Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2021; 11(1):304-315. doi:10.7150/thno.45540 This issue Cite
Research Paper
PET imaging facilitates antibody screening for synergistic radioimmunotherapy with a 177Lu-labeled αPD-L1 antibody
Jingyun Ren1, Mengxin Xu2, Junyi Chen2, Jie Ding1, Peipei Wang1, Li Huo1 ![]() , Fang Li1
, Fang Li1 ![]() , Zhibo Liu2,3
, Zhibo Liu2,3 ![]()
1. Department of Nuclear Medicine, Peking Union Medical College Hospital, Chinese Academy of Medical Science & Peking Union Medical College, Beijing Key Laboratory of Molecular Targeted Diagnosis and Therapy in Nuclear Medicine, Beijing, China.
2. Radiochemistry and Radiation Chemistry Key Laboratory of Fundamental Science, Beijing National Laboratory for Molecular Sciences, College of Chemistry and Molecular Engineering, Peking University, Beijing, 100871, China.
3. Peking University-Tsinghua University Center for Life Sciences, Beijing, 100871, China.
Received 2020-3-2; Accepted 2020-9-21; Published 2021-1-1
Abstract

Rationale: The low response rate of immunotherapy, such as anti-PD-L1/PD-1 and anti-CTLA4, has limited its application to a wider population of cancer patients. One widely accepted view is that inflammation within the tumor microenvironment is low or ineffective for inducing the sufficient infiltration and/or activation of lymphocytes. Here, a highly tumor-selective anti-PD-L1 (αPD-L1) antibody was developed through PET imaging screening, and it was radiolabeled with Lu-177 for PD-L1-targeted radioimmunotherapy (RIT) and radiation-synergized immunotherapy.
Methods: A series of αPD-L1 antibodies were radiolabeled with zirconium-89 for PET imaging to screen the most suitable antibodies for RIT. Mice were divided into an immunotherapy group, a RIT group and a radiation-synergized immunotherapy group to evaluate the therapeutic effect. Alterations in the tumor microenvironment after treatment were assessed using flow cytometry and immunofluorescence microscopy.
Results: Radiation-synergistic RIT can achieve a significantly better therapeutic effect than immunotherapy or RIT alone. The dosages of the radiopharmaceuticals and αPD-L1 antibodies were reduced, the infiltration of CD4+ and CD8+ T cells in the tumor microenvironment was increased, and no side effects were observed. This radiation-synergistic RIT strategy successfully showed a strong synergistic effect with αPD-L1 checkpoint blockade therapy, at least in the mouse model.
Conclusions: PET imaging of 89Zr-labeled antibodies is an effective method for antibody screening. RIT with a 177Lu-labeled αPD-L1 antibody could successfully upregulate antitumor immunity in the tumor microenvironment and turn “cold” tumors “hot” for immunotherapy.
Keywords: Immune checkpoint blockade (ICB), αPD-L1, Lutetium-177 (177Lu), Radioimmunotherapy (RIT), CD8+ T cell
Introduction
Immune checkpoint blockade therapy, such as anti-PD1/PD-L1 and anti-CTLA4, has been highly successful in the clinical treatment of many cancers [1, 2]. However, this emerging cancer therapy suffers from a low response rate, limiting its application to a wider population of cancer patients [3, 4]. The exact reasons for the general resistance of cancer patients to checkpoint blockade therapy are still unclear and probably vary among different cancers, and preliminary studies have shown that resistance seems to be related to the tumor PD-L1 expression level and immune infiltration status [5-7]. Although some studies have found that the treatment response seems to be related to PD-L1 expression [8], it has been reported that patients with PD-L1-negative tumors can also respond to treatment, which may be related to limited tissue sampling or the temporal and spatial heterogeneity of the tumor [9]. As one of the mainstream cancer treatment strategies, external radiotherapy could induce DNA damage in rapidly dividing cancer cells, resulting in tumor antigen release and creating a focal inflammatory response [10], which is often considered a key factor that could upregulate the immune response of tumors.
The relationship between radiation and the immune system was proposed 100 years ago [11], yet the effect on bystander cells has been largely ignored for decades. The discovery of immunogenic cell death (ICD) and in vitro effects provides formal evidence for the immune effect of radiation [12, 13]. The nonpersistent and limited response to checkpoint blockade among patients is a key challenge for cancer immunotherapy [14]. The direct and indirect effects of radiotherapy on tumor cells and tumor-related immune cells together determine the extent to which radiotherapy increases tumor immunogenicity and the synergistic effect between radiotherapy and immunotherapy. Sharverdian et al. reported that in the cohort of patients enrolled in the KEYNOTE-001 trial (NCT01295827), non-small cell lung cancer (NSCLC) patients who received radiotherapy before pembrolizumab treatment showed better progression-free survival (PFS) and overall survival (OS) than those who did not receive radiotherapy [15]. Recently, Liniker et al. reported that radiotherapy and αPD-1 antibodies can be safely combined and well tolerated, with no detectable excess toxicity [16].
However, a limitation of external radiotherapy is the limited number of foci lesions that can be targeted, and its practicability is reduced when multiple systemic metastases occur. Therefore, we wondered whether αPD-L1 antibody can be radiolabeled with potent isotopes for internal targeted radioimmunotherapy (RIT). Ideally, the following radiotherapy-induced inflammation could turn “cold” tumors “hot” and then synergize with the checkpoint blockade agent in triggering robust antitumor immunity [17]. Though monoclonal antibodies are characterized by a well-defined structure, high binding affinity and long half-life in serum, which make them suitable for targeting tumors [18], they often show high liver accumulation that hampers their application in targeted RIT. An ideal antibody for RIT should have the characteristics of high tumor uptake, long tumor retention and low uptake in the liver, kidney and other major organs. In this paper, we propose an antibody screening strategy based on PET images and performed a systematic PET imaging study of a series of αPD-L1 antibodies, screening the antibody with high tumor-specific uptake and labeling it with the β-emitting radionuclide Lu-177 for RIT and further radiation-synergistic RIT.
Methods
Materials
All starting materials were purchased from commercial suppliers (J&K, Sigma-Aldrich, Beijing, China) and were used as received unless otherwise indicated. 11-(4-isothiocyanatophenyl)-3-[6,17-dihydroxy-7,10,18,21-tetraoxo-27-(N-acetylhydroxylamino)-6,11,17, 22-tetraazaheptaeicosine] thiourea (p-SCN-Bn-DFO) and S-2-(4-isothiocyanatobenzyl)-1,4,7,10-tetraazacyclododecane tetraacetic acid (p-SCN-Bn-DOTA) were purchased from Macrocyclics, Inc. (Dallas, TX). An Amicon 50K cut-off ultrafiltration centrifuge was purchased from Millipore Corp., Billerica, MA. The PD-10 column (dead volume 2.5 mL) was purchased from GE Healthcare. Zirconium-89 (3.7 MBq/μL) was purchased from the China Institute of Atomic Energy. Lutetium-177 (40 MBq/μL) was purchased from the ITM Group (Germany). IgG1 isotype control antibody (clone MOPC-21) was purchased from BioLegend.
