Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2021; 11(2):540-554. doi:10.7150/thno.49517 This issue Cite
Research Paper
Boiling histotripsy and in-situ CD40 stimulation improve the checkpoint blockade therapy of poorly immunogenic tumors
Mohit Pratap Singh1, Sri Nandhini Sethuraman1, Craig Miller2, Jerry Malayer1, Ashish Ranjan1 ![]()
1. Department of Physiological Sciences, College of Veterinary Medicine, Oklahoma State University, Stillwater, OK 74078.
2. Department of Pathobiology, College of Veterinary Medicine, Oklahoma State University, Stillwater, OK 74078.
Received 2020-6-15; Accepted 2020-9-17; Published 2021-1-1
Abstract

Background: Advanced stage cancers with a suppressive tumor microenvironment (TME) are often refractory to immune checkpoint inhibitor (ICI) therapy. Recent studies have shown that focused ultrasound (FUS) TME-modulation can synergize ICI therapy, but enhancing survival outcomes in poorly immunogenic tumors remains challenging. Here, we investigated the role of focused ultrasound based boiling histotripsy (HT) and in-situ anti-CD40 agonist antibody (αCD40) combinatorial therapy in enhancing therapeutic efficacy against ICI refractory murine melanoma.
Methods: Unilateral and bilateral large (~330-400 mm3) poorly immunogenic B16F10 melanoma tumors were established in the flank regions of mice. Tumors were exposed to single local HT followed by an in-situ administration of αCD40 (HT+ αCD40: HT40). Inflammatory signatures post treatment were assessed using pan-cancer immune profiling and flow cytometry. The ability of HT40 ± ICI to enhance local and systemic effects was determined by immunological characterization of the harvested tissues, and by tumor growth delay of local and distant untreated tumors 4-6 weeks post treatment.
Results: Immune profiling revealed that HT40 upregulated a variety of inflammatory markers in the tumors. Immunologically, HT40 treated tumors showed an increased population of granzyme B+ expressing functional CD8+ T cells (~4-fold) as well as an increased M1 to M2 macrophage ratio (~2-3-fold) and CD8+ T: regulatory T cell ratio (~5-fold) compared to the untreated control. Systemically, the proliferation rates of the melanoma-specific memory T cell population were significantly enhanced by HT40 treatment. Finally, the combination of HT40 and ICI therapy (anti-CTLA-4 and anti-PD-L1) caused superior inhibition of distant untreated tumors, and prolonged survival rates compared to the control.
Conclusions: Data suggest that HT40 reprograms immunologically cold tumors and sensitizes them to ICI therapy. This approach may be clinically useful for treating advanced stage melanoma cancers.
Keywords: Boiling histotripsy, αCD40, Checkpoint blockade, Cold tumors, Anti-tumor immunity
Introduction
Immune checkpoint inhibitors (ICIs) targeting CTLA-4, PD-1, and PD-L1 proteins have revolutionized the treatment of melanoma and other tumor types in patients [1-4]. Although promising, the immunosuppressive tumor microenvironment (TME) can influence ICI outcomes in a large proportion of treated patients [5-10]. This occurs due to masking of tumor antigens and proliferation of suppressive immune cells (e.g., regulatory T cells and M2 macrophages), which directly influence the functions of cytotoxic T cells [11-15]. Thus, there is a critical need to develop novel means for efficient activation of innate and adaptive immunity in the TME for superior ICI outcomes [16-20]. Herein, we evaluated the role of anti-CD40 agonistic antibody (αCD40) combined with focused ultrasound (FUS)-induced boiling histotripsy (HT) in TME activation and ICI therapy of melanoma tumors.
Focused ultrasound (FUS) is a non-invasive treatment modality that utilizes sonic energy to treat at an unlimited depth from the body surface. We and others have shown that FUS thermal therapy has an immunomodulatory effect in melanoma tumors [21-23]. However, tumor-heating approaches require accurate temperature feedback, and off-target effects can damage vital structures (e.g. nerves, blood vessels) adjacent to treated tissues. Recently, mechanical FUS was also shown to cause immune-modulations [24]. In particular mechanical tissue homogenization using FUS based boiling histotripsy was found to be particularly efficient in enhancement of tumor inflammation [24-27], and anti-tumor immune effects [28, 29]. HT can be attained by both cavitation or boiling means [30-33]. However, boiling HT achieves instantaneous, predictable and controllable homogenization and heating of focal regions to 100 ºC by using millisecond-long pulses with lower pulse repetition frequency (PRF) [34]. Boiling HT occurs for milliseconds, so it induces little or no damage to nearby structures due to minimal heat diffusion, while also achieving rapid accumulation of tumor-associated and neo-antigens in targeted tumor regions, enhancing immune cell infiltration by chemotaxis [35, 36]. The activation of infiltrated antigen-presenting cells (APCs) and their subsequent migration to lymphoid tissues improve tumor antigen presentation to naïve T cells, thus causing antigen-specific tumor destruction [24, 37, 38].
Although the feasibility of HT in murine models has increasingly been reported [39], its ability to reprogram advanced stage poorly immunogenic tumors (e.g., B16F10) that lack major histocompatibility complex (MHC) and co-stimulatory molecules is not known. In general, “immunologically cold” tumors such as B16F10 exhibit minimal APC functions, failure to accumulate cytotoxic infiltrating lymphocytes, dominant expression of PDL1 on tumor cells, and poor response to ICIs in advanced stages, thereby evading antitumor immunity [40, 41]. To overcome this barrier, recently we combined FUS-induced tumor heating (40-45 ºC) with in-situ αCD40 to achieve remissions of treated and untreated melanomas in vivo in a B16F10 mice melanoma model [21]. We found that this combined treatment administered 3-4 times over 2 weeks increased the population and quality of T-cells (rich in Granzyme B and poor in PD-1 expression), and generated sufficient systemic antitumor immunity to significantly reduce growth of untreated contralateral tumors [21]. αCD40 attaches to the CD40 receptor on APCs, enhancing CD40 signaling as well as expression of CD80, IL-12, and CCR7. These cause efficient APC activity and T cell-based cytotoxic responses [42-45]. Since HT exerts its immunogenic actions by creating an in-situ depot of tumor antigens/debris and immunogenic cell death, with high spatial precision inside the tumor, and without significantly heating neighboring untreated tumor tissues, the objective of this study was to understand whether the local application of single HT and αCD40 (HT40) attains remission of advance stage melanoma tumors, addressing the current limitations of hyperthermia-based immunotherapy approaches. To investigate our hypothesis, we established late stage ICI refractory B16F10 melanoma and determine the mechanisms involved in APC infiltration and T cell homing with HT40 in treated and untreated remote tumors. Our data suggested that HT40 sensitized poorly immunogenic B16F10 melanoma to ICIs and improved the survival outcomes in melanoma bearing mice.
Materials
B16F10 murine melanoma cells were provided by Dr. Mary Jo Turk at the Geisel School of Medicine at Dartmouth College (Hanover, NH, USA). They were cultured in DMEM supplemented with 10% fetal bovine serum and 1% streptomycin/penicillin. αCD40 (FGK45), anti-PDL-1 antibody (10F.9G2), and anti-CTLA-4 antibody (9H10) were purchased from BioXCell (West Lebanon, NH, USA). Fluorochrome-conjugated monoclonal antibodies (mAbs) purchased from BioLegend (San Diego, CA, USA) and BD Biosciences (San Jose, CA, USA) for flow cytometry were as follows: FITC, APC-Cy7 or PE-Cy7 anti-CD45.2 (104 and 30-F11), APC-Cy7 anti-CD11c (1A8), APC or BV786 anti-CD4 (GK1.5 and RM4-5), PE, PERCP, or BV510 anti-CD3 (145-2C11), BB515 anti-MHCII (2G9), PE anti-Granzyme B (QA16A02), APC anti-CD206 (C068C2), AF700 anti-IFN-γ (XMG1.2), BB700 anti-CD11b (M1/70), PE-Cy7 anti-IL-2 (JES6-5H4), APC anti-CD44 (IM7), AF488 anti-CD62L (MEL-14), BV711 anti-F4/80 (T45-2342), PE-Cy7 anti-CD8a (53-6.7), and Alexa Fluor 488 anti-Foxp3 (MF23). Quick-RNA Miniprep Kits were purchased from Zymo Research (Tustin, CA, USA). The nCounter PanCancer Immune Profiling Panel was purchased from NanoString Technologies, Inc. (Seattle, WA).
