Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(1):426-444. doi:10.7150/thno.50281 This issue Cite
Research Paper
Oscillating lncRNA Platr4 regulates NLRP3 inflammasome to ameliorate nonalcoholic steatohepatitis in mice
Yanke Lin1#, Shuai Wang1,2#, Lu Gao1, Ziyue Zhou1, Zemin Yang1, Jingpan Lin1, Shujing Ren1, Huijie Xing3 ![]() , Baojian Wu1
, Baojian Wu1 ![]()
1. College of Pharmacy, Jinan University, Guangzhou 510632, China.
2. Integrated Chinese and Western Medicine Postdoctoral research station, Jinan University, Guangzhou, 510632, China.
3. Institution of Laboratory Animal, Jinan University, 601 Huangpu Avenue West, Guangzhou, China.
#These authors contributed equally to this work.
Received 2020-7-4; Accepted 2020-9-30; Published 2021-1-1
Abstract

Background: Understanding the molecular events and mechanisms underlying development and progression of nonalcoholic steatohepatitis is essential in an attempt to formulating a specific treatment. Here, we uncover Platr4 as an oscillating and NF-κB driven lncRNA that is critical to the pathological conditions in experimental steatohepatitis
Methods: RNA-sequencing of liver samples was used to identify differentially expressed lncRNAs. RNA levels were analyzed by qPCR and FISH assays. Proteins were detected by immunoblotting and ELISA. Luciferase reporter, ChIP-sequencing and ChIP assays were used to investigate transcriptional gene regulation. Protein interactions were evaluated by Co-IP experiments. The protein-RNA interactions were studied using FISH, RNA pull-down and RNA immunoprecipitation analyses
Results: Cyclic expression of Platr4 is generated by the core clock component Rev-erbα via two RevRE elements (i.e., -1354/-1345 and -462/-453 bp). NF-κB transcriptionally drives Platr4 through direct binding to two κB sites (i.e., -1066/-1056 and -526/-516 bp), potentially accounting for up-regulation of Platr4 in experimental steatohepatitis. Intriguingly, Platr4 serves as a circadian repressor of Nlrp3 inflammasome pathway by inhibiting NF-κB-dependent transcription of the inflammasome components Nlrp3 and Asc. Loss of Platr4 down-regulates Nlrp3 inflammasome activity in the liver, blunts its diurnal rhythm, and sensitizes mice to experimental steatohepatitis, whereas overexpression of Platr4 ameliorates the pathological conditions in an Nlrp3-dependent manner. Mechanistically, Platr4 prevents binding of the NF-κB/Rxrα complex to the κB sites via a physical interaction, thereby inhibiting the transactivation of Nlrp3 and Asc by NF-κB.
Conclusions: Platr4 functions to inactivate Nlrp3 inflammasome via intercepting NF-κB signaling. This lncRNA might be an attractive target that can be modulated to ameliorate the pathological conditions of steatohepatitis.
Keywords: Platr4, NLRP3 inflammasome, NF-κB, RXR, Steatohepatitis
Introduction
Nonalcoholic fatty liver disease (NAFLD) is the most common chronic liver disorder, with a high incidence in developed countries (20-30% prevalence) and an increasing incidence in developing countries [1]. Nonalcoholic steatohepatitis (NASH) is a severe form of NAFLD, characterized by fat accumulation in the liver and varying degrees of inflammation and fibrosis [2]. NASH may progress to liver cirrhosis and hepatocellular carcinoma, and affected patients tend to have an increased liver-related mortality [3]. Notably, NASH is becoming a leading indication for liver transplantation in the United States [4,5]. The pathogenesis of NASH is poorly understood and there are no specific drugs for disease treatment, although some risk factors (e.g., oxidative stress and metabolic morbidities such as dyslipidemia, obesity, insulin resistance and type 2 diabetes) have been defined [4]. Accordingly, the goal of current interventions for NAFLD and NASH is to mitigate the associated metabolic comorbidities [4]. Nevertheless, “two-hit” and “multiple-hit” hypotheses have been proposed to account for the pathogenesis of NASH [6,7]. In both theories, aberrant inflammation is implicated the development and progression of NASH. Therefore, studies to clarify the specific mechanisms for deregulated inflammation are critical for development of new therapeutic strategies to prevent and manage steatohepatitis.
The innate immunity recognizes not only the pathogen products (pathogen-associated molecular patterns, PAMPs) but also endogenous danger signals (damage-associated molecular patterns, DAMPs), and provides immediate defense by mounting strong inflammatory responses [8]. NLRP3 (NOD-, LRR- and pyrin domain-containing protein 3) inflammasome plays a central role in these responses. It is a multi-protein complex consisting of NLRP3 (a sensor), ASC (apoptosis-associated speck-like protein containing a CARD, an adaptor), and pro-caspase-1 (an effector). NLRP3 inflammasome activation is a two-step process, namely, it must be first primed and then can be activated [9]. The priming step, triggered by the signal 1 (e.g., PAMPs and cytokines), mainly serves to up-regulate the expression of the inflammasome components via NF-κB activation and gene transcription [9]. Signal 2 (e.g., PAMPs and DAMPs such as particulates, crystals and adenosine triphosphate (ATP)) triggers various upstream signaling events including potassium efflux, calcium flux, mitochondrial dysfunction and lysosomal disruption which induce the complex formation of and activation of NLRP3 inflammasome [9]. The inflammasome complex promotes the cleavage of pro-caspase-1 (to form caspase-1) and ensuing maturation of the proinflammatory cytokines IL-1β and IL-18. Although NLRP3 inflammasome is essential for immune defense against microbial infection and cellular damage, excessive and deregulated activation can lead to the development of pathological inflammation that may be a cause of a wide range of diseases, including gout, atherosclerosis, diabetes, rheumatoid arthritis and Alzheimer's disease [10]. Additionally, recent years of studies have revealed a tight association of NASH pathogenesis with aberrant activation of NLRP3 inflammasome [11]. Inhibition of NLRP3 inflammasome activity is shown to reduce liver inflammation and fibrosis in experimental NASH [12]. Thus, there has been a great interest to understand how the NLRP3 inflammasome is regulated in NASH.
Most facets of physiology and behaviors in mammals are governed by circadian rhythms (with a period of about 24-h). The immune functions are no exception as manifested by circadian rhythmicity in immune cell counts and cytokine levels as well as diurnal variations in the response to endotoxin and the circadian control of allergic reactions [13]. Notably, NLRP3 transcription and expression are oscillating, accounting for circadian rhythmicity in NLRP3 inflammasome pathway [14]. Circadian rhythms are driven and maintained by the circadian clock system with a hierarchical organization. The central clock (pacemaker) is located at the suprachiasmatic nucleus of the hypothalamus and peripheral clocks are present in the peripheral tissues. The central clock synchronizes the peripheral clocks via nervous and hormonal pathways [15]. On the other hand, local clocks in peripheral tissues such as liver and kidney can be entrained by external stimuli (e.g., food) independent of the central clock [16]. Strikingly, approximately 43% of all protein coding genes are clock-controlled genes [17]. Molecular components of circadian clock maintain their oscillations using a negative feedback mechanism (so-called “transcriptional-translational feedback loop”) [18]. In the main feedback loop, the positive components [BMAL1 (brain and muscle ARNT-like 1) and CLOCK (circadian locomotor output cycles kaput)] form a heterodimer to activate gene transcription of negative components [period (PER) 1,2 and cryptochrome (CRY) 1,2] via direct binding to the E-box cis-regulatory element (CACGTG) [19]. Following protein accumulation to a high level, PERs and CRYs in turn inhibit the activity of the positive components, thereby down-regulating their own expression levels. Once the proteins of negative components are degraded, their inhibitory actions no longer exist. Positive components can bind to target genes to start a new cycle of transcription and translation [18]. The nuclear heme receptor REV-REBα is another clock component that regulates circadian rhythms via inhibiting the transcription and expression of BMAL1 [18].
Retinoid X receptors [including RXRα (NR2B1), RXRβ (NR2B2) and RXRγ (NR2B3)] are essential members of the nuclear receptor (NR) superfamily. RXRs function as transcription factors and regulate gene transcription by forming homodimers or heterodimers with other NRs. Like many other NRs, RXRs are ligand-responsive and can be activated by endogenous 9-cis-retinoic acid and fatty acids as well as synthetic retinoids [20]. Of three isotypes, RXRα perhaps plays a more important role in biology and physiology because disruption of RXRα leads to embryonic lethality, while deficiency of RXRβ or RXRγ is less severe [20]. RXRs display a tissue-specific difference in expression. Notably, RXRα is predominantly expressed in the liver, intestine and kidney, as well as in myeloid cells [21]. RXRs have been implicated in regulation of a variety of biological processes and are attractive drug targets for cancer and metabolic diseases [22]. Intriguingly, NF-κB may exist constitutively with RXR (an NF-κB active complex); however, this complex becomes inactive in the presence of an RXR ligand [23].
A dominant portion of mammalian genome is transcribed as ncRNAs (non-coding RNAs). LncRNAs (long non-coding RNAs) are a class of ncRNAs that have over 200 nucleotides. They have emerged as major regulators of the transcriptional process by interacting physically with DNA, other RNA or proteins [24]. Many lncRNAs have been implicated in the development of liver diseases and in regulation of inflammation and immunity [25-28]. For instance, MALAT1 lncRNA may contribute to development of fibrosis in NAFLD through a mechanism involving the chemokine CXCL5 [29]. In addition, lncRNAs have potential impact on circadian biology, and in turn they may be under the control of circadian clock [30,31]. It is thus hypothesized that circadian NLRP3 inflammasome activity and steatohepatitis (an NLRP3-related disease) may be regulated by an oscillating lncRNA. Platr (pluripotency-associated transcript) family consists of 32 members (i.e., Platr1 to -32), which were identified in an RNA-seq screen of embryonic stem cells (ESCs) and implicated in maintenance of the ESC gene expression profile [32]. In MMTV-Neu-NDL breast cancer model, Platr4 was upregulated more than 4-fold and may be associated with tumor progression [33]. In this study, we uncover Platr4 as an oscillating lncRNA deregulated in mice with steatohepatitis. Oscillation of Platr4 is driven by the circadian clock component Rev-erbα. We found that Platr4 serves as a circadian repressor of Nlrp3 inflammasome activity by inhibiting transcription and expression of the inflammasome components Nlrp3 and Asc. Loss of Platr4 sensitizes mice to experimental steatohepatitis, whereas overexpression of Platr4 ameliorates the pathological conditions. Mechanistically, Platr4 prevents binding of the NF-κB/Rxrα complex to the κB sites via a physical interaction, thereby inhibiting the transactivation of Nlrp3 and Asc by NF-κB. We thus propose that Platr4 might be an attractive target for management of steatohepatitis.
Results
Identification of Platr4 as an oscillating and steatohepatitis-related lncRNA
Mice were fed a methionine-choline-deficient diet (MCD) to induce steatohepatitis. Disease model was confirmed by histopathological examinations (Figure 1A) and biochemical analyses (Figures S1-2). According to transcriptome analysis, Platr4 (Gencode ID: ENSMUSG00000097639.1) was up-regulated (a > 5-fold increase) in mice with MCD-induced steatohepatitis (Figure 1B). As expected, MCD-associated differentially expressed genes converged on inflammation- and nutrition metabolism-related pathways (Figure S3). Platr4 was also deregulated in the livers of Rev-erbα-/- mice (Figure 1C). Rev-erbα-/- mice were used to identify oscillating (or circadian clock-controlled) lncRNAs because Rev-erbα is a core clock component. Quantitative polymerase chain reaction (qPCR) assays confirmed that Platr4 was rhythmically expressed in the liver with a peak level at ZT14, and indeed up-regulated in MCD mice (Figure 1D). Coding potential analyses indicated Platr4 as a “real” noncoding transcript (i.e., does not code a protein) (Figure S4). Notably, Platr4 was predominantly expressed in the liver among various major tissues (Figure 1E). All three liver cells (i.e., Kupffer cells, hepatocytes and hepatic stellate cells) expressed Platr4 (Figure 1F). For Kupffer cells, the copy number of Platr4 was determined to be 40 (i.e., 40 copies per cell). Moderate expression of Platr4 was also seen in bone marrow-derived macrophages (BMDMs) (Figure 1E). In addition, like Neat1 (a known nuclear lncRNA), Platr4 was primarily localized in the nuclei of Kupffer cells and BMDMs (Figure 1G-H). Altogether, the oscillating lncRNA Platr4 is liver-specific and may have a regulatory role in development of steatohepatitis.
