Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(18):8298-8314. doi:10.7150/thno.46934 This issue Cite
Research Paper
ATF6 aggravates acinar cell apoptosis and injury by regulating p53/AIFM2 transcription in Severe Acute Pancreatitis
Jie-Hui Tan1#, Rong-Chang Cao1#, Lei Zhou1, Zhi-Tao Zhou2, Huo-Ji Chen3, Jia Xu4, Xue-Mei Chen5, Yang-Chen Jin6, Jia-Yu Lin6, Jun-Ling Zeng7, Shu-Ji Li8, Min Luo9, Guo-Dong Hu10, Xiao-Bing Yang11, Jin Jin12, Guo-Wei Zhang1 ![]()
1. Division of Hepatobiliopancreatic Surgery, Department of General Surgery, Nanfang Hospital, Southern Medical University, Guangzhou, China.
2. Department of the Electronic Microscope Room, Central Laboratory, Southern Medical University, Guangzhou, China.
3. School of Traditional Chinese Medicine, Southern Medical University, Guangzhou, China.
4. Department of Pathophysiology, Southern Medical University, Guangzhou, China.
5. Department of Occupational Health and Medicine, Guangdong Provincial Key Laboratory of Tropical Disease Research, School of Public Health, Southern Medical University, Guangzhou, China.
6. The First Clinical Medical College, Southern Medical University, Guangzhou, China.
7. Laboratory Animal Research Center of Nanfang Hospital, Southern Medical University, Guangzhou, China.
8. Guangdong-Hong Kong-Macao Greater Bay Area Center for Brain Science and Brain-Inspired Intelligence, Southern Medical University, Guangzhou, China.
9. Department of Laboratory Medicine, Nanfang Hospital, Southern Medical University, Guangzhou, China.
10. Department of Respiratory and Crit Care Medicine, Nanfang Hospital, Southern Medical University, Guangzhou, China.
11. Division of Nephrology, Nanfang Hospital, Southern Medical University, National Clinical Research Center for Kidney Disease, State Key Laboratory of Organ Failure Research, Guangdong Institute, Guangzhou, China.
12. Department of Gynaecology and Obstetrics, Nanfang Hospital, Southern Medical University, Guangzhou, China.
#These authors contributed equally to this work.
Received 2020-4-12; Accepted 2020-5-22; Published 2020-7-9
Abstract

Background: There is no curative therapy for severe acute pancreatitis (SAP) due to poor understanding of its molecular mechanisms. Endoplasmic reticulum (ER) stress is involved in SAP and increased expression of ATF6 has been detected in SAP patients. Here, we aimed to investigate the role of ATF6 in a preclinical SAP mouse model and characterize its regulatory mechanism.
Methods: Pancreatic tissues of healthy and SAP patients were collected during surgery. Humanized PRSS1 transgenic mice were treated with caerulein to mimic the SAP development, which was crossed to an ATF6 knockout mouse line, and pancreatic tissues from the resulting pups were screened by proteomics. Adenovirus-mediated delivery to the pancreas of SAP mice was used for shRNA-based knockdown or overexpression. The potential functions and mechanisms of ATF6 were clarified by immunofluorescence, immunoelectron microscopy, Western blotting, qRT-PCR, ChIP-qPCR and luciferase reporter assay.
Results: Increased expression of ATF6 was associated with elevated apoptosis, ER and mitochondrial disorder in pancreatic tissues from SAP patients and PRSS1 mice. Knockout of ATF6 in SAP mice attenuated acinar injury, apoptosis and ER disorder. AIFM2, known as a p53 target gene, was identified as a downstream regulatory partner of ATF6, whose expression was increased in SAP. Functionally, AIFM2 could reestablish the pathological disorder in SAP tissues in the absence of ATF6. p53 expression was also increased in SAP mice, which was downregulated by ATF6 knockout. p53 knockout significantly suppressed acinar apoptosis and injury in SAP model. Mechanistically, ATF6 promoted AIFM2 transcription by binding to p53 and AIFM2 promoters.
Conclusion: These results reveal that ATF6/p53/AIFM2 pathway plays a critical role in acinar apoptosis during SAP progression, highlighting novel therapeutic target molecules for SAP.
Keywords: severe acute pancreatitis, ER stress, AIFM2, ATF6, p53, apoptosis
Introduction
Acute pancreatitis (AP) with persistent single or multiple organ failure (MOF) and/or local pancreatic complications is characterized as severe acute pancreatitis (SAP) [1,2]. SAP patients with persistent organ failure have a high risk of early mortality within the first 2 weeks after diagnosis, with an overall mortality of over 40%. However, none of the pharmacological treatment studies decrease short-term mortality (in-hospital mortality or mortality within six months) in people with SAP [3]. Therefore, the treatment of SAP patients with organ failure currently remains largely supportive [4,5] due to a lack of understanding about SAP-specific regulatory mechanisms.
Premature trypsinogen activation, ER stress and apoptosis are the main contributors to AP pathogenesis [6]. In the past two decades, numerous studies using experimental models have suggested that prematurely activated intra-acinar trypsinogen, coded by PRSS1, is sufficient to initiate AP [7]. Furthermore, increased trypsin activity, can cause ER stress, is the key mechanism that makes mice more susceptible to pancreatitis [8]. In human, this notion was strongly supported by the observation that gain-of-function mutant PRSS1 genes are associated with hereditary pancreatitis [9]. In view of this, we have constructed the humanized PRSS1 transgenic mouse for next study.
The ER is critical for the proper folding, maturation and secretion of transmembrane and secreted proteins [10]. Activating transcription factor 6 (ATF6) is a key sensor protein on the ER membrane orchestrating the unfolded protein response (UPR), which helps the cell adapt to ER stress [11]. ATF6-induced disturbance of fatty acid metabolism caused mitochondrial dysfunction, which led to apoptosis during the development of tubulointerstitial fibrosis in patients with chronic kidney disease [12]. ATF6, but not other ER stress sensors, was specifically activated during apoptosis in myoblasts [13]. ATF6-mediated signaling pathway of the UPR was involved in nonsteroidal anti-inflammatory drug-induced apoptosis [14]. Moreover, ATF6 promoted inflammation during chronic pancreatitis (CP) progression [15]. However, whether and how ATF6 participates in ER stress-mediated acinar apoptosis during SAP progression remains undiscovered.
In present study, PRSS1Tg mice were treated with caerulein to establish a preclinical trypsin-induced SAP model, and then analyzed by crossing to ATF6 knockout mice, in addition to using a proteomics-based approach for identifying regulatory partners of ATF6. Apoptosis-inducing factor mitochondria-associated 2 (AIFM2), a p53-inducing protein localized in the mitochondrial outer membrane or freely distributed in the cytoplasm [16,17], as an important downstream regulatory partner of ATF6. AIFM2 protein expression was upregulated when ATF6 was activated and augmented apoptotic signaling pathway activity in response to ER stress. Moreover, AIFM2 was downregulated upon ATF6 or p53 knockout, resulting in decreased apoptosis and inflammation. Consistent with the above regulatory relationship, the structures of the ER, mitochondria and nucleus changed noticeably upon SAP induction. These results point to the ATF6-p53-AIFM2 pathway as a critical factor in ER-induced apoptosis during SAP development, and this knowledge will contribute to the enhancement of clinical management strategies.
Materials and Methods
Human pancreatic tissues and study approval
Human pancreatic tissues were collected from 6 SAP patients (mean age = 48.33 ± 4.61) and 12 patients with benign pancreatic tumours or peritumoural normal pancreatic tissues (mean age = 38.50 ± 11.54) as controls. All human samples and matching clinical data were obtained from Nanfang Hospital, Southern Medical University, between 2014 and 2019 (Supplemental Table S1). Written consent was obtained from each patient before the study, which were approved by the Ethics Committee of the Southern Medical University.
Mouse models
The generation of preclinical PRSS1 (GenBank Accession Number: NM_002769.4) transgenic (PRSS1Tg) mice and ATF6 (NCBI Reference Sequence: NM_001081304; Ensembl: ENSMUSG00000026663) knockout mice has been described previously [15]. TRp53 (NCBI reference sequence: NM_011640.3; Ensembl: ENSMUSG00000059552) knockout mice were purchased from Cyagen (Guangzhou, China). The tails of pups were genotyped by PCR followed by sequence analysis.
Sex- and age-matched mice (20-25g) were used for assays. Single transgenic littermates subjected to the same treatment were used as controls. All mouse colonies were maintained in a specific pathogen-free barrier facility at the laboratory animal center of Nanfang Hospital, Southern Medical University. Experimental procedures were carried out in strict accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and were approved by the Institutional Animal Care and Use Committee of Southern Medical University.
ATF6- and shAIFM2-expressing adenovirus
To investigate the effect of ATF6 on apoptosis and inflammation, the pancreases of PRSS1Tg mice were infected with adenoviral vectors harboring full-length ATF6 for ATF6 overexpression or a scrambled adRNA for the negative control, respectively. Similarly, to investigate the effect of AIFM2 on apoptosis and inflammation, the pancreases of PRSS1Tg mice were infected with adenoviral vectors harboring full-length AIFM2 for AIFM2 overexpression, shAIFM2 fragments for AIFM2 silencing or a scrambled shRNA for the negative control. All adenoviral vectors were purchased from VectorBuilder Inc. (Guangzhou, China). The sequences of the primers and the adenoviruses used are listed in Supplemental Table S2.
SAP induction and treatment
To establish a SAP model, PRSS1Tg mice were intraperitoneally injected with 15 μg/mL caerulein dissolved in phosphate-buffered saline (PBS) at 50 μg/kg every hour for a total of 8 injections and sacrificed 24 hours later. For ATF6 or AIFM2 overexpression and AIFM2 inhibition, the PRSS1Tg mice were injected with adenovirus three days prior to caerulein injection. Mice were intraperitoneally injected with 2.2 mg/kg pifithrin-α (PFT-α, a p53 inhibitor) each day before the caerulein injection to inhibit p53 expression.