Methods
Mouse melanoma study design and ICI treatments
All the animal related procedures were approved by the Oklahoma State University Animal Care and Use Committee. For tumor inoculation, B16F10 cells at 80- 90% confluency were harvested, washed, and diluted with sterile cold PBS. Male C57/BL-6 mice (n=5/group, 6-8 weeks old), were subcutaneously implanted with 0.5 × 106 cells (50 μL) in the right flank for flow cytometry and gene expression assessment. To measure the therapeutic effects (abscopal effect and survival rates), mice (n=5) were injected subcutaneously in the right flank on day 0 with 0.5 × 106 cells and in the left flank on day 4 with 0.125 × 106 cells [21, 46]. Tumor volume of mice was measured every day using a serial caliper (General Tools Fraction™, New York, NY, USA); volumes were calculated using the formula (length × width2)/2, where length was the largest dimension and width was the smallest dimension perpendicular to the length. Treatments were initiated once the mice tumor volumes reached 330-400 mm3. For all studies, boiling HT treatment of tumors covered 40-50% of the tumor volume. αCD40 at a dose of 50 µg was injected by intratumoral injection within 2 h of HT. For pan-cancer immune profiling and flow cytometry, we compared the following groups: 1) Untreated Control, 2) HT, 3) αCD40, and 4) HT40. Mice tumors (n=3-5) and spleens (n=3-5) from surviving mice were harvested 1wk post treatment. For flow cytometry, harvested tissues were processed on the same day. For gene expression analysis, tumor tissues were snap-frozen in liquid nitrogen and stored at -80 °C until further use. For therapeutic assessments, we compared the following groups: 1) Untreated Control, 2) HT, 3) αCD40, and 4) HT40, each with and without the combination of anti-CTLA-4 and anti-PDL-1. Anti-CTLA-4 (100 µg/dose) and anti-PD-1(200 µg/dose) were injected intraperitoneally following HT, αCD40, or HT40 treatment, and two subsequent ICI dose were given every third day. Mice were sacrificed for survival studies when the tumors reached ~2 cm in any dimension.
Boiling HT set-up and tumor exposures
We utilized the Alpinion FUS transducer with a 1.5 MHz central frequency, 45 mm radius, and 64 mm aperture diameter with a central opening of 40 mm in diameter for HT exposures. For ultrasound exposure, the tumor was aligned at a fixed focal depth to cover voxel size of 1 x 1 x 10 mm. VIFU-2000 software was used to define the target boundary and slice distance in x, y, and z directions for automatic rastering of the transducer during treatment. The focal points were rastered to cover 40-50% of the tumor. HT was performed by using boiling histotripsy regimen (1 Hz PRF, 1% duty cycle, 450 W acoustic power) and were adapted from prior publications that used a similar device [39, 47]. Each focal spot was treated for 10 s. Mice were administered buprenorphine (0.05 mg/kg; SC) daily for 3 days post HT treatment.
Histopathological analysis of treated and untreated tumors
HT was confirmed by histopathology. HT exposed tumor tissues (n=3) were fixed in 10% neutral buffered formalin, processed, and embedded in paraffin [48]. Histopathological examinations of 4 μm sections stained with hematoxylin and eosin (H&E) were performed by a veterinary pathologist. To examine the tumor immune environments, the tumor sections from mice bearing bilateral tumors were stained for CD3 antibody for immunohistochemical analysis. Briefly, 10% neutral buffered formalin fixed treated and untreated (abscopal) tumors were embedded in paraffin and cut into 4 μm sections. Antigen retrieval was performed in a decloaking chamber. Normal goat serum was used for blocking. Incubation with the anti-rat CD3 primary antibody (Abcam, Cambridge, MA) was performed at 4°C overnight. Biotinylated goat anti-rat (Vector Laboratories, Burlingame, CA) was used as the secondary antibody and detected with ImmPACT DAB HRP Substrate (Vector Laboratories, Burlingame, CA).
Pan-cancer immune profiling of tumors
Total RNA extracted from snap-frozen tumors (n = 3/group) using the Quick-RNA Miniprep Kit (Zymo Research) was profiled using the nCounter® PanCancer Immune Profiling Panel (NanoString Technologies, Inc., Seattle, WA, USA). This panel contains 770 genes involved in the cancer immune response. Gene expression profiling was performed using the following steps: (i) Hybridization: 25 ng of total RNA were hybridized with the mouse PanCancer immune profiling code set having 770 unique pairs of 35-50 base pair biotin-labeled capture probes and reporter probes with internal reference controls. Hybridization was performed overnight at 65 °C. (ii) Washing: Excess probes were removed with magnetic bead purification on the nCounter® Prep Station (software v4.0.11.2). Unbound probes were washed away, the tripartite structure was bound to the streptavidin-coated cartridge by the biotin capture probe, aligned by an electric current (negative to positive), and immobilized. Degradation of fluorophore and photobleaching were prevented by adding SlowFade. Read counts from the raw data output were assessed for differential gene expression and cell type scoring after normalization using NanoString nSolver (version 3.0) [49]. Briefly, Log2 counts were represented as z-scores in heat map to indicate alterations in gene expression and immune cell profile for each sample. Additionally, the relative differences in gene signatures between treated and control tumors were represented as volcano plots (log2 fold change vs log10 P-value).
Immune profiling of melanoma tumors by flow cytometry
Tumors were mechanically disrupted and digested with 200 U/mL collagenase IV (Life Technologies, NY, USA) followed by filtration through a 70 μm cell strainer (Corning Inc., Corning, NY, USA) to obtain a single cell suspension. Fixable Viability Stain 575V (BD Biosciences) was used to stain cell suspensions to exclude dead cells from analysis as per the manufacturer's instructions. To block FcγIII/II receptor-mediated unspecific binding, anti-CD16/CD32 antibody was used. Cells were stained with indicated anti-mouse fluorochrome-conjugated antibody combinations for 30 min on ice in the dark using the following panel: CD45+ (tumor infiltrating leukocytes; TILs), CD11b+, F4/80+ (macrophages), CD11b+, F4/80+, MHCIIhi (M1 macrophages), CD11b+, F4/80+ MHCII lo/neg, CD206+ (M2 macrophages), CD11b+ CD11c+, F4/80-, MHCII+ (dendritic cells), CD3+, CD4+ (CD4+ T or helper Th cells), CD3+, CD4+, CD44hi CD62lo (CD4+ T effector/memory cells), and CD3+, CD8+ (CD8+ T cells). To detect IFNγ, IL-2, Granzyme-B, and Foxp3 positive T cells, cells (without stimulation) were washed after surface marker staining, fixed and permeabilized with a transcription factor buffer set (BD Biosciences), and incubated with Pe-Cy7 anti-IL-2, BV650 or APC-Cy7 anti- IFNγ, PE anti-Granzyme-B, or Alexa Fluor 488 anti-Foxp3 antibody for 30 min in the dark on ice [23, 46, 50]. Stained cells were run in an LSRII flow cytometer (BD Biosciences) within 24 h. Compensations were performed with single-stained UltraComp eBeads or cells. FlowJo software v.10.2 (Treestar Inc., Ashland, OR, USA) was used for data analysis. For all channels, positive and negative cells were gated based on a fluorescence minus one control.
Evaluation of the melanoma-specific systemic T cell response
Single cell suspension of splenocytes were stimulated ex-vivo with the melanoma-specific differentiation antigen tyrosinase-related protein 2 (TRP-2) peptide for 8 h to determine generation of TRP-2 melanoma antigen specific immunity in mice [51, 52]. Briefly, 1-2 x 106 splenocytes were incubated at 37 °C and 5% CO2 with 2.5 µg of TRP-2 peptide for 8 h in the presence of Brefeldin A (eBioscience, San Diego, CA; 1000X solution). Treated cells were washed with PBS and stained with CD45, CD3, CD4, CD8, IFNγ and IL-2 antibodies for flow cytometry analysis. The number of T effector cells responding to TRP-2 stimulation was calculated as CD45+ CD3+ CD4+ or CD8+ T cells that were positive for IFNγ or IL-2, and results were expressed as percentage of total splenocytes.
Tumor regression and survival rate evaluations in murine melanoma
Tumor regression in the treated and untreated sites were determined by computing the difference in the tumor volumes for the various groups relative to untreated control. For survival studies, tumor bearing mice were followed for 40 days post inoculation, and the median survival for each treatment group was assessed by the Kaplan-Meier survival curve.
