Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Glioblastoma intratumoral...
Hypoxia and acidic stress as...
Hypoxia and/or HIF signaling...
The relationship between acidic...
Hypoxia-induced angiogenesis and...
Hypoxia and GSC invasion
Hypoxia and acidic stress...
Interactions of the immune...
Concluding Statement
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(2):665-683. doi:10.7150/thno.41692 This issue Cite
Review
Glioma stem cells and their roles within the hypoxic tumor microenvironment
Nathaniel H. Boyd1, Anh Nhat Tran2, Joshua D. Bernstock3, Tina Etminan4, Amber B. Jones1, G. Yancey Gillespie5, Gregory K. Friedman6, Anita B. Hjelmeland1,5 ![]()
1. Department of Cell, Developmental, and Integrative Biology, University of Alabama at Birmingham, Birmingham, AL.
2. Department of Neurosurgery, Northwestern University, Chicago, IL.
3. Department of Neurosurgery, Brigham and Women's Hospital, Harvard Medical School, Boston, MA, USA.
4. Department of Chemistry, University of Alabama at Birmingham, Birmingham, AL.
5. Department of Neurosurgery, University of Alabama at Birmingham, Birmingham, AL.
6. Division of Pediatric Hematology and Oncology, Department of Pediatrics, University of Alabama at Birmingham, Birmingham, AL.
Received 2019-10-31; Accepted 2020-8-4; Published 2021-1-1
Abstract

Tumor microenvironments are the result of cellular alterations in cancer that support unrestricted growth and proliferation and result in further modifications in cell behavior, which are critical for tumor progression. Angiogenesis and therapeutic resistance are known to be modulated by hypoxia and other tumor microenvironments, such as acidic stress, both of which are core features of the glioblastoma microenvironment. Hypoxia has also been shown to promote a stem-like state in both non-neoplastic and tumor cells. In glial tumors, glioma stem cells (GSCs) are central in tumor growth, angiogenesis, and therapeutic resistance, and further investigation of the interplay between tumor microenvironments and GSCs is critical to the search for better treatment options for glioblastoma. Accordingly, we summarize the impact of hypoxia and acidic stress on GSC signaling and biologic phenotypes, and potential methods to inhibit these pathways.
Keywords: acidic stress, glioma, hypoxia, cancer stem cells, tumor microenvironment
Introduction
Glioblastoma (GBM), also known as a World Health Organization grade IV astrocytoma, is the most common and aggressive primary brain tumor in adults. From 2012-2016, the average incidence of malignant brain tumors in the U.S. was 7.08 per 100,000, and GBM accounted for about 15% of all central nervous system tumors, and close to half of all malignant brain tumors diagnosed. GBMs have a disproportionate incidence rate by sex and race, occurring 1.58 times more often in males than females, 1.95 times more often in whites than blacks, and 2.39 times more often in whites than Asian or Pacific Islanders [1]. Although past studies have significantly increased our understanding of the signaling pathways and molecular processes involved in gliomagenesis, the prognosis remains dismal despite a multimodal approach utilizing maximal surgical resection, adjuvant radiation and chemotherapy using the DNA alkylating agent, temozolomide (TMZ) [2]. Patients receiving standard of care have a median survival of 14.6 months, with only around 10% of patients surviving 5 years or longer [3]. The failure of current treatments in substantially prolonging life expectancy highlights the importance of increasing our understanding of the pathobiology promoting/driving GBM growth, recurrence and therapeutic resistance.
Glioblastoma intratumoral heterogeneity
A characteristic that tumor cells share with non-neoplastic stem cells is their ability to proliferate indefinitely. Accordingly, many tumors appear to be maintained via a hierarchical organization that consists of slowly-dividing stem cells, precursor cells, and differentiated cells [4, 5]. In line with such evidence, only a subset of cancer cells has the ability to form tumors within immunodeficient mouse models when derived from multiple cancers [6]. In brain tumors, this subset of cells, known glioma stem cells (GSCs), exhibit self-renewal that can be measured in vitro via neurosphere formation assays and the expression of molecular markers (e.g. SOX2, NANOG, CD15, CD133) in symmetric and asymmetric division studies [7]. Indeed, GBMs have long been known to express both neural and glial markers, which suggest a cell subset with neural stem cell-like characteristics [4, 8]. While a number of methods have been used to isolate GSCs, fluorescence-activated cell sorting (FACS) using the cell surface marker CD133 remains one of the most characterized [9-11]. Isolation of CD133 positive and negative populations in GSCs first revealed differences in tumor propagation in xenograft mouse models [9, 12]. Subsequent investigation revealed co-expression of CD133 with Nestin and other canonical neural stem cell markers, and increases of CD133 expressing GSCs post-irradiation, indicative of a therapy-resistant, stem-like fraction [13]. In addition to self-renewal and a multilineage differentiation capacity, GSCs exhibit invasive and angiogenic potential, as well as therapeutic resistance, which will be discussed in more detail.
Through genetic and epigenetic characterization, a number of adult GBM subtypes have been defined [14]. Of note, single cell RNA-sequencing has clearly determined that multiple subtypes exist within one GBM tumor [4, 15], suggesting that the vast heterogeneity within these tumors may complicate the ultimate goal of preventing recurrence of GBMs. Heterogeneity is further complicated by the presence of GSCs that are capable of propagating tumors in immunocompromised mice, as well as maintaining the expression of neural stem cell markers and/or dividing asymmetrically to generate more differentiated progeny [9-11]. Critically, GSCs resist radiation- and chemotherapy-induced cell death to a greater extent than the bulk tumor, with data from mouse models indicating that a quiescent GSC fraction is directly associated with therapeutic resistance and tumor recurrence. These GSCs reside in multiple niches that include those located in tumor microenvironments (e.g. low oxygen tension [hypoxia], acidic stress, and/or nutrient restriction) that promote the characteristics mentioned above and contribute to intratumoral heterogeneity, which leads to major challenges in treatment [16]. Thus, understanding GSC molecular signaling pathways in the context of the tumor microenvironment is of paramount importance for developing novel treatment paradigms for this intractable central nervous system neoplasm [17, 18].
GSCs assist in establishing the tumor microenvironment through complex crosstalk within their niche. The two most commonly described niches in which GSCs have been characterized are the perivascular and the perinecrotic niches [19-21]. Both of these niches deliver instructive cues that serve to maintain GSCs and stimulate cellular plasticity towards a stem-like phenotype [22, 23]. Perinecrotic niches are enriched for cells expressing molecular markers of both hypoxia and GSCs (e.g. SOX2, NANOG, CD133) [24, 25], suggesting a connection between the tumor microenvironment and differentiation state of cells. Similarities between GSC-regulated and hypoxia-induced biology strengthen these apparent connections. For example, angiogenesis and invasion are well-established pro-tumorigenic cellular behaviors induced by hypoxia, while tumors that arise from GSCs are highly vascular and more invasive as compared to tumors generated from non-GSCs. As GSCs are responsible for tumor propagation and invasion, promote angiogenesis, are resistant to therapy, and contribute to tumor recurrence, it is essential to develop therapeutic agents capable of targeting GSCs.
Other GSC-associated niches, the peri-hypoxic, peri-immune, and extracellular matrix niche are reviewed in-depth in Aderetti et al. [26]. Briefly, the peri-hypoxic niche promotes the stemness capability of GSCs, as well as promotes the acidification of the tumor microenvironment, which stabilizes HIF [27]. Acidosis can also be induced through elevated carbonic anhydrase, lactate, and ion transporter activity [28]. The peri-immune niche is maintained through upregulated activity of tumor associated macrophages with enhanced immunosuppressive activity [29], and the ECM niche has immense interaction with the other distinct niches, and its components can influence GSCs ability to synthesize their own ECM in both a direct and indirect manner [30]. These physiologic niches comprise an integrated network that can promote overall GSC maintenance, and each component will be described in-depth in the current review.