Cell lines and experimental animals
Murine colon adenocarcinoma MC38 cells were obtained from the National Infrastructure of Cell Line Resources (Beijing, China). MC38 cells were cultured in RPMI 1640 supplemented with 10% FBS, penicillin (100 IU/mL) and streptomycin sulfate (100 mg/mL) in a humidified atmosphere containing 5% CO2 at 37 °C. C57BL/6 male mice (six- to eight-week-old, 18-22 g) were provided by Vital River (Beijing, China).
Tumor models
We complied with all relevant ethical regulations for animal testing and research. Six- to eight-week-old male C57BL/6 mice were subcutaneously injected in the shoulder with 1 × 106 cells suspended in 100 µL of PBS. The mice underwent imaging and biodistribution studies when the tumors grew to a diameter of ~500 mm3, and studies on treatment were initiated when tumor size reached ~100 mm3.
Preparation of 89Zr-DFO-αPD-L1/177Lu-DOTA-αPD-L1 and radiochemistry
The αPD-L1 antibody was purified using an ultrafiltration centrifuge tube and PBS (pH = 7.4) to remove the L-histidine in the original buffer and stored at 4 °C [19]. An aliquot of the antibody stock was then transferred to a 1.5 mL microcentrifuge tube, and the pH of the final solution was adjusted to 8.5-9.0 with sodium tartrate buffer (pH = 9). Finally, 4.0 equivalents of p-SCN-Bn-DOTA or p-SCN-Bn-DFO were added to the solution, which was previously dissolved in DMSO. After incubating for 1 h at 37 °C, the antibody conjugate was purified twice with PBS (pH = 7.4) using an ultrafiltration centrifuge tube. The antibody complex (DOTA-αPD-L1 or DFO-αPD-L1) stock solution was stored at 4 °C.
The conjugated DFO-αPD-L1 solution was transferred to 1.5 mL microcentrifuge tubes and adjusted to the final pH of the resulting solution of 7.0 by adding sodium acetate buffer (pH = 7.0, 100 mM), followed by the addition of an aliquot of 89Zr (37 MBq) solution. After incubation at 37 °C for 1 h, the antibody conjugate was purified through PD-10 chromatography. 89Zr-DFO-αPD-L1 was eluted with 25 mL fractions of PBS at pH = 7.4 (shown in Figure S1). 150 μg of DOTA-αPD-L1 was incubated with 1 mCi of Lu-177 in sodium acetate buffer (pH = 5.5, 200 mM) for 1 h at 37 °C and purified through PD-10 chromatography (shown in Figure 2A).
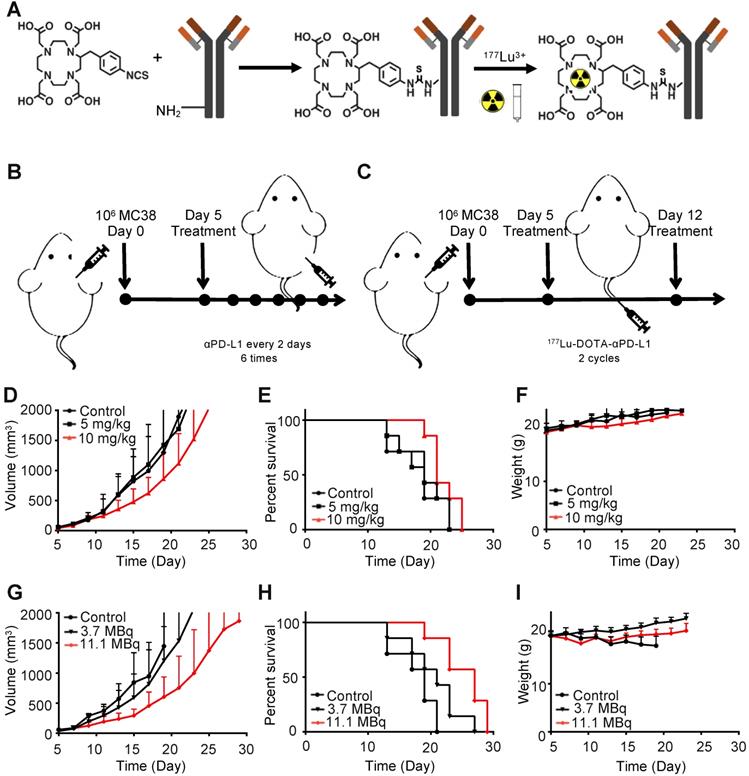
Antitumor treatment study of 177Lu-DOTA-Y003 and Y003 in MC38 tumor-bearing mice. A. Synthesis and radiolabeling of 177Lu-DOTA-Y003. B-C. Treatment flow chart of Y003 (B) and 177Lu-DOTA-Y003 (C). D. Tumor growth curve of Y003 immunotherapy. E. Survival curve of mice in the Y003 immunotherapy groups. F. Record of body weight in the Y003 immunotherapy groups. G. Tumor growth curve of the 177Lu-DOTA-Y003 therapy groups. H. Survival curve of mice in the 177Lu-DOTA-Y003 therapy groups. I. Record of body weight in the 177Lu-DOTA-Y003 therapy groups (n = 7/group).
In vivo biodistribution and small animal PET imaging
C57BL/6 mice were subcutaneously injected in the right shoulder with 1 × 106 MC38 cells. When the tumors reached ~500 mm3, the mice were intravenously (i.v.) injected with 89Zr-DFO-αPD-L1 (3.7 ± 0.1 MBq) and imaged at 2, 12, 24, 48, 72, 96 and 120 h. Approximately 5 min prior to PET/CT image acquisition, the mice were anesthetized through inhalation of a 2% isoflurane/oxygen gas mixture and placed on the scanner bed; a reduced 1.5% isoflurane/oxygen mixture was used to maintain anesthesia during imaging. PET scans were performed on the NanoScan PET-CT scanner (Mediso Medical Solutions, Inc., Hungary). PET data were reconstructed by a Tera-Tomo 3D method. Variance-reduced DW reconstructed PET images were analyzed by Nucline NanoScan software (InterView FUSION, Mediso Medical Solutions, Inc., Hungary) [20, 21].