Statistical analyses
Statistical analyses were performed using GraphPad Prism 8.4.2 software (GraphPad Software Inc, La Jolla, CA, USA). The differences between the treatments compared to the untreated control were analyzed by multiple t-tests without multiple comparisons correction. The nanostring data were represented as mean of log2 fold change relative to control. All other data were presented as mean ± standard error of the mean (SEM) unless otherwise indicated. For analysis of three or more groups, one-way analysis of variance was performed followed by Tukey's multiple comparison tests. The overall P value for Kaplan-Meier analysis was calculated using the log-rank test. Analysis of differences between two normally distributed test groups was performed using an unpaired t-test assuming unequal variance and multiple t-tests. P < 0.05 was considered to be statistically significant.
Results
Local HT achieved precise homogenizations of the treated regions
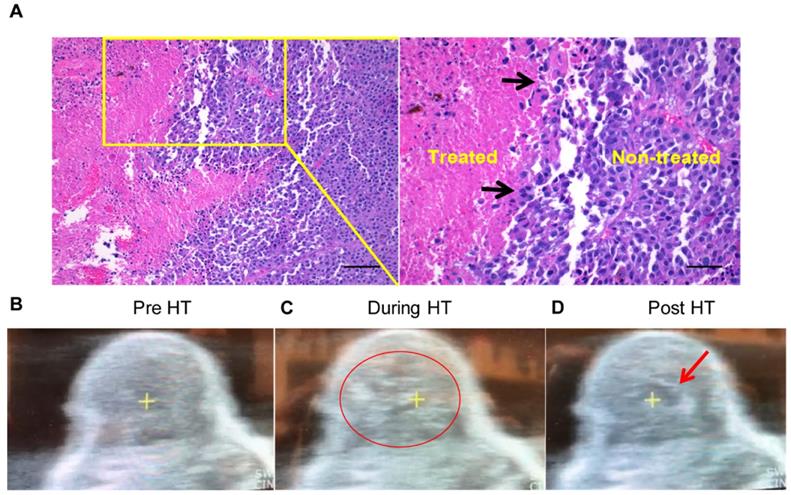
H&E showed that HT created a core of homogenized tumor tissue covering 40-50% of the total volume and this was surrounded by intact tumor tissue (Figure 2A). There was a clear transition zone between the HT-treated and non-treated tumor regions such that viable tumor tissue was negligible in the area treated with HT. These were also verified by real-time US imaging during HT treatment in those regions, whereby hyperechoic regions during each pulse at the focal point followed by hypoechoic contrast at the end of the pulse was noted (Figure 2B-D).
Local HT achieved precise melanoma homogenizations. (A) The H&E stained tumor sections showing sharp transition zone (black arrows) between histotripsy treated and untreated tumor region (n=3). Scale bar: 200μm (left image) and 100μm (right image). (B-D) HT therapy induced homogenizations of treated tumor regions (B) Pre-treatment image of a mouse tumor. (C) HT treatment produced hyperechoic regions during each pulse (indicated by the red circle). (D) Hypoechoic contrast at the end of the pulse was visible adjacent to the focal point (indicated by red arrow).
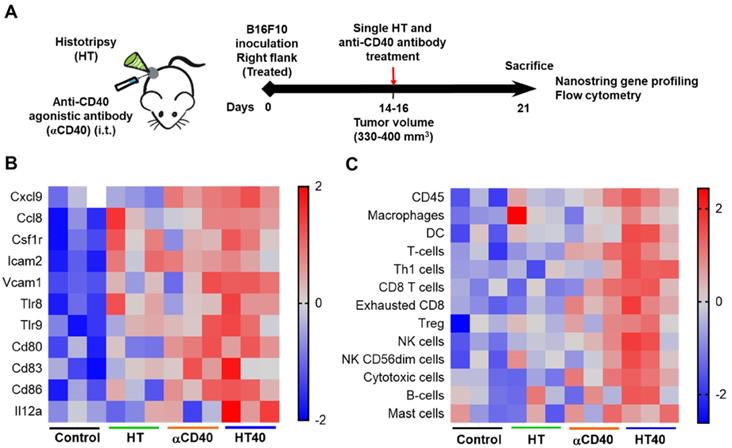
HT40 induced inflammation and checkpoint expression in established melanoma
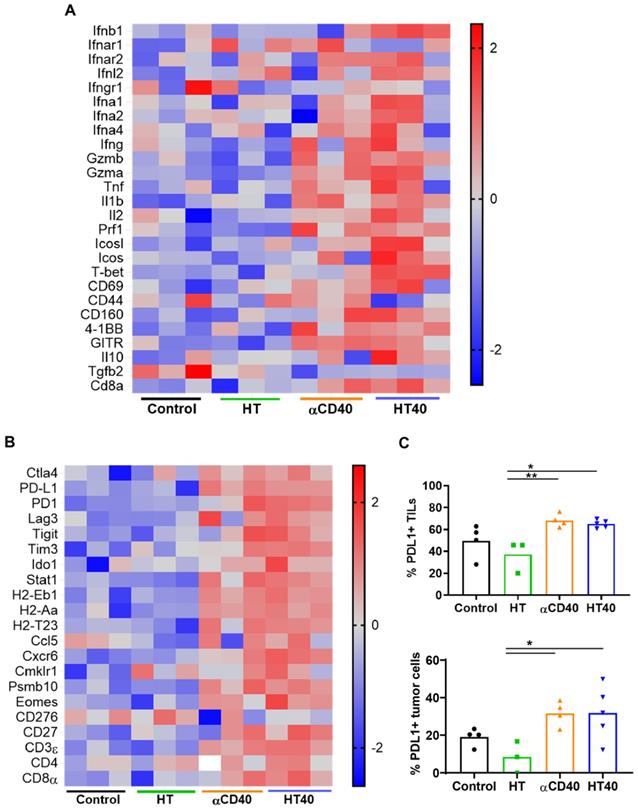
HT40 was performed in B16F10 melanoma tumors established unilaterally (Figure 3A). Screening of immune related genes (n=3/treatment group) in the tumor microenvironment using nanostring technique suggested an increased expression of inflammatory genes associated with phagocytosis, cell adhesion, cytokine, and antigen processing and presentation for HT, αCD40 and HT40 compared to the control, but this profile was most significant and dominant in HT40-treated tumors (Figure 1 and S1). HT alone increased immune infiltration markers (1.26 log2 fold for ICAM-2 and 0.71 log2 fold for VCAM-1), and APC chemo attractants (CCL8: ~2.6- and CSF1R: ~1.78-log2 fold) compared to control (Figure 3B; also see Figure S2 volcano plots for quantitative changes in gene expression). αCD40 and HT40 upregulated the expressions of the genes associated with CD45, T cells, and NK cell activations (Figure 3C). Also, HT40 tumors had enhanced dendritic, Th1, CD8+ T, cytotoxic, and NK CD56 dim cell markers. HT40 increased the expression of CXCL9 (~4.23 log2 fold), TLR-8, TLR-9 (~2 log2 fold), IL12-α and STAT1 (~1 log2 fold) genes (Figure 3B). Further, it upregulated the T cell activation genes (IFNβ1, IFNL2, granzyme α, granzyme β, IL1b, IL2, ICOSL, ICOS, TBET, CD69, CD44, CD160, and 4-1BB) and downregulated TGFβ2 (Figure 4A). Consistent with T cell activation, the checkpoint marker genes (CTLA4, PDL1, PD1, TIM3, and LAG3) were enhanced with αCD40 and HT40 treatment (Figure 4B). In particular, immune activation markers such as TIGIT, IDO1, STAT1, and EOMES were significantly expressed in HT40-treated tumors relative to controls (Figure 4B). Finally, to test, whether the gene expression results correlated with flow cytometry findings, we isolated the CD45+ and CD45- cells harvested from the tumors and assessed the PDL1 expressions. A 1.3-1.5-fold enhanced expression of PDL1 in TILs for αCD40 and HT40 treated tumors were noted, demonstrating strong associations between assays (Figure 4C).