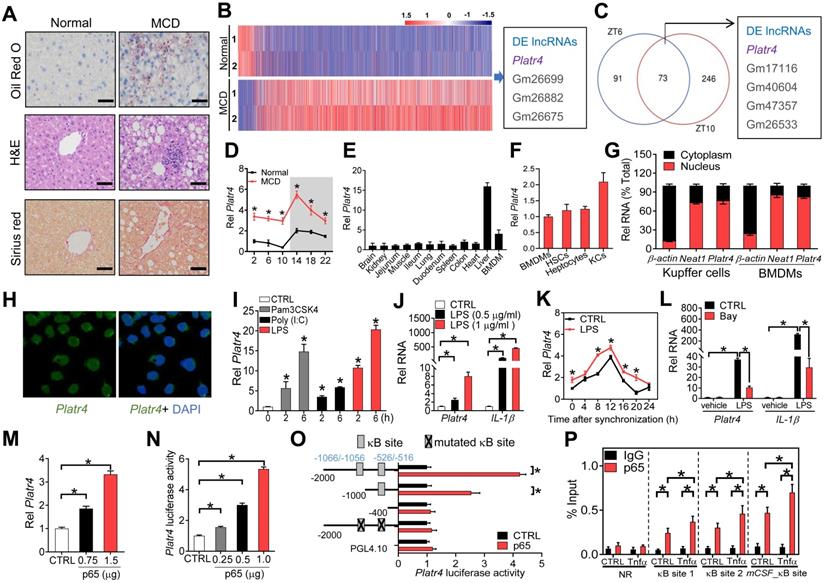
Platr4 is an oscillating and steatohepatitis-related lncRNA whose expression is driven by NF-κB. (A) Representative micrographs of Oil Red O, H&E and Sirius red staining of liver sections from normal mice and mice with MCD-induced steatohepatitis. Scale bar = 50 μm. (B) Heatmap diagram for differentially expressed (DE) genes in livers from normal mice and mice with MCD-induced steatohepatitis. (C) Venn diagram showing the numbers of differentially expressed lncRNAs (DE lncRNAs) in the livers of wild-type and Rev-erbα-/- mice at ZT6 and ZT10. Data are available upon request. (D) Diurnal hepatic Platr4 expression in normal mice and mice with MCD-induced steatohepatitis. Data are mean ± SD (n = 8). *p <0.05 versus normal mice at individual time points (two-way ANOVA and Bonferroni post hoc test). (E) Expression comparisons of Platr4 in the various tissues and BMDMs of wild-type mice. Data are mean ± SD (n = 3). (F) Expression comparisons of Platr4 in BMDMs, hepatic stellate cells (HSCs), hepatocytes and Kupffer cells (KCs) derived from wild-type mice. Data are mean ± SD (n = 3). (G) Relative cytoplasm and nuclear expression of Platr4 in Kupffer cells and BMDMs of wild-type mice. Data are mean ± SD (n = 3). (H) FISH analysis of subcellular Platr4 (green) location in BMDMs from wild-type mice. (I) Effects of Pam3CSK4 (a TLR1/2 agonist, 100 nM), Poly (I:C) (a TLR3 agonist, 25 µg/ml) and LPS (a TLR4 agonist, 500 ng/ml) on Platr4 expression in BMDMs. Data are mean ± SD (n = 3). *p <0.05 versus CTRL (one-way ANOVA and Bonferroni post hoc test). (J) Dose-dependent effects of LPS (0.5 and 1 μg/ml) on Platr4 and IL-1β mRNA in BMDMs. Data are mean ± SD (n = 3). *p <0.05 (one-way ANOVA and Bonferroni post hoc test). (K) Effects of LPS (500 ng/ml) on Platr4 expression in synchronized BMDMs. Data are mean ± SD (n = 3). *p <0.05 versus control at individual time points (two-way ANOVA and Bonferroni post hoc test). (L) Effects of Bay 11-7082 on Platr4 and IL-1β mRNA in unstimulated or LPS-stimulated BMDMs. Data are mean ± SD (n = 3). *p <0.05 (two-way ANOVA and Bonferroni post hoc test). (M) Dose-dependent effects of p65 plasmid (0.75 or 1 μg) on Platr4 expression in BMDMs. Data are mean ± SD (n = 3). *p <0.05 (one-way ANOVA and Bonferroni post hoc test). (N) Dose-dependent effects of p65 plasmid (0.25, 0.5 or 1 μg) on 2.1 kb Platr4 reporter activity in BMDMs. Data are mean ± SD (n = 6). *p <0.05 (one-way ANOVA and Bonferroni post hoc test). (O) Effects of p65 on the activities of different versions of Platr4-Luc reporters. Data are mean ± SD (n = 6). *p <0.05 (t-test). (P) Recruitment of p65 protein to the κB sites (-1066/-1056 bp and -526/-516 bp) of Platr4 promoter in unstimulated or Tnfα-stimulated BMDMs. Data are mean ± SD (n = 3). *p <0.05 (two-way ANOVA and Bonferroni post hoc test). NR, non-binding region.
NF-κB activation promotes Platr4 expression
NF-κB activation is implicated in development of steatohepatitis [34]. We wondered whether up-regulation of Platr4 is associated with NF-κB activation in experimental steatohepatitis. Expression of Platr4 in BMDMs was induced by Pam3CSK4 (an activator of NF-κB mediated by TLR1/2 receptors), Poly (I:C) (an activator of NF-κB mediated by TLR3) and lipopolysaccharide (LPS, an activator of NF-κB mediated by TLR4) in a time-dependent manner (Figure 1I). Also, induction of expression of Platr4 and IL-1β (a known NF-κB target gene) by LPS was dose-dependent (Figure 1J). Similar inductive effects of LPS on Platr4 were observed in synchronized BMDMs (Figure 1K). In contrast, Bay 11-7082 (a specific NF-κB inhibitor) inhibited LPS-stimulated expression of Platr4 and IL-1β (Figure 1L). These findings suggested a positive role of NF-κB in controlling Platr4 expression. Supporting this, overexpression of p65 (an NF-κB subunit) enhanced the expression of Platr4 in BMDMs (Figure 1M). In line with a positive regulation effect, p65 dose-dependently induced the promoter activity of a 2.1 kb Platr4-Luc reporter (Figure 1N). We further searched for κB sites to which NF-κB binds and thereafter activates target gene transcription. Promoter sequence analysis identified two κB sites (i.e., -1066/-1056 bp and -526/-516 bp) in Platr4 promoter. Truncation and mutation experiments indicated that the two κB sites were essential for NF-κB binding and activity (Figure 1O). Recruitment of p65 to these two κB sites was confirmed by ChIP (chromatin immunoprecipitation) assays (Figure 1P). Of note, p65 recruitment was enhanced by recombinant TNFα protein (Figure 1P). Altogether, NF-κB drives expression of Platr4 via a transcriptional activation mechanism, and up-regulation of Platr4 in experimental steatohepatitis may be driven by activated NF-κB.
Clock component Rev-erbα regulates rhythmic expression of Platr4 in normal mice
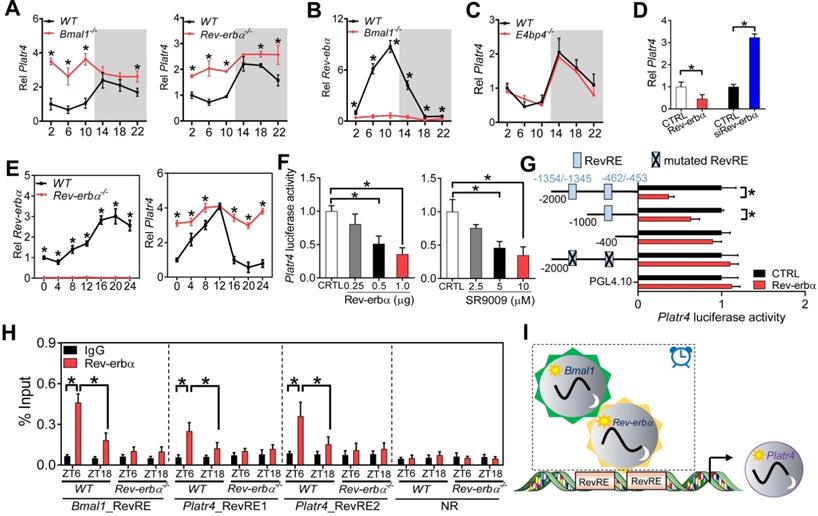
Circadian expression of most genes is driven by molecular components of circadian clock via transcriptional actions on their cis-elements (E-box, D-box and RevRE or RORE) [35,36]. We thus investigated the roles of these three cis-elements in rhythmic expression of Platr4 using Bmal1-/-, E4bp4-/- and Rev-erbα-/- mice. Bmal1, E4bp4 and Rev-erbα are representative clock components that bind to E-box, D-box and RevRE, respectively. Bmal1 functions as a transcriptional activator, while both E4bp4 and Rev-erbα (also known to be activated by Bmal1) are repressors [36]. Hepatic Platr4 was up-regulated and its rhythm was blunted (with a decrease in amplitude) in Bmal1-/- and in Rev-erbα-/- mice (Figure 2A). As expected, Rev-erbα (as a Bmal1 target gene) was markedly suppressed and its rhythm was lost in Bmal1-/- mice (Figure 2B). In contrast, hepatic Platr4 was unaffected in E4bp4-/- mice (Figure 2C). These data suggested that Rev-erbα directly represses circadian expression of Platr4, and that the repressive effect of Bmal1 on Platr4 is attained indirectly via its target Rev-erbα. Supporting a negative regulation mechanism, Rev-erbα overexpression in BMDMs led to reduced Platr4 expression, while Rev-erbα knockdown promoted the expression of Platr4 (Figure 2D). Consistently, Rev-erbα ablation up-regulated Platr4, and dampened its rhythm in synchronized BMDMs (Figure 2E). Furthermore, Rev-erbα dose-dependently inhibited the 2.1-kb Platr4 promoter activity (Figure 2F). Similar dose-dependent inhibitory effects were observed for SR9009 (a Rev-erbα agonist) (Figure 2F). Two RevRE elements (located at -1354/-1345 bp and -462/-453 bp) in Platr4 promoter were identified to be critical for Rev-erbα action based on promoter analysis, truncation and mutation experiments (Figure 2G). ChIP assays confirmed that hepatic Rev-erbα protein was recruited to the RevRE elements of Platr4 in wild-type mice in a circadian time-dependent manner (Figure 2H). Notably, Rev-erbα recruitment was more extensive at ZT6 than at ZT18 (Figure 2H). However, Rev-erbα recruitment was reduced and its time-dependency was abolished in Rev-erbα-/- mice (Figure 2H). Altogether, Rev-erbα periodically trans-represses Platr4, contributing to its diurnal rhythmicity in normal mice (Figure 2I).
Rev-erbα regulates rhythmic expression of Platr4 in normal mice. (A) Hepatic Platr4 expression in the Rev-erbα-/-, Bmal1-/- and wild-type (WT) mice at 6 circadian time points. Data are mean ± SD (n = 5). *p <0.05 versus WT at individual time points (two-way ANOVA and Bonferroni post hoc test). (B) Hepatic Rev-erbα expression in the Bmal1-/- and WT mice at 6 circadian time points. Data are mean ± SD (n = 5). *p <0.05 versus WT at individual time points (two-way ANOVA and Bonferroni post hoc test). (C) Platr4 expression in the livers of E4bp4-/- and WT mice at 6 circadian time points. Data are mean ± SD (n = 5). (D) Effects of Rev-erbα overexpression and knockdown on Platr4 expression in BMDMs. Data are mean ± SD (n = 3). *p <0.05 (t-test). (E) Temporal expression of Rev-erbα (left panel) and Platr4 (right panel) in synchronized BMDMs derived from Rev-erbα-/- and WT mice. Data are mean ± SD (n = 3). *p <0.05 versus WT at individual time points (two-way ANOVA and Bonferroni post hoc test). (F) Effects of Rev-erbα overexpression (left panel) and SR9009 (a Rev-erbα agonist, right panel) on the 2.1-kb Platr4 promoter activity in BMDMs. Data are mean ± SD (n = 6). *p <0.05 (one-way ANOVA and Bonferroni post hoc test). (G) Effects of Rev-erbα on the activities of different versions of Platr4-Luc reporters. Data are mean ± SD (n = 6). *p <0.05 (t-test). (H) Recruitment of Rev-erbα protein to the two RevRE sites (-1354/-1345 and -462/-453 bp) of Platr4 promoter in livers derived from WT and Rev-erbα-/- mice at ZT6 and ZT18. Data are mean ± SD (n = 3). *p <0.05 (two-way ANOVA and Bonferroni post hoc test). (I) Schematic diagram showing the potential mechanism for rhythmic expression of Platr4. NR, non-binding region.