89Zr-DFO-αPD-L1 (0.22 ± 0.01 MBq) was injected intravenously into MC38 tumor-bearing mice. Blood, small intestine, large intestine, pancreas, liver, spleen, kidney, stomach (without content), brain, bone, lung, heart, muscle, fat and tumor samples were collected at 24 and 96 h post injection (n = 4), and then these items were weighed and measured with a gamma counter (FH-421).
Immunofluorescence microscopy
The representative tissues were fixed in 4% paraformaldehyde and embedded in paraffin. Deparaffinization was carried out with xylene and ethanol gradients, and antigens were recovered with EDTA buffer (pH = 8.0). Then tissues were incubated with BSA for 30 min and added the anti-CD4 mAb (Servicebio, China; GB13064-2) and incubated overnight at 4 ° C. The slides were then washed in PBS buffer for 5 min and then treated with anti-rabbit horseradish peroxidase (HRP)-conjugated secondary mAb (Servicebio, China; GB23303) for 50 min at room temperature (RT). The slides were then washed in PBS buffer for 5 min, added CY3 (Servicebio, China) and incubate for 10 min, transferred into EDTA buffer and heat-treated using a microwave, then cooled in the same solution to RT. The same process was repeated for the following mAbs and fluorescent dyes, in order: anti-CD8 mAb (Servicebio, China; GB13429)/HRP-conjugated secondary mAb (Servicebio, China; GB22303)/FITC (Servicebio, China). Each slide was then treated with two drops of DAPI (Servicebio, China; G1012), washed in distilled water. A coverslip was applied to each slide in the end. All images were acquired by a laser scan confocal microscope (Nikon Eclipse C1, Japan).
Flow cytometry analysis
Mouse tumor tissue was obtained, and necrotic tissues and fat were removed.
The tumor tissue was cut into small pieces (approximately 2 mm) and treated with 1 mg/mL collagenase I (Gibco, USA) for 1 h and then ground with the rubber end of a syringe. Cells were filtered through nylon mesh filters and washed with PBS. PBS containing 5% FBS was added for resuspension, and the cell concentration was adjusted to 107 cells/mL for use. The cells were further stained with the following fluorochrome-conjugated antibodies: FITC anti-mouse CD3 (100204), PE anti-mouse CD8a (100708), APC anti-mouse CD4 (100412), PerCP-Cy5.5 anti-mouse CD45 (103132), APC anti-mouse PD-L1 (124312), FITC anti-mouse CD4 (11-0041-81), APC anti-mouse CD25 (17-0251-82), and PE anti-mouse Foxp3 (12-4771-82) (all from BioLegend).
FACS Diva 6.0 Software (BD LSRII) was used for cell acquisition, and data analysis was carried out using FlowJo software (TreeStar, Ashland, OR). All antibodies were diluted 1:200 for use.
In vivo therapy regimen
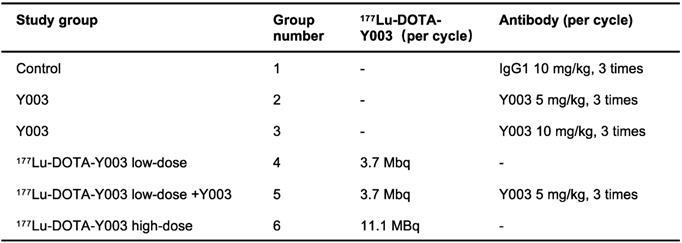
MC38 cells (1 × 106) were subcutaneously injected into the right shoulder of six- to eight-week-old C57BL/6 mice. The body weights of these tumor-bearing mice were monitored for systemic radiotherapy-related toxicity. Tumor growth was monitored by measurement with a digital caliper, where tumor volumes were calculated as follows: (width2 × length)/2. For the immunotherapy group, the mice were divided into a control group (injected with 10 mg/kg IgG1) and an αPD-L1 treatment group (injected with 5 mg/kg αPD-L1 or 10 mg/kg αPD-L1). For the radiotherapy group, the mice were divided into a control group (injected with 11.1 MBq of 177Lu-DOTA-IgG1) and a 177Lu-DOTA-αPD-L1 group (injected with 3.7 MBq or 11.1 MBq of 177Lu-DOTA-αPD-L1). The mice in the radiation-synergized immunotherapy group were injected with 3.7 MBq of 177Lu-DOTA-αPD-L1, followed by injection of 5 mg/kg αPD-L1. The treatment methods corresponding to the different groups are shown in Table 2.
Therapeutic study groups. Mice bearing MC38 colorectal tumors received 2 cycles of injections of the given agent (n = 7-9/group)
Five days after tumor inoculation, the αPD-L1 treatment groups were intraperitoneally (i.p.) injected with the given agent once every 2 days for up to 6 times (Figure 2B). Five days after tumor inoculation, the 177Lu-DOTA-αPD-L1 treatment groups were intravenously (i.v.) injected with the given agent, which was repeated once 7 days later (Figure 2C). For the radiation-synergized immunotherapy group, five days after tumor inoculation, the group was i.v. injected with 3.7 MBq of 177Lu-DOTA-αPD-L1, followed by i.p. injection of 5 mg/kg αPD-L1 3 times. The treatment cycle was repeated two times in total (Figure 3A). When the tumor volume reached 1000 mm3, the tumor-bearing mouse was also regarded as dead.
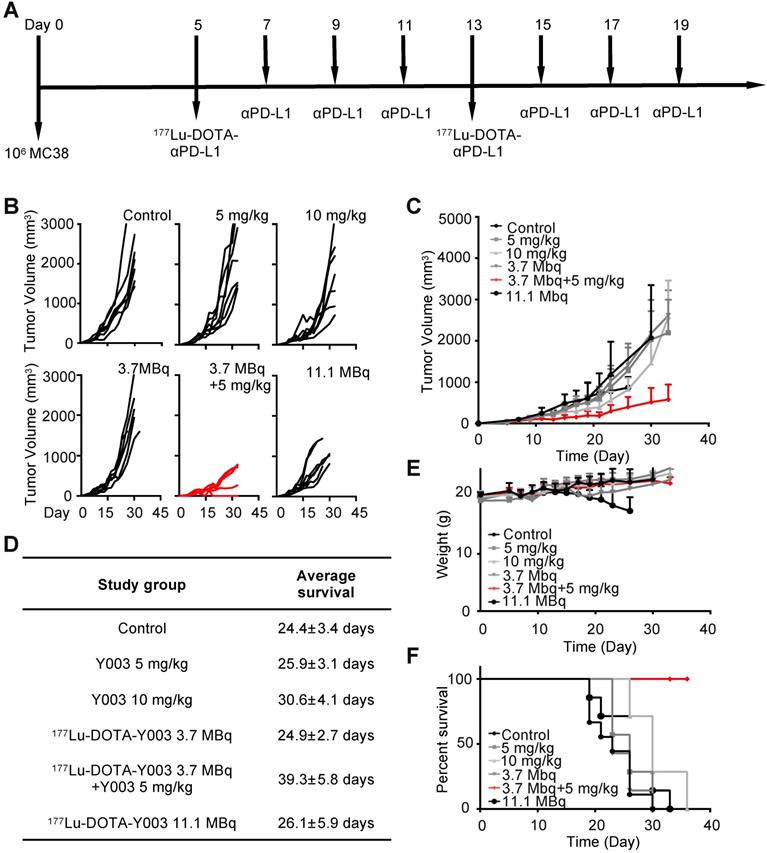
177Lu-DOTA-Y003+Y003 treatment triggers tumor regression in mice. A. Treatment scheme of 177Lu-DOTA-Y003+Y003 in MC38 tumor-bearing mice. B. Individual tumor volume at the indicated time point after tumor implantation. C. Tumor growth curve of the indicated treatment groups. D. Summary of average survival time. E. Record of body weight of the indicated treatment groups. F. Summary of average survival time (n = 7-9/group).
