Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
PS biology
PS as an immune therapy target...
PS binding molecules and cancer...
PS exposure and CD47 for cancer...
Perspective and Conclusion
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(20):9214-9229. doi:10.7150/thno.45125 This issue Cite
Review
Targeting phosphatidylserine for Cancer therapy: prospects and challenges
Wenguang Chang1 ![]() , Hongge Fa1,2, Dandan Xiao1,2, Jianxun Wang2
, Hongge Fa1,2, Dandan Xiao1,2, Jianxun Wang2
1. Institute for Translational Medicine, The Affiliated Hospital, College of medicine, Qingdao University, Qingdao, China.
2. School of Basic Medical Sciences, College of medicine, Qingdao University, Qingdao, China.
Received 2020-2-20; Accepted 2020-7-13; Published 2020-7-23
Abstract

Cancer is a leading cause of mortality and morbidity worldwide. Despite major improvements in current therapeutic methods, ideal therapeutic strategies for improved tumor elimination are still lacking. Recently, immunotherapy has attracted much attention, and many immune-active agents have been approved for clinical use alone or in combination with other cancer drugs. However, some patients have a poor response to these agents. New agents and strategies are needed to overcome such deficiencies. Phosphatidylserine (PS) is an essential component of bilayer cell membranes and is normally present in the inner leaflet. In the physiological state, PS exposure on the external leaflet not only acts as an engulfment signal for phagocytosis in apoptotic cells but also participates in blood coagulation, myoblast fusion and immune regulation in nonapoptotic cells. In the tumor microenvironment, PS exposure is significantly increased on the surface of tumor cells or tumor cell-derived microvesicles, which have innate immunosuppressive properties and facilitate tumor growth and metastasis. To date, agents targeting PS have been developed, some of which are under investigation in clinical trials as combination drugs for various cancers. However, controversial results are emerging in laboratory research as well as in clinical trials, and the efficiency of PS-targeting agents remains uncertain. In this review, we summarize recent progress in our understanding of the physiological and pathological roles of PS, with a focus on immune suppressive features. In addition, we discuss current drug developments that are based on PS-targeting strategies in both experimental and clinical studies. We hope to provide a future research direction for the development of new agents for cancer therapy.
Keywords: phosphatidylserine, cancer, T lymphocytes, immunotherapy, bavituximab
Introduction
Cancer is a leading cause of mortality and morbidity worldwide [1]. Currently, the main treatments for cancer are surgery, chemotherapy and radiation therapy, which are designed to eliminate tumor cells by directly removing or killing them [2-4]. With extensive research based on tumor biology, various genes and proteins have demonstrated potential inhibitory effects on tumor growth and proliferation, such as noncoding RNA, certain transcription factors and apoptotic pathway activators associated with tumor growth [5-8]. Moreover, a new therapeutic strategy, which eliminates tumor cells by enhancing the immunity of patients, called immunotherapy, is becoming a popular therapeutic method for mono- or polytherapy of malignant tumors [9, 10]. Immunotherapy fights cancer by helping the immune system recognize and target tumor cells. Until now, several types of immunotherapy have been used for cancer treatment, including T cell transfer therapy, immune checkpoint inhibitors, monoclonal antibodies and vaccines. For instance, chimeric antigen receptor T-cell immunotherapy (CAR-T), a T cell transfer therapy in which T cells from patients are modified and reinfused to better recognize tumor antigens, has been successfully used in some hematologic malignancies [11, 12]. Additionally, some agents targeting the immune system have been developed for cancer treatment. Nivolumab and pembrolizumab are immune checkpoint inhibitors that enhance T cell immunity by blocking programmed cell death protein 1 (PD-1). Atezoizumab and avelumab are PD-1 ligand (PD-L1) inhibitors that were approved by the US Food and Drug Administration (FDA) in 2014 for certain cancers [13-16]. Alemtuzumab, an antibody that binds to CD52 antigen on lymphocytes and attracts immune cells to destroy these cells, is used in chronic lymphocytic leukemia therapy [17, 18]. Nonetheless, antibodies targeting CD47 [19], OX40 [20], CD20 [21] and cytotoxic T-lymphocyte antigen 4 (CTLA-4) all show immune activation effects. However, many patients in the clinic do not respond well [22, 23]. For example, some patients have poor responses to PD-L1 inhibitors, which may be due to impaired T cell infiltration via upregulation of PD-L1, indoleamine-2,3-dioxygenase (IDO), and FoxP3(+) regulatory T cells (Tregs), termed primary resistance [24] or due to loss of T cell function via expression of different immune checkpoint proteins [25], defects in interferon signaling and antigen presentation, termed acquired resistance [26]. Thus, new immunostimulatory targets are urgently needed to compensate for the deficiencies in cancer therapy.
Phospholipids compose the asymmetric bilayer membrane in eukaryotic cells [27]. Among all the phospholipids, phosphatidylserine (PS) is a negatively charged amino-phospholipid and is predominately localized in the inner membrane leaflet [28]. PS exposed on the outer leaflet of the plasma membrane responds to various stimuli; alternatively, PS present in certain vesicle membranes during vesicle generation participates in the progression of various diseases [29-31]. In tumor microenvironments, PS exposure on tumor cells and immune cells leads to immune suppression and the promotion of tumor growth. PS exposure on blood cells, microparticles and neutrophil extracellular traps affects procoagulant activity in pancreatic cancer patients [32]. Therefore, the location of PS on membranes is important for cell survival, growth, proliferation and cancer-related symptoms [33, 34]. In this review, we summarize recent research on the roles of PS in physical and cancer biology, as well as related current clinical pharmacological trials, and we hope to provide new insights into future applications of PS in cancer therapy.
PS biology
PS synthesis
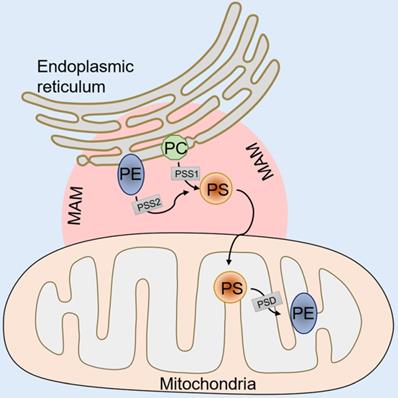
In mammalian cells, PS is synthesized in a specific domain of the endoplasmic reticulum called the mitochondria-associated membrane (MAM). The MAM facilitates the molecular exchange between the endoplasmic reticulum and mitochondria, and it plays a pivotal role in maintaining cellular health [35-37]. PS synthesis in the MAM is from either phosphatidylcholine (PC) or phosphatidylethanolamine (PE) by phosphatidylserine synthase-1 (PSS-1) (from PC) or phosphatidylserine synthase-2 (PSS-2) (from PE) via a base-exchange reaction with serine (Figure 1). After synthesis, some of the PS is transported into the mitochondria by physical contact between the MAM and mitochondrial outer membranes [38, 39]. Then, the PS in the mitochondria is decarboxylated and PE is synthesized by phosphatidylserine decarboxylase (PSD), an enzyme restricted to the mitochondrial inner membranes (Figure 1). This PE synthesis pathway from PS in the mitochondria is essential for the maintenance of mitochondrial integrity and cell growth, and a deficiency in the PSD gene results in embryonic lethality in mice [40, 41]. The remaining synthesized PS in the MAM is transported to other organelles, such as the plasma membrane and the Golgi (Figure 1). The transportation mechanism is mostly through nonspontaneous diffusion mechanisms, including soluble transport proteins or vesicles [42]. The proportion of synthesized PS that enters the mitochondria versus other organelles remains elusive. Previous phospholipid composition analysis of different organelles has shown that the highest PS content can be found in the plasma membrane and the lowest content is in mitochondria in cells of the rat liver and kidney [43, 44]. However, the steady state levels of PS may not reflect actual synthesized PS transport because the mitochondrial PS would be rapidly converted to PE by PSD after entering the mitochondria. Additionally, recent studies have shown that the disruption of PS transport to the plasma membrane by knockdown gradient required protein rescue of the mitochondrial deficiency induced by PSS knockdown [45], suggesting a coordinated regulation of PS distribution from the ER into the plasma membrane or mitochondria.
An illustration of PS synthesis. PS synthesis in the MAM from PC by PSS-1 or PE by PSS-2. After synthesis, some of the PS transported into mitochondria is decarboxylated to PE by PSD. The remaining synthesized PS in the MAM is transported to other organelles, such as the plasma membrane and the Golgi. MAM, mitochondria associated membranes; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PSS-1/2, phosphatidylserine synthase 1/2; PSD, phosphatidylserine decarboxylase.
PS exposure regulation
Flippases and scramblases coordinate the exposure of PS [46]. The type IV subfamily of P-type ATPases (P4-ATPase) are eukaryotic flippases that translocate phospholipids from the outer leaflet to the inner leaflet of biological membranes by ATP-dependent active transport [47]. The human gene encodes five classes of P4-ATPases: Class 1a (ATP8A1, ATP8A2); Class 1b (ATP8B1, ATP8B2, ATP8B3, ATP8B4); Class 2 (ATP9A, ATP9B); Class 5 (ATP10A, ATP10B, ATP10D); and Class 6 (ATP11A, ATP11B, ATP11C). ATP8A1, ATP8A2, ATP11A and ATP11C are known to flip PS from the exoplasmic to the cytoplasmic leaflet [48-50]. Mechanistic studies have shown that the activities of ATP11A and ATP11C are affected by the cytosolic Ca2+ flux. In normal growing cells, ATP11A and ATP11C persistently transport PS from outer to inner membranes. When cytosolic Ca2+ increases due to various stimuli, such as platelet activation, ATP11A and ATP11C are inactivated, which enables exposure of PS on the cell surface. In addition, CDC50 family proteins (CDC50A, CDC50B, and CDC50C) complexed with P4-ATPase are required for normal flippase activity [51]. CDC50A deficiency results in increasing PS exposure in cell membranes. It is worth noting that different variants of ATP11C are not functionally equivalent. ATP11C-a participates in PKC-mediated endocytosis, but not ATP11C-b or ATP11C-c, while ATP11C-b regulates the PS distribution in distinct regions of the polarized cell plasma membrane [52].
Scramblases catalyze another kind of phospholipid movement, which normally occurs in apoptotic cells or activated platelets [53]. Scramblases mediate nonspecific, bidirectional, and ATP-independent phospholipid movement. TMEM16 and Xk-related protein 8 (Xkr8) are two proteins that have been identified as scramblases for PS transport. TMEM16 mediates Ca2+-activated scrambling activity and catalyzes PS exposure on the platelet surface. Xkr8 has been shown to increase PS exposure in response to apoptotic stimuli such as DNA degradation and oxidative stress. The molecular mechanism of Xkr8 activation depends on either a caspase-dependent signaling pathway [54, 55] or regulation via phosphorylation [56]. The PS exposure mediated by Xkr8 is slow and irreversible, thus facilitating phagocytosis.
Apoptotic function of PS
The cells in our body constantly renew each day [57]; thus, the “replaced” cells must be cleared out efficiently to prevent inflammation or autoimmunity [58]. PS provides a recognizable and distinguished signal for cell phagocytic processes, also known as “apoptosis” [59, 60]. Apoptotic cell death can be triggered by the intrinsic (via mitochondria) pathway or the extrinsic (via cell death factor) pathway. Caspase activation is the common signature of these two apoptotic pathways. Cytochrome C activates caspase 3 or caspase 7, which are released from mitochondria and induce the intrinsic apoptotic pathway. Alternatively, caspase 8 is activated by death factors such as tumor necrosis factor (TNF) and Fas ligand (FasL), subsequently activating caspase 3 and inducing apoptosis via the extrinsic pathway [61]. PS exposure on the cell surface is a hallmark of apoptosis activation, presenting a recognition signal for macrophage recruitment and cell engulfment [62]. Caspase 3 and caspase 7 directly activate Xkr8 by cleavage and execute scrambling activity. Mouse and human cancer cells with repressed Xkr8 expression via hypermethylation fail to expose PS during apoptosis [63]. In addition to Xkr8, Xkr4 and Xkr9 possess a caspase recognition site and aid in PS exposure during apoptosis [64]. Additional studies have shown that a complex consisting of Xkr8 with basigin (BSG) and neuroplastin (NPTN), which are type I membrane proteins of the Ig superfamily, is essential for PS exposure. Cells with a knockdown of BSG and NPTN failed to expose PS in response to apoptotic stimuli, though Xkr8 localized intracellularly [65], suggesting the involvement of a more complicated mechanism in caspase-dependent phospholipid scrambling activity. Conversely, caspases also recognize ATP11C sites and inactivated ATP11C on cell membranes. Mutation of the caspase recognition site of ATP11C leads to no exposure of PS during apoptosis and no engulfment by macrophages, indicating that apoptosis-related PS exposure is coordinated by both scramblase and flippase activity.