HT40 therapy increased pro-inflammatory immune markers in tumors. (A) C57BL/6J mice were implanted subcutaneously in the right flank with B16F10 cells unilaterally and single treatments of HT, αCD40 or HT40 were administered (n=5 per group). Tumors were harvested 7 days post treatments. Total RNA (n = 3 samples per treatment group) was isolated, and immune profiling was performed using the NanoString PanCancer Immune panel. (B) Heat maps showing gene markers of cell adhesion molecules, chemokines, innate sensors, and activation status of APCs was higher for HT40 tumors relative to the corresponding controls. (C) Total tumor infiltrating leucocytes, dendritic cells, Th1 cells, cytotoxic cells and activated NK cell expression markers were significantly higher with HT40 therapy compared to the controls. Please see supplemental figure S2 for mean log2 fold changes for individual gens compared to the control. Statistical analysis was performed using multiple t-tests without correction for multiple comparisons. p < 0.05 is considered significant.
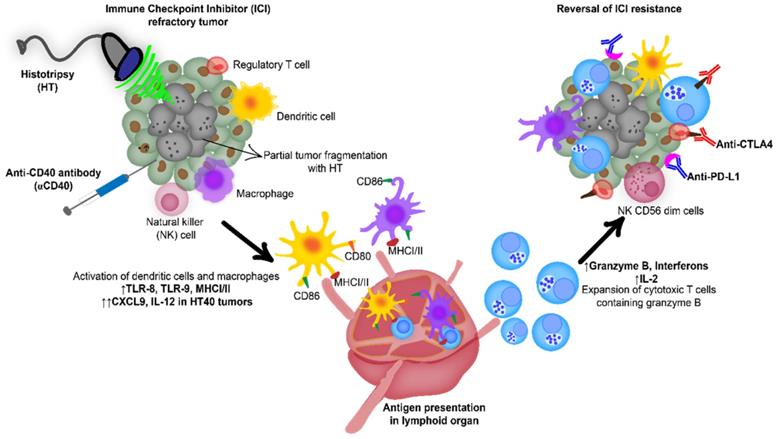
Combination of anti-CD40 agonistic antibody (αCD40) with focused ultrasound (FUS) induced boiling histotripsy (HT) activates the tumor microenvironment to enhance the efficacy of immune checkpoint inhibitors in poorly immunogenic tumors.
HT40 and αCD40 therapy enhanced T-cell activation and checkpoint expressions in the melanoma tumors. (A) Heat maps showed an enhanced expression of T-cell activation genes in the treated tumors compared to the control. (B) The checkpoint marker genes (e.g. CTLA4, PDL1, PD1, TIM3, and LAG3) were enhanced with CD40 and HT40 treatment. (C) PD-L1+ CD45+ (tumor infiltrating leukocytes; TILs) and PD-L1+ CD45- (tumor cells) cells assessed using flow cytometry (n=3-5 per group). Gene expression statistical analysis was performed using multiple t-tests without correction for multiple comparisons (n=3 per group). p < 0.05 is considered significant. For flow cytometry (C), data were presented as mean ± SEM and the statistical differences between groups were measured by ANOVA followed by Tukey's multiple comparisons. * p < 0.05, ** p < 0.01.
Local treatments suppressed tumor progression by enhancing melanoma immunogenicity
Mice with unilateral B16F10 tumors in the flank regions were established. HT treatment alone slightly inhibited the tumor growth rate 1-week post treatment, but its combination with anti-CD40 antibody reduced tumor growth by > 70% compared to the control. This reduction was 30-50% greater than that of respective monotherapies (Figure 5A). The reduction in tumor volumes accompanied a significant reduction in tumor weights for the HT40 cohort compared to the other groups (Figure 5B). Local and systemic evaluation of the immune responses of harvested tumors from the surviving mice revealed an increase (~1.2-2-fold) in the populations of CD45+ TILs and CD3+ T cells in the HT-treated group compared to the untreated control. The TIL increase was not accompanied by a significant increase in CD8+ subtypes in HT-treated tumors. In contrast, HT40 enhanced the CD3+ CD8+ T cell population by 2-3-fold relative to HT post treatment (Figure 5C-E). The populations of effector CD8+ T cells exhibited an increased level of IFNγ and granzyme B expression, suggesting an activated cytotoxic phenotype (Figure 6A-B). We also found that the T cell activation was not accompanied by a concurrent increase in the Foxp3+ CD4+ Tregs. Overall, a 2.5 to 5-fold increase in the granzyme B+ CD8+ T cell to Treg ratio in αCD40 and HT40-treated tumors compared to the untreated control was noted, which reflects enhanced mobilization of cytotoxic cells in the treated tumor (Figure 6C).
Local HT40 suppressed tumor progression and improved the infiltration of T lymphocytes. (A) Time course of treatment effects on tumor volumes through 21 days post-inoculation and (B) Tumor weights at time of harvest in mice unilaterally inoculated with B16F10 cells in the right flank regions. HT40 inhibited tumor growth significantly vs. that of the respective controls (C) HT, αCD40, and HT40 enhanced the populations of tumor infiltrating leucocytes (TILs) compared to control in the harvested tumors of surviving mice. Overall, HT40 demonstrated the highest infiltration rates compared to the other groups. (D) HT40 induced a higher percentage of CD3+ T cell population than the control. (E) Frequency of CD8+ T cells in HT40 group was 2-folds higher compared to the HT and control group. Results are shown as mean ± SEM, n=3-5 per group. One-way ANOVA followed by Tukey's multiple comparison was used for data analysis. * p < 0.05, ** p<0.01.
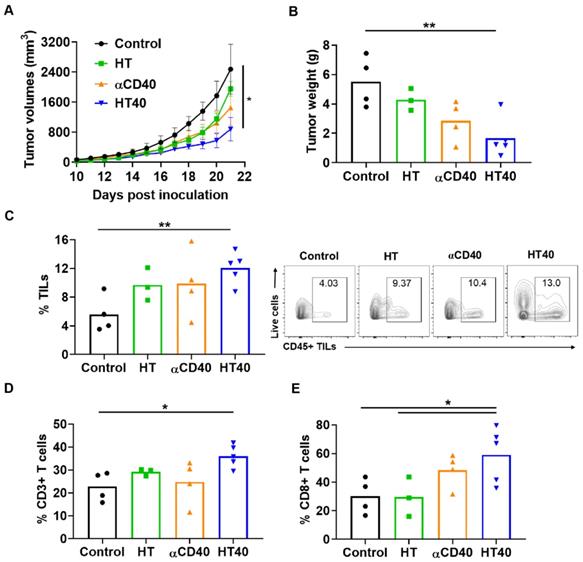
HT40 augmented the T cell functions in treated tumors. (A and B) HT40 promoted IFNγ (~2-fold) and Granzyme B (~4-fold) secretion from CD8+ T cells in the treated tumors. (C) Ratio of cytotoxic CD8+ T cells and immunosuppressive regulatory T (Treg) cells in tumors increased by 2.5 and 5-fold with αCD40 and HT40 compared to the untreated controls, respectively. Data are shown as mean ± SEM, n=3-5 per treatment group, * p < 0.05, ** p < 0.01. Data were analyzed by One-way ANOVA followed by Tukey's multiple comparisons; changes between control and treatments in Figure 6C were analyzed using an unpaired t test assuming unequal variance.
HT40 promoted melanoma specific immunological memory
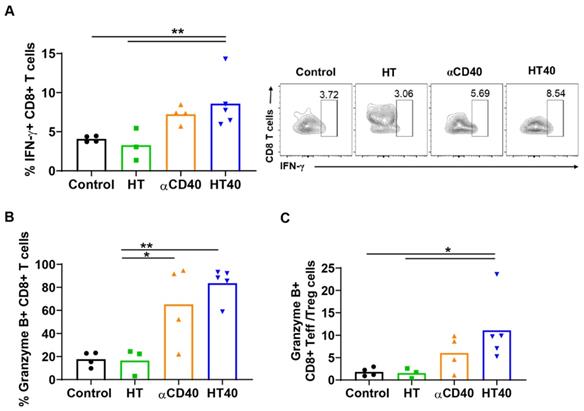
A significant increase in CD44+ CD62lo CD4+ T cells, which represent the CD4+ effector-memory T cell population, was observed for the HT- and HT40-treated tumors (1.5-2-fold). Additionally, an increased population of M1 macrophages along with a concurrent decrease of M2 macrophages was noted for HT40-treated tumors. αCD40 alone did not increase CD4+ effector cells, but it did enhance the populations of M1 macrophages, which suggested APC activation (Figure 7A-C). HT, αCD40, and HT40 also increased M1 macrophages and reduced the M2 phenotype in the spleen tissues, with the HT40 having the greatest effect (Figure 7D-E). To assess antigen specificity, splenocytes stimulated ex vivo with TRP-2 were assessed for IL2 production. A significant (1.3-1.7-fold) increase in TRP-2 specific IL2+ CD4+ T cells in the spleen of HT40-treated mice compared to the control was noted, and this number was relatively higher compared to that of the other therapies (Figure 7F). Thus, we posited that the HT40 induced a potent melanoma memory response.