Hypoxia and acidic stress as important factors in the brain tumor microenvironment
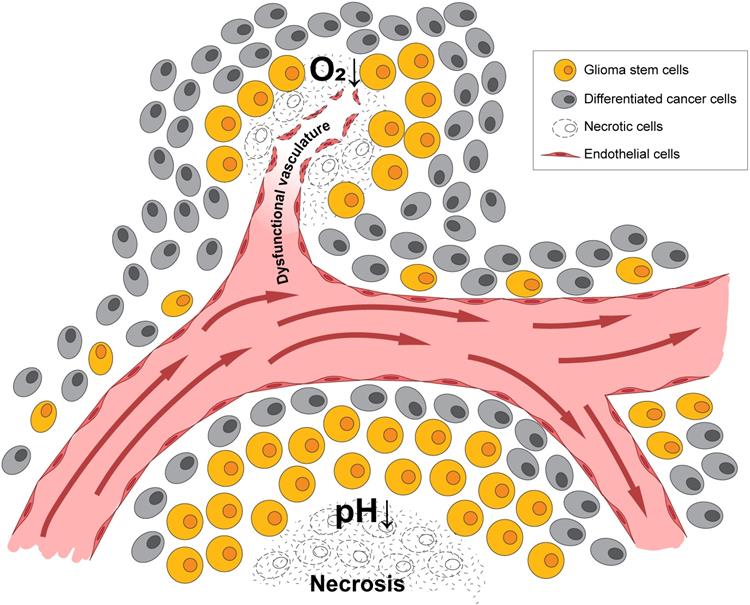
A pathologic hallmark of GBMs, pseudopalisading necrosis, often occurs near a collapsed blood vessel, and tends to be surrounded by cells surviving in a hypoxic and often, acidic zone [31]. In patients with GBM and in murine xenograft models of GBMs, it has been revealed that a median partial pressure of oxygen (pO2) of 5-9 mm Hg and an acidic pH of 6.8 or lower [32-34] often existed within these tumors. In contrast, oxygen tension in arterial blood is approximately 70-100 mm Hg, while the normal brain often measures 25-40 mm Hg with some niches being even lower. Thus, there is a significant difference between physiologic oxygen tension and pH, and that of the tumor. While hypoxia and low pH often occur simultaneously, studies in xenograft models using microscopy with phosphorescence quenching to monitor pO2, and ratio imaging to measure pH, have demonstrated these microenvironments occur independently [35], as illustrated in Figure 1. As hypoxia and/or low pH correlate with many aspects of tumorigenicity, including therapeutic resistance, patient survival, and tumor invasion, understanding tumor microenvironmental effects on GBM growth and recurrence is critical [36].
Hypoxia and acidic stress exist in microenvironmental niches for brain tumor initiating cells. Normally represented together as necrotic zones, they are also found separately in brain tumors and can independently affect biologies and gene expression patterns.
The presence of hypoxia promotes the use of anaerobic glycolysis to generate energy and essential precursors (nucleic and amino acids as well as lipids) required for cell growth [37]. Anaerobic glycolysis results in the production of acidic metabolites, including lactic acid, and facilitates an altered pH balance in solid tumors wherein extracellular pH is lower than intracellular pH. There is a marked reduction in median solid tumor partial pressure of oxygen (pO2) and extracellular pH compared to non-neoplastic tissue, as well as the development of regions of extreme hypoxia and low pH [38]. However, oxygen tensions vary significantly depending on the non-neoplastic tissue, indicating the importance of establishing a physiologic normoxia control when designing experiments to uncover differences in these microenvironmental conditions. While there are some innovative studies attempting to address this problem, the overwhelming majority of in vitro experiments continue to be performed in atmospheric oxygen (~21% O2 or 159 mmHg) using buffered media to minimize pH changes (typically pH 7.4) [39]. Recognizing the limitations of these in vitro approaches, a shift towards improved modeling of physiologic microenvironments is desperately needed.
Hypoxia-responsive signaling in both tumors and nonmalignant tissue is mediated through transcription factors called hypoxia-inducible factors (HIFs). HIFs exist as heterodimers, consisting of an alpha and a beta subunit. The beta subunit is constitutively expressed in all cells, while the alpha subunit exists in three isoforms, which are unstable and rapidly degraded in the presence of oxygen [40, 41]. Under well-oxygenated conditions, prolyl hydroxylases (e.g. PHD1, 2, and 3) hydroxylate proline residues on HIFα subunits. The hydroxylation site acts as a substrate for Von Hippel Lindau factor (VHL), which ubiquitinates the HIFα subunit and targets it for proteasomal degradation. However, under hypoxic conditions, the interaction between VHL and the HIF alpha subunit is disrupted. Free iron and α-ketoglutarate are required for the hydroxylation of the proline residues. In hypoxia, free iron is chelated, and the alpha subunit cannot be hydroxylated and is therefore stabilized [42, 43]. The HIFα subunit can then translocate to the nucleus, bind to the beta subunit, and subsequently recognize hypoxia-responsive elements (HREs), which are consensus sequences (5'-ACGTG-3') in the promoter region of identified target genes. The binding of HIFs to these sequences upregulates the transcription of genes controlling cell survival, glycolysis, pH regulation, angiogenesis, migration, and invasion [40, 44]. This becomes particularly important in secondary GBMs, as many possess a mutation in the Isocitrate Dehydrogenase (IDH) enzyme that is responsible for the conversion of isocitrate to α-ketoglutarate [45]. IDH mutations disrupt enzyme functionality, causing the production of D-2-hydroxyglutarate, a competitive inhibitor of α-ketoglutarate. Through this mechanism, HIF levels may be increased [46] and the mutation is associated with global methylation changes [47]. Furthermore, hypoxia or low pH in normoxia in glioma cells increased levels of L-2-hydroxyglutarate (a mirror-image enantiomer) and acidic stress increased HIF stabilization [48, 49].
Two well-characterized HIF alpha isoforms are HIF1α and HIF2α. Suggesting that these two isoforms program distinct responses to changes in the microenvironment, HIF2α expression can be stabilized at higher oxygen levels (approximately 5%) than the more severely hypoxic conditions (less than or equal to 1%), where HIF1α is induced [50-52]. These two isoforms both bind to HRE sequences but can have distinct target genes. For example, genes preferentially induced by HIF2α have ETS binding elements adjacent to HREs [53]. HIF2α can also stabilize MYC and myc associated protein (MAX) interactions to enhance myc transcription [54, 55]. In contrast, HIF1α appears to bind to MAX to prevent MYC signaling. Thus, the isoforms of HIFα can function to activate distinct signaling compartments, although there are many common, HRE containing HIF targets such as Vascular Endothelial Growth Factor (VEGF), a well-known proangiogenic protein.
Hypoxia and/or HIF signaling promote GSC maintenance
GSCs are identified using cell surface markers or cell selection techniques that take advantage of phenotypes increased in this cellular subset, with sorting or validation using GSC markers that can include CD133, OCT4, and SOX2 among others [7, 9, 12, 13, 52, 56, 57]. Exposure to hypoxia upregulates these same canonical stem cell genes and facilitates plasticity towards a stem-like state (as indicated by increased expression of stem cell markers including CD133) [52, 58-64]. Furthermore, the hypoxic microenvironment may help GBM cells under certain inhibitor treatments to maintain their stem-like phenotype, while normoxia does not [65]. Hypoxia was also shown to promote glycosylation of CD133, which could play a role in the process of anti-hypoxia-mediated apoptosis [66]. This is notable as antibodies used to isolate CD133 during flow cytometry are often specific to the glycosylated form of the protein. Other GSC markers that increased in expression under relatively low oxygen levels for GBM include podoplanin, BMI-1, and Nestin [62]. However, Sox2 expression increased only in in vivo-like multicellular tumor spheroids derived from GBM short-term culture with tumor stem cell properties, indicating that tumor cell phenotypes associated with stemness and chemoresistance may depend on the oxygen tension surrounding that particular tumor cell as well as cellular interactions [62]. Thus, hypoxia increases expression of many GSC markers, which may contribute to the growth of the tumor.
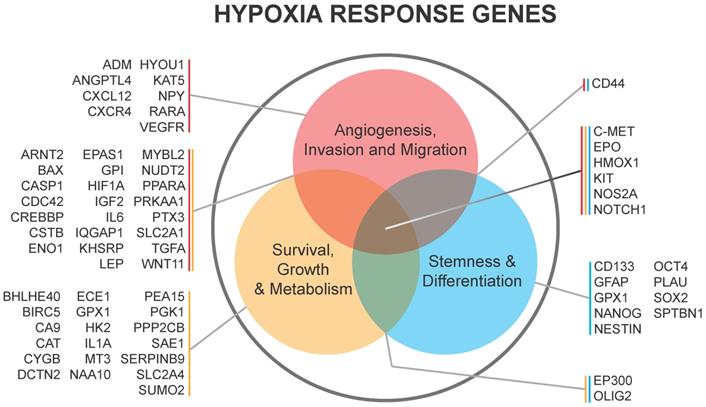
HIF1α and HIF2α are both critical for GSC function and can be expressed in different patterns dependent upon the level of hypoxia as mentioned above. The stabilization of HIF1α leads to the expansion of the GSC population within the bulk of the tumor, which is, in part, mediated by the extracellular signaling related kinase (ERK) and the PI3K/AKT pathways [58]. Conversely, RNA interference-mediated silencing of HIF1α depleted the self-renewal capacity of GSCs and led to a reduction of tumorigenic potential in vivo [58]. HIF1α is also a negative regulator of bone morphogenetic proteins [67], which are known to signal differentiation of GSCs towards an astrocyte lineage [68]. HIF1α/STAT3 co-activator complex induces the transcription of Vasorin, which, in turn, stabilizes Notch1 and augments Notch signaling, promoting GSC maintenance [69]. Additional evidence supports the importance of HIF1α activation of the JAK1/2-STAT3 transcriptional program, including via VEGF as an important autocrine factor, to enhance GSC maintenance [70]. Thus, HIF1α promotes GSC self-renewal and represses differentiation to increase GBM growth. A list of genes regulated by hypoxia in GSCs is shown in Figure 2.
