Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(2):602-613. doi:10.7150/thno.48353 This issue Cite
Research Paper
Cytosolic Ca2+ transients during pulsed focused ultrasound generate reactive oxygen species and cause DNA damage in tumor cells
Robert B. Rosenblatt1, Joseph A. Frank1,2, Scott R. Burks1, ![]()
1. Frank Laboratory, Department of Radiology and Imaging Sciences, NIH Clinical Center, Bethesda, MD, 20892
2. National Institute of Biomedical Imaging and Bioengineering, Bethesda, MD 20892
Received 2020-5-18; Accepted 2020-9-1; Published 2021-1-1
Abstract

Mechanical forces from non-ablative pulsed focused ultrasound (pFUS) generate pro-inflammatory tumor microenvironments (TME), marked by increased cytokines, chemokines, and trophic factors, as well as immune cell infiltration and reduced tumor growth. pFUS also causes DNA damage within tumors, which is a potent activator of immunity and could contribute to changes in the TME. This study investigated mechanisms behind the mechanotransductive effects of pFUS causing DNA damage in several tumor cell types.
Methods: 4T1 (murine breast tumor), B16 (murine melanoma), C6 (rat glioma), or MDA-MB-231 (human breast tumor) cells were sonicated in vitro (1.1MHz; 6MPa PNP; 10ms pulses; 10% duty cycle; 300 pulses). DNA damage was detected by TUNEL, apoptosis was measured by immunocytochemistry for cleaved caspase-3. Calcium, superoxide, and H2O2 were detected by fluorescent indicators and modulated by BAPTA-AM, mtTEMPOL, or Trolox, respectively.
Results: pFUS increased TUNEL reactivity (range = 1.6-2.7-fold) in all cell types except C6 and did not induce apoptosis in any cell line. All lines displayed cytosolic Ca2+ transients during sonication. pFUS increased superoxide (range = 1.6-2.0-fold) and H2O2 (range = 2.3-2.8-fold) in all cell types except C6. BAPTA-AM blocked increased TUNEL reactivity, superoxide and H2O2 formation, while Trolox also blocked increased TUNEL reactivity increased after pFUS. mtTEMPOL allowed H2O2 formation and did not block increased TUNEL reactivity after pFUS. Unsonicated C6 cells had higher baseline concentrations of cytosolic Ca2+, superoxide, and H2O2, which were not associated with greater baseline TUNEL reactivity than the other cell lines.
Conclusions: Mechanotransduction of pFUS directly induces DNA damage in tumor cells by cytosolic Ca2+ transients causing formation of superoxide and subsequently, H2O2. These results further suggest potential clinical utility for pFUS. However, the lack of pFUS-induced DNA damage in C6 cells demonstrates a range of potential tumor responses that may arise from physiological differences such as Ca2+ or redox homeostasis.
Keywords: focused ultrasound, calcium, DNA damage, tumor, reactive oxygen species
Introduction
Non-invasive image-guided focused ultrasound (FUS) is a therapeutic modality that has potential to precisely target and treat solid tumors with minimal bystander effects on intervening tissues [1]. FUS has received FDA approval for thermal ablation of uterine fibroids and prostate tumors, and is currently in clinical trials as a potential adjuvant in tumor treatment, including immunotherapy [2, 3]. Nonthermal pulsed FUS (pFUS) at high intensities mechanically ablates tissue (i.e., histotripsy) and has shown promise in preclinical studies to both ablate tumor volumes and induce antigen release that could stimulate innate and adaptive immune responses [4]. Nondestructive sonication of tissues with pFUS at lower intensities can induce a wide range of molecular responses that occur through mechanotransduction of pFUS acoustic radiation forces (ARF) and cavitation [5].
pFUS, with or without microbubbles (MB), has been shown to activate inflammatory signaling pathways such as nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB), tumor necrosis factor-α (TNFα), and cyclo-oxygenase 2 (COX2) signaling pathways [6-9] that can result in release of cytokines, chemokines, and trophic factors (CCTF) from cells in tissues into the surrounding microenvironments of normal and diseased tissues [9-20]. Non-ablative pFUS alters CCTF and vascular cell adhesion molecule (CAM) expression for 24-48 h post-sonication in muscle [9-12], kidney [7, 13-15], heart [16], pancreas [17], brain [8, 18], and various malignancies [19, 20]. Microenvironmental changes from nondestructive pFUS prior to infusion of cellular products increases tropism to targeted sites to prevent or improve tissue damage [7, 9, 11-15, 21-24]. We recently reported that pFUS at a peak negative pressure (PNP) of 6 MPa (f0 = 1.15 MHz; no MB contrast agents) to murine flank tumor models of B16 melanoma and 4T1 breast tumors resulted in numerous proteomic changes to the tumor microenvironment (TME) that were accompanied by infiltration of immune cells into tumors [19, 20]. Moreover, non-ablative pFUS generated a shift toward an anti-tumor TME and reduced growth rates of B16 and 4T1 flank tumors over time [20]. Histological evaluation of possible mechanisms behind the retarded growth rates of the flank tumors following pFUS revealed increased nuclear DNA damage using terminal deoxynucleotidyl transferase dUTP end-nick labeling (TUNEL) reactions [19, 20]. This observation was of particular interest as DNA damage and repair process can stimulate the innate immune system and alter the TME [25]. Further, cellular release of DNA after apoptosis is a damage associated molecular pattern (DAMP) that can contribute to a pro-inflammatory TME and anti-tumor immune responses [26, 27].
The importance of DNA damage as a potential immune modulator following pFUS led us to investigate the mechanisms underlying sonication-induced DNA damage. Mechanical pFUS forces (ARF or cavitation) generates cytosolic Ca2+ transients in several cell types [6, 28-33] by activating mechanically-sensitive ion channels, such as the transient receptor potential (TRP) channels [6, 32, 34]. Changes in cytosolic Ca2+ alter mitochondrial biology and generate reactive oxygen species (ROS) that can ultimately induce DNA damage [35]. Our previous tumor studies [19, 20] did not mechanistically investigate how pFUS caused DNA damage or how it might contribute to slowing tumor growth. This study investigated the biological mechanisms underpinning pFUS-induced DNA damage in vitro in multiple tumor cell lines: B16-F10 (B16), a murine melanoma line; 4T1, a murine breast tumor line; C6, a rat glioma line; and MDA-MB-231BRL a luciferase-transfected human breast tumor line with high brain metastatic potential. Measurements of cytosolic Ca2+, ROS production, DNA damage, and apoptosis following pFUS were performed with and without various physiological manipulations to elucidate biological mechanisms. The results of this study also revealed different responses to pFUS across cell lines that were associated with underlying differences in cell biology and physiology that could ultimately influence how non-ablative pFUS should be incorporated into the treatment of malignancy.
Materials and Methods
Cell Culture
4T1, MDA-MB-231, B16, and C6 were all cultured in Roswell Park Memorial Institute (RPMI) 1640 medium without phenol red and supplemented with 10% FBS and 1% penicillin/streptomycin. Cells were grown at 37 ºC under 5% CO2 atmosphere. 4T1, B16, and C6 cells were from ATCC (Manassas, VA). MDA-MB-231 were the brain-metastasizing subtype previously transfected with a luciferase reporter gene and were a gift from Dr. Patricia Steeg (National Cancer Institute, Bethesda, MD).