HT40 increased melanoma specific antitumor immunity. (A) A significant increase in CD44hi CD62lo CD4+ effector T memory cells (percentage out of total leukocytes) in HT and HT40 treated tumors was noted. (B and C) αCD40 and HT40 enhanced the percent of M1 macrophages by 2-fold and decreased M2 macrophages by 1.5-fold compared to controls. (D and E) HT, αCD40 and HT40 increased M1 macrophages (~1.3-1.7-fold) and decreased M2 macrophages (~1.5-2-fold) in splenic tissues compared to the control. (F) IL-2 production from CD4+ T cells was significantly improved by αCD40 and HT40 treatments compared to untreated controls. Amongst all the treatments, HT40 showed the most dominant effect upon TRP-2 stimulation ex-vivo. Data are shown as mean ± SEM, n=3-5 per group. Data were analyzed by ANOVA followed by Tukey's multiple comparisons. * p < 0.05, ** p < 0.01.
HT40 therapy sensitized mice bearing bilateral melanoma tumors to Immune checkpoint inhibitors
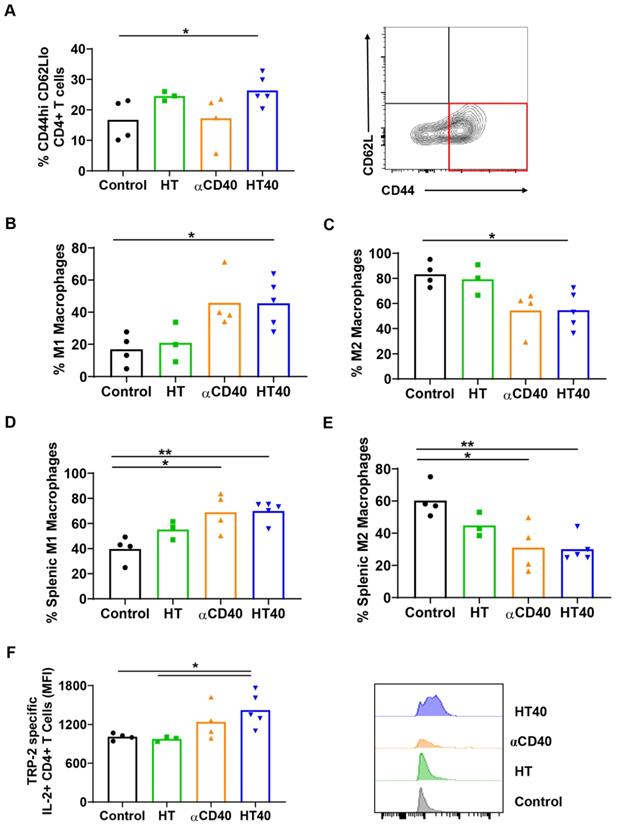
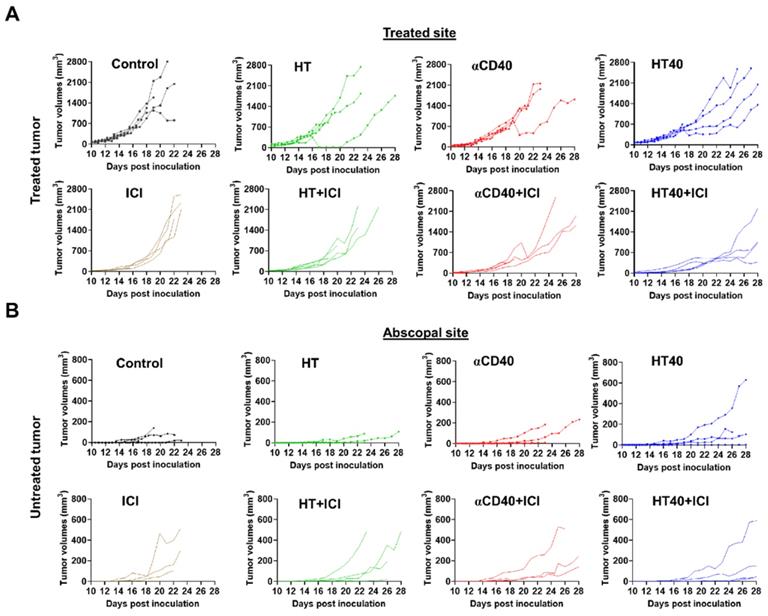
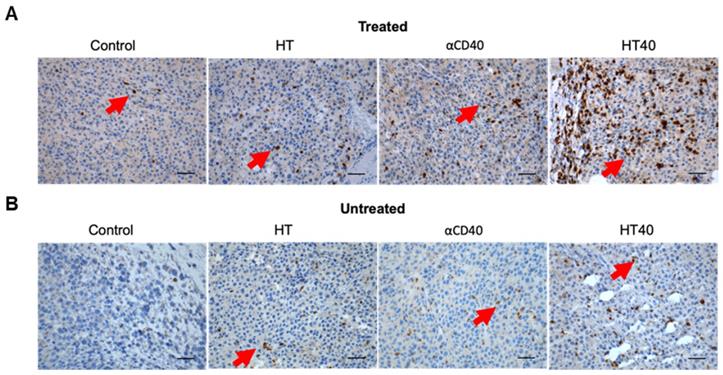
B16F10 melanoma are known to demonstrate a modest response to ICIs [53, 54]. Also, ICIs are most effective when administered immediately post tumor inoculation [55]. Since the goal of this study was to assess the feasibility of our combinatorial approach in immune-resistant tumors, we performed treatments in B16F10 tumors when they reached a volume of 330-400 mm3. For assessing ICI effect in a bilateral melanoma model, unilateral HT40 treatment of the right flank tumor was followed by intraperitoneal injection of ICIs (n=5 per group, Figure 8A). ICI by themselves were ineffective in inducing tumor growth suppression and survival rates compared to the control, suggesting that the B16F10 melanoma was refractory to the checkpoint blockade therapy at the time point tested (Figure 8B and Figure 9). Also, HT or αCD40 alone moderately enhanced ICI efficacy and survival compared to ICI and untreated controls. In contrast, HT40 significantly improved outcomes vs. HT or αCD40 alone. Also, when primed with HT40, ICI therapy was most effective in delaying tumor growth rates, and in enhancing survival responses vs. all other treatments. In general, untreated control mice that were bilaterally inoculated didn't survive beyond day 23 post inoculation, suggesting an absence of abscopal effect. For other treatments (especially for the HT40), the presence of abscopal effect induced a prophylactic effect, enhancing the survival rates presumably by decreasing the overall tumor burden in the treated mice (Figure 9). We found that 40% (2 of 5) of HT40+ICI-treated mice showed abscopal tumor suppression for the entire treatment period (40 days; not shown). In contrast, other treatments were relatively less effective, and mice reached euthanasia endpoints before the end of study. To understand the mechanisms, and examine the change in tumor microenvironments in treated and untreated tumors, we processed both treated and abscopal tumors for CD3 immunohistochemical analysis. The treated and untreated tumors from HT and αCD40 alone showed mild T cell infiltration. In contrast, the treated and untreated HT40 tumors showed a prominent increase in the density of CD3+ T cells within the tumor compared to monotherapies (Figure 10), correlating with the improved therapeutic outcomes in HT40 cohorts.
HT40 priming enhanced the therapeutic effects in ICI refractory melanoma. (A) Mice bearing B16F10 melanoma in the left and right flank regions were treated unilaterally followed by ICI therapy. (B) ICI combinations alone were ineffective in enhancing survival rates vs. those of controls, suggesting that at the time-point tested, the B16F10 melanoma was refractory to ICIs. HT40 improved ICI survival outcome compared to other groups by reversing resistance to ICIs. Differences in median survival (n=5) were determined by the Kaplan-Meier method and the log-rank test used to determine P value. Differences in the median survival (n=5 per group) were determined by the Kaplan-Meier method and the log-rank test was used to determine P value. p < 0.05: HT40+ICI vs αCD40+ICI; p < 0.1: HT40+ICI vs HT+ICI, HT40.