Non-apoptotic functions of PS
Coagulation
Blood coagulation consists of initiation, amplification and propagation phases [66]. Formation of the prothrombin activator complex initiates coagulation. The process involves the activation of several coagulation cascades, including tissue factor (TF), activated factor VII (FVIIa), activated factors IX and X (FIXa and FXa) and the cofactor activated factor V (FVa) [67]. PS present in the exofacial leaflet facilitates assembly of the complex and promotes thrombin formation [68, 69]. Specifically, PS interacts with the 9-12-carboxyglutamic acid (Gla) domain at the NH2-terminus of the coagulation cascade (FVII, FIX, FX, FII) via Ca2+ [70, 71]. PS exposure on platelets is mediated by TMEM16F activation. TMEM16F exerts its scrambling activity in response to Ca2+ and inactivates ATP11A and ATP11C, thereby exposing PS on limited regions of the cell surface [55, 72]. However, this exposure of PS is transient, as the PS distribution will resume when the Ca2+ concentration returns to a normal level. This may explain why under physiological conditions, the constant flipping of PS prevents cells from being engulfed by macrophages [73].
Myoblast fusion
Myotube formation by myoblast fusion is essential for skeletal muscle development and regeneration. PS exposure is an initial fusion signal in myoblasts [74]. Studies have shown that ATP11A and CDC50A-mediated PS exposure is required for myotube formation by myoblasts. In addition, the activity of a mechanosensitive Ca2+ channel that is predominantly expressed during myotube formation, PIEZO1, is affected by flippase-mediated inward translocation of PS from the cell surface. Ca2+ influx via PIEZO1 is impaired in CDC50A-deficient or ATP11A-deficient C2C12 cells, while this impairment can be recovered by exogenous expression of CDC50A or a PS flippase [75].
Apoptotic myoblasts also expose PS. The differences between the myoblast fusion-related PS exposure and myoblast apoptosis-related PS exposure are still elusive. One report has shown that differentiating muscle cells appear normal in terms of mitochondria potential and negative caspase 3 protein expression. Additionally, myotube formation and exposure of PS cannot be blocked by the caspase inhibitor zVAD(OMe)-FMK [74]. However, other reports show that apoptosis blocked by a caspase inhibitor impaired myoblast fusion. They also found that a fraction of the myoblasts underwent apoptosis during myoblast fusion and that this small fraction induced PS exposure to promote myoblast fusion in the presence of caspase inhibitors [76, 77]. How apoptotic myoblasts affect healthy myoblast fusion is still under investigation. Xkr8 may play a central role in myoblast differentiation. Studies have shown that Xkr8 knockdown myoblasts exhibit impaired differentiation and more apoptotic cells during differentiation. Moreover, Xkr8 accelerates myoblast differentiation via a mechanism unrelated to PS exposure and caspase-dependent cleavage [78].
T lymphocyte activation
Immunotherapies rely on boosting the preexisting or inducing a new tumor-resident T cell pool to eliminate tumor cells. CD4+ and CD8+ T cells are two types of T cells that are closely related to cancer immunotherapies. CD8+ T cells are cytotoxic T cells that are considered major drivers of antitumor immunity, while CD4+ T cells, including Th1, Th2, Th17, and Treg cells, play a prominent role in controlling tumor growth [79]. PS exposure on T lymphocytes is normally associated with dead cell clearance. However, Elliott et al. reported that a subpopulation of activated/memory CD4+ T cells, which express low levels of the RB isoform of CD45 (CD4+CD45RBlo), express high levels of PS but are not apoptotic. In this study, CD45 expression levels inversely correlated with the proportion of T cells that exposed PS following P2X7 stimulation induced by benzoyl ATP (BzATP). This specific PS exposure in a subpopulation of T lymphocytes affected T cell migration, infiltration and rapid inflammation responses [80]. Furthermore, human CD8+ cytotoxic T lymphocytes (CTLs) with antigen (Ag) recognition-induced PS exposure are also not apoptotic. The exposed PS on Ag-specific T cells is concentrated at the immunologic synapse, and a blockade of PS exposure by the annexin V protein during Ag recognition diminishes cytokine secretion [81], indicating that PS exposure on CTLs is related to cell-cell communication and T cell activation. The mechanism of PS exposure on lymphoma cells is related to TMEM16F activation, and this exposure level is comparable to that observed in apoptotic cells [82], again indicating that PS exposure alone is not sufficient to initiate phagocytosis.
PS receptors
PS receptors function as mediators to invoke immune suppression [83]. Multiple proteins and protein families have been identified as PS receptors, including brain angiogenesis inhibitor 1 (BAI1), stabilin-1/2, integrin, TIM family proteins (T cell/transmembrane, immunoglobulin, and mucin), and TAM family proteins (Tyro3, AXL, and the MerTK receptor tyrosine kinase family). BAI1 [84], stabilin-1/2 [85] and the TIM family proteins [86] directly bind to PS, while TAM and integrin receptors indirectly bind to PS via ligand proteins. TAM receptors bind PS via the vitamin K-dependent proteins growth arrest-specific 6 (Gas6) and protein S (Pros1) [87] and integrin receptors bind to PS via Mfge8 [88]. Here, we briefly introduce the most studied PS receptors and their expression on immune cells. For more details on each PS receptor, please see other published reviews [89-92].
Stabilin-1/2
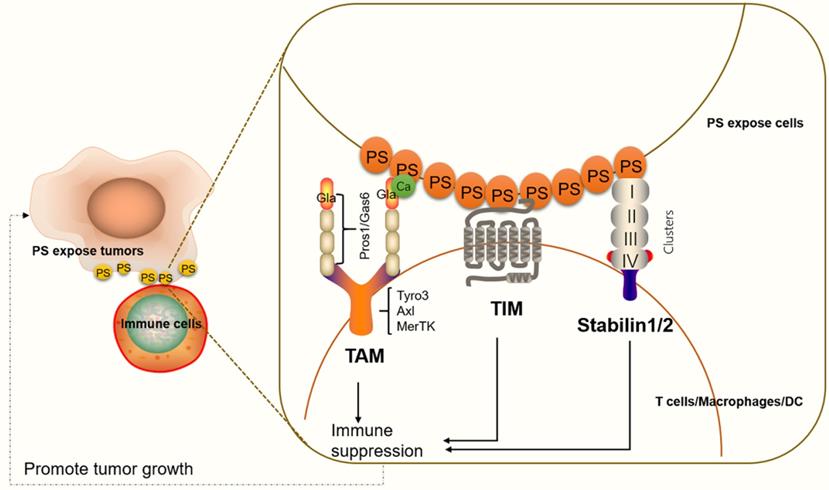
Stabilin-1 and stabilin-2 are widely expressed in endothelial cells in different organs, such as the liver, spleen, lymph nodes, bone marrow (stabilin-2), and adrenal cortex (stabilin-1) [92, 93]. In apoptotic cells, stabilin-1 and stabilin-2 directly bind to PS on the cell surface and initiate cell engulfment by activation of Rac family small GTPase 1 (Rac1) via a phosphotyrosine-binding domain-containing engulfment adaptor protein (Gulp1)-dependent mechanism or a Gulp1-independent pathway [92]. On immune cells, stabilin-1 is expressed in alternatively activated macrophages, also known as M2-like macrophages. M2 macrophages usually activate responses for healing or repair and provide a promoting environment for cancer growth [94, 95]. In macrophages cocultured with apoptotic cells, stabilin-1 is recruited to sites of recognition and mediates clearance of dead cell in a PS-dependent manner [85]. Additionally, stabilin-1-deficient macrophages in mice show reduced growth of primary tumors compared with controls. Anti-stabilin-1 antibody treatment leads to diminished numbers of immunosuppressive leukocytes in tumors [95], indicating that stabilin-1 on macrophages participates in tumor-related immune responses. Stabilin-2 has also been shown to be expressed on macrophages and to participate in TGF-β production. TGF-β is a key immune suppressive cytokine, which controls the adaptive and innate immune systems by regulating the function and generation of immune cells [96, 97] (Figure 2).
An illustration of PS receptors and their functions in immune suppression. TAM interacts with PS through Gla domain-containing proteins, Gas6 or Pros1. Ca2+ also participates in effective PS binding and receptor activation. PS binds to TAM to regulate the feedback inhibition of the innate immune response in immune cells. The TIM protein forms a narrow cavity or pocket that PS binds to and plays a critical role in regulating immune responses. Stabilin-1/2 interacts with PS through four clusters (each cluster includes an EGF-like domain, an atypical EGF-like domain, a FAS domain and/or a link domain), which in turn activate a series of signals that lead to immune suppression.
TIM family
TIM proteins are type I cell surface glycoproteins and share common structural features, including Ig-like, mucin, transmembrane, and cytoplasmic domains [98]. Structural studies have shown that PS binds to a narrow cavity formed by the CC and FG loops of IgV domains [99]. TIM-1, TIM-3, and TIM-4 belong to the human TIM family and have been identified as PS receptors [100]. TIM proteins are expressed on various immune cells and play critical roles in regulating immune responses and viral infections [101, 102]. TIM-1 is expressed on CD4+ T cells [103], mast cells [104] and regulatory B cells [105]. TIM-1 on Th2 cells functions as a potent costimulatory molecule for T cell activation [99]. TIM-1-deficient B cells promote Th1 and Th17 responses but inhibit the generation of regulatory T cells [105]. Moreover, TIM-1 is a binding site for Ebola virus on T lymphocytes and blocking TIM1-PS interactions reduces viral binding, T-cell activation, and cytokine production [106]. TIM-4 is highly expressed on dendritic cells and macrophages, and TIM-4 on macrophages participates in inflammatory responses [107]. TIM-3 is not expressed on naïve T cells, but it is expressed on fully differentiated Th1 cells [108]. TIM-3 is also expressed on cytotoxic CD8+ T cells, Th2 cells, Th17 cells and regulatory T cells [109, 110], as well as on dendritic cells (DCs) and a subpopulation of macrophages [111]. Targeting TIM-3 is known to suppress tumor growth (Figure 2). We will discuss these details in the section “Targeting PS receptors and cancer therapy”.
TAM family
The TAM family members indirectly bind to PS via the Gla domain of Gas6 or Pros1 ligand. Previous research has shown that Gas6 binds to and activates all three TAM receptors (Tyro3, AXL, and MerTK), while Pros1 is a ligand only for Tyro3 and MerTK [112]. However, recent evidence has shown that Pros1 does bind to and activate AXL in glioma sphere cultures [113], and it modulates AXL expression in oral squamous cell carcinoma cell lines [114], suggesting that Pros1 induces receptor activity function differently in different tumor microenvironments. Of all three receptors, MerTK is the most studied receptor regarding immune responses. MerTK is expressed on dendritic cells, nature killer cells, B cells, and macrophages [115, 116]. Regarding T cells, MerTK is expressed both on CD4+ T cells and CD8+ T cells. MerTK on dendritic cells competes for Pros1 interaction with MerTK in CD4+ T cells to control T cell activation [117] (Figure 2).
