity to HDAC Inhibitors by Liberating BH3-Only Proteins. Cancer Res. 2018;78:4704-15
19. Chen D, Ming L, Zou F, Peng Y, Van Houten B, Yu J. et al. TAp73 promotes cell survival upon genotoxic stress by inhibiting p53 activity. Oncotarget. 2014;5:8107-22
20. Leibowitz BJ, Yang L, Wei L, Buchanan ME, Rachid M, Parise RA. et al. Targeting p53-dependent stem cell loss for intestinal chemoprotection. Sci Transl Med. 2018;10:eaam7610. doi: 10.1126/scitranslmed.aam7610
21. Wang X, Wei L, Cramer JM, Leibowitz BJ, Judge C, Epperly M. et al. Pharmacologically blocking p53-dependent apoptosis protects intestinal stem cells and mice from radiation. Sci Rep. 2015;5:8566
22. Tan X, Tong J, Wang YJ, Fletcher R, Schoen RE, Yu J. et al. BET inhibitors potentiate chemotherapy and killing of SPOP-mutant colon cancer cells via induction of DR5. Cancer Res. 2019;79:1191-203
23. Chen D, Wei L, Yu J, Zhang L. Regorafenib inhibits colorectal tumor growth through PUMA-mediated apoptosis. Clin Cancer Res. 2014;20:3472-84
24. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF. et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378-90
25. Castle JC, Loewer M, Boegel S, de Graaf J, Bender C, Tadmor AD. et al. Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genomics. 2014;15:190
26. Chu E. An update on the current and emerging targeted agents in metastatic colorectal cancer. Clin Colorectal Cancer. 2012;11:1-13
27. Lee JJ, Chu E. Recent Advances in the Clinical Development of Immune Checkpoint Blockade Therapy for Mismatch Repair Proficient (pMMR)/non-MSI-H Metastatic Colorectal Cancer. Clin Colorectal Cancer. 2018;17:258-73
28. Jin K, Teng L, Shen Y, He K, Xu Z, Li G. Patient-derived human tumour tissue xenografts in immunodeficient mice: a systematic review. Clin Transl Oncol. 2010;12:473-80
29. Poston GJ, Figueras J, Giuliante F, Nuzzo G, Sobrero AF, Gigot JF. et al. Urgent need for a new staging system in advanced colorectal cancer. J Clin Oncol. 2008;26:4828-33
30. Sun L, Fang Y, Wang X, Han Y, Du F, Li C. et al. miR-302a Inhibits Metastasis and Cetuximab Resistance in Colorectal Cancer by Targeting NFIB and CD44. Theranostics. 2019;9:8409-25
31. Demetri GD, Reichardt P, Kang YK, Blay JY, Rutkowski P, Gelderblom H. et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:295-302
32. Henderson-Jackson EB, Helm J, Ghayouri M, Hakam A, Nasir A, Leon M. et al. Correlation between Mcl-1 and pAKT protein expression in colorectal cancer. Int J Clin Exp Pathol. 2010;3:768-74
33. Peddaboina C, Jupiter D, Fletcher S, Yap JL, Rai A, Tobin RP. et al. The downregulation of Mcl-1 via USP9X inhibition sensitizes solid tumors to Bcl-xl inhibition. BMC Cancer. 2012;12:541
34. Lee WS, Park YL, Kim N, Oh HH, Son DJ, Kim MY. et al. Myeloid cell leukemia-1 is associated with tumor progression by inhibiting apoptosis and enhancing angiogenesis in colorectal cancer. Am J Cancer Res. 2015;5:101-13
35. Backus HH, van Riel JM, van Groeningen CJ, Vos W, Dukers DF, Bloemena E. et al. Rb, mcl-1 and p53 expression correlate with clinical outcome in patients with liver metastases from colorectal cancer. Ann Oncol. 2001;12:779-85
36. Tong J, Tan S, Nikolovska-Coleska Z, Yu J, Zou F, Zhang L. FBW7-Dependent Mcl-1 Degradation Mediates the Anticancer Effect of Hsp90 Inhibitors. Mol Cancer Ther. 2017;16:1979-88
37. Tahir SK, Yang X, Anderson MG, Morgan-Lappe SE, Sarthy AV, Chen J. et al. Influence of Bcl-2 family members on the cellular response of small-cell lung cancer cell lines to ABT-737. Cancer Res. 2007;67:1176-83
38. van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE. et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389-99
39. O'Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C. et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med. 2007;204:1813-24
40. He L, Torres-Lockhart K, Forster N, Ramakrishnan S, Greninger P, Garnett MJ. et al. Mcl-1 and FBW7 control a dominant survival pathway underlying HDAC and Bcl-2 inhibitor synergy in squamous cell carcinoma. Cancer Discov. 2013;3:324-37
41. Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ. et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471:110-4
42. Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399-434
43. Chan SL, Ma BB. An update on the safety and efficacy of regorafenib in the treatment of solid cancers. Expert Opin Drug Metab Toxicol. 2014;10:1607-14
44. Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF. et al. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol. 2009;27:2091-6
45. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK. et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409-13
Author contact
![]() Corresponding authors: Lin Zhang, the UPCI Research Pavilion, Room 2.42a, 5117 Centre Ave., Pittsburgh, PA 15213, USA. Phone: (412) 623-1009. Fax: (412) 623-7778. E-mail: zhanglxedu; or Haiping Pei, the Department of Gastrointestinal Surgery, Xiangya Hospital, Central South University, Changsha, Hunan, P.R. China, 410008. E-mail: php1966edu.cn.
Corresponding authors: Lin Zhang, the UPCI Research Pavilion, Room 2.42a, 5117 Centre Ave., Pittsburgh, PA 15213, USA. Phone: (412) 623-1009. Fax: (412) 623-7778. E-mail: zhanglxedu; or Haiping Pei, the Department of Gastrointestinal Surgery, Xiangya Hospital, Central South University, Changsha, Hunan, P.R. China, 410008. E-mail: php1966edu.cn.