Tumor growth rates in mice bearing melanoma in left and right flank regions shown till day 28 post inoculation. (A) HT40 and HT40+ICI delayed growth of treated tumors compared to HT and αCD40 alone. (B) Tumor growth rates at distant untreated sites were relatively slower with HT40+ICI and HT40 compared to other treatments. Untreated and HT treated mice succumbed to the disease between day 22-26, preventing the estimation of tumor growth rates at the untreated/abscopal sites.
HT40 therapy enhanced infiltration of T cells in the treated and distant untreated (abscopal) tumors. (A & B) Representative examples of the immunohistochemical staining of treated (A) and untreated tumor (B) sections for CD3 marker. Both treated and untreated tumors from HT40 cohort showed an increased population of CD3+T cells in the tumor (arrows) compared to monotherapies. Scale bar: 100μm.
Discussion
The objective of this study was to understand the ability of HT40 to reprogram the immunologically cold melanoma tumor such that it becomes more receptive to ICI therapy. HT has been utilized to debulk tumor tissue, release damage associated molecular patterns (DAMPs), and improve immune sensitization in various tumor models [24, 27, 36, 56]. We and others have also shown that local αCD40 therapy activates APCs and improves the functional status of TILs in melanoma [21, 57, 58]. This is likely via enhanced antigen presentation by APCs through improved CD40L binding with CD40 receptor on APCs, and by the upregulation of costimulatory molecules such as MHC class II, CD80, CD86, and CD58 on the cell membrane [59]. Particularly, we have found that that αCD40 efficacy is enhanced with local FUS-heating of B16F10 melanoma in mice [21]. Tumor heating is highly challenging to perform in highly perfused organs, and can cause collateral damage to nearby healthy tissues [60]. To overcome this barrier, in this study we combined boiling HT and αCD40 for anti-tumor immunity induction in immunologically cold melanoma tumors. Boiling HT is a non-invasive mechanical homogenization technology that rapidly generates tumor antigen depots with sharp boundaries in solid cancers. Since HT achieves heating to 100 ºC by using millisecond-long pulses with lower pulse repetition frequency (PRF) [34], it avoids denaturation of tumor antigens in the focal regions [36]. Thus, its combination with αCD40 can hypothetically improve tumor immune environment, and immunotherapeutic response.
To investigate the potential of the HT and αCD40 combination, we utilized an ICI refractory and poorly immunogenic B16F10 model. B16F10 tumors exhibit reduced expression of the MHC class I and co-stimulatory molecules (e.g. CD80, CD86 etc.) [40]. Its self-antigen (TRP-2) also shows poor affinity to T cell receptors, thereby making it an excellent poorly immunogenic model for immunotherapy studies [61, 62]. High intensity, low duty cycle, and short ultrasound HT pulses were used to fractionate ~40-50% of the tumor mass (Figure 2A-B). Pan-cancer immune profiling suggested that the selected HT parameters elevated the expression of chemo-attractants (CCL8 and CSF1R) and cell adhesion molecules (ICAM and VCAM). These markers are essential for cell-cell interaction and leukocyte migration into tumors (Figure 3) [63-65].
HT treatment also lowered the immunosuppressive cytokine TGFβ2 in tumors, and the addition of αCD40 caused upregulation of several immune-activation markers, including CXCL9. Chemokines such as CCL3-5, CCL8, CCL11-12, CXCL9 and CXCL10 produced from mature APCs play a crucial role in recruiting CD8+ T cells, CD4+ helper T cells, and natural killer cells into TME [64, 66]. CXCL9 also positions tumor infiltrating T cells in APC rich regions to remove T cell anergy [67]. CXCL9 is constitutively produced from myeloid cells following stimulation of IFN secreting T cells [67, 68]. IFN-γ can induce additional production of this chemokines via STAT1 signaling to enhance CD8+ T cells recruitment into tumors [69-71]. Our tumor immune analysis suggested that HT40 treatment induced an influx of CD8+ IFN-γ expressing T cells (Figure 5 and 6), indicating a CXCL9 mediated amplification of cytotoxic T cell-based antitumor immunity [72, 73]. In addition, increased accumulation of M1 macrophages and granzyme B+ activated CD8+ T cells without alteration of Tregs was noted in tumors treated with HT40 (Figure 7). Also, the population of TRP-2 specific CD4+ T cells and CD44hi CD62lo CD4+ T cells that help with the memory T cell response was enhanced. These were also verified in IHC analysis where relatively enhanced populations of CD3+ T cells were observed in the treated and untreated tumors of HT cohorts (Figure 10). HT also increased PD-L1, CTLA4, and other immune checkpoints within the tumor microenvironment (Figure 3). These phenotypic alterations are typically an adaptive mechanism to suppress T cell function [74]. However, enhanced expression of checkpoint proteins can also be a positive prognostic marker of ICI outcomes in melanoma patients [75-77]. To investigate whether this was true in our model system, ICIs were added to the HT40 regimen, and this resulted in improved efficacy and mice survival rates (Figure 8 and 9). Thus, we believe that HT40 may have significant clinical value, especially when combined with ICIs or other immune activators such as TLR and chemokine/cytokine agonists.
Our study had some limitations. First, HT40 therapy improved survival but did not eliminate the melanoma tumors. We do not know the reasons for this outcome, but the response of melanoma to HT40 may depend on the degree of mechanical damage, dosing, sequence, and schedule of the HT and αCD40 therapies. Studies are currently underway to further investigate these mechanisms. These include first enhancing CD40 stimulation in smaller tumors, followed by HT40 treatment of larger tumors to provide sufficient priming. Alternatively, combining FUS parameters (e.g., mild hyperthermia + HT) with CD40 stimulation might be more insightful. Second, although the addition of HT40 to ICI improved the response of refractory melanoma, local recurrence and emergence of distant metastasis may still be possible [78]. Future re-challenge studies and histopathological evaluations of lung tissues may shed more light on such mechanisms. Third, only a single B16F10 model and single HT40 treatment was investigated. Future studies employing multiple models with multiple HT40 doses would elucidate the differences in clinical efficacies of various therapies. Lastly, mechanical homogenization of tumors using HT can induce metastasis. This aspect was not studied, although recent studies from other groups suggest that it is highly unlikely [27, 29].
In summary, boiling HT40 therapy augmented innate and adaptive immunity in the B16F10 model. An inflamed TME with an active interaction of CXCL9-cytotoxic T cell axis was the likely mechanism responsible for sensitization to ICI and improved survival rates of mice. Combining HT40 with ICIs may enhance outcomes in advanced stage cancer patients.
Supplementary Material
Supplementary figures.
Acknowledgements
We thank the seed grant from the Center for Veterinary Health Sciences, National Cancer Institute of the National Institutes of Health under Award Number R37 CA239150-02, Focused Ultrasound Foundation, PETCO and the Kerr (Ranjan) and McCasland Foundation (Malayer) Endowed Chair at Oklahoma State University for supporting the immunotherapy research.
Data availability
The data supporting the findings of this study are available within the article and from the corresponding author upon request.