Platr4 inhibits Nlrp3 inflammasome activation in macrophages
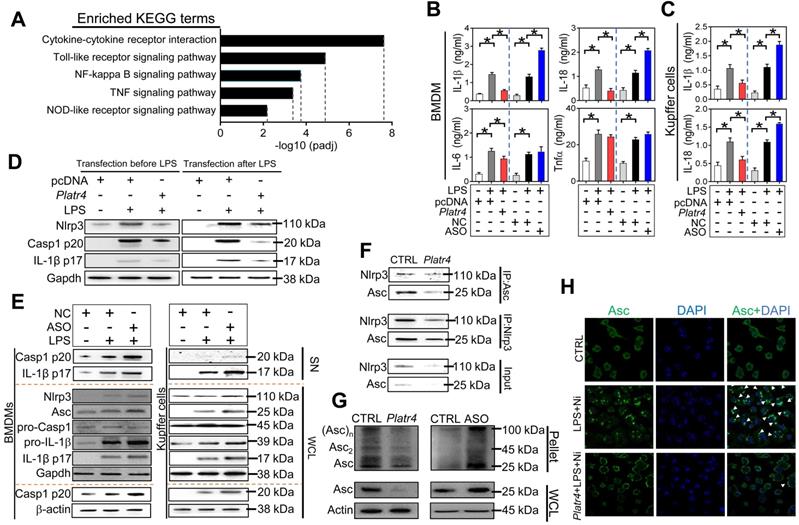
To explore the immune function of Platr4, Platr4-overexpressed and control BMDMs were subjected RNA-seq and KEGG pathway analyses. Differentially expressed genes were enriched in NF-κB activation-related pathways (i.e., NF-κB, Toll-like receptor, TNF, NOD-like receptor signaling pathways, and cytokine-cytokine receptor interaction), suggesting potential involvement of Platr4 in regulation of NF-κB activation and related immune responses (Figures 3A-S5). Overexpression of Platr4 decreased, whereas knockdown of Platr4 (by an antisense oligonucleotide, ASO) increased, the protein levels of the inflammatory cytokines IL-1β and IL-18 in LPS-stimulated BMDMs (Figure 3B). By contrast, Platr4 had a milder effect on IL-6 or Tnfα (Figure 3B). Negative regulation of both IL-1β and IL-18 by Platr4 was confirmed in LPS-stimulated Kupffer cells (hepatic macrophages) (Figure 3C). IL-1β and IL-18 production are mainly dependent on caspase-1 activity which is under the control of NLRP3 inflammasome [37,38]. We thus tested whether Platr4 can regulate NLRP3 inflammasome activation. Activation of NLRP3 inflammasome involves two critical processes, namely, a priming step to increase NLRP3 expression (via NF-κB activation) and an assembling step to form the inflammasome assembly [39]. Platr4 overexpression prior to LPS priming inhibited the generation of active caspase-1 (p20) and mature IL-1β (p17) in BMDMs (Figure 3D). Similar inhibitory effects of Platr4 on production of active caspase-1 and mature IL-1β were also observed in LPS-primed BMDMs (Figure 3D). This suggested potential actions of Platr4 on both priming and assembling stages of inflammasome activation. We went on to examine the effects of Platr4 on three components of Nlrp3 inflammasome (i.e., Nlrp3, Asc and pro-caspase-1). Platr4 knockdown did not affect the expression of pro-caspase-1, but induced the proteins of Nlrp3, pro-IL-1β and Asc accompanied by elevated cleaved caspase 1 and mature IL-1β in LPS-stimulated BMDMs and Kupffer cells (Figure 3E). After priming, NLRP3 and ASC are assembled via oligomerization to form a large protein aggregate (termed “speck”), a hallmark of inflammasome assembly and activation [40]. Overexpression of Platr4 led to a reduced protein interaction between Nlrp3 and Asc in BMDMs (Figure 3F). Further, Platr4 suppressed the formation of Asc oligomer, and Platr4 deficiency promoted Asc oligomerization (Figure 3G). Moreover, overexpression of Platr4 resulted in a marked reduction in the number of Asc specks upon LPS/nigericin stimulation (Figure 3H). Taken together, Platr4 functions as a negative regulator of Nlrp3 inflammasome activation probably through repressing the expression of Nlrp3 and Asc.
Platr4 inhibits Nlrp3 inflammasome activation in macrophages. (A) KEGG analysis of Platr4-induced differentially expressed genes in LPS-primed BMDMs. (B) Effects of Platr4 overexpression and knockdown on production of inflammatory cytokines (IL-1β, IL-18, IL-6 and Tnfα) in LPS-stimulated BMDMs. Data are mean ± SD (n = 3). *p <0.05 (one-way ANOVA and Bonferroni post hoc test). (C) Effects of Platr4 overexpression and knockdown on production of IL-1β and IL-18 in LPS-stimulated Kupffer cells. Data are mean ± SD (n = 3). *p <0.05 (one-way ANOVA and Bonferroni post hoc test). (D) Effects of Platr4 on protein levels of Nlrp3, active caspase 1 (p20) and mature IL-1β (p17) in LPS/nigericin-stimulated BMDMs. LPS was added before or after Platr4 transfection for 8 h, followed by nigericin addition for 30 min (added last). (E) Effects of Platr4 knockdown (by ASO) on Nlrp3 inflammasome-related proteins in LPS/nigericin-stimulated BMDMs (left panel) and Kupffer cells (right panel). (F) Effects of Platr4 overexpression on interactions between Nlrp3 and Asc proteins in LPS/nigericin-stimulated BMDMs. (G) Effects of Platr4 overexpression and knockdown on Asc oligomerization in LPS/nigericin-stimulated BMDMs. (H) Effects of Platr4 on formation of Asc specks in LPS/nigericin-stimulated BMDMs. The concentrations of LPS and nigericin for macrophage treatment were 500 ng/ml and 10 μM, respectively. SN, supernatant. WCL, whole cell lysate. NC, negative control. Ni, nigericin.
Loss of Platr4 blunts the oscillation of Nlrp3 inflammasome and sensitizes mice to experimental steatohepatitis
We next tried to determine whether Platr4 regulates Nlrp3 inflammasome in vivo. To this end, we established Platr4-deficient (Platr4-/-) mice by deleting the two exons of Platr4 gene (Figure S6). Platr4 knockout did not affect the expression of its neighbor gene Jade1 (Figure S7). Mouse liver samples were collected every 4-h over a 24-h light-dark cycle to assess the regulatory effects of Platr4 on Nlrp3 inflammasome considering that Nlrp3 inflammasome pathway displays a diurnal rhythm [14]. Hepatic Nlrp3 and Asc expression were up-regulated and their rhythms were blunted in Platr4-/- mice (Figure 4A). Likewise, IL-1β and IL-18 mRNAs were elevated and their oscillations were dampened (Figure 4A). Accordingly, Platr4 ablation led to increased levels of and altered rhythmicity of produced IL-1β and IL-18 in the liver (Figure 4B). These data indicated that Platr4 is essential to drive diurnal oscillations in Nlrp3 inflammasome expression and activity.
Loss of Platr4 blunts the oscillation of Nlrp3 inflammasome and sensitizes mice to experimental steatohepatitis. (A) Hepatic Nlrp3, Asc, IL-1β and IL-18 mRNA levels in wild-type (WT) and Platr4-/- mice at 6 circadian time points. Data are mean ± SD (n = 5). *p <0.05 versus WT at individual time points (two-way ANOVA and Bonferroni post hoc test). (B) IL-1β and IL-18 proteins in the livers derived from WT or Platr4-/- mice at 6 circadian time points. Data are mean ± SD (n = 5). *p <0.05 versus WT at individual time points (two-way ANOVA and Bonferroni post hoc test). (C) H&E staining of livers from WT and Platr4-/- mice fed on normal or MCD diet (top panel). Scale bar = 50 μm. NAS and inflammation scores are shown in the bottom panel. *p <0.05 (two-way ANOVA and Bonferroni post hoc test). (D) Plasma ALT/AST and hepatic MPO/MCP-1 activities in WT and Platr4-/-mice fed on normal or MCD diet. Data are mean ± SD (n = 8). *p <0.05 (two-way ANOVA and Bonferroni post hoc test). (E) Hepatic α-SMA, Col1a1 and Tgf-β1 mRNAs in WT and Platr4-/- mice fed on normal or MCD diet. Data are mean ± SD (n = 8). *p <0.05 (two-way ANOVA and Bonferroni post hoc test). (F) Hepatic Nlrp3, Asc, IL-1β and IL-18 mRNAs in WT and Platr4-/- mice fed on normal or MCD diet. Data are mean ± SD (n = 8). *p <0.05 (two-way ANOVA and Bonferroni post hoc test). (G) Caspase-1 activity in livers of WT and Platr4-/- mice fed on normal or MCD diet. Data are mean ± SD (n = 8). *p <0.05 (two-way ANOVA and Bonferroni post hoc test). (H) Hepatic IL-1β (left panel) and IL-18 (right panel) levels in livers of WT and Platr4-/- mice fed on normal or MCD diet. Data are mean ± SD (n = 8). *p <0.05 (two-way ANOVA and Bonferroni post hoc test).
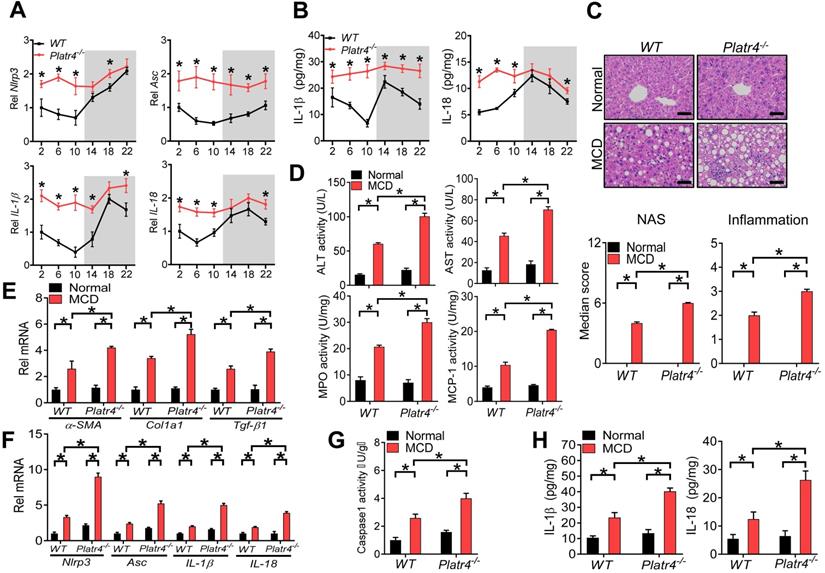
Nlrp3 inflammasome plays a central role in innate immune responses and in development of nonalcoholic steatohepatitis [41]. We thus wondered whether deficiency of Platr4 is associated with exacerbated steatohepatitis. Both Platr4-/- and wild-type mice were challenged with an MCD diet to induce steatohepatitis. Loss of Platr4 increased the sensitivity of mice to MCD-induced steatohepatitis as evidenced by a higher overall NAS (NAFLD activity score) value and more severe inflammation in Platr4-/- mice than in wild-type mice as derived from histopathological examinations (Figure 4C). Supporting this, MCD-challenged Platr4-/- mice showed higher levels of plasma ALT (alanine aminotransferase) and AST (aspartate aminotransferase) as well as higher activities of liver MPO (myeloperoxidase) and MCP-1 (chemoattractant protein-1) as compared with wild-type mice (Figure 4D). Also, hepatic levels of the pro-fibrotic markers α-SMA, Col1a1 and Tgf-β1 were higher in Platr4-/- mice with steatohepatitis than in wild-type controls (Figure 4E). More severe steatohepatitis in Platr4-/- mice was associated with higher hepatic levels of Nlrp3, Asc, IL-1β and IL-18 mRNAs, accompanied by an elevated caspase-1 activity and higher levels of IL-1β and IL-18 proteins (Figure 4F-H). Collectively, our data suggested a regulatory role of Platr4 in experimental steatohepatitis via controlling Nlrp3 inflammasome activity.