Statistical analysis
The data were analyzed by IBM SPSS version 23.0 (IBM, Armonk, NY, USA).
Continuous variables with a normal distribution are presented as the mean and standard deviation [SD]. The comparisons of the tumor growth volume and body weight of the mice in different therapeutic study groups were performed by two-way repeated measures ANOVA. The differences in CD4+ and CD8+ T cells, PD-L1+ neoplastic cells and regulatory T cells between different treatment groups were compared by independent Student's t-test.
For survival analysis, the periods at risk of tumor volume oversize (reaching 1000 mm3 or death) were defined in days for each mouse. Each observation was separated by 2 days. The risk of an event that did not end in tumor oversize or death was due to the end of the observation period. Exploratory analyses revealed that the relationship between the type of therapies and rate of tumor oversize was consistent across time. Survival curves of different therapeutic study groups were compared by the log-rank test. Statistical significance was set at p < 0.05 (*p < 0.05, **p < 0.01, ***p < 0.001).
Results
PET imaging-guided antibody screening in MC38 tumor-bearing mice
The 89Zr-DFO-Y001, 89Zr-DFO-Y002 and 89Zr-DFO-Y003 were radiolabeled at an average radiochemical yield of 90.2 ± 2.3%, 89.3 ± 3.5% and 88.2 ± 3.0%, respectively, with greater than 95% for radiochemical purity.
The in vivo biodistributions of 89Zr-DFO-Y001, 89Zr-DFO-Y002 and 89Zr-DFO-Y003 were investigated in MC38 tumor-bearing mice. Static PET-CT imaging at 2, 12, 24, 48, 72, 96 and 120 h post administration was recorded and is shown in Figure 1A-C. The pharmacokinetics were further interpreted with time-activity curves, which are presented in Figure 1D-F.
PET-CT imaging of 89Zr-labeled αPD-L1 antibodies identifies Y003 for subsequent RIT because of its remarkable tumor selectivity. A-C. Representative PET imaging of 89Zr-DFO-Y001 (A), 89Zr-DFO-Y002 (B) and 89Zr-DFO-Y003 (C) at a series of time points in MC38 tumor-bearing mice. D-E. Time-activity curves (TACs) of 89Zr-DFO-Y001 (D), 89Zr-DFO-Y002 (E) and 89Zr-DFO-Y003 (F) in the tumor, blood, liver and muscle (n = 4/group).
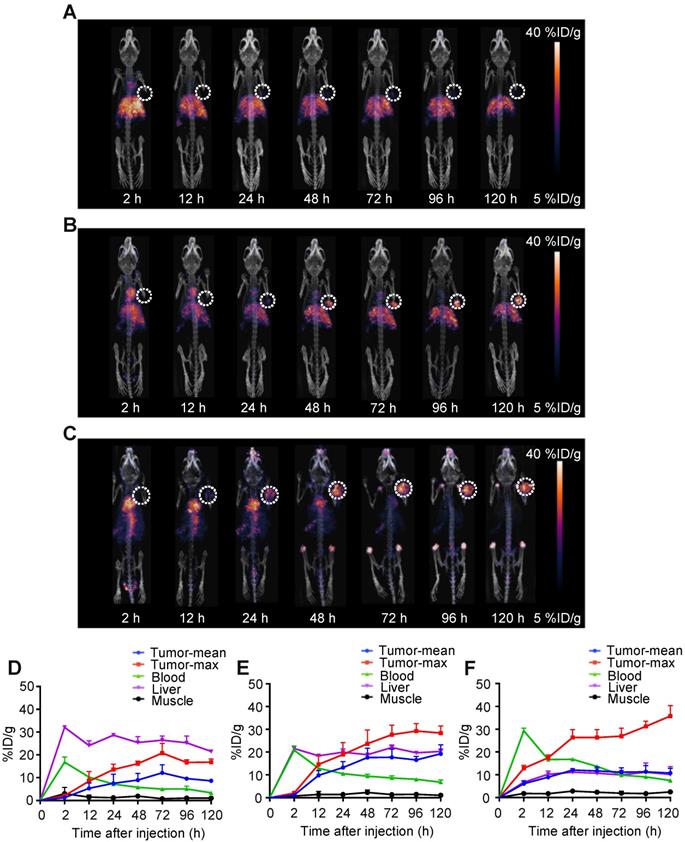
According to PET imaging, the tumor uptake of 89Zr-DFO-Y001 was low (Figure 1A). Mild tumor uptake was observed at 24 h after injection. The tumor uptake increased slightly at 48 and 72 h and then decreased at 96 h. At 120 h postinjection, the tumor uptake became negligible. We reason that the low tumor uptake was due to the rapid clearance of 89Zr-DFO-Y001 from circulation. At 2 h postinjection, the blood pool uptake could be seen but was minimal. Meanwhile, the liver uptake of 89Zr-DFO-Y001 was high at 2 h postinjection and then gradually increased until 120 h after injection, indicating strong evidence of fast hepatobiliary clearance, which is corroborative with the rapid decline of blood uptake. In summary, the poor tumor uptake and unfavorable pharmacokinetics indicate that Y001 is not an optimal candidate for PD-L1-targeted radiotherapy.
