Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
CSC definition and key features
The transcriptional regulation...
OCT4
SOX2
KLF4
c-MYC
NANOG
Wnt/TCF4
STAT3
NF-κB
Post-transcriptional regulation...
RNA methylation in CSCs
RNA editing in CSCs
Dysregulation of RNA splicing in...
CSC epigenetics
DNA methylation in CSCs
Chromatin remodeling in CSCs
Noncoding RNAs in CSCs
CSC metabolism
CSC microenvironment
Cancer associated fibroblasts...
Tumor vasculature
Immune cells in the TME
Cytotoxic T lymphocytes and...
CSC in therapy resistance
Quiescence
Epithelial-to-mesenchymal...
Multi-drug resistance
Resistance to DNA damage-induced...
Therapies targeting CSC
Targeting of key CSC signaling...
Viral therapy
Awakening quiescent CSCs
Immunotherapy
Conclusions and future...
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(19):8721-8743. doi:10.7150/thno.41648 This issue Cite
Review
Stem cell programs in cancer initiation, progression, and therapy resistance
Tianzhi Huang1* ![]() , Xiao Song1*, Dandan Xu2*, Deanna Tiek1, Anshika Goenka1, Bingli Wu1, Namratha Sastry1, Bo Hu1, Shi-Yuan Cheng1
, Xiao Song1*, Dandan Xu2*, Deanna Tiek1, Anshika Goenka1, Bingli Wu1, Namratha Sastry1, Bo Hu1, Shi-Yuan Cheng1 ![]()
1. The Ken & Ruth Davee Department of Neurology, Lou and Jean Malnati Brain Tumor Institute, The Robert H. Lurie Comprehensive Cancer Center, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA
2. Department of Pathology, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA
*These authors contributed equally to this work
Received 2019-10-30; Accepted 2020-4-9; Published 2020-7-9
Abstract

Over the past few decades, substantial evidence has convincingly revealed the existence of cancer stem cells (CSCs) as a minor subpopulation in cancers, contributing to an aberrantly high degree of cellular heterogeneity within the tumor. CSCs are functionally defined by their abilities of self-renewal and differentiation, often in response to cues from their microenvironment. Biological phenotypes of CSCs are regulated by the integrated transcriptional, post-transcriptional, metabolic, and epigenetic regulatory networks. CSCs contribute to tumor progression, therapeutic resistance, and disease recurrence through their sustained proliferation, invasion into normal tissue, promotion of angiogenesis, evasion of the immune system, and resistance to conventional anticancer therapies. Therefore, elucidation of the molecular mechanisms that drive cancer stem cell maintenance, plasticity, and therapeutic resistance will enhance our ability to improve the effectiveness of targeted therapies for CSCs. In this review, we highlight the key features and mechanisms that regulate CSC function in tumor initiation, progression, and therapy resistance. We discuss factors for CSC therapeutic resistance, such as quiescence, induction of epithelial-to-mesenchymal transition (EMT), and resistance to DNA damage-induced cell death. We evaluate therapeutic approaches for eliminating therapy-resistant CSC subpopulations, including anticancer drugs that target key CSC signaling pathways and cell surface markers, viral therapies, the awakening of quiescent CSCs, and immunotherapy. We also assess the impact of new technologies, such as single-cell sequencing and CRISPR-Cas9 screening, on the investigation of the biological properties of CSCs. Moreover, challenges remain to be addressed in the coming years, including experimental approaches for investigating CSCs and obstacles in therapeutic targeting of CSCs.
Keywords: cancer stem cells, transcriptional and posttranslational regulation, epigenetics, metabolism, tumor microenvironment, therapy resistance, CSC-targeting therapies
Introduction
Tumors are complex systems that include cancerous cells as well as the associated tumor microenvironment (TME), which is comprised of cancer-associated fibroblasts (CAFs), vasculature, infiltrating immune cells, and other components. Considerable efforts have been made to model the complexity of cancer by integrating the TME to interrogate tumor heterogeneity based on epigenetic and genetic variations. The concept of cancer stem cells (CSCs) states that tumors, like their normal tissue counterparts, have a unidirectional cellular hierarchy with CSCs at the apex, which are responsible for sustaining tumorigenicity and recapitulating the cellular heterogeneity inherent within the original tumor [1]. The CSC model stimulated intense interest when evidence emerged supporting the model in human acute myeloid leukemia (AML) and breast cancer. It was found that a small subpopulation of cancer cells were capable of initiating leukemia when transplanted into immune-deficient mice. Moreover, these AML cells could be enriched by using a combination of the cell surface markers CD34+CD38- [2], while breast cancer-propagating cells were marked with CD44+CD24-/low [3]. There is now convincing evidence for CSCs in a variety of solid tumors, including brain [4, 5], prostate [6, 7], colon [8, 9], pancreatic [10], ovarian [11], and lung [12]. Although the existence of CSCs has been evident in various cancer types, it remains uncertain whether the CSC model applies to all or only some cancers due to issues concerning the robustness of CSC markers and the extent to which existing assays underestimate the tumorigenic cell frequencies.
The CSC model has received wide attention because it explains the clinical observation that many treatments seem to initially eradicate cancer cells, but later the cancer will often return. Therefore, it is of considerable importance in the clinic to target CSCs within the tumor to prevent tumor relapse. CSCs are often more resistant to currently available anticancer therapies than the rapidly dividing bulk tumor cells surrounding the CSC subpopulation. Sporadic genetic mutations and epigenetic changes within the tumor cells, as well as interactions with the TME, influence both CSCs and the tumors response to therapies and overall prognosis [13, 14]. In the following sections, we provide an update on intrinsic and extrinsic regulators of CSCs, including transcriptional and post-transcriptional regulation, epigenetics, metabolism, and the TME that have shaped our understanding of how CSCs function to drive tumor growth and therapeutic resistance. We also highlight recent developments of CSC-based therapies and clinical trials.
CSC definition and key features
Various methods have been developed to characterize CSCs and determine the degree to which a cell possesses the ability to self-renew [15]. CSCs form spheres in vitro that are maintained through serial passages, while progenitor or differentiated cells lack this ability [16]. Moreover, unlike differentiated cells, in vivo xenografts with CSCs yield sizable tumors in immunocompromised mice, and these can be faithfully recapitulated with serial transplantations. In addition, cell surface markers have been a useful tool to characterize CSCs, as many of these markers are present on CSCs and normal stem cells but are not expressed on differentiated tumor cells [17]. For instance, CD133 is a marker for hematopoietic stem cells (HSCs), but has been widely acknowledged as a CSC marker in breast, prostate, colon, glioma, liver, lung, and ovarian cancers. Finally, lineage tracing studies are able to use markers (e.g. GFP) to monitor the ability of a cell that gives rise to and maintains clonal progeny containing the parental marker [1]. CSCs that can grow and maintain these colonies demonstrate a hierarchical organization structure.
There is growing evidence indicating that a tumor mass composed of CSCs, differentiated cancer cells, and the non-malignant stromal cell network all work together to allow the tumor to adapt and thrive in the harsh TME [18]. A well-characterized example of cellular plasticity in normal cells is the intestinal stem cell population [19], in which certain differentiated endocrine cells modulate their genetic profiles to resemble intestinal stem cells after tissue injury [20]. Moreover, in colorectal cancer with genetic ablation of Leucine Rich Repeat Containing G Protein-Coupled Receptor 5+ (LGR5+) CSCs, differentiated keratin 20+ (KRT20+) cancer cells become dedifferentiated upon entering the niche previously occupied by the ablated LRG5+ CSCs [21]. Such functional plasticity is also seen in glioma stem-like cells (GSCs). Upon treatment with receptor tyrosine kinase (RTK) inhibitors, GSCs can adopt a slow cell cycling state that is dependent upon Notch signaling and is associated with chromatin remodeling using H3K27 demethylases [22]. This epigenetic modulation allows GSCs to persist when confronted with therapeutic insults, thereby providing an avenue for therapeutic resistance. In breast cancer, differentiated basal and luminal cells can revert to a stem cell-like state at a low but significant rate [23]. Given sufficient time, subpopulations of stem, basal, or luminal cells cultured individually can eventually recapitulate phenotypic proportions that include the other two cell types, thereby mirroring the heterogeneity of clinical breast cancer. The ability of cancer cells to endure therapeutic stress is once again evident in this situation [1, 23]. Unlike stem cells, basal and luminal breast cancer cells are normally unable to give rise to tumors in mice. However, upon co-inoculation with irradiated cells, all three subpopulations are effectively tumorigenic.
The transcriptional regulation of CSCs
CSCs have the ability to self-renew and differentiate which allows them to not only be tumorigenic, but also possess the plasticity to promote drug/radiation resistance following treatment. These processes involve multiple critical and highly regulated transcription factors (TFs), which govern CSC homeostasis. CSCs also express several critical TFs that play a key role in inducing pluripotency in somatic cells, including octamer-binding transcription factor 4 (OCT4), Sry-related HMG box 2 (SOX2), Kruppel-like factor 4 (KLF4), NANOG, and c-MYC [24-26]. In addition, many intracellular signaling pathways [27], such as Wnt/TCF, Signal transducer and activator of transcription 3 (STAT3), and NF-κB also have important roles in the regulation of CSC phenotypes (Table 1). In particular, the central stemness-associated TFs, OCT4, SOX2, KLF4, c-MYC, and NANOG are expressed in both CSCs and normal stem cells, such as embryonic stem cells (ESCs) [28]. Accumulating evidence shows that the overexpression of these central stemness-associated TFs occurs in various types of human cancers, including breast cancer [29], prostate cancer [30], and oral squamous cell carcinoma [31]. The aberrant expression of these TFs is associated with tumor initiation, progression [29], and therapy resistance [32]. The central stemness-associated TFs play critical roles in maintaining the pluripotency and self-renewal properties of CSCs and ESCs, but with distinct mechanistic functions between them [28]. Although CSCs and ESCs share common properties such as self-renewal, they have different phenotypic features and proliferative potentials [33]. The central stemness-associated TFs play a vital role in the maintenance of self-renewal and pluripotency in ESCs as well as in the control of early cell fate decisions in ESCs [34]. However, the overexpression of these central stemness-associated TFs in CSCs regulates signaling pathways to promote tumorigenicity and cell survival in response to cancer treatments [28].
Key transcriptional factors of CSCs
| Transcription factor | Cancer type | Effects | Refs |
|---|---|---|---|
| OCT4 | Pancreatic | Chemoresistance, and tumorigenesis | [38-41] |
| SOX2 | Glioma, breast and others | Self-renewal, tumor growth and therapy resistance | [44, 45] |
| KLF4 | Breast, glioma, and osteosarcoma | Metastasis, migration and drug resistance | [50-52] |
| MYC | Glioma, breast | Drug resistance | [54, 55] |
| NANOG | Hepatic, prostate, colorectal, and brain cancers | Tumorigenesis and therapy resistance | [56, 62, 63] |
| Wnt/TCF | Breast and glioma | Metastasis and stemness maintenance | [66, 67] |
| STAT3 | Breast, liver, colon leukemia and prostate | Proliferation, stemness maintenance and immunosuppressive | [69-75] |
| NF-κB | Breast, prostate, ovarian and pancreatic cancer | Pro-inflammatory, angiogenesis and invasion | [76-80] |
OCT4
OCT4, encoded by the POU5F1 gene, is implicated in multiple processes, including stem cell maintenance and embryogenesis [35]. OCT4 is also overexpressed in CSCs of various types of human cancers, and has a positive association with tumorigenesis, therapy resistance, and a worse prognosis in cancers [36, 37]. Suppression of OCT4 increases sensitivity to irradiation and chemotherapy in lung cancer [38], glioma [39], and oral squamous cancer cells [40]. In pancreatic CSCs (PCSCs), combined inhibition of OCT4 and NANOG suppressed the proliferation, migration, invasion of CSCs in vitro, and the tumorigenicity of CSCs in immunocompromised mice, but increased the chemosensitivity of CSCs [41]. OCT4-regulated expression of EMT-related genes, CXCR4, MMP2, MMP9, and TIMP1 is associated with tumor growth, metastasis, and drug resistance of PCSCs [41]. Mouse breast cancer cells with elevated expression of OCT4 have an increased ability of forming tumor spheres, and a worse high levels of stemness-associated genes such as ATXN1, PROM1, CD34, and ALDH1, thereby displaying higher tumorigenic potential in vivo [42].
SOX2
SOX2 plays an essential role in embryonic development and the maintenance of stemness in embryonic and adult stem cells [43]. Dysregulated expression of SOX2 is associated with cancer pathogenesis and several traits of cancer cells such as proliferation, EMT, CSC formation, resistance to apoptosis, and chemotherapy [44]. Moreover, accumulating evidence shows that SOX2 is involved in the regulation of self-renewal, tumor growth, and therapy resistance of CSCs in cancer [44, 45]. In glioblastoma (GBM), suppression of SOX2 in GSCs resulted in cell cycle arrest and markedly reduced their abilities of cell growth, migration, invasion, and tumorigenicity [46]. In breast cancer, SOX2 is required for the proper functioning of CSCs, and mediates resistance to the estrogen receptor antagonist tamoxifen [47, 48]. Given the roles of SOX2 in cancer biology and CSCs, it is of high importance to investigate the downstream regulatory pathways either directly or indirectly mediated by SOX2 and the development of specific SOX2-targeting cancer therapies.
KLF4
KLF4 is a zinc finger transcription factor that is involved in diverse cellular processes, including regulation of the cell cycle and maintenance of cellular pluripotency [49]. Since the discovery of KLF4 in 2006 as one of four key factors required for the induction of pluripotent stem cells (iPSCs) [24], there has been an increase in research to determine the function of KLF4. MicroRNA-7 (miR-7) suppresses the expression of KLF4 by directly targeting its 3'-untranslated region, which leads to a decrease in brain metastasis [50] of breast CSCs. In glioma, KLF4 was found to directly bind to the promoter region of Integrin β4 (ITGB4) and increase ITGB4 expression, resulting in an increase in GSC self-renewal and enhanced glioma cell migration and proliferation both in vitro and in vivo [51]. Furthermore, KLF4 was shown to enhance the sphere-formation ability, drug resistance, and metastatic potential in osteosarcoma CSCs through activation of the p38 mitogen-activated protein kinase (MAPK) signaling pathway [52]. Taken together, these studies reveal an essential role for KLF4 in the regulation of CSC properties and suggest KLF4 as a potential therapeutic target for cancer treatments.
c-MYC
MYC proteins, including c-MYC, N-MYC, and L-MYC have important roles in tumorigenesis and therapeutic resistance. Among these MYC members, c-MYC is perhaps the most frequently dysregulated protein in human tumorigenesis, thereby serving as a promising therapeutic target for cancer treatments [53]. c-MYC amplification in patient-derived GSCs generates sensitivity to PARP inhibition via c-MYC-mediated transcriptional repression of CDK18, CCNE2, and CDKN1A [54]. Similarly, in triple-negative breast cancer, c-MYC interacts with a lncRNA, LINC01638, which prevent its ubiquitination and degradation. In turn, c-MYC transcriptionally regulates MTDH expression and then stimulates Twist1 signaling to maintain an EMT signature and CSC-like state [55].
NANOG
As one of the central stemness-associated TFs, NANOG plays a critical role in embryonic development and cellular reprogramming [24]. NANOG is broadly expressed in various types of cancers [56]. Recently, the involvement of NANOG in tumorigenesis and cancer progression has drawn significant attention. Aberrant overexpression of NANOG has been described in hepatic, prostate, colorectal, and brain CSCs [4, 57-59]. Correlative expression between NANOG and CSC markers has also been reported. For example, NANOG is enriched in CD133+ or CD44+ cancer cells as compared to CD133- or CD44- cells [60-62]. NANOG regulates the fundamental properties of CSCs such as cell proliferation, cell cycle, self-renewal, EMT, tumorigenicity, and chemoresistance [56, 62, 63]. Therefore, NANOG represents a critical molecular nexus underlying tumor initiation and progression and may prove to be a novel therapeutic target for CSC elimination.
Wnt/TCF4
Wnt signaling is one of the key cascades in control of embryonic development and is also involved in self-renewal, tumorigenesis, and metastasis of CSCs [64]. Several components in the Wnt signaling pathway, such as LEF1, cyclin D1, β-catenin, and TCF-4 are expressed at markedly higher levels in the CSC population as compared to non-CSCs [65]. The decreased Wnt1 in breast CSCs caused the down-regulation of the stemness-associated genes CD44, ALDH1, and ATXN1, as well as reductions in the CSC subpopulation and tumor sphere formation [66]. Additionally, Wnt signaling activity is enriched in the proneural subtype of GSCs. Wnt signaling is tightly associated with CSC properties through the induction of miR-20b and miR-125b transcription: these miRs function as suppressors of two negative regulators of Wnt signaling, FZD6 and APC [67].