Platr4 regulates expression of Nlrp3 and Asc in an NF-κB-dependent manner
NF-κB is a known upstream transcriptional activator of Nlrp3 and a potential regulator of Asc [42,43]. We next investigated whether NF-κB plays a role in regulation of Nlrp3 and Asc by Platr4. Platr4 overexpression decreased, whereas Platr4 knockdown increased, the mRNA levels of both Nlrp3 and Asc in LPS-stimulated BMDMs and Kupffer cells (Figure 5A-B). The alterations in Nlrp3 and Asc mRNAs paralleled those in their proteins (Figure 5C), suggesting involvement of transcriptional repression in Platr4-mediated regulation. Bay 11-7082 (a specific inhibitor of NF-κB) attenuated the mRNA and protein expressions of both Nlrp3 and Asc in LPS-stimulated BMDMs (Figure 5D-E). Tnfα is an activator of NF-κB signaling pathway [44]. Overexpression of Platr4 inhibited, whereas knockdown of Platr4 increased, the expression levels of Nlrp3 and Asc in Tnfα-treated BMDMs (Figure 5F-G). Furthermore, overexpression of p65 (an NF-κB subunit) induced the expression of Nlrp3 and Asc (Figure 5H-I). However, the induction effects of p65 were diminished in the presence of Platr4 (Figure 5H-I). These findings supported NF-κB-dependent regulation of Nlrp3 and Asc by Platr4. Nlrp3 has been previously shown to be a direct target gene of NF-κB [45]. We confirmed that NF-κB p65 indeed activated Nlrp3 transcription (Figure 5J). We further tested whether NF-κB directly regulated Asc gene transcription. In luciferase reporter assay, p65 induced the transcriptional activity of Asc (Figure 5K). Promoter analysis identified two potential κB sites (NF-κB-binding sites, located at +52/+62 bp and +85/+95 bp, respectively) in Asc promoter. The activation effect of p65 was attenuated when single site was mutated, and was completely lost when both sites were mutated (Figure 5K). Supporting this, p65 protein was significantly recruited to the κB sites of Asc according to ChIP-seq analysis and ChIP assays (Figure 5L-M), and the recruitment was enhanced in the presence of Tnfα (Figure 5N). Therefore, NF-κB trans-activated Asc via direct binding to two κB sites in gene promoter (Figure 5O). Altogether, regulation of Nlrp3 and Asc expression by Platr4 was probably through inhibition of their transcriptional activator NF-κB.
Platr4 regulates expression of Nlrp3 and Asc in an NF-κB-dependent manner. (A) Effects of Platr4 overexpression (left panel) and knockdown (right panel) on Nlrp3 and Asc mRNAs in LPS-stimulated BMDMs. Data are mean ± SD (n = 3). *p <0.05 (one-way ANOVA and Bonferroni post hoc test). (B) Effects of Platr4 overexpression (left panel) and knockdown (right panel) on Nlrp3 and Asc mRNAs in LPS-stimulated Kupffer cells. Data are mean ± SD (n = 3). *p <0.05 (one-way ANOVA and Bonferroni post hoc test). (C) Effects of Platr4 overexpression on Nlrp3 and Asc proteins in LPS-stimulated BMDMs. (D) Effects of Bay 11-7082 (Bay, 5 μM) on Nlrp3 and Asc mRNAs in LPS-stimulated BMDMs. Data are mean ± SD (n = 3). *p <0.05 (one-way ANOVA and Bonferroni post hoc test). (E) Effects of Bay 11-7082 (5 μM) on Nlrp3 and Asc proteins in LPS-stimulated BMDMs. Data are mean ± SD (n = 3). (F) Effects of Platr4 overexpression (left panel) and knockdown (right panel) on Nlrp3 and Asc mRNAs in Tnfα-stimulated BMDMs. Data are mean ± SD (n = 3). *p <0.05 (t-test). (G) Effects of Platr4 overexpression on Nlrp3 and Asc proteins in Tnfα-stimulated BMDMs. (H) Effects of Platr4 on Nlrp3 and Asc mRNAs in p65-overexpressed BMDMs. Data are mean ± SD (n = 3). *p <0.05 (one-way ANOVA and Bonferroni post hoc test). (I) Effects of Platr4 on Nlrp3 and Asc proteins in p65-overexpressed BMDMs. (J) p65 activates Nlrp3 transcription in luciferase reporter assay with BMDMs. Data are mean ± SD (n = 6). *p <0.05 (t-test). (K) Effects of p65 on the activities of various versions of Asc-Luc reporters in BMDMs. Data are mean ± SD (n = 6). *p <0.05 (t-test). (L) p65 binding peaks at the Nlrp3, IL-1β, and Asc promoters derived from published ChIP-seq data (GSM2522473). (M) ChIP assays showing recruitment of p65 protein to the κB sites of Asc promoter in mouse liver. Data are mean ± SD (n = 3). *p <0.05 (t-test). NR, non-binding region. (N) Effects of Tnfα (20 ng/ml) on recruitment of p65 protein to Asc promoter. Data are mean ± SD (n = 3). *p <0.05 (two-way ANOVA and Bonferroni post hoc test). (O) Schematic diagram showing transcriptional regulation of Asc by NF-κB.
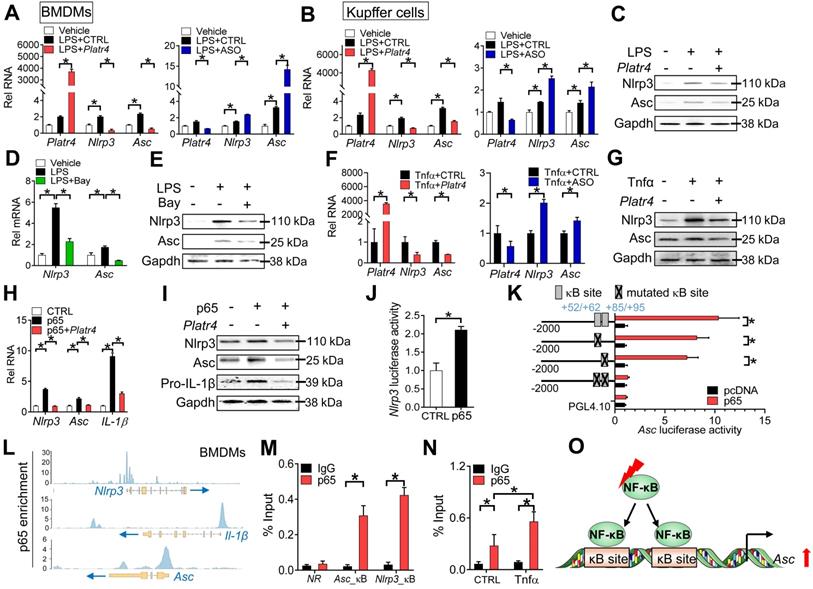
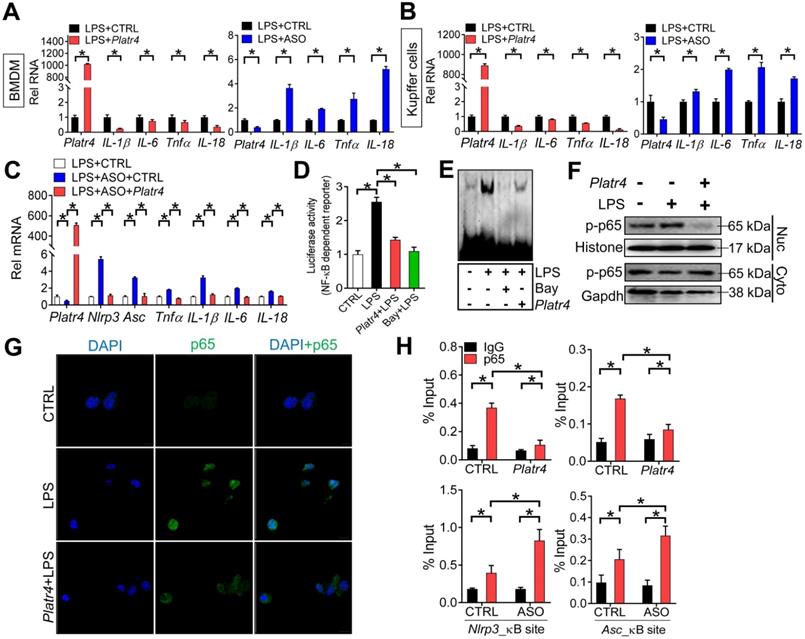
Platr4 inhibits NF-κB activity via interacting with Rxrα
We next tested whether Platr4 affects the activity of NF-κB. Overexpression of Platr4 decreased the mRNA levels of known NF-κB target genes (e.g., IL-1β, IL-6, Tnfα and IL-18) in LPS-stimulated BMDMs and Kupffer cells, while Platr4 knockdown increased the expressions of these genes (Figure 6A-B) [46]. Platr4 knockdown-induced changes in expressions of NF-κB target genes (i.e., IL-1β, IL-6, Tnfα, IL-18, Nlrp3 and Asc) can be restored by Platr4 overexpression, confirming a specific inhibitory effect of Platr4 on NF-κB activity (Figure 6C). Supporting this, Platr4 inhibited the transcriptional activity of NF-κB according to the luciferase reporter and EMSA assays, similar effects as observed for the specific NF-κB inhibitor Bay 11-7082 (Figure 6D-E). Phosphorylation of p65 is implicated in optimal NF-κB activation and in controlling NF-κB directed transactivation [47]. We found that Platr4 was able to decrease the levels of both phosphorylated p65 (p-p65) and p65 (measured by immunofluorescence analysis) in the nuclei (Figure 6F-G). Moreover, recruitment of p65 onto the promoters of NF-κB target genes (Nlrp3 and Asc) was inhibited by overexpression of Platr4, while Platr4 knockdown led to enhanced recruitment of p65 (Figure 6H) [48]. Overall, these findings indicated that Platr4 inhibits NF-κB activity via reducing NF-κB binding to target genes.
Platr4 inhibits NF-κB activity. (A) Effects of Platr4 overexpression and knockdown on NF-κB target genes (IL-1β, IL-6, Tnfα and IL-18) in LPS-stimulated BMDMs. Data are mean ± SD (n = 3). *p <0.05 (t-test). (B) Effects of Platr4 overexpression and knockdown on NF-κB target genes (IL-1β, IL-6, Tnfα and IL-18) in LPS-stimulated Kupffer cells. Data are mean ± SD (n = 3). *p <0.05 (t-test). (C) Rescue experiments showing that Platr4 knockdown-induced changes in expressions of NF-κB target genes can be restored by Platr4 overexpression. BMDMs were transfected with ASO or negative control. 48 h later, the medium was replaced with fresh medium and BMDMs were transfected with Platr4 or pcDNA blank plasmid for 24 h. Cells were collected for qPCR assays. Data are mean ± SD (n = 3). *p <0.05 (one-way ANOVA and Bonferroni post hoc test). (D) Effects of Platr4 and Bay (Bay 11-7082, 5 μM) on NF-κB-dependent reporter activity in LPS-stimulated BMDMs. Data are mean ± SD (n = 6). *p <0.05 (one-way ANOVA and Bonferroni post hoc test). (E) Effects of Platr4 and Bay (5 μM) on binding of NF-κB DNA probe to nuclear proteins derived from LPS-stimulated BMDMs. (F) Effects of Platr4 on the level of phosphorylated (p)-p65 in nuclear (Nuc) and cytoplasmic (Cyto) fractions of LPS-stimulated BMDMs. (G) Immunofluorescence images showing that Platr4 suppresses nuclear translocation of p65 in LPS-stimulated BMDMs. (H) ChIP assays showing that Platr4 overexpression reduces, whereas Platr4 knockdown enhances, the recruitment of p65 protein to κB sites in Nlrp3 and Asc promoters. Data are mean ± SD (n = 3). *p <0.05 (two-way ANOVA and Bonferroni post hoc test).
Nuclear lncRNAs may interact with target proteins to regulate their functions [49]. Platr4 may be one of such lncRNAs due to the presence of five stem loop structures (i.e., 1-153 nt, 234-606 nt, 694-815 nt, 856-1132 nt and 1501-1565 nt) (Figure 7A). RNA pull-down assays followed by mass spectrometric analysis and Western blotting identified retinoid X receptor α (Rxrα, ~50 kDa) as a nuclear protein interacting with Platr4 in mouse liver (Figure 7B and Table S1). A direct interaction of Platr4, but not Malat1 (another lncRNA mainly distributed in the nucleus), with Rxrα was further confirmed by a RIP (RNA immunoprecipitation) assay (Figure 7C). Moreover, Platr4 and Rxrα co-localized in the nuclei of BMDMs (Figure 7D). Truncation analyses indicated that the fragment of 1-153 nt was responsible for Rxrα binding (Figure 7B), that agreed with the highest predicted interaction score for this fragment (Figure 7E). Supporting this, Platr4 carrying a mutation of 1-153 nt fragment failed to inhibit the expressions of NF-κB target genes (i.e., Nlrp3, Asc, IL-1β and IL-18) in LPS-stimulated BMDMs (Figure 7F).