Proteomics
PRSS1Tg mice were treated with caerulein (group A) or saline (group B) and PRSS1Tg/ATF6-/- mice were treated with caerulein (group C). Caerulein or saline treatment was performed for 8 hours and mice were sacrificed 24 hours later.
For proteomic analysis, tandem mass spectrometry data was collected for peptide identification through automated data-dependent acquisition (DDA). Using the DDA method, mass information on intact peptides in a full-scan mass spectrum was used to determine which subset of mass signals should be targeted for further acquisition of fragmentation spectra to identify peptide sequences.
Protein extraction, quality control of the extraction process and proteolysis were conducted. Fractions from each sample were analyzed by both DDA and data-independent acquisition (DIA) methods on a Q Exactive HF X mass spectrometer (Thermo Fisher Scientific) coupled to an Ultimate 3000 RSLC Nano system (Thermo Fisher Scientific). DDA data were identified using the Andromeda search engine within MaxQuant, and the identification results were used in Spectronaut™ for spectral library construction. For large-scale DIA data, the constructed spectral library information was used in Spectronaut™ to complete deconvolution and extraction, and the mProphet algorithm was used to complete analytical quality control, thus generating a large number of reliable quantitative results. This pipeline was also used to perform Gene Ontology (GO), Clusters of Orthologous Groups (COG), pathway functional annotation and time series analyses.
Transmission electron microscopy (TEM) and immunoelectron microscopy (IEM)
Ultrastructural examination was performed using a transmission electron microscope according to a previously described protocol [18]. Human pancreatic tissues (1 mm3) and mice tissues (1 mm3) from the pancreas, lung, liver, kidney, duodenum, heart, and spleen were fixed with 2.5% glutaraldehyde at room temperature for 1 hour and then at 4°C overnight. After fixation, the samples were rinsed in PBS three times for 10 min each.
For immunogold staining, the experimental methods has been adjusted, referring to the previous literatures [19,20]. Mice pancreatic tissues (1 mm3) were fixed with a mixture of 4% paraformaldehyde and 0.1% glutaraldehyde dissolved in 0.1 mol/L phosphate buffer (PB) at 4°C overnight. Then, the specimens in PBS were sliced into 50-μm sections on a vibrating microtome (Lecia VT1200S). The sections were then processed following standard procedures in Supplemental Methods. The resin-embedded sections were cut into ultrathin sections (70-80 nm thickness) and were collected on nickel grids (Nisshin EM). Finally, the sections were analyzed with an electron microscope (Hitachi H-7500, Japan) operated at 60 kV.
Quantitative real-time PCR (qRT-PCR)
qRT-PCR was performed to measure mRNA expression in pancreatic tissues. The sequences of the primers used are listed in Supplemental Table S2. Please see the Supplementary Methods for more details.
Western blotting
Total protein was extracted from the mice pancreatic tissues. Primary antibodies used in this study include anti-ATF6 (Abcam; diluted 1: 500), anti-p53 (Abcam; diluted 1: 500), anti-AIFM2 (Biorbyt; diluted 1: 500), and anti-GAPDH (Abcam; diluted 1:1000). The protein abundance was evaluated by immunoblotting with at least three biological replicates. Please refer to the Supplemental Methods for more details.
Immunohistochemistry and Immunofluorescence assay
H&E staining and immunohistochemistry analysis of pancreatic tissue slides for ATF6, p53, AIFM2, Caspase-3, PARP and MPO were performed according to a previously described protocol [21]. To visualize ATF6, p53 and AIFM2 colocalization in pancreatic tissue, immunofluorescence assay was also performed. Experimental details can be obtained by the Supplemental Methods.
Transferase-mediated d-UTP nick-end-labeling (TUNEL) assay
Terminal deoxynucleotidyl TUNEL was performed to detect apoptosis. For more details, please refer to the Supplemental Methods.
Enzyme-Linked Immunosorbent Assay (ELISA)
Supernatants were collected and stored at -80℃ after removal of serum by centrifugation. The protein concentration of IL-1β, IL-6, and TNF-α in the serum were detected using specific ELISA kits (ROCHE COBAS8000 E602, Switzerland) according to the manufacturer's instructions.
Chromatin immunoprecipitation (ChIP) assay
Chromatin immunoprecipitation (ChIP) assays were performed using the Pierce Magnetic ChIP Kit (Thermo Fisher Scientific) according to the manufacturer's instructions. Briefly, pancreatic acinar cells were collected from PRSS1Tg mice and treated with caerulein or untreated. Cells were cross-linked with 1% formaldehyde for 10 minutes at room temperature, followed by nucleus separation and sonication in lysis buffer. After centrifugation, the supernatant was separated and used for western blotting to detect the expression of ATF6. ChIP grade anti-ATF6 antibody (Abcam) and protein G magnetic beads were used for immunoprecipitation of chromatin. Normal rabbit IgG served as negative control. Finally, PCR analysis using the PCR Kit (Promega) was performed on purified input and immunoprecipitated DNA. The sequences of the primers for promoter region used are listed in Supplemental Table S3.
Luciferase assay
A series of deletion mutants of p53 promoter (chr11:69578359-69580458) and AIFM2 promoter (chr10:61713263-61715362) regions were respectively generated into promoter luciferase constructs, which were then inserted into the pGL3 vector. Subsequently, ATF6 CDS sequence (1971bp) was cloned into pcDNA3.1 plasmid. 293T cells were grown in 24-well plates and co-transfected with plasmids containing a series of deletion mutants of p53 promoter or AIFM2 promoter and either ATF6 overexpression plasmids or the empty control plasmids using the Lipofectamine 2000 Reagent (Invitrogen). Cellular extracts were measured for luciferase activity using the luciferase reporter assay system at 48h. Each experiment was repeated three times.
Statistical analysis
Statistical analysis was carried out using GraphPad Prism 8.0. The data are expressed as mean ± standard error of the mean. Significant differences between two groups were analyzed by Student's t-test, and one-way analysis of variance was performed to investigate the differences among more than two groups. Significant differences were defined by a P value of < 0.05.
Results
Elevated ATF6 expression in AP is associated with increased apoptosis and structural damage to the ER and mitochondria
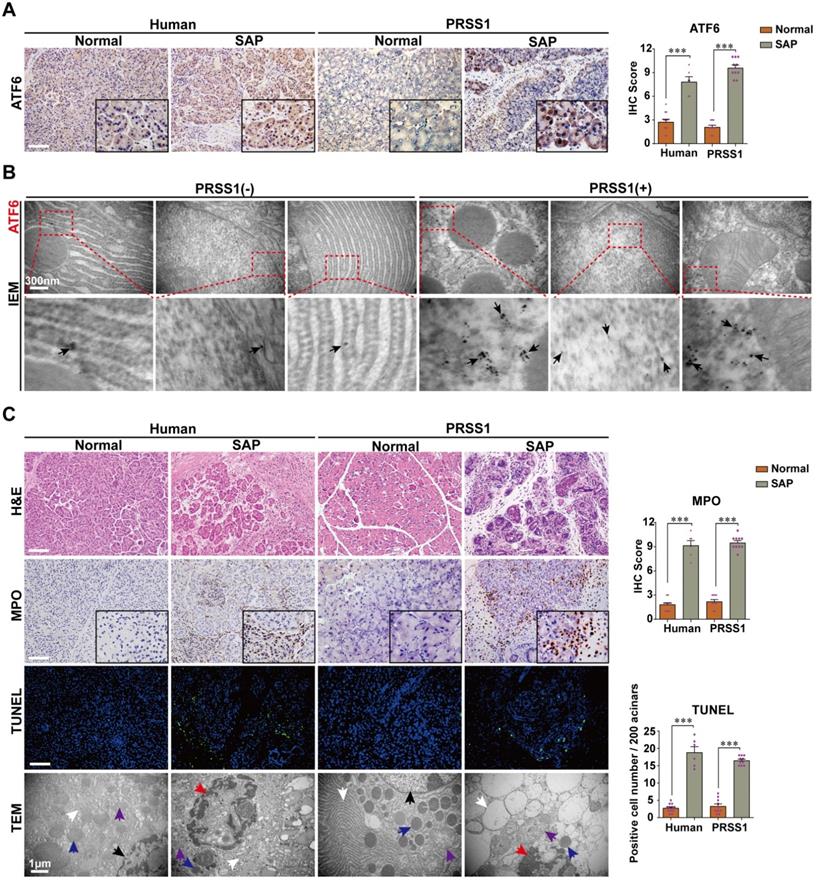
To elucidate the role that increased ATF6 expression plays in SAP pathogenesis, we first sought to identify the phenotypes associated with elevated levels of this protein in SAP patients or human volunteers (detailed in Supplemental Table S1) and a transgenic mouse model. Immunohistochemistry analysis showed that the expression levels of ATF6 in human and PRSS1Tg mouse SAP samples was highly elevated compared with control samples (Figure 1A). Additionally, immunoelectron microscopy (IEM) analysis showed that large amounts of ATF6 localized mainly in the ER and nucleus in SAP tissues from PRSS1Tg mice, while this was rarely observed in pancreatic tissues from untreated PRSS1Tg mice (Figure 1B). Histological analysis showed that myeloperoxidase (MPO) expression, pancreatic acinar cell deformation and inflammatory cell infiltration were markedly increased in human and mouse SAP tissues compared with control tissues (Figure 1C). Ultrastructural analysis by transmission electron microscopy (TEM) revealed extensively irregular and dilated ERs, mitochondrial swelling, nuclear fragmentation and apoptotic bodies were observed in human and PRSS1Tg mouse SAP tissues; however, no obvious abnormal alterations were observed in normal acinar cells (Figure 1C). An increase in the number of apoptotic cells (Figure 1C) and in the expression of the apoptotic cell death markers Caspase-3 and poly ADP-ribose polymerase (PARP) was seen in SAP tissues from PRSS1Tg mice (Figure S1A). These findings suggest a correlation of ATF6 with apoptosis during SAP progression.