STAT3
STAT3 is dysregulated in a number of human cancers, acting as a key molecular driver in multiple signaling processes. Phosphorylated STAT3 at tyrosine (Tyr) 705 by Janus kinases (JAK), regulates its dimerization, nuclear accumulation, and DNA binding, thereby initiating its transcriptional regulation [68]. STAT3 is centrally important for the maintenance of CD44+/CD24- CSCs in breast cancer, suppression of genes involved in cell proliferation correspondingly reduce STAT3 activation [69]. Furthermore, in colon cancer, the internalized CD44 and acetyltransferase p300 induce STAT3 acetylation at Lysine (Lys) 685, which in turn promotes the expression of cell cycle regulators cyclin D1 [70], MYC, and Twist1 in colon cancer cells [71]. Moreover, in hepatocellular carcinoma, activated STAT3 can increase CD133 expression through functional cooperation with NF-κB and hypoxia inducible factor 1 alpha (HIF-1α) [72]. STAT3 is also a key regulator of GSCs. STAT3 inhibition suppresses the expression of stemness-associated genes OLIG2 and NES, but enhances expression of the differentiation marker TUBB3 [73]. Additionally in leukemia, inhibition of STAT3 with its inhibitor AZD9150 leads to deceased expression of leukemic drivers including IL1RAP, MSI2, CXCR2, and IL8, as well as MCL1 [74]. Similarly, the inactivation of STAT3 in PTEN-deficient tumors activates immunosurveillance in prostate cancer by reducing the expression of cytokines including CXCL2, GM-CSF, M-CSF, C5a, IL10, and IL13, thereby displaying the immunostimulatory features of the senescence-associated secretory phenotype of tumor cells [75].
NF-κB
NF-κB is critical in the toll-like receptor 2 (TLR2)- initiated TLR2-MyD88-NF-κB signaling pathway, which creates a pro-inflammatory microenvironment and supports self-renewal of epithelial ovarian CSCs, through up-regulation of CD44, NANOG, SOX2, as well as IL-6 [76]. NF-κB co-operates with β-catenin to enhance the expression of CD44, ALDH1, MYC, and OCT4, known markers of breast CSCs, which results in an enriched CD44high/CD24-/low sub-population post poly(I:C) treatment [77]. On the other hand, NF-κB plays a critical role in the expansion of breast CSCs through heterotypic signals that promote macrophage recruitment and angiogenesis [78]. TLR9 is essential for the propagating potential of prostate cancer cells via activation of NF-κB and STAT3, which induces the expression of key stem cell-related genes, including NKX3.1, KLF-4, BMI-1, and COL1A1 by directly interacting with their promoters [79]. In PCSCs, NF-κB induces SOX9 expression, which is critical in controlling the CSC population and invasion ability of the pancreatic cancer cells [80]. Finally in GSCs, the STAT3/NF-κB signaling pathway is constitutively activated in both adherent and spheroid GSCs, and helps to up-regulate the positive regulators of the Notch pathway, including NOTCH1, NOTCH3, NOTCH4, HES5, HEY1, and JAG1, but also down-regulates the negative regulators, such as CTBP1 and RBPJ [81].
Post-transcriptional regulation in CSCs
Advancements in technology and methodology have broadened our understanding of post-transcriptional modifications as an additional regulatory mechanism of gene expression in cancer. The term “epitranscriptome” [82] was coined to refer to diverse post-transcriptional modifications on RNA, including various RNA processing events such as RNA methylation, editing, and alternative splicing. Emerging data suggest that epitranscriptome regulation plays a central role in CSC maintenance and cancer development [83].
RNA methylation in CSCs
N6-methyladenosine (m6A) represents the most widely distributed internal mRNA modification in mammals, with around 25% of mRNAs containing at least one m6A site [84]. As a dynamic process, m6A is catalyzed by RNA methyltransferases known as “writers” and can be removed by RNA demethylases called “erasers” (Figure 1A). A number of proteins have been identified in the m6A writer complex, including methyltransferase-like 3 (METTL3, catalytic component), Wilms tumor 1-associated protein (WTAP, key adaptor for METTL3), methyltransferase-like 14 (METTL14, RNA adaptor needed for METTL3 activity), RNA binding motif protein 15 (RBM15, mediator of methylation specificity through binding to U-rich regions in mRNAs), and Vir Like M6A Methyltransferase Associated (VIRMA) [85]. Two different enzymes were identified as m6A demethylases: fat mass and obesity-associated protein (FTO) and alkylated DNA repair protein AlkB homolog 5 (ALKBH5). However, recent studies using methylated RNA immunoprecipitation sequencing (MeRIP-Seq), and several other findings have shown that FTO physiologically targets m6Am (N6,2′-O-dimethyladenosine), which is indistinguishable from m6A, suggesting that m6A might not be the correct substrate of FTO [86]. Additionally, several m6A-binding proteins, such as YTH domain containing protein family (YTHDF1/2/3 and YTHDC1/2) and insulin-like growth factor 2 mRNA-binding proteins (IGF2BP1/2/3), have been identified as m6A “readers”. By recruiting various “readers”, m6A exerts diverse effects on RNA metabolism, stability (YTHDF2), splicing (YTHDC1 and hnRNPG), nuclear export (YTHDC1), translation efficiency (YTHDF1, IGF2BPs, eukaryotic initiation factor 3, eIF3), and phase separation potential (YTHDF1/2/3) of targeted mRNAs [87-93].
Mechanism of N6-methyladenosine (m6A) modification and its roles in CSCs. (A) m6A is regulated by writers, erasers, and readers. “Writers” refer to the m6A methyltransferase complex including METTL3, METTL14, WTAP, and RMB15. “Erasers” are m6A demethylases including ALKBH5 and FTO. “Readers” are proteins that recognize m6A, including YTH domain containing proteins (YTHDF1/2/3 and YTHDC1/2), IGF2BP1/2/3, and other factors such as hnRNPG and eIF3. The binding of these “readers” to m6A mediates downstream RNA processes, including stability, splicing, nuclear export, translation, and phase separation potential of targeted mRNAs. (B) The m6A modification and its regulators play critical roles in cancer stem cell maintenance and tumorigenicity.
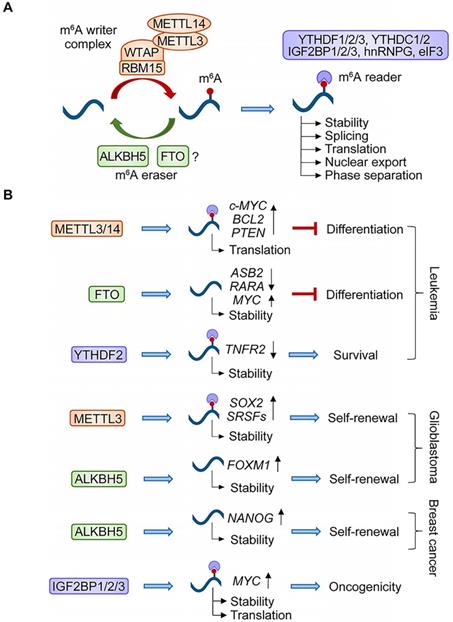
Given the critical role of m6A modifications in regulating cellular pluripotency and differentiation [94], their linkage with CSC maintenance and tumorigenesis is not surprising. Studies strongly support the critical requirement for the m6A “writer” complex in AML (Figure 1B). Depletion of METTL3, METTL14, or WTAP results in cell-cycle arrest, differentiation of leukemic cells, and delayed leukemogenesis in vivo, through m6A modifications of target mRNAs, such as c-MYC, BCL2, PTEN, SP1, SP2, and MYB [95-97]. FTO also exerts an oncogenic role in AMLs, which suppresses leukemia cell differentiation and enhances leukemogenesis by modulating the expression of targets such as ASB2, RARA, and MYC [98, 99]. Targeting YTHDF2 stabilizes m6A-modified transcripts, such as TNFR2, and selectively compromises AML development without dampening normal hematopoiesis [100]. In glioma (Figure 1B), studies addressing the roles of m6A in GSCs have generated divergent results. For example, treatment of serum and retinoic acid elevated the level of m6A in GSCs, inducing GSC differentiation, while knockdown of METTL3 or METTL14 promoted growth, self-renewal, and tumorigenesis [101]. Controversially, other studies showed that global m6A levels were decreased during serum-induced GSC differentiation and METTL3-mediated m6A modifications play essential roles in the maintenance and tumorigenicity of GSCs through stabilizing SOX2 and SRSF transcripts [102, 103]. ALKBH5 is overexpressed in GSCs and plays a critical role in GSC self-renewal and tumorigenesis through demethylating FOXM1 nascent transcripts and enhancing FOXM1 expression [104]. In breast cancer (Figure 1B), hypoxia-induced ALKBH5 expression in breast cancer cells demethylated and stabilized NANOG mRNA, thereby promoting the self-renewal of breast CSCs [105]. IGF2BPs play oncogenic roles in cancers as m6A readers by enhancing mRNA stability and translation of MYC transcripts [92]. Taken together, RNA methylation has been recognized as a critical regulator of CSC generation and maintenance by modulating the expression of various oncoproteins.
RNA editing in CSCs
Comprehensive RNA sequencing has revealed extensive post-transcriptional modifications in the human transcriptome with the most prevalent form being adenosine-to-inosine (A-to-I) conversion [106]. These modifications are catalyzed by adenosine deaminases acting on double-stranded RNA (ADAR) enzymes that include three ADAR members, ADAR1, ADAR2, and ADAR3. Following deamination of adenosine to inosine, the residue is interpreted as a guanosine (G) by the splicing and translational machinery, producing various functional results, such as changes in translated amino acids, biosynthesis and target recognition by small noncoding RNA, alternative RNA splicing, and lncRNA functions [107].
Although the overall biological functions of ADARs are still under investigation, aberrant activity of ADAR and dysregulated A-to-I RNA editing has been found in many human cancers [107, 108]. In various tumor tissues, the level of A-to-I RNA editing is increased, probably due to the upregulation of ADAR1 in tumors [108]. Several A-to-I RNA editing events have been found to correlate with cancer development and progression. In human chronic myeloid leukemia (CML), ADAR1 activation enhances self-renewal capacity of leukemia stem cells (LSCs) by editing let-7 pri-microRNA (pri-miRNA), resulting in impaired let-7 biogenesis and increased LIN28B pluripotency gene expression [109, 110]. In hepatocellular carcinoma (HCC), ADAR1-induced amino acid substitution in antizyme inhibitor 1 (AZIN1) enhances its activity and promotes tumor initiation and development [111]. Moreover, SLC22A3 RNA editing is required for early tumor invasion and cell migration in familial esophageal cancer [112]. In oral squamous cell carcinoma, ADAR1 promotes the EMT and stem-like cell phenotype by facilitating oncogenic onco-miRNA maturation. A systematic multi-cancer miRNA analysis identified A-to-I editing in miR-200b, where increased levels were found associated with a worse prognosis for cancer patients [113]. Mechanistically, the edited miR-200b switches its role from suppressing to enhancing tumor invasion through targeting LIFR, an established inhibitor of cancer metastasis [113]. ADAR2 is an essential enzyme for brain development [114] and is downregulated in gliomas [115]. Overexpression of ADAR2 inhibits glioma cell proliferation and tumor growth by editing and reducing the expression of several onco-miRNAs, including miR-222/221 and miR-21 [116, 117]. Taken together, these studies suggest that aberrant regulation of RNA editing significantly contributes to the malignant phenotypes of CSCs.
Dysregulation of RNA splicing in CSCs
Comprehensive transcriptomic analyses across cancer types have revealed widespread RNA alternative splicing (AS) alterations in tumors, which can be derived from somatic mutations in splicing-related genes, altered expression or activity of splicing factors, or mutations in cis-regulatory elements [118, 119]. As an important regulator of embryonic stem cell pluripotency and reprogramming [120], RNA splicing also contributes to CSC generation and maintenance. For example, specific isoform expression signatures distinguish LSCs from normal human HSCs where the mis-spliced gene products include a pro-survival isoform of BCL-XL and splice variants of SHP-1 and PTK2B, which have been associated with hematological malignancies. More importantly, treatment with a spliceosome modulatory drug, 17S-FD-895, impairs LSC maintenance while sparing normal hematopoietic cells in humanized pre-clinical models [121]. Moreover, specific splice variants of different stem cell regulatory RNAs, including CD44v3 and exon 8, 9-deleted GSK3β enhances LSC self-renewal [122, 123]. Protein arginine methyltransferase 5 (PRMT5) regulates the alternative splicing of genes involved in DNA repair, which maintain the genomic integrity of HSCs [124]. Mutations affecting key spliceosome components, frequently occurring in hematologic malignancies, contribute to HSC maintenance and tumorigenic potential [125]. Loss of SETD2/H3K36me3 mediates splicing modulation, regulates intestinal self-renewal, and aggravates Wnt/β-catenin-dependent colorectal tumorigenesis [126]. The splicing factor, SRSF3, governs alternative splicing programs and promotes the self-renewal and tumorigenicity of GSCs [127]. CD44 splice isoform switching was also shown to modulate the plasticity of breast CSC [128].
Together, recent studies revealed widespread alterations of RNA processing events in various types of human cancers. However, the events that have been functionally demonstrated as relevant to tumorigenesis or tumor development are still limited. Future studies should elucidate the extent to which epitranscriptomic alterations detected by next-generation sequencing technology can be incorporated into functionally relevant gene products and contribute to tumor development and malignancy. Moreover, other RNA processing events, such as alternative cleavage and polyadenylation, have been demonstrated to control cell fate decisions and pluripotency in iPSCs [129], but their involvement in CSC generation and maintenance remains largely unknown. Future studies aimed at comprehensively deciphering the epitranscriptome in CSCs and the crosstalk between different RNA processing events should unveil novel biomarkers and therapeutic targets in CSCs.
CSC epigenetics
Tumor development and progression is regulated by both genetic and epigenetic modifications. The complex phases of tumorigenesis require discrete genetic changes in neoplastic cells and epigenetic alterations. Epigenetic changes within a cell do not involve primary DNA sequence alterations but rather accessibility of genetic loci to transcriptional machinery and chromatin remodeling, thus modulating DNA accessibility and transcription. Epigenetic mechanisms such as changes in DNA methylation, histone modifications, as well as noncoding RNAs, have been shown to play critical roles in cancer progression which influence cellular states at multiple steps in carcinogenesis. In the initial stage of cancer, changes in DNA methylation and chromatin caused by genetic mutations lead to oncogenic cellular reprogramming and acquisition of undifferentiated phenotypes [130]. Convincing evidence in support of this view may come from findings in GBM, a lethal form of primary brain tumor characterized by high genetic heterogeneity. H3K27M mutations, which are found in over 70% of diffuse intrinsic pontine gliomas (DIPG), lead to a global reduction of the repressive H3K27me3 mark induced by the polycomb repressive complex 2 (PRC2), and drive neoplastic transformation in neural precursor cells and the induction of stemness properties [131, 132]. As cancers grow and progress, additional epigenetic changes triggered by cell-extrinsic signaling from the TME affect behaviors and features of cells and help to establish tumor architecture [130].
DNA methylation in CSCs
The complex phases of tumorigenesis cannot only be accounted for by discrete genetic alterations in neoplastic cells alone, but also involve epigenetic alterations. Proteins implicated in the regulation of DNA methylation are critical regulators of oncogenic self-renewal capacity. DNA methyltransferases, including DNMT1, DNMT3A, and DNMT3B, transfer a methyl-group to cytosines followed by guanine residues (CpG), while methylcytosine dioxygenases (TET1 and TET2) initiate a demethylation process by converting 5-methylcytosine (5mC) to 5-hydroxymethylcytosine. DNMT3A is a commonly mutated gene in ∼20% of patients with AML [133], resulting in inhibition of the methyltransferase activity of DNMT3A and expansion of preleukemic HSCs. Mutations in the TET proteins suppress the function of DNMTs [134]. Furthermore, isocitrate dehydrogenase (IDH) is a gene that encodes the protein responsible for converting isocitrate to α-ketoglutarate, a required co-factor for TET and other dioxygenases [135]. Interestingly, mutations in both TET and IDH proteins are associated with the onset and progression of myeloid malignancy. These data indicate that dysregulation of DNA methylation resulting from different genetic mutations can have similar results [133]. In addition to leukemia, IDH mutations also frequently occur in gliomas. IDH1 and/or IDH2 mutations have been observed in a majority of patients with low-grade primary gliomas and secondary high-grade gliomas, which generate a genome-wide hypermethylation of CpG islands, known as the glioma-CpG island methylator phenotype (G-CIMP) [136]. IDH mutations can reprogram committed cells and promote the acquisition of self-renewal capabilities. In normal human astrocytes, IDH1R132H overexpression can increase cell proliferation and promote acquisition of stem cell characteristics [137]. In addition to the canonical DNA methylation modification (5mC), noncanonical DNA methylation events occur on the sixth position of adenine bases (N6-methyladenine, N6-mA) where regulation by the DNA demethylase ALKBH1 plays an important role in GSC growth, self-renewal, and tumor formation capacity [138].