PS as an immune therapy target for cancer
PS exposure on tumor cells and induction of immune suppression
Immunosuppression and inflammation contribute to the creation of an environment that facilitates tumor growth and proliferation [118-120]. Tumor cells can escape immune system surveillance by disrupting any step of T cell activation, or some tumor cells escape immune elimination by recruiting immunosuppressive leukocytes and orchestrating an antitumor microenvironment [121]. PS is naturally exposed on tumor cells, the immature tumor vasculature and tumor-derived microvesicles [122-124]. In addition, radiation therapy also causes an increase in the expression of PS on the surface of viable immune infiltrates in mouse B16 melanoma [125]. The PS exposure on the surface of tumor cells prevents immune reaction by ligation of PS to receptors present on dendritic cells, macrophages and T cells [126]. The ligation of PS to receptors on macrophages promotes macrophage polarization from a proinflammatory M1-like phenotype towards a protumor M2-like phenotype, allowing the secretion of the anti-inflammatory cytokines interleukin-10 (IL-10) and TGF-β [127]. IL-10 and TGF-β are immunosuppressive cytokines that inhibit T cell activation by inhibiting tumor antigen presentation by dendritic cells and inducing regulatory T cells [128, 129] (Figure 3). Additionally, ligation of PS to receptors on T cells inhibits T cell activation via G protein-coupled receptor 174 (GPR174). GPR174-deficient Treg cells also promote the polarization of macrophages towards M2 and elevated expression of IL-10 [130] (Figure 3).
An illustration of PS exposure on tumor cells and vesicles inducing immune suppression. PS exposure on tumor cells induces immune suppression by ligation of PS to receptors on macrophages and T cells. PS binds to PSR on macrophages to promote maturation of M2-like macrophages, which are able to secrete the anti-inflammatory cytokines IL-10 and TGF-β. IL-10 and TGF-β are immunosuppressive cytokines that inhibit T cell activation. Additionally, ligation of PS to receptors on T cells inhibits T cell activation via GPR174-mediated M2 macrophage maturation. Conversely, PS exposed on tumor-derived microvesicles (externalized PS on microvesicles) increase the removal of apoptotic cells via phagocytes to prevent an undesirable inflammatory response and maintain an anti-inflammatory status in tumor microenvironments. GPR174, G protein-coupled receptor 174; M1Mф, M1-like macrophages; M2Mф, M2-like macrophages.
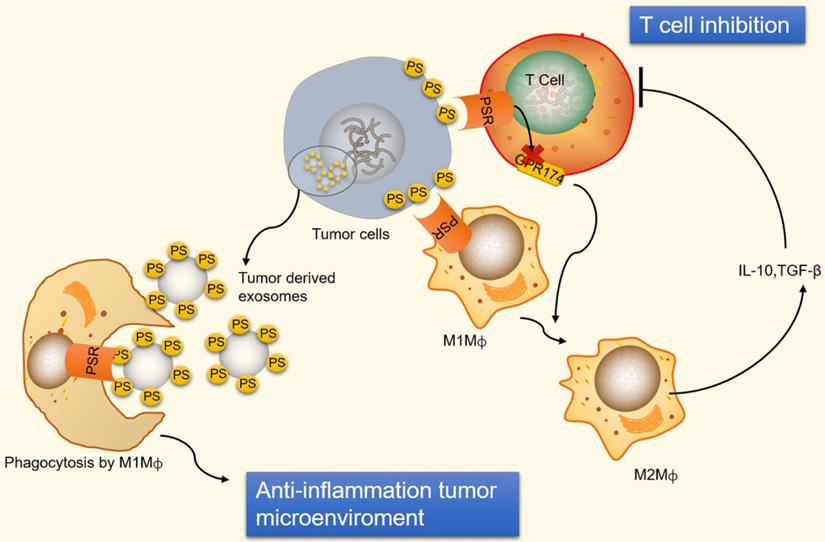
Conversely, PS is exposed on tumor-derived microvesicles, and microvesicles externalizing PS show increased removal of apoptotic cells via phagocytes, preventing an undesirable inflammatory response and maintaining an anti-inflammatory status in tumor microenvironments (Figure 3). Indeed, PS exposure on microvesicles (exosomes) derived from patient tumor samples has been shown to suppress the activation of T cell responses [131]. However, PS exposure on tumor cells also favors antitumor effects by mediating long-lived inflammation. A recent study has shown that the exposure of PS on tumor cells is necessary for IFNγ and IL-12 binding and the conversion of transient cytokine stimuli into long-lived inflammation, thus mitigating immunosuppressive functions [132]. Therefore, a complex mechanism underlies the immune response to PS exposure and its immune suppressive effects in tumor microenvironments.
PS and PS species as an imaging tool and biomarkers for cancer, respectively
Tumor cells expose PS on their surface, and therefore, methods for labeling PS are useful for tumor imaging [133]. A liposomal nanoprobe PGN-L-IO/DiR, which binds specifically to PS and is subsequently internalized into cells, was shown to be a good imaging contrast agent for mice bearing MDA-MB231 breast tumors [134]. Similarly, using a cyanine dye, indocyanine green, bound to PS antibodies helped to track and image apoptosis in triple-negative breast cancer cells [135], facilitating an effective treatment plan. In addition, PS-recognizing peptide 1 (PSP1) selectively binds to apoptosis-induced tumors in a radiation dose-dependent manner, which allows it to be used as an index probe to determine whether to continue radiation therapy in colorectal cancer [136].
Moreover, PS species can be biomarkers for cancer diagnosis. A clinical study conducted in 15 prostate cancer patients and 13 healthy controls examined the 36 most abundant lipid species. The results showed that a certain species of PS, PS (18:1/18:1), showed high significance between control and prostate cancer patients; furthermore, combinations of PS (18:1/18:1), lactosylceramide (d18:1/16:0) and PS (18:0/18:2) distinguished the two groups with 93% sensitivity and 100% specificity [137]. More recently, a study compared lipids in surgical aerosols between tumor and adjacent normal tissues in lung cancer patients. Overexpression of PS (34:2), phosphatidylcholine (36:4), and triacylglycerol (46:2) and decreased phosphatidylcholine (34:3) were observed in cancerous aerosols [138], indicating that PS combined with other indices could be potential diagnostic biomarkers for lung cancer. In addition, species of PS may correlate with cancer proliferation and progression. The fatty acyl chain of PS has been shown to determine signal transduction efficiency. Studies have shown that nanoclusters of K-Ras, an important transduction signal, colocalize with markers of PS [139], and these nanoclusters are associated with PS species with one saturated and one unsaturated fatty acyl group (PS 18:0/18:1 or PS 16:0/18:1) [140]. K-Ras is an isoform of Ras GTPase, in which mutations are commonly found in many cancers.
PS binding molecules and cancer therapy
As we discussed above, PS exposure on tumor and immune cells regulates immune responses in tumor microenvironments. Blocking PS on tumor cells restricts phagocytosis and T cell-mediated killing. Thus, targeting PS is considered a promising strategy for cancer therapy. Both preclinical and early phase clinical trials using PS targeting agents, including monoclonal antibodies, antibody-drug conjugations, liposomal carriers and natural products, have shown potential antitumor activities. Synergistic effects of PS targeting antibodies in combination with traditional cancer drugs have been observed in clinical trials. The therapeutic strategy of blocking PS is based mainly on two methods: 1) Disrupting PS on tumor cells. 2) Disrupting receptor signaling by targeting PS receptors.
Disrupting direct PS binding in cancer therapy
2aG4 and bavituximab
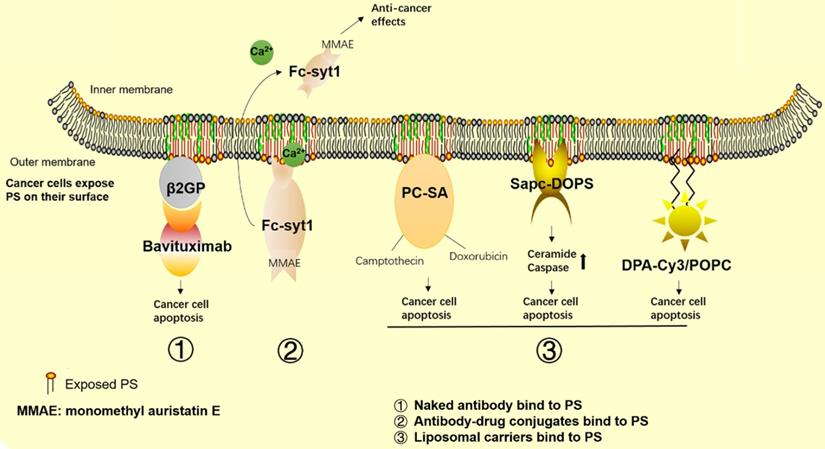
In recent years, a number of agents targeting PS have been developed for cancer therapy. Among these agents, bavituximab, a human-mouse chimeric monoclonal antibody that binds to PS indirectly by linking to β2GP1 with high affinity (Figure 4), has been the most studied. Mechanistic studies have shown that bavituximab blocks PS and exerts its antitumor immunity effects by promoting M1 macrophage maturation, as well as by inducing cellular cytotoxicity of tumor-associated endothelial cells. In experimental studies, 2aG4, the murine version of bavituximab, has been shown to exert a superior therapeutic effect in combination with the hepatocellular carcinoma therapy drug sorafenib compared with its use alone [141]. Further mechanistic studies have shown increased PS exposure in response to sorafenib treatment in the tumor vasculature, while 2aG4 targets PS and significantly increases the amount of M1 macrophages, exerting its antitumor effects by secreting TNF-α, IL-12 and promoting the Th1 response. These findings indicate that 2aG4 targets PS to regulate the immune function of cells. In addition, the effects of transforming M2 to M1 macrophages are observed in prostate tumor-bearing mice by treatment with 2aG4 in combination with docetaxel [142]. A recent study also showed that a manmade immunocytokine, 2aG4-IL2, which genetically links IL-2 to 2aG4, blocks PS induced immunosuppression in lung cancer. The vaccine was generated by coating tumor cells with 2aG4-IL2, and it reduced the incidence and number of spontaneous lung metastases [143]. In addition, the combination of 2aG4 and APR-246, which is a small molecule drug that restores p53 function, effectively inhibited tumor growth in advanced hormone-dependent breast cancer [144].
Cancer therapeutics related to PS. The graph shows the approved and promising drugs designed to target PS for cancer therapy. 1) Naked antibodies bind to PS. 2) Antibody-drug conjugates bind to PS. 3) Liposomal carriers bind to PS.
In clinical trials, bavituximab is used as a single agent or in combination with other traditional therapies in the treatment of lung cancer, hepatocellular carcinoma, breast cancer, pancreatic cancer, and solid tumors. By summarizing all the clinical results (Table 1), we found that as a monotherapy or in combination with chemotherapy or radiation therapy, bavituximab shows minimal side effects in the treatment of patients with lung cancer, hepatocellular carcinoma, breast cancer, solid tumors and rectal adenocarcinoma [141, 145-148]. In phase Ⅰb and Ⅱ clinical trials, as a front line drug, bavituximab showed an overall response rate (ORR) of 28% and a progression-free survival rate (PFR) of 4.8% in combination with carboplatin and pemetrexed, and an ORR of 40.8% and PFR of 6.0% in combination with carboplatin and paclitaxel in non-small cell lung cancer therapy [149, 150]. As a second-line drug, bavituximab also showed good ORRs and PFRs in combination with docetaxel in non-small cell lung cancer treatment [151]. Similar results were observed in phase I and II clinical trials in the treatment of metastatic breast cancer; as a front-line drug, bavituximab combined with paclitaxel showed an ORR of 85% and a PFR of 4.8% in HER2-negative breast cancer [147]. As a second-line drug, bavituximab combined with docetaxel showed an ORR of 60.9% and a PFR of 7.4%. In other phase II trials, bavituximab also showed good therapeutic results in the treatment of hepatocellular carcinoma in combination with sorafenib [152] and in the treatment of pancreatic cancers in combination with gemcitabine [153]. However, a recent phase III clinical trial in non-small cell lung cancer patients showed that bavituximab was not sufficient to improve overall survival compared to treatment with docetaxel alone [154]. This result is frustrating for the pharmaceutical company developing bavituximab; however, a retrospective case study analysis showed that this failure was due to poor quality data in phase II clinical trials and commercial consideration, so the phase III clinical trials should have been stopped earlier [155]. To date, there are no other phase III clinical trial results for bavituximab in other cancer treatments, and whether this drug can be used in cancer treatments outside of non-small cell lung cancer or should be abandoned is still unknown. Concurrently, other strategies targeting PS are in development for tumor suppression.