ATF6 is upregulated in SAP pancreatic tissues from patients and PRSS1 transgenic (PRSS1Tg) mice accompanying with elevated apoptosis. (A) ATF6 expression was assessed by immunohistochemistry. (B) The localization and expression of ATF6 were examined by immunoelectron microscopy (IEM); black arrows (↑): representative gold nanoparticles. (C) Histological alterations, myeloperoxidase (MPO) activity, acinar cell apoptosis, and microstructural changes in pancreatic tissues from human and PRSS1Tg mice were assessed by hematoxylin and eosin (H&E) staining, immunohistochemistry, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assays and transmission electron microscopy (TEM) as indicated. black arrows (↑): cell nuclei; white arrows (↑): endoplasmic reticula; blue arrows (↑): zymogen granules; purple arrows (↑): mitochondria; red arrows (↑): apoptotic bodies. (-), not treated with caerulein; (+), treated with caerulein. The data are presented as the means ± SDs; * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001. Scale bars = 100 µm.
ATF6 promotes acinar cell injury and multiple organ injury in PRSS1Tg SAP model
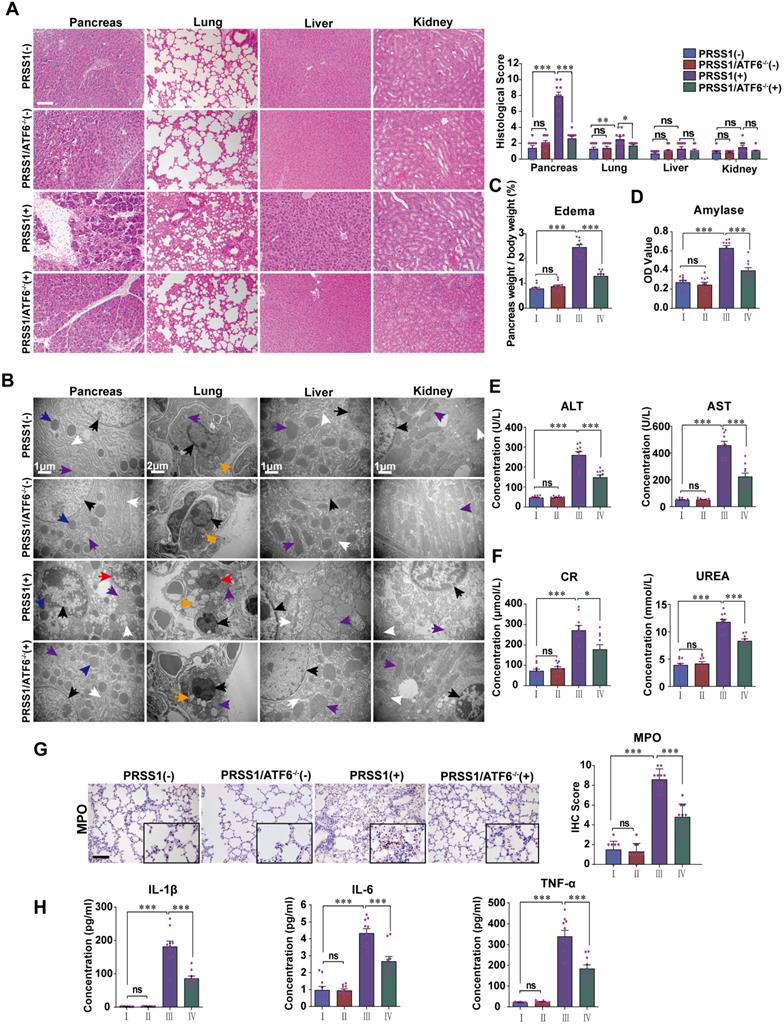
To analyze if increased expression in SAP tissues is indicative of a role for ATF6 during SAP progression, PRSS1Tg mice were crossed with ATF6 knockout (ATF6-/-) mice. Caerulein treatment of PRSS1Tg mice ideally mimicked human SAP with multiple organ injury and could serve as a suitable preclinical model for mechanistic research on pancreatic inflammatory diseases. SAP was induced in PRSS1Tg and PRSS1Tg/ATF6-/- mice by caerulein treatment, followed by analysis of histology, ultrastructure, and function of pancreas, lung, liver, kidney, duodenum, heart, and spleen. Histological analysis showed that the cell deformation and inflammatory cell infiltration seen in the pancreas and lung upon treatment with caerulein were negated in PRSS1Tg/ATF6-/- mice compared to PRSS1Tg mice (Figure 2A). Ultrastructural analysis showed that disorder of ER, swelling of mitochondria, and fragmentation of cell nuclei in pancreatic and liver tissues, swelling and deformation of lamellar bodies in lung tissues, swelling of mitochondria in kidney tissue, destruction of microvilli in duodenum tissue, rupture of myofibrils in heart tissue, and apoptosis of lymphocytes in spleen upon caerulein treatment in PRSS1Tg mice were attenuated in PRSS1Tg/ATF6-/- mice, indicating inhibition of caerulein-induced acinar injury in the absence of ATF6 (Figure 2B and Figure S2B). However, no significant changes upon caerulein treatment were observed in the other tissues examined (Figure 2A and Figure S2B). Caerulein-induced increases in the degree of pancreatic edema (Figure 2C and Figure S2A), levels of pancreas-related serum amylase (Figure 2D), liver-related alanine aminotransferase (ALT) and aspartate aminotransferase (AST) (Figure 2E), kidney-related creatinine (CR) and urea (Figure 2F), MPO activity in lung tissue (Figure 2G), heart-related creatine kinase-MB (CK-MB) (Figure S2C), and the inflammatory cytokines IL-1β, IL-6, and TNF-α in serum (Figure 2H) seen in PRSS1Tg mice were negated upon ATF6 knockout. However, no significant changes were observed in the level of heart-related lactate dehydrogenase (LDH) (Figure S2C).
ATF6 promotes apoptosis, inflammation, and multiple organ injury in the PRSS1Tg SAP mouse model. (A) Histological and (B) microstructural changes in pancreas, lung, liver, and kidney tissues collected from caerulein-treated PRSS1Tg mice and PRSS1Tg/ATF6-/- mice were analyzed by H&E staining and TEM, respectively; black arrows (↑): cell nuclei; white arrows (↑): endoplasmic reticula; blue arrows (↑): zymogen granules; purple arrows (↑): mitochondria; red arrows (↑): apoptotic bodies; orange arrows (↑): lamellar bodies. Degree of edema (C) and serum amylase level (D) in caerulein-treated PRSS1Tg mice and PRSS1Tg/ATF6-/- mice. Expression of liver-related ALT and AST (E), kidney-related CR and urea (F) in serum from caerulein-treated PRSS1Tg mice and PRSS1Tg/ATF6-/- mice. The MPO expression in lung tissues (G) from caerulein-treated PRSS1Tg mice and PRSS1Tg/ATF6-/- mice. (H) The levels of IL-1β, IL-6, and TNF-α in serum were measured by ELISA from caerulein-treated PRSS1Tg mice and PRSS1Tg/ATF6-/- mice. (-), not treated with caerulein; (+), treated with caerulein. Group I, PRSS1Tg (-); group II, PRSS1Tg/ATF6-/- (-); group III, PRSS1Tg (+); group IV, PRSS1Tg/ATF6-/- (+). The data are presented as the means ± SDs; ns, no significant difference; * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001. Scale bars = 100 µm.
To ensure that the inhibition of caerulein-induced effects in PRSS1Tg/ATF6-/- mice was specific to ATF6, complementation was performed in the pancreas using ATF6-expressing adeno-associated virus, with high transfection efficiency. Caerulein-induced cell deformation and inflammatory cell infiltration in pancreatic and lung tissues were reestablished in PRSS1Tg/ATF6-/- mice upon restoration of ATF6 expression (Figure S3A). Caerulein-induced ER dilatation, mitochondrial swelling, and peripheral chromatin condensation in pancreatic and liver tissues, lamellar bodies deformation in lung tissues, mitochondrial swelling in kidney tissues, microvilli destruction in duodenum tissue, myofibrils rupture in heart tissues, and lymphocytes apoptosis in spleen tissues were reestablished in PRSS1Tg/ATF6-/- mice upon restoration of ATF6 expression (Figure S3B). These effects were not significant in the other tissues examined (Figure S3A, B). Caerulein-induced increases in the degree of pancreatic edema (Figure S3C), levels of serum amylase (Figure S3D), ALT and AST (Figure S3E), CR and urea (Figure S3F), MPO activity in lung tissue (Figure S3G), and the inflammatory cytokines IL-1β, IL-6, and TNF-α in serum (Figure S3I) in PRSS1Tg mice that were inhibited upon ATF6 knockout, were reestablished after ATF6 expression was restored. No significant changes were observed in the CK-MB and LDH levels (Figure S3H) upon restoration of ATF6 expression. Collectively, these results indicate that ATF6 promotes acinar cell injury in PRSS1Tg SAP mouse model.