Chromatin remodeling in CSCs
Chromatin remodelers and chromatin marks are frequently altered in human tumors. In AML CSCs, genes implicated in stemness maintenance, proliferation, or metabolism are marked with both H3K4me3 and H3K27me3. Moreover, during differentiation from CSCs to progenitor cells, genes related to stem cell identity were repressed via loss of the H3K4me3 mark alone [139]. Similarly, DIPGs with H3K27M display a global reduction of H3K27me3, with specific enrichment at the loci of tumor suppressor genes. In these tumors, H3K27M binds to the catalytic site of the PRC2 and inhibits its methyltransferase activity [131, 140]. Gene mutations in the subunits of PRC2 are frequently observed in cancer. Elevated expression of EZH2 is found to be associated with poor clinical prognosis and tumor invasiveness in various types of cancers [141]. In addition, increased EZH2 expression is also associated with tumor progression and poor prognosis of glioma, wherein both genetic and pharmacological EZH2 inhibition eradicate self-renewal and tumorigenicity of GSCs [131, 142, 143]. Additionally, EZH2 loss in combination with JAK2 V617F, RUNX1, TET2, or NRas G12D mutations can initiate myeloid or lymphoid malignancies [144-148]. Both gain- and loss-of-function PRC2 mutations can be tumorigenic [149, 150], indicating that chromatin remodeling factors drive cancer initiation and progression in a context-dependent manner. Moreover, expression of EZH2 is not correlated with the abundance of H3K27me3 across breast cancer subtypes [151]. Therefore, it is important to consider several aspects of chromatin remodelers in future studies, including the expression of chromatin remodelers to evaluate chromatin state, histone marks as a consequence of epigenetic regulators, and the undefined functions of chromatin remodelers beyond chromatin [151].
Noncoding RNAs in CSCs
Noncoding RNA such as long noncoding RNAs (lncRNAs) and miRNAs play important roles in epigenetic modulations. LncRNA of transcription factor 7 (lncTCF7), which is overexpressed in liver CSCs, promotes self-renewal and tumor propagation of human liver CSCs through activation of Wnt signaling. This results from the recruitment of the Switch/sucrose nonfermentable (SWI/SNF) complex to the promoter of TCF7 which enhances TCF7 expression [152]. Furthermore in glioma, epidermal growth factor receptor (EGFR) signaling-regulated lncRNA NEAT1 regulates the Wnt/β-Catenin pathway by scaffolding EZH2 [153]. Moreover, LINC00339 is upregulated in glioma tumors and cells, and promotes cell proliferation, migration, invasion, and vascular mimicry formation by modulating the miR-539-5p/TWIST1/MMPs axis [154]. Finally, lncRNA-Low Expression in Tumor (lncRNA-LET) participates in the development of chemo-resistance in urinary bladder cancers [155].
Over 2,500 miRNAs have been identified in humans, forming complicated regulatory networks in which each miRNA can simultaneously control the expression of various genes based on sequence homology, while each mRNA can be regulated by various miRNAs. Malignancy and stemness-associated miRNAs have been identified in GSCs, and their dysregulation is related with a poor prognosis of glioma patients, as well as cancer initiation and therapeutic resistance of CSCs [67, 156]. As cancer is a disease with multiple gene and miRNA aberrations, miRNA-based strategies have the potential to be employed in combination with conventional therapies for cancer treatment [157]. Overall, recent advances in epigenetics offer a better understanding of the epigenetic alterations underlying malignant transformation and provide novel opportunities for therapeutic strategies to target CSCs.
CSC metabolism
Metabolic plasticity is a hallmark of cancer [158]. Most cancer cells and other rapidly proliferating cells predominantly produce their energy through glycolysis followed by lactic acid fermentation, even in the presence of sufficient oxygen, a phenomenon termed the Warburg effect. Such aerobic glycolysis generates ATP less efficiently but more rapidly, and provides the building blocks for macromolecule synthesis, maintains redox homeostasis, and generates a tumor-supporting acidic microenvironment [159]. Unlike bulk tumor cells which depend on glycolysis, CSCs demonstrate a unique metabolic flexibility. Several reports suggest that CSCs are primarily glycolytic and exhibit decreased mitochondrial function [160-165], whereas other studies, especially examination of patient-derived low-passage CSCs and chemoresistant CSCs, report an increased dependence on mitochondrial function and oxidative phosphorylation [166-173]. Possible explanations for these discrepancies may be related to tumor types, environmental stimuli in the experimental system, and dynamic cellular phenotypes, including the transition from quiescent to proliferative CSCs. Several studies show that CSCs can switch between glycolysis and oxidative phosphorylation to survive in malleable, sometimes hostile environments such as hypoxia, starvation, or metastatic sites [167, 174-176].
In addition to the glucose-related metabolic reprogramming, lipid metabolism also regulates the functionality of CSCs in terms of self-renewal and tumorigenic abilities by building the plasma membranes, supplying bioenergy through fatty acid oxidation, and activating signal pathways as second messengers [160, 177-184]. Additionally, increased amino acid metabolism, especially glutamine, fuels oxidative phosphorylation and favors survival in LSCs [185]. Metabolism of lysine, serine, and branched-chain amino acids may also support CSC features [186-188]. GSCs upregulate de novo purine synthesis under the transcriptional control of MYC, which maintains self-renewal, proliferation, and glioma sphere forming capacity [189].
Apart from providing substrates for cellular biosynthesis, metabolites could also actively affect stemness and lineage differentiation through epigenetic modulation. Histone acetylation is under the control of processes which influence the local acetyl-CoA pools, such as hypoxia, nutrient limitation, and intracellular acidification [190-192]. The balance of DNA and histone methylation relies on the dynamics of methyl mark deposition and removal, under the control of S-adenosylmethionine metabolism and α-ketoglutarate/D-2-hydroxyglutarate metabolites that provide methyl groups and control the activity of demethylase enzymes, respectively [193]. Small-molecule perturbation of the S-adenosylmethionine metabolism impacts the tumorigenicity of CSCs [194]. Cancer-associated IDH mutations generate the oncometabolite, D-2-hydroxyglutarate, a competitive suppressor of α-ketoglutarate-dependent dioxygenases, and lock IDH-mutant cancer cells in a stem-like state [195-197].
CSC microenvironment
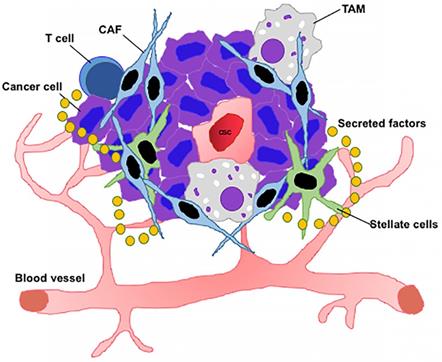
The TME constitutes a heterogeneous population of neoplastic and non-transformed cells driven by both extrinsic and intrinsic factors (Figure 2). It is initiated by the expansion of neoplastic cells comprised of CSCs which create the tumor niche. CSCs continuously remodel the TME to maintain an amenable niche. To maintain this architecture, CSCs continuously interact with other TME components including CAFs, immune cells, tumor vasculature, other differentiated cells, and extracellular cues, thus establishing a favorable environment [198, 199]. The origin of intra-tumor heterogeneity is based on two different theories. The first, known as the CSC model, states that the cancer “stem cell” is the only tumorigenic fraction capable of indefinite self-renewal and differentiation, thus initiating and maintaining tumor growth. The second, a clonal evolution model, emphasizes that genomic/genetic instability arising from stochastic mutations in individual tumor cells results in a clonal diversity. Thus, as a result of natural selection, clones that acquire an advantageous mutation outgrow those that lack such mutations [200]. While ambiguity remains, CSCs are believed to be source of tumor heterogeneity and progression in various cancers [10, 201].
The TME consists of a heterogeneous population of cells including CSCs, dormant cancer cells, TAMs, T cells, other immune cells, and various secretory factors such as cytokines and growth factors.
Cancer associated fibroblasts (CAFs)
Mesenchymal stromal cells (MSCs) are an integral component of the TME and include undifferentiated MSCs along with fibroblasts, pericytes, and vascular or lymphatic endothelial cells [202]. MSCs present in solid tumors are called CAFs and are pro-tumorigenic as opposed to the normal fibroblasts which typically suppress tumor formation. The abundance of these stromal cells is correlated with poor prognosis in certain cancers due to increased tissue remodeling through expression of matrix associated proteolytic enzymes, extracellular matrix (ECM) deposition, and dysregulated angiogenesis [199, 203]. Moreover, MSCs affect tumor growth by secreting growth factors that bind to surface receptors on tumor cells and pro-angiogenic factors, such as VEGF and PDGF, which promote tumor niche neovascularization. Furthermore, CAFs are involved in the resistance to drug treatment and therapy. For instance, in preclinical models, fibroblast-associated secreted factors, such as WNT-16b, enhanced tumor cell proliferation and their depletion augmented chemotherapy response [204]. MSCs are known to be involved in various immunosuppressive mechanisms mediated by metabolites such as indoleamine 2, 3 dioxygenase (IDO), arginase 1 and 2, nitric oxide synthase 2, TGF-β, prostaglandin E2, and adenosine. Specifically, high levels of CD73 expressed on the surface of MSCs catalyze the hydrolysis of adenosine monophosphate (AMP). Increased levels of adenosine in the TME lead to activation of the immunosuppressive A2A adenosine receptor on CD8+ anti-tumor T cells and NK cells, resulting in a dampened immune response [202, 205, 206]. Thus, attempts are being made to relieve the immunosuppression caused by MSCs in addition to other members of the immunosuppressive microenvironment. One such target is fibroblast activation protein (FAP), a type of serine protease that is expressed at abnormally high levels by CAFs. Inhibition of FAP using an anti-tumor vaccine, monoclonal antibody, or chimeric antigen receptor (CAR) T-cell therapy, affects tumor cell growth and increases the CD8+ T cell response [207-209]. Moreover, the signaling pathways that affect the tumor mesenchyme are being targeted. For instance, tyrosine kinase inhibitors (TKIs) are known to inhibit MSC proliferation and differentiation. Hedgehog signaling inhibition reduces the fibrous tissue in the stroma, thereby increasing vessel formation and facilitating drug delivery. Also, secretion of high amounts of the chemokine CXCL12 results in increased TGF-β secretion through binding to its receptor CXCR4, which in turn causes EMT, a key step in metastasis. Thus, targeting this signaling axis has shown anti-metastatic potential in vivo [210-214]. However, no clinically relevant data is available for the aforementioned targets and new therapeutic strategies are thus required.
A reciprocal crosstalk exists between CSCs and CAFs in the TME that proves essential for self-renewal of CSCs. CSCs secrete various cytokines and ligands, such as such as hepatocyte growth factor (HGF) and CCL2, to reprogram normal fibroblasts into CAFs, thereby contributing to cancer cell stemness. These factors induce various stemness regulators in CSCs such as Wnt and NOTCH, which contribute to CSC proliferation and self-renewal [201]. Further, in lung and breast cancer, the CD10+/GPR77+ specific population of CAFs has been correlated with poor patient survival. Driven by active NF-κB signaling, these cells secrete IL6 and IL8 to induce cancer stemness [215]. Thus, targeting CAFs would indirectly provide the therapeutic benefit of inhibiting CSCs.
Tumor vasculature
Development of tumor vasculature, or angiogenesis, is primarily driven by the condition of hypoxia [216]. Hypoxic cancer cells secrete VEGF-A which binds VEGF receptor 2 (VEGFR2) on the surface of endothelial cells (ECs) of nearby blood vessels and initiates tumor angiogenesis [217]. In pre-malignant epithelial tumors, a basal lamina separates the tumor from the surrounding vascular tissue along with angiostatic signals from the ECM and relatively low pro-angiogenic factors, thus preventing the pre-malignant lesions from developing a vasculature. However, during malignant transformation, there is an angiogenic switch wherein pro-angiogenic factors such as growth factors, cytokines, ECM proteins, and ECM remodeling enzymes regulate the vascular ECs to establish an infiltrative and actively growing vascular network [218]. Furthermore, the tumor-associated blood vessels develop an aberrant morphology involving excessive branching, abnormal bulges, defective basement membrane, and discontinuous EC lining, thus depicting impaired vascular maturation [219]. Poorly organized tumor vasculature results in regions of hypoxia and acidity in the tumor, creating gradients based on the distance from the vascular bed, which in turn affects the distribution and availability of chemotherapeutic drugs to all cancer cells [220]. The process of vessel normalization using low dose anti-VEGF therapy can alleviate tumor hypoxic condition and enhance anti-cancer immunity, indicating that the tumor vasculature directly regulates the immune microenvironment [221, 222].
CSCs reciprocally interact with members of the perivascular niche including ECs and ECM components. ECs promote the self-renewal of CSCs through signaling pathways such as Sonic hedgehog, NOTCH, nitric oxide, Jagged-1 [223-227], and VEGF-neuropilin 1 (Nrp1, a receptor for VEGF and other molecules) [227]. Furthermore the nutrient-rich environment and pro-angiogenic factors such as VEGF and MYC, promote CSC proliferation [201]. Reciprocally, CSCs drive the tumor vascularization by stimulating endogenous ECs and creating blood vessel-like structures in melanoma, glioma, breast cancer, and colorectal cancer [228-230]. Furthermore, pericytes derived from CSCs modulate the blood-tumor barrier (BTB) by modulating tight junctions. The BTB acts as a barrier for effective drug delivery against GBM. Thus, selective elimination of the CSC-derived pericytes in xenograft murine models disrupt tight junctions of the BTB and increase vesicular transport, thereby enhancing drug effusion into the tumors [92, 231]. Therefore, targeting the supportive cross talk of CSCs and the tumor vasculature can be therapeutically valuable.
Immune cells in the TME
Immune cell infiltration is a complex phenomenon in solid tumors serving pro-tumorigenic functions. Profiles of immune cells substantially differ among various types of cancers. However, the interplay between cancer cells and immune cells remains constant in solid tumors. The tumor infiltrating immune cells are comprised of both lymphoid and myeloid lineages recruited to the tumor from the bone marrow through systemic circulation, which largely consists of various types of leukocytes such as neutrophils, lymphocytes, monocytes, and macrophages and their immature precursors [232]. While tumor-associated macrophages (TAMs) form the dominant population of immune cells in different tumor types (up to ~50%), T-cells constitute a very low fraction of tumor infiltrating immune cells [233].
The heterogeneity of TAMs is a major barrier to tumor therapy. Cancer cells recruit TAMs to the TME and reprogram them, thereby using the innate arm of the immune system for their own benefit [234]. TAMs are derived from circulating monocytes that arrive at the TME in response to signaling molecules such as chemokines, pro-inflammatory signals, and damage-associated molecule patterns (DAMPs) containing high mobility group box 1 (HMGB1) [235, 236]. DAMPs bind to their specific pattern-recognition receptors on macrophages, such as TLR4 for HMGB1, triggering pro-inflammatory signaling [237], typically representing an M1 phenotype. M1 TAMs are classically activated macrophages with an enhanced ability to engulf pathogens, thus possessing anti-tumorigenic properties. However, upon their arrival into the TME, monocytes differentiate and are polarized to an alternatively activated state of macrophage called 'M2'. M2-polarized TAMs have a pro-tumorigenic potential that supports and maintains the CSC population by secreting chemokines and ligands activating cell stemness pathways such as Sonic hedgehog. For example, Milk-fat globule-epidermal growth factor-VIII (MFG-E8) secreted from TAMs activates STAT3 and Sonic hedgehog signaling in CSCs, thus increasing resistance to therapies [238]. Furthermore, TAMs secrete higher amounts of TGF-β1 which promotes EMT and CSC properties in many types of cancers [239, 240]. Additionally, CSCs reprogram the immune cells in the TME by secreting immunosuppressive proteins such as IL4, which otherwise mitigates the anti-cancer immune response [241]. Moreover, GSCs secrete periostin (POSTN) to recruit macrophages/monocytes and accelerate tumor growth; while M2 macrophages physically interact with mouse breast CSCs through ligand-receptor interactions of EphA4-Ephrin and CD90-CD11b [242, 243]. Lastly, lung CSCs promote the polarization of myeloid cells to an M2-like phenotype through an IFN-regulated transcription factor IRF5 that is critical for producing macrophage colony-stimulating factor (M-CSF) required for generation of tumorigenic myeloid cells [244].