Platr4 interacts with Rxrα to modulate the activity of NF-κB. (A) Secondary structure for Platr4 with minimum free energy predicted by Vienna RNA web server. (B) Western blotting analyses of interactions of Rxrα protein with various versions of Platr4 probes following RNA-pull down assays. Full-length (FL), antisense (AS) and truncated fragments are labeled. (C) RIP assay showing an interaction between Rxrα protein and Platr4 in mouse liver. Data are mean ± SD (n = 3). *p <0.05 (t-test). (D) Immunofluorescence assays showing colocalization of Platr4 and Rxrα protein in BMDMs. (E) Prediction of interaction propensity between Rxra and full length Platr4 (catRAPID). Positive interaction score predicts increased propensity of binding. (F) Effects of a Platr4 mutant (1-153 nt fragment was mutated) on expression of NF-κB target genes (Nlrp3, Asc, IL-1β and IL-18) in LPS-stimulated BMDMs. Data are mean ± SD (n = 3). (G) Co-IP assays showing an interaction between Rxrα and p65 proteins. (H) Effects of Rxrα knockdown on the Platr4-mediated inhibition of NF-κB reporter activity in p65-overexpressed BMDMs. Data are mean ± SD (n = 6). *p <0.05 (one-way ANOVA and Bonferroni post hoc test). (I) Effects of Rxrα knockdown on Platr4-mediated inhibition of Nlrp3, Asc, IL-1β and IL-18 mRNAs in LPS-stimulated BMDMs. Data are mean ± SD (n = 3). *p <0.05 (one-way ANOVA and Bonferroni post hoc test). (J) Effects of Platr4 overexpression and knockdown on binding of NF-κB to its DNA probe in Rxrα- and p65- ovexpressed BMDMs. (K) ChIP assays showing that Rxrα protein is recruited to the κB sites of Nlrp3 and Asc promoters, and that the recruitment is inhibited by overexpression of Platr4 but enhanced when Platr4 is knocked-down. Data are mean ± SD (n = 3). *p <0.05 (two-way ANOVA and Bonferroni post hoc test).
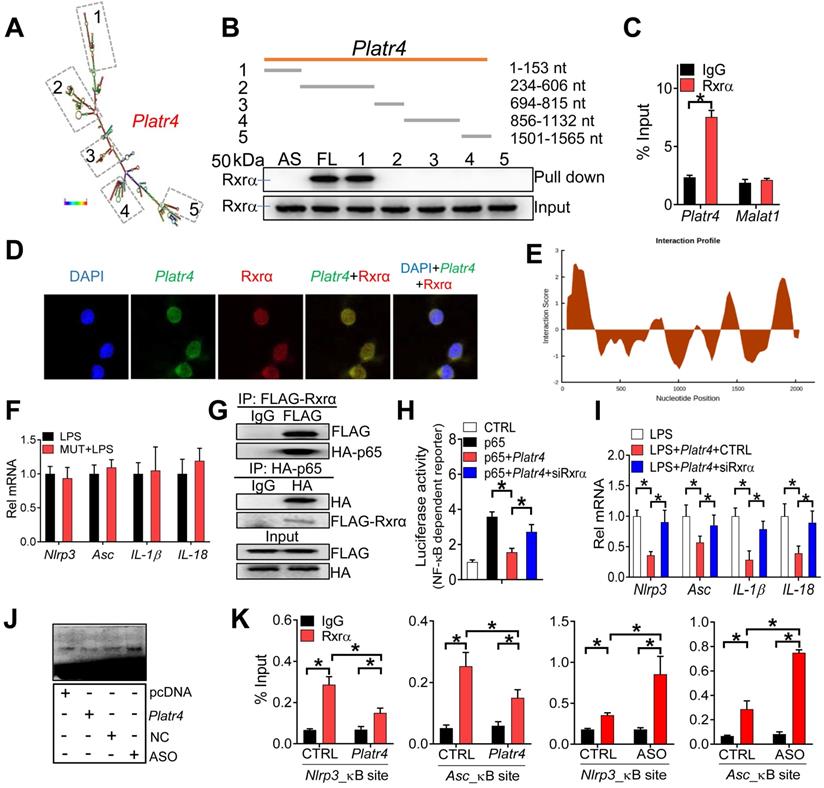
NF-κB constitutively binds to RXR receptor through the N-terminal ABC domains of RXR and the Rel homology domains of p50 and p65 [23]. The NF-κB/RXR complex is κB site-binding competent and transcriptionally active [23]. We confirmed a physical interaction between murine Rxrα and p65 based on Co-IP (co-immunoprecipitation) assays (Figure 7G). Knockdown of Rxrα decreased the expression levels of Nlrp3 and other NF-κB target genes in LPS-stimulated BMDMs (Figure S8). Also, Rxrα enhanced the promoter activity of an NF-κB reporter in BMDMs stimulated with Tnfα, and promoted the promoter activity of Nlrp3 reporter in the presence or absence of p65 (Figure S9). Moreover, Rxrα knockdown was associated with reduced recruitment of p65 to the κB sites of Nlrp3 and mCSF (a known NF-κB target gene) (Figure S10). All these supported a role of Rxrα in regulating NF-κB activity via a physical interaction.
Since Rxrα interacts with both Platr4 and NF-κB, we thus examined whether Platr4 regulates NF-κB activity through Rxrα. The effects of Platr4 on NF-κB-dependent reporter activity and NF-κB target genes (i.e., Nlrp3 and Asc) were attenuated when Rxrα was silenced (Figure 7H-I). Furthermore, according to the EMSA assays, overexpression of Platr4 inhibited, while Platr4 knockdown enhanced, the DNA-binding activity of NF-κB in the presence of Rxrα (Figure 7J). ChIP assays showed significant recruitment of both Rxrα and p65 to the κB sites of Nlrp3 and Asc promoters (Figure 6H-7K). Recruitment of these two proteins was inhibited by overexpression of Platr4, but enhanced when Platr4 was knocked-down (Figure 6H-7K). Additionally, we observed unaffected expression of Rxrα in Platr4 overexpressed or knocked-down cells (Figure S11). These findings indicated that Platr4 prevents binding of the NF-κB/Rxrα complex to κB sites, thereby inhibiting the transactivation activity of NF-κB. By referring to the main archetypes of lncRNA mechanisms [49], we postulated that Platr4 may act as a decoy and titrate away Rxrα and its partner NF-κB from the promoters of target genes such as Nlrp3 and Asc.
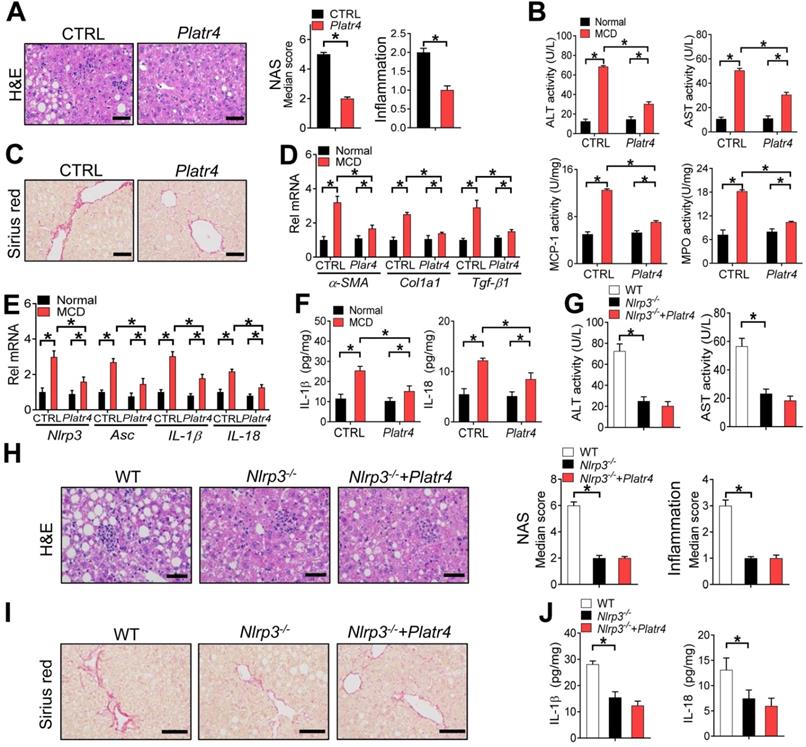
Overexpression of Platr4 ameliorates experimental steatohepatitis in an Nlrp3-dependent manner
The role of Platr4 in inhibition of Nlrp3 inflammasome prompted us to test whether targeted overexpression of Platr4 in the liver can prevent steatohepatitis. We thus generated a recombinant adeno-associated virus serotype 8 (AAV8) vector encoding Platr4 driven by a liver-specific thyroxine-binding globulin (TBG) promoter (named “AAV8.TBG.Platr4”). AAV8.TBG.Platr4 was able to increase hepatic expression of Platr4 by over 100-fold on day 15 after a single vector injection (Figure S12). According to histopathological examinations, overexpression of Platr4 protected mice from MCD-induced steatohepatitis because of lower NAS and inflammation scores in AAV8.TBG.Platr4-treated mice than in control mice (Figure 8A). Supporting this, MCD-challenged and AAV8.TBG.Platr4-treated mice showed lower levels of plasma ALT and AST as well as reduced activities of liver MPO and MCP-1 as compared with control mice (Figure 8B). Overexpression of Platr4 also retarded the development of liver fibrosis in steatohepatitis mice as revealed by the Sirius red staining (Figure 8C). This was in accordance with lower hepatic levels of the pro-fibrotic markers α-SMA, Col1a1 and Tgf-β1 in Platr4-overexpressed mice (Figure 8D). Intriguingly, alleviation of steatohepatitis in Platr4-overexpressed mice was associated with lower hepatic levels of Nlrp3, Asc, IL-1β and IL18 mRNAs, and with reduced production of IL-1β and IL18 proteins, suggesting a role of Nlrp3 inflammasome inactivation in Platr4 prevention of steatohepatitis (Figure 8E-F). To corroborate this finding, we determined the effects of Platr4 overexpression on steatohepatitis development in Nlrp3-deficient (Nlrp3-/-) mice. In line with previous reports, Nlrp3 ablation in mice reduced the severity of MCD-induced steatohepatitis [12]. The protective effect of Nlrp3 ablation on disease was similar to that elicited by Platr4 overexpression in wild-type mice (Figure 8G-J). However, Platr4 overexpression showed no effects on steatohepatitis development in Nlrp3-/- mice (Figure 8G-J). Altogether, our data indicated that targeted overexpression of Platr4 in the liver ameliorates experimental steatohepatitis in an Nlrp3-dependent manner.
Overexpression of Platr4 ameliorates experimental steatohepatitis in an Nlrp3-dependent manner. (A) H&E staining of livers from Platr4-overexpressed (AAV8.TBG.Platr4) and control mice fed on an MCD diet (left panel). Scale bar = 50 μm. NAS and inflammation scores are shown in the right panel. Data are mean ± SD (n = 8). * p <0.05 (t-test). (B) Plasma ALT/AST and hepatic MPO/MCP-1 activities in Platr4-overexpressed and control mice fed on normal or MCD diet. Data are mean ± SD (n = 8). *p <0.05 (two-way ANOVA and Bonferroni post hoc test). (C) Sirius red staining of livers from Platr4-overexpressed and control mice fed on a MCD diet. Scale bar = 50 μm. (D) Hepatic α-SMA, Col1a1 and Tgf-β1 mRNAs in Platr4-overexpressed and control mice fed on normal or MCD diet. Data are mean ± SD (n = 8). *p <0.05 (two-way ANOVA and Bonferroni post hoc test). (E) Hepatic Nlrp3, Asc, IL-1β and IL-18 mRNAs in Platr4-overexpressed and control mice fed on normal or MCD diet. Data are mean ± SD (n = 8). *p <0.05 (two-way ANOVA and Bonferroni post hoc test). (F) IL-1β and IL-18 in livers of Platr4-overexpressed and control mice fed on normal or MCD diet. Data are mean ± SD (n = 8). *p <0.05 (two-way ANOVA and Bonferroni post hoc test). (G) Plasma ALT and AST activities in Platr4-overexpressed Nlrp3-/- mice and control mice fed on a MCD diet. Data are mean ± SD (n = 8). *p <0.05 (one-way ANOVA with Bonferroni post hoc test). (H) H&E staining of livers from Platr4-overexpressed Nlrp3-/- mice and control mice fed on a MCD diet. Scale bar = 50 μm. NAS and inflammation scores are shown in the rightmost panel. Data are mean ± SD (n = 8). *p <0.05 (one-way ANOVA with Bonferroni post hoc test). (I) Sirius red staining of livers from Platr4-overexpressed Nlrp3-/- mice and control mice fed a MCD diet. Scale bar = 50 μm. (J) Hepatic IL-1β and IL-18 levels in Platr4-overexpressed Nlrp3-/- mice and control mice fed on an MCD diet. Data are mean ± SD (n = 8). *p <0.05 (one-way ANOVA with Bonferroni post hoc test).