AIFM2 is a regulatory partner of ATF6 in SAP
To elucidate the mechanism underlying ATF6-mediated promotion of acinar cell injury, pancreatic tissues from PRSS1Tg/ATF6-/- mice treated with caerulein were subjected to proteomics analysis, and differentially expressed proteins (DEPs) were identified compared to control animals (Figure 3A). DEPs, with the same changing trends (up or down), from group A versus group B that were also differentially expressed in group A versus group C would be potential ATF6 partners. A total of 73 DEPs were identified. Due to increased apoptosis and structural damage to the ER in SAP, we screened out 14 DEPs, of which functions strongly correlated with apoptosis and ER stress, from the 73 DEPs by performing an analysis with the UniProt database. The data-independent acquisition (DIA) quantification results from the proteomic analysis ultimately identified AIFM2 (an apoptosis-inducing factor (AIF)-homologous mitochondrion-associated protein 2), an inducer of caspase-independent apoptosis [22,23], as a potential downstream regulatory partner of ATF6 (Figure 3B). The upregulated and downregulated proteins were classified by their GO function (Figure S4A-C). A protein-protein interaction (PPI) network of the 14 DEPs was constructed (Figure S4D).
Proteomic analysis to identify potential regulatory proteins related to SAP progression. (A) Schematic representation of the protocol followed for proteomic analysis. (B) Schematic diagram of proteomic screening process to identify target proteins related to SAP progression. (C) Representative images of immunohistochemical staining of AIFM2 in pancreatic tissues from humans and PRSS1Tg mice. (D) Immunofluorescence staining to detect the localization of ATF6 and AIFM2 in human and PRSS1Tg mouse pancreatic tissues. (E) IEM analysis of AIFM2 localization and expression; black arrows (↑): representative gold nanoparticles. The data are presented as the means ± SDs; ns, no significant difference; * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001. Scale bars = 100 µm.
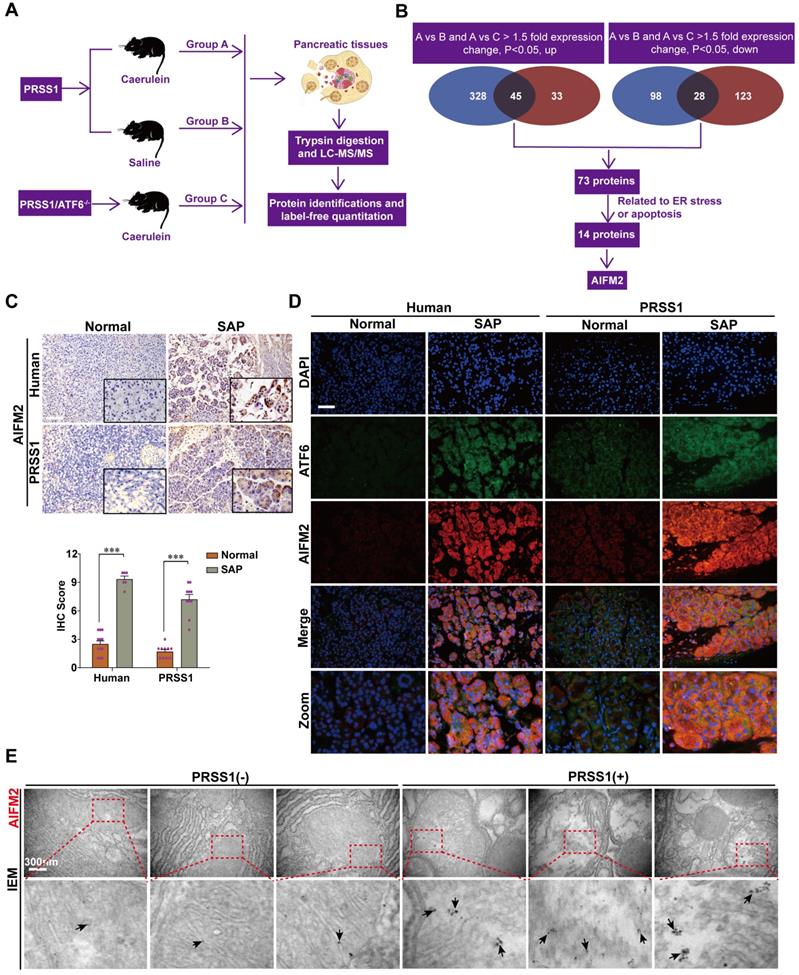
To validate the proteomic identification of AIFM2, we examined its expression in SAP tissues from patients and PRSS1Tg mice by immunohistochemistry and immunofluorescence. Compared with control pancreatic tissues, SAP tissues of humans and PRSS1Tg mice exhibited significantly increased expression of AIFM2 (Figure 3C, D). IEM analysis showed large concentrations of AIFM2 in SAP tissues from PRSS1Tg mice that were localized mainly in mitochondria, but such concentrations were rarely seen in normal pancreatic tissues (Figure 3E). The increased mitochondrial expression of AIFM2 in SAP suggests a role in acinar apoptosis.
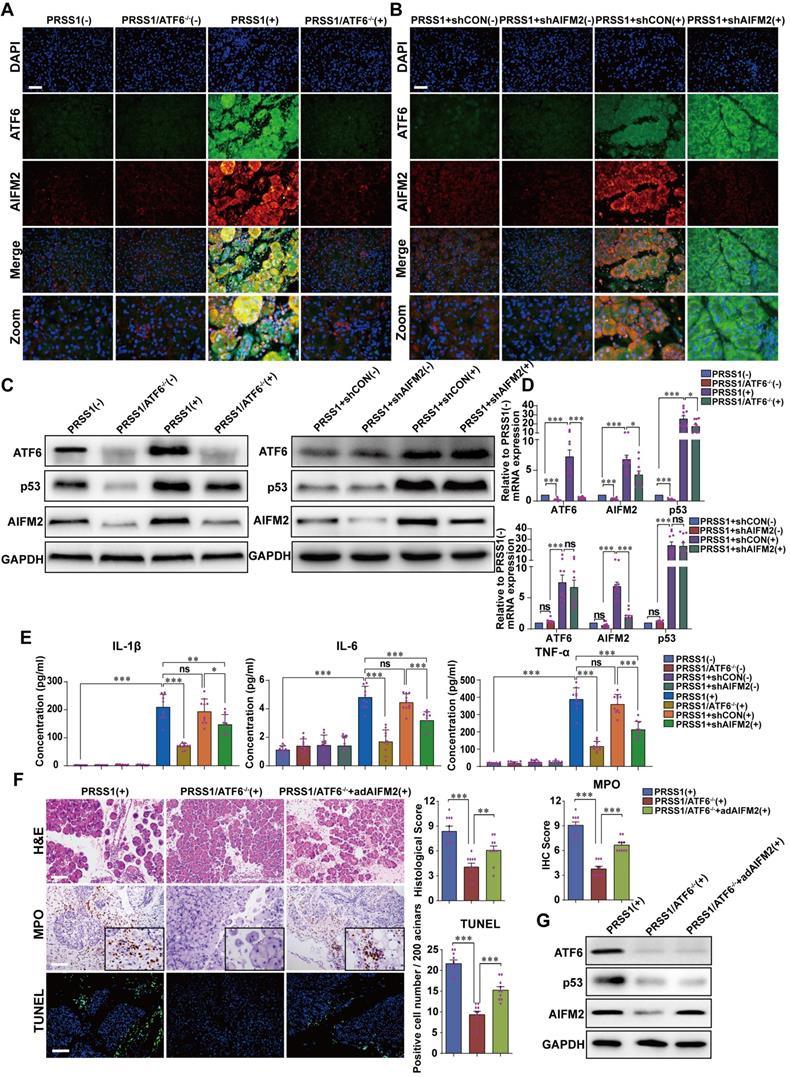
To evaluate the regulatory relationship between ATF6 and AIFM2, the expression of AIFM2 upon ATF6 knockout in SAP tissues from PRSS1Tg mice was assessed. Immunofluorescence (Figure 4A, B), western blotting (Figure 4C) and qRT-PCR (Figure 4D) analysis revealed that the increase in mRNA and protein expression of AIFM2 upon SAP induction was attenuated upon knockout of ATF6 in pancreatic tissues of PRSS1Tg mice. However, upon AIFM2 silencing, the increase in the mRNA and protein expression of ATF6 remained unchanged (Figure 4A-D). Moreover, the levels of the inflammatory cytokines IL-1β, IL-6, and TNF-α in serum were significantly increased in caerulein-treated PRSS1Tg mice but significantly reduced in these SAP model mice with ATF6 knockout or AIFM2 silencing (Figure 4E). To further investigate if AIFM2 is regulated by ATF6 in SAP, AIFM2 was overexpressed in the pancreas of caerulein-treated PRSS1Tg/ATF6-/- mice using adenovirus-based delivery. The caerulein-induced increases in inflammatory cell infiltration, MPO activity, apoptotic cell number, pancreatic edema, and the level of serum amylase in PRSS1Tg mice that were inhibited upon ATF6 knockout, were partially reestablished after AIFM2 overexpression (Figure 4F and Figure S5A-C). The result of western blotting also revealed that the attenuation in protein expression of AIFM2 upon knockout of ATF6 in pancreatic tissues of caerulein-treated PRSS1Tg mice. However, upon restoration of AIFM2 expression, the increase in the protein expression of ATF6 remained unchanged (Figure 4G). Due to AIFM2 is a p53-inducing protein, p53 expression was also measured by western blotting and qRT-PCR which indicated that the increase in p53 expression was attenuated upon knockout of ATF6 in PRSS1Tg mice SAP tissues, but that remained unchanged when silencing or restoration of AIFM2 expression (Figure 4C, D, G). Collectively, these results suggest a regulatory correlation between AIFM2 and ATF6.