As shown in Figure 1B, the PET imaging of 89Zr-DFO-Y002 exhibited more favorable tumor accumulation than that of 89Zr-DFO-Y001. At 2 h postinjection, notable uptake in the cardiac blood pool and abdominal aorta was observed, indicating a remarkably longer blood circulation than that of 89Zr-DFO-Y001. The uptake in the blood pool decreased gradually and was almost negligible at 72 h and later time points. Tumor uptake was perceived at 24 h postinjection and inclined gradually until reaching a peak (29 %ID/g) at 96 h. The liver uptake reached the apex (21.5 %ID/g) at the earliest time point and then slowly declined to 20 %ID/g at 120 h postinjection, which is 50% less than that of 89Zr-DFO-Y001. According to PET imaging, the most intensive uptake of 89Zr-DFO-Y002 was found in the tumor, showing that Y002 is a prospective candidate for immunotherapy but still not good enough for RIT.
Encouraged by the aforementioned progress, we continued the screening until finding Y003. At 2 h postinjection, we found that most of the radioactivity remained in the blood circulation, and almost no liver uptake was observed (Figure 1C). Tumor uptake was visible at 12 h postinjection and then continually increased to 40 %ID/g at 120 h postinjection. Compared to 89Zr-DFO-Y001 and 89Zr-DFO-Y002, the liver uptake of 89Zr-DFO-Y003 was almost negligible in PET images at all time points. It is worth noting that the bone uptake became intense at later time points, which could be from the metabolized 89Zr-DFO-Y003 that had been internalized in the tumor cells. In fact, except for the bone uptake, the radioactivity could only be found in tumors at time points later than 72 h. Therefore, we chose Y003 as a highly tumor-selective candidate for Lu-177 labeling and subsequent synergistic RIT.
Biodistribution studies of 89Zr-DFO-Y003
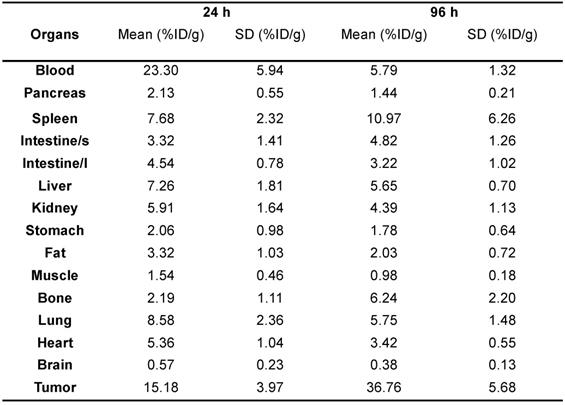
The ex vivo biodistribution of 89Zr-DFO-Y003 was performed at 24 h and 96 h after the injection of 89Zr-DFO-Y003. As summarized in Table 1, the tumor uptake at 24 and 96 h after injection was 15.18 ± 3.97 %ID/g and 36.76 ± 5.68 %ID/g, respectively (n = 4). The blood uptake of 89Zr-DFO-Y003 was high at early time points (up to 23.30 ± 5.94 %ID/g at 24 h after injection), followed by a gradual decrease, and the uptake at 96 h after injection was 5.79 ± 1.32 %ID/g, which is corroborative with PET scans.
Ex vivo biodistribution of 89Zr-DFO-Y003 in MC38 tumor-bearing mice at 24 and 96 h postinjection (n = 4/group)
Characterization and radiochemistry of 177Lu-DOTA-Y003
The conjugation scheme and structure of 177Lu-DOTA-Y003 is illustrated in Figure 2A. The radiochemical yield of 177Lu-DOTA-Y003 was 62.7 ± 5.2%, and the purity was > 99% according to instant thin-layer chromatography (iTLC) analysis. The specific activity was calculated to be 250 ± 10 MBq/mg, 177Lu-DOTA-Y003 showed good stability in vitro, and no decomposition was observed after incubation in PBS for 168 h (Figure S3).
Immunotherapy with Y003 only
The tumor-bearing mice were injected intraperitoneally with 10 mg/kg of IgG1, 5 mg/kg of Y003 or 10 mg/kg of Y003. The treatment was repeated every two days, and each mouse was treated 6 times in total. As shown in Figure 2D-E, no difference was observed in either the tumor growth curve or survival curve between the control group and the 5 mg/kg group. The tumor growth of the 10 mg/kg group was slightly slower than that of the former two groups, and the survival period of the mice was also slightly longer than that of the former two groups, indicating that Y003 immunotherapy had a certain therapeutic effect, but it was not good enough to suppress tumor growth by itself. There was no significant difference (P = 0.126) in body weight between the treatment group and the control group (Figure 2F).
Radiotherapy with 177Lu-DOTA-Y003
Then, we examined whether 177Lu-DOTA-Y003 could promote tumor clearance. The tumor-bearing mice were intravenously injected with 11.1 MBq of 177Lu-DOTA-IgG1 or 3.7 MBq or 11.1 MBq of 177Lu-DOTA-Y003. The treatment was repeated once 7 days after the first treatment. Encouragingly, the low-dose 177Lu-DOTA-Y003 treatment group showed slower tumor growth than the control group (Figure 2G), though there was no significant difference (P > 0.05) in the survival curve (Figure 2H). For the high-dose treatment group, the treatment efficacy was remarkable, especially at the early stage of tumor growth. The survival period of the high-dose group was significantly longer than that of the first two groups (P = 0.032), but the weight loss of the mice (Figure 2I) was also noteworthy (> 10%, P < 0.001), indicating that the high-dose 177Lu-DOTA-Y003 treatment could have side effects.
Synergistic radiotherapy with 177Lu-DOTA-Y003+Y003
We reason the poor efficacy of Y003 to the insufficient infiltration and/or inactivation of lymphocytes, and for this reason, checkpoint blockade-resistant tumors are considered to be “cold”. Some colon cancers with high CD8+ T cell infiltration and overexpression of regulatory immune checkpoints had a higher response rate to αPD-1 checkpoint blocking immunotherapy [22-24]. We wondered whether 177Lu-DOTA-Y003-induced inflammation within the tumor microenvironment could “heat” some “cold” tumors and therefore synergize with the checkpoint blockade agent in triggering robust antitumor immunity. To test this idea, the tumor-bearing mice were intravenously injected with 177Lu-DOTA-Y003 at day 5 followed by three sequential intraperitoneal injections of Y003 on days 7, 9, and 11 (Figure 3A). The treatment cycle was repeated once on day 13 to augment the extent of radiotherapy induction. In control mice treated with 10 mg/kg IgG1, the volume of the MC38 tumors increased by approximately 50-fold in 3 weeks (Figure 3B). Strikingly, upon treatment with 177Lu-DOTA-Y003+Y003, most of the tumors became reddish in the first few days, and notable tumor shrinkage was observed in some tumors (Figure 3B). A pronounced growth delay of approximately 15 days was seen in 177Lu-DOTA-Y003+Y003 mice compared with control mice. In contrast, mice treated with 177Lu-DOTA-Y003 or Y003 alone behaved similarly to IgG1-treated mice and showed normal and aggressive tumor growth, respectively (Figure 3B-C). The survival curve (Figure 3F) showed that the survival of the 177Lu-DOTA-Y003+Y003 group was significantly prolonged (P < 0.001), and the survival rate was 100% at 36 days, while the mice in all the other groups died. The average survival time of the 177Lu-DOTA-Y003+Y003 group was also significantly prolonged (P < 0.001, Figure 3D).