Pulsed Focused Ultrasound
pFUS was delivered with a focused transducer (H101, Sonic Concepts, Bothell WA) operating at 1.1 MHz and driven with hardware controlled by LabView (National Instruments, Austin TX). All suspended-cell samples were treated using 6 MPa peak negative pressure (PNP), 10 ms pulse length, 10 Hz pulse repetition frequency, and 300 pulses. These parameters were previously used in vivo [19, 20] and PNP were measured in degassed water. Treatments were performed with cells suspended in RPMI-1640 in 500 μL centrifuge tubes. Cell suspensions were placed at the focus of the transducer submerged in degassed water. Untreated cell groups were received sham sonications (power=0 W). For live-cell Ca2+ imaging, the transducer was placed in a coupling cone filled with degassed water and aligned over the objective of an inverted epifluorescence microscope (Axio Observer 5, Zeiss, Oberkochen, Germany). Cells were adhered to 35mm dishes with #1 glass bottoms and the transducer cone was coupled directly into the imaging medium during sonication. Sonication for live-cell Fluo-4 imaging (Figure 2A-B) was performed at 3 MPa to minimize detachment or movement of cells during image acquisition.
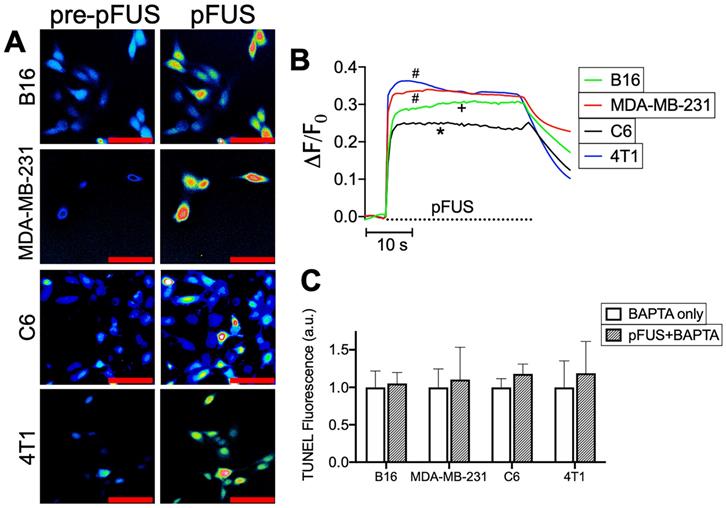
pFUS activates cytosolic Ca2+ transients in tumor cells and intracellular Ca2+ chelation suppresses pFUS-induced TUNEL reactivity. A) Representative imaging of Fluo-4 fluorescence before and during sonication (psuedocolor; scale bars = 50 μm) and B) fluorometric traces of Fluo-4 intensity before and during sonication (n = 69-130 per cell type; dashed line represents sonication time). Groups with like symbols are statistically similar to each other and significantly different from groups denoted by other symbols following ANOVA comparing peak magnitudes of each group. C) Quantification of TUNEL reactivity in cells with or without pFUS in the presence of intracellular BAPTA (n = 9 per group per cell type). Statistical significance was tested between control and treated groups using t-tests for each cell line.
Intracellular Loading with Chemicals to Modulate Physiology and Fluorescent Indicators
For some experiments, cells were incubated at room temperature with various agents including: 1,2-bis(o-aminophenoxy) ethane-N,N,N',N'-tetraacetic acid-acetoxymethyl ester (BAPTA-AM) to chelate intracellular calcium at 1μM for 30 min; 2-(2,2,6,6-Tetramethylpiperidin-1-oxyl-4-ylamino)-2-oxoethyl) triphenylphosphonium chloride (mtTEMPOL) to neutralize superoxide at a 20 μM for 1 h; or (±)-6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Trolox), a peroxyl radical scavenger at 5 μM for 30 minutes. All incubations were performed prior to pFUS.
Fluorescent indicators were also loaded into cells. Cells cultured in glass-bottomed imaging dishes were loaded with Fluo-4 AM to detect intracellular calcium (ThermoFisher, Waltham, MA) according to manufacturer instructions prior to pFUS. Suspended cells were incubated with either MitoSOX (mitochondrial superoxide indicator) (ThermoFisher) at 5 μM immediately prior to pFUS or a proprietary intracellular H2O2 indicator (Cat# MAK164; MilliporeSigma, St. Louis, MO) immediately after pFUS according to manufacturer instructions. Cells loaded with MitoSOX or the H2O2 indicator were re-plated into glass-bottomed dishes 2 h prior to imaging.
TUNEL and Immunocytochemistry (ICC)
Cells for TUNEL measurements were re-plated following treatment and incubated for 6 hr. Cells were fixed in 4% paraformaldehyde for 1 h at 4 ºC and permeabilized with 0.1% Triton X-100 for 10 minutes. For TUNEL quantification, ~105 cells in 96-well plates were measured using fluorescence-based In Situ Cell Death Detection Kit (Millipore Sigma) according to manufacturer guidelines. Cells were incubated with 4′,6-diamidino-2-phenylindole (DAPI) and fluorescence of TUNEL substrates and DAPI were measured using a plate reader. TUNEL reactivity was quantified as the ratio of fluorescent TUNEL intensity to DAPI intensity. For TUNEL imaging, cells were spun onto glass slides using the Thermo Shandon Cytospin 3 and then fixed and permeabilized. TUNEL reactions were performed on slides and imaged with an upright fluorescent microscope (Axio, Zeiss).
Immunocytochemistry (ICC) for activated caspase-3 was performed on fixed and permeabilized cells. Slides were blocked with SuperBlock (ThermoFisher) for 30 minutes and then cells were incubated for 1 h with an anti-Caspase 3 (cleaved form) antibody (Cat# PC679; Millipore Sigma) at a dilution of 1:100. The secondary antibody was an AlexaFluor 555-conjugated donkey anti-rabbit antibody (Invitrogen) incubated at a dilution of 1:1000 for 30 minutes. Cells were then imaged using an upright fluorescent microscope. Positive control groups for both TUNEL and activated caspase-3 measurements consisted of re-plated cells being incubated for 6 h in the presence of 1 mM H2O2. This reflects the duration that H2O2 formation would occur for following sonication of pFUS-treated groups.
Cytosolic and Intracellular Calcium Measurements
Suspensions containing 2 × 105 cells/mL were permeabilized in divalent-ion-free phosphate buffered saline (PBS) containing 20 μM digitonin or 0.1% (v/v) Triton X-100. Cellular debris were centrifuged and Fluo-4, Pentapotassium Salt (1 μM), was added to the supernatant. Fluorescence was measured on a fluorometric plate reader and compared to a standard curve of CaCl2. Cell diameters were measured for each cell line by microscopy (Figure S1) and spherical intracellular volumes were calculated. Ca2+ concentrations from supernatants were adjusted based on intracellular volumes to approximate cytosolic Ca2+ concentrations.