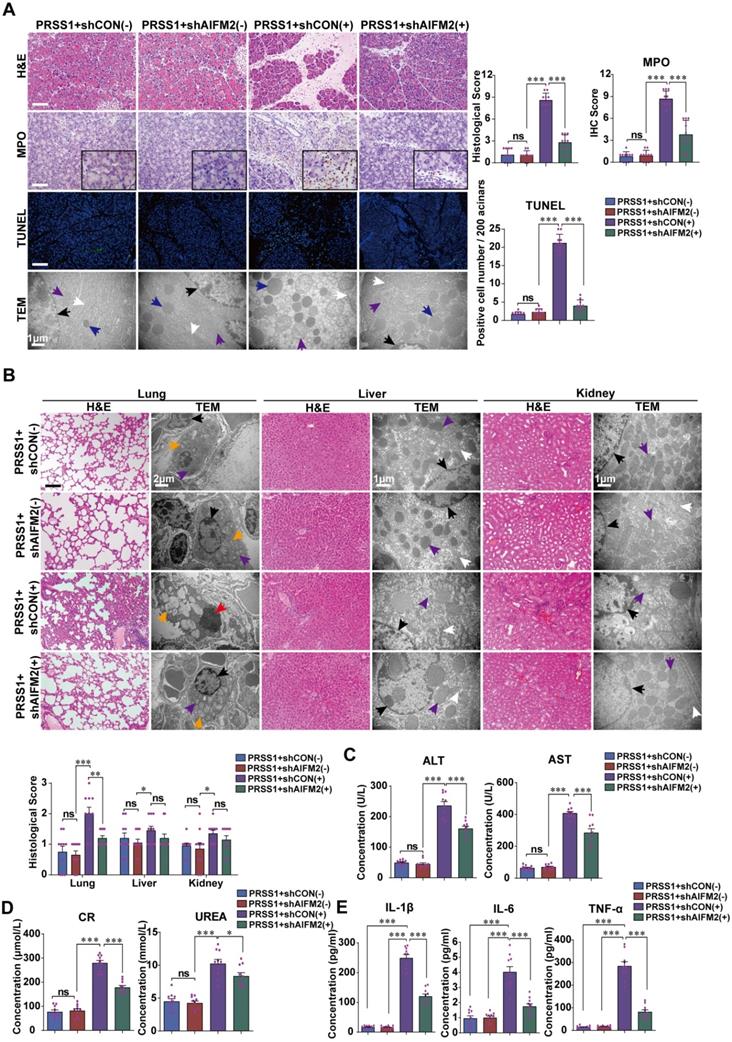
AIFM2 is regulated by ATF6 in the PRSS1Tg SAP mouse model. (A) Immunofluorescence staining to examine the colocalization of ATF6 (green fluorescence) and AIFM2 (red fluorescence) in pancreatic tissues from caerulein-treated PRSS1Tg mice and PRSS1Tg/ATF6-/- mice. (B) shAIFM2 for AIFM2 silencing or control shRNA (shCON) for the negative control were delivered to the pancreas of PRSS1Tg mice. The colocalization of ATF6 (green fluorescence) and AIFM2 (red fluorescence) in pancreatic tissues from PRSS1Tg SAP mice with AIFM2 silencing was examined by immunofluorescence staining. Western blot (C) and qRT-PCR (D) analyses of ATF6, p53, and AIFM2 expression in pancreatic tissues from caerulein-treated PRSS1Tg/ATF6-/- and PRSS1Tg mice with AIFM2 silencing; GAPDH served as the internal loading control. (E) Expression of IL-1β, IL-6, and TNF-α in serum were measured by ELISA from caerulein-treated PRSS1Tg/ATF6-/- and PRSS1Tg mice with AIFM2 silencing. (F) AIFM2-expressing adenovirus (adAIFM2) for AIFM2 overexpression was delivered to the pancreas of PRSS1Tg/ATF6-/- mice. Histological alterations, MPO expression, and acinar cell apoptosis in pancreatic tissues from caerulein-treated PRSS1Tg mice, PRSS1Tg/ATF6-/- mice, and PRSS1Tg/ATF6-/-+adAIFM2 mice were measured by H&E staining, immunohistochemistry, and TUNEL, respectively. (G) Western blot analyses of ATF6, p53, and AIFM2 expression in pancreatic tissues from caerulein-treated PRSS1Tg mice, PRSS1Tg/ATF6-/- mice, and PRSS1Tg/ATF6-/-+adAIFM2 mice; GAPDH served as the internal loading control. The data are presented as the means ± SDs; ns, no significant difference; * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001. Scale bars = 100 µm.
AIFM2 promotes acinar apoptosis, injury and inflammation in SAP
To ascertain the role of AIFM2 in SAP development, we performed shRNA-mediated silencing of AIFM2 through adenoviral transduction in pancreas of PRSS1Tg mice, followed by assessment of pathological changes. The increases in inflammatory cell infiltration, MPO activity, apoptotic cells numbers, ER dilatation, mitochondrial swelling, peripheral chromatin condensation, and levels of inflammatory cytokines observed in PRSS1Tg SAP mice were significantly ameliorated upon AIFM2 silencing (Figure 5A, E). The degree of pancreatic edema, serum amylase level (Figure S6A-C), ALT and AST level (Figure 5C), CR and urea level (Figure 5D) in PRSS1Tg SAP mice that were ameliorated upon AIFM2 silencing. No significant change was observed in the CK-MB and LDH levels (Figure S6F). Consistent with the observations of apoptotic cell numbers, the increase in the expression of Caspase-3 and PARP in PRSS1Tg mouse pancreatic tissues upon SAP induction was markedly negated upon AIFM2 silencing (Figure S6D). Similarly, results of ultrastructural analysis showed that lamellar bodies deformation in lung tissues, mitochondrial swelling in liver and kidney tissues, microvilli destruction in duodenum tissues, myofibrils rupture in heart tissues, and lymphocytes apoptosis in spleen tissues upon SAP induction were markedly negated upon AIFM2 silencing, but no significant differences were observed in the histological analysis of these tissues (Figure 5B and Figure S6E). Collectively, these results indicate that AIFM2 promotes acinar apoptosis during SAP.
AIFM2 silencing ameliorates inflammation and multiple organ injury in SAP. (A) Histological alterations, MPO expression, acinar cell apoptosis, and microstructural changes in pancreatic tissues from PRSS1Tg SAP mice with shAIFM2-mediated AIFM2 silencing were measured by H&E, immunohistochemical staining, TUNEL assays, and TEM, respectively. (B) Histological and ultrastructural evaluation of lung, liver, and kidney tissues from PRSS1Tg SAP model mice treated with shAIFM2. Black arrows (↑): cell nuclei; white arrows (↑): endoplasmic reticula; blue arrows (↑): zymogen granules; purple arrows (↑): mitochondria; red arrows (↑): apoptotic bodies; orange arrows (↑): lamellar bodies. The levels of ALT, AST (C), CR and urea (D) in serum from PRSS1Tg mice treated with shAIFM2 or shCON. (E) Expression of IL-1β, IL-6, and TNF-α in serum were measured by ELISA from PRSS1Tg mice treated with shAIFM2. The data are presented as the means ± SDs; ns, no significant difference; * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001. Scale bars (H&E, MPO) = 100 µm; scale bars (TUNEL) = 200 µm.
ATF6 regulates the p53-AIFM2 pathway in SAP
Given that AIFM2 is a p53-inducible gene, we next ask whether ATF6 regulates AIFM2 and induces apoptosis in SAP by modulating p53 expression. Here, we identified AIFM2 as a regulatory partner of ATF6 in SAP. So, we hypothesize that, during SAP development, ATF6 regulates the p53-AIFM2 pathway via apoptosis. To test this hypothesis, we first examined the expression of p53 in SAP. IEM revealed large concentrations of p53 in PRSS1Tg mouse SAP tissues that were localized mainly in the nucleus and cytoplasm, but such concentrations were rarely seen in normal pancreatic tissues (Figure 6A). Consistent with these results, immunofluorescence analysis revealed that the increased expression of ATF6 and AIFM2 upon SAP induction was associated with increased expression of p53 (Figure S7A, B).
AIFM2 is regulated by p53 and p53 knockout ameliorates acinar apoptosis and inflammation in SAP. (A) Microscopic localization and expression of p53 was examined by IEM; black arrows (↑): representative gold nanoparticles. (B) Representative images of H&E staining, TUNEL, and MPO activity in caerulein-treated PRSS1Tg/p53-/- mice. Western blot (C) and qRT-PCR (D) analyses of ATF6, AIFM2, and p53 expression in pancreatic tissues from PRSS1Tg/p53-/- mice; GAPDH served as the internal loading control. (E) Histological alterations, MPO expression, and acinar cell apoptosis in pancreatic tissues from caerulein-treated PRSS1Tg mice, PRSS1Tg/p53-/- mice, and PRSS1Tg/p53-/-+adATF6 mice were assessed by H&E staining, immunohistochemistry, and TUNEL, respectively. (F) Western blot analyses of ATF6, p53, and AIFM2 expression in pancreatic tissues from caerulein-treated PRSS1Tg mice, PRSS1Tg/p53-/- mice, and PRSS1Tg/p53-/-+adATF6 mice. The data are presented as the means ± SDs; * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001. Scale bars = 100 µm.