Hypoxia response genes in glioma and their subsequent downstream biologies relevant to BTICs in Li et al. 2009 and Keith et al. 2011.
Under less extreme hypoxia, when HIF2α is preferentially expressed in GSC populations, HIF2α levels were regulated not only via post-translational modification but also through increased transcription [50]. HIF2α activity is repressed by the DEAD box protein DDX28 [71], while stability is also regulated by the transcriptional regulator Inhibitor of DNA Binding 2 (ID2) [72]. In normoxia, dual specificity tyrosine-phosphorylation-regulated kinases (DYRK1A and DYRK1B) phosphorylated ID2. Phosphorylated ID2 can no longer interact with the VHL ubiquitin ligase complex, so HIF2α is ubiquitinated and degraded. In hypoxia, phosphorylation of DYRKs and ID2 is decreased, resulting in HIF2α expression. HIF2α stabilization upregulates some stem cell factors, including Oct4, Sox2, and Nanog, while knockdown of HIF2α reduced the self-renewal capacity of GSCs in vitro and decreased tumor growth in vivo [50, 58, 59, 63, 73-75]. Recently, CD44 was shown to be cleaved into an intracellular domain that interacts with HIF2α and promotes HIF dependent hypoxic signaling [76]. As CD44 is associated with mesenchymal GSCs, this data highlights the potential for heterogeneous hypoxic responses in GBM. Together, the data indicate that hypoxia and HIFs play important roles in maintaining the GSC phenotype through multiple mechanisms [50].
The relationship between acidic stress and GSCs
Although low pH is recognized to be an important component of the tumor microenvironment and has been shown to exist in the absence of hypoxia in vivo, relatively few studies focus on the biological effects of acidic stress in GBM or other solid tumor cells. The extracellular pH of solid tumors including GBMs has been measured as low as 5.9 with an average pH of 6.8, whereas the normal brain pH is approximately pH 7.1 [77]. Considering the protumorigenic properties of tumor acidification and the altered pH gradient in cancer (pHExtracellular< pHIntracellular), drugs altering proton export or production, and buffer therapies are being explored as novel treatments [78-86], as well as metabolic enzymes that contribute to the production of lactic acid as a result of a glycolytic shift [87]. Sodium bicarbonate buffer therapies have decreased tumor growth and metastasis in breast and prostate cancers [81, 85, 88, 89], and carbonic anhydrase IX inhibition effectively targeted breast cancer initiating cells in vivo and improved the efficacy of immunotherapy [90, 91]. Thus, there are multiple strategies that could be employed to modulate pH with only a limited number having been explored in GBM, particularly in the context of standard of care.
Applying acidic stress to GBM cells upregulates VEGFA mRNA via the ERK1/2 and MAPK pathway, which enhances AP-1 binding to the VEGF promoter [92]. Importantly, VEGF can be upregulated by hypoxia or acidic stress independently, including in GSCs [11, 35, 93]. These data suggest that both regions are involved in GBM neovascularization and could be targeted for anti-angiogenic therapies. The VEGF antibody bevacizumab is already approved for treatment of GBM [92, 94-97], but bevacizumab is ineffective as a standalone therapy for newly diagnosed GBM patients [98, 99]. However, studies have yet to assess the efficacy of combinatorial treatments of bevacizumab with tumor pH gradient-targeting drugs.
Acidic stress also promotes glioma stem cell phenotypes independently of hypoxia, but these may still involve HIF2α expression. Exposure of GBM cells to acidic stress increased GSC marker expression as well as self-renewal and tumor growth [93, 100]. Acidosis also functions in concert with hypoxia to upregulate HIFs in GSCs through an alternative PHD/VHL-independent pathway involving the stress-induced HSP90 chaperone protein [27]. Importantly, acidic stress is also likely to shape the epigenetic response within cellular programs, as it has been shown that acidic stress represses the epigenetic reader chromodomain helicase DNA binding domain protein 7 (CHD7) in GSCs [101].
CAIX and CAXII contribute to cellular growth by maintaining extracellular acidification and an alkaline intracellular pH in response to tumor acidosis [102]. CAIX is upregulated in glioma, and acidosis leads to an increase in CAIX in GBM cells involving HIF transcriptional machinery that is independent of hypoxia [103, 104]. CAIX protein expression is an independent poor prognostic factor in GBM patients [105], suggesting the importance of understanding its function and the potential of successfully targeting the pH regulator. After CAIX knockdown, cell attachment, migration, and chemotherapeutic resistance are reduced, while apoptosis increases [106]. CAIX si/shRNA inhibits GBM growth and enhances anti-VEGF therapy, while the CAIX/XII small molecule inhibitor SLC-0111 in combination with TMZ prevents GSC enrichment and increases survival [82, 107, 108]. CAXII inhibitors also reduced the growth of TMZ-resistant GSCs by inhibiting P-glycoprotein-mediated drug efflux [109]. TMZ resistance in GSCs was further shown to be CAII mediated, with the broader CA inhibitor acetazolamide showing efficacy in combination with TMZ in xenograft models [110, 111]. Furthermore, CAIX is suggested as a target in GBM for CAR T-cells that could be effective against GSCs [112]. A recent modeling study also suggested modulating pH to increase glioma TMZ sensitivity [113]. Importantly, regions of acidic stress can cause GBM tumor cells to exhibit differential growth patterns depending on their genetics, as well as differential responses to chemotherapeutics [114]. For example, induction of WAF1 by acidosis, which results in cell cycle arrest, does not occur in p53 mutated cells and represents a mechanism by which acidosis could select for certain advantageous mutations in GBMs [115]. GBM cells with loss of p53 also had reduced responses to the CAIX/XII inhibitor SLC-0111 [107]. These reports suggest the roles for acidic stress and carbonic anhydrases in clonal selection and therapeutic resistance.
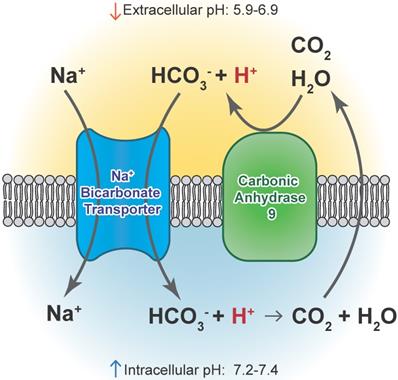
Acidic stress-induced changes in metabolism occur in GBM, and GSCs can adapt to different tumor microenvironments via shifts between glycolysis and oxidative respiration [116-118]. GBM cells adapted to low pH exhibited increased activation of AMPK, leading to a higher rate of glycolysis and inhibited oxygen consumption, indicative of the common glycolytic phenotype in brain tumor cells [119]. Conversely, Hu et al. observed increased respiration under acidic conditions that was driven by an increase in CYP24A1 expression. CYP2A1 metabolizes the active form of Vitamin D, an inhibitor of GSC phenotypes in these experiments. The authors did not report measurements of extracellular acidification rate that would describe glycolytic metabolism, which could explain the discrepancy. The authors also observed a co-localization of CYP24A1 with CAIX in vivo, suggesting that this enzyme is active in the necrotic zones of GBM tumors [100]. In our own experiments, treatment with the CAIX inhibitor SLC-0111 decreased tumor growth in association with a reduced metabolic state [107]. An overview of CAIX function is illustrated in Figure 3.
Carbonic anhydrase 9 functions to modulate extracellular and intracellular pH by generating protons and bicarbonate via hydrolysis of carbon dioxide and water. This enzyme works in tandem with sodium bicarbonate transporters that import bicarbonate into the cell to buffer intracellular pH.