Image and Data Analyses
Image analyses for activated caspase-3, Fluo-4, and the H2O2 and superoxide (MitoSOX) indicators were processed using ImageJ (v2.0). Using controls (i.e., no added dye), thresholding was applied to remove background and the mean cellular fluorescence was measured in images. Regions-of-interest (ROI) were drawn around individual cells for Fluo-4 imaging while mean cellular fluorescence of activated caspase-3, the H2O2 and superoxide indicators were measured in the entire microscopic frames. Statistical analyses and data presentation were performed in Prism (v8, GraphPad, La Jolla, CA). All figures present means and standard deviations. Statistical testing used t-tests for pairwise comparisons and one-way analyses of variance (ANOVA) for multiple comparisons. All statistical tests were two-sided and p values <0.05 were considered significant.
Results
pFUS increases TUNEL reactivity in cancer cells without inducing apoptosis
Cells were sonicated with pFUS operating at 1.1 MHz using 6 MPa PNP, 10 ms pulse length, 10 Hz pulse repetition frequency, and 300 pulses. Sonication increased nuclear TUNEL reactivity at 6 h in the B16, MDA-MB-231 and 4T1 cells (p < 0.05 by ANOVA) (N = 9 per per cell line) (Figure 1A-B). There was no increase in nuclear TUNEL reactivity detected following pFUS to C6 cells. A positive control incubating cells with H2O2 (1 mM) for 6 h prior to fixation revealed increased DNA damage in all cell lines (p < 0.05 by ANOVA). Although increased TUNEL reactivity was observed in some cell lines, ICC revealed that pFUS did not increase activation of caspase-3, an indicator of apoptosis, however apoptosis was significantly induced in all lines following incubation with H2O2 (p < 0.05 by ANOVA) (Figure 1C).
pFUS increases TUNEL reactivity without apoptosis in tumor cells. A) Representative imaging (TUNEL-positive nuclei in red) and B) quantification of TUNEL reactivity in tumor cells with or without pFUS (n = 9 per group per cell line) at 6 h post-pFUS. C) Representative imaging of ICC for cleaved (activated) caspase-3 (green) in tumor cells with or without pFUS at 6 h post-pFUS. D) Quantification of activated caspase-3 in each group (n = 9 per group per cell line). H2O2 group in each panel represents a positive controls for each measurement where cells were incubated with H2O2 (1 mM) for 6 h. Asterisks represent p < 0.05 by ANOVA comparisons performed on all groups for each cell line. Scale bars = 50 μm.
pFUS generates cytosolic Ca2+ transients and increased TUNEL reactivity is suppressed by intracellular Ca2+ chelation
To delineate the potential relationship between pFUS and TUNEL reactivity, cytosolic Ca2+ entry during sonication was determined by incubating cells with the Ca2+ ionophore Fluo-4-AM. Live cell imaging revealed cytosolic Ca2+ influxes in all cell lines (n = 64-130 cells per cell line) that persisted through the duration of pFUS treatment (Figure 2A-B). Multiple comparisons of maximal ΔF/F0 values by ANOVA revealed that MDA-MB-231 and 4T1 lines exhibited the greatest mean changes in fluorescence intensities and were statistically similar to each other (p > 0.05). The B16 line was significantly lower (p < 0.05) than either MDA-MB-231 or 4T1 cells. C6 cells exhibited significantly smaller changes (p < 0.05) than all other cell lines. In order to define the influence of cytosolic Ca2+ transients on TUNEL reactivity, cells were incubated with the Ca2+ chelator BAPTA-AM prior to pFUS treatments. BAPTA-loaded cells were measured for TUNEL reactivity at 6 h (n = 9 per cell line). BAPTA prevented pFUS from increasing TUNEL reactivity and were statistically similar to BAPTA -loaded sham-sonicated cells (p > 0.05 by t-tests) (Figure 2C).
pFUS induces Ca2+-dependent mitochondrial superoxide formation, but increased TUNEL reactivity is not suppressed by a superoxide dismutase (SOD) mimetic
To investigate the role of cytosolic Ca2+ influx and mitochondrial superoxide formation generated by pFUS, cells were incubated with MitoSOX immediately before sonication. Fluorescence quantification from live-cell imaging (n = 15 per cell line) at 2 h post-pFUS revealed significantly increased (p < 0.05 by ANOVA) superoxide production in the B16, MDA-MB-231, and 4T1 cell lines, but not in the C6 line (p > 0.05 by ANOVA) (Figure 3A-B). Chelation of cytosolic Ca2+ by BAPTA did not affect baseline levels of superoxide production (p > 0.05 by ANOVA) but, did prevent pFUS from significantly increasing superoxide production in any cell line compared to controls (p > 0.05 by ANOVA).
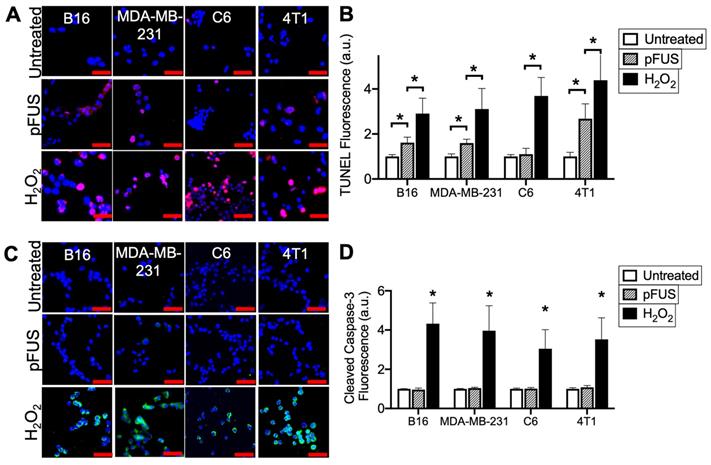
pFUS increases Ca2+-dependent superoxide formation in cells, but neutralization of superoxide by SOD mimetic does not reduce pFUS-induced increases in TUNEL reactivity. A) Representative imaging of Mitosox fluorescence intensity (red; scale bars = 100 μm) and B) quantification with or without pFUS and in the presence or absence of intracellular BAPTA at 2 h post-sonication (n = 15 per group per cell line). Mitosox was loaded immediately prior to pFUS. Asterisks represent p < 0.05 from ANOVA comparisons performed on all groups from each cell line. C) Quantification of TUNEL reactivity with or without pFUS following incubation with the SOD mimetic mtTEMPOL (n = 9 per group per cell line). Asterisks represent p < 0.05 by t-tests between treated and control groups for each cell line.