Discussion
It is imperative to understand the pathogenesis of NALFD and NASH and to search for specific treatments due to a high incidence and prevalence. Given that the NLRP3 inflammasome plays a central role in chronic inflammatory diseases such as NASH and displays a circadian rhythmicity, it was hypothesized that NLRP3 inflammasome and steatohepatitis may be regulated by an oscillating lncRNA (i.e., rhythmically expressed). In an attempt to identify a potential oscillating lncRNA associated with steatohepatitis, we applied two screening criteria, namely, steatohepatitis-regulated and Rev-erbα (a core clock component)-controlled lncRNA (Figures 1-2). Platr4 with a liver-specific expression was screened as a cycling lncRNA that up-regulated the most in mice with MCD-induced steatohepatitis. This lncRNA was initially identified in an RNA-seq screen of embryonic stem cells but the exact functions remain underexplored [32]. We have further defined an anti-Nlrp3 inflammasome function of Platr4 and established a negative relationship between the abundance of Platr4 and the severity of experimental steatohepatitis. Therefore, Platr4 may be an attractive target for nonalcoholic steatohepatitis. However, there is a limitation for the translational potential because it is necessary to increase the expression of this lncRNA for its effect, which is not the common way the therapeutic approaches are currently addressing.
Our findings may suggest a self-protection mechanism against steatohepatitis, which involves auto-inhibition of NF-κB. Activated NF-κB pathway in steatohepatitis transcriptionally drives the expression of Platr4 that in turn inhibits NF-κB activity and inactivates Nlrp3 inflammasome by preventing binding of NF-κB to κB sites in the promoters of target genes including Nlrp3 and Asc. Mechanistically, Platr4 physically interacts with Rxrα protein and titrates it and its constitutive partner NF-κB p65 away from the κB binding site, thereby reducing the transcriptional activity of NF-κB and suppressing Nlrp3 inflammasome activation. This conforms to a general RNA-protein interaction mechanism for lncRNA actions [24]. We argue that Platr4 may bring the NF-κB/Rxrα complex away from the nucleus (probably directed to the cytoplasm) because of a reduction in nuclear accumulation of NF-κB in the presence of Platr4, although the mechanisms underlying this process remain to be determined (Figure 6). The proposed self-protection mechanism may support the notion that the body has certain ability to self-heal diseases [50]. However, the protective effect of Platr4 may be overwhelmed by MCD-induced inflammation insult. In other words, Platr4 (a several-fold increase) may be insufficient to reverse the massive and destructive inflammatory responses caused by MCD feeding, otherwise, MCD cannot induce steatohepatitis at all.
Circadian gene expression is mainly driven by the molecular components of circadian clock via direct transcriptional actions on their cis-elements (E-box, D-box and RevRE or RORE) [35,36]. Alternatively, an indirect mechanism has been proposed for generation of rhythmic gene expression by circadian clock [35]. This mechanism involves cycling transcriptional factors (TFs) (including nuclear receptors) as the intermediates. The rhythms of clock-output cycling TFs are propagated to their downstream target genes. Examples of such cycling TFs are aryl hydrocarbon receptor, peroxisome proliferator-activated receptor gamma (Ppar-γ), hepatocyte nuclear factor 4α (Hnf4α), and Hnf1α [35,51]. Our finding that Platr4 regulates the circadian rhythmicity in Nlrp3 inflammasome pathway herein leads to a proposal of an additional mechanism for circadian regulation of immune genes. That is rhythmic gene expression can be partially accounted for by clock-controlled oscillating lncRNAs. This type of lncRNAs periodically interact with target proteins (or other molecules), thereby generating rhythms in transcriptional activities and in target gene expression.
We have revealed a physical interaction between Platr4 and Rxrα in the liver, that leads to a reduction in the transcriptional activity of NF-κB as an Rxrα heterodimer partner. It is well-known that many NRs (e.g., constitutive androstane receptor, pregnane X receptor, farnesoid X receptor, vitamin D receptor, and PPARs) function as a heterodimer with RXR. This raises a possibility that Platr4 may modulate the transcriptional activities of these RXR-interacting NRs in a similar manner as it does to the NF-κB. The functions of Platr4 (as a NR modifier) thus may be extended to many other aspects of biology and physiology. It is noteworthy that the transcriptional activity (for NF-κB) of the NF-κB/RXR complex depends on the RXR ligands (e.g., retinoids) [23]. In the absence of retinoids, the complex is transcriptionally active and favorable as evidenced by the observations that (1) overexpression of Rxrα increases, whereas knockdown of Rxrα decreases, the expression levels of Nlrp3 and other NF-κB target genes (Figure S8); (2) Rxrα enhances the promoter activity of an NF-κB reporter in macrophages, and promotes the promoter activity of Nlrp3 reporter in the presence or absence of p65 (Figure S9); and (3) Rxrα knockdown was associated with reduced recruitment of p65 to the κB sites of Nlrp3 (Figure S10). However, the protein complex becomes inactive in the presence of a retinoid [23,52]. Na and coworkers propose that retinoid binding may induce conformational changes in RXR protein, leading to inhibition of the interactions between NF-κB and κB site [23]. Alternatively, co-activators (e.g., SRC-1 and CBP/p300) may constitutively bind NF-κB but recognizes RXR only in the presence of a retinoid [23].
We have demonstrated that Platr4 inactivates Nlrp3 inflammasome via inhibiting the transcription of both Nlrp3 and Asc in an NF-κB-dependent manner. It is a previously unreported finding that NF-κB trans-activates Asc through direct binding to two κB sites (i.e., +52/+62 bp and +85/+95 bp) in the gene promoter (Figure 5). Our finding sheds light on previously unexplained observations that ASC is up-regulated by inflammation in human neutrophils, and that ASC is up-regulated by TNFα in breast epithelial cells and the induction effect is attenuated when p65 is knocked-down [43,53]. Contrasting with the general notion that the purpose of inflammasome priming is to increase the expression levels of Nlrp3 and IL-1β via NF-κB activation [9], inflammasome activation may also require an increase in ASC expression occurring in the priming stage.
The lncRNA (Platr4) may represent a new layer of molecular mechanisms for the regulation of inflammation by circadian clock. Prior studies have indicated that regulation of inflammation is attained through the core components of circadian clock such as Clock, Rev-erbα and Cry1/2, accounting for diurnal rhythmicity in the severity (symptom) of inflammatory diseases (e.g., colitis and rheumatoid arthritis) and increased susceptibility to inflammation-related diseases (e.g., obesity and NAFLD) [54-59]. Platr4 appears to act as an integrator of circadian clock and inflammation through the NF-κB/Nlrp3 inflammasome axis. Defining this integrator role for Platr4 enhances our understanding of the crosstalk between circadian clock and inflammation, and highlights a complexity in circadian modifiers of inflammation. We argue for an essential role of potential alternative factors beyond the clock components in controlling the rhythmicity of Nlrp3 inflammasome pathway because loss of Rev-erbα fails to cause elevations over the entire light/dark cycle in Nlrp3 expression as observed for a typical target gene such as Bmal1 [14,54,60]. We propose that Platr4 may act as one of such “alternative factors” in the liver that has a significant contributing role in circadian behaviors of hepatic inflammation (Figure 4). Regulation by oscillating Platr4 (as an NF-κB repressor) may contribute to previously reported diurnal variations in transcriptional activity of NF-κB with a zenith at ZT6 that corresponds to the trough expression of Platr4 [57].
MCD diet model was selected herein to study the regulation mechanisms of Nlrp3 inflammasome and steatohepatitis as noted in prior studies [12,61]. The deficiency in choline and methionine, two essential nutrients, results in impaired β-oxidation and compromised production and secretion of very low-density lipoprotein particles, and therefore in hepatic fat accumulation, ballooning, inflammation and early development of fibrosis, which are typical characteristics of steatohepatitis [62]. The MCD model is considered adequate to study the intrahepatic events in relation to steatohepatitis and the therapeutic interventions [62]. However, this model may not reflect the metabolic changes (e.g., insulin resistance and obesity) associated with typical human steatohepatitis [62].
It is our future work to identify the equivalent lncRNA of Platr4 in humans. It may be not a practical approach using evolutional history to predict a function of lncRNA in different species because the current comparative genomic tools cannot easily detect homologs among lncRNAs from different species [63,64]. Therefore, it remains a challenge to identify a human “homolog” of Platr4, which has a similar structure or function to mouse Platr4. Considering this, we plan to use anti-RXRα antibody to reciprocally pull-down the lncRNAs that are associated with RXRα in livers from human samples. By using RNA-seq techniques, we would identify the lncRNAs that might modulate RXRα function and examine their functions in regulation of hepatic protein turnover.
In summary, the oscillating lncRNA Platr4 functions as a circadian repressor of Nlrp3 inflammasome by inhibiting NF-κB-dependent transcription of Nlrp3 and Asc. Mechanistically, Platr4 prevents binding of the NF-κB/Rxrα complex to κB sites via a physical interaction. Accordingly, loss of Platr4 sensitizes mice to experimental steatohepatitis, whereas overexpression of Platr4 ameliorates the pathological conditions. Therefore, Platr4 may be an attractive target that can be modulated to ameliorate the pathological conditions of steatohepatitis.
Methods
Materials
Murine macrophage colony-stimulating factor (M-CSF) was purchased from Peprotech (Rocky Hill, NJ). Pam3CSK4 was purchased from InvivoGen (San Diego, CA). Poly (I:C), nigericin, LPS and collagenase IV were purchased from Sigma-Aldrich (St. Louis, MO). Pronase E was purchased from Yuanye Bio-Technology Company (Shanghai, China). ChIP kit was purchased from Cell Signaling Technology (Beverly, MA). Bay 11-7082, murine Tnfα, 4',6-diamidino-2-phenylindole (DAPI), BCA assay kit, cytoplasmic/nuclear protein extraction kit and EMSA kit were purchased from Beyotime (Shanghai, China). Cytoplasmic/nuclear RNA purification kit was purchased from Norgen Biotek (Belmont, CA). InRcute lncRNA First-Strand cDNA Synthesis kit (with gDNase) and FastQuant RT kit (with gDNase) were purchased from Tiangen (Beijing, China). RNAiso Plus reagent and PrimeScript RT Master Mix were purchased from Vazyme (Nanjing, China). JetPrime transfection kit was purchased from POLYPLUS Transfection (Ill kirch, France). Anti-Nlrp3, anti-caspase-1 and anti-Asc antibodies were purchased from AdipoGen (San Diego, CA). Anti-IL-1β antibody was purchased from R&D systems (Minneapolis, MN). Anti-p65, anti-Rev-erbα, anti-p-p65 and anti-rabbit IgG antibodies were obtained from Cell Signaling Technology (Danvers, MA). Anti-Gapdh antibody was purchased from Abcam (Cambridge, UK). Anti-Rxrα, anti-FLAG and anti-HA antibodies were obtained from Proteintech Group (Chicago, IL). Asc luciferase reporters (-2000/+100 bp and κB site-mutated versions), Platr4 luciferase reporters (-2000/+100 bp, -1000/+100 bp, -400/+100 bp, and κB or RevRE site-mutated versions), Nlrp3 luciferase reporter (2.1 kb), pcDNA3.1, pcDNA3.1-p65, pcDNA3.1-Rev-erbα, pcDNA3.1-Platr4 (the Platr4-203 isoform, transcript ID: ENSMUST00000199155.1), siRev-erbα, siBmal1, siRxrα and ASO [targeting the sequenece (GCACUGAGCCAUCUUACUUG) in Platr4-203] were obtained from Transheep Technologies (Shanghai, China). Platr4 probes for RNA pull-down were purchased from GenePharma (Shanghai, China). FISH probe for Platr4 was obtained from BersinBio (Guangzhou, China). The sequences of siRNAs and ASOs are shown in Table S2.
Animals
Wild-type C57BL/6 mice were obtained from HFK Bio-Technology (Beijing, China). Rev-erbα-/-, Bmal1-/- and Nlrp3-/- mice (a C57BL/6 background) have been established and validated in our laboratory [54,65,66]. E4bp4-/- mice (a C57BL/6 background) was obtained from Dr. Masato Kubo at RIKEN Institute in Japan [67]. Platr4-/- mice (on a C57BL/6 background) were generated by deleting the DNA fragment (exons 1 and 2) of Platr4 gene and using the CRISPR/Cas9 technique with the aid of Cyagen Biosciences Inc (Guangzhou, China). All mice were maintained under a 12-h light/12-h dark cycle [lights on at 7 AM (= ZT0) and lights off at 7 PM (= ZT12)] and fed ad libitum. For characterization of diurnal gene expression, wild-type and gene knockout (Rev-erbα-/-, Bmal1-/-- and E4bp4-/-) mice (male, aged 8-10 weeks) were sacrificed at a 4-h interval around the clock (ZT2, ZT6, ZT10, ZT14, ZT18 and ZT22). Liver samples were collected and snap frozen in liquid nitrogen, and stored at -80 °C until use.
Steatohepatitis model
Wild-type and gene knockout (Platr4-/- and Nlrp3-/-) mice (male, aged 8-10 weeks) were fed a methionine-choline-deficient (MCD) diet (A02082002B, Research Diets) to induce steatohepatitis as previously described [68]. Control groups of mice were fed a normal diet. After 6 weeks, mice were sacrificed at ZT2, and blood and liver samples were collected for biochemical, expression and/or histological analyses. To evaluate the effects of Platr4 overexpression on steatohepatitis, wild-type and Nlrp3-/- mice were pretreated with AAV8.TBG.Platr4 (2.5×1011 virus particles per mouse, Transheep Technologies, Shanghai, China) or control vector via the tail vein injection at ZT10. On day 15, mice were fed an MCD diet to induce steatohepatitis. Six weeks later, mice were sacrificed, and blood and liver samples were collected for biochemical, expression and/or histological analyses.
Histological analysis
Formalin-fixed liver samples were embedded in paraffin and cut into 4-μm-thick sections. The liver sections were stained with hematoxylin and eosin (H&E). The NAFLD activity score (NAS) were derived based on the severity of hepatic steatosis (with a score of 0-3), lobular inflammation (with a score of 0-3), and hepatocellular ballooning (with a score of 0-2) [69]. For assessment of lipid accumulation, 10-μm paraffin sections of liver samples were stained with Oil Red O and counterstained with hematoxylin. Additionally, liver paraffin-sections (5 µm in thickness) were stained with Sirius Red to visualize the collagen content.
Biochemical analysis
Plasma ALT/AST and hepatic triglyceride levels were measured using enzymatic assay kits (Jiancheng Bioengineering Institute, Nanjing, China). Hepatic IL-1β, IL-6, Tnfα and IL-18 levels, and caspase 1/MCP-1/MPO activities were measured using enzyme linked immunosorbent assay kits (Neobioscience, Shenzhen, China).
Isolation of BMDMs
BMDMs were isolated from mice as previously described [54]. In brief, tibias and femurs were disinfected using 75% ethanol. Bone marrow cavities were rinsed with RPMI 1640 medium. The rinsing solution was collected and centrifuged at 1,000 rpm for 5 min. The pellet cells (BMDMs) were collected and cultured in RPMI 1640 medium containing 10% FBS, 1% penicillin-streptomycin and 20 ng/ml M-CSF. After 7 days, BMDMs were washed using sterile PBS and used for further experiments.
Isolation of Kupffer cells
Kupffer cells were isolated from mice as previously described [70]. In brief, livers were perfused with saline solution for 10 min followed by in vivo digestion with liberase enzyme for 5 min and in vitro digestion for 30 min. The nonhepatocyte content was subjected to Percoll gradient centrifugation. The intercushion fraction was washed and adhered to plastic in DMEM containing 5% FBS. The nonadherent fraction was washed and the adherent Kupffer cells were collected for further experiments.
Isolation and culture of primary mouse hepatocytes
Mouse hepatocytes were isolated using a collagenase perfusion method as previously reported [71]. In brief, livers from wild-type mice were perfused with Hanks' balanced salt solution (HBSS) buffer through the portal vein and digested with collagenase IV. After washing with HBSS, hepatocytes were seeded into collagen type I-coated plates and cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. 3 h later, the medium was replaced with serum-free DMEM. On the next day, cells were collected for qPCR assays.
Isolation of hepatic stellate cells
Hepatic stellate cells (HSCs) were isolated from wild-type mice as previously described [72]. In brief, mouse liver was perfused with pronase E and 0.1% collagenase IV (dissolved in phosphate buffer saline) through the inferior vena cava. The liver was then digested with 0.2% collagenase IV, and filtered through a 70-µm mesh. Cells were separated using a Nycodenz gradient centrifugation. HSCs were collected, seeded into polylysine-coated plates, and cultured in DMEM containing 10% FBS and 1% penicillin/streptomycin.
Synchronized cells
Synchronization (serum shock) experiments with cultured cells were performed as previously described [14]. BMDMs were cultured in RPMI medium containing 10% FBS and 1% penicillin-streptomycin. On the next day, culture medium was replaced with serum-free medium. 12 h later, 50% horse serum was added for 2 h and the medium was changed back to serum-free medium. Cells were harvested for RNA extraction at specific time points (0, 4, 8, 12, 16, 20 and 24 h). Notably, BMDMs were primed with LPS (500 ng/mL) for 3 h before cell harvest.
ASC oligomerization assay
Transfected and control BMDMs were lysed in AO buffer (containing 150 mM KCl, 20 mM HEPES-KOH, 1% Triton-X 100 and 1% protease inhibitor). The lysate was centrifugated at 5000 g for 15 min. The pellets (Triton-insoluble fraction) were collected in PBS, and cross-linked with 2 mM disuccinimidyl suberate for 30 min. After centrifugation (5000 g for 15 min), the pellets were subjected to Western blotting analysis.
FISH (fluorescence in situ hybridization)
Subcellular distribution of Platr4 was assessed using a FISH kit according to the manufacturer's instructions (GenePharma, Shanghai, China). In brief, BMDMs were fixed in 4% paraformaldehyde, and hybridized with 10 nM Platr4 probe in a hybridization buffer. Cells were washed with saline sodium citrate and counterstained with DAPI. Images were obtained using a laser scanning microscope (Carl Zeiss, Oberkochen, Germany). The forward and reverse sequences of FISH probe were 5'-CTGTGACTTCTCCAGGGCAG and 5'-GGACAATCTCACGTGCTCCA, respectively.
RIP (RNA immunoprecipitation)
RIP assay was performed using a Magna RIP kit (Millipore, Bedford, MA). In brief, livers were homogenized and lysed in lysis buffer containing an RNase inhibitor and a protease inhibitor cocktail. After centrifugation (14000 rpm for 10 min), the supernatant was incubated with magnetic beads bound with anti-Rxrα or normal rabbit IgG antibody in immunoprecipitation buffer overnight at 4 °C. Beads were then washed with RIP washing buffer containing proteinase K. The immunoprecipitates were subjected to RNA extraction and qPCR assays.
RNA pull-down
Nuclear proteins were prepared from mouse liver using a nuclear protein extraction kit according to the manufacturer's protocol (Beyotime, Shanghai, China). Full-length Platr4 and five truncated versions of Platr4 were cloned into T7 promoter-based vector and transcribed using TranscriptAid T7 High Yield Transcription kit (Thermo Scientific, Madison, WI), followed by RNA purification. Purified RNA was biotinylated using a Pierce Magnetic RNA-Protein Pull-down kit (Thermo Scientific, Madison, WI). Biotinylated RNA was incubated with streptavidin magnetic beads at room temperature. After 30 min, beads were incubated with 5 mg nuclear proteins at 4 °C for 1 h. RNA binding proteins were eluted from the beads with elution buffer. An aliquot of pull-down products were subjected to mass spectrometric analysis, while the remaining were analyzed by Western blotting.
Immunofluorescence analysis
Immunofluorescence analysis was performed as described in our previous publication [54]. Cells were fixed in 4% paraformaldehyde, permeated in 0.1% Triton X-100, and blocked with 5% BSA. Then, cells were sequentially incubated with primary antibody and Alexa Fluor 488-conjugated or Alexa Fluor 594-conjugated anti-rabbit secondary antibody, followed by DAPI staining. Images were obtained using a laser scanning microscope (Carl Zeiss, Oberkochen, Germany).
RNA-seq (RNA-sequencing)
Three normal mice and three steatohepatitis mice were sacrificed at ZT2, and the livers were isolated. Total RNA was extracted using RNAiso Plus reagent. Two RNA pools (i.e., experimental samples) for normal and steatohepatitis groups were assembled by mixing equal amounts of RNA from three animals. The RNA pools were subjected to library construction and RNA-seq as described in our previous publication [54]. In the second set of experiments, livers were collected from three wild-type and three Rev-erbα-/- mice at each of circadian time points (ZT6 and ZT10). RNA samples were pooled from three animals, and subjected to RNA-seq analysis. Additionally, RNA-seq was performed with BMDM samples (Platr4-transfected and LPS-stimulated BMDMs versus control BMDMs). Transcriptomics data analysis was performed as described [54].
qPCR, Western blotting, luciferase reporter assay, EMSA, ChIP and Co-IP
Standard methods were used for qPCR, Western blotting, luciferase reporter assay, EMSA, ChIP and Co-IP, and experimental procedures have been described in our previous publications [54,65,73]. The sequences for oligonucleotides (primers) used in qPCR and ChIP are provided in Tables S3-4. The amplified sequences in qPCR analysis of Platr4 are specifically located within the Platr4-203 (an amplified product from 1211 to 1288 bp in this isoform, Platr4 primers shown in Table S3). Copy number quantification was performed using in vitro-synthesized Platr4 for the creation of a standard curve.
Statistical analysis
Data are presented as mean ± SD. Statistical significance was determined using Student's t test or ANOVA (one-way or two-way with Bonferroni post hoc test). The level of significance was set at p <0.05 (*).
Abbreviations
AAV8: adeno-associated virus serotype 8; ALT: alanine aminotransferase; Asc: apoptosis-associated speck-like protein containing a CARD; α-SMA: α-smooth muscle actin; ASO: antisense oligonucleotide; AST: aspartate transaminase; ATP: adenosine triphosphate; BMAL1: brain and muscle ARNT-like 1; BMDMs: bone marrow derived macrophages; Col1a1: collagen, type I, alpha 1; ChIP: chromatin immunoprecipitation; CLOCK: circadian locomotor output cycles kaput; Co-IP: co-immunoprecipitation; CRY: cryptochrome; DAPI: 4',6-diamidino-2-phenylindole; Dbp: D-box binding protein; E4bp4: E4 promoter-binding protein 4; EMSA: electrophoretic mobility shift assay; FISH: fluorescence in situ hybridization; HSCs: hepatic stellate cells; LncRNA: long non-coding RNAs; LPS: lipopolysaccharide; MCD: methionine-choline-deficient; MCP-1: monocyte chemoattractant protein-1; MPO: myeloperoxidase; NAFLD: nonalcoholic fatty liver disease; NASH: nonalcoholic steatohepatitis; NAS: NAFLD activity score; NF-κB: nuclear factor kappa-B; NLRP3: NOD- LRR- and pyrin domain-containing protein 3; IL: interleukin; PER: period; qPCR: quantitative polymerase chain reaction; RevRE: REV-ERB response element; RIP: RNA immunoprecipitation; RNA-seq: RNA sequencing; Rxrα: retinoid X receptor alpha; TBG: liver-specific thyroxine-binding globulin; Tgf-β1: transforming growth factor beta 1; Tnf: tumor necrosis factor; ZT: zeitgeber time.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81722049), Natural Science Foundation of Guangdong Province (No. 2017A03031387), Guangzhou Science and Technology Project (No. 201904010472), and China Postdoctoral Science Foundation (No. 2019TQ0120).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Milić S, Stimac D. Nonalcoholic fatty liver disease/steatohepatitis: epidemiology, pathogenesis, clinical presentation and treatment. Dig Dis. 2012;30:158-62
2. Neuschwander-Tetri BA, Caldwell SH. Nonalcoholic steatohepatitis: summary of an AASLD single topic conference. Hepatology. 2003;37:1202-19
3. Rafiq N, Bai C, Fang Y, Srishord M, McCullough A, Gramlich T. et al. Long-term follow-up of patients with nonalcoholic fatty liver. Clin Gastroenterol Hepatol. 2009;7:234-8
4. Younossi ZM. Nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: Implications for liver transplantation. Liver Transpl. 2018;24:166-170
5. Wong RJ, Cheung R, Ahmed A. Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carcinoma in the U.S. Hepatology. 2014;59:2188-95
6. Day CP, James OF. Steatohepatitis: a tale of two "hits"? Gastroenterology. 1998;114:842-5
7. Takaki A, Kawai D, Yamamoto K. Molecular mechanisms and new treatment strategies for non-alcoholic steatohepatitis (NASH). Int J Mol Sci. 2014;15:7352-79
8. Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochem Biophys Res Commun. 2009;388:621-5
9. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19:477-489
10. Menu P, Vince JE. The NLRP3 inflammasome in health and disease: the good, the bad and the ugly. Clin Exp Immunol. 2011;166:1-15
11. Wan X, Xu C, Yu C, Li Y. Role of NLRP3 Inflammasome in the progression of NAFLD to NASH. Can J Gastroenterol Hepatol. 2016;2016:6489012
12. Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM. et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol. 2017;66:1037-46
13. Cermakian N, Lange T, Golombek D, Sarkar D, Nakao A, Shibata S. et al. Crosstalk between the circadian clock circuitry and the immune system. Chronobiol Int. 2013;30:870-88
14. Pourcet B, Zecchin M, Ferri L, Beauchamp J, Sitaula S, Billon C. et al. Nuclear receptor subfamily 1 group D member 1 regulates circadian activity of NLRP3 inflammasome to reduce the severity of fulminant hepatitis in mice. Gastroenterology. 2018;154:1449-1464.e20
15. Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature. 2002;418:935-41
16. Tahara Y, Shibata S. Entrainment of the mouse circadian clock: Effects of stress, exercise, and nutrition. Free Radic Biol Med. 2018;119:129-138
17. Zhang R, Lahens NF, Ballance HI, Hughes ME, Hogenesch JB. A circadian gene expression atlas in mammals: implications for biology and medicine. Proc Natl Acad Sci U S A. 2014;111:16219-24
18. King DP, Takahashi JS. Molecular genetics of circadian rhythms in mammals. Annu Rev Neurosci. 2000;23:713-42
19. Shearman LP, Sriram S, Weaver DR, Maywood ES, Chaves I, Zheng B. et al. Interacting molecular loops in the mammalian circadian clock. Science. 2000;288:1013-9
20. Szanto A, Narkar V, Shen Q, Uray IP, Davies PJ, Nagy L. Retinoid X receptors: X-ploring their (patho)physiological functions. Cell Death Differ. 2004;11:S126-43
21. Núñez V, Alameda D, Rico D, Mota R, Gonzalo P, Cedenilla M. et al. Retinoid X receptor alpha controls innate inflammatory responses through the up-regulation of chemokine expression. Proc Natl Acad Sci U S A. 2010;107:10626-31
22. Altucci L, Leibowitz MD, Ogilvie KM, de Lera AR, Gronemeyer H. RAR and RXR modulation in cancer and metabolic disease. Nat Rev Drug Discov. 2007;6:793-810
23. Na SY, Kang BY, Chung SW, Han SJ, Ma X, Trinchieri G. et al. Retinoids inhibit interleukin-12 production in macrophages through physical associations of retinoid X receptor and NFkappaB. J Biol Chem. 1999;274:7674-80
24. Long Y, Wang X, Youmans DT, Cech TR. How do lncRNAs regulate transcription? Sci Adv. 2017;3:eaao2110
25. Chen J, Ao L, Yang J. Long non-coding RNAs in diseases related to inflammation and immunity. Ann Transl Med. 2019;7:494
26. Quagliata L, Terracciano LM. Liver Diseases and long non-Coding RNAs: new insight and perspective. Front Med (Lausanne). 2014;1:35
27. Sookoian S, Rohr C, Salatino A, Dopazo H, Fernandez Gianotti T, Castaño GO. et al. Genetic variation in long noncoding RNAs and the risk of nonalcoholic fatty liver disease. Oncotarget. 2017;8(14):22917-26
28. Sookoian S, Flichman D, Garaycoechea ME, San Martino J, Castaño GO, Pirola CJ. Metastasis-associated lung adenocarcinoma transcript 1 as a common molecular driver in the pathogenesis of nonalcoholic steatohepatitis and chronic immune-mediated liver damage. Hepatol Commun. 2018;2(6):654-65
29. Leti F, Legendre C, Still CD, Chu X, Petrick A, Gerhard GS. et al. Altered expression of MALAT1 lncRNA in nonalcoholic steatohepatitis fibrosis regulates CXCL5 in hepatic stellate cells. Transl Res. 2017;190:25-39.e21
30. Coon SL, Munson PJ, Cherukuri PF, Sugden D, Rath MF, Møller M. et al. Circadian changes in long noncoding RNAs in the pineal gland. Proc Natl Acad Sci U S A. 2012;109:13319-24
31. Fan Z, Zhao M, Joshi PD, Li P, Zhang Y, Guo W. et al. A class of circadian long non-coding RNAs mark enhancers modulating long-range circadian gene regulation. Nucleic Acids Res. 2017;45:5720-38
32. Bergmann JH, Li J, Eckersley-Maslin MA, Rigo F, Freier SM, Spector DL. Regulation of the ESC transcriptome by nuclear long noncoding RNAs. Genome Res. 2015;25:1336-46
33. Diermeier SD, Chang KC, Freier SM, Song J, El Demerdash O, Krasnitz A. et al. Mammary tumor-associated RNAs impact tumor cell proliferation, invasion, and migration. Cell Rep. 2016;17:261-74
34. Dela Peña A, Leclercq I, Field J, George J, Jones B, Farrell G. NF-kappaB activation, rather than TNF, mediates hepatic inflammation in a murine dietary model of steatohepatitis. Gastroenterology. 2005;129:1663-74
35. Lu D, Zhao M, Chen M, Wu B. Circadian clock-controlled drug metabolism: implications for chronotherapeutics. Drug Metab Dispos. 2020;120:090472
36. Takahashi JS. Transcriptional architecture of the mammalian circadian clock. Nat Rev Genet. 2017;18:164-79
37. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013-22
38. Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821-32
39. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677-87
40. Broderick L, De Nardo D, Franklin BS, Hoffman HM, Latz E. The inflammasomes and autoinflammatory syndromes. Annu Rev Pathol. 2015;10:395-424
41. Arrese M, Cabrera D, Kalergis AM, Feldstein AE. Innate immunity and inflammation in NAFLD/NASH. Dig Dis Sci. 2016;61:1294-303
42. Boaru SG, Borkham-Kamphorst E, Van de Leur E, Lehnen E, Liedtke C, Weiskirchen R. NLRP3 inflammasome expression is driven by NF-κB in cultured hepatocytes. Biochem Biophys Res Commun. 2015;458:700-6
43. Parsons MJ, Vertino PM. Dual role of TMS1/ASC in death receptor signaling. Oncogene. 2006;25:6948-58
44. Dhingra S, Sharma AK, Arora RC, Slezak J, Singal PK. IL-10 attenuates TNF-alpha-induced NF kappaB pathway activation and cardiomyocyte apoptosis. CardiovAsc Res. 2009;82:59-66
45. Qiao Y, Wang P, Qi J, Zhang L, Gao C. TLR-induced NF-κB activation regulates NLRP3 expression in murine macrophages. FEBS Lett. 2012;586:1022-6
46. Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017;2:pii 17023
47. Viatour Merville MP, Bours V Chariot A. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005;30:43-52
48. Brach MA, Henschler R, Mertelsmann RH, Herrmann F. Regulation of M-CSF expression by M-CSF: role of protein kinase C and transcription factor NF kappa B. Pathobiology. 1991;59:284-8
49. Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43:904-14
50. Goudie DR, D'Alessandro M, Merriman B, Lee H, Szeverényi I, Avery S. et al. Multiple self-healing squamous epithelioma is caused by a disease-specific spectrum of mutations in TGFBR1. Nat Genet. 2011;43:365-9
51. Tanimura N, Kusunose N, Matsunaga N, Koyanagi S, Ohdo S. Aryl hydrocarbon receptor-mediated Cyp1a1 expression is modulated in a CLOCK-dependent circadian manner. Toxicology. 2011;290:203-7
52. Lee HC, Headley MB, Iseki M, Ikuta K, Ziegler SF. Cutting edge: inhibition of NF-kappaB-mediated TSLP expression by retinoid X receptor. J Immunol. 2008;181:5189-93
53. Shiohara M, Taniguchi S, Masumoto J, Yasui K, Koike K, Komiyama A. et al. ASC, which is composed of a PYD and a CARD, is up-regulated by inflammation and apoptosis in human neutrophils. Biochem Biophys Res Commun. 2002;293:1314-8
54. Wang S, Lin Y, Yuan X, Li F, Guo L, Wu B. REV-ERBα integrates colon clock with experimental colitis through regulation of NF-κB/NLRP3 axis. Nat Commun. 2018;9:42-46
55. Hand LE, Hopwood TW, Dickson SH, Walker AL, Loudon AS, Ray DW. et al. The circadian clock regulates inflammatory arthritis. FASEB J. 2016;30:3759-70
56. Zhou Z, Lin Y, Gao L, Yang Z, Wang S, Wu B. Circadian pharmacological effects of berberine on chronic colitis in mice: role of the clock component Rev-erbα. Biochem Pharmacol. 2020;172:113773
57. Spengler ML, Kuropatwinski KK, Comas M, Gasparian AV, Fedtsova N, Gleiberman AS. et al. Core circadian protein CLOCK is a positive regulator of NF-κB-mediated transcription. Proc Natl Acad Sci U S A. 2012;109:E2457-65
58. Sookoian S, Castaño G, Gemma C, Gianotti TF, Pirola CJ. Common genetic variations in CLOCK transcription factor are associated with nonalcoholic fatty liver disease. World J Gastroenterol. 2007;13(31):4242-8
59. Sookoian S, Gemma C, Gianotti TF, Burgueño A, Castaño G, Pirola CJ. Genetic variants of Clock transcription factor are associated with individual susceptibility to obesity. Am J Clin Nutr. 2008;87(6):1606-15
60. Cho H, Zhao X, Hatori M, Yu RT, Barish GD, Lam MT. et al. Regulation of circadian behaviour and metabolism by REV-ERB-α and REV-ERB-β. Nature. 2012;485:123-7
61. Tang Y, Cao G, Min X, Wang T, Sun S, Du X. et al. Cathepsin B inhibition ameliorates the non-alcoholic steatohepatitis through suppressing caspase-1 activation. J Physiol Biochem. 2018;74:503-10
62. Van Herck MA, Vonghia L, Francque SM. Animal Models of Nonalcoholic Fatty Liver Disease-A Starter's Guide. Nutrients. 2017;9:pii E1072
63. Hezroni H, Koppstein D, Schwartz MG, Avrutin A, Bartel DP, Ulitsky I. Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Rep. 2015;11(7):1110-22
64. Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154(1):26-46
65. Zhang T, Chen M, Guo L, Yu F, Zhou C, Xu H. et al. Reverse Erythroblastosis Virus α Antagonism promotes homocysteine catabolism and ammonia clearance. Hepatology. 2019;70:1770-84
66. Lin Y, Wang S, Zhou Z, Guo L, Yu F, Wu B. Bmal1 regulates circadian expression of cytochrome P450 3a11 and drug metabolism in mice. Commun Biol. 2019;2:378
67. Motomura Y, Kitamura H, Hijikata A, Matsunaga Y, Matsumoto K, Inoue H. et al. The transcription factor E4BP4 regulates the production of IL-10 and IL-13 in CD4(+) T cells. Nat Immunol. 2011;12:450-9
68. Ye J, Lv L, Wu W, Li Y, Shi D, Fang D. et al. Butyrate protects mice against methionine-choline-deficient diet-induced non-alcoholic steatohepatitis by improving gut barrier function, attenuating inflammation and reducing endotoxin levels. Front Microbiol. 2018;9:1967
69. Fan JG, Jia JD, Li YM, Wang BY, Lu LG, Shi JP. et al. Chinese association for the study of liver disease. guidelines for the diagnosis and management of nonalcoholic fatty liver disease: update 2010: (published in Chinese on Chinese Journal of Hepatology 2010; 18:163-166). J Dig Dis. 2011;12:38-44
70. Hritz I, Mandrekar P, Velayudham A, Catalano D, Dolganiuc A, Kodys K. et al. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology. 2008;48:1224-31
71. Zhang T, Yu F, Guo L, Chen M, Yuan X, Wu B. Small Heterodimer Partner regulates circadian Cytochromes p450 and drug-Induced Hepatotoxicity. Theranostics. 2018;8(19):5246-58
72. Inzaugarat ME, Johnson CD, Holtmann TM, McGeough MD, Trautwein C, Papouchado BG. et al. NLR Family Pyrin Domain-Containing 3 inflammasome activation in hepatic stellate cells induces liver fibrosis in Mice. Hepatology. 2019;69(2):845-59
73. Wang S, Yuan X, Lu D, Guo L, Wu B. Farnesoid X receptor regulates SULT1E1 expression through inhibition of PGC1α binding to HNF4α. Biochem Pharmacol. 2017;145:202-9
Author contact
![]() Corresponding authors: Baojian Wu, Ph.D. E-mail: bj.wucom; College of Pharmacy, Jinan University; or Huijie Xing, E-mail: huijie920326com; Institution of Laboratory Animal, Jinan University.
Corresponding authors: Baojian Wu, Ph.D. E-mail: bj.wucom; College of Pharmacy, Jinan University; or Huijie Xing, E-mail: huijie920326com; Institution of Laboratory Animal, Jinan University.