Hypoxia-induced angiogenesis and GSCs
One of the most well-defined biological effects of hypoxia and HIFs in solid tumors including GBMs is the promotion of new blood vessel formation, or angiogenesis. While gliomas first support their growth by taking advantage of existing brain blood vessels in a process called vascular co-option, hypoxia will still ultimately occur as the tumor grows. A hypoxic microenvironment has also been linked to defective neovasculature, resulting in significantly poorer prognosis [120]. GSC-derived tumors have increased vessel density and blood perfusion in comparison to tumors that form from non-GSCs [50, 121, 122], suggesting differences in angiogenesis. Although controversial, hypoxia, including that induced by chemotherapy, may promote the transdifferentiation of GSCs to endothelial cells, creating tumor-derived blood vessels (see additional details below) [123-126]. The increase in angiogenic properties of GSCs is, at least to a certain degree, due to the upregulation of VEGF in GSCs in comparison to non-GSCs isolated from the same tumor [11, 50, 121, 122]. VEGF protein production is regulated by both HIF1α and HIF2α in GSCs, whereas only HIF1α impacts VEGF levels in non-GSCs [50]. Furthermore, VEGF production is regulated by OCT4 that is expressed in GSCs through the AKT-HIF1 pathway, where AKT is an oncogenic signaling factor and HIF1 is a transcription factor that is upregulated under hypoxia [127]. VEGF produced by GSCs can be secreted in extracellular vesicles called exosomes [128], suggesting a mechanism for more distant VEGF delivery that may regulate tumor angiogenesis and blood-brain barrier permeability [129]. Indeed, VEGF has been shown to be induced via delivery of miR-21 in GSC-derived exosomes [130].
GSCs express VEGF receptor 2 (VEGFR2), permitting an autocrine loop of VEGF/VEGFR signaling that can become further activated with increased VEGF under hypoxia [131]. VEGF/VEGFR2 signaling may, in turn, support the self-renewal of GSCs: VEGF was shown to activate STAT3 and subsequently induce MYC and Sox2 expression [132]. GSCs transdifferentiation towards an endothelial or pericyte lineage has also been suggested to involve VEGFR2 [125, 126, 133, 134]. GSCs exposed to hypoxia and/or endothelial cell media expressed vascular markers including CD31 and formed tubes similar to endothelial cells in vitro. Experiments with fluorescently labeled GSCs demonstrated that GSCs can incorporate into the vasculature of GBM xenografts. However, the lack of endothelial cells with common driver mutations present in GSCs suggests that transdifferentiation is very rare in human GBMs [125, 126, 134].
VEGF production may also be regulated by the pro-angiogenic chemokine CXCL12, also known as stromal cell-derived factor 1, in GSCs. CXCL12 is highly expressed in GSCs [122] and stimulates GSC VEGF production via the PI3K/AKT signaling pathway [135]. The CXCL12 receptors, CXCR4 and CXCR7, are also overexpressed within GSCs [135, 136], and the CXCR4 inhibitor AMD3100 reduced GSC-mediated tumor growth and angiogenesis in association with lower VEGF production [135]. GSCs expressing CXCR4 have been identified in close proximity to tumor vascular capillaries, strengthening the idea that these cells are involved in vascular remodeling in the tumor. Recent evidence also strongly implicates the CXCL12/CXCR4 pathway in the growth of GBM cells and GSCs under hypoxia [137] through maintaining GSC self-renewal. CD133-positive GSCs expressed higher levels of CXCR4 mRNA and protein compared to CD133-negative cells, indicating that chemokines target GSCs inducing a migratory response. These data highlight evidence that GSC mediated angiogenesis is regulated by VEGF and CXCL12 signaling.
Hypoxia and GSC invasion
Solid tumor cells in a hypoxic niche contribute to tumor aggressiveness and metastases, with specific roles for cancer stem cells that can have epithelial-to-mesenchymal transition (EMT) phenotypes [121, 138]. While cancer stem cell-mediated invasion and metastasis are more commonly studied in epithelial tumors, GSCs have been shown to be more migratory and invasive than their non-GSC counterparts [139]. Understanding the mechanisms responsible for this effect is important as the invasive nature of GBMs makes them very difficult to completely resect.
Hypoxia is an important modulator of GSCs in the context of epithelial to mesenchymal transition, which often leads to greater migration and invasion and ultimately, tumor recurrence [121]. When non-mesenchymal GBM lines were exposed to hypoxia, a mesenchymal shift resulted in greater invasive capacity. The morphological change was inhibited by knockdown of HIF1α and the EMT transcription factor ZEB1. Further evidence of a hypoxia-induced mesenchymal shift in GBM was seen through the co-localization of GLUT1, ZEB1, and the mesenchymal marker YKL40 in hypoxic areas [140]. Furthermore, hypoxia-activated A3 Adenosine Receptor was demonstrated to promote the migration/invasion of GSCs via a HIF-2-dependent mechanism [141].
The increased invasiveness of GBMs in hypoxic condition has also been linked to their enhanced hyaluronic acid production [142]. Hyaluronic acid (HA) is one of the main extracellular matrix (ECM) components in the brain, and higher levels of HA have been linked to the invasive edge of GBMs [143]. GSCs have been identified by their distinct expression of stem-marker and HA receptor, CD44, which, in addition to maintenance of stem-like properties [144], contributes to the migratory and invasive capability of GSCs [145]. The integration of these observations has led to experimental targeting of HA in GSCs to investigate the effect on therapy-resistant GSCs [146].
A recent study focused on the importance of recombination signal binding protein for immunoglobulin kappa J (RBPJ, CBF1). CBF1 is a master transcriptional regulator of the notch signaling pathway and contributor of GSC maintenance as well as an important regulator of EMT in GSCs [147]. In patient tissue, CBF1 is a clinically predictive biomarker, but its expression is heterogeneous within the tumor tissue and likely marks those cells that have undergone EMT and likely to be more invasive and resistant to chemotherapeutics. Structural changes within cells at the invasive edge are influenced by hypoxia-induced Cyclin G2 that facilitates membrane ruffles for directing cellular movement. Cyclin G2 is seen in vivo in abundance in areas of pseudopalisades, where glioma cells are actively migrating [148]. Although additional research is needed, hypoxia-induced changes in migration may also be a consequence of alterations to mitochondrial dynamics. More recent evidence specifically implicates Drp1 in GSC maintenance, with higher levels of an activating phosphorylation in the GSC fraction [149]. Together, these data suggest important links between GSC invasion and hypoxia, mesenchymal phenotypes, and the extracellular matrix.
Hypoxia and acidic stress regulation of the GBM epigenome
Chromatin remodeling regulates the GSC state as well as therapeutic resistance, and a rapidly developing area of interest is regulation of the epigenetic landscape by the hypoxic tumor microenvironment. While it is known that there are many epigenetic alterations that are involved in the initiation and progression of brain tumors, much less is known about how hypoxia contributes to these mechanisms. The discovery that expression of mixed-lineage leukemia 1 (MLL1), a histone methyltransferase specific for the lysine 4 methylation of histone H3, is increased in GSCs by hypoxic conditions has driven interest in this field. MLL1 enhances hypoxia response gene expression, including VEGF, by enforcing expression of HIFs, mainly HIF2α, in hypoxia [150]. Recently, it was also shown that the induction of the Ten-eleven Translocation (TET) family of DNA demethylases by hypoxia promotes the expression of the stem cell genes OCT4 and NANOG in glioma cells. TET1 and TET3 were shown to bind specifically to the genomic regulatory regions of these genes and actively demethylate these regions, which led to increased expression of these pluripotency genes and ultimately increased formation of GSCs [151].
In GSCs, there are also established roles for some Jumonji Domain-Containing proteins that modify histones and are known to be oxygen dependent. GSCs that survived kinase inhibitor treatment had differential H3K27me3 profiles, and the Jumonji Domain-Containing Protein 3 (JMJD3)/Lysine Demethylase 6B (KDM6B) was implicated in cellular maintenance [152]. JMJD3/KDM6B was also important for maintaining GSC neurosphere formation potential due in part to regulation of STAT3 activity [153]. GSK-J4, a JMJD3 inhibitor, was recently shown to inhibit glioma cell growth in association with elevation of H3K27me3 [154], and targeting KDM4A reduced glioma cell survival via increased autophagy [155]. Thus, there are multiple lines of evidence that hypoxia regulates DNA and histone methylation that is important for GSC maintenance.
Hypoxia and acidic stress also repress the epigenetic modifier chromodomain-helicase-DNA-binding protein 7 (CHD7) [101]. CHD7 is one of a family of CHD proteins involved in transcription, chromosomal stability, and DNA repair [156, 157], which binds methylated histone H3 lysine 4 (H3K4me) [158]. In mice, CHD7 appears to have the capability of both enhancing and inhibiting embryonic stem cell genes by co-localizing with OCT4, SOX2, and NANOG on enhancer regions [159]. CHD7 is mutated in a congenital disorder called CHARGE syndrome [160, 161], but very little is known about CHD7 and cancer [162-164]. However, studies showed CHD7 binding within 10kb of VEGF as well as increases in VEGF transcription upon loss of CHD7 in mice [159, 165], suggesting a potential role in angiogenesis. We recently reported that CHD7 was repressed by acidic stress and that CHD7 targeting in GBM cells promotes angiogenesis as determined by increased tube formation [101]. These data suggest that additional investigation of hypoxia and acidic stress effects on epigenetic modifiers may identify further mechanisms through which these tumor microenvironments may impact cell state.