The effect of mitochondrial superoxide on TUNEL reactivity post- pFUS was investigated. Cells were incubated with the mitochondrial-targeting SOD mimetic mtTEMPOL (20 μM) prior to pFUS (n = 9 per cell line). mtTEMPOL effectively neutralized superoxide formation following pFUS treatment (Figure S1). However, mtTEMPOL did not significantly (p < 0.05 by t-test) block pFUS-induced increases in TUNEL reactivity for the B16, MDA-MB-231, and 4T1 cell lines after 6 h compared to control cells (Figure 3C). TUNEL reactivity for C6 cells did not increase following pFUS in the presence of mtTEMPOL (p > 0.05 by t-test).
pFUS induces Ca2+-dependent H2O2 formation and increased TUNEL reactivity is suppressed by peroxyl radical scavengers
SOD catalyzes the formation H2O2 radicals and therefore, H2O2 formation and its potential role in pFUS-induced TUNEL reactivity was investigated. Intracellular H2O2 was detected using a fluorescent indicator loaded into cells immediately after pFUS (n = 15 per cell type). The B16, MDA-MB-231, and 4T1 cells showed significantly increased H2O2 formation at 2 h post-pFUS compared to controls (p < 0.05 by ANOVA) (Figure 4A-B). The C6 line showed no significant increase in H2O2 formation following pFUS (p > 0.05 by ANOVA).
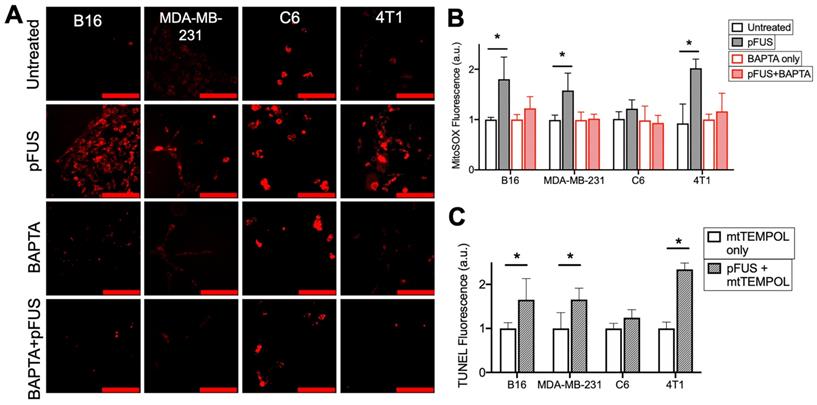
pFUS increases Ca2+-dependent formation of H2O2 and neutralization of H2O2 with Trolox suppresses pFUS-induced increases in TUNEL reactivity. A) Representative images of intracellular H2O2 indicator (MAK-164, MilliporeSigma) fluorescence (green; scale bars = 100 μm) and B) quantification with or without pFUS and in the presence or absence of intracellular BAPTA or mtTEMPOL at 2 h post-sonication. The H2O2 indicator was loaded immediately after pFUS. Asterisks represent p < 0.05 by ANOVA comparisons performed on all groups from each cell line. C) Quantification of TUNEL reactivity in cells with or with pFUS in the presence of intracellular Trolox (n = 9 per group per cell type). Statistical significance was tested between control and treated groups using t-tests for each cell line.
Chelating cytosolic Ca2+ with BAPTA (n = 15 per cell type) did not affect baseline H2O2 formation in any cell line (p > 0.05 by ANOVA), but it did suppress increased H2O2 formation following pFUS treatment in the B16, MDA-MB-231 and 4T1 lines (p < 0.05 by ANOVA) (Figure 4A-B). No significant increase in H2O2 formation was observed in sonicated C6 cells loaded with BAPTA (p < 0.05 by ANOVA). pFUS-induced H2O2 formation was also investigated in the presence of mtTEMPOL (20 μM) (n = 15 per cell type). The B16, MDA-MB-231 and 4T1 lines incubated with mtTEMPOL still demonstrated significant increases in H2O2 formation following pFUS (p < 0.05 by ANOVA) while the C6 line showed no increase in H2O2 formation following pFUS (p > 0.05).
Lastly, each cell line was incubated with the peroxyl radical scavenger Trolox (5 μM) prior to pFUS treatment and assayed for TUNEL reactivity at 6 h post-sonication (n = 9 per cell line). The presence of Trolox inhibited increased TUNEL reactivity in the B16, MDA-MB-231 and 4T1 lines following pFUS. Additionally, the C6 line exhibited no statistical increase in TUNEL reactivity in the presence of Trolox (p > 0.05 by ANOVA).
Physiological differences between tumor cell lines
The C6 cell line demonstrated markedly different responses to pFUS than other tumor cell lines assayed in this study. Although cytosolic Ca2+ transients were observed during pFUS, it was unable to increase superoxide, H2O2, or TUNEL reactivity in C6 cells. C6 cells demonstrated the smallest relative increase in Fluo-4 fluorescence during pFUS, but those relative measurements provide little information regarding the resting cytosolic Ca2+ concentrations. Therefore, the cytosolic fractions from each cell line in the absence of pFUS were collected following permeabilization with digitonin (20 μM) in a divalent-ion-free solution. Lysates were incubated with Fluo-4 (pentapotassium salt; 1 μM) (n = 8-9 per cell line) and fluorescence intensities were compared to standard curves obtained with CaCl2 solutions. Cytosolic Ca2+ concentrations were estimated by adjusting values to average intracellular volumes measured for each cell line (Figure S2). C6 cells were revealed to have statistically higher resting cytosolic concentration of Ca2+ compared to the other cell lines (Figure 5A; p < 0.05 by ANOVA). All cell lines had similar intracellular Ca2+ concentrations when measuring lysates following permeabilization with Triton X-100 (0.1% v/v) to obtain organellular fractions (n = 5 per cell line).
Unsonicated C6 cells have elevated concentrations of cytosolic Ca2+, superoxide, and H2O2 compared to other unsonicated cells types, not higher levels of TUNEL reactivity. A) Ca2+ quantification in cytosolic volumes by Fluo-4 following cell permeabilization with digitonin (20 μM) (n = 8-9 per cell type). B) Ca2+ quantification in intracellular volumes by Fluo-4 following cell permeabilization with Triton X-100 (0.1% v/v) (n = 5 per cell type). C) Quantification of Mitosox fluorescence following 2 h intracellular incubation (n = 15 per cell type). D) Quantification of intracellular H2O2 indicator (MAK-164, MilliporeSigma) fluorescence following 2 h intracellular incubation (n = 15 per cell type). E) Quantification of TUNEL reactivity in all cell types without sonication (n = 9 per cell type). Asterisks in all graphs represent p < 0.05 by ANOVA for each measurement. Groups with asterisks in (E) were statistically similar (p < 0.05).