Cytotoxic T lymphocytes and their interaction with other members of the TME
Studies from animal models of cancer have shown that CAFs, the tumor vasculature, and TAMs restrict the accumulation of CD8+ cytotoxic T lymphocytes (CTLs) in the TME [245]. The apoptosis inducer Fas ligand (FasL) is expressed in the vasculature of different types of cancers such as breast, ovarian, bladder, colon, and prostate cancer, but not in the normal vasculature. Cancer cell-derived FasL expression in endothelial cells is associated with scarce CD8+ T cell infiltration, while facilitating enhanced infiltration of immunosuppressive regulatory T cells (Tregs) that express high level of apoptosis suppressor, c-FLIP [246]. CAF cells with membrane protein fibroblast activation protein-α (FAP), hinder T cell infiltration by producing either a dense collagen matrix or the chemokine CXCL12. Inhibition of CXCR4, a CXCL12 receptor, promotes T cell accumulation and cancer regression [247, 248]. TAMs can directly or indirectly inhibit CTLs through different mechanisms. TAMs directly inhibit CTLs through immune checkpoint engagement by expressing programmed cell death ligand 1 (PD-L1) and B7-H4, secreting inhibitory cytokines IL-10 and TGF-β, and depleting metabolites such as L-arginine, which is essential for T cell fitness and anti-tumor activity [249]. TAMs also inhibit T cells indirectly by regulating the immune microenvironment. TAMs in human ovarian cancer produce CCL22 to recruit immunosuppressive Treg cells which suppress CTLs, stimulate TAMs to produce immunosuppressive cytokines IL-6 and IL-10, and enhance B7-H4 expression, which in turn suppresses IL-2 production and T cell proliferation [249]. Furthermore, inhibition of the CSF1 receptor (CSF1R) pathway or CC-chemokine receptor 2 (CCR2), attenuates macrophage recruitment and enhances T cell infiltration in the tumor. CSF1R inhibition suppresses murine glioma. However, clinical trials of CSF1R inhibition have failed to increase overall survival for patients with gliomas [250, 251]. Additionally, TAMs restrict the intra-tumoral localization of T cells by producing reactive nitrogen species, increasing fibrosis, and inducing TGF-β signaling [249]. TGF-β produced by CSCs inhibits the proliferation of active T cells while inducing immunosuppressive Treg cells through both Foxp3-dependent and independent pathways [252, 253].
CD8+ cytotoxic T lymphocytes are the key players involved in killing cancer cells. For this reason, T cells need to sufficiently accumulate in the TME, efficiently infiltrate, and physically contact CSCs. Furthermore, T cells should adequately respond to tumor antigens and activation signals from cells. However, T cells become dysfunctional after arriving at the tumor milieu. This acquired dysfunction of T cells is due to the induction of multiple inhibitory receptors (IRs), including cytotoxic T lymphocyte antigen 4 (CTLA-4), programmed cell death 1 (PD-1), T-cell immunoglobulin domain and mucin domain-3 (Tim-3), T cell immunoreceptor with Ig and ITIM domains (TIGIT), lymphocyte activation gene 3 (LAG-3) and others, wherein the severity of dysfunction depends on the type and number of IR co-expression. These IRs bind to their respective ligands, which are typically expressed on antigen presenting cells or tumor cells. These ligands then deliver an inhibitory signal to T cells attenuating their proliferation and effector functions [254]. Several other factors cause the dysfunction of T cells in the TME including presence of inhibitory cells, suppressive soluble mediators, metabolic pathways, and epigenetic and transcriptional regulation which also cause T cell dysfunction in the TME [254] (Figure 2). Various treatment strategies are aimed at inhibiting the repression on T cell activity in the TME [255]. The key function of T cells in tumor immunology is demonstrated by a positive correlation between T cell infiltration and better clinical outcome as described in melanoma, colorectal, and breast cancers [256]. Cytotoxic T cell activity in tumors is inhibited directly by CSCs through engagement of IRs such as PD-1 and CTLA-4 on T cells, and by up-regulation of their ligands on antigen-presenting cells [257]. Checkpoint inhibitor drugs which target CTLA-4, PD-1, and its ligand PD-L1 have been successful in clinical trials for treating metastatic melanoma, non-small cell lung cancer, and renal cancer, among others. However, mixed results are emerging for the expression of PD-L1 on CSCs, with high expression in CSCs in head and neck, breast, and colon cancers, and low or undetectable expression in other types of cancers [201]. In low-grade glioma, GBM, prostate adenocarcinoma, and lung squamous cell carcinoma, stemness features identified from TCGA datasets correlated negatively with PD-L1 expression [201]. Thus, checkpoint inhibitors might be less efficient for targeting CSCs and investigation of other immune invasive mechanisms is required.
CSC in therapy resistance
Resistance to broad chemotherapeutic agents and selective targeted therapies is a leading cause of cancer deaths [14]. Cells that survive treatment are able to expand and can lead to recurrence of disease or metastatic spread, both of which dramatically decrease patient overall survival [258, 259]. CSCs have been suggested to be the major part of this therapy-resistant cell population within the tumor due to their defined phenotype of quiescence, EMT, multi-drug resistance (MDR), and resistance to DNA damage-induced apoptosis [260, 261].
Quiescence
CSCs have been thought to exist in a dormant state, or G0 phase, after tumors have formed [262]. A cell lineage study showed that serial xenograft passages of colorectal cancer cells in mice gave rise to distinct subpopulations of cells whereas chemotherapy largely eliminated the rapidly proliferating clones, but enhanced the dominance of the dormant clones [184]. This depicts the challenge of eradicating a dormant cell with common chemotherapeutic agents that target rapidly dividing cells. In a mouse glioma model, a Nestin-∆TK-IRES-GFP transgenic mouse was created to label quiescent stem cells and glioma tumor cells. In these animals, temozolomide (TMZ) treatment efficiently ablated dividing cells, but increased the expansion of the GFP-labeled quiescent cells [263]. In a bladder cancer model, combination of gemcitabine and cisplatin also induced rapid cell division of previously quiescent cells to repopulate the tumor in a way similar to that of normal stem cells, post wound formation [184].
Nevertheless, research is being conducted to specifically target these quiescent CSCs. Genetic ablation of the F-box protein Fbxw7 induced LSC proliferation, which could then be successfully targeted by imatinib [264]. Quiescent cells have also shown a dependence on autophagy in colorectal [265], liver [156], brain [266], and melanoma [267] CSCs. Therefore, recent studies targeting both autophagy [268] and the overarching metabolic state have shown pre-clinical promise [105]. Moreover, the opposing idea of maintaining cells in a quiescent state has been tested as well. A recent study shows that retinoic acid and NR2F1 signaling work together to induce a dormancy-like epigenetic state [269]. Fenretinide treatment in lung and colorectal cancers also induced a quiescent state along with inhibition of the mTOR pathway, cell cycle block, and a mixed death pathway with both autophagic and apoptotic qualities [270].
Epithelial-to-mesenchymal transition (EMT)
CSCs express many of the markers of normal stem cells, including their ability to survive in a de-differentiated state [271]. Experimental evidence shows that induction of EMT or de-differentiation of immortalized human mammary epithelial cells leads to an increase in their local invasion and metastatic burden [272, 273]. Further characterization of breast epithelial cells that have been induced to undergo EMT mimic characteristics of mesenchymal stem cells. Specifically, they show enhanced wound-homing abilities and are able to differentiate into multiple distinct lineages [274]. There is also a relationship between a more EMT-like phenotype and multi-drug resistance [275]. Markers of stemness, like OCT4, NANOG, and SOX2, have been associated with resistance to chemotherapy [276-279]. Another example is ZEB1, a transcription factor associated with EMT. ZEB1 is able to regulate both self-renewal and therapy resistance in GSCs through regulation of O-6-methylguanine methyltransferase (MGMT) via miR-200c and c-MYB [280]. Additionally, cancer cells that undergo EMT can go into a dormant state. The Wnt pathway also plays a dominant role in both EMT and stemness since Wnt-1 stimulation induces EMT in breast cancer [281, 282]. In lung cancer cells, gefitinib treatment activates NOTCH-1 signaling, which allows for an acquired EMT phenotype and resistance to the therapy [283]. Among a list of other factors including those of invasive potential and tumorigenicity, EMT markers have often been used to predict resistance to the anti-EGFR antibody cetuximab in urothelial carcinoma cells [284].
As EMT is an important feature of stem cells, and a pivotal stage in metastasis, inhibitors of EMT have been developed and exploited [285]. TGF-β signaling is one of the major pathways associated with EMT, as one and a half days of TGF-β treatment induced the reprogramming of mouse embryonic fibroblasts (MEFs) into iPSCs [286]. For this reason, inhibitors targeting TGF-β signaling have been developed and are currently undergoing clinical trials [287]. Lastly, natural compounds have also been tested to inhibit EMT. Curcumin, an abundant compound in turmeric and an HGF inhibitor, induced the epithelial marker E-cadherin and inhibited tumor growth [287].
Multi-drug resistance
Aldehyde dehydrogenase (ALDH) is a potential selective marker for CSCs in breast, bladder, embryonal rhabdomyosarcoma, head and neck squamous cell carcinoma, and lung cancer [288, 289]. As a cytosolic enzyme that oxidizes intracellular aldehydes, ALDH protects cells from elevated reactive oxygen species (ROS) levels. Decreasing or maintaining a low ROS level is critical to a cell, as ROS accumulation causes cell death [290]. Higher expression of ALDH, as compared to ALDH-negative lung cancer cells, has been shown to confer resistance to multiple chemotherapeutic agents like cisplatin, etoposide, fluorouracil, and gefitinib [291]. ALDH+ pancreatic CSCs also showed therapy resistance, but also an increase in phosphorylated STAT3 which could be targeted with STAT3 inhibitors [292]. In ovarian CSCs, ALDH1A2 was shown to be directly regulated by NF-κB signaling via the transcription factor RelB. Loss of RelB inhibited CSC spheroid formation, ALDH expression, tumorigenesis, and chemoresistance [293]. As expected, an inhibitor of ALDH, diethylaminobenzaldehyde, has been shown to re-sensitize breast CSCs to chemotherapy [294].
The ATP-binding cassette (ABC) transporter family has been implicated in the multi-drug resistance of CSCs. This family is made up of 49 family members that have 7 gene sub-families [295]. The major function of ABC transporters is to pump substrate proteins and drugs across the plasma membrane powered by ATP hydrolysis [296]. However, considerable numbers of ABC transporters are often overexpressed in cancers, particularly in CSCs [297]. Moreover, OCT4 has been shown to control the gene expression of multiple ABC proteins [298]. Many oncogenes have been implicated in regulating ABC functions as well. For examples, MYC plays a dual role in ABC expression. MYC activation can increase the expression of ABCC1 and ABCC4, while attenuating levels of ABCC3 [299]. The PI3K/AKT pathway, but not mTOR, regulates ABCG2 in GSCs via its localization to the plasma membrane. This phenotype is exaggerated with PTEN loss and TMZ treatment, which is representative of clinical presentation and treatment of gliomas [300]. Inhibition of BCR-ABL and its downstream target pathway PI3K/AKT can also lead to downregulation of ABCG2 in CML cells [301]. Treatment of breast cancer cells with an EGFR/HER2 inhibitor lapatinib, resulted in a decrease in ABCB1 and ABCG2 expression, thereby sensitizing breast cancer cells to a chemotherapeutic agent doxorubicin [302]. Although ABC transporters have been implicated in CSC resistance to therapies, therapeutically targeting ABC transporters will target normal stem cells and brain ECs that make up the blood brain barrier [229]. Therefore, identification of CSC-specific ABC transporters is necessary. One example is the breast cancer resistance protein (BCRP), which is overexpressed in mitoxantrone-resistant cancer cells. Treatment with fumitremorgin C (FTC) efficiently reverses drug resistance in these mitoxantrone-resistant breast cancer cells and in other types of cancer cells transfected with exogenous BCRP [303].
Resistance to DNA damage-induced death
CSCs have an altered DNA damage response (DDR) and repair pathways which closely mimic that of normal stem cells [304]. As normal stem cells are needed to repopulate normal tissue post-damage, their DDR must be error-free to preserve normal tissue DNA [305]. In CSCs, this efficient DDR leads to radio- and chemoresistance [272]. In GSCs, multiple, sometimes contradicting, studies have described CD133+ progenitor cells and DNA repair. CD133+ cells were shown to efficiently repair DNA damage [306]. However, changes in γ-H2AX foci resolution, DNA base excision, or single-strand break repair in radiation-treated GSCs were not found [307]. However, CD133+ GSCs enriched for the polycomb group protein BMI1 have increased radio-resistance, in which BMI1 recruits both double-stranded break repair and nonhomologous end joining proteins [308]. CD133+ GSCs also had an increase in Chk 1-dependent DNA repair response, and increased expression of the Mre11, Rad50, and Nbs1 (MRN) complex component NBS1 [309]. CD133+ CSCs in breast, lung, and non-small-cell lung cancers also showed an increase in DNA damage response and repair genes [310]. The homologous repair (HR) pathway of DNA damage is vitally important to CSCs, as this is less error-prone process for DNA repair. HR takes place in S phase, when a homologous chromosome template is present [310]. GSCs have been shown to overexpress RAD51, the major HR DNA repair protein [311]. In head and neck squamous cell carcinoma, overexpression of Fanconi anemia DNA repair proteins is observed specifically in ALDH1+ cells [312]. Genes inducing cell death via DDR pathway such as p53 are often dysregulated in CSCs. p53 acts as a sensor when detrimental DNA damage has occurred. However, p53 is frequently mutated or downregulated in CSCs. Therefore, restoration of normal p53 function in p53-mutated GSCs may provide a new treatment avenue [313].
Therapies targeting CSC
The promise of the CSC hypothesis is that an in-depth understanding of CSC biology will allow us to develop more effective approaches to eradicate CSCs in patients. Although the inherent plasticity of CSCs presents major challenges for design of anti-CSC therapies, some strategies have been tested to interfere with CSCs in preclinical models and patients, including inhibition of key CSC signaling pathways, viral therapy, awakening quiescent CSCs, and immunotherapy.
Targeting of key CSC signaling pathways
The lysine-specific demethylase (LSD1), also known as lysine-specific histone demethylase 1A (KDM1A), is a histone demethylase that demethylates H3K4me1/2 and has been extensively studied [314]. LSD1 represents an attractive therapeutic target for LSCs, because LSD1 plays a critical role in maintenance of LSCs. Tranylcypromine analogs have been used for LSD1 inhibition and induce CSC differentiation and suppress LSC expansion and acute leukemia development without any notable side-effects [315]. Several LSD1 inhibitors are currently in phase I/II clinical trials in AML patients [316]. Accumulating evidence indicates that autophagy is implicated in CSC maintenance and therapy resistance [317]. Suppression of autophagy also holds promise for the therapeutic elimination of CSCs. In GBM, targeting a key autophagy regulator, ATG4B, with a small molecule inhibitor, NSC185058, enhanced the efficacy of radiation therapy in orthotopic xenograft models [268]. Additionally, targeting BMI1 by using a small molecule, PTC-209, has also demonstrated efficacy against CSCs in models of colorectal cancer through decreasing BMI1 protein levels [318].
Viral therapy
With the success of modern immunotherapy, oncolytic viral therapy has become a novel and promising strategy to target cancers and CSCs. Oncolytic viruses can effectively replicate within cancer cells rather than in normal cells, resulting in lysis of the tumor mass [319]. In addition to this primary effect, oncolytic viruses are also able to stimulate the immune system against cancer cells, in which the immune system is silenced by the TME [320]. The use of improved oncolytic adenovirus treatment regimens has demonstrated therapeutic activity in immunocompetent C57BL/6 mouse models by stimulating an influx of CD8+ T cells specific to tumor-associated antigens [321, 322]. Moreover, in human patients, viral treatments have been shown to induce a tumor macrophage phenotypic shift [323]. Oncolytic viruses also represent attractive combination strategies with PD-1/PD-L1 blockade therapy, and initial clinical studies have suggested promising results [324]. Phase I trials of intratumoral inoculation of the recombinant nonpathogenic polio-rhinovirus chimera (PVSRIPO) have demonstrated some efficacy in the treatment of patients with recurrent GBM, with evidence that the survival rate among patients treated with PVSRIPO immunotherapy gain relatively long-term survival benefit [325]. The highly neurotropic flavivirus Zika virus expressing an exogenous Endostatin‐Angiostatin fusion gene (VAE) can infect and suppress GSCs in organoid and mouse models, indicating that VAE-based gene oncolytic viral therapy is a promising strategy for the treatment of brain tumors [326]. Similarly, oncolytic retroviruses have the potential to inhibit the growth of CSC xenograft tumors [327]. Although several clinical trials performed in a small population of breast cancer patients have demonstrated oncolytic reovirus safety [328-330], further investigations with a heterogeneous population are necessary for reliable prediction of oncolytic viral therapy in cancer patients.
Awakening quiescent CSCs
The presence of CSCs is a well-recognized concept, wherein poorly differentiated and quiescent cells within a tumor mass are thought to be the major cause of chemotherapy resistance. Thus, targeting quiescent CSCs is emerging as a viable treatment option [261]. Genetic ablation of the ubiquitin ligase FBXW7, a negative regulator of MYC, forces quiescent LSCs to re-enter the cell cycle and increases their sensitivity to the tyrosine kinase inhibitor (TKI) imatinib [331]. CSCs can also reprogram metabolic pathways to modulate cancer growth. It has been demonstrated that CSCs can switch between oxidative phosphorylation and glycolysis even in the presence of oxygen to support tumor growth [332]. Several studies have shown that quiescent CSCs are dependent on oxidative metabolism. Therefore, suppression of oxidative phosphorylation reduces the ability of self-renewal and tumor initiation of CSCs and enhances the response to targeted therapies in preclinical mouse cancer models [168, 170, 172, 333].