Interactions of the immune system and GSCs in the hypoxic niche
Although we have thus far focused on direct effects of hypoxia on GBM cells to regulate tumor growth, the tumor microenvironment also impacts GBM cells indirectly via paracrine effects mediated through nearby non-neoplastic cells. Under a hypoxic microenvironment, macrophages undergo phenotypic changes that activate the expression of mitogenic and proangiogenic cytokines and enzymes [166]. This enhances tumor progression, angiogenesis, and metastasis [166]. The immunosuppressive activity of tumor-associated macrophages (TAMs) is enhanced in solid tumors when HIF1α is upregulated [167]. GSCs promote TAM immunosuppressive phenotypes via mechanisms involving cytokines such macrophage inhibitory cytokine-1 (MIC-1) and Transforming Growth Factor β (TGF-β) and the transcription factor STAT3 [168]. GSCs also efficiently recruit TAMs including through secreting periostin [169], a protein that serves as an integrin ligand [29]. Periostin secretion levels positively correlated with TAM numbers and silencing periostin in GSCs resulted in decreased TAM density, decreased tumor growth, and increased survival of mice bearing GSC-derived xenografts [29]. While this model suggested periostin-mediated GSC and TAM co-localization in the perivascular niche, hypoxia is known to increase periostin in glioma cells to promote macrophage recruitment through mechanisms involving TGF-β [170].
Macrophage migration inhibitory factor (MIF) is a type of cytokine released by leukocytes. MIF levels have been associated with immunosuppression as well as angiogenesis, cell differentiation, and cell proliferation in tumor cell lines [171]. Immunohistological analysis of MIF in GBM tissues demonstrated a large accumulation of MIF protein in necrotic areas and tumor cells surrounding blood vessels [171]. Under hypoxic stress, the MIF gene was transcriptionally upregulated, leading to elevated MIF mRNA as determined in Northern analysis [171]. While experiments have largely focused on a GBM-cell-intrinsic role for MIF signaling, additional data demonstrated MIF promoted mast cell migration to GBMs [172]. Recent evidence also indicated that GSC-derived MIF increased the activity of myeloid derived suppressor cells (MDSCs) [173] to suppress the immune system, and it has been shown that these cells accumulate in GBM patients [174]. In contrast, the MIF receptor CD74 was shown to be restricted to TAMs where it appeared to promote a proinflammatory phenotype and was associated with improved patient outcomes [175]. LGALS1 (galectin-1) and IGFBP2, which are upregulated in GBM and correlate with poor patient outcomes, have been identified with a subset of genes involved with immunosuppression. They have also been shown to be positive regulators of MDSC and immunosuppressive macrophages, which could provide an explanation for MDSC accumulation in GBM [176, 177]. Interestingly, IGFBP2 increases neural stem cell and GSC maintenance [178, 179], providing additional links between stem cell and immune phenotypes.
Gliomas have been characterized as immunologically “cold” tumors that have a highly immunosuppressive microenvironment, particularly in terms of adaptive immunity. Using immunogenomic characterization data compiled by The Cancer Genome Atlas (TCGA), Thorsson et al. characterized these tumors as immunologically quiet and lymphocyte depleted, as the current understanding of immune activity in these tumors is that it is largely driven by monocytes and innate immunity [180]. HIFs play a role in the immunosuppressive environment as it has been shown that HIF-1α encourages the migration of T regulatory cells (Tregs) in the presence of hypoxia, and HIF-1α knockout in Tregs enhanced survival in a murine model of glioma, indicating that this response is important for immunosuppression and tumor progression [181]. Considering the increasing importance of immunotherapy-based approaches, these data highlight the critical need to better model the effects of hypoxia on tumor-associated immune populations.
Hypoxia promotes GSC therapeutic resistance
The hypoxic microenvironment of GBMs has many effects that result in tumor cell resistance to chemotherapy and radiation. These include effects on DNA repair, DNA stability, ABC transporter expression, cell cycle checkpoint protein expression, and vasculature function. As a broad indicator of GSC survival in the hypoxic microenvironment, the CD133-positive GSC fraction are more resistant to apoptosis under hypoxia [182]. GSCs are also less sensitive to irradiation and chemotherapy induced cell death, suggesting that tumor recurrence is mediated by GSCs [10], due in part to hypoxia-mediated GSC maintenance [183].
Changes in the ability to repair DNA in the hypoxia microenvironment lead to therapeutic resistance [184]. Short-term hypoxic conditions activate DNA damage signaling pathways, with therapeutic resistance possibly due to increased activation of checkpoint proteins [185]. Long term effects of hypoxia lead to downregulation of DNA repair pathways that may promote genetic instability: these include DNA double-strand break repair, mismatch repair, and nucleotide excision repair [186]. The lack of oxidation of DNA free radicals that occurs when oxygen tensions are low also prevents the damage and breakage of DNA [187, 188]. While these pathways have not all been investigated in the context of hypoxia in GBMs, GSCs are known to have increased activating phosphorylation of ATM and checkpoint proteins as well as increased levels of some DNA repair proteins, which enables the cells to more rapidly repair damaged DNA [10, 189]. However, the fidelity of this repair is unclear: once arrested cells continue though the cell cycle, secondary tumors may arise from the damaged cells [185]. Furthermore, elevation of proliferating cell nuclear antigen associated factor (PAF) in GSCs may facilitate DNA damage tolerance via translesion DNA synthesis [190]. Quiescent populations of GSCs that are not replicating are also relatively insensitive to DNA damaging agents [191]. The suppression of DNA repair under a hypoxic microenvironment could be a potential cause for the genetic instability of cancer cells that drives the progression of brain tumors [184]. These results support the hypothesis that hypoxia induces drug resistance by preventing drugs from damaging the DNA of tumor cells [188].
Cell surface transporter proteins are considered important mechanisms of drug resistance and are affected by hypoxia in GSCs. The ATP-binding cassette (ABC) transporters are a class of proteins that have a wide range of biological effects. These transporters promote therapeutic resistance by removing chemotherapy from the tumor cells, minimizing the time that the drugs have to be effective [192]. The stem cell transcription factor OCT4, which is known to be HIF2α-regulated, increases expression of ABCG2 in GBM cells [193]. The cyclic hypoxic microenvironment (as opposed to chronic hypoxia) in GBM cells has also been shown to increase ABCB1 expression via HIF1α promoter binding, enhancing its expression [192]. The greater expression of the ABC transporters, in turn, strengthens chemoresistance [192]. Furthermore, ABCB5, which has been suggested to be a CSC marker in some tumors, is expressed in GSCs and mediated TMZ-resistance in GBM cells [194].
Hypoxic regions form when tumor cells lack proximity to a functional blood vessel leading to low oxygen tensions. Cells located at least 70 µm away from the nearest functional blood vessel do not receive adequate amounts of oxygen, leading to a conversion to a hypoxic cell state [195]. Because chemotherapies reach tumor cells via the circulatory system, the distance cells are to the nearest blood vessel has an important effect on the efficacy of the drug [196, 197]. This, combined with the need for drugs in the brain to cross the blood brain barrier, often results in drug penetration that is lowest in brain tumors especially compared to cancers of the heart, kidney, and liver [198]. While the blood brain barrier is disrupted in GBM, the blood brain barrier can remain intact in brain adjacent to tumor where the cells responsible for recurrences have dispersed, preventing chemotherapies from reaching critical tumor cells.
Diagnostic and therapeutic modalities targeting acidic stress and hypoxia
Diagnostic monitoring of tumor progression post-resection can be complicated by ischemia, and there are a number of clinical trials (see Table 1 and Figure 4) focused on improved imaging modalities that identify tumors by regions of hypoxia or acidosis. An innovative study synergized radiomic features in MRI scans and RNA expression data from hypoxia markers to generate a hypoxia enrichment score (HES) that was highly predictive of the survival of GBM patients [199]. Therapeutic interventions targeting acidic stress are rare, and diagnostic procedures to measure the acidic microenvironments in vivo have proven challenging to develop but have recently gained traction. Cutting-edge MRI techniques that highlight regions of acidosis are being developed as novel imaging tools to improve patient care and explore the pathophysiology of brain tumors [200-202]. Using this technique, rat brain tumors treated with TMZ, the first-line chemotherapy approved for standard of care in human GBM patients, were shown to have a normalized intratumoral pH, indicating the ability of TMZ to modulate pH in solid brain tumors [203]. However, there has been little follow-up on the concept of extracellular pH modulation by TMZ in GBM. In the authors' opinion, acidic stress is an important contributor to the GBM tumor microenvironment and should be considered as a therapeutic target. Diagnostic procedures that are developed to incorporate estimation of tumor hypoxia and/or acidity could prove useful in stratification of patients for personalized therapies in the future.