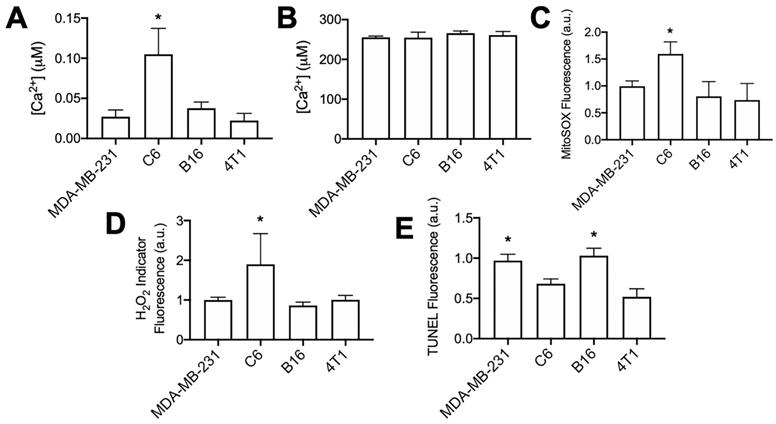
Furthermore, experiments directly comparing each cell line at baseline (no pFUS) were performed to determine relative superoxide and H2O2 production among the cell lines (n = 15 per cell line). C6 cells demonstrated statistically elevated levels (p < 0.05 by ANOVA) of both superoxide (Figure 5B) and H2O2 (Figure 5C) compared to the three other cell lines. Lastly, samples of each cell line (n = 8 per cell line) were directly compared for TUNEL reactivity without pFUS treatment. Increased cytosolic Ca2+, superoxide, and H2O2 levels in C6 cells did not correlate with higher baseline TUNEL reactivity (Figure 5D) compared to other cell lines. The B16 and MDA-MB-231 lines exhibited the highest baseline levels of TUNEL reactivity and were statistically similar to each other (p > 0.05 by ANOVA). Moreover, TUNEL reactivities in the C6 and 4T1 lines were statistically similar to each other (p > 0.05 by ANOVA) and lower than B16 and MDA-MB-231 (p < 0.05 by ANOVA).
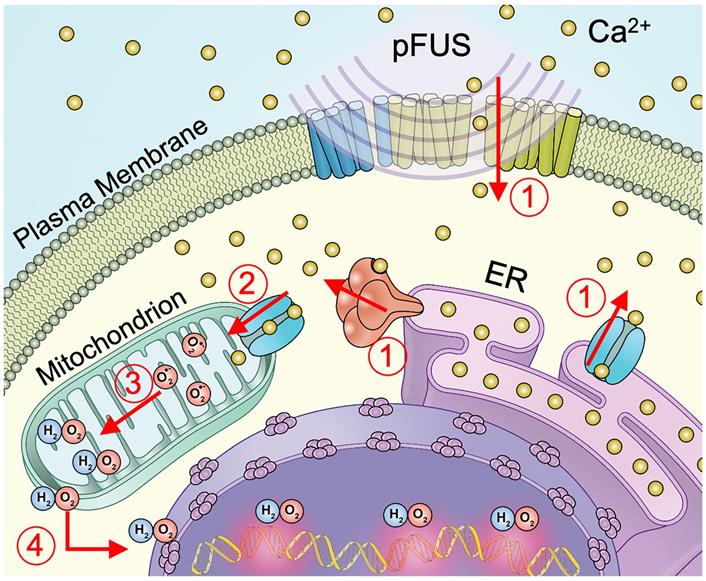
Schematic representation of cellular Ca2+ and ROS dynamics from pFUS to tumor cells. 1) pFUS causes increases in cytosolic Ca2+. Both plasma-membrane and store-release mechanisms (i.e., CICR and IP3-mediated release) are hypothesized to be involved. 2) Elevated cytosolic Ca2+ levels increase mitochondrial Ca2+ influx through the MCU leading to increased superoxide formation. 3) Reduction of superoxide forms H2O2 which 4) diffuses from the mitochondria to induce nuclear DNA damage.
Discussion
This study demonstrates that pFUS can induce DNA damage in some tumor cell lines such as murine 4T1 breast tumor, B16 melanoma and human MDA-MB-231 breast tumors, but not rat C6 glioma. DNA damage occurred after pFUS without initiating apoptosis. DNA damage resulted from pFUS eliciting cytosolic Ca2+ transients that increased mitochondrial superoxide formation and subsequent H2O2 formation. The lack of pFUS effects and inherent physiological differences of the C6 demonstrated that differences in susceptibility to pFUS exist among different tumor cell lines.
In this study, the in vitro culture systems allowed physiological measurements that are difficult in complex in vivo tumor models. Furthermore, sonicating tumor cells alone can determine whether the DNA damage observed in previous in vivo studies was a direct result of pFUS or a bystander effect from the anti-tumor shift in the TME and immune system activation. This study did not rule out potential contributions from the CCTF and immune cell populations in the TME contributing to DNA damage observed in non-ablative flank tumor studies [19, 20]. However, it demonstrated that pFUS alone at the described parameters was sufficient to cause DNA damage by TUNEL assays without apoptosis. Due to the complexity of DNA damage/repair mechanisms, apoptosis is not always the consequence of DNA damage [36]. Due to these complexities and potential differences between cell lines, this study measured direct DNA damage by TUNEL as opposed to other methods (e.g. γ-H2AX staining) that are associated DNA repair process which could confound interpretations.
pFUS generated cytosolic Ca2+ transients during sonication which were consistent with previous studies in normal tissues [6]. Live-cell Fluo-4 imaging was performed at 3 MPa to permit high-quality image acquisition and sonicated cells cultured on a glass substrate rather than in suspension. Thus, there is some difficulty extrapolating relative differences in cytosolic Ca2+ transients at 3 MPa to cells treated at 6 MPa in all other experiments. However, the necessity of increased cytosolic Ca2+ during pFUS at 6 MPa was clearly demonstrated by chelating intracellular Ca2+ with BAPTA. Increased DNA damage was not apparent in C6 glioma despite generating cytosolic Ca2+ transients. pFUS-induced DNA damage depended on cytosolic Ca2+ entry, but it remains unclear specifically how pFUS affects intracellular Ca2+ dynamics. Cytosolic Ca2+ increases during sonoporation by ultrasound with MB contrast agents have been widely reported [37-39]. Sonoporation allows diffusion of Ca2+ into the cytosol through indiscriminate openings of the plasma membrane and can induce Ca2+-dependent bioeffects [38]. However, we previously demonstrated [6] that pFUS at 4 MPa without MB opened mechano-sensitive transient receptor potential C1 (TRPC1) channels in the plasma membranes of normal kidney and skeletal muscle cells. Na+- and Ca2+-containing currents through TRPC1 channels activate complexed voltage-gated Ca2+ channels (VGCC) to amplify Ca2+ entry from the extracellular space. Future studies should examine specific mechanisms of cytosolic Ca2+ influx into tumor cells and investigate entry from both the plasma membrane and endoplasmic reticulum (ER) Ca2+ release through mechanisms such as Ca2+-induced Ca2+ release (CICR) or inositol triphosphate (IP3)-mediated Ca2+ release [40].
Increased cytosolic Ca2+ following pFUS initiates a cascade of biological processes. Cytosolic Ca2+ can generate mitochondrial Ca2+ flux through the mitochondrial Ca2+ uniporter (MCU) [41]. Mitochondrial Ca2+ influx augments O2 metabolism and drives formation of ROS, including superoxide [35]. Increased superoxide formation was observed in cell lines that exhibited pFUS-induced DNA damage and found to be dependent on elevated cytosolic Ca2+. To investigate the role of superoxide formation in pFUS-induced DNA damage, cells were loaded with a mitochondria-targeting antioxidant (mtTEMPOL) that acts as a SOD mimetic [42]. mtTEMPOL effectively scavenged mitochondrial superoxide (see Figure S2). However, mtTEMPOL did not reduce the incidence of DNA damage in pFUS-treated cells, which would presumably indicate a lack of involvement of contributing to DNA damage. SOD and mtTEMPOL catalyze a single-electron reduction of superoxide leading to the formation of the ROS H2O2 [43]. Superoxide, while thus a necessary intermediate, is membrane impermeant and would not escape the mitochondria to interact with nuclear DNA. Therefore, we investigated the role for pFUS to increase H2O2 formation via superoxide dismutation and its potential relationship to DNA damage.