Immunotherapy
Immunotherapy has gained significant attention for its potential to treat and cure various types of cancers and represents an alternative option to target CSCs. Given that the combination of surface expression markers that are used to identify the CSC populations in different tumor types, CSC markers are therefore an attractive target for cancer therapeutics [334]. For example, transmembrane glycoprotein CD44 is overexpressed in CSCs of various cancer types, including breast, prostate, bladder, gastric cancer, and others [335]. A novel therapeutic strategy employing near-infrared photo immunotherapy (NIR-PIT) targeting CD133 in GBM has also been shown to be highly specific and efficient for eliminating GSCs [336]. The monoclonal antibody (mAb) H90 was first shown to efficiently eradicate CD44+ human AML CSCs [337]. Additionally, anti-tumor vaccines could be developed to target tumor-associated antigens and elicit tumor-specific T cell responses. In two early clinical trials, administration of personalized vaccines containing tumor neoepitopes elicited sustained responses of central memory T cells with evidence of immunologic memory and tumor-infiltrating capacity [338, 339]. T cell receptor (TCR) gene therapy is another kind of adoptive cellular immunotherapy where patient-derived T cells are engineered to produce a CAR selective for a tumor antigen, with subsequent ex vivo cell expansion and adoptive transfer back into the patient. CAR-T cell therapy against tumor-specific targets in CSCs, including CD33 for AML [340] and EGFRvIII for GBM [341], displayed some efficacy in treating patients in phase I clinical trials. The association of increased expression of immune checkpoint ligands with CSC function inspired investigators to use inhibitors of these checkpoint molecules to target CSCs for cancer treatments. Moreover, a preclinical study showed that combined inhibition of CTLA-4, PD-L1, and a CSC vaccine remarkably suppressed tumor growth in multiple mouse models [342]. Therefore, targeting CSCs with the immune system for treating cancers remains an intriguing and fruitful field of ongoing investigation.
Conclusions and future perspectives
Decades of accumulating evidence shows the significant progress that has been made in our understanding of CSCs at the transcriptional, posttranscriptional, epigenetic, metabolic, and microenvironmental regulatory levels. These findings have been driven by the implementation of new technologies, such as single cell multi-omics technology, and have provided further insight into our understanding of the evolution and heterogeneity of human cancers, as well as the TME [343]. The advent of CRISPR-Cas9 screening technologies has also greatly accelerated cancer research in many aspects, including robust site-specific gene editing, generation of animal cancer models, and functional genetic screening. The potential role of CRISPR-Cas9-based gene editing has received a lot of attention and has become a critical tool in the development of cancer therapeutics [344, 345]. The role of the TME as a critical regulator of CSC properties and an essential target for CSC elimination has now come into sharper focus. With the advances in the immunotherapy and TME fields that have been made in recent years, we can also expect to see big landmark developments of more efficient approaches to eliminate these highly tumorigenic and therapy-resistant cells.
Despite the optimism resulting from these marked advances, intense ongoing research is attempting to address many of the remaining unanswered questions. In this review, we focused on several outstanding challenges, which include approaches for investigating and the therapeutic targets of CSCs. First, a large proportion of cancer research continues to use in vitro sphere formation in serum-free medium as a surrogate CSC assay. These studies are frequently performed in hypoxic, hyperglycemic, and non-physiologic conditions. In addition, the in vitro culture conditions used for CSCs may not include key factors that are essential for the in vivo fundamental properties of most CSCs. Therefore, these experiments likely fail to recapitulate the clinical presentation seen in human cancers, illustrating the need for direct CSC detection approaches that better mimic physiological conditions. Recently, organoid model systems and other tools have gained appreciation in CSC research and their use in high-throughput and high-fidelity tumor modeling may be beneficial in overcoming these challenges [346]. Second, immunodeficient mouse models have been used effectively to identify the capacity of CSCs to reconstruct tumor heterogeneity that resembles the parent tumor in vivo. This powerful strategy is very useful for detection and quantification of CSCs in various types of human cancers. However, a major shortcoming of the immunodeficient mouse model is the lack of an intact TME. This is an important consideration, given that components of TME have an important role in supporting tumor generation, progression, and therapeutic response [347]. Human-severe combined immunodeficient (SCID) mouse chimeric models have been developed for the engraftment of human tumor cells that recapitulate the native tumor microenvironment, allowing researchers to evaluate the role of microenvironment components, such as tumor-infiltrating leukocytes and other human stromal cells [348]. Third, increasing evidence has revealed that both CSCs and non-CSCs represent a very plastic and dynamic population, which is capable of changing cell types in response to certain environmental stimuli. This is exemplified by a study in which subpopulations of cells purified for a given phenotypic state (i.e. stem cell, basal-, or luminal-like phenotypes) have equal tumorigenic potency. Each subpopulation of cancer cells effectively initiated tumor growth in mouse models, and eventually returned to equilibrium proportions with sufficient time [23]. Given that the plasticity of CSCs may present major challenges in the development of efficient therapies, it is essential to improve our insights into the molecular mechanisms underlying tumor cell plasticity and develop more effective therapies to target these cells.
Finally, despite the intense focus on uncovering important molecular targets as potential strategies for therapeutic intervention of CSC function, very few targets have been effectively translated into clinical care, and the failure rate of clinical trials remains high. It is likely that inefficient drug delivery and drug intervention in advanced stages of disease may be impeding their impact on tumor growth. Improving approaches of delivery through advanced biomaterials and drug delivery systems represent another important space to explore and may improve the efficacy of targeted therapies for cancer treatments.
Abbreviations
ABC: ATP-binding cassette; ADAR: adenosine Deaminases acting on double-stranded RNA; ALDH: aldehyde dehydrogenase; AML: acute myeloid leukemia; CAF: cancer associated fibroblast; CAR: chimeric antigen receptor; CML: chronic myeloid leukemia; CSC: cancer stem cell; CTL: cytotoxic T lymphocyte; CTLA-4: cytotoxic T-lymphocyte-associated antigen-4; DAMP: damage-associated molecule pattern; DDR: DNA damage response; DIPG: diffuse intrinsic pontine glioma; DNMT: DNA methyltransferase; EC: endothelial cell; ECM: extracellular matrix; EGFR: epidermal growth factor receptor; EMT: epithelial-mesenchymal transition; FTO: fat mass and obesity-associated protein; GBM: glioblastoma; GSC: glioma stem like cell; G-CIMP: glioma-CpG island methylator phenotype; H3K27me3: histone H3 trimethylated lysine 27; HSC: hematopoietic stem cell; HIF-1α: hypoxia inducible factor 1 alpha; ITGB4: Integrin β4; IDH: isocitrate dehydrogenase; IR: inhibitory receptor; iPSCs: induced pluripotent stem cells; JAK: Janus kinase; KLF4: Kruppel-like factor 4; LncRNA: long noncoding RNA; LSC: leukemia stem cell; LSD1: lysine-specific demethylase; m6A: N6-methyladenosine; MAPK: mitogen-activated protein kinase; MDR: multi-drug resistance; MEF: mouse embryonic fibroblast; MGMT: O-6-methylguanine Methyltransferase; miRNA: microRNA; Mesenchymal: MES; MSC: mesenchymal stromal cell; OCT4: octamer-binding transcription factor 4; OXPHOS: oxidative phosphorylation; PD-1: programmed death- 1; PD-L1: programmed cell death ligand 1; PRC2: polycomb repressive complex 2; Proneural: PN; RTK: receptor tyrosine kinase; ROS: reactive oxygen species; SCID: severe combined immunodeficient; STAT3: signal transducer and activator of transcription 3; SOX2: Sry-related HMG box 2; TAM: tumor associated macrophage; TCR: T cell receptor; TET: methylcytosine dioxygenase; TF: transcription factor; TKI: tyrosine kinase inhibitor; TME: tumor microenvironment; TMZ: temozolomide; VEGF: vascular endothelial growth factor; WT: wild-type.
Acknowledgements
This work was supported by US NIH grants NS093843, NS095634, NS115403, P50CA221747-Project 4 (S.Y.C.), F31 CA232630 (N.S); K00 CA234799 (D.T.), Fishel Family Fellowship in Cancer Research, the Robert H. Lurie Comprehensive Cancer Center, Northwestern University Feinberg School of Medicine (N.S.), and Lou and Jean Malnati Brain Tumor Institute at Northwestern Medicine (S.-Y.C., B.H.). S.-Y.C. is a Zell Scholar at Northwestern University.
Author Contributions
S.Y.C. and T.H. conceived, organized, and supervised this work. All authors contributed to the writing and editing of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017;23:1124-34
2. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730-7
3. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983-8
4. Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M. et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A. 2003;100:15178-83
5. Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J. et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821-8
6. Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65:10946-51
7. Li C, Liu S, Yan R, Han N, Wong KK, Li L. CD54-NOTCH1 axis controls tumor initiation and cancer stem cell functions in human prostate cancer. Theranostics. 2017;7:67-80
8. O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106-10
9. Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C. et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111-5
10. Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V. et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030-7
11. Zhang S, Balch C, Chan MW, Lai HC, Matei D, Schilder JM. et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008;68:4311-20
12. Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A. et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008;15:504-14
13. Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 2012;21:283-96
14. Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012;10:717-28
15. Beck B, Blanpain C. Unravelling cancer stem cell potential. Nat Rev Cancer. 2013;13:727-38
16. Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29:1203-17
17. Kim WT, Ryu CJ. Cancer stem cell surface markers on normal stem cells. BMB Rep. 2017;50:285-98
18. Behnan J, Stangeland B, Hosainey SA, Joel M, Olsen TK, Micci F. et al. Differential propagation of stroma and cancer stem cells dictates tumorigenesis and multipotency. Oncogene. 2017;36:570-84
19. Yousefi M, Li L, Lengner CJ. Hierarchy and plasticity in the intestinal stem cell compartment. Trends Cell Biol. 2017;27:753-64
20. Jadhav U, Saxena M, O'Neill NK, Saadatpour A, Yuan GC, Herbert Z. et al. Dynamic reorganization of chromatin accessibility signatures during dedifferentiation of secretory precursors into Lgr5+ intestinal stem cells. Cell Stem Cell. 2017;21:65-77 e5
21. Shimokawa M, Ohta Y, Nishikori S, Matano M, Takano A, Fujii M. et al. Visualization and targeting of LGR5(+) human colon cancer stem cells. Nature. 2017;545:187-92
22. Liau BB, Sievers C, Donohue LK, Gillespie SM, Flavahan WA, Miller TE. et al. Adaptive chromatin remodeling drives glioblastoma stem cell plasticity and drug tolerance. Cell Stem Cell. 2017;20:233-46 e7
23. Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C. et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146:633-44
24. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663-76
25. Huang CF, Xu XR, Wu TF, Sun ZJ, Zhang WF. Correlation of ALDH1, CD44, OCT4 and SOX2 in tongue squamous cell carcinoma and their association with disease progression and prognosis. J Oral Pathol Med. 2014;43:492-8
26. van Schaijik B, Davis PF, Wickremesekera AC, Tan ST, Itinteang T. Subcellular localisation of the stem cell markers OCT4, SOX2, NANOG, KLF4 and c-MYC in cancer: a review. J Clin Pathol. 2018;71:88-91
27. Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang J. et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. 2020;5:8
28. Liu A, Yu X, Liu S. Pluripotency transcription factors and cancer stem cells: small genes make a big difference. Chin J Cancer. 2013;32:483-7
29. Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A. et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40:499-507
30. Gu G, Yuan J, Wills M, Kasper S. Prostate cancer cells with stem cell characteristics reconstitute the original human tumor in vivo. Cancer Res. 2007;67:4807-15
31. Chiou SH, Yu CC, Huang CY, Lin SC, Liu CJ, Tsai TH. et al. Positive correlations of Oct-4 and Nanog in oral cancer stem-like cells and high-grade oral squamous cell carcinoma. Clin Cancer Res. 2008;14:4085-95
32. Herreros-Villanueva M, Bujanda L, Billadeau DD, Zhang JS. Embryonic stem cell factors and pancreatic cancer. World J Gastroenterol. 2014;20:2247-54
33. Shackleton M. Normal stem cells and cancer stem cells: similar and different. Semin Cancer Biol. 2010;20:85-92
34. Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP. et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947-56
35. Radzisheuskaya A, Silva JC. Do all roads lead to Oct4? the emerging concepts of induced pluripotency. Trends Cell Biol. 2014;24:275-84
36. Mohiuddin IS, Wei SJ, Kang MH. Role of OCT4 in cancer stem-like cells and chemotherapy resistance. Biochim Biophys Acta Mol Basis Dis. 2020;1866:165432
37. Villodre ES, Kipper FC, Pereira MB, Lenz G. Roles of OCT4 in tumorigenesis, cancer therapy resistance and prognosis. Cancer Treat Rev. 2016;51:1-9
38. Cortes-Dericks L, Yazd EF, Mowla SJ, Schmid RA, Karoubi G. Suppression of OCT4B enhances sensitivity of lung adenocarcinoma A549 cells to cisplatin via increased apoptosis. Anticancer Res. 2013;33:5365-73
39. Ikushima H, Todo T, Ino Y, Takahashi M, Saito N, Miyazawa K. et al. Glioma-initiating cells retain their tumorigenicity through integration of the Sox axis and Oct4 protein. J Biol Chem. 2011;286:41434-41
40. Samardzija C, Luwor RB, Volchek M, Quinn MA, Findlay JK, Ahmed N. A critical role of Oct4A in mediating metastasis and disease-free survival in a mouse model of ovarian cancer. Mol Cancer. 2015;14:152
41. Lu Y, Zhu H, Shan H, Lu J, Chang X, Li X. et al. Knockdown of Oct4 and Nanog expression inhibits the stemness of pancreatic cancer cells. Cancer Lett. 2013;340:113-23
42. Kim RJ, Nam JS. OCT4 expression enhances features of cancer stem cells in a mouse model of breastcancer. Lab Anim Res. 2011;27:147-52
43. Novak D, Huser L, Elton JJ, Umansky V, Altevogt P, Utikal J. SOX2 in development and cancer biology. Semin Cancer Biol. 2019;18:30185-8
44. Huser L, Novak D, Umansky V, Altevogt P, Utikal J. Targeting SOX2 in anticancer therapy. Expert Opin Ther Targets. 2018;22:983-91
45. Weina K, Utikal J. SOX2 and cancer: current research and its implications in the clinic. Clin Transl Med. 2014;3:19
46. Garros-Regulez L, Garcia I, Carrasco-Garcia E, Lantero A, Aldaz P, Moreno-Cugnon L. et al. Targeting SOX2 as a therapeutic strategy in glioblastoma. Front Oncol. 2016;6:222
47. Piva M, Domenici G, Iriondo O, Rabano M, Simoes BM, Comaills V. et al. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol Med. 2014;6:66-79
48. Das T, Nair RR, Green R, Padhee S, Howell M, Banerjee J. et al. Actinomycin D down-regulates SOX2 expression and induces death in breast cancer stem cells. Anticancer Res. 2017;37:1655-63
49. Ghaleb AM, Yang VW. Kruppel-like factor 4 (KLF4): What we currently know. Gene. 2017;611:27-37
50. Okuda H, Xing F, Pandey PR, Sharma S, Watabe M, Pai SK. et al. miR-7 suppresses brain metastasis of breast cancer stem-like cells by modulating KLF4. Cancer Res. 2013;73:1434-44
51. Ma B, Zhang L, Zou Y, He R, Wu Q, Han C. et al. Reciprocal regulation of integrin beta4 and KLF4 promotes gliomagenesis through maintaining cancer stem cell traits. J Exp Clin Cancer Res. 2019;38:23