In terms of the side-effect assessment, there were no significant differences (P > 0.05) in body weight among the groups treated with 10 mg/kg of IgG1, 5 mg/kg of Y003 only, 10 mg/kg of Y003 only, 3.7 MBq of 177Lu-DOTA-Y003 only and radiation-synergized immunotherapy. As observed previously, the body weight of the mice treated with 11.1 MBq of 177Lu-DOTA-Y003 decreased significantly (P < 0.01, Figure 3E), and HE staining also confirmed that the livers of the mice in the high-dose 177Lu-DOTA-Y003 group had anatomical structural changes; a large number of hepatocytes showed focal necrosis, nuclear fragmentation or lysis, accompanied by a small number of lymphocytes and neutrophil infiltration (Figure S5).
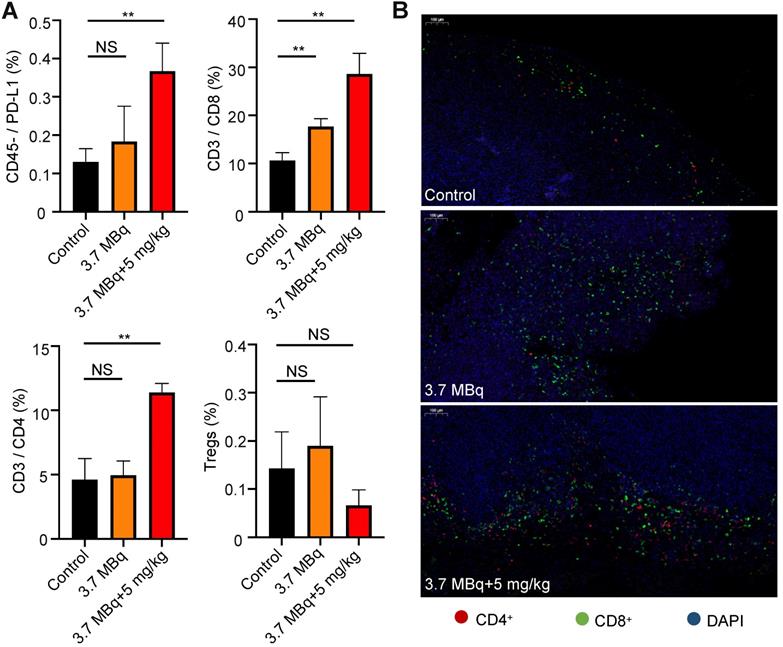
Treatment with 177Lu-DOTA-Y003 increases tumor-infiltrating lymphocytes and upregulates PD-L1 expression in tumors. A. Percentage of PD-L1 neoplastic cells (CD45-/PD-L1), CD8+ T cells, CD4+ T cells and regulatory T cells in tumors after the indicated treatment. B. Representative fluorescent images of the immunofluorescence staining of tumors.
Radiotherapy with 177Lu-DOTA-Y003 successfully tunes the tumor microenvironment
Using flow cytometry, we analyzed PD-L1 neoplastic cells (CD45-/PD-L1) and T cells after the administration of combined RIT and radiation-synergized immunotherapy. The tumors in the 3.7 MBq 177Lu-DOTA-Y003 group had increased CD8+ T cells and CD45-/PD-L1 cells compared with those in the control group. Importantly, the addition of αPD-L1 to 3.7 MBq 177Lu-DOTA-Y003 resulted in a significant increase in CD8+ T cells and CD45-/PD-L1 cells compared with the control and 3.7 MBq 177Lu-DOTA-Y003 groups. Compared with the control group, the proportion of CD4+ T cells in the radiation-synergized immunotherapy group was significantly increased, whereas no significant difference was observed in the 3.7 MBq 177Lu-DOTA-Y003 group. There was no significant difference in Treg cells between the control group and the therapeutic groups.
At the end of the treatment, we collected the tumors of mice for immunofluorescence staining to further confirm the effect of the combined treatment on mouse immunity. Anti-CD4 and anti-CD8 fluorescence imaging of the tumor tissue confirmed the remarkably increased T cell infiltration. The therapeutic effect of the 177Lu-DOTA-Y003+Y003 group confirmed our hypothesis: the immune response could be enhanced after the injection of 177Lu-DOTA-Y003 to achieve the purpose of RIT. In the 177Lu-DOTA-Y003+Y003 group, the expression of PD-L1 and CD4+ and CD8+ T cells increased after 177Lu-DOTA-Y003+Y003 treatment. This finding shows that radiotherapy has a certain effect on immunity, and the combination of αPD-L1 therapy can achieve a better immunotherapeutic effect.
Discussion
As a logical biomarker for predicting the response to αPD-1/PD-L1 immunotherapy, the expression of PD-L1 has been extensively studied in clinical trials since the early development of immunotherapy [5, 25]. Currently, the most commonly used predictive biomarker is PD-L1 expression as evaluated in tumor biopsies through immunohistochemical (IHC) staining; however, limitations are present, for example, PD-L1 expression is not always correctly assessed by core needle biopsy and this method cannot reflect the changes in PD-L1 expression over time [26, 27]. It is contended that as a probe, the αPD-L1 antibody is more informative, as it provides an image of the entire tumor, both primary and metastatic [28-31]. Nevertheless, antibodies could be more limited in their tissue-penetrating capacity and longer circulation time due to their size. Antibodies need to be matched with radiopharmaceuticals with a long half-life (such as Zr-89), and the consequent problem is the radiation dose to healthy tissues and organs. To reduce side effects, we performed a systematic PET imaging study of a series of αPD-L1 antibodies. The PET images and time-activity curves of three representative candidates, denoted as Y001, Y002 and Y003, are presented in this work. Among them, Y003 exhibited remarkably high tumor uptake (up to 40 %ID/g) and particularly rapid clearance from major organs. Thus, it was chosen as the antibody for Lu-177 radiolabeling and subsequent animal studies.