Details of hypoxia or HIF-based diagnostics and therapeutics currently on clinicaltrials.gov.
| Intervention | Mechanism | Trial ID | Title | Phase | Start year | Status |
|---|---|---|---|---|---|---|
18F-FMISO PET | PET radio-tracer for imaging hypoxia | NCT00902577 | Multicenter, Phase II Assessment of Tumor Hypoxia in Glioblastoma Using 18F-Fluoromisonidazole (FMISO) With PET and MRI | Phase 2 | 2009 | Completed |
| NCT00906893 | Methodological Evaluation of Fluor 18 Labelled Fluoromisonidazole ([18F]-FMISO) Positon Emission Tomography-Computed Tomography (PET-CT) for Non Operated Glioblastoma | Phase 2 | 2009 | Completed | ||
| NCT01200134 | Hypoxia Diagnosis and Evaluation Using F-MISO PET and Biomarkers in Brain Tumors | Phase 2 | 2010 | Completed | ||
| NCT01246869 | Assessment of Primary and Metastatic Brain Tumor Hypoxia With 18F-Fluoromisonidazole, [18F]Fluoro-2-deoxy-D-glucose (FDG) and [15O]Water (H215O) | N/A | 2010 | Recruiting | ||
| NCT02076152 | A Study to Evaluate Vascular Normalization in Patients With Recurrent Glioblastoma Treated With Bevacizumab Using FMISO PET and Vascular MRI | N/A | 2014 | Completed | ||
| NCT03649880 | Feasibility of FMISO in Brain Tumors | Phase 1I | 2018 | Recruiting | ||
| Ferumoxytol-based qBOLD MRI  | Iron-based Quantitative BOLD MRI for hypoxia detection | NCT02466828 | Quantitative Blood Oxygenation Level Dependent (qBOLD) MR Imaging of Glioblastoma Multiforme for Assessment of Tumor Hypoxia | Early Phase 1 | 2015 | Completed |
| SatO2-MRI | Mapping tissue oxygenation with MRI | NCT03716986 | Multimodal Imaging of Hypoxia in Gliomas | 2018 | Not yet recruiting | |
PT2977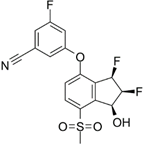 | Selective HIF-2α inhibitor | NCT02974738 | A Phase 1, Multiple-Dose, Dose-Escalation and Expansion Trial of PT2977, a HIF-2α Inhibitor, in Patients With Advanced Solid Tumors | Phase 1 | 2016 | Recruiting |
PT2385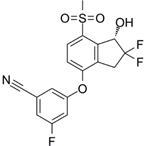 | Selective HIF-2α inhibitor | NCT03216499 | Single-Arm, Open-Label Phase II Efficacy Study of First-in-Class HIF2-Alpha Inhibitor, PT2385, for Patients With Recurrent Glioblastoma | Phase 2 | 2017 | Active, not recruiting |
Acetazolamide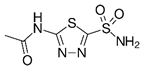 | Carbonic anhydrase inhibitor | NCT03011671 | A Phase I Study of Safety and Tolerability of Acetazolamide With Temozolomide in Adults With Newly Diagnosed MGMT Promoter-Methylated Malignant Glioma | Phase 1 | 2018 | Recruiting |
Hypoxia or HIF-based diagnostics and therapeutics currently in clinical trial for gliomas. Numbers in each section correspond to studies listed as current on clinicaltrials.gov.
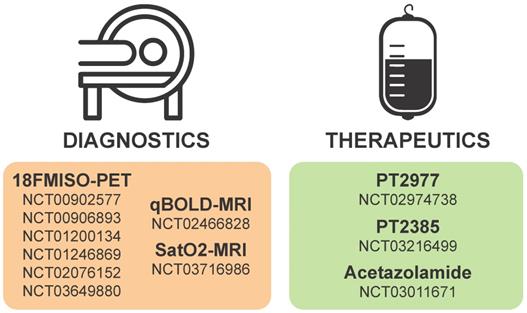
Considering the critical roles of HIF1α and HIF2α in cancer biology, many strategies have been considered to target HIFα signaling in solid tumors including GBMs. Targeting hypoxia/HIFα signaling is thought to be a viable strategy to sensitize GSCs to radiation and chemotherapy as well as to inhibit the pro-tumorigenic biology induced when blood vessel collapse occurs with anti-angiogenics [44, 204]. Inhibition could be mediated by therapies that promote oxygenation, decrease HIFα stability, prevent HIFα DNA binding, or inhibit the downstream mediators of pro-tumorigenic hypoxia/HIF effects. A current list of ongoing clinical trials targeting hypoxia in brain tumors is illustrated in Figure 4 and additional details are provided in Table 1.
HIF-1α has been shown to reach maximal expression levels in the brain after 5 hours of hypoxia exposure, but returns to basal levels at 12 hours [205]. The activators responsible for HIF1 transcription are CREB binding protein and p300, both of which interact with the carboxy-terminal transactivation domain of HIF1 [206]. As a result, HIF1α activation promotes the expression of numerous gene products. These include pluripotency-associated transcription factors like OCT3/4, NANOG, and SOX2; glycolysis- and EMT-associated molecules like CXCR4, SNAIL, and TWIST; microRNAs; and angiogenic factors such as VEGF [207]. These gene products lead to increased self-renewal ability, tumor survival, distorted energy metabolism, invasion, angiogenesis, and treatment resistance [207]. Because HIF1α activates these gene products, targeting HIF1 is a potential therapeutic strategy to target GSCs [207]. However, it is important to consider the potential for side effects against neoplastic neural stem cells when targeting GSCs, as hypoxia is present in neural stem cell niches and HIF1α regulates neural stem cell proliferation and differentiation [208].
Targeting of HIF expression or activity could occur through multiple mechanisms. In GBM, the cardiac glycoside digoxin has been shown to inhibit HIF1α expression in hypoxia [209]. Digoxin treatment reduced expression of CD133 and decreased neurosphere formation, suggesting this clinically utilized drug may be able to target GSCs. Importantly, a clinical trial (clinicaltrials.gov identifier NCT03216499) is currently recruiting recurrent GBM patients for treatment with the HIF2α specific inhibitor PT2385, which was effective against renal cell carcinomas in preclinical studies [210, 211]. Of note, recent work has also shown that the FDA approved drug topotecan may in fact be capable of targeting HIF1α via perturbations in levels of SUMOylation thereby altering the stability/degradation of HIF [212].
The canonical hypoxia-induced gene is the proangiogenic factor VEGF that is elevated in GSCs [11]. Therapies have been developed to target VEGF activity by binding the ligand (the VEGF antibody bevacizumab) and inhibiting the receptor with varying degrees of specificity (sorafenib, sunitinib, etc.) to normalize the tumor vasculature [213]. Neutralizing anti-VEGF antibody was previously shown to extend the survival of GBM bearing mice [11, 214]. While these initial studies suggested that treated tumors had reduced vasculature and increased apoptosis [214], subsequent results demonstrated increased tumor cell invasion [215]. Furthermore, treatment of GBM patients with bevacizumab did not improve patient survival [216]. Another potent angiogenic chemokine, SDF-1a, and its cognate receptor, CXCR4, are highly expressed in hypoxic regions and have been used to develop hypoxia-targeted drug delivery mechanisms. Nanoparticle induced CXCR4-overexpressing human adipose cells have been tested in GSC organoid models and murine models: they effectively home to the necrotic core of tumors and could serve as an effective delivery mechanism for nanoparticle-based drug treatments [217]. CXCR4 inhibition in combination with anti-VEGF therapies has been tested in animal models with some success, and is another possible therapeutic modality to target angiogenic responses within the tumor microenvironment [218]. Another innovative study combined engineered liposomal delivery that targets brain microvascular endothelial cells. Using the low-density lipoprotein receptor-related protein-1 and a hypoxic prodrug radiosensitizer in animal models, radiosensitivity was increased by enhancing the DNA damaged caused by ionizing radiation [219].
Mammalian target of rapamycin (mTOR) is an intracellular kinase that regulates cell growth and cell cycle progression via signaling from nutrients and growth factors [220]. Previous studies have shown that mTOR is deregulated in GBM contributing to radiosensitization. To combat this problem, Kahn et al. demonstrated that exposing the tumor cells to AZD2014, an mTORC1 inhibitor, increased the radiosensitivity of GSCs. Additionally, clonogenic survival analysis showed that CD133-positive and CD15-positive GSC cells exposed to this mTORC1 inhibitor at least one hour before irradiation increased sensitivity to radiation [221]. These data suggest that mTORC1 inhibition could target GSCs, which serve as a reservoir for radioresistance.