52. Qi XT, Li YL, Zhang YQ, Xu T, Lu B, Fang L. et al. KLF4 functions as an oncogene in promoting cancer stem cell-like characteristics in osteosarcoma cells. Acta Pharmacol Sin. 2019;40:546-55
53. Kress TR, Sabo A, Amati B. MYC: connecting selective transcriptional control to global RNA production. Nat Rev Cancer. 2015;15:593-607
54. Ning JF, Stanciu M, Humphrey MR, Gorham J, Wakimoto H, Nishihara R. et al. Myc targeted CDK18 promotes ATR and homologous recombination to mediate PARP inhibitor resistance in glioblastoma. Nat Commun. 2019;10:2910
55. Luo L, Tang H, Ling L, Li N, Jia X, Zhang Z. et al. LINC01638 lncRNA activates MTDH-Twist1 signaling by preventing SPOP-mediated c-Myc degradation in triple-negative breast cancer. Oncogene. 2018;37:6166-79
56. Jeter CR, Yang T, Wang J, Chao HP, Tang DG. Concise review: NANOG in cancer stem cells and tumor development: an update and outstanding questions. Stem Cells. 2015;33:2381-90
57. Jeter CR, Badeaux M, Choy G, Chandra D, Patrawala L, Liu C. et al. Functional evidence that the self-renewal gene NANOG regulates human tumor development. Stem Cells. 2009;27:993-1005
58. Chiou SH, Wang ML, Chou YT, Chen CJ, Hong CF, Hsieh WJ. et al. Coexpression of Oct4 and Nanog enhances malignancy in lung adenocarcinoma by inducing cancer stem cell-like properties and epithelial-mesenchymal transdifferentiation. Cancer Res. 2010;70:10433-44
59. Zhang J, Espinoza LA, Kinders RJ, Lawrence SM, Pfister TD, Zhou M. et al. NANOG modulates stemness in human colorectal cancer. Oncogene. 2013;32:4397-405
60. He A, Qi W, Huang Y, Feng T, Chen J, Sun Y. et al. CD133 expression predicts lung metastasis and poor prognosis in osteosarcoma patients: A clinical and experimental study. Exp Ther Med. 2012;4:435-41
61. Leung EL, Fiscus RR, Tung JW, Tin VP, Cheng LC, Sihoe AD. et al. Non-small cell lung cancer cells expressing CD44 are enriched for stem cell-like properties. PLoS One. 2010;5:e14062
62. Xu F, Dai C, Zhang R, Zhao Y, Peng S, Jia C. Nanog: a potential biomarker for liver metastasis of colorectal cancer. Dig Dis Sci. 2012;57:2340-6
63. Wang ML, Chiou SH, Wu CW. Targeting cancer stem cells: emerging role of Nanog transcription factor. Onco Targets Ther. 2013;6:1207-20
64. Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36:1461-73
65. Matsui WH. Cancer stem cell signaling pathways. Medicine (Baltimore). 2016;95:S8-S19
66. Jang GB, Kim JY, Cho SD, Park KS, Jung JY, Lee HY. et al. Blockade of Wnt/beta-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci Rep. 2015;5:12465
67. Huang T, Alvarez AA, Pangeni RP, Horbinski CM, Lu S, Kim SH. et al. A regulatory circuit of miR-125b/miR-20b and Wnt signalling controls glioblastoma phenotypes through FZD6-modulated pathways. Nat Commun. 2016;7:12885
68. Johnson DE, O'Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. 2018;15:234-48
69. Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR. et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(-) stem cell-like breast cancer cells in human tumors. J Clin Invest. 2011;121:2723-35
70. Lee JL, Wang MJ, Chen JY. Acetylation and activation of STAT3 mediated by nuclear translocation of CD44. J Cell Biol. 2009;185:949-57
71. Su YJ, Lai HM, Chang YW, Chen GY, Lee JL. Direct reprogramming of stem cell properties in colon cancer cells by CD44. EMBO J. 2011;30:3186-99
72. Won C, Kim BH, Yi EH, Choi KJ, Kim EK, Jeong JM. et al. Signal transducer and activator of transcription 3-mediated CD133 up-regulation contributes to promotion of hepatocellular carcinoma. Hepatology. 2015;62:1160-73
73. Sherry MM, Reeves A, Wu JK, Cochran BH. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells. 2009;27:2383-92
74. Shastri A, Choudhary G, Teixeira M, Gordon-Mitchell S, Ramachandra N, Bernard L. et al. Antisense STAT3 inhibitor decreases viability of myelodysplastic and leukemic stem cells. J Clin Invest. 2018;128:5479-88
75. Toso A, Revandkar A, Di Mitri D, Guccini I, Proietti M, Sarti M. et al. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 2014;9:75-89
76. Chefetz I, Alvero AB, Holmberg JC, Lebowitz N, Craveiro V, Yang-Hartwich Y. et al. TLR2 enhances ovarian cancer stem cell self-renewal and promotes tumor repair and recurrence. Cell Cycle. 2013;12:511-21
77. Jia D, Yang W, Li L, Liu H, Tan Y, Ooi S. et al. beta-Catenin and NF-kappaB co-activation triggered by TLR3 stimulation facilitates stem cell-like phenotypes in breast cancer. Cell Death Differ. 2015;22:298-310
78. Liu M, Sakamaki T, Casimiro MC, Willmarth NE, Quong AA, Ju X. et al. The canonical NF-kappaB pathway governs mammary tumorigenesis in transgenic mice and tumor stem cell expansion. Cancer Res. 2010;70:10464-73
79. Moreira D, Zhang Q, Hossain DM, Nechaev S, Li H, Kowolik CM. et al. TLR9 signaling through NF-κB_RELA and STAT3 promotes tumor-propagating potential of prostate cancer cells. Oncotarget. 2015;6:17302-13
80. Sun L, Mathews LA, Cabarcas SM, Zhang X, Yang A, Zhang Y. et al. Epigenetic regulation of SOX9 by the NF-kappaB signaling pathway in pancreatic cancer stem cells. Stem Cells. 2013;31:1454-66
81. Garner JM, Fan M, Yang CH, Du Z, Sims M, Davidoff AM. et al. Constitutive activation of signal transducer and activator of transcription 3 (STAT3) and nuclear factor kappaB signaling in glioblastoma cancer stem cells regulates the Notch pathway. J Biol Chem. 2013;288:26167-76
82. Saletore Y, Meyer K, Korlach J, Vilfan ID, Jaffrey S, Mason CE. The birth of the epitranscriptome: deciphering the function of RNA modifications. Genome Biol. 2012;13:175
83. Jiang Q, Crews LA, Holm F, Jamieson CHM. RNA editing-dependent epitranscriptome diversity in cancer stem cells. Nat Rev Cancer. 2017;17:381-92
84. Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12:767-72
85. Shi H, Wei J, He C. Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol Cell. 2019;74:640-50
86. Meyer KD, Jaffrey SR. Rethinking m(6)A readers, writers, and erasers. Annu Rev Cell Dev Biol. 2017;33:319-42
87. Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D. et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117-20
88. Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF. et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61:507-19
89. Roundtree IA, Luo GZ, Zhang Z, Wang X, Zhou T, Cui Y. et al. YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. Elife. 2017 6
90. Zhou KI, Shi H, Lyu R, Wylder AC, Matuszek Z, Pan JN. et al. Regulation of co-transcriptional Pre-mRNA splicing by m(6)A through the low-complexity protein hnRNPG. Mol Cell. 2019;76:70-81 e9
91. Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H. et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161:1388-99
92. Huang H, Weng H, Sun W, Qin X, Shi H, Wu H. et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20:285-95
93. Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O. et al. 5' UTR m(6)A promotes cap-independent translation. Cell. 2015;163:999-1010
94. Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M. et al. Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science. 2015;347:1002-6
95. Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G. et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. 2017;23:1369-76
96. Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millan-Zambrano G, Robson SC. et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature. 2017;552:126-31
97. Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L. et al. METTL14 Inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)A modification. Cell Stem Cell. 2018;22:191-205 e9
98. Li Z, Weng H, Su R, Weng X, Zuo Z, Li C. et al. FTO plays an oncogenic role in acute myeloid leukemia as a N(6)-methyladenosine RNA demethylase. Cancer Cell. 2017;31:127-41
99. Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y. et al. R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell. 2018;172:90-105 e23
100. Paris J, Morgan M, Campos J, Spencer GJ, Shmakova A, Ivanova I. et al. Targeting the RNA m(6)A reader YTHDF2 selectively compromises cancer stem cells in acute myeloid leukemia. Cell Stem Cell. 2019;25:137-48 e6
101. Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G. et al. m(6)A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18:2622-34
102. Li F, Yi Y, Miao Y, Long W, Long T, Chen S. et al. N(6)-methyladenosine modulates nonsense-mediated mRNA decay in human glioblastoma. Cancer Res. 2019;79:5785-98
103. Visvanathan A, Patil V, Arora A, Hegde AS, Arivazhagan A, Santosh V. et al. Essential role of METTL3-mediated m(6)A modification in glioma stem-like cells maintenance and radioresistance. Oncogene. 2018;37:522-33
104. Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z. et al. m(6)A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31:591-606 e6
105. Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I. et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m(6)A-demethylation of NANOG mRNA. Proc Natl Acad Sci U S A. 2016;113:E2047-56
106. Peng Z, Cheng Y, Tan BC, Kang L, Tian Z, Zhu Y. et al. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat Biotechnol. 2012;30:253-60
107. Xu X, Wang Y, Liang H. The role of A-to-I RNA editing in cancer development. Curr Opin Genet Dev. 2018;48:51-6
108. Han L, Diao L, Yu S, Xu X, Li J, Zhang R. et al. The genomic landscape and clinical relevance of A-to-I RNA editing in human cancers. Cancer Cell. 2015;28:515-28
109. Jiang Q, Crews LA, Barrett CL, Chun HJ, Court AC, Isquith JM. et al. ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Proc Natl Acad Sci U S A. 2013;110:1041-6
110. Zipeto MA, Court AC, Sadarangani A, Delos Santos NP, Balaian L, Chun HJ. et al. ADAR1 activation drives leukemia stem cell self-renewal by impairing Let-7 biogenesis. Cell Stem Cell. 2016;19:177-91
111. Chen L, Li Y, Lin CH, Chan TH, Chow RK, Song Y. et al. Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nat Med. 2013;19:209-16
112. Fu L, Qin YR, Ming XY, Zuo XB, Diao YW, Zhang LY. et al. RNA editing of SLC22A3 drives early tumor invasion and metastasis in familial esophageal cancer. Proc Natl Acad Sci U S A. 2017;114:E4631-E40
113. Wang Y, Xu X, Yu S, Jeong KJ, Zhou Z, Han L. et al. Systematic characterization of A-to-I RNA editing hotspots in microRNAs across human cancers. Genome Res. 2017;27:1112-25
114. Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N. et al. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature. 2000;406:78-81
115. Paz N, Levanon EY, Amariglio N, Heimberger AB, Ram Z, Constantini S. et al. Altered adenosine-to-inosine RNA editing in human cancer. Genome Res. 2007;17:1586-95
116. Galeano F, Rossetti C, Tomaselli S, Cifaldi L, Lezzerini M, Pezzullo M. et al. ADAR2-editing activity inhibits glioblastoma growth through the modulation of the CDC14B/Skp2/p21/p27 axis. Oncogene. 2013;32:998-1009
117. Tomaselli S, Galeano F, Alon S, Raho S, Galardi S, Polito VA. et al. Modulation of microRNA editing, expression and processing by ADAR2 deaminase in glioblastoma. Genome Biol. 2015;16:5
118. Kahles A, Lehmann KV, Toussaint NC, Huser M, Stark SG, Sachsenberg T. et al. Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer Cell. 2018;34:211-24 e6
119. Sebestyen E, Singh B, Minana B, Pages A, Mateo F, Pujana MA. et al. Large-scale analysis of genome and transcriptome alterations in multiple tumors unveils novel cancer-relevant splicing networks. Genome Res. 2016;26:732-44
120. Gabut M, Samavarchi-Tehrani P, Wang X, Slobodeniuc V, O'Hanlon D, Sung HK. et al. An alternative splicing switch regulates embryonic stem cell pluripotency and reprogramming. Cell. 2011;147:132-46
121. Crews LA, Balaian L, Delos Santos NP, Leu HS, Court AC, Lazzari E. et al. RNA splicing modulation selectively impairs leukemia stem cell maintenance insecondary human AML. Cell Stem Cell. 2016;19:599-612
122. Abrahamsson AE, Geron I, Gotlib J, Dao KH, Barroga CF, Newton IG. et al. Glycogen synthase kinase 3beta missplicing contributes to leukemia stem cell generation. Proc Natl Acad Sci U S A. 2009;106:3925-9
123. Holm F, Hellqvist E, Mason CN, Ali SA, Delos-Santos N, Barrett CL. et al. Reversion to an embryonic alternative splicing program enhances leukemia stem cell self-renewal. Proc Natl Acad Sci U S A. 2015;112:15444-9
124. Tan DQ, Li Y, Yang C, Li J, Tan SH, Chin DWL. et al. PRMT5 modulates splicing for genome integrity and preserves proteostasis of hematopoietic stem cells. Cell Rep. 2019;26:2316-28 e6
125. Saez B, Walter MJ, Graubert TA. Splicing factor gene mutations in hematologic malignancies. Blood. 2017;129:1260-9
126. Yuan H, Li N, Fu D, Ren J, Hui J, Peng J. et al. Histone methyltransferase SETD2 modulates alternative splicing to inhibit intestinal tumorigenesis. J Clin Invest. 2017;127:3375-91
127. Song X, Wan X, Huang T, Zeng C, Sastry N, Wu B. et al. SRSF3-regulated RNA alternative splicing promotes glioblastoma tumorigenicity by affecting multiple cellular processes. Cancer Res. 2019;79:5288-301
128. Zhang H, Brown RL, Wei Y, Zhao P, Liu S, Liu X. et al. CD44 splice isoform switching determines breast cancer stem cell state. Genes Dev. 2019;33:166-79
129. Brumbaugh J, Di Stefano B, Wang X, Borkent M, Forouzmand E, Clowers KJ. et al. Nudt21 controls cell fate by connecting alternative polyadenylation to chromatinsignaling. Cell. 2018;172:629-31
130. Wainwright EN, Scaffidi P. Epigenetics and cancer stem cells: unleashing, hijacking, and restricting cellular plasticity. Trends Cancer. 2017;3:372-86
131. Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA. et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340:857-61
132. Funato K, Major T, Lewis PW, Allis CD, Tabar V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science. 2014;346:1529-33
133. Cancer Genome Atlas Research N, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ. et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059-74
134. Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016;30:733-50
135. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y. et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930-5
136. Turcan S, Makarov V, Taranda J, Wang Y, Fabius AWM, Wu W. et al. Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat Genet. 2018;50:62-72
137. Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S. et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483:484-8
138. Xie Q, Wu TP, Gimple RC, Li Z, Prager BC, Wu Q. et al. N(6)-methyladenine DNA modification in glioblastoma. Cell. 2018;175:1228-43 e20
139. Yamazaki J, Estecio MR, Lu Y, Long H, Malouf GG, Graber D. et al. The epigenome of AML stem and progenitor cells. Epigenetics. 2013;8:92-104
140. Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K. et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226-31
141. Takawa M, Masuda K, Kunizaki M, Daigo Y, Takagi K, Iwai Y. et al. Validation of the histone methyltransferase EZH2 as a therapeutic target for various types of human cancer and as a prognostic marker. Cancer Sci. 2011;102:1298-305
142. Crea F, Hurt EM, Farrar WL. Clinical significance of polycomb gene expression in brain tumors. Mol Cancer. 2010;9:265
143. Orzan F, Pellegatta S, Poliani PL, Pisati F, Caldera V, Menghi F. et al. Enhancer of Zeste 2 (EZH2) is up-regulated in malignant gliomas and in glioma stem-like cells. Neuropathol Appl Neurobiol. 2011;37:381-94
144. Gu Z, Liu Y, Cai F, Patrick M, Zmajkovic J, Cao H. et al. Loss of EZH2 Reprograms BCAA metabolism to drive leukemic transformation. Cancer Discov. 2019;9:1228-47
145. Sashida G, Wang C, Tomioka T, Oshima M, Aoyama K, Kanai A. et al. The loss of Ezh2 drives the pathogenesis of myelofibrosis and sensitizes tumor-initiating cells to bromodomain inhibition. J Exp Med. 2016;213:1459-77
146. Muto T, Sashida G, Oshima M, Wendt GR, Mochizuki-Kashio M, Nagata Y. et al. Concurrent loss of Ezh2 and Tet2 cooperates in the pathogenesis of myelodysplastic disorders. J Exp Med. 2013;210:2627-39
147. Shimizu T, Kubovcakova L, Nienhold R, Zmajkovic J, Meyer SC, Hao-Shen H. et al. Loss of Ezh2 synergizes with JAK2-V617F in initiating myeloproliferative neoplasms and promoting myelofibrosis. J Exp Med. 2016;213:1479-96
148. Booth CAG, Barkas N, Neo WH, Boukarabila H, Soilleux EJ, Giotopoulos G. et al. Ezh2 and Runx1 mutationscollaborate to initiate lympho-myeloid leukemia in early thymic progenitors. Cancer Cell. 2018;33:274-91 e8
149. Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG. et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624-9
150. Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R. et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42:181-5
151. Holm K, Grabau D, Lovgren K, Aradottir S, Gruvberger-Saal S, Howlin J. et al. Global H3K27 trimethylation and EZH2 abundance in breast tumor subtypes. Mol Oncol. 2012;6:494-506