Author Contributions
A.R conceived and designed the project study goals, and was assisted by M.P.S; M.P.S. and S.N.S. conducted various experiments under the supervision of A.R.; A.R., S.N.S., C.M., J.M. and M.P.S. analyzed data; and A.R. and M.P.S. wrote the paper.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hargadon KM, Johnson CE, Williams CJ. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. International immunopharmacology. 2018;62:29-39
2. Massard C, Gordon MS, Sharma S, Rafii S, Wainberg ZA, Luke J. et al. Safety and efficacy of durvalumab (MEDI4736), an anti-programmed cell death ligand-1 immune checkpoint inhibitor, in patients with advanced urothelial bladder cancer. Journal of Clinical Oncology. 2016;34:3119
3. McDermott DF, Sosman JA, Sznol M, Massard C, Gordon MS, Hamid O. et al. Atezolizumab, an Anti-Programmed Death-Ligand 1 Antibody, in Metastatic Renal Cell Carcinoma: Long-Term Safety, Clinical Activity, and Immune Correlates From a Phase Ia Study. J Clin Oncol. 2016;34:833-42
4. Ott PA, Hodi FS, Robert C. CTLA-4 and PD-1/PD-L1 blockade: new immunotherapeutic modalities with durable clinical benefit in melanoma patients. AACR. 2013
5. Gajewski TF. The next hurdle in cancer immunotherapy: Overcoming the non-T-cell-inflamed tumor microenvironment. Seminars in oncology: Elsevier. 2015 p. 663-71
6. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480-9
7. Smyth MJ, Ngiow SF, Ribas A, Teng MW. Combination cancer immunotherapies tailored to the tumour microenvironment. Nature reviews Clinical oncology. 2016;13:143
8. Janco JMT, Lamichhane P, Karyampudi L, Knutson KL. Tumor-infiltrating dendritic cells in cancer pathogenesis. The Journal of Immunology. 2015;194:2985-91
9. Bertrand F, Montfort A, Marcheteau E, Imbert C, Gilhodes J, Filleron T. et al. TNFα blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nature communications. 2017;8:1-13
10. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L. et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015;372:2521-32
11. Lee C, Jeong H, Bae Y, Shin K, Kang S, Kim H. et al. Targeting of M2-like tumor-associated macrophages with a melittin-based pro-apoptotic peptide. Journal for immunotherapy of cancer. 2019;7:147
12. Bodey B, Bodey Jr B, Siegel SE, Kaiser HE. Failure of cancer vaccines: the significant limitations of this approach to immunotherapy. Anticancer research. 2000;20:2665-76
13. Gujar SA, Marcato P, Pan D, Lee PW. Reovirus virotherapy overrides tumor antigen presentation evasion and promotes protective antitumor immunity. Molecular cancer therapeutics. 2010;9:2924-33
14. Garris CS, Arlauckas SP, Kohler RH, Trefny MP, Garren S, Piot C. et al. Successful anti-PD-1 cancer immunotherapy requires T cell-dendritic cell crosstalk involving the cytokines IFN-γ and IL-12. Immunity. 2018;49:1148-61 e7
15. Daud AI, Loo K, Pauli ML, Sanchez-Rodriguez R, Sandoval PM, Taravati K. et al. Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. J Clin Invest. 2016;126:3447-52
16. Gaudino SJ, Kumar P. Cross-Talk Between Antigen Presenting Cells and T Cells Impacts Intestinal Homeostasis, Bacterial Infections, and Tumorigenesis. Frontiers in Immunology. 2019 10
17. Ferris RL, Whiteside TL, Ferrone S. Immune Escape Associated with Functional Defects in Antigen-Processing Machinery in Head and Neck Cancer. Clinical Cancer Research. 2006;12:3890-5
18. Palucka K, Banchereau J. Dendritic cells: a link between innate and adaptive immunity. J Clin Immunol. 1999;19:12-25
19. Beatty GL, Gladney WL. Immune Escape Mechanisms as a Guide for Cancer Immunotherapy. Clinical Cancer Research. 2015;21:687-92
20. Ting Koh Y, Luz García-Hernández M, Martin Kast W. Tumor Immune Escape Mechanisms. In: Teicher BA, editor. Cancer Drug Resistance. Totowa, NJ: Humana Press. 2006 p. 577-602
21. Singh MP, Sethuraman SN, Ritchey J, Fiering S, Guha C, Malayer J. et al. In-situ vaccination using focused ultrasound heating and anti-CD-40 agonistic antibody enhances T-cell mediated local and abscopal effects in murine melanoma. Int J Hyperthermia. 2019;36:64-73
22. Rosberger DF, Coleman DJ, Silverman R, Woods S, Rondeau M, Cunningham-Rundles S. Immunomodulation in choroidal melanoma: reversal of inverted CD4/CD8 ratios following treatment with ultrasonic hyperthermia. Biotechnol Ther. 1994;5:59-68
23. Sethuraman SN, Singh MP, Patil G, Li S, Fiering S, Hoopes PJ. et al. Novel calreticulin-nanoparticle in combination with focused ultrasound induces immunogenic cell death in melanoma to enhance antitumor immunity. Theranostics. 2020;10:3397
24. Qu S, Worlikar T, Felsted AE, Ganguly A, Beems MV, Hubbard R. et al. Non-thermal histotripsy tumor ablation promotes abscopal immune responses that enhance cancer immunotherapy. Journal for ImmunoTherapy of Cancer. 2020;8:e000200
25. Hu Z, Yang XY, Liu Y, Sankin GN, Pua EC, Morse MA. et al. Investigation of HIFU-induced anti-tumor immunity in a murine tumor model. Journal of translational medicine. 2007;5:34
26. Eranki A, Farr N, Partanen A, Sharma KV, Rossi CT, Rosenberg AZ. et al. Mechanical fractionation of tissues using microsecond-long HIFU pulses on a clinical MR-HIFU system. International Journal of Hyperthermia. 2018;34:1213-24
27. Eranki A, Srinivasan P, Ries M, Kim A, Lazarski CA, Rossi CT. et al. High Intensity Focused Ultrasound (HIFU) Triggers Immune Sensitization of Refractory Murine Neuroblastoma to Checkpoint Inhibitor Therapy. Clinical Cancer Research. 2019: clincanres.1604. 2019
28. Simon JC, Sapozhnikov OA, Khokhlova VA, Wang Y-N, Crum LA, Bailey MR. Ultrasonic atomization of tissue and its role in tissue fractionation by high intensity focused ultrasound. Physics in Medicine & Biology. 2012;57:8061
29. Khokhlova TD, Canney MS, Khokhlova VA, Sapozhnikov OA, Crum LA, Bailey MR. Controlled tissue emulsification produced by high intensity focused ultrasound shock waves and millisecond boiling. The Journal of the Acoustical Society of America. 2011;130:3498-510
30. Vlaisavljevich E, Maxwell A, Warnez M, Johnsen E, Cain CA, Xu Z. Histotripsy-induced cavitation cloud initiation thresholds in tissues of different mechanical properties. IEEE Trans Ultrason Ferroelectr Freq Control. 2014;61:341-52
31. Xu J, Bigelow TA, Whitley EM. Assessment of ultrasound histotripsy-induced damage to ex vivo porcine muscle. J Ultrasound Med. 2013;32:69-82
32. Khokhlova VA, Fowlkes JB, Roberts WW, Schade GR, Xu Z, Khokhlova TD. et al. Histotripsy methods in mechanical disintegration of tissue: towards clinical applications. Int J Hyperthermia. 2015;31:145-62
33. Xu Z, Hall TL, Fowlkes JB, Cain CA. Effects of acoustic parameters on bubble cloud dynamics in ultrasound tissue erosion (histotripsy). J Acoust Soc Am. 2007;122:229-36
34. Eranki A, Srinivasan P, Ries M, Kim A, Lazarski CA, Rossi CT. et al. High-Intensity Focused Ultrasound (HIFU) Triggers Immune Sensitization of Refractory Murine Neuroblastoma to Checkpoint Inhibitor Therapy. Clin Cancer Res. 2020;26:1152-61
35. Ziadloo A, Burks SR, Gold EM, Lewis BK, Chaudhry A, Merino MJ. et al. Enhanced homing permeability and retention of bone marrow stromal cells by noninvasive pulsed focused ultrasound. Stem Cells. 2012;30:1216-27
36. Pahk KJ, Shin C-H, Bae IY, Yang Y, Kim S-H, Pahk K. et al. Boiling Histotripsy-induced Partial Mechanical Ablation Modulates Tumour Microenvironment by Promoting Immunogenic Cell Death of Cancers. Scientific Reports. 2019;9:9050
37. Gardner A, Ruffell B. Dendritic cells and cancer immunity. Trends in immunology. 2016;37:855-65
38. Petersen TR, Dickgreber N, Hermans IF. Tumor antigen presentation by dendritic cells. Crit Rev Immunol. 2010;30:345-86
39. Schade GR, Wang YN, D'Andrea S, Hwang JH, Liles WC, Khokhlova TD. Boiling Histotripsy Ablation of Renal Cell Carcinoma in the Eker Rat Promotes a Systemic Inflammatory Response. Ultrasound Med Biol. 2019;45:137-47
40. Lechner MG, Karimi SS, Barry-Holson K, Angell TE, Murphy KA, Church CH. et al. Immunogenicity of murine solid tumor models as a defining feature of in vivo behavior and response to immunotherapy. J Immunother. 2013;36:477-89
41. Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E. et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520:373-7
42. Long KB, Gladney WL, Tooker GM, Graham K, Fraietta JA, Beatty GL. IFNgamma and CCL2 Cooperate to Redirect Tumor-Infiltrating Monocytes to Degrade Fibrosis and Enhance Chemotherapy Efficacy in Pancreatic Carcinoma. Cancer Discov. 2016;6:400-13
43. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W. et al. CD40 Agonists Alter Tumor Stroma and Show Efficacy Against Pancreatic Carcinoma in Mice and Humans. Science (New York, NY). 2011;331:1612
44. Hunter TB, Alsarraj M, Gladue RP, Bedian V, Antonia SJ. An agonist antibody specific for CD40 induces dendritic cell maturation and promotes autologous anti-tumour T-cell responses in an in vitro mixed autologous tumour cell/lymph node cell model. Scandinavian journal of immunology. 2007;65:479-86
45. Thompson EA, Liang F, Lindgren G, Sandgren KJ, Quinn KM, Darrah PA. et al. Human Anti-CD40 Antibody and Poly IC:LC Adjuvant Combination Induces Potent T Cell Responses in the Lung of Nonhuman Primates. The Journal of Immunology. 2015;195:1015
46. Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P. et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med. 2014;6:226ra32-ra32
47. Chevillet JR, Khokhlova TD, Giraldez MD, Schade GR, Starr F, Wang YN. et al. Release of Cell-free MicroRNA Tumor Biomarkers into the Blood Circulation with Pulsed Focused Ultrasound: A Noninvasive, Anatomically Localized, Molecular Liquid Biopsy. Radiology. 2017;283:158-67
48. Breshears MA, Eberle R, Ritchey JW. Temporal Progression of Viral Replication and Gross and Histological Lesions in Balb/c Mice Inoculated Epidermally with Saimiriine herpesvirus 1 (SaHV-1). Journal of Comparative Pathology. 2005;133:103-13
49. Newton JM, Hanoteau A, Liu H-C, Gaspero A, Parikh F, Gartrell-Corrado RD. et al. Immune microenvironment modulation unmasks therapeutic benefit of radiotherapy and checkpoint inhibition. Journal for ImmunoTherapy of Cancer. 2019;7:216
50. Silvestrini MT, Ingham ES, Mahakian LM, Kheirolomoom A, Liu Y, Fite BZ. et al. Priming is key to effective incorporation of image-guided thermal ablation into immunotherapy protocols. JCI Insight. 2017;2:e90521-e
51. Fan Y, Kuai R, Xu Y, Ochyl LJ, Irvine DJ. Immunogenic Cell Death Amplified by Co-localized Adjuvant Delivery for Cancer Immunotherapy. 2017; 17: 7387-93.