Mitogen-activated protein kinase (MAPK) can be activated by stress signaling such as that resulting from hypoxia [222]. In general, the RAS-MAPK pathway plays a role in cell development, cell cycle regulation, tumor formation, and metastasis [223]. When activated, RAS then activates RAF kinase, which then activates downstream MAPK signaling [224]. The altered activity of the RAS/MAPK signaling pathway leads to abnormal cell growth and proliferation, as well as initiating other abnormal cellular behaviors like invasion and apoptosis [224]. Expression of constitutively active Ras in the mouse subventricular zone led to the development of gliomas through Ets transcription factor-dependent mechanisms [225] (and Ets binding elements are in HIF2a target genes). However, rather than RAS mutations, activation of this pathway in GBM is frequently due to amplification or constitutive activation of receptor tyrosine kinases including Epidermal Growth Factor Receptor (EGFR) and Platelet Derived Growth Factor Receptor (PDGFR): further elevation of ligands and/or receptors under hypoxia influences even greater activation of the RAS/MAPK signaling pathway [224, 226, 227]. Thus, targeting the RAS protein with chemotherapy in GBM may be useful because of its high expression and its association with tumorigenesis [224]. Further downstream of MAPK, ERK signaling can regulate AMP-activated Protein Kinase (AMPK), which is an important regulator of cellular bioenergetics via activation of catabolism to generate ATP [228]. The AMPK stress-induced pathway is hijacked by GSCs for their adaptation to tumor-related stressors in the microenvironment through the Cyclic AMP-Responsive Element-Binding Protein 1 (CREB1) transcriptional program that controls both HIF-1 and GA Binding Protein Transcription Factor Subunit Alpha (GABPA) expression. This pathway has been successfully targeted in mouse models with AMPK tissue-specific and whole-animal knockouts, which prompts interest in developing specific AMPK inhibitors for glioma therapy [229].
Nitro compounds, N-oxides, and quinones are bioreductive prodrugs that target the hypoxic tumor cells by being reduced by intracellular oxidoreductases in an oxygen-sensitive manner to form cytotoxins [230]. While hypoxia causes tumors to become resistant to radiation, bioreductive drugs are used as antimicrobials, chemotherapeutic agents, and radiation sensitizers [231]. AQ4N is an N-oxide that has been shown to have an anti-tumor effect on hypoxic tumor cells [232] and has been used in phase I clinical trial in GBM patients [233]. AQ4N metabolizes to AQ4 that binds non-covalently to DNA to initiate anti-tumor effects [230, 232]. AQ4 can then inhibit topoisomerase activity as tumor cells begin to re-enter the cell cycle [232]. Tirapazamine (TPZ) is another N oxide which was shown to selectively kill cells in hypoxic environments [234]. TPZ has been used in a phase II clinical trial in GBM patients, unfortunately providing no significant survival advantage [235]. As TPZ has poor extravascular penetration [236], more optimized analogues including SN30000 have been developed. While SN30000 has not been extensively studied in GBM, the compound can cross the blood-brain-barrier [237], suggesting possible efficacy in patients.
The quinone mitomycin C (MMC) is activated under hypoxic conditions in tumor cells [238]. In glioma, MMC combined with recombinant adeno-associated virus II resulted in reduced tumor growth in vivo [239]. MMC-mediated GBM cell death was also increased when cells were pretreated with the DT-diaphorase inducer, dimethyl fumarate [240]. As a reductase, DT-diaphorase is known to activate quinones like MMC and is elevated in GBMs [241]. Thus, MMC treatment may take advantage of the elevated DT-diaphorase levels in GBM to provide a greater therapeutic window and molecular analysis of DT-diaphorase levels may provide a biomarker for MMC therapeutic response.
Concluding Statement
Glioblastoma remains a formidable tumor that is notoriously difficult to prevent from recurrence, which contributes to abysmal survival in patients. Compared to solid tumors outside the brain, glioblastoma treatment is also more complex due to the presence of the blood-brain-barrier: development of certain types of treatment modalities is precluded without novel delivery methods that are able to circumvent the restrictions of the blood-brain-barrier. For example, many conventional chemotherapies and antibodies that target antigens in and on the surface of tumor cells have difficulty crossing an intact blood-brain-barrier, although there are antibodies that work well in the bloodstream without having to cross into the tumor (bevacizumab, nivolumab, pembrolizumab and ipilumimab for example). While the tumor associated blood-brain-barrier is remarkably fenestrated and is constantly being remodeled, the blood-brain-barrier in regions where invading tumor cells reside may be intact preventing eradication of the disease. Continual failures with novel treatment strategies suggest that there are major characteristics of these tumors that are not adequately being modeled for drug testing in vitro or that we still do not fully understand. It is of our opinion that more accurately modeling the tumor microenvironment in vitro and consideration of its links to differentiation state and therapeutic resistance will allow for more efficient transfer of novel therapeutics from in vitro studies to the clinic. In addition, added emphasis on targeting these microenvironments during therapeutic design may lead to desirable improvements in standard of care. Considering this, hypoxia and acidic stress are major micro-environmental stresses that occur commonly in GBM solid tumors. This is especially important when considering the GSC niche that can be found within hypoxic and acidic zones. Establishing efficient models of these microenvironments in vitro and targeting these in vivo to exploit resistant cell populations, i.e. GSCs, is of substantial importance when identifying novel treatment strategies to synergize with standard of care.
Abbreviations
GSC: glioma stem cell; GBM: glioblastoma; TMZ: temozolomide; FACs: fluorescence activated cell sorting; VEGF: vascular endothelial growth factor; AKT: protein kinase B; HIF: hypoxia inducible factor; ARNT: aryl hydrocarbon receptor nuclear translocator; VHL: von Hippel Lindau factor; HRE: hypoxia response elements; MAX: Myc associated protein; EPO: erythropoietin; EDN1: endothelin 1; GLUT1: glucose transporter 1; MYC: MYC proto-oncogene; HGF: hepatocyte growth factor; CAIX: carbonic anhydrase IX; PHD: prolyl hydroxylase domain protein; WAF1: cyclin-dependent kinase inhibitor p21; EMT: epithelial to mesenchymal transition; RBPJ, CBF1: recombination signal binding protein for immunoglobulin kappa J region; DRP1: dynamin related protein 1; MLL1: mixed lineage leukemia 1; TET: ten-eleven translocation methylcytosine dioxygenase; MAPK: mitogen-activated protein kinase; MTOR: mammalian target of rapamycin; CREB1: CAMP responsive element binding protein 1; TPZ: tirapazamine; MMC: mitomycin C; EGF: epidermal growth factor; PDGF: platelet derived growth factor; ECM: extracellular matrix; AMPK: AMP-activated protein kinase; HA: hyaluronic acid.
Acknowledgements
We appreciate the support of the National Institutes of Health (R01NS104339, A.B.H; U01CA223976, A.B.H and G.Y.G.; R21CA252382, G.Y.G; F31CA200085 to N.H.B.) and the National Science Foundation Bridge to the Doctorate Grant (A.B.J). G.K.F. is supported by Hyundai Hope on Wheels, the Rally Foundation for Childhood Cancer Research, and the Andrew McDonough B+ Foundation.
Competing Interests
Dr. Bernstock has positions/equity in CITC Ltd and Avidea Technologies and is member of the POCKiT Diagnostics Board of Scientific Advisors. Dr. Gillespie has positions/equity in Treovir, LLC and Aettis, Inc. The remaining authors declare that they have no conflict(s) of interest.