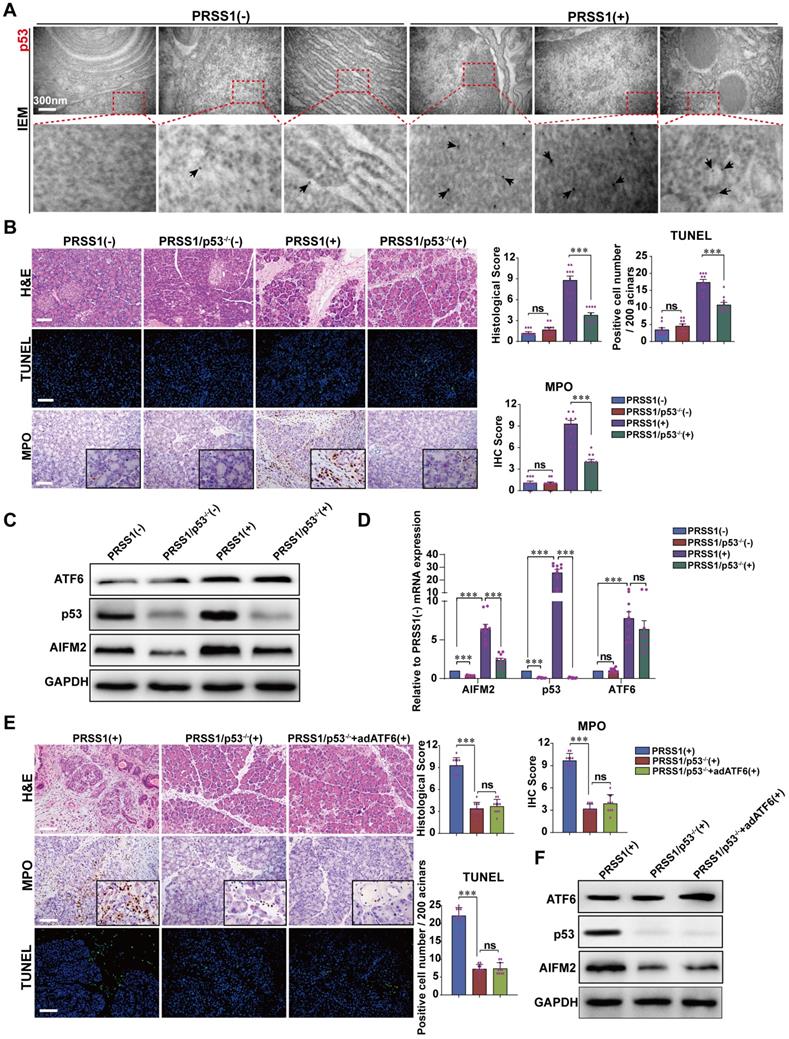
To confirm that p53 is involved in SAP, PRSS1Tg mice were crossed to p53 knockout (p53-/-) mice. Compared with caerulein-treated PRSS1Tg mice, caerulein-treated PRSS1Tg/p53-/- mice exhibited significantly attenuated pancreatic acinar cell deformation, inflammatory cell infiltration, apoptotic cell numbers, and MPO activity (Figure 6B). Similar results were seen upon p53 inhibition using PFT-α in caerulein-treated PRSS1Tg mice (Figure S8A, B). The reestablishment of caerulein-induced increases in inflammatory cell infiltration, MPO activity, apoptotic cell numbers, ER disorder (Figure S8A), and levels of inflammatory cytokines (Figure S8B) upon ATF6 complementation in PRSS1Tg/ATF6-/- mice was also attenuated upon p53 inhibition. ATF6 overexpression in pancreas of caerulein-treated PRSS1Tg/p53-/- mice using adenoviral delivery of ATF6 did not affect the attenuation in inflammatory cell infiltration, MPO activity, apoptotic cell numbers (Figure 6E), p53 and AIFM2 expression (Figure 6F), pancreatic edema, and serum amylase level (Figure S7C-E).
Western blotting (Figure 6C) and qRT-PCR (Figure 6D) analyses revealed that following p53 knockout in SAP tissues, the expression of AIFM2 was decreased but higher than control, while the expression of ATF6 did not change. Additionally, immunofluorescence analysis revealed that the increase in p53 expression upon SAP induction was attenuated upon ATF6 knockout (Figure S8C). These data indicate that p53 involves in acinar apoptosis and ATF6 regulates acinar apoptosis via p53-AIFM2 pathway during SAP development.
ATF6 transcriptionally regulates p53 and AIFM2 expressions in acinar cells
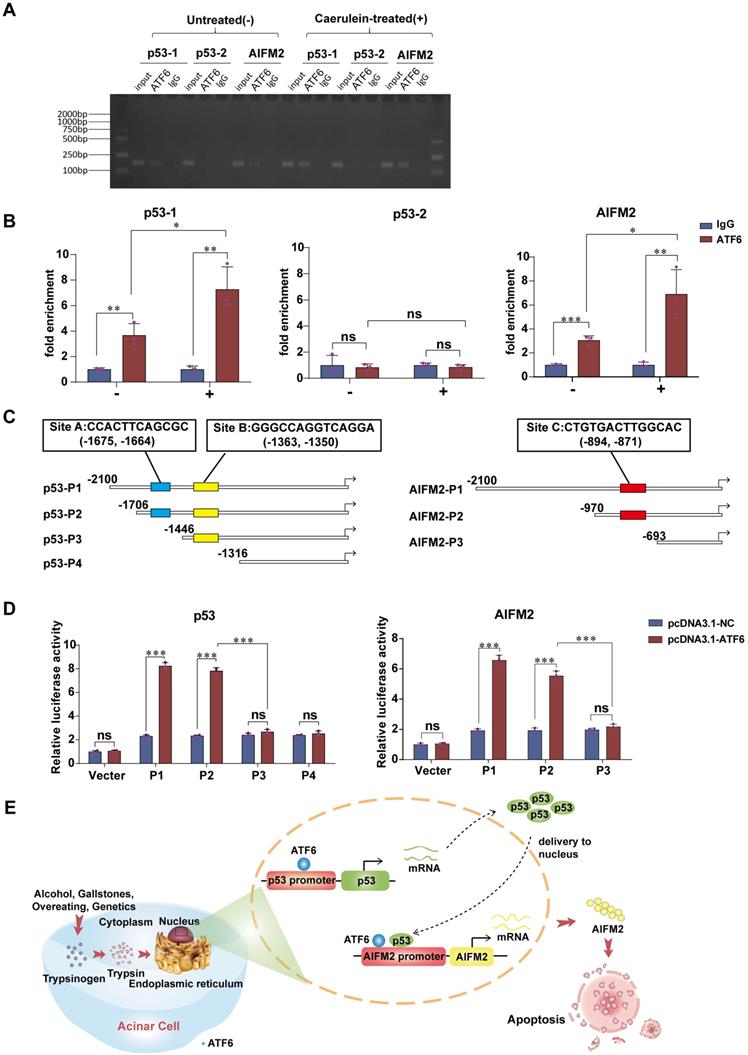
Due to its role of transcription factor, we speculate that ATF6 regulates the expression of AIFM2 by binding to the p53 and/or AIFM2 promoter in SAP. ChIP-qPCR (Figure 7A) was conducted to verify the prediction that there were two potential binding sites of ATF6 on the p53 promoter and one binding site on the AIFM2 promoter by Jaspar and hTFtarget. ChIP assay showed substantial increase in the binding of ATF6 to chromatin region of the p53-1 and AIFM2 promoter upon use of caerulein in pancreatic acinar cells, while p53-2 promoter exhibited no difference (Figure 7B), which indicated that ATF6 could bind to both the p53 promoter and the AIFM2 promoter, but ATF6 only bound to prediction site p53-1, not prediction site p53-2 in SAP. Based on the predicted binding sites of ATF6 to p53 and AIFM2 promoter (marked as Site A, B and C), four deletion mutants of p53 promoter (p53-P1, p53-P2, p53-P3, p53-P4) and three deletion mutants of AIFM2 promoter (AIFM2-P1, AIFM2-P2, AIFM2-P3) (Figure 7C) were respectively cloned into promoter-reporter vectors. Their activities in 293T cells were measured 48h after co-transfection by luciferase reporter system. Compared to p53-P2, p53-P3 and p53-P4, higher luciferase activities of p53-P1 suggested that ATF6 regulated p53 transcription by binding to the A site of the p53 promoter rather than the B site. ATF6-induced promoter activity of AIFM2-P2 was hardly declined in comparison with AIFM2-P1, indicating that ATF6 regulated the transcriptional activity of AIFM2 by binding to the C site of the AIFM2 promoter (Figure 7D). Together, using ChIP and luciferase assays, we conclude that ATF6 regulates the transcriptional activity of p53 by interacting with the p53 promoter through the consensus sites between -1706 and -1446, and ATF6 regulates the transcription of AIFM2 by interacting with the AIFM2 promoter at the consensus sites between -970 and -693 in SAP.
ATF6 binds to the promoters of p53 and AIFM2 in acinar cells. (A) Binding of ATF6 to p53 and AIFM2 promoter region in vitro by chromatin immunoprecipitation (ChIP) using anti-ATF6 or anti-IgG antibodies in untreated (-) or caerulein-treated (+) pancreatic acinar cells of PRSS1Tg mice. (B) Input and immunoprecipitated DNA purified by ChIP were measured using qRT-PCR. (C) Schematic of deletion mutants from p53 and AIFM2 promoters. The predicted ATF6 binding sites were marked as Site A, B and C. (D) Luciferase reporter studies on the p53 and AIFM2 promoter in 293T cells. Cells were co-transfected with a series of mutants of p53 or AIFM2 promoter plasmid and either Atf6 overexpression plasmids or the control plasmids. Luciferase activities was measured 48h later. (E) A mechanistic figure for regulation of ATF6 on p53-AIFM2 pathway through apoptosis in SAP. The data are presented as the means ± SDs; ns, no significant difference; * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001.
Discussion
In the present study, we focused on understanding the importance of ATF6 for SAP development using a preclinical SAP mouse model which could mimic the “real world” performance of SAP. Our data identified ATF6 as a critical molecule in trypsin-induced acinar ER stress and apoptosis. Transcription factor ATF6 expression was elevated in pancreatic tissues of SAP and promoted acinar apoptosis. AIFM2, which is involved in the p53-induced apoptosis pathway, was upregulated by ATF6 in SAP. In turn, AIFM2 was downregulated when ATF6 was silenced, and this downregulation was accompanied with decreased apoptosis and inflammation. Moreover, ATF6 promoted p53 and AIFM2 transcription to induce acinar apoptosis in SAP (Figure 7E). These results provide the first indication that the AIFM2 regulates SAP with multiple organ injury via ATF6/p53 mediated apoptosis. Theoretically, ATF6 could be translated to a valuable therapeutic target to improve the dilemma of therapeutic limitations of SAP.