Summary of clinical trials evaluating the combination of PS-targeting antibodies with chemotherapy or radiation
| Clinical trial phase | Tumor type | Drug name targeting PS | N | Duration (months) | Chemotherapy orradiation combination | Tumor growth inhibition | Side effects | Reference |
|---|---|---|---|---|---|---|---|---|
| Phase Ib | Front line-advanced non-small-cell lung cancer | Bavituximab (0.3, 1 or 3 mg/kg) | 26 | Once every 3 weeks for 6 cycles | Carboplatin pemetrexed | RR, 28% PFS, 4.8 OS, 12.2 | Well tolerate | [149] |
| Phase II | Front line-Advanced non-small-cell lung cancer | Bavituximab (3 mg/kg) | 49 | Once every 3 weeks for 6 cycles then monotherapy | Carboplatin Paclitaxel | RR, 40.8% PFS, 6.0 OS,12.4 | 40.8% | [150] |
| Phase II | Second line-Advanced nonsquanous non-small-cell lung cancer | Bavituximab (1 or 3 mg/kg) or placebo | 121 | Once every 3 weeks for 6 cycles | Docetaxel | 1mg-RR,11.3%; PFS, 4.5 3mg-RR,17.1%; PFS, 4.5 | Well tolerate | [151] |
| Phase III | Second line-Advanced non-small-cell lung cancer | Bavituximab (3 mg/kg) or placebo | 597 | Once a week | Docetaxel | Not superior to Docetaxel monotherapy | Well tolerate | [154] |
| Phase I | Front line-HER-2 negative metastatic breast cancer | Bavituximab (3 mg/kg) | 14 | Once every 4 weeks for 4 cycles | Paclitaxel | RR, 85%; PFS, 7.3 | -- | [147] |
| Phase II | Second line-advanced or metastatic breast cancer. | Bavituximab (3 mg/kg) | 46 | Once a week for first 3 weeks in a 28-day cycle of 6 cycles | Docetaxel | RR, 60.9%; PFR, 7.4 | Well tolerate | [194] |
| Phase I | Preoperative treatment of Rectal adenocarcinoma | Bavituximab (0.3, 1 or 3 mg/kg) | 14 | Once a week for 8 weeks | Radiation and capecitabine | -- | Well tolerate | [146] |
| Phase I | Hepatocellular carcinoma | Bavituximab (0.3, 1 or 3 mg/kg) | 9 | Once a week for 8 weeks | Sorafenib or monotherapy | -- | Well tolerate | [195] |
| Phase II | Front line-advanced hepatocellular carcinoma | Bavituximab (3 mg/kg) | 38 | Once a week | Sorafenib | PFS, 6.7; OS, 6.2 | [152] | |
| Phase I | Advanced solid tumors | Bavituximab (0.3, 1 or 3 mg/kg) | 26 | Once a week for 8 weeks | -- | -- | Well tolerate | [145] |
| Phase Ib | Front line-advanced solid tumors | Bavituximab (3 mg/kg) | 14 | Once a week for 8 weeks | Docetaxel or gemcitabine or paclitaxel plus carboplatin | -- | Well tolerate | [148] |
| Phase II | Front line-Stage IV Pancreatic Cancer | Bavituximab (3 mg/kg) vs Gemcitabine alone | 70 | Once a week for first 3 weeks in a 28-day cycle | Gemcitabine | ORR, 28.1 vs 12.9%; OS, 5.6 vs 5.2 months | Well tolerate | [153] |
| Phase II | advanced gastric or gastroesophageal junction cancer | Bavituximab | 80 | Pembrolizumab | Still in progress |
OS, median overall survival time; ORR, overall response rate; PFR, Progression-free survival rate.
Antibody-drug conjugates
Antibody-drug conjugates are another branch of developing PS-targeting drugs. Antibody-drug conjugates combine a PS-targeting antibody to a cytotoxic drug to exert tumor killing effects. Experimental studies have shown that some antibody-drug conjugates have better tumor suppressive effects than “naked” antibodies. For example, Fc-Syt1 is a PS-targeting agent generated by fusing PS-binding domains to a human IgG1-derived Fc fragment C2A. Use of a Fc-Syt1 conjugated to monomethyl auristatin E, a cytotoxic drug, showed good antitumor effects in mouse breast and prostate cancer models [156]. In addition, recent researchers have developed a new method in which a PS binding peptide, PSBP-6, is conjugated to pH-sensitive mixed micelles (PEG-PDLLA and PEG-PHIS) and then loaded onto the chemotherapeutic agent paclitaxel (PTX) in prepared micelles. These pH sensitive micelles represent an acid triggered drug release system that is suitable for the acidic tumor environment. An in vivo study showed that the conjugated agents had improved cytotoxicity and uptake by tumor cells, as well as accumulation at tumor sites [157].
Moreover, fusion proteins consisting of L-methionase linked to human Annexin-V, an antibody with high affinity for PS, have shown good effects in tumor cell killing compared with L-methionase with no fusion protein present [158]. Moreover, the fusion protein shows almost no effects on normal cells, indicating that this strategy is a promising approach for new drug development.
Liposomal carriers
Liposomes are used as drug carriers because of their drug protecting and specific targeting characteristics. Targeting PS is a main antitumor mechanism of some liposomal carriers. Those liposomal carriers bind to PS on tumor cells and exert antitumor effects either by metabolic interference or synergistic effects with drug loading.
SapC-DOPS is a protein/lipid nanovesicle composed of the sphingolipid-activating protein Saposin C (SapC), which functions to catabolize glycosphingolipids and dioleoylphosphatidylserine (DOPS). SapC-DOPS selectively binds to PS on cancer cells and induces cell apoptosis via ceramide accumulation and caspase activation [159] (Figure 4). Research groups have used SapC-DOPS to treat different cancer cells, such as brain tumor cells [160], skin cancer cell lines [161] and pancreatic cancer cells [162]. The results showed that SapC-DOPS has good antitumor effects in a number of primary and metastatic tumors. The same group later found that during irradiation therapy, cells with high levels of surface PS had a higher survival rate, which inversely correlated with sensitivity to radiation therapy and some chemotherapy. However, these cells are sensitive to SapC-DOPS treatment, suggesting that SapC-DOPS can be used as a combination therapy for cancer cells with high PS exposure during radiation therapy [163]. A recent review focusing on cancer therapy treatments with SapC-DOPS is available [164].
In addition, phosphatidylcholine-stearylamine (PC-SA) is a cationic liposomal carrier that specifically binds to PS on cancer cells and tumors. A preclinical study showed that PC-SA has anticancer effects as a single agent and has higher efficacy when loaded with traditional antitumor drugs (Figure 4). In this study, using PC-SA alone or the anticancer drugs camptothecin and/or doxorubicin entrapped in PC-SA liposomes inhibited tumor growth and decreased tumor microvasculature formation in three tumor models. PC-SA enhances the half-life of antitumor drugs and shows no signs of obvious toxicity to other organs, suggesting its potential as a new drug or drug carrier for cancer combination therapy [165].
DPA-Cy3 is a lipid-soluble zinc(II)-bis-dipicolylamine derivative that contains the fluorophore cyanine 3 (Cy3) and two 22-carbon chains that can be anchored into liposomal membrane bilayers. Use of DPA-CY3 and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) liposomes resulted in selective binding to PS-enriched cancer cells. That study demonstrated that DPA-CY3/POPC could exert antitumor effects without any drugs loaded. DPA-CY3/POPC prefers to bind to the surface of breast cancer cells (MCF-7) versus noncancer cells (MCF-12A) due to the different levels of PS exposure. Additionally, internalization of DPA-CY3/POPC by MCF-7 is more stable than that by MCF-12A, and thus, the cytotoxic effects to tumor cells are more robust in tumor cells compared to normal cells [166].
Natural plant extracts targeting PS
It is worth noting that some natural products also show anticancer effects by targeting PS. For example, Chalepin is a compound extracted from the plant Ruta angustifolia that has been shown to exert cytotoxic activity against breast cancer cells but not normal cells. Mechanistic studies have shown that the induction of apoptosis by Chalepin is associated with PS externalization [167].
Targeting PS receptors and cancer therapy
As discussed in the “PS receptors” section, previous research has shown that PS receptors participate in the progression of tumor cells and control immune responses in the tumor microenvironment. Agents targeting TIM and TAM receptors have been developed for cancer treatment. TIM-3 antibody suppresses tumor growth via T cell regulation, especially in combination with anti-PD-1 drugs, in the treatment of fibrosarcomas [168]. Blockade of TIM-3 enhances the antitumor immune response in head and neck cancer [169]. Not surprisingly, a few pharmaceutical companies are now developing anti-TIM-3 antibodies as new antitumor agents, such as TSR-022 (NCT02817633; Tesaro), MBG543 (NCT02608268; Novartis), BMS-986258 (NCT03446040; MBS), and LY3321367 (NCT03099109; Eli Lilly and Company), which are all in phase I/II clinical trials [170-172]. Most of those trials have not yet provided results. However, some results from the TSR-022 phase I clinical trial were released at the 2018 annual meeting of the Society for Immunotherapy of Cancer (SITC). The results showed that the combination of TSR-022 with PD-1 antibody showed good tolerance in both non-small cell lung cancer and melanoma patients, and a high dose of TSR-022 (300 mg) showed observed activity, with an ORR of 15% (3/20) and 40% stable disease (8/20) [173]. The results showed the promise of anti-TIM-3 drugs in cancer treatment.
Agents that target TAM have also attracted much attention. TAM is abnormally expressed in various cancer cells, including acute myeloid leukemia, gastric and colorectal adenocarcinomas, non-small cell lung, breast and prostate cancers, and it is considered an oncogene. In this case, PS receptor inhibitors were developed to help enhance the innate immune response and kill cancer cells. Multiple drugs are being developed to selectively or nonselectively target TAM receptors. For example, Sitravatinib is a multikinase inhibitor that targets all three TAM receptors, and the combination of Sitravatinib with the PD-1 inhibitor Nivolumab is currently in phase Ⅱ (NCT02954991) and phase Ⅲ (NCT03906071) clinical trials for non-small cell lung cancer treatment. Additionally, the use of Sitravatinib for other cancer treatments, such as in combination with Tislelizumab for advanced or metastatic hepatocellular carcinoma (HCC) or gastric/gastroesophageal junction cancer (GC/GEJC) (NCT03941873), for advanced solid tumors (NCT03666143), and in combination with Nivolumab for advanced or metastatic kidney cancer (NCT03015740), urothelial carcinoma (NCT03606174) and clear cell renal cell carcinoma (NCT03680521), are in the recruiting phase for phase I or phase II clinical trials. Additionally, some agents that were initially not designed for TAM targeting were found to suppress TAM concurrently with their main targets. Those drugs, such as bosutinib [175, 176], crizotinib [177, 178], and cabozantinib [179, 180], have been approved for clinical use [174] as nonselective drugs. Moreover, many drugs have been developed to selectively target one of the TAM receptors, including selective AXL inhibitors (R428, TP0903, BMS777607 and NPS1304), selective MerTk inhibitors (MRX2843, UNC2025, UNC3133 and ONO7475) and Tyro3 inhibitors (Pfizer compounds 11, 12, 19, 21 and KRCT-6j) [91]. Most of these agents have demonstrated synergistic effects to overcome the resistance of classical antitumor drugs. BMS777607 recovers Sunitinib sensitivity in advanced renal cell carcinoma [181], and NPS1034 restores gefitinib or erlotinib sensitivity in an EGFR-resistant lung cancer cell model [182]. These selective TAM receptor drugs are designed to lower the side effects of nonselective TAM receptor drugs. Most of these agents do not specifically target TAM but also suppress other kinase families. For example, BMS777607 suppresses c-MET and AXL, and MRX2843 suppresses MerTK and Flit.