52. De Palma R, Marigo I, Del Galdo F, De Santo C, Serafini P, Cingarlini S. et al. Therapeutic effectiveness of recombinant cancer vaccines is associated with a prevalent T-cell receptor alpha usage by melanoma-specific CD8+ T lymphocytes. Cancer research. 2004;64:8068-76
53. Saida Y, Brender JR, Yamamoto K, Mitchell JB, Krishna MC, Kishimoto S. Detecting Early Response to Immune Checkpoint Blockade by Multimodal Molecular Imaging. bioRxiv. 2020. 2020 04.30.070508
54. Singh M, Vianden C, Cantwell MJ, Dai Z, Xiao Z, Sharma M. et al. Intratumoral CD40 activation and checkpoint blockade induces T cell-mediated eradication of melanoma in the brain. Nature communications. 2017;8:1447 -
55. Zamarin D, Ricca JM, Sadekova S, Oseledchyk A, Yu Y, Blumenschein WM. et al. PD-L1 in tumor microenvironment mediates resistance to oncolytic immunotherapy. The Journal of clinical investigation. 2018;128:1413-28
56. Schade GR, Wang Y-N, D'Andrea S, Hwang JH, Liles WC, Khokhlova TD. Boiling Histotripsy Ablation of Renal Cell Carcinoma in the Eker Rat Promotes a Systemic Inflammatory Response. Ultrasound in Medicine & Biology. 2019;45:137-47
57. Yasmin-Karim S, Bruck PT, Moreau M, Kunjachan S, Chen GZ, Kumar R. et al. Radiation and Local Anti-CD40 Generate an Effective in situ Vaccine in Preclinical Models of Pancreatic Cancer. Frontiers in immunology. 2018;9:2030 -
58. Vonderheide RH. CD40 Agonist Antibodies in Cancer Immunotherapy. Annual review of medicine. 2020;71:47-58
59. Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev. 2009;229:152-72
60. Chaussy C, Thuroff S. Results and side effects of high-intensity focused ultrasound in localized prostate cancer. J Endourol. 2001;15:437-40 discussion 47-8
61. Bellone M, Cantarella D, Castiglioni P, Crosti MC, Ronchetti A, Moro M. et al. Relevance of the Tumor Antigen in the Validation of Three Vaccination Strategies for Melanoma. The Journal of Immunology. 2000;165:2651-6
62. Bloom MB, Perry-Lalley D, Robbins PF, Li Y, el-Gamil M, Rosenberg SA. et al. Identification of tyrosinase-related protein 2 as a tumor rejection antigen for the B16 melanoma. The Journal of experimental medicine. 1997;185:453-9
63. Sabel MS, Skitzki J, Stoolman L, Egilmez NK, Mathiowitz E, Bailey N. et al. Intratumoral IL-12 and TNF-α-loaded microspheres lead to regression of breast cancer and systemic antitumor immunity. Annals of surgical oncology. 2004;11:147-56
64. Tiberio L, Del Prete A, Schioppa T, Sozio F, Bosisio D, Sozzani S. Chemokine and chemotactic signals in dendritic cell migration. Cellular & molecular immunology. 2018;15:346-52
65. Messina JL, Fenstermacher DA, Eschrich S, Qu X, Berglund AE, Lloyd MC. et al. 12-Chemokine gene signature identifies lymph node-like structures in melanoma: potential for patient selection for immunotherapy? Scientific reports. 2012;2:765
66. Sokol CL, Luster AD. The chemokine system in innate immunity. Cold Spring Harbor perspectives in biology. 2015;7:a016303
67. Kastenmüller W, Brandes M, Wang Z, Herz J, Egen Jackson G, Germain Ronald N. Peripheral Prepositioning and Local CXCL9 Chemokine-Mediated Guidance Orchestrate Rapid Memory CD8+ T Cell Responses in the Lymph Node. Immunity. 2013;38:502-13
68. Böttcher JP, Reis e Sousa C. The Role of Type 1 Conventional Dendritic Cells in Cancer Immunity. Trends in Cancer. 2018;4:784-92
69. Nakanishi Y, Lu B, Gerard C, Iwasaki A. CD8+ T lymphocyte mobilization to virus-infected tissue requires CD4+ T-cell help. Nature. 2009;462:510-3
70. Wei SC, Anang N-AAS, Sharma R, Andrews MC, Reuben A, Levine JH. et al. Combination anti-CTLA-4 plus anti-PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proceedings of the National Academy of Sciences. 2019;116:22699-709
71. Vilgelm AE, Richmond A. Chemokines Modulate Immune Surveillance in Tumorigenesis, Metastasis, and Response to Immunotherapy. Frontiers in immunology. 2019 10
72. Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY. et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41:830-42
73. Rahman AH, Taylor DK, Turka LA. The contribution of direct TLR signaling to T cell responses. Immunol Res. 2009;45:25-36
74. Bommareddy PK, Aspromonte S, Zloza A, Rabkin SD, Kaufman HL. MEK inhibition enhances oncolytic virus immunotherapy through increased tumor cell killing and T cell activation. Sci Transl Med. 2018;10:eaau0417
75. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR. et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. The Journal of clinical investigation. 2017;127:2930-40
76. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L. et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568
77. Nakamura Y. Biomarkers for Immune Checkpoint Inhibitor-Mediated Tumor Response and Adverse Events. Frontiers in Medicine. 2019 6
78. Izadifar Z, Izadifar Z, Chapman D, Babyn P. An Introduction to High Intensity Focused Ultrasound: Systematic Review on Principles, Devices, and Clinical Applications. J Clin Med. 2020 9
Author contact
![]() Corresponding author: Ashish Ranjan, B.V.Sc., Ph.D., Professor & Kerr Chair, 169 McElroy Hall, College of Veterinary Medicine, Oklahoma State University, Stillwater, Oklahoma 74078. Email: ashish.ranjanedu; Phone: 4057446292
Corresponding author: Ashish Ranjan, B.V.Sc., Ph.D., Professor & Kerr Chair, 169 McElroy Hall, College of Veterinary Medicine, Oklahoma State University, Stillwater, Oklahoma 74078. Email: ashish.ranjanedu; Phone: 4057446292