References
1. Ostrom QT, Cioffi G, Gittleman H, Patil N, Waite K, Kruchko C. et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012-2016. Neuro Oncol. 2019;21:v1-v100
2. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987-96
3. Stupp R, Hegi ME, Neyns B, Goldbrunner R, Schlegel U, Clement PM. et al. Phase I/IIa study of cilengitide and temozolomide with concomitant radiotherapy followed by cilengitide and temozolomide maintenance therapy in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28:2712-8
4. Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ. et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell. 2019;178(e21):835-49
5. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730-7
6. Vescovi AL, Galli R, Reynolds BA. Brain tumour stem cells. Nat Rev Cancer. 2006;6:425-36
7. Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29:1203-17
8. Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL. et al. Cancer stem cells-perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339-44
9. Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J. et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821-8
10. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB. et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756-60
11. Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB. et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006;66:7843-8
12. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T. et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396-401
13. Lathia JD, Hitomi M, Gallagher J, Gadani SP, Adkins J, Vasanji A. et al. Distribution of CD133 reveals glioma stem cells self-renew through symmetric and asymmetric cell divisions. Cell Death Dis. 2011;2:e200
14. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR. et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462-77
15. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H. et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396-401
16. Tejero R, Huang Y, Katsyv I, Kluge M, Lin JY, Tome-Garcia J. et al. Gene signatures of quiescent glioblastoma cells reveal mesenchymal shift and interactions with niche microenvironment. EBioMedicine. 2019;42:252-69
17. Li P, Zhou C, Xu L, Xiao H. Hypoxia enhances stemness of cancer stem cells in glioblastoma: an in vitro study. Int J Med Sci. 2013;10:399-407
18. Garnier D, Renoult O, Alves-Guerra MC, Paris F, Pecqueur C. Glioblastoma Stem-Like Cells, Metabolic Strategy to Kill a Challenging Target. Frontiers in oncology. 2019;9:118
19. Prager BC, Xie Q, Bao S, Rich JN. Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell. 2019;24:41-53
20. Colwell N, Larion M, Giles AJ, Seldomridge AN, Sizdahkhani S, Gilbert MR. et al. Hypoxia in the glioblastoma microenvironment: shaping the phenotype of cancer stem-like cells. Neuro-oncology. 2017;19:887-96
21. Filatova A, Acker T, Garvalov BK. The cancer stem cell niche(s): the crosstalk between glioma stem cells and their microenvironment. Biochim Biophys Acta. 2013;1830:2496-508
22. Heddleston JM, Hitomi M, Venere M, Flavahan WA, Yang K, Kim Y. et al. Glioma stem cell maintenance: the role of the microenvironment. Curr Pharm Des. 2011;17:2386-401
23. Bouwens van der Vlis TAM, Kros JM, Mustafa DAM, van Wijck RTA, Ackermans L, van Hagen PM. et al. The complement system in glioblastoma multiforme. Acta Neuropathol Commun. 2018;6:91
24. Ishii A, Kimura T, Sadahiro H, Kawano H, Takubo K, Suzuki M. et al. Histological Characterization of the Tumorigenic "Peri-Necrotic Niche" Harboring Quiescent Stem-Like Tumor Cells in Glioblastoma. PLoS One. 2016;11:e0147366
25. Jung J, Zhang Y, Celiku O, Zhang W, Song H, Williams BJ. et al. Mitochondrial NIX Promotes Tumor Survival in the Hypoxic Niche of Glioblastoma. Cancer Res. 2019;79:5218-32
26. Aderetti DA, Hira VVV, Molenaar RJ, van Noorden CJF. The hypoxic peri-arteriolar glioma stem cell niche, an integrated concept of five types of niches in human glioblastoma. Biochim Biophys Acta Rev Cancer. 2018;1869:346-54
27. Filatova A, Seidel S, Bogurcu N, Graf S, Garvalov BK, Acker T. Acidosis Acts through HSP90 in a PHD/VHL-Independent Manner to Promote HIF Function and Stem Cell Maintenance in Glioma. Cancer Res. 2016;76:5845-56
28. Chiche J, Brahimi-Horn MC, Pouyssegur J. Tumour hypoxia induces a metabolic shift causing acidosis: a common feature in cancer. J Cell Mol Med. 2010;14:771-94
29. Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J. et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol. 2015;17:170-82
30. Gilbertson RJ, Rich JN. Making a tumour's bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer. 2007;7:733-6
31. Rong Y, Durden DL, Van Meir EG, Brat DJ. 'Pseudopalisading' necrosis in glioblastoma: a familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J Neuropathol Exp Neurol. 2006;65:529-39
32. Rampling R, Cruickshank G, Lewis AD, Fitzsimmons SA, Workman P. Direct measurement of pO2 distribution and bioreductive enzymes in human malignant brain tumors. Int J Radiat Oncol Biol Phys. 1994;29:427-31
33. Collingridge DR, Piepmeier JM, Rockwell S, Knisely JP. Polarographic measurements of oxygen tension in human glioma and surrounding peritumoural brain tissue. Radiother Oncol. 1999;53:127-31
34. Kallinowski F, Schlenger KH, Runkel S, Kloes M, Stohrer M, Okunieff P. et al. Blood flow, metabolism, cellular microenvironment, and growth rate of human tumor xenografts. Cancer Res. 1989;49:3759-64
35. Fukumura D, Xu L, Chen Y, Gohongi T, Seed B, Jain RK. Hypoxia and acidosis independently up-regulate vascular endothelial growth factor transcription in brain tumors in vivo. Cancer Res. 2001;61:6020-4
36. Evans SM, Jenkins KW, Chen HI, Jenkins WT, Judy KD, Hwang WT. et al. The relationship among hypoxia, proliferation, and outcome in patients with de novo Glioblastoma: A Pilot Study. Transl Oncol. 2010;3:160-9
37. Garcia-Bermudez J, Baudrier L, La K, Zhu XG, Fidelin J, Sviderskiy VO. et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nature cell biology. 2018;20:775-81
38. Tannock IF, Rotin D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. 1989;49:4373-84
39. Gilbert AN, Walker K, Tran AN, Boyd NH, Gillespie GY, Singh RK. et al. Modeling Physiologic Microenvironments in Three-Dimensional Microtumors Maintains Brain Tumor Initiating Cells. J Cancer Stem Cell Res. 2017 5
40. Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010;40:294-309
41. Kaur B, Khwaja FW, Severson EA, Matheny SL, Brat DJ, Van Meir EG. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro Oncol. 2005;7:134-53
42. Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ. et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468-72
43. Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M. et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464-8
44. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721-32
45. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W. et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765-73
46. Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P. et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324:261-5
47. Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E. et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479-83
48. Intlekofer AM, Wang B, Liu H, Shah H, Carmona-Fontaine C, Rustenburg AS. et al. L-2-Hydroxyglutarate production arises from noncanonical enzyme function at acidic pH. Nat Chem Biol. 2017;13:494-500
49. Intlekofer AM, Dematteo RG, Venneti S, Finley LW, Lu C, Judkins AR. et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metab. 2015;22:304-11
50. Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S. et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15:501-13
51. Holmquist-Mengelbier L, Fredlund E, Lofstedt T, Noguera R, Navarro S, Nilsson H. et al. Recruitment of HIF-1alpha and HIF-2alpha to common target genes is differentially regulated in neuroblastoma: HIF-2alpha promotes an aggressive phenotype. Cancer Cell. 2006;10:413-23
52. McCord AM, Jamal M, Shankavaram UT, Lang FF, Camphausen K, Tofilon PJ. Physiologic oxygen concentration enhances the stem-like properties of CD133+ human glioblastoma cells in vitro. Molecular cancer research: MCR. 2009;7:489-97
53. Aprelikova O, Wood M, Tackett S, Chandramouli GV, Barrett JC. Role of ETS transcription factors in the hypoxia-inducible factor-2 target gene selection. Cancer Res. 2006;66:5641-7
54. Gordan JD, Bertout JA, Hu CJ, Diehl JA, Simon MC. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell. 2007;11:335-47
55. Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2011;12:9-22
56. Bar EE. Glioblastoma, cancer stem cells and hypoxia. Brain Pathol. 2011;21:119-29
57. Stoltz K, Sinyuk M, Hale JS, Wu Q, Otvos B, Walker K. et al. Development of a Sox2 reporter system modeling cellular heterogeneity in glioma. Neuro Oncol. 2015;17:361-71
58. Soeda A, Park M, Lee D, Mintz A, Androutsellis-Theotokis A, McKay RD. et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene. 2009;28:3949-59
59. Heddleston JM, Li Z, McLendon RE, Hjelmeland AB, Rich JN. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle. 2009;8:3274-84
60. Bar EE, Lin A, Mahairaki V, Matsui W, Eberhart CG. Hypoxia increases the expression of stem-cell markers and promotes clonogenicity in glioblastoma neurospheres. Am J Pathol. 2010;177:1491-502
61. Griguer CE, Oliva CR, Gobin E, Marcorelles P, Benos DJ, Lancaster JR Jr. et al. CD133 is a marker of bioenergetic stress in human glioma. PLoS One. 2008;3:e3655
62. Kolenda J, Jensen SS, Aaberg-Jessen C, Christensen K, Andersen C, Brunner N. et al. Effects of hypoxia on expression of a panel of stem cell and chemoresistance markers in glioblastoma-derived spheroids. J Neurooncol. 2011;103:43-58
63. Seidel S, Garvalov BK, Wirta V, von Stechow L, Schanzer A, Meletis K. et al. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha. Brain. 2010;133:983-95
64. Wang P, Lan C, Xiong S, Zhao X, Shan Y, Hu R. et al. HIF1alpha regulates single differentiated glioma cell dedifferentiation to stem-