Increased H2O2 formation following pFUS was detected in the three cell lines that exhibited DNA damage following pFUS. H2O2 formation was suppressed by chelating intracellular Ca2+, but not by the SOD mimetic. Since the SOD mimetic did not prevent pFUS-induced H2O2 formation it is possible that the changes in H2O2 could be involved in DNA damage. To test this hypothesis, cells were incubated with Trolox to neutralize H2O2 radicals [44] and assessed DNA damage following pFUS. Trolox suppressed increases in TUNEL reactivity following pFUS and demonstrated that H2O2 contributed to DNA damage in sonicated tumor cells. H2O2 is freely diffusible throughout the cell and H2O2 can cause nuclear DNA damage after undergoing Fenton reactions [45]. The TUNEL assay detects double-strand DNA breaks and while controversial, H2O2 can result in double-strand DNA breaks [46]. Additionally, DNA replication is frequently upregulated in tumor cells [47] and single-strand breaks from H2O2 on a complementary strand during replication would be detectable by TUNEL.
In the current study, C6 glioma cells appeared resistant to pFUS-induced increases in ROS production or DNA damage. pFUS increased cytosolic Ca2+ in C6 cells, but this did not translate into increased superoxide or H2O2 formation. Physiological profiling of the different cell types in their native states revealed that C6 cells had significantly higher levels of cytosolic Ca2+, superoxide, and H2O2. We hypothesize that elevated baseline levels of ROS would be due to elevated cytosolic Ca2+ concentrations causing increased mitochondrial Ca2+ through MCU transport. One explanation for the inability of pFUS to increase ROS through cytosolic Ca2+ transients is potentially abnormal MCU expression in C6 cells. This study did not examine mitochondrial expression of MCU in these cell lines, but human gliomas have lower MCU expression compared to other tumor types [48, 49]. It is feasible that elevated baseline cytosolic Ca2+ concentrations combined with lower MCU expression causes mitochondrial Ca2+ transport to be at or near saturation in C6 cells. Thus, the elevation in cytosolic Ca2+ from pFUS does not substantially increase mitochondrial Ca2+ flux and thus does not stimulate additional ROS formation or DNA damage. Interestingly, BAPTA alone did not reduce baseline ROS concentrations in C6 cells. This could result from the 2 h incubation period not being long enough to markedly reduce baseline ROS production. Previous studies have shown C6 cells to be sensitive to sonodynamic therapies [50, 51], which rely on Ca2+ overload. However, we hypothesize that magnitude and/or duration of increased Ca2+ between SDT and pFUS would be vastly different.
An additional observation to consider in C6 cells is that higher baseline ROS concentrations did not correlate with higher baseline TUNEL reactivity. It is possible that C6 glioma cells have inherent protection mechanisms from ROS-induced DNA damage aside from neutralizing ROS such as enhanced DNA repair processes. Previous studies in alveolar epithelium have demonstrated resistance to DNA damage in spite of chronically elevated H2O2 concentrations [52]. Further studies are warranted on the unique nature of C6 cells to further understand their resistance to pFUS-induced DNA damage. These physiological observations may help explain the resistance of gliomas to radiation therapy [53] which causes tumor cell damage via ROS production and serve as indicators to identify tumors that have altered sensitivity to pFUS-induced DNA damage.
While the lack of C6 response to pFUS is notable from a potential therapeutic standpoint, the remaining cell lines showed desired responses to pFUS, including two breast tumor lines from different species. It is unclear if the similarities in breast cancer (and also melanoma) responses are simply coincidental, but they could be related to metastatic potential of those tumor lines. Previous research has demonstrated a role for the mechanostretch receptor TRPC1 in the invasion and metastatic potential of numerous tumors [54]. While TRPC1 expression was not specifically assayed in this study, C6 gliomas have been demonstrated not to upregulate TRPC1 [55]. The potential necessity for TRPC1 to mediate pFUS-induced Ca2+ signaling at similar intensities to those used here [6] open the possibility of a correlation between pFUS responses and expression of mechanostrech receptors like TRPC1. Further investigations are warranted to examine the expression of TRPC1 or similar stretch-sensitive cationic channels and pFUS-induced DNA damage in different types of cancers and cell lines from different species.
The primary goal of this study was to investigate biological mechanisms behind DNA damage from pFUS, however the results have wide implications for potential clinical translation as an anti-tumor therapy for three main reasons. First, DNA damage may play a critical role in activating the immune system. While extracellular DNA is a DAMP [26, 27], DNA damage and repair processes in non-apoptotic cells can stimulate the immune system [25]. Interestingly, this investigation also revealed H2O2 as a mediator of DNA damage. However, H2O2 also induces neutrophil recruitment as the first phase of innate immune response and could directly contribute to a shift towards a pro-inflammatory TME [56]. Second, the lack of apoptosis is an initial demonstration that non-ablative pFUS may be possible to incorporate as an immunological adjuvant approach to treat peripheral tissues containing radiologically undetected local micro-metastatic disease. Non-destructive pFUS could be used to alter the microenvironment of normal-appearing tissue to trigger innate and adaptive immune responses in areas containing microscopic tumors while preserving the architecture and function of nonmalignant tissues. Our previous work has extensively demonstrated pFUS at 4 MPa does not have deleterious effects on normal and diseased muscle [9-12] or kidney [7, 13, 15] tissue, yet augments a pro-inflammatory microenvironment to increase homing of stem cells. Future studies would need to be performed at different pFUS intensities to see if optimal parameters could be identified to mount anti-tumor responses in regions containing radiologically undetectable disease without negatively impacting surrounding normal tissues. Lastly, not all tumor types may be responsive to the mechanical effects of pFUS. The results of this study suggest that inherent biological differences between tumor cell types could render some tumors more or less sensitive to pFUS.
This study has several limitations. First, in vivo studies will be necessary to confirm that mechanotransductive effects of pFUS observed in suspended or glass-plated cells are similar in solid tumors and also to clarify the explicit roles of both H2O2 formation and DNA damage in the shift toward proinflammatory TME in vivo. Our previous studies [19, 20] did not examine H2O2 formation or neutrophil infiltration, but the role of H2O2 in pro-inflammatory TME shifts after pFUS could be due to neutrophil recruitment in addition to DNA damage. Another limitation is that only four tumor cell lines were evaluated. It is unclear how numerous other cancer cell types would respond to pFUS-induced Ca2+ fluxes regarding oxidative stress and DNA damage. Investigating additional tumor lines may provide further understanding of the mechanobiology induced by pFUS and its therapeutic potential. Lastly, pFUS in this study did not initiate apoptosis in the tumor cells. Future studies will need to examine the effects of pFUS on cell cycle progression (e.g. inducing senescence) and proliferation which could also reduce tumor growth rates in vivo.