Current knowledge indicates that inflammation induces protein misfolding in the ER, which compromises cellular function and promotes apoptosis and the development of inflammatory diseases [10,11]. To better understand the pathogenesis of SAP, we constructed a preclinical PRSS1Tg mouse model that could mimic the development of SAP. Ultrastructural analysis of human pancreatic tissues and SAP model tissues from the pancreas, lung, liver, kidney, duodenum, heart, and spleen conducted using TEM revealed that the structures of cellular organelles, such as the ER, mitochondria and nucleus, changed significantly in multiple organ injury during the pathogenesis of SAP. Importantly, we observed significant increases in the destruction of ER and mitochondria, and in apoptosis (nucleus) in pancreatic tissues from both SAP model mice and patients. This manifestation suggests ER-mitochondrial-nuclear crosstalk involves and probably plays a role in SAP progression.
ATF6, a transmembrane-type glycoprotein on the ER membrane, functions as not only a UPR sensor/transducer, but also a transcription factor. When misfolded proteins accumulate in the ER, ATF6 transits to the Golgi apparatus, where it is processed to produce a cytosolic fragment that is the primary mediator of the adaptive response to ER protein misfolding [11,24]. While the core function of ATF6 is to restore homeostasis, it can also induce apoptosis [25,26]. In the present study, we found that ATF6 expression was elevated in pancreatic tissues from SAP patients and PRSS1Tg mice and that it regulated acinar injury through apoptosis. The number of ATF6-linked gold beads was significantly higher in areas of ER rupture and nucleus, and mitochondrial swelling in SAP acinar cells compared to normal, as visualized by IEM. Interestingly, we found that ATF6-linked gold beads also surrounded the membrane of zymogen granules. This manifestation suggests an interaction between PRSS1 and ATF6. Importantly, we revealed that during SAP development, ATF6 promoted acinar apoptosis through binding p53 and AIFM2 promoter sites, which had never been reported. The above results confirm that ATF6 plays an important transcriptional role during SAP.
ER-mitochondrial interconnections form specific microdomains that play important roles in mitochondrial dynamics, inflammation, autophagy, and apoptosis [27]. The physical and functional crosstalk between these two dynamic organelles emphasizes the outcome of altered ER-mitochondrial interconnections in SAP [28]. Considering this interrelationship, we focused on identifying proteins that partner with ATF6 in roles related to mitochondrial functions and apoptosis. AIFM2 was identified as a functional ATF6-downregulating partner in SAP.
AIFM2, also named apoptosis-inducing factor-like mitochondrion-associated inducer of death (AMID) or PRG3, exhibits 90% similarity between mice and humans, is located on chromosome 10 in humans. It plays a role in mediating the p53-dependent apoptosis response and has been demonstrated to be an apoptosis-inducing factor-homologous mitochondrion-associated protein that induces caspase-independent apoptosis in humans and mice [22,23]. AIFM2 expression is regulated by p53, which binds to p53-responsive elements in the AIFM2 promoter region [29]. AIFM2 plays a key role in promoting cell proliferation, invasion, and migration [30,31]. Also, AIFM2 was identified as a NADH oxidase, which regenerates NAD to promote glycolysis for thermogenesis [32]. In human breast [33], gastric [34] and lung [35] cancer cells, apoptosis is mediated by the upregulation of AIFM2 expression and accumulation of AIFM2 in the mitochondria. In our SAP mouse model, AIFM2 accumulated mainly in the mitochondria. AIFM2 silencing significantly abated acinar apoptosis and injury; moreover, ATF6 knockdown downregulated AIFM2 expression, which was accompanied by reduced apoptosis and acinar injury in SAP. These results suggest a functional role for the ATF6-AIFM2 pathway in ER-mitochondrial-nuclear crosstalk in SAP. In addition to its role in apoptosis, AIFM2 was recently identified as a potent ferroptosis resistance factor (and is also called ferroptosis suppressor protein 1, FSP1), which functions as an oxidoreductase that reduces coenzyme Q10, generating a lipophilic radical-trapping antioxidant. FSP1 expression positively correlates with ferroptosis resistance across hundreds of cancer cell lines and FSP1 mediates resistance to ferroptosis in lung cancer both in vivo and in vitro [36,37]. Therefore, AIFM2 may play different roles in different cell death pathways.
Under physiological conditions, p53 is expressed at low levels. Following DNA damage, p53 rapidly accumulates and becomes an active transcription factor for a set of downstream genes. Transcription of the human AMID gene is regulated by p53, and overexpression of AMID causes chromatin condensation at the nuclear periphery and apoptotic body formation [16,38], consistent with our results in SAP (Figure 6). Apoptosis induced by AMID is regulated by p53, is caspase-independent and is not affected by overexpression of the Bcl-2 protein [22,39]. In addition to apoptotic functions, alterations in the mitochondrial membrane potential, mitochondrial ROS production and/or cytochrome C release can also result from p53-mediated transcriptional activation of mitochondrial proteins such as Noxa, p53, AIP1 and Bax [40]. Our data also showed that p53 knockout in the PRSS1Tg SAP mouse model significantly downregulated the expression of AIFM2 and alleviated acinar apoptosis and injury. Combined with our previous results (Figure 6, 7 and Figure S8), this experimental evidence demonstrates that the p53-AIFM2 pathway regulates acinar apoptosis and injury mediated by ATF6 in SAP, thus confirming the functional role of the ATF6-p53-AIFM2 pathway in SAP.
Coordination between the mitochondria and the nucleus is essential for maintaining cellular homeostasis and ensuring an effective response to stress. Under physiological or various cellular stress conditions, p53 can regulate diverse cellular processes, including DNA repair, the cell cycle, apoptosis, redox homeostasis and metabolism [41]. In addition to its nuclear role, p53 can be localized to mitochondria, where it executes various transcription-independent functions [42]. In our SAP mouse model, we found that p53 was localized mainly in the nucleus and cytoplasm (Figure 6A). Our ChIP and luciferase studies provide evidence that ATF6 binds to the p53 promoter and subsequently activates the p53-AIFM2 pathway. Moreover, the directly binding of ATF6 with the AIFM2 promoter synergistically regulates acinar apoptosis in SAP development (Figure 7). Thus, ATF6 activates the p53-AIFM2 pathway by binding to the p53 and AIFM2 promoter, leading to apoptosis.
In conclusion, our study sheds new light on a mechanism by which the p53-AIFM2 pathway regulates SAP with multiple organ injury via ATF6-mediated apoptosis, suggesting that ER-mitochondrial-nuclear crosstalk plays an important role in SAP development. These findings provide novel insights into ATF6-triggered signaling as a promising targeted therapy for pancreatitis and other common human diseases.
Abbreviations
ATF6: activating transcription factor 6; AIFM2: apoptosis-inducing factor mitochondria-associated 2; SAP: severe acute pancreatitis; PRSS1: serine protease 1; PRSS1Tg: PRSS1 transgenic; qRT-PCR: quantitative real-time PCR; ChIP: chromatin immunoprecipitation; AP: acute pancreatitis; MOF: multiple organ failure; ER: endoplasmic reticulum; UPR: unfolded protein response; CP: chronic pancreatitis; PFT-α: pifithrin-α; DDA: data-dependent acquisition; DIA: data-independent acquisition; GO: Gene Ontology; COG: Clusters of Orthologous Groups; TEM: transmission electron microscopy; IEM: immunoelectron microscopy; PBS: phosphate-buffered saline; PB: phosphate buffer; PARP: poly ADP-ribose polymerase; MPO: myeloperoxidase; TUNEL: transferase-mediated d-UTP nick-end-labeling; ELISA: enzyme-linked immunosorbent assay; IL-1β: interleukin 1β; IL-6: interleukin 6; TNF-α: tumor necrosis factor α; ALT: alanine aminotransferase; AST: aspartate aminotransferase; CR: creatinine; CK-MB: creatine kinase-MB; LDH: lactate dehydrogenase; DEPs: differentially expressed proteins; AIF: apoptosis-inducing factor; PPI: protein-protein interaction; AMID: apoptosis-inducing factor-like mitochondrion-associated inducer of death; PRG3: p53-responsive gene 3; FSP1: ferroptosis suppressor protein 1.
Supplementary Material
Supplementary methods, figures and tables.
Acknowledgements
We thank Chen-Yu Yang in the Center of Cryo-Electron Microscopy (CCEM), Zhejiang University for her technical assistance on Immunolabeled Transmission Electron Microscopy; thank Yan-Meng Lu in the Electronic Microscope Room, Central Laboratory, Southern Medical University for her technical assistance on Transmission Electron Microscopy; thank Jin Su in the State Key Lab of Respiratory Disease, Guangzhou Institute of Respiratory Disease for providing her laboratory on immunohistochemistry; thank Ya-Ping Ye and Ling-Jie Zhang in the Department of Pathology, School of Basic Medical Sciences, Southern Medical University for their assistance in pathological section.
Access to data
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD016703.
Funding
The article is supported by Guangdong Science and Technology Planning Project (2019A030317018) & Scientific Research Startup Program of Southern Medical University by High-level University Construction Funding of Guangdong Provincial Department of Education (CX2018N012) & Clinical Research Program of Nanfang Hospital, Southern Medical University (2018CR046) & College Students' Innovative Entrepreneurial Training Plan Program (201812121128, 201812121263) & Clinical Research Startup Program of Southern Medical University by High-level University Construction Funding of Guangdong Provincial Department of Education (LC2016PY011) & National Natural Science Foundation of China (81801487).