Radioisotope-labeled monoclonal antibodies for RIT are effective cancer treatment strategies because tumor-associated mAbs linked to cytotoxic nuclides can selectively bind tumor antigens and release targeted cytotoxic radiation [17, 32-34]. Therefore, these antibodies are an attractive tool to use the immune modulatory function of radiotherapy to transform the immune “cold” environment into a "hot" environment to improve the response rate of immunotherapy. Although the concept of RIT is simple, in practice, it is difficult to achieve substantial clinical success, especially in solid tumors, due to the limited delivery of mAbs in tumors [35, 36]. The expression of PD-L1 on tumor cells inactivates CD4+ and CD8+ T cells through the interaction with PD-1 on their surface [37], and the immune response is stopped [38]. Therefore, αPD-1 and αPD-L1 antibodies can reverse the inactivated interaction between tumor cells and T cells, leading to a sufficient immune response and tumor cell killing. In the study of Schaue et al. [39], it was observed that tumor-specific T cells increased significantly (P < 0.01) in most colorectal cancer (CRC) patients after the completion of radiotherapy. Different doses or fractionations are thought to result in different forms of cell death [40], thus regulating the response of downstream cells. As a result, radiotherapy regimens that induce cell death in the form of immune silencing (i.e., apoptotic cell death) are not expected to synergize with ICB, while the dose that triggers the inflammatory response can be used as an immune modulator to induce the additional effects of radiotherapy and immunotherapy [41]. Chen et al. [42] proposed a novel therapeutic regimen that combined αPD-L1 immunotherapy with peptide-based targeted radionuclide therapy and confirmed that this combination therapy can stimulate the infiltration of CD8+ T cells in the tumor microenvironment. In our study, the therapeutic effect of Y003 alone and 177Lu-DOTA-Y003 RIT on CRC was not satisfactory, but the therapeutic effect of 177Lu-DOTA-Y003 radiation-synergized immunotherapy was much better than that of Y003 alone or 177Lu-DOTA-Y003 RIT alone, and the combination therapy used a low (50%) radiation dose and antibody dose, with no detectable excess toxicity. Flow cytometry confirmed that after 177Lu-DOTA-Y003 injection, CD8+ T cell upregulation occurred in the tumor microenvironment, but there was no obvious change in CD4+ T cells. More interestingly, after combination therapy, CD4+ and CD8+ T cells were significantly upregulated in the tumor microenvironment, and no significant increase in Treg cells was observed. Immunofluorescence staining showed that radiation-synergized immunotherapy significantly increased CD4+ and CD8+ T cells in the tumor microenvironment, which indicated that 177Lu-DOTA-Y003 radiation-synergized immunotherapy could achieve double effects, especially at low radiation and antibody doses. It is worth noting that our research not only considers the expression level of PD-L1 in tumor cells, but also considers the state of T cells infiltrating in tumors. As described in a recent review [43], “hot”, “altered” and “cold” tumors were defined based on immune cells in tumor microenvironment, and the tumors are classified according to their immune cells infiltration rather than tumor types, this also explains the superior antitumor effect of the 177Lu-DOTA-Y003 radiation-synergized immunotherapy.
It should be noted that our study also has some limitations. First, the study was only tested in a mouse MC38 colon cancer tumor model, which is generally an immune "hot" tumor model that is sensitive to anti-PD-1/PD-L1 therapy. In addition, determining the appropriate dose and timing of RIT is also essential for maximizing the therapeutic efficacy of radiation-synergized immunotherapy, and more studies are needed to find the optimal combination.
Conclusion
A highly tumor-selective antibody, Y003, was achieved through PET imaging screening as a candidate for Lu-177 labeling and subsequent RIT. We found that the combination of 3.7 MBq of 177Lu-DOTA-Y003 and 5 mg/kg Y003 showed remarkably better therapeutic efficacy than 10 mg/kg Y003 immunotherapy or 11.1 MBq 177Lu-DOTA-Y003 RIT. The dosages of 177Lu-DOTA-Y003 and Y003 were reduced by > 50%, and almost no side effects were observed. 177Lu-DOTA-Y003 RIT-induced inflammation within the TME can turn “cold” tumors into “hot” ones. This further indicates that radioisotope (e.g., Lu-177, Ac-225)-labeled antibodies have the potential of improving the efficacy of cancer immunotherapy. Compared to external radiation-induced therapy, this therapeutic combination is also a promising approach for treating metastatic tumors.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wei SC, Duffy CR, Allison JP. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018;8:1069-86
2. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM. et al. Nivolumab plus Ipilimumab in Advanced Melanoma. New Engl J Med. 2013;369:122-33
3. van de Donk PP, Kist de Ruijter L, Lub-de Hooge MN, Brouwers AH, van der Wekken AJ, Oosting SF. et al. Molecular imaging biomarkers for immune checkpoint inhibitor therapy. Theranostics. 2020;10:1708-18
4. Lecocq Q, De Vlaeminck Y, Hanssens H, D'Huyvetter M, Raes G, Goyvaerts C. et al. Theranostics in immuno-oncology using nanobody derivatives. Theranostics. 2019;9:7772-91
5. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF. et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443-54
6. Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US. et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother. 2010;33:828-33
7. Teng MW, Ngiow SF, Ribas A, Smyth MJ. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res. 2015;75:2139-45
8. Daud AI, Wolchok JD, Robert C, Hwu WJ, Weber JS, Ribas A. et al. Programmed Death-Ligand 1 Expression and Response to the Anti-Programmed Death 1 Antibody Pembrolizumab in Melanoma. J Clin Oncol. 2016;34:4102-09
9. Bensch F, van der Veen EL, Lub-de Hooge MN, Jorritsma-Smit A, Boellaard R, Kok IC. et al. (89)Zr-atezolizumab imaging as a non-invasive approach to assess clinical response to PD-L1 blockade in cancer. Nat Med. 2018;24:1852-58
10. Zhang B, Bowerman NA, Salama JK, Schmidt H, Spiotto MT, Schietinger A. et al. Induced sensitization of tumor stroma leads to eradication of established cancer by T cells. J Exp Med. 2007;204:49-55