In conclusion, in vitro cell culture models have demonstrated that pFUS generates downstream biological effects through cytosolic Ca2+ influx that resulted in mitochondrial ROS formation and double-strand DNA damage in the absence of apoptosis. This study investigated several tumor types and observed heterogenous pFUS responses between cell lines. Furthermore, it suggests differences in pFUS effects arise from specific physiological differences and could provide direction for optimal implementation and translation of pFUS into clinical trials.
Supplementary Material
Supplementary figures.
Acknowledgements
This study was funded by the Intramural Research Programs at the National Institutes of Health Clinical Center and the National Institute of Biomedical Imaging and Bioengineering.
Author Contributions
RBR, JAF, and SRB conceptualized study and designed experiments; RBR performed experiments; RBR and SRB analyzed data; RBR, JAF, and SRB wrote the paper. All authors have read and approved the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Izadifar Z, Izadifar Z, Chapman D, Babyn P. An Introduction to High Intensity Focused Ultrasound: Systematic Review on Principles, Devices, and Clinical Applications. J Clin Med. 2020 9
2. Elhelf IAS, Albahar H, Shah U, Oto A, Cressman E, Almekkawy M. High intensity focused ultrasound: The fundamentals, clinical applications and research trends. Diagn Interv Imaging. 2018;99:349-59
3. Sheybani ND, Price RJ. Perspectives on Recent Progress in Focused Ultrasound Immunotherapy. Theranostics. 2019;9:7749-58
4. Schade GR, Wang YN, D'Andrea S, Hwang JH, Liles WC, Khokhlova TD. Boiling Histotripsy Ablation of Renal Cell Carcinoma in the Eker Rat Promotes a Systemic Inflammatory Response. Ultrasound Med Biol. 2019;45:137-47
5. O'Brien WD Jr. Ultrasound-biophysics mechanisms. Prog Biophys Mol Biol. 2007;93:212-55
6. Burks SR, Lorsung RM, Nagle ME, Tu TW, Frank JA. Focused ultrasound activates voltage-gated calcium channels through depolarizing TRPC1 sodium currents in kidney and skeletal muscle. Theranostics. 2019;9:5517-31
7. Burks SR, Nguyen BA, Bresler MN, Nagle ME, Kim SJ, Frank JA. Anti-inflammatory drugs suppress ultrasound-mediated mesenchymal stromal cell tropism to kidneys. Sci Rep. 2017;7:8607
8. Kovacs ZI, Kim S, Jikaria N, Qureshi F, Milo B, Lewis BK. et al. Disrupting the blood-brain barrier by focused ultrasound induces sterile inflammation. Proc Natl Acad Sci U S A. 2017;114:E75-E84
9. Tebebi PA, Burks SR, Kim SJ, Williams RA, Nguyen BA, Venkatesh P. et al. Cyclooxygenase-2 or tumor necrosis factor-alpha inhibitors attenuate the mechanotransductive effects of pulsed focused ultrasound to suppress mesenchymal stromal cell homing to healthy and dystrophic muscle. Stem Cells. 2015;33:1173-86
10. Burks SR, Ziadloo A, Hancock HA, Chaudhry A, Dean DD, Lewis BK. et al. Investigation of cellular and molecular responses to pulsed focused ultrasound in a mouse model. PLoS One. 2011;6:e24730
11. Burks SR, Ziadloo A, Kim SJ, Nguyen BA, Frank JA. Noninvasive pulsed focused ultrasound allows spatiotemporal control of targeted homing for multiple stem cell types in murine skeletal muscle and the magnitude of cell homing can be increased through repeated applications. Stem Cells. 2013;31:2551-60
12. Tebebi PA, Kim SJ, Williams RA, Milo B, Frenkel V, Burks SR. et al. Improving the therapeutic efficacy of mesenchymal stromal cells to restore perfusion in critical limb ischemia through pulsed focused ultrasound. Sci Rep. 2017;7:41550
13. Burks SR, Nagle ME, Bresler MN, Kim SJ, Star RA, Frank JA. Mesenchymal stromal cell potency to treat acute kidney injury increased by ultrasound-activated interferon-gamma/interleukin-10 axis. J Cell Mol Med. 2018;22:6015-25
14. Burks SR, Nguyen BA, Tebebi PA, Kim SJ, Bresler MN, Ziadloo A. et al. Pulsed focused ultrasound pretreatment improves mesenchymal stromal cell efficacy in preventing and rescuing established acute kidney injury in mice. Stem Cells. 2015;33:1241-53
15. Ziadloo A, Burks SR, Gold EM, Lewis BK, Chaudhry A, Merino MJ. et al. Enhanced homing permeability and retention of bone marrow stromal cells by noninvasive pulsed focused ultrasound. Stem Cells. 2012;30:1216-27
16. Jang KW, Tu TW, Nagle ME, Lewis BK, Burks SR, Frank JA. Molecular and histological effects of MR-guided pulsed focused ultrasound to the rat heart. J Transl Med. 2017;15:252
17. Razavi M, Zheng F, Telichko A, Ullah M, Dahl J, Thakor AS. Effect of Pulsed Focused Ultrasound on the Native Pancreas. Ultrasound Med Biol. 2020;46:630-8
18. McMahon D, Hynynen K. Acute Inflammatory Response Following Increased Blood-Brain Barrier Permeability Induced by Focused Ultrasound is Dependent on Microbubble Dose. Theranostics. 2017;7:3989-4000
19. Aydin O, Chandran P, Lorsung RR, Cohen G, Burks SR, Frank JA. The Proteomic Effects of Pulsed Focused Ultrasound on Tumor Microenvironments of Murine Melanoma and Breast Cancer Models. Ultrasound Med Biol. 2019;45:3232-45
20. Cohen G, Chandran P, Lorsung RM, Tomlinson LE, Sundby M, Burks SR. et al. The Impact of Focused Ultrasound in Two Tumor Models: Temporal Alterations in the Natural History on Tumor Microenvironment and Immune Cell Response. Cancers (Basel). 2020 12
21. Liu DD, Ullah M, Concepcion W, Dahl JJ, Thakor AS. The role of ultrasound in enhancing mesenchymal stromal cell-based therapies. Stem Cells Transl Med. 2020
22. Ren G, Rezaee M, Razavi M, Taysir A, Wang J, Thakor AS. Adipose tissue-derived mesenchymal stem cells rescue the function of islets transplanted in sub-therapeutic numbers via their angiogenic properties. Cell Tissue Res. 2019;376:353-64
23. Ullah M, Liu DD, Rai S, Dadhania A, Jonnakuti S, Concepcion W. et al. Reversing Acute Kidney Injury Using Pulsed Focused Ultrasound and MSC Therapy: A Role for HSP-Mediated PI3K/AKT Signaling. Mol Ther Methods Clin Dev. 2020;17:683-94