It should be noted that targeting TIM does not fully represent inhibition of PS because TIM can bind to other proteins; for example, TIM-3 has been identified to interact with galectin-9 (gal-9) and high-mobility group protein box 1 (HMGB-1) [183, 184], which also participate in immune responses. Moreover, conflicting results were reported in studies of TAM receptor targeting. MerTK-deficient tumors show increase leukocyte proliferation and higher infiltration of CD8+ T lymphocytes in a mouse model [185]. Additionally, MerTK deficiency shows better tumor control after radiotherapy in colorectal and pancreatic adenocarcinoma mice [186]. However, another study showed that MerTK and AXL-deficient mice exhibit exacerbated tumor growth and inflammation associated cancer [186]. In addition, a recent study showed that MerTK expression on CD8+ T cells improves tumor-infiltrating lymphocyte expansion [187]. The discrepancy between different studies may result from the different tumor types and PS exposure levels studied, but these results provide a hint that blindly inhibiting TAM, at least in the case of MerTK inhibitory agents, should be used with caution due to its potential for inducing inhibition of tumor killing.
PS exposure and CD47 for cancer therapy
PS exposure on the cell surface provides a phagocytic signal for macrophages, while CD47 expression on cells inhibits phagocytosis [124]. CD47 is widely expressed on all cells but has high expression levels on various tumor cells [188]. CD47 is a ligand of signal regulatory protein-α (SIRPα), a protein expressed on macrophages and dendritic cells. Upon binding CD47, SIRPα initiates a signaling cascade that results in the inhibition of phagocytosis [189], through which tumor cells can escape from immune surveillance of macrophages and T cells [190]. In erythrocytes, CD47 ligation induces PS expression as part of a death pathway [191], suggesting that CD47 could affect PS exposure. Indeed, knockout of CDC50A, a subunit of ATP11C that participates in PS flipping, increases tumor-associated macrophages and enhances the effect of anti-CD47 blockade on limiting tumor growth [192]. Knockout of CDC50A also increases PS exposure on Jurkat cells, which may affect T cell function, as we mentioned earlier. Thus, blockade of CD47 inhibits phagocytosis, however, PS exposure has been utilized to target tumor cells for macrophage clearance. In clinical trials, the combination of anti-CD47 agents has shown synergistic antitumor effects. The anti-CD47 inhibitor 5F9 combined with rituximab (anti-CD20 antibody) has shown a good response with minimal side effects in patients with aggressive and indolent lymphoma [193].
Perspective and Conclusion
Currently, immunotherapy that enhances T cell immunity has become a hot research area. PS exposure is known to be an immune suppressor in tumor microenvironments. In general, agents that directly or indirectly target PS rescue immune suppression, enhance antitumor drug activity and are accompanied by good tolerance, suggesting that PS is promising as a new drug for mono- or polytherapy for cancer. However, we notice that some risks arise during treatment with this kind of drug. First, inhibition of PS receptors, such as MerTk on T cells, may reduce tumor killing effects. Second, PS exposure on the cell surface provides a phagocytic signal for macrophages, and some immune therapies have utilized this mechanism to target tumor cells for macrophage clearance, such as CD47 inhibitors; thus, using anti-PS agents may modulate those drug therapy effects. Third, anti-PS therapy is mainly utilized in combination with other cancer drugs, and the safety and unknown side effects, such as the risk of developing thromboembolic events, is largely unknown. Therefore, although targeting PS shows promise in cancer therapy, many obstacles must be overcome for its successful application in the clinic. Ideally, agents that specifically target PS on tumor cells but do not affect PS on normal cells, and agents that sense the quantity of PS exposure when in combination with other cancer drugs, merit further investigation. Future research could focus on these obstacles and challenges to develop new kinds of drugs.
Acknowledgements
This work was supported by a grant from the National Natural Science Foundation of China (81700704, 81622005), and the Natural Science Foundation of Shandong province (ZR2017BH087).
Author contributions
W Chang, Hongge Fa and Dandan Xiao collected all of the data, and W Chang and J Wang drafted and wrote the manuscript. All authors have read and approved the final version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394-424
2. Takeyama H, Beppu T, Higashi T, Kaida T, Arima K, Taki K. et al. Impact of surgical treatment after sorafenib therapy for advanced hepatocellular carcinoma. Surg Today. 2018;48(4):431-8
3. Smith BD, Bellon JR, Blitzblau R, Freedman G, Haffty B, Hahn C. et al. Radiation therapy for the whole breast: Executive summary of an american society for radiation oncology (ASTRO) evidence-based guideline. Pract Radiat Oncol. 2018;8(3):145-52
4. Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F. et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med. 2018;378(22):2078-92
5. Liu Y, Ding W, Ge H, Ponnusamy M, Wang Q, Hao X. et al. Foxk transcription factors: Regulation and critical role in cancer. Cancer Lett. 2019;458:1-12
6. Liu Y, Ao X, Ding W, Ponnusamy M, Wu W, Hao X. et al. Critical role of foxo3a in carcinogenesis. Mol Cancer. 2018;17(1):104
7. Xu T, Ding W, Ji X, Ao X, Liu Y, Yu W. et al. Molecular mechanisms of ferroptosis and its role in cancer therapy. J Cell Mol Med. 2019;23(8):4900-12
8. Zhou Z, Lin Z, He Y, Pang X, Wang Y, Ponnusamy M. et al. The long noncoding RNA d63785 regulates chemotherapy sensitivity in human gastric cancer by targeting mir-422a. Mol Ther Nucleic Acids. 2018;12:405-19
9. Gamboa L, Zamat AH, Kwong GA. Synthetic immunity by remote control. Theranostics. 2020;10(8):3652-67
10. Tong L, Li J, Li Q, Wang X, Medikonda R, Zhao T. et al. Act001 reduces the expression of PD-L1 by inhibiting the phosphorylation of stat3 in glioblastoma. Theranostics. 2020;10(13):5943-56
11. Golubovskaya V. Wu L. Different subsets of T cells, memory, effector functions, and CAR-T immunotherapy. Cancers (Basel). 2016;8(3):36
12. June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359(6382):1361-5
13. Ravandi F, Assi R, Daver N, Benton CB, Kadia T, Thompson PA. et al. Idarubicin, cytarabine, and nivolumab in patients with newly diagnosed acute myeloid leukaemia or high-risk myelodysplastic syndrome: A single-arm, phase 2 study. Lancet Haematol. 2019;6(9):e480-8
14. Larkin J, Chiarion SV, Gonzalez R, Grob JJ, Rutkowski P, Lao CD. et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2019;381(16):1535-46
15. Usmani SZ, Schjesvold F, Oriol A, Karlin L, Cavo M, Rifkin RM. et al. Pembrolizumab plus lenalidomide and dexamethasone for patients with treatment-naive multiple myeloma (keynote-185): A randomised, open-label, phase 3 trial. Lancet Haematol. 2019;6(9):e448-58
16. Ascierto PA, Ferrucci PF, Fisher R, Del Vecchio M, Atkinson V, Schmidt H. et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat Med. 2019;25(6):941-6
17. Sellar RS, Mehra V, Fox TA, Grigg A, Kulasekararaj A, Sarma A. et al. Comparative analysis of melphalan versus busulphan T-cell deplete conditioning using alemtuzumab in unrelated donor stem cell transplantation for acute myeloid leukaemia. Br J Haematol. 2019;187(1):e20-4
18. Ishizawa K, Fukuhara N, Nakaseko C, Chiba S, Ogura M, Okamoto A. et al. Safety, efficacy and pharmacokinetics of humanized anti-CD52 monoclonal antibody alemtuzumab in Japanese patients with relapsed or refractory B-cell chronic lymphocytic leukemia. Jpn J Clin Oncol. 2017;47(1):54-60
19. Zhang W, Huang Q, Xiao W, Zhao Y, Pi J, Xu H. et al. Advances in anti-tumor treatments targeting the cd47/sirpalpha axis. Front Immunol. 2020;11:18
20. Aspeslagh S, Postel-Vinay S, Rusakiewicz S, Soria JC, Zitvogel L, Marabelle A. Rationale for anti-OX40 cancer immunotherapy. Eur J Cancer. 2016;52:50-66
21. Nakamura K, Casey M, Oey H, Vari F, Stagg J, Gandhi MK. et al. Targeting an adenosine-mediated "don't eat me signal" augments anti-lymphoma immunity by anti-CD20 monoclonal antibody. Leukemia. 2020 [Epub ahead of print]
22. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L. et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568-71
23. Mooradian MJ. Sullivan RJ. What to do when anti-PD-1 therapy fails in patients with melanoma. Oncology (Williston Park). 2019;33(4):141-8
24. Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT. et al. Up-regulation of PD-L1, Ido, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med. 2013;5(200):200ra116
25. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG. et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501
26. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S. et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375(9):819-29
27. Bretscher MS. Asymmetrical lipid bilayer structure for biological membranes. Nat New Biol. 1972;236(61):11-2
28. Yeung T, Gilbert GE, Shi J, Silvius J, Kapus A, Grinstein S. Membrane phosphatidylserine regulates surface charge and protein localization. Science. 2008;319(5860):210-3
29. Ale A, Siebenhaar F, Kosanke K, Aichler M, Radrich K, Heydrich S. et al. Cardioprotective c-kit(+) bone marrow cells attenuate apoptosis after acute myocardial infarction in mice- in-vivo assessment with fluorescence molecular imaging. Theranostics. 2013;3(11):903-13
30. Chen D, Zhao X, Sui Z, Niu H, Chen L, Hu C. et al. A multi-omics investigation of the molecular characteristics and classification of six metabolic syndrome relevant diseases. Theranostics. 2020;10(5):2029-46
31. Chen J, Yang J, Liu R, Qiao C, Lu Z, Shi Y. et al. Dual-targeting theranostic system with mimicking apoptosis to promote myocardial infarction repair via modulation of macrophages. Theranostics. 2017;7(17):4149-67
32. Yu M, Li T, Li B, Liu Y, Wang L, Zhang J. et al. Phosphatidylserine-exposing blood cells, microparticles and neutrophil extracellular traps increase procoagulant activity in patients with pancreatic cancer. Thromb Res. 2020;188:5-16