152. Wang Y, He L, Du Y, Zhu P, Huang G, Luo J. et al. The long noncoding RNA lncTCF7 promotes self-renewal of human liver cancer stem cells through activation of Wnt signaling. Cell Stem Cell. 2015;16:413-25
153. Chen Q, Cai J, Wang Q, Wang Y, Liu M, Yang J. et al. Long noncoding RNA NEAT1, regulated by the EGFR pathway, contributes to glioblastoma progression through the WNT/beta-catenin pathway by scaffolding EZH2. Clin Cancer Res. 2018;24:684-95
154. Guo J, Cai H, Liu X, Zheng J, Liu Y, Gong W. et al. Long non-coding RNA LINC00339 stimulates glioma vasculogenic mimicry formation by regulating the miR-539-5p/TWIST1/MMPs axis. Mol Ther Nucleic Acids. 2018;10:170-86
155. Zhuang J, Shen L, Yang L, Huang X, Lu Q, Cui Y. et al. TGFbeta1 promotes gemcitabine resistance through regulating the lncRNA-let/NF90/miR-145 signaling axis in bladder cancer. Theranostics. 2017;7:3053-67
156. Huang T, Wan X, Alvarez AA, James CD, Song X, Yang Y. et al. MIR93 (microRNA -93) regulates tumorigenicity and therapy response of glioblastoma by targeting autophagy. Autophagy. 2019;15:1100-11
157. Anthiya S, Griveau A, Loussouarn C, Baril P, Garnett M, Issartel JP. et al. MicroRNA-based drugs for brain tumors. Trends Cancer. 2018;4:222-38
158. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
159. Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 2016;41:211-8
160. Chen CL, Uthaya Kumar DB, Punj V, Xu J, Sher L, Tahara SM. et al. NANOG metabolically reprograms tumor-initiating stem-like cells through tumorigenic changes in oxidative phosphorylation and fatty acid metabolism. Cell Metab. 2016;23:206-19
161. Dong C, Yuan T, Wu Y, Wang Y, Fan TW, Miriyala S. et al. Loss of FBP1 by snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell. 2013;23:316-31
162. Saito Y, Chapple RH, Lin A, Kitano A, Nakada D. AMPK protects leukemia-initiating cells in myeloid leukemias from metabolic stress in the bone marrow. Cell Stem Cell. 2015;17:585-96
163. Wang YH, Israelsen WJ, Lee D, Yu VWC, Jeanson NT, Clish CB. et al. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell. 2014;158:1309-23
164. Simsek T, Kocabas F, Zheng J, Deberardinis RJ, Mahmoud AI, Olson EN. et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. 2010;7:380-90
165. Liu PP, Liao J, Tang ZJ, Wu WJ, Yang J, Zeng ZL. et al. Metabolic regulation of cancer cell side population by glucose through activation of the Akt pathway. Cell Death Differ. 2014;21:124-35
166. Janiszewska M, Suva ML, Riggi N, Houtkooper RH, Auwerx J, Clement-Schatlo V. et al. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012;26:1926-44
167. Vlashi E, Lagadec C, Vergnes L, Matsutani T, Masui K, Poulou M. et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc Natl Acad Sci U S A. 2011;108:16062-7
168. Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M. et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12:329-41
169. Kuntz EM, Baquero P, Michie AM, Dunn K, Tardito S, Holyoake TL. et al. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat Med. 2017;23:1234-40
170. Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D. et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell. 2013;23:811-25
171. Feng W, Gentles A, Nair RV, Huang M, Lin Y, Lee CY. et al. Targeting unique metabolic properties of breast tumor initiating cells. Stem Cells. 2014;32:1734-45
172. Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M. et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514:628-32
173. Pasto A, Bellio C, Pilotto G, Ciminale V, Silic-Benussi M, Guzzo G. et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget. 2014;5:4305-19
174. Flavahan WA, Wu Q, Hitomi M, Rahim N, Kim Y, Sloan AE. et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat Neurosci. 2013;16:1373-82
175. Dupuy F, Tabaries S, Andrzejewski S, Dong Z, Blagih J, Annis MG. et al. PDK1-dependent metabolic reprogramming dictates metastatic potential in breast cancer. Cell Metab. 2015;22:577-89
176. Tamada M, Nagano O, Tateyama S, Ohmura M, Yae T, Ishimoto T. et al. Modulation of glucose metabolism by CD44 contributes to antioxidant status and drug resistance in cancer cells. Cancer Res. 2012;72:1438-48
177. Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M. et al. Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell. 2016;19:23-37
178. Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS. et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature. 2017;541:41-5
179. Li J, Condello S, Thomes-Pepin J, Ma X, Xia Y, Hurley TD. et al. Lipid desaturation is a metabolic marker and therapeutic target of ovarian cancer stem cells. Cell Stem Cell. 2017;20:303-14 e5
180. Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE. et al. A PML-PPAR-delta pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med. 2012;18:1350-8
181. Hale JS, Otvos B, Sinyuk M, Alvarado AG, Hitomi M, Stoltz K. et al. Cancer stem cell-specific scavenger receptor CD36 drives glioblastoma progression. Stem Cells. 2014;32:1746-58
182. Wang T, Fahrmann JF, Lee H, Li YJ, Tripathi SC, Yue C. et al. JAK/STAT3-regulated fatty acid beta-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab. 2018;27:136-50 e5
183. Hirata N, Yamada S, Shoda T, Kurihara M, Sekino Y, Kanda Y. Sphingosine-1-phosphate promotes expansion of cancer stem cells via S1PR3 by a ligand-independent Notch activation. Nat Commun. 2014;5:4806
184. Kurtova AV, Xiao J, Mo Q, Pazhanisamy S, Krasnow R, Lerner SP. et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature. 2015;517:209-13
185. Jones CL, Stevens BM, D'Alessandro A, Reisz JA, Culp-Hill R, Nemkov T. et al. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell. 2018;34:724-40 e4
186. Wu Z, Wei D, Gao W, Xu Y, Hu Z, Ma Z. et al. TPO-induced metabolic reprogramming drives liver metastasis of colorectal cancer CD110+ tumor-initiating cells. Cell Stem Cell. 2015;17:47-59
187. Samanta D, Park Y, Andrabi SA, Shelton LM, Gilkes DM, Semenza GL. PHGDH expression is required for mitochondrial redox homeostasis, breast cancer stem cell maintenance, and lung metastasis. Cancer Res. 2016;76:4430-42
188. Raffel S, Falcone M, Kneisel N, Hansson J, Wang W, Lutz C. et al. BCAT1 restricts alphaKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature. 2017;551:384-8
189. Wang X, Yang K, Xie Q, Wu Q, Mack SC, Shi Y. et al. Purine synthesis promotes maintenance of brain tumor initiating cells in glioma. Nat Neurosci. 2017;20:661-73
190. Bulusu V, Tumanov S, Michalopoulou E, van den Broek NJ, MacKay G, Nixon C. et al. Acetate recapturing by nuclear acetyl-CoA synthetase 2 prevents loss of histone acetylation during oxygen and serum limitation. Cell Rep. 2017;18:647-58
191. Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK. et al. Acetate dependence of tumors. Cell. 2014;159:1591-602
192. McBrian MA, Behbahan IS, Ferrari R, Su T, Huang TW, Li K. et al. Histone acetylation regulates intracellular pH. Mol Cell. 2013;49:310-21
193. Intlekofer AM, Finley LWS. Metabolic signatures of cancer cells and stem cells. Nat Metab. 2019;1:177-88
194. Wang Z, Yip LY, Lee JHJ, Wu Z, Chew HY, Chong PKW. et al. Methionine is a metabolic dependency of tumor-initiating cells. Nat Med. 2019;25:825-37
195. Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O. et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474-8
196. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A. et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553-67
197. Saha SK, Parachoniak CA, Ghanta KS, Fitamant J, Ross KN, Najem MS. et al. Mutant IDH inhibits HNF-4alpha to block hepatocyte differentiation and promote biliary cancer. Nature. 2014;513:110-4
198. Heppner GH. Tumor heterogeneity. Cancer Res. 1984;44:2259-65
199. Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501:346-54
200. Welch DR. Tumor heterogeneity-a 'contemporary concept' founded on historical insights and predictions. Cancer Res. 2016;76:4-6
201. Prager BC, Xie Q, Bao S, Rich JN. Cancer stem cells: the architects of the tumor Ecosystem. Cell Stem Cell. 2019;24:41-53
202. Poggi A, Varesano S, Zocchi MR. How to hit mesenchymal stromal cells and make the tumor microenvironment immunostimulant rather than immunosuppressive. Front Immunol. 2018;9:262
203. Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K. et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2:737-44
204. Sun Y, Campisi J, Higano C, Beer TM, Porter P, Coleman I. et al. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat Med. 2012;18:1359-68
205. Turcotte M, Spring K, Pommey S, Chouinard G, Cousineau I, George J. et al. CD73 is associated with poor prognosis in high-grade serous ovarian cancer. Cancer Res. 2015;75:4494-503
206. Allard D, Allard B, Gaudreau PO, Chrobak P, Stagg J. CD73-adenosine: a next-generation target in immuno-oncology. Immunotherapy. 2016;8:145-63
207. Loeffler M, Kruger JA, Niethammer AG, Reisfeld RA. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J Clin Invest. 2006;116:1955-62
208. Scott AM, Wiseman G, Welt S, Adjei A, Lee FT, Hopkins W. et al. A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin Cancer Res. 2003;9:1639-47
209. Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V. et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. 2014;2:154-66
210. Borriello A, Caldarelli I, Bencivenga D, Stampone E, Perrotta S, Oliva A. et al. Tyrosine kinase inhibitors and mesenchymal stromal cells: effects on self-renewal, commitment and functions. Oncotarget. 2017;8:5540-65
211. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D. et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457-61
212. Rettig MP, Ansstas G, DiPersio JF. Mobilization of hematopoietic stem and progenitor cells using inhibitors of CXCR4 and VLA-4. Leukemia. 2012;26:34-53
213. Ide M, Jinnin M, Tomizawa Y, Wang Z, Kajihara I, Fukushima S. et al. Transforming growth factor beta-inhibitor repsox downregulates collagen expression of scleroderma dermal fibroblasts and prevents bleomycin-induced mice skin fibrosis. Exp Dermatol. 2017;26:1139-43
214. Scala S. Molecular Pathways: Targeting the CXCR4-CXCL12 axis-untapped potential in the tumor microenvironment. Clin Cancer Res. 2015;21:4278-85
215. Su S, Chen J, Yao H, Liu J, Yu S, Lao L. et al. CD10(+)GPR77(+) cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell. 2018;172:841-56 e16
216. LaGory EL, Giaccia AJ. The ever-expanding role of HIF in tumour and stromal biology. Nat Cell Biol. 2016;18:356-65
217. Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:873-87
218. Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3:401-10
219. De Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer. 2017;17:457-74
220. Tredan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst. 2007;99:1441-54
221. Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J. et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci U S A. 2012;109:17561-6
222. Johansson-Percival A, He B, Ganss R. Immunomodulation of tumor vessels: it takes two to tango. Trends Immunol. 2018;39:801-14
223. Charles N, Ozawa T, Squatrito M, Bleau AM, Brennan CW, Hambardzumyan D. et al. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell. 2010;6:141-52
224. Varnat F, Duquet A, Malerba M, Zbinden M, Mas C, Gervaz P. et al. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol Med. 2009;1:338-51
225. Zhu TS, Costello MA, Talsma CE, Flack CG, Crowley JG, Hamm LL. et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res. 2011;71:6061-72
226. Lu J, Ye X, Fan F, Xia L, Bhattacharya R, Bellister S. et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged-1. Cancer Cell. 2013;23:171-85
227. Beck B, Driessens G, Goossens S, Youssef KK, Kuchnio A, Caauwe A. et al. A vascular niche and a VEGF-Nrp1 loop regulate the initiation and stemness of skin tumours. Nature. 2011;478:399-403
228. Maniotis AJ, Folberg R, Hess A, Seftor EA, Gardner LM, Pe'er J. et al. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol. 1999;155:739-52
229. Fan J, Kamphorst JJ, Mathew R, Chung MK, White E, Shlomi T. et al. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol Syst Biol. 2013;9:712
230. Shangguan W, Fan C, Chen X, Lu R, Liu Y, Li Y. et al. Endothelium originated from colorectal cancer stem cells constitute cancer blood vessels. Cancer Sci. 2017;108:1357-67
231. Zhou W, Chen C, Shi Y, Wu Q, Gimple RC, Fang X. et al. Targeting glioma stem cell-derived pericytes disrupts the blood-tumor barrier and improves chemotherapeutic efficacy. Cell Stem Cell. 2017;21:591-603 e4
232. Fang S, Salven P. Stem cells in tumor angiogenesis. J Mol Cell Cardiol. 2011;50:290-5
233. van Ravenswaay Claasen HH, Kluin PM, Fleuren GJ. Tumor infiltrating cells in human cancer. On the possible role of CD16+ macrophages in antitumor cytotoxicity. Lab Invest. 1992;67:166-74
234. Finn OJ. A believer's overview of cancer immunosurveillance and immunotherapy. J Immunol. 2018;200:385-91
235. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39-51
236. Lahmar Q, Keirsse J, Laoui D, Movahedi K, Van Overmeire E, Van Ginderachter JA. Tissue-resident versus monocyte-derived macrophages in the tumor microenvironment. Biochim Biophys Acta. 2016;1865:23-34
237. Kalbasi A, June CH, Haas N, Vapiwala N. Radiation and immunotherapy: a synergistic combination. J Clin Invest. 2013;123:2756-63
238. Jinushi M, Chiba S, Yoshiyama H, Masutomi K, Kinoshita I, Dosaka-Akita H. et al. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc Natl Acad Sci U S A. 2011;108:12425-30
239. Fan QM, Jing YY, Yu GF, Kou XR, Ye F, Gao L. et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014;352:160-8
240. Li S, Xu F, Zhang J, Wang L, Zheng Y, Wu X. et al. Tumor-associated macrophages remodeling EMT and predicting survival in colorectal carcinoma. Oncoimmunology. 2018;7:e1380765
241. Nappo G, Handle F, Santer FR, McNeill RV, Seed RI, Collins AT. et al. The immunosuppressive cytokine interleukin-4 increases the clonogenic potential of prostate stem-like cells by activation of STAT6 signalling. Oncogenesis. 2017;6:e342
242. Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J. et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol. 2015;17:170-82
243. Lu H, Clauser KR, Tam WL, Frose J, Ye X, Eaton EN. et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat Cell Biol. 2014;16:1105-17
244. Yamashina T, Baghdadi M, Yoneda A, Kinoshita I, Suzu S, Dosaka-Akita H. et al. Cancer stem-like cells derived from chemoresistant tumors have a unique capacity to prime tumorigenic myeloid cells. Cancer Res. 2014;74:2698-709
245. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348:74-80
246. Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS. et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. 2014;20:607-15
247. Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean MC, Validire P, Trautmann A. et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest. 2012;122:899-910
248. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS. et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110:20212-7
249. DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. 2019;19:369-82
250. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF. et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19:1264-72
251. Butowski N, Colman H, De Groot JF, Omuro AM, Nayak L, Wen PY. et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy foundation early phase clinical trials consortium phase II study. Neuro Oncol. 2016;18:557-64
252. Fantini MC, Becker C, Monteleone G, Pallone F, Galle PR, Neurath MF. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25- T cells through Foxp3 induction and down-regulation of Smad7. J Immunol. 2004;172:5149-53
253. Oh SA, Liu M, Nixon BG, Kang D, Toure A, Bivona M. et al. Foxp3-independent mechanism by which TGF-beta controls peripheral T cell tolerance. Proc Natl Acad Sci U S A. 2017;114:E7536-E44
254. Xia A, Zhang Y, Xu J, Yin T, Lu XJ. T cell dysfunction in cancer immunity and immunotherapy. Front Immunol. 2019;10:1719
255. Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C. et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc Natl Acad Sci U S A. 2018;115:E4041-E50
256. Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12:298-306
257. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252-64
258. Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nat Rev Cancer. 2004;4:448-56
259. Chang JC. Cancer stem cells: Role in tumor growth, recurrence, metastasis, and treatment resistance. Medicine (Baltimore). 2016;95:S20-5
260. Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K. et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature. 2015;526:131-5
261. Phi LTH, Sari IN, Yang YG, Lee SH, Jun N, Kim KS. et al. Cancer stem cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells Int. 2018;2018:5416923