Author Contributions
Guo-Wei Zhang and Jin Jin conceived and designed the study. Jie-Hui Tan, Rong-Chang Cao, Lei Zhou, Zhi-Tao Zhou, Yang-Chen Jin, Jia-Yu Lin, Jun-Ling Zeng, Shu-Ji Li and Min Luo contributed to carry out the experiments. Jie-Hui Tan and Rong-Chang Cao provided clinical samples and clinical information. Rong-Chang Cao, Huo-Ji Chen, Jia Xu, Xue-Mei Chen, Guo-Dong Hu and Xiao-Bing Yang participated in data analysis. Jie-Hui Tan drafted the manuscript. Guo-Wei Zhang and Jin Jin revised the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Leppäniemi A, Tolonen M, Tarasconi A, Segovia-Lohse H, Gamberini E, Kirkpatrick AW. et al. 2019 WSES guidelines for the management of severe acute pancreatitis. World J Emerg Surg. 2019;14:27
2. Wolbrink DRJ, Kolwijck E, Ten Oever J, Horvath KD, Bouwense SAW, Schouten JA. Management of infected pancreatic necrosis in the intensive care unit: a narrative review. Clin Microbiol Infect. 2020;26:18-25
3. Moggia E KRBA. Pharmacological interventions for acute pancreatitis. Cochrane Database Syst Rev. 2017;4:D11384
4. Garg PK, Singh VP. Organ Failure Due to Systemic Injury in Acute Pancreatitis. Gastroenterology. 2019;156:2008-23
5. Yin T, Peeters R, Liu Y, Feng Y, Zhang X, Jiang Y. et al. Visualization, Quantification and Characterization of Caerulein-Induced Acute Pancreatitis in Rats by 3.0T Clinical MRI, Biochemistry and Histomorphology. Theranostics. 2017;7:285-94
6. Sah RP, Garg P, Saluja AK. Pathogenic mechanisms of acute pancreatitis. Curr Opin Gastroenterol. 2012;28:507-15
7. RP S, RK D, AK S. New insights into the pathogenesis of pancreatitis. Curr Opin Gastroenterol. 2013;29:523-30
8. Gui F, Zhang Y, Wan J, Zhan X, Yao Y, Li Y. et al. Trypsin activity governs increased susceptibility to pancreatitis in mice expressing human PRSS1R122H. The Journal of clinical investigation. 2020;130:189-202
9. Whitcomb DC. Hereditary pancreatitis: new insights into acute and chronic pancreatitis. Gut. 1999;45:317-22
10. Wu JS, Li WM, Chen YN, Zhao Q, Chen QF. Endoplasmic reticulum stress is activated in acute pancreatitis. J Dig Dis. 2016;17:295-303
11. Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature. 2016;529:326-35
12. Jao TM, Nangaku M, Wu CH, Sugahara M, Saito H, Maekawa H. et al. ATF6α downregulation of PPARα promotes lipotoxicity-induced tubulointerstitial fibrosis. Kidney Int. 2019;95:577-89
13. Nakanishi K, Sudo T, Morishima N. Endoplasmic reticulum stress signaling transmitted by ATF6 mediates apoptosis during muscle development. J Cell Biol. 2005;169:555-60
14. Tsutsumi S, Gotoh T, Tomisato W, Mima S, Hoshino T, Hwang HJ. et al. Endoplasmic reticulum stress response is involved in nonsteroidal anti-inflammatory drug-induced apoptosis. Cell Death Differ. 2004;11:1009-16
15. Zhou L, Tan JH, Cao RC, Xu J, Chen XM, Qi ZC. et al. ATF6 regulates the development of chronic pancreatitis by inducing p53-mediated apoptosis. Cell Death Dis. 2019;10:662
16. Ohiro Y, Garkavtsev I, Kobayashi S, Sreekumar KR, Nantz R, Higashikubo BT. et al. A novel p53-inducible apoptogenic gene, PRG3, encodes a homologue of the apoptosis-inducing factor (AIF). FEBS Lett. 2002;524:163-71
17. Wu M, Xu LG, Su T, Tian Y, Zhai Z, Shu HB. AMID is a p53-inducible gene downregulated in tumors. Oncogene. 2004;23:6815-9
18. Esrefoglu M, Gul M, Ates B, Selimoglu MA. Ultrastructural clues for the protective effect of melatonin against oxidative damage in cerulein-induced pancreatitis. J Pineal Res. 2006;40:92-7
19. Reinshagen K. Autophagocytosis of the apical membrane in microvillus inclusion disease. Gut. 2002;51:514-21
20. Tambasco De Oliveira P, Nanci A. Nanotexturing of titanium-based surfaces upregulates expression of bone sialoprotein and osteopontin by cultured osteogenic cells. Biomaterials. 2004;25:403-13
21. Foygel K, Wang H, Machtaler S, Lutz AM, Chen R, Pysz M. et al. Detection of Pancreatic Ductal Adenocarcinoma in Mice by Ultrasound Imaging of Thymocyte Differentiation Antigen 1. Gastroenterology. 2013;145:885-94
22. Wu M, Xu L, Li X, Zhai Z, Shu H. AMID, an Apoptosis-inducing Factor-homologous Mitochondrion-associated Protein, Induces Caspase-independent Apoptosis. J Biol Chem. 2002;277:25617-23
23. Bilyy R, Kit Y, Hellman U, Stoika R. AMID: new insights on its intracellular localization and expression at apoptosis. Apoptosis. 2008;13:729-32
24. Yatchenko Y, Horwitz A, Birk R. Endocrine and exocrine pancreas pathologies crosstalk: Insulin regulates the unfolded protein response in pancreatic exocrine acinar cells. Exp Cell Res. 2019;375:28-35
25. Xiong Y, Chen H, Lin P, Wang A, Wang L, Jin Y. ATF6 knockdown decreases apoptosis, arrests the S phase of the cell cycle, and increases steroid hormone production in mouse granulosa cells. Am J Physiol Cell Physiol. 2017;312:C341-53
26. Iurlaro R, Muñoz Pinedo C. Cell death induced by endoplasmic reticulum stress. The FEBS Journal. 2016;283:2640-52
27. Veeresh P, Kaur H, Sarmah D, Mounica L, Verma G, Kotian V. et al. Endoplasmic reticulum-mitochondria crosstalk: from junction to function across neurological disorders. Ann NY Acad Sci. 2019;1457:41-60
28. Biczo G, Vegh ET, Shalbueva N, Mareninova OA, Elperin J, Lotshaw E. et al. Mitochondrial Dysfunction, Through Impaired Autophagy, Leads to Endoplasmic Reticulum Stress, Deregulated Lipid Metabolism, and Pancreatitis in Animal Models. Gastroenterology. 2018;154:689-703
29. Marshall KR, Gong M, Wodke L, Lamb JH, Jones DJL, Farmer PB. et al. The Human Apoptosis-inducing Protein AMID Is an Oxidoreductase with a Modified Flavin Cofactor and DNA Binding Activity. J Biol Chem. 2005;280:30735-40
30. Chen W, Liu H, Wang T, Bao G, Wang N, Li R. Downregulation of AIF-2 Inhibits Proliferation, Migration, and Invasion of Human Glioma Cells via Mitochondrial Dysfunction. J Mol Neurosci. 2019;68:304-10
31. Kojima N, Tanaka Y, Kulkeaw K, Nakanishi Y, Shirasawa S, Sugiyama D. Apoptosis-inducing Factor, Mitochondrion-associated 2, Regulates Klf1 in a Mouse Erythroleukemia Cell Line. Anticancer Res. 2015;35:4493-9
32. Nguyen HP, Yi D, Lin F, Viscarra JA, Tabuchi C, Ngo K. et al. Aifm2, a NADH Oxidase, Supports Robust Glycolysis and Is Required for Cold- and Diet-Induced Thermogenesis. Mol Cell. 2020;77:600-17
33. Tsuchiya A, Kanno T, Saito M, Miyoshi Y, Gotoh A, Nakano T. et al. Intracellularly transported adenosine induces MCF-7 human breast cancer cells by accumulating AMID in the nucleus. Cancer Lett. 2012;321:65-72
34. Ohyama M, Tsuchiya A, Kaku Y, Kanno T, Shimizu T, Tanaka A. et al. Phosphatidylinositol Derivatives Induce Gastric Cancer Cell Apoptosis by Accumulating AIF and AMID in the Nucleus. Anticancer Res. 2015;35:6563-71
35. Lu J, Chen J, Xu N, Wu J, Kang Y, Shen T. et al. Activation of AIFM2 enhances apoptosis of human lung cancer cells undergoing toxicological stress. Toxicol Lett. 2016;258:227-36
36. Doll S, Freitas FP, Shah R, Aldrovandi M, Da Silva MC, Ingold I. et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693-698
37. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH. et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688-692
38. Mei J, Webb S, Zhang B, Shu H. The p53-inducible apoptotic protein AMID is not required for normal development and tumor suppression. Oncogene. 2006;25:849-56
39. Vařecha M, Amrichová J, Zimmermann M, Ulman V, Lukášová E, Kozubek M. Bioinformatic and image analyses of the cellular localization of the apoptotic proteins endonuclease G, AIF, and AMID during apoptosis in human cells. Apoptosis. 2007;12:1155-71
40. Moll UM, Zaika A. Nuclear and mitochondrial apoptotic pathways of p53. FEBS Lett. 2001;493:65-9
41. Park JH, Zhuang J, Li J, Hwang PM, Just W. p53 as guardian of the mitochondrial genome. FEBS Lett. 2016;590:924-34
42. Wickramasekera NT, Das GM. Tumor suppressor p53 and estrogen receptors in nuclear-mitochondrial communication. Mitochondrion. 2014;16:26-37
Author contact
![]() Corresponding authors: Guo-Wei Zhang, MD, PhD, Address: No.1838, North Guangzhou Avenue, Guangzhou 510515, P.R. China. Mobile: +86-13600039982; E-mail: guoweizhang77com.
Corresponding authors: Guo-Wei Zhang, MD, PhD, Address: No.1838, North Guangzhou Avenue, Guangzhou 510515, P.R. China. Mobile: +86-13600039982; E-mail: guoweizhang77com.