33. Zargarian S, Shlomovitz I, Erlich Z, Hourizadeh A, Ofir-Birin Y, Croker BA. et al. Phosphatidylserine externalization, "necroptotic bodies" release, and phagocytosis during necroptosis. PLoS Biol. 2017;15(6):e2002711
34. Vance JE. Tasseva G. Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochim Biophys Acta. 2013;1831(3):543-54
35. Annunziata I, Sano R, D'Azzo A. Mitochondria-associated ER membranes (MAMs) and lysosomal storage diseases. Cell Death Dis. 2018;9(3):328
36. Missiroli S, Patergnani S, Caroccia N, Pedriali G, Perrone M, Previati M. et al. Mitochondria-associated membranes (MAMs) and inflammation. Cell Death Dis. 2018;9(3):329
37. Stacchiotti A. Exploring cellular stress response and chaperones. Cells. 2019;8(5):408
38. Kainu V, Hermansson M, Hanninen S, Hokynar K, Somerharju P. Import of phosphatidylserine to and export of phosphatidylethanolamine molecular species from mitochondria. Biochim Biophys Acta. 2013;1831(2):429-37
39. Vance JE. Phospholipid synthesis and transport in mammalian cells. Traffic. 2015;16(1):1-18
40. Selathurai A, Kowalski GM, Mason SA, Callahan DL, Foletta VC, Della Gatta PA. et al. Phosphatidylserine decarboxylase is critical for the maintenance of skeletal muscle mitochondrial integrity and muscle mass. Mol Metab. 2019;27:33-46
41. Steenbergen R, Nanowski TS, Beigneux A, Kulinski A, Young SG, Vance JE. Disruption of the phosphatidylserine decarboxylase gene in mice causes embryonic lethality and mitochondrial defects. J Biol Chem. 2005;280(48):40032-40
42. Moser von Filseck J, Copic A, Delfosse V, Vanni S, Jackson CL, Bourguet W. et al. Intracellular transport. Phosphatidylserine transport by ORP/OSH proteins is driven by phosphatidylinositol 4-phosphate. Science. 2015;349(6246):432-6
43. Ruggiero FM, Landriscina C, Gnoni GV, Quagliariello E. Lipid composition of liver mitochondria and microsomes in hyperthyroid rats. Lipids. 1984;19(3):171-8
44. Zambrano F, Fleischer S, Fleischer B. Lipid composition of the golgi apparatus of rat kidney and liver in comparison with other subcellular organelles. Biochim Biophys Acta. 1975;380(3):357-69
45. Yang X, Liang J, Ding L, Li X, Lam SM, Shui G. et al. Phosphatidylserine synthase regulates cellular homeostasis through distinct metabolic mechanisms. PLoS Genet. 2019;15(12):e1008548
46. Kay JG. Fairn GD. Distribution, dynamics and functional roles of phosphatidylserine within the cell. Cell Commun Signal. 2019;17(1):126
47. Nagata S, Sakuragi T, Segawa K. Flippase and scramblase for phosphatidylserine exposure. Curr Opin Immunol. 2020;62:31-8
48. Coleman JA, Kwok MC, Molday RS. Localization, purification, and functional reconstitution of the p4-ATPase ATP8A2, a phosphatidylserine flippase in photoreceptor disc membranes. J Biol Chem. 2009;284(47):32670-9
49. Lee S, Uchida Y, Wang J, Matsudaira T, Nakagawa T, Kishimoto T. et al. Transport through recycling endosomes requires ehd1 recruitment by a phosphatidylserine translocase. EMBO J. 2015;34(5):669-88
50. Takatsu H, Tanaka G, Segawa K, Suzuki J, Nagata S, Nakayama K. et al. Phospholipid flippase activities and substrate specificities of human type iv p-type atpases localized to the plasma membrane. J Biol Chem. 2014;289(48):33543-56
51. Coleman JA. Molday RS. Critical role of the beta-subunit CDC50A in the stable expression, assembly, subcellular localization, and lipid transport activity of the p4-ATPase ATP8A2. J Biol Chem. 2011;286(19):17205-16
52. Takayama M, Takatsu H, Hamamoto A, Inoue H, Naito T, Nakayama K. et al. The cytoplasmic c-terminal region of the ATP11c variant determines its localization at the polarized plasma membrane. J Cell Sci. 2019;132(17):jcs231720
53. Williamson P. Phospholipid scramblases. Lipid Insights. 2015;8(Suppl 1):41-4
54. Sivagnanam U, Palanirajan SK, Gummadi SN. The role of human phospholipid scramblases in apoptosis: An overview. Biochim Biophys Acta Mol Cell Res. 2017;1864(12):2261-71
55. Bevers EM. Williamson PL. Getting to the outer leaflet: Physiology of phosphatidylserine exposure at the plasma membrane. Physiol Rev. 2016;96(2):605-45
56. Sakuragi T, Kosako H, Nagata S. Phosphorylation-mediated activation of mouse xkr8 scramblase for phosphatidylserine exposure. Proc Natl Acad Sci U S A. 2019;116(8):2907-12
57. Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L. et al. Cell death modalities: Classification and pathophysiological implications. Cell Death Differ. 2007;14(7):1237-43
58. Raff MC, Barres BA, Burne JF, Coles HS, Ishizaki Y, Jacobson MD. Programmed cell death and the control of cell survival: Lessons from the nervous system. Science. 1993;262(5134):695-700
59. Shlomovitz I, Speir M, Gerlic M. Flipping the dogma-phosphatidylserine in non-apoptotic cell death. Cell Commun Signal. 2019;17(1):139
60. Chua BA, Ngo JA, Situ K, Morizono K. Roles of phosphatidylserine exposed on the viral envelope and cell membrane in hiv-1 replication. Cell Commun Signal. 2019;17(1):132
61. Green DR. Llambi F. Cell death signaling. Cold Spring Harb Perspect Biol. 2015;7(12):a006080
62. Hayes SC, James Wilcox CR, Forbes White HS, Vanicek N. The effects of robot assisted gait training on temporal-spatial characteristics of people with spinal cord injuries: A systematic review. J Spinal Cord Med. 2018;41(5):529-43
63. Suzuki J, Denning DP, Imanishi E, Horvitz HR, Nagata S. Xk-related protein 8 and ced-8 promote phosphatidylserine exposure in apoptotic cells. Science. 2013;341(6144):403-6
64. Suzuki J, Imanishi E, Nagata S. Exposure of phosphatidylserine by xk-related protein family members during apoptosis. J Biol Chem. 2014;289(44):30257-67
65. Suzuki J, Imanishi E, Nagata S. Xkr8 phospholipid scrambling complex in apoptotic phosphatidylserine exposure. Proc Natl Acad Sci U S A. 2016;113(34):9509-14
66. Ten Cate H, Hackeng TM, Garcia de Frutos P. Coagulation factor and protease pathways in thrombosis and cardiovascular disease. Thromb Haemost. 2017;117(7):1265-71
67. Tanaka KA, Key NS, Levy JH. Blood coagulation: Hemostasis and thrombin regulation. Anesth Analg. 2009;108(5):1433-46
68. Hussain JF. Mahaut-Smith MP. Reversible and irreversible intracellular Ca2+ spiking in single isolated human platelets. J Physiol. 1999;514( Pt 3):713-8
69. Carafoli E. Calcium pump of the plasma membrane. Physiol Rev. 1991;71(1):129-53
70. Srivasatava KR, Majumder R, Kane WH, Quinn-Allen MA, Lentz BR. Phosphatidylserine and Fva regulate Fxa structure. Biochem J. 2014;459(1):229-39
71. Malvezzi M, Chalat M, Janjusevic R, Picollo A, Terashima H, Menon AK. et al. Ca2+-dependent phospholipid scrambling by a reconstituted TMEM16 ion channel. Nat Commun. 2013;4:2367
72. Muratori C, Pakhomov AG, Gianulis E, Meads J, Casciola M, Mollica PA. et al. Activation of the phospholipid scramblase TMEM16F by nanosecond pulsed electric fields (NSPEF) facilitates its diverse cytophysiological effects. J Biol Chem. 2017;292(47):19381-91
73. Nagata S, Suzuki J, Segawa K, Fujii T. Exposure of phosphatidylserine on the cell surface. Cell Death Differ. 2016;23(6):952-61
74. Van den Eijnde SM, Van den Hoff MJ, Reutelingsperger CP, Van Heerde WL, Henfling ME, Vermeij-Keers C. et al. Transient expression of phosphatidylserine at cell-cell contact areas is required for myotube formation. J Cell Sci. 2001;114(Pt 20):3631-42
75. Tsuchiya M, Hara Y, Okuda M, Itoh K, Nishioka R, Shiomi A. et al. Cell surface flip-flop of phosphatidylserine is critical for piezo1-mediated myotube formation. Nat Commun. 2018;9(1):2049
76. Hochreiter-Hufford AE, Lee CS, Kinchen JM, Sokolowski JD, Arandjelovic S, Call JA. et al. Phosphatidylserine receptor BAI1 and apoptotic cells as new promoters of myoblast fusion. Nature. 2013;497(7448):263-7
77. Hochreiter-Hufford AE, Arandjelovic S, Ravichandran KS. Using phosphatidylserine exposure on apoptotic cells to stimulate myoblast fusion. Methods Mol Biol. 2015;1313:141-8
78. Kim GW, Nam GH, Kim IS, Park SY. Xk-related protein 8 regulates myoblast differentiation and survival. FEBS J. 2017;284(21):3575-88
79. Van der Leun AM, Thommen DS, Schumacher TN. Cd8(+) T cell states in human cancer: Insights from single-cell analysis. Nat Rev Cancer. 2020;20(4):218-32
80. Elliott JI, Surprenant A, Marelli-Berg FM, Cooper JC, Cassady-Cain RL, Wooding C. et al. Membrane phosphatidylserine distribution as a non-apoptotic signalling mechanism in lymphocytes. Nat Cell Biol. 2005;7(8):808-16
81. Fischer K, Voelkl S, Berger J, Andreesen R, Pomorski T, Mackensen A. Antigen recognition induces phosphatidylserine exposure on the cell surface of human CD8+ T cells. Blood. 2006;108(13):4094-101
82. Segawa K, Suzuki J, Nagata S. Constitutive exposure of phosphatidylserine on viable cells. Proc Natl Acad Sci U S A. 2011;108(48):19246-51
83. Dayoub AS. Brekken RA. TIMs, TAMs, and PS-antibody targeting: Implications for cancer immunotherapy. Cell Commun Signal. 2020;18(1):29
84. Park D, Tosello-Trampont AC, Elliott MR, Lu M, Haney LB, Ma Z. et al. BAI1 is an engulfment receptor for apoptotic cells upstream of the elmo/dock180/rac module. Nature. 2007;450(7168):430-4
85. Park SY, Jung MY, Lee SJ, Kang KB, Gratchev A, Riabov V. et al. Stabilin-1 mediates phosphatidylserine-dependent clearance of cell corpses in alternatively activated macrophages. J Cell Sci. 2009;122(Pt 18):3365-73
86. Schweigert O, Dewitz C, Moller-Hackbarth K, Trad A, Garbers C, Rose-John S. et al. Soluble T cell immunoglobulin and mucin domain (TIM)-1 and -4 generated by a disintegrin and metalloprotease (adam)-10 and -17 bind to phosphatidylserine. Biochim Biophys Acta. 2014;1843(2):275-87
87. Geng K, Kumar S, Kimani SG, Kholodovych V, Kasikara C, Mizuno K. et al. Requirement of gamma-carboxyglutamic acid modification and phosphatidylserine binding for the activation of Tyro3, Axl, and Mertk receptors by growth arrest-specific 6. Front Immunol. 2017;8:1521
88. Akakura S, Singh S, Spataro M, Akakura R, Kim JI, Albert ML. et al. The opsonin mfg-e8 is a ligand for the alphavbeta5 integrin and triggers dock180-dependent rac1 activation for the phagocytosis of apoptotic cells. Exp Cell Res. 2004;292(2):403-16
89. Birge RB, Boeltz S, Kumar S, Carlson J, Wanderley J, Calianese D. et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016;23(6):962-78