24. Ullah M, Liu DD, Thakor AS. Mesenchymal Stromal Cell Homing: Mechanisms and Strategies for Improvement. iScience. 2019;15:421-38
25. Pateras IS, Havaki S, Nikitopoulou X, Vougas K, Townsend PA, Panayiotidis MI. et al. The DNA damage response and immune signaling alliance: Is it good or bad? Nature decides when and where. Pharmacol Ther. 2015;154:36-56
26. Lamphier MS, Sirois CM, Verma A, Golenbock DT, Latz E. TLR9 and the recognition of self and non-self nucleic acids. Ann N Y Acad Sci. 2006;1082:31-43
27. Ishii KJ, Suzuki K, Coban C, Takeshita F, Itoh Y, Matoba H. et al. Genomic DNA released by dying cells induces the maturation of APCs. J Immunol. 2001;167:2602-7
28. Razavi M, Zheng F, Telichko A, Wang J, Ren G, Dahl J. et al. Improving the Function and Engraftment of Transplanted Pancreatic Islets Using Pulsed Focused Ultrasound Therapy. Sci Rep. 2019;9:13416
29. Hu M, Lee W, Jiao J, Li X, Gibbons DE, Hassan CR. et al. Mechanobiological modulation of in situ and in vivo osteocyte calcium oscillation by acoustic radiation force. Ann N Y Acad Sci. 2020;1460:68-76
30. Liao D, Li F, Lu D, Zhong P. Activation of Piezo1 mechanosensitive ion channel in HEK293T cells by 30MHz vertically deployed surface acoustic waves. Biochem Biophys Res Commun. 2019;518:541-7
31. Yoon CW, Jung H, Goo K, Moon S, Koo KM, Lee NS. et al. Low-Intensity Ultrasound Modulates Ca(2+) Dynamics in Human Mesenchymal Stem Cells via Connexin 43 Hemichannel. Ann Biomed Eng. 2018;46:48-59
32. Ibsen S, Tong A, Schutt C, Esener S, Chalasani SH. Sonogenetics is a non-invasive approach to activating neurons in Caenorhabditis elegans. Nat Commun. 2015;6:8264
33. Hwang JY, Lim HG, Yoon CW, Lam KH, Yoon S, Lee C. et al. Non-contact high-frequency ultrasound microbeam stimulation for studying mechanotransduction in human umbilical vein endothelial cells. Ultrasound Med Biol. 2014;40:2172-82
34. Qi L, Zhang Q, Tan Y, Lam KH, Zheng H, Qian M. Non-Contact High-Frequency Ultrasound Microbeam Stimulation: A Novel Finding and Potential Causes of Cell Responses. IEEE Trans Biomed Eng. 2020;67:1074-82
35. Delierneux C, Kouba S, Shanmughapriya S, Potier-Cartereau M, Trebak M, Hempel N. Mitochondrial Calcium Regulation of Redox Signaling in Cancer. Cells. 2020 9
36. De Zio D, Cianfanelli V, Cecconi F. New insights into the link between DNA damage and apoptosis. Antioxid Redox Signal. 2013;19:559-71
37. Beekers I, Mastik F, Beurskens R, Tang PY, Vegter M, van der Steen AFW. et al. High-Resolution Imaging of Intracellular Calcium Fluctuations Caused by Oscillating Microbubbles. Ultrasound Med Biol. 2020
38. Hassan MA, Campbell P, Kondo T. The role of Ca(2+) in ultrasound-elicited bioeffects: progress, perspectives and prospects. Drug Discov Today. 2010;15:892-906
39. Park J, Fan Z, Deng CX. Effects of shear stress cultivation on cell membrane disruption and intracellular calcium concentration in sonoporation of endothelial cells. J Biomech. 2011;44:164-9
40. Bootman MD, Berridge MJ, Roderick HL. Calcium signalling: more messengers, more channels, more complexity. Curr Biol. 2002;12:R563-5
41. Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360-4
42. Iannone A, Bini A, Swartz HM, Tomasi A, Vannini V. Metabolism in rat liver microsomes of the nitroxide spin probe tempol. Biochem Pharmacol. 1989;38:2581-6
43. Srnec M, Aquilante F, Ryde U, Rulisek L. Reaction mechanism of manganese superoxide dismutase studied by combined quantum and molecular mechanical calculations and multiconfigurational methods. J Phys Chem B. 2009;113:6074-86
44. Scott JW, Cort WM, Harley H, Parrish DR, Saucy G. 6-Hydroxychroman-2-Carboxylic Acids - Novel Antioxidants. J Am Oil Chem Soc. 1974;51:200-3
45. Imlay JA, Chin SM, Linn S. Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science. 1988;240:640-2
46. Driessens N, Versteyhe S, Ghaddhab C, Burniat A, De Deken X, Van Sande J. et al. Hydrogen peroxide induces DNA single- and double-strand breaks in thyroid cells and is therefore a potential mutagen for this organ. Endocr Relat Cancer. 2009;16:845-56
47. Lange SS, Takata K, Wood RD. DNA polymerases and cancer. Nat Rev Cancer. 2011;11:96-110
48. Uhlen M, Zhang C, Lee S, Sjostedt E, Fagerberg L, Bidkhori G. et al. A pathology atlas of the human cancer transcriptome. Science. 2017 357
49. Ponten F, Jirstrom K, Uhlen M. The Human Protein Atlas-a tool for pathology. J Pathol. 2008;216:387-93
50. Song D, Yue W, Li Z, Li J, Zhao J, Zhang N. Study of the mechanism of sonodynamic therapy in a rat glioma model. Onco Targets Ther. 2014;7:1801-10
51. Hao D, Song Y, Che Z, Liu Q. Calcium overload and in vitro apoptosis of the C6 glioma cells mediated by sonodynamic therapy (hematoporphyrin monomethyl ether and ultrasound). Cell Biochem Biophys. 2014;70:1445-52
52. Fujii Y, Tomita K, Sano H, Yamasaki A, Hitsuda Y, Adcock IM. et al. Dissociation of DNA damage and mitochondrial injury caused by hydrogen peroxide in SV-40 transformed lung epithelial cells. Cancer Cell Int. 2002;2:16
53. Osuka S, Van Meir EG. Overcoming therapeutic resistance in glioblastoma: the way forward. J Clin Invest. 2017;127:415-26
54. Elzamzamy OM, Penner R, Hazlehurst LA. The Role of TRPC1 in Modulating Cancer Progression. Cells. 2020 9
55. Grimaldi M, Maratos M, Verma A. Transient receptor potential channel activation causes a novel form of [Ca 2+]I oscillations and is not involved in capacitative Ca 2+ entry in glial cells. J Neurosci. 2003;23:4737-45
56. Klyubin IV, Kirpichnikova KM, Gamaley IA. Hydrogen peroxide-induced chemotaxis of mouse peritoneal neutrophils. Eur J Cell Biol. 1996;70:347-51
Author contact
![]() Corresponding author: Scott R. Burks, Ph.D., 10 Center Dr., RmB1N256, Bethesda, MD 20892. Ph: (301) 594-2368; scott.burksgov
Corresponding author: Scott R. Burks, Ph.D., 10 Center Dr., RmB1N256, Bethesda, MD 20892. Ph: (301) 594-2368; scott.burksgov