11. Schaue D. A Century of Radiation Therapy and Adaptive Immunity. Front Immunol. 2017;8:431
12. Demaria S, Ng B, Devitt ML, Babb JS, Kawashima N, Liebes L. et al. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol. 2004;58:862-70
13. Tesniere A, Panaretakis T, Kepp O, Apetoh L, Ghiringhelli F, Zitvogel L. et al. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ. 2008;15:3-12
14. Saeed M, Gao J, Shi Y, Lammers T, Yu H. Engineering Nanoparticles to Reprogram the Tumor Immune Microenvironment for Improved Cancer Immunotherapy. Theranostics. 2019;9:7981-8000
15. Shaverdian N, Lisberg AE, Bornazyan K, Veruttipong D, Goldman JW, Formenti SC. et al. Previous radiotherapy and the clinical activity and toxicity of pembrolizumab in the treatment of non-small-cell lung cancer: a secondary analysis of the KEYNOTE-001 phase 1 trial. Lancet Oncol. 2017;18:895-903
16. Liniker E, Menzies AM, Kong BY, Cooper A, Ramanujam S, Lo S. et al. Activity and safety of radiotherapy with anti-PD-1 drug therapy in patients with metastatic melanoma. Oncoimmunology. 2016 5
17. Ozpiskin OM, Zhang L, Li JJ. Immune targets in the tumor microenvironment treated by radiotherapy. Theranostics. 2019;9:1215-31
18. Dammes N, Peer D. Monoclonal antibody-based molecular imaging strategies and theranostic opportunities. Theranostics. 2020;10:938-55
19. Price EW, Zeglis BM, Cawthray JF, Ramogida CF, Ramos N, Lewis JS. et al. H(4)octapa-Trastuzumab: Versatile Acyclic Chelate System for In-111 and Lu-177 Imaging and Therapy. J Am Chem Soc. 2013;135:12707-21
20. Liu H, Han YX, Li JY, Qin M, Fu QF, Wang CH. et al. F-18-Alanine Derivative Serves as an ASCT2 Marker for Cancer Imaging. Mol Pharm. 2018;15:947-54
21. Duan DB, Liu H, Xu Y, Han YX, Xu MX, Zhang ZC. et al. Activating TiO2 Nanoparticles: Gallium-68 Serves as a High-Yield Photon Emitter for Cerenkov-Induced Photodynamic Therapy. ACS Appl Mater Interfaces. 2018;10:5278-86
22. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD. et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372:2509-20
23. Overman MJ, Kopetz S, McDermott RS, Leach J, Lonardi S, Lenz HJ. et al. Nivolumab +/- ipilimumab in treatment (tx) of patients (pts) with metastatic colorectal cancer (mCRC) with and without high microsatellite instability (MSI-H): CheckMate-142 interim results. J Clin Oncol. 2016 34 (15)
24. Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA. et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18:1182-91
25. Reck M, Rodriguez-Abreu D, Robinson AG, Hui RN, Csoszi T, Fulop A. et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. New Engl J Med. 2016;375:1823-33
26. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS. et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563-7
27. Patel SP, Kurzrock R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol Cancer Ther. 2015;14:847-56
28. Josefsson A, Nedrow JR, Park S, Banerjee SR, Rittenbach A, Jammes F. et al. Imaging, Biodistribution, and Dosimetry of Radionuclide-Labeled PD-L1 Antibody in an Immunocompetent Mouse Model of Breast Cancer. Cancer Res. 2016;76:472-9
29. Kikuchi M, Clump DA, Srivastava RM, Sun L, Zeng D, Diaz-Perez JA. et al. Preclinical immunoPET/CT imaging using Zr-89-labeled anti-PD-L1 monoclonal antibody for assessing radiation-induced PD-L1 upregulation in head and neck cancer and melanoma. Oncoimmunology. 2017;6:e1329071
30. Truillet C, Oh HLJ, Yeo SP, Lee CY, Huynh LT, Wei J. et al. Imaging PD-L1 Expression with ImmunoPET. Bioconjug Chem. 2018;29:96-103
31. Hettich M, Braun F, Bartholoma MD, Schirmbeck R, Niedermann G. High-Resolution PET Imaging with Therapeutic Antibody-based PD-1/PD-L1 Checkpoint Tracers. Theranostics. 2016;6:1629-40
32. Erdi AK, Erdi YE, Yorke ED, Wessels BW. Treatment planning for radio-immunotherapy. Phys Med Biol. 1996;41:2009-26
33. Allen TM. Ligand-targeted therapeutics in anticancer therapy. Nat Rev Cancer. 2002;2:750-63
34. Larson SM, Carrasquillo JA, Cheung NK, Press OW. Radioimmunotherapy of human tumours. Nat Rev Cancer. 2015;15:347-60
35. Jang BS, Lee SM, Kim HS, Shin IS, Razjouyan F, Wang S. et al. Combined-modality radioimmunotherapy: synergistic effect of paclitaxel and additive effect of bevacizumab. Nucl Med Biol. 2012;39:472-83
36. Wang S, Shin IS, Hancock H, Jang BS, Kim HS, Lee SM. et al. Pulsed high intensity focused ultrasound increases penetration and therapeutic efficacy of monoclonal antibodies in murine xenograft tumors. J Control Release. 2012;162:218-24
37. Latchman Y, Wood C, Chemova T, Iwai Y, Malenkovich N, Long A. et al. PD-L2, a novel B7 homologue, is a second ligand for PD-1 and inhibits T cell activation. Faseb J. 2001;15:A345-A45
38. Dong HD, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB. et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat Med. 2002;8:793-800
39. Schaue D, Comin-Anduix B, Ribas A, Zhang L, Goodglick L, Sayre JW. et al. T-cell responses to survivin in cancer patients undergoing radiation therapy. Clin Cancer Res. 2008;14:4883-90
40. Hellevik T, Martinez-Zubiaurre I. Radiotherapy and the tumor stroma: the importance of dose and fractionation. Front Oncol. 2014;4:1
41. Brown JS, Sundar R, Lopez J. Combining DNA damaging therapeutics with immunotherapy: more haste, less speed. Brit J Cancer. 2018;118:312-24
42. Chen H, Zhao L, Fu K, Lin Q, Wen X, Jacobson O. et al. Integrin alphavbeta3-targeted radionuclide therapy combined with immune checkpoint blockade immunotherapy synergistically enhances anti-tumor efficacy. Theranostics. 2019;9:7948-60
43. Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. 2019;18:197-218
Author contact
![]() Corresponding author: Zhibo Liu, Peking University-Tsinghua University Center for Life Sciences, Beijing, 100871, China. Email: zbliuedu.cn Or Li Huo & Fang Li, Department of Nuclear Medicine, Peking Union Medical College Hospital, Chinese Academy of Medical Science & Peking Union Medical College, Beijing Key Laboratory of Molecular Targeted Diagnosis and Therapy in Nuclear Medicine, Beijing, China.
Corresponding author: Zhibo Liu, Peking University-Tsinghua University Center for Life Sciences, Beijing, 100871, China. Email: zbliuedu.cn Or Li Huo & Fang Li, Department of Nuclear Medicine, Peking Union Medical College Hospital, Chinese Academy of Medical Science & Peking Union Medical College, Beijing Key Laboratory of Molecular Targeted Diagnosis and Therapy in Nuclear Medicine, Beijing, China.