90. Kerr D, Tietjen GT, Gong Z, Tajkhorshid E, Adams EJ, Lee KYC. Sensitivity of peripheral membrane proteins to the membrane context: A case study of phosphatidylserine and the tim proteins. Biochim Biophys Acta Biomembr. 2018;1860(10):2126-33
91. Park M. Kang KW. Phosphatidylserine receptor-targeting therapies for the treatment of cancer. Arch Pharm Res. 2019;42(7):617-28
92. Park SY. Kim IS. Stabilin receptors: Role as phosphatidylserine receptors. Biomolecules. 2019;9(8):387
93. Zhou B, McGary CT, Weigel JA, Saxena A, Weigel PH. Purification and molecular identification of the human hyaluronan receptor for endocytosis. Glycobiology. 2003;13(5):339-49
94. Mills CD. Anatomy of a discovery: M1 and M2 macrophages. Front Immunol. 2015;6:212
95. Karikoski M, Marttila-Ichihara F, Elima K, Rantakari P, Hollmen M, Kelkka T. et al. Clever-1/stabilin-1 controls cancer growth and metastasis. Clin Cancer Res. 2014;20(24):6452-64
96. Park SY, Jung MY, Kim HJ, Lee SJ, Kim SY, Lee BH. et al. Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ. 2008;15(1):192-201
97. Batlle E. Massague J. Transforming growth factor-beta signaling in immunity and cancer. Immunity. 2019;50(4):924-940
98. DeKruyff RH, Bu X, Ballesteros A, Santiago C, Chim YL, Lee HH. et al. T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J Immunol. 2010;184(4):1918-30
99. Freeman GJ, Casasnovas JM, Umetsu DT, DeKruyff RH. Tim genes: A family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev. 2010;235(1):172-89
100. McIntire JJ, Umetsu SE, Akbari O, Potter M, Kuchroo VK, Barsh GS. et al. Identification of tapr (an airway hyperreactivity regulatory locus) and the linked TIM gene family. Nat Immunol. 2001;2(12):1109-16
101. Li Z, Ju Z, Frieri M. The T-cell immunoglobulin and mucin domain (TIM) gene family in asthma, allergy, and autoimmunity. Allergy Asthma Proc. 2013;34(1):e21-6
102. Evans JP. Liu SL. Multifaceted roles of TIM-family proteins in virus-host interactions. Trends Microbiol. 2020;28(3):224-35
103. Wang S, Zhu X, Xu Y, Zhang D, Li Y, Tao Y. et al. Programmed cell death-1 (PD-1) and T-cell immunoglobulin mucin-3 (TIM-3) regulate CD4+ T cells to induce type 2 helper T cell (Th2) bias at the maternal-fetal interface. Hum Reprod. 2016;31(4):700-11
104. Corredera E, Phong BL, Moore JA, Kane LP, Lee SE. Tim-3-expressing mast cells are present in chronic rhinosinusitis with nasal polyps. Otolaryngol Head Neck Surg. 2018;159(3):581-6
105. Xiao S, Brooks CR, Sobel RA, Kuchroo VK. TIM-1 is essential for induction and maintenance of IL-10 in regulatory b cells and their regulation of tissue inflammation. J Immunol. 2015;194(4):1602-8
106. Younan P, Iampietro M, Nishida A, Ramanathan P, Santos RI, Dutta M. et al. Ebola virus binding to TIM-1 on T lymphocytes induces a cytokine storm. mBio. 2017;8(5):e00845-17
107. Liu W, Bai F, Wang H, Liang Y, Du X, Liu C. et al. TIM-4 inhibits NLRP3 inflammasome via the LKB1/AMPKalpha pathway in macrophages. J Immunol. 2019;203(4):990-1000
108. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T. et al. Th1-specific cell surface protein TIM-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415(6871):536-41
109. Sakhdari A, Mujib S, Vali B, Yue FY, MacParland S, Clayton K. et al. TIM-3 negatively regulates cytotoxicity in exhausted CD8+ T cells in HIV infection. PLoS One. 2012;7(7):e40146
110. Fu R, Li L, Hu J, Wang Y, Tao J, Liu H. et al. Elevated TIM3 expression of T helper cells affects immune system in patients with myelodysplastic syndrome. J Investig Med. 2019;67(8):1125-30
111. Das M, Zhu C, Kuchroo VK. TIM-3 and its role in regulating anti-tumor immunity. Immunol Rev. 2017;276(1):97-111
112. Tsou WI, Nguyen KQ, Calarese DA, Garforth SJ, Antes AL, Smirnov SV. et al. Receptor tyrosine kinases, Tyro3, Axl, and Mer, demonstrate distinct patterns and complex regulation of ligand-induced activation. J Biol Chem. 2014;289(37):25750-63
113. Sadahiro H, Kang KD, Gibson JT, Minata M, Yu H, Shi J. et al. Activation of the receptor tyrosine kinase Axl regulates the immune microenvironment in glioblastoma. Cancer Res. 2018;78(11):3002-13
114. Abboud-Jarrous G, Priya S, Maimon A, Fischman S, Cohen-Elisha M, Czerninski R. et al. Protein S drives oral squamous cell carcinoma tumorigenicity through regulation of Axl. Oncotarget. 2017;8(8):13986-4002
115. Seitz HM, Camenisch TD, Lemke G, Earp HS, Matsushima GK. Macrophages and dendritic cells use different Axl/Mertk/Tyro3 receptors in clearance of apoptotic cells. J Immunol. 2007;178(9):5635-42
116. Paolino M, Choidas A, Wallner S, Pranjic B, Uribesalgo I, Loeser S. et al. The E3 ligase CBL-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature. 2014;507(7493):508-12
117. Cabezon R, Carrera-Silva EA, Florez-Grau G, Errasti AE, Calderon-Gomez E, Lozano JJ. et al. Mertk as negative regulator of human T cell activation. J Leukoc Biol. 2015;97(4):751-60
118. Wang F, Li B, Fu P, Li Q, Zheng H, Lao X. Effects on tumor growth and immunosuppression of a modified talpha1 peptide along with its circular dichroism spectroscopy data. Data Brief. 2018;20:126-31
119. Farhad M, Rolig AS, Redmond WL. The role of galectin-3 in modulating tumor growth and immunosuppression within the tumor microenvironment. Oncoimmunology. 2018;7(6):e1434467
120. Rajan R, Sabnani MK, Mavinkurve V, Shmeeda H, Mansouri H, Bonkoungou S. et al. Liposome-induced immunosuppression and tumor growth is mediated by macrophages and mitigated by liposome-encapsulated alendronate. J Control Release. 2018;271:139-48
121. Beatty GL. Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res. 2015;21(4):687-92
122. Lima LG, Chammas R, Monteiro RQ, Moreira ME, Barcinski MA. Tumor-derived microvesicles modulate the establishment of metastatic melanoma in a phosphatidylserine-dependent manner. Cancer Lett. 2009;283(2):168-75
123. Ran S. Thorpe PE. Phosphatidylserine is a marker of tumor vasculature and a potential target for cancer imaging and therapy. Int J Radiat Oncol Biol Phys. 2002;54(5):1479-84
124. Sharma B. Kanwar SS. Phosphatidylserine: A cancer cell targeting biomarker. Semin Cancer Biol. 2018;52(Pt 1):17-25
125. Budhu SaG, Rachel G, Aditi Z, Roberta S, Sara HC, Daniel C. et al. Targeting phosphatidylserine enhances the anti-tumor response to tumor-directed radiation therapy in a preclinical model of melanoma. Cell Rep. 2019 D-19-02869
126. Myers KV, Amend SR, Pienta KJ. Targeting Tyro3, Axl and Mertk (TAM receptors): Implications for macrophages in the tumor microenvironment. Mol Cancer. 2019;18(1):94
127. Tse E. Kwong YL. T-cell lymphoma: Microenvironment-related biomarkers. Semin Cancer Biol. 2015;34:46-51
128. Thepmalee C, Panya A, Junking M, Chieochansin T, Yenchitsomanus PT. Inhibition of IL-10 and TGF-beta receptors on dendritic cells enhances activation of effector T-cells to kill cholangiocarcinoma cells. Hum Vaccin Immunother. 2018;14(6):1423-31
129. Taylor A, Verhagen J, Blaser K, Akdis M, Akdis CA. Mechanisms of immune suppression by interleukin-10 and transforming growth factor-beta: The role of T regulatory cells. Immunology. 2006;117(4):433-42
130. Qiu D, Chu X, Hua L, Yang Y, Li K, Han Y. et al. GPR174-deficient regulatory T cells decrease cytokine storm in septic mice. Cell Death Dis. 2019;10(3):233
131. Kelleher RJ Jr, Balu-Iyer S, Loyall J, Sacca AJ, Shenoy GN, Peng P. et al. Extracellular vesicles present in human ovarian tumor microenvironments induce a phosphatidylserine-dependent arrest in the T-cell signaling cascade. Cancer Immunol Res. 2015;3(11):1269-78
132. Oyler-Yaniv J, Oyler-Yaniv A, Shakiba M, Min NK, Chen YH, Cheng SY. et al. Catch and release of cytokines mediated by tumor phosphatidylserine converts transient exposure into long-lived inflammation. Mol Cell. 2017;66(5):635-47
133. Kawakami T, Takeuchi S, Soma Y. Elevated levels of serum Igm anti-phosphatidylserine-prothrombin complex antibodies in patients with cancer-associated vasculitis. Int J Dermatol. 2017;56(10):e203-4
134. Zhang L, Zhou H, Belzile O, Thorpe P, Zhao D. Phosphatidylserine-targeted bimodal liposomal nanoparticles for in vivo imaging of breast cancer in mice. J Control Release. 2014;183:114-23
135. Kannadorai RK, Udumala SK, Sidney YW. Noninvasive in vivo multispectral optoacoustic imaging of apoptosis in triple negative breast cancer using indocyanine green conjugated phosphatidylserine monoclonal antibody. J Biomed Opt. 2016;21(12):126002
136. Bae SM, Park SJ, Choi M, Song M, Cho YE, Do EJ. et al. Psp1, a phosphatidylserine-recognizing peptide, is useful for visualizing radiation-induced apoptosis in colorectal cancer in vitro and in vivo. Transl Oncol. 2018;11(4):1044-52
137. Skotland T, Ekroos K, Kauhanen D, Simolin H, Seierstad T, Berge V. et al. Molecular lipid species in urinary exosomes as potential prostate cancer biomarkers. Eur J Cancer. 2017;70:122-32
138. Zhang J, Zheng Q, Zhang W, Wang N, Xu J, Cheng X. et al. Lipids in surgical aerosol as diagnosis biomarkers for discrimination of lung cancer. Cancer Manag Res. 2019;11:5537-43
139. Zhou Y. Hancock JF. Deciphering lipid codes: K-RAS as a paradigm. Traffic. 2018;19(3):157-65
140. Zhou Y, Prakash P, Liang H, Cho KJ, Gorfe AA, Hancock JF. Lipid-sorting specificity encoded in K-RAS membrane anchor regulates signal output. Cell. 2017;168(1-2):239-51
141. Cheng X, Li L, Thorpe PE, Yopp AC, Brekken RA, Huang X. Antibody-mediated blockade of phosphatidylserine enhances the antitumor effect of sorafenib in hepatocellular carcinomas xenografts. Ann Surg Oncol. 2016;23(Suppl 5):S583-91
142. Yin Y, Huang X, Lynn KD, Thorpe PE. Phosphatidylserine-targeting antibody induces M1 macrophage polarization and promotes myeloid-derived suppressor cell differentiation. Cancer Immunol Res. 2013;1(4):256-68
143. Huang X, Ye D, Thorpe PE. Enhancing the potency of a whole-cell breast cancer vaccine in mice with an antibody-IL-2 immunocytokine that targets exposed phosphatidylserine. Vaccine. 2011;29(29-30):4785-93
144. Liang Y, Mafuvadze B, Besch-Williford C, Hyder SM. A combination of p53-activating APR-246 and phosphatidylserine-targeting antibody potently inhibits tumor development in hormone-dependent mutant p53-expressing breast cancer xenografts. Breast Cancer (Dove Med Press). 2018;10:53-67
145. Gerber DE, Stopeck AT, Wong L, Rosen LS, Thorpe PE, Shan JS. et al. Phase I safety and pharmacokinetic study of bavituximab, a chimeric phosphatidylserine-targeting monoclonal antibody, in patients with advanced solid tumors. Clin Cancer Res. 2011;17(21):6888-96