262. Schmidt-Kittler O, Ragg T, Daskalakis A, Granzow M, Ahr A, Blankenstein TJ. et al. From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. Proc Natl Acad Sci U S A. 2003;100:7737-42
263. Chen Y, Li D, Wang D, Liu X, Yin N, Song Y. et al. Quiescence and attenuated DNA damage response promote survival of esophageal cancer stem cells. J Cell Biochem. 2012;113:3643-52
264. Takeishi S, Matsumoto A, Onoyama I, Naka K, Hirao A, Nakayama KI. Ablation of Fbxw7 eliminates leukemia-initiating cells by preventing quiescence. Cancer Cell. 2013;23:347-61
265. Ou J, Peng Y, Yang W, Zhang Y, Hao J, Li F. et al. ABHD5 blunts the sensitivity of colorectal cancer to fluorouracil via promoting autophagic uracil yield. Nat Commun. 2019;10:1078
266. Moeckel S, LaFrance K, Wetsch J, Seliger C, Riemenschneider MJ, Proescholdt M. et al. ATF4 contributes to autophagy and survival in sunitinib treated brain tumor initiating cells (BTICs). Oncotarget. 2019;10:368-82
267. Ojha R, Leli NM, Onorati A, Piao S, Verginadis II, Tameire F. et al. ER translocation of the MAPK pathway drives therapy resistance in BRAF-mutant melanoma. Cancer Discov. 2019;9:396-415
268. Huang T, Kim CK, Alvarez AA, Pangeni RP, Wan X, Song X. et al. MST4 phosphorylation of ATG4B regulates autophagic activity, tumorigenicity, and radioresistance in glioblastoma. Cancer Cell. 2017;32:840-55 e8
269. Sosa MS, Parikh F, Maia AG, Estrada Y, Bosch A, Bragado P. et al. NR2F1 controls tumour cell dormancy via Sox9- and RARbeta-driven quiescence programmes. Nat Commun. 2015;6:6170
270. Orienti I, Francescangeli F, De Angelis ML, Fecchi K, Bongiorno-Borbone L, Signore M. et al. A new bioavailable fenretinide formulation with antiproliferative, antimetabolic, and cytotoxic effects on solid tumors. Cell Death Dis. 2019;10:529
271. Gupta PB, Pastushenko I, Skibinski A, Blanpain C, Kuperwasser C. Phenotypic plasticity: driver of cancer initiation, progression, and therapy resistance. Cell Stem Cell. 2019;24:65-78
272. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY. et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704-15
273. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871-90
274. Battula VL, Evans KW, Hollier BG, Shi Y, Marini FC, Ayyanan A. et al. Epithelial-mesenchymal transition-derived cells exhibit multilineage differentiation potential similar to mesenchymal stem cells. Stem Cells. 2010;28:1435-45
275. Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265-73
276. Chou MY, Hu FW, Yu CH, Yu CC. Sox2 expression involvement in the oncogenicity and radiochemoresistance of oral cancer stem cells. Oral Oncol. 2015;51:31-9
277. Huang CE, Yu CC, Hu FW, Chou MY, Tsai LL. Enhanced chemosensitivity by targeting Nanog in head and neck squamous cell carcinomas. Int J Mol Sci. 2014;15:14935-48
278. Ji J, Yu Y, Li ZL, Chen MY, Deng R, Huang X. et al. XIAP limits autophagic degradation of Sox2 and is a therapeutic target in nasopharyngeal carcinoma stem cells. Theranostics. 2018;8:1494-510
279. Koo BS, Lee SH, Kim JM, Huang S, Kim SH, Rho YS. et al. Oct4 is a critical regulator of stemness in head and neck squamous carcinoma cells. Oncogene. 2015;34:2317-24
280. Siebzehnrubl FA, Silver DJ, Tugertimur B, Deleyrolle LP, Siebzehnrubl D, Sarkisian MR. et al. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol Med. 2013;5:1196-212
281. Yook JI, Li XY, Ota I, Hu C, Kim HS, Kim NH. et al. A Wnt-Axin2-GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol. 2006;8:1398-406
282. Kim K, Lu Z, Hay ED. Direct evidence for a role of beta-catenin/LEF-1 signaling pathway in induction of EMT. Cell Biol Int. 2002;26:463-76
283. Xie M, Zhang L, He CS, Xu F, Liu JL, Hu ZH. et al. Activation of Notch-1 enhances epithelial-mesenchymal transition in gefitinib-acquired resistant lung cancer cells. J Cell Biochem. 2012;113:1501-13
284. Black PC, Brown GA, Inamoto T, Shrader M, Arora A, Siefker-Radtke AO. et al. Sensitivity to epidermal growth factor receptor inhibitor requires E-cadherin expression in urothelial carcinoma cells. Clin Cancer Res. 2008;14:1478-86
285. Aguirre-Ghiso JA, Sosa MS. Emerging topics on disseminated cancer cell dormancy and the paradigm of metastasis. Ann Rev Can Biol. 2018;2:377-93
286. Liu X, Sun H, Qi J, Wang L, He S, Liu J. et al. Sequential introduction of reprogramming factors reveals a time-sensitive requirement for individual factors and a sequential EMT-MET mechanism for optimal reprogramming. Nat Cell Biol. 2013;15:829-38
287. Neuzillet C, Tijeras-Raballand A, Cohen R, Cros J, Faivre S, Raymond E. et al. Targeting the TGFbeta pathway for cancer therapy. Pharmacol Ther. 2015;147:22-31
288. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M. et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555-67
289. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755-68
290. Raha D, Wilson TR, Peng J, Peterson D, Yue P, Evangelista M. et al. The cancer stem cell marker aldehyde dehydrogenase is required to maintain a drug-tolerant tumor cell subpopulation. Cancer Res. 2014;74:3579-90
291. Huang CP, Tsai MF, Chang TH, Tang WC, Chen SY, Lai HH. et al. ALDH-positive lung cancer stem cells confer resistance to epidermal growth factor receptor tyrosine kinase inhibitors. Cancer Lett. 2013;328:144-51
292. Wang JY, Ma J, Lin YN, Wang J, Shen H, Gui FM. et al. [Mutational analysis of RNA splicing machinery genes SF3B1, U2AF1 and SRSF2 in 118 patients with myelodysplastic syndromes and related diseases]. Zhonghua Xue Ye Xue Za Zhi. 2017;38:192-7
293. House CD, Jordan E, Hernandez L, Ozaki M, James JM, Kim M. et al. NFkappaB promotes ovarian tumorigenesis via classical pathways that support proliferative cancer cells and alternative pathways that support ALDH(+) cancer stem-like cells. Cancer Res. 2017;77:6927-40
294. Croker AK, Allan AL. Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiation resistance of stem-like ALDHhiCD44(+) human breast cancer cells. Breast Cancer Res Treat. 2012;133:75-87
295. DeGorter MK, Xia CQ, Yang JJ, Kim RB. Drug transporters in drug efficacy and toxicity. Annu Rev Pharmacol Toxicol. 2012;52:249-73
296. Begicevic RR, Falasca M. ABC transporters in cancer stem cells: beyond chemoresistance. Int J Mol Sci. 2017;18:E2362
297. Zheng X, Zhan Z, Naren D, Li J, Yan T, Gong Y. Prognostic value of SRSF2 mutations in patients with de novo myelodysplastic syndromes: A meta-analysis. PLoS One. 2017;12:e0185053
298. Oliveira BR, Figueiredo MA, Trindade GS, Marins LF. OCT4 mutations in human erythroleukemic cells: implications for multiple drug resistance (MDR) phenotype. Mol Cell Biochem. 2015;400:41-50
299. Porro A, Haber M, Diolaiti D, Iraci N, Henderson M, Gherardi S. et al. Direct and coordinate regulation of ATP-binding cassette transporter genes by Myc factors generates specific transcription signatures that significantly affect the chemoresistance phenotype of cancer cells. J Biol Chem. 2010;285:19532-43
300. Bleau AM, Hambardzumyan D, Ozawa T, Fomchenko EI, Huse JT, Brennan CW. et al. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell. 2009;4:226-35
301. Nakanishi T, Shiozawa K, Hassel BA, Ross DD. Complex interaction of BCRP/ABCG2 and imatinib in BCR-ABL-expressing cells: BCRP-mediated resistance to imatinib is attenuated by imatinib-induced reduction of BCRP expression. Blood. 2006;108:678-84
302. Chun SY, Kwon YS, Nam KS, Kim S. Lapatinib enhances the cytotoxic effects of doxorubicin in MCF-7 tumorspheres by inhibiting the drug efflux function of ABC transporters. Biomed Pharmacother. 2015;72:37-43
303. Rabindran SK, Ross DD, Doyle LA, Yang W, Greenberger LM. Fumitremorgin C reverses multidrug resistance in cells transfected with the breast cancer resistance protein. Cancer Res. 2000;60:47-50
304. Maugeri-Sacca M, Bartucci M, De Maria R. DNA damage repair pathways in cancer stem cells. Mol Cancer Ther. 2012;11:1627-36
305. Schulz A, Meyer F, Dubrovska A, Borgmann K. Cancer stem cells and radioresistance: DNA repair and beyond. Cancers (Basel). 2019;11:E862
306. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB. et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756-60
307. Ropolo M, Daga A, Griffero F, Foresta M, Casartelli G, Zunino A. et al. Comparative analysis of DNA repair in stem and nonstem glioma cell cultures. Mol Cancer Res. 2009;7:383-92
308. Facchino S, Abdouh M, Chatoo W, Bernier G. BMI1 confers radioresistance to normal and cancerous neural stem cells through recruitment of the DNA damage response machinery. J Neurosci. 2010;30:10096-111
309. Cheng L, Wu Q, Huang Z, Guryanova OA, Huang Q, Shou W. et al. L1CAM regulates DNA damage checkpoint response of glioblastoma stem cells through NBS1. EMBO J. 2011;30:800-13
310. Desai A, Webb B, Gerson SL. CD133+ cells contribute to radioresistance via altered regulation of DNA repair genes in human lung cancer cells. Radiother Oncol. 2014;110:538-45
311. King HO, Brend T, Payne HL, Wright A, Ward TA, Patel K. et al. RAD51 is a selective DNA repair target to radiosensitize glioma stem cells. Stem Cell Reports. 2017;8:125-39
312. Wu J, Mu Q, Thiviyanathan V, Annapragada A, Vigneswaran N. Cancer stem cells are enriched in Fanconi anemia head and neck squamous cell carcinomas. Int J Oncol. 2014;45:2365-72
313. Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG. et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522-6
314. Wang Y, Zhang H, Chen Y, Sun Y, Yang F, Yu W. et al. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell. 2009;138:660-72
315. Harris WJ, Huang X, Lynch JT, Spencer GJ, Hitchin JR, Li Y. et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell. 2012;21:473-87
316. Magliulo D, Bernardi R, Messina S. Lysine-specific demethylase 1A as a promising target in acute myeloid leukemia. Front Oncol. 2018;8:255
317. Huang T, Song X, Yang Y, Wan X, Alvarez AA, Sastry N. et al. Autophagy and hallmarks of cancer. Crit Rev Oncog. 2018;23:247-67
318. Kreso A, van Galen P, Pedley NM, Lima-Fernandes E, Frelin C, Davis T. et al. Self-renewal as a therapeutic target in human colorectal cancer. Nat Med. 2014;20:29-36
319. Harrington K, Freeman DJ, Kelly B, Harper J, Soria JC. Optimizing oncolytic virotherapy in cancer treatment. Nat Rev Drug Discov. 2019;18:689-706
320. Marelli G, Howells A, Lemoine NR, Wang Y. Oncolytic viral therapy and the immune system: a double-edged sword against cancer. Front Immunol. 2018;9:866
321. Jiang H, Shin DH, Nguyen TT, Fueyo J, Fan X, Henry V. et al. Localized treatment with oncolytic adenovirus delta-24-RGDOX induces systemic immunity against disseminated subcutaneous and intracranial melanomas. Clin Cancer Res. 2019;25:6801-14
322. Jiang H, Rivera-Molina Y, Gomez-Manzano C, Clise-Dwyer K, Bover L, Vence LM. et al. Oncolytic adenovirus and tumor-targeting immune modulatory therapy improve autologous cancer vaccination. Cancer Res. 2017;77:3894-907
323. van den Bossche WBL, Kleijn A, Teunissen CE, Voerman JSA, Teodosio C, Noske DP. et al. Oncolytic virotherapy in glioblastoma patients induces a tumor macrophage phenotypic shift leading to an altered glioblastoma microenvironment. Neuro Oncol. 2018;20:1494-504
324. LaRocca CJ, Warner SG. Oncolytic viruses and checkpoint inhibitors: combination therapy in clinical trials. Clin Transl Med. 2018;7:35
325. Desjardins A, Gromeier M, Herndon JE 2nd, Beaubier N, Bolognesi DP, Friedman AH. et al. Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med. 2018;379:150-61
326. Zhu G, Su W, Jin G, Xu F, Hao S, Guan F. et al. Glioma stem cells targeted by oncolytic virus carrying endostatin-angiostatin fusion gene and the expression of its exogenous gene in vitro. Brain Res. 2011;1390:59-69
327. Cheema TA, Wakimoto H, Fecci PE, Ning J, Kuroda T, Jeyaretna DS. et al. Multifaceted oncolytic virus therapy for glioblastoma in an immunocompetent cancer stem cell model. Proc Natl Acad Sci U S A. 2013;110:12006-11
328. Sahin TT, Kasuya H, Nomura N, Shikano T, Yamamura K, Gewen T. et al. Impact of novel oncolytic virus HF10 on cellular components of the tumor microenviroment in patients with recurrent breast cancer. Cancer Gene Ther. 2012;19:229-37
329. Hemminki O, Diaconu I, Cerullo V, Pesonen SK, Kanerva A, Joensuu T. et al. Ad3-hTERT-E1A, a fully serotype 3 oncolytic adenovirus, in patients with chemotherapy refractory cancer. Mol Ther. 2012;20:1821-30
330. Kimata H, Imai T, Kikumori T, Teshigahara O, Nagasaka T, Goshima F. et al. Pilot study of oncolytic viral therapy using mutant herpes simplex virus (HF10) against recurrent metastatic breast cancer. Ann Surg Oncol. 2006;13:1078-84
331. Xi Z, Yao M, Li Y, Xie C, Holst J, Liu T. et al. Guttiferone K impedes cell cycle re-entry of quiescent prostate cancer cells via stabilization of FBXW7 and subsequent c-MYC degradation. Cell Death Dis. 2016;7:e2252
332. Sancho P, Barneda D, Heeschen C. Hallmarks of cancer stem cell metabolism. Br J Cancer. 2016;114:1305-12
333. Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M. et al. MYC/PGC-1alpha balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. 2015;22:590-605
334. Guo Y, Feng K, Wang Y, Han W. Targeting cancer stem cells by using chimeric antigen receptor-modified T cells: a potential and curable approach for cancer treatment. Protein Cell. 2018;9:516-26
335. Negi LM, Talegaonkar S, Jaggi M, Ahmad FJ, Iqbal Z, Khar RK. Role of CD44 in tumour progression and strategies for targeting. J Drug Target. 2012;20:561-73
336. Jing H, Weidensteiner C, Reichardt W, Gaedicke S, Zhu X, Grosu AL. et al. Imaging and selective elimination of glioblastoma stem cells with theranostic near-infrared-labeled CD133-specific antibodies. Theranostics. 2016;6:862-74
337. Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12:1167-74
338. Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S. et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. 2019;565:234-9
339. Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanovic S, Gouttefangeas C. et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature. 2019;565:240-5
340. Wang QS, Wang Y, Lv HY, Han QW, Fan H, Guo B. et al. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol Ther. 2015;23:184-91
341. O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD. et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9:399
342. Zheng F, Dang J, Zhang H, Xu F, Ba D, Zhang B. et al. Cancer stem cell vaccination with PD-L1 and CTLA-4 blockades enhances the eradication of melanoma stem cells in a mouse tumor model. J Immunother. 2018;41:361-8
343. Ren X, Kang B, Zhang Z. Understanding tumor ecosystems by single-cell sequencing: promises and limitations. Genome Biol. 2018;19:211
344. Xiao-Jie L, Hui-Ying X, Zun-Ping K, Jin-Lian C, Li-Juan J. CRISPR-Cas9: a new and promising player in gene therapy. J Med Genet. 2015;52:289-96
345. Jiang C, Meng L, Yang B, Luo X. Application of CRISPR/Cas9 gene editing technique in the study of cancer treatment. Clin Genet. 2020;97:73-88
346. van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A. et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015;161:933-45
347. Valent P, Bonnet D, De Maria R, Lapidot T, Copland M, Melo JV. et al. Cancer stem cell definitions and terminology: the devil is in the details. Nat Rev Cancer. 2012;12:767-75
348. Bankert RB, Egilmez NK, Hess SD. Human-SCID mouse chimeric models for the evaluation of anti-cancer therapies. Trends Immunol. 2001;22:386-93
Author contact
![]() Corresponding authors: Shi-Yuan Cheng, PhD, The Ken & Ruth Davee Department of Neurology, Lou and Jean Malnati Brain Tumor Institute, The Robert H. Lurie Comprehensive Cancer Center, Northwestern University Feinberg School of Medicine, 303 E. Superior, Lurie 6-119, Chicago, IL 60611. Phone: (312)503-3043; Fax: (312)503-5603; Email: shiyuan.chengedu or Tianzhi Huang, PhD, The Ken & Ruth Davee Department of Neurology, Lou and Jean Malnati Brain Tumor Institute, The Robert H. Lurie Comprehensive Cancer Center, Northwestern University Feinberg School of Medicine, 303 E. Superior, Lurie 6-115, Chicago, IL 60611. Phone: (312)503-5034; Fax: (312)503-5603; Email: tianzhi.huangedu
Corresponding authors: Shi-Yuan Cheng, PhD, The Ken & Ruth Davee Department of Neurology, Lou and Jean Malnati Brain Tumor Institute, The Robert H. Lurie Comprehensive Cancer Center, Northwestern University Feinberg School of Medicine, 303 E. Superior, Lurie 6-119, Chicago, IL 60611. Phone: (312)503-3043; Fax: (312)503-5603; Email: shiyuan.chengedu or Tianzhi Huang, PhD, The Ken & Ruth Davee Department of Neurology, Lou and Jean Malnati Brain Tumor Institute, The Robert H. Lurie Comprehensive Cancer Center, Northwestern University Feinberg School of Medicine, 303 E. Superior, Lurie 6-115, Chicago, IL 60611. Phone: (312)503-5034; Fax: (312)503-5603; Email: tianzhi.huangedu