146. Meyer J, Arriaga Y, Anandam J, Karri S, Syed S, Verma U. et al. A phase I clinical trial of the phosphatidylserine-targeting antibody bavituximab in combination with radiation therapy and capecitabine in the preoperative treatment of rectal adenocarcinoma. Am J Clin Oncol. 2018;41(10):972-6
147. Chalasani P, Marron M, Roe D, Clarke K, Iannone M, Livingston RB. et al. A phase I clinical trial of bavituximab and paclitaxel in patients with Her2 negative metastatic breast cancer. Cancer Med. 2015;4(7):1051-9
148. Digumarti R, Bapsy PP, Shan JS. A phase Ib safety and pharmacokinetic study of bavituximab plus chemotherapy in patients with refractory advanced solid tumor malignancies. J Clin Oncol. 2008;26(Suppl 15):S3038
149. Grilley-Olson JE, Weiss J, Ivanova A, Villaruz LC, Moore DT, Stinchcombe TE. et al. Phase Ib study of bavituximab with carboplatin and pemetrexed in chemotherapy-naive advanced nonsquamous non-small-cell lung cancer. Clin Lung Cancer. 2018;19(4):e481-7
150. Digumarti R, Bapsy PP, Suresh AV, Bhattacharyya GS, Dasappa L, Shan JS. et al. Bavituximab plus paclitaxel and carboplatin for the treatment of advanced non-small-cell lung cancer. Lung Cancer. 2014;86(2):231-6
151. Gerber DE, Spigel DR, Giorgadze D, Shtivelband M, Ponomarova OV, Shan JS. et al. Docetaxel combined with bavituximab in previously treated, advanced nonsquamous non-small-cell lung cancer. Clin Lung Cancer. 2016;17(3):169-76
152. Yopp AC, Singal AG, Arriaga YE, Verma UN, Shan J, Kallinteris N. et al. A phase II study of bavituximab and sorafenib in advanced hepatocellular carcinoma (HCC). J Clin Oncol. 2015;33(Suppl 3):S345
153. Pandya SS, Wong L, Bullock AJ, Grabelsky SA, Shum MK, Shan J. et al. Randomized, open-label, phase II trial of gemcitabine with or without bavituximab in patients with nonresectable stage IV pancreatic adenocarcinoma. J Clin Oncol. 2013;31(Suppl 15):S4054
154. Gerber DE, Horn L, Boyer M, Sanborn R, Natale R, Palmero R. et al. Randomized phase III study of docetaxel plus bavituximab in previously treated advanced non-squamous non-small-cell lung cancer. Ann Oncol. 2018;29(7):1548-53
155. Sun A. Benet LZ. Late-stage failures of monoclonal antibody drugs: A retrospective case study analysis. Pharmacology. 2020;105(3-4):145-63
156. Li R, Chiguru S, Li L, Kim D, Velmurugan R, Kim D. et al. Targeting phosphatidylserine with calcium-dependent protein-drug conjugates for the treatment of cancer. Mol Cancer Ther. 2018;17(1):169-82
157. Guan S, Zhang Q, Bao J, Duan T, Hu R, Czech T. et al. Phosphatidylserine targeting peptide-functionalized ph sensitive mixed micelles for enhanced anti-tumor drug delivery. Eur J Pharm Biopharm. 2020;147:87-101
158. Van Rite BD, Lazrak YA, Pagnon ML, Palwai NR, Neves LF, McFetridge PS. et al. Enzyme prodrug therapy designed to target L-methioninase to the tumor vasculature. Cancer Lett. 2011;301(2):177-84
159. Blanco VM, Curry R, Qi X. Sapc-dops nanovesicles: A novel targeted agent for the imaging and treatment of glioblastoma. Oncoscience. 2015;2(2):102-10
160. Wojton J, Chu Z, Mathsyaraja H, Meisen WH, Denton N, Kwon CH. et al. Systemic delivery of Sapc-dops has antiangiogenic and antitumor effects against glioblastoma. Mol Ther. 2013;21(8):1517-25
161. Abu-Baker S, Chu Z, Stevens AM, Li J, Qi X. Cytotoxicity and selectivity in skin cancer by Sapc-dops nanovesicles. J Cancer Ther. 2012;3(4):321-6
162. Blanco VM, Latif T, Chu Z, Qi X. Imaging and therapy of pancreatic cancer with phosphatidylserine-targeted nanovesicles. Transl Oncol. 2015;8(3):196-203
163. Davis HW, Vallabhapurapu SD, Chu Z, Vallabhapurapu SL, Franco RS, Mierzwa M. et al. Enhanced phosphatidylserine-selective cancer therapy with irradiation and Sapc-dops nanovesicles. Oncotarget. 2019;10(8):856-68
164. N'Guessan KF, Patel PH, Qi X. Sapc-dops-a phosphatidylserine-targeted nanovesicle for selective cancer therapy. Cell Commun Signal. 2020;18(1):6
165. De M, Ghosh S, Sen T, Shadab M, Banerjee I, Basu S. et al. A novel therapeutic strategy for cancer using phosphatidylserine targeting stearylamine-bearing cationic liposomes. Mol Ther Nucleic Acids. 2018;10:9-27
166. Ayesa U, Gray BD, Pak KY, Chong PL. Liposomes containing lipid-soluble Zn(II)-bis-dipicolylamine derivatives show potential to be targeted to phosphatidylserine on the surface of cancer cells. Mol Pharm. 2017;14(1):147-56
167. Schuck-Paim C, Taylor RJ, Alonso WJ, Weinberger DM, Simonsen L. Effect of pneumococcal conjugate vaccine introduction on childhood pneumonia mortality in Brazil: A retrospective observational study. Lancet Glob Health. 2019;7(2):e249-56
168. Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer Res. 2011;71(10):3540-51
169. Liu JF, Wu L, Yang LL, Deng WW, Mao L, Wu H. et al. Blockade of TIM3 relieves immunosuppression through reducing regulatory T cells in head and neck cancer. J Exp Clin Cancer Res. 2018;37(1):44
170. Greenberger PA, Saltoun CA, Grammer LC. Northwestern university allergy-immunology syllabus. Allergy Asthma Proc. 2019;40(6):361
171. Murtaza A, Laken H, Da Silva Correia J, McNeeley P, Altobell L, Zhang J. et al. Discovery of TSR-022, a novel, potent anti-human TIM-3 therapeutic antibody. Eur J Cancer. 2016;69:S102
172. Rotte A, Jin JY, Lemaire V. Mechanistic overview of immune checkpoints to support the rational design of their combinations in cancer immunotherapy. Ann Oncol. 2018;29(1):71-83
173. Chen X, Song X, Li K, Zhang T. Fcgammar-binding is an important functional attribute for immune checkpoint antibodies in cancer immunotherapy. Front Immunol. 2019;10:292
174. Wu G, Ma Z, Cheng Y, Hu W, Deng C, Jiang S. et al. Targeting Gas6/TAM in cancer cells and tumor microenvironment. Mol Cancer. 2018;17(1):20
175. Rabbani SA, Valentino ML, Arakelian A, Ali S, Boschelli F. SKI-606 (bosutinib) blocks prostate cancer invasion, growth, and metastasis in vitro and in vivo through regulation of genes involved in cancer growth and skeletal metastasis. Mol Cancer Ther. 2010;9(5):1147-57
176. Hebbard L, Cecena G, Golas J, Sawada J, Ellies LG, Charbono A. et al. Control of mammary tumor differentiation by SKI-606 (bosutinib). Oncogene. 2011;30(3):301-12
177. Liu C, Yu H, Long Q, Chen H, Li Y, Zhao W. et al. Real world experience of crizotinib in 104 patients with ALK rearrangement non-small-cell lung cancer in a single chinese cancer center. Front Oncol. 2019;9:1116
178. Mattar MS, Chang J, Benayed R, Halpenny D, Powers A, Kleiner DE. et al. Complete pathological response to crizotinib in a patient with ALK-rearranged lung adenocarcinoma. Clin Lung Cancer. 2020;21(1e):25-9
179. Kurzrock R, Sherman SI, Ball DW, Forastiere AA, Cohen RB, Mehra R. et al. Activity of XL184 (cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J Clin Oncol. 2011;29(19):2660-6
180. Choueiri TK, Pal SK, McDermott DF, Morrissey S, Ferguson KC, Holland J. et al. A phase I study of cabozantinib (XL184) in patients with renal cell cancer. Ann Oncol. 2014;25(8):1603-8
181. Qu L, Ding J, Chen C, Wu ZJ, Liu B, Gao Y. et al. Exosome-transmitted lncarsr promotes sunitinib resistance in renal cancer by acting as a competing endogenous RNA. Cancer Cell. 2016;29(5):653-68
182. Rho JK, Choi YJ, Kim SY, Kim TW, Choi EK, Yoon SJ. et al. Met and Axl inhibitor NPS-1034 exerts efficacy against lung cancer cells resistant to EGFR kinase inhibitors because of Met or Axl activation. Cancer Res. 2014;74(1):253-62
183. Tang D. Lotze MT. Tumor immunity times out: TIM-3 and HMGB1. Nat Immunol. 2012;13(9):808-10
184. Wang Y, Zhao E, Zhang Z, Zhao G, Cao H. Association between TIM3 and Gal9 expression and gastric cancer prognosis. Oncol Rep. 2018;40(4):2115-26
185. Cook RS, Jacobsen KM, Wofford AM, DeRyckere D, Stanford J, Prieto AL. et al. Mertk inhibition in tumor leukocytes decreases tumor growth and metastasis. J Clin Invest. 2013;123(8):3231-42
186. Tormoen GW, Blair TC, Bambina S, Kramer G, Baird J, Rahmani R. et al. Targeting mertk enhances adaptive immune responses after radiation therapy. Int J Radiat Oncol Biol Phys. 2020 [Epub ahead of print]
187. Peeters MJW, Dulkeviciute D, Draghi A, Ritter C, Rahbech A, Skadborg SK. et al. Mertk acts as a costimulatory receptor on human CD8(+) T cells. Cancer Immunol Res. 2019;7(9):1472-84
188. Oldenborg PA. CD47: A cell surface glycoprotein which regulates multiple functions of hematopoietic cells in health and disease. ISRN Hematol. 2013;2013:614619
189. Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS. et al. The CD47-signal regulatory protein alpha (SIRPα) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U S A. 2012;109(17):6662-7
190. Unanue ER. Perspectives on anti-CD47 antibody treatment for experimental cancer. Proc Natl Acad Sci U S A. 2013;110(27):10886-7
191. Haubelt H, Anders C, Vogt A, Hoerdt P, Seyfert UT, Hellstern P. Variables influencing platelet function analyzer-100 closure times in healthy individuals. Br J Haematol. 2005;130(5):759-67
192. Ennishi D, Healy S, Bashashati A, Saberi S, Hother C, Mottok A. et al. TMEM30A loss-of-function mutations drive lymphomagenesis and confer therapeutically exploitable vulnerability in B-cell lymphoma. Nat Med. 2020;26(4):577-88
193. Advani R, Flinn I, Popplewell L, Forero A, Bartlett NL, Ghosh N. et al. CD47 blockade by Hu5f9-G4 and rituximab in non-Hodgkin's lymphoma. N Engl J Med. 2018;379(18):1711-21
194. Tabagari D, Nemsadze G, Janjalia M, Jincharadze M, Shan J. Phase Ⅱ study of bavituximab plus docetaxel in locally advanced or metastatic breast cancer. J Clin Oncol. 2010;28(Suppl 15):S1042
195. Bonar EE, Goldstick JE, Collins RL, Cranford JA, Cunningham RM, Chermack ST. et al. Daily associations between cannabis motives and consumption in emerging adults. Drug Alcohol Depend. 2017;178:136-42
Author contact
![]() Corresponding author: Dr. Wenguang Chang, Institute for Translational Medicine, The Affiliated Hospital, College of medicine, Qingdao University, Qingdao, 266021, China. Tel.: +86-0532-82991791, E-mail: changsubmitcom.
Corresponding author: Dr. Wenguang Chang, Institute for Translational Medicine, The Affiliated Hospital, College of medicine, Qingdao University, Qingdao, 266021, China. Tel.: +86-0532-82991791, E-mail: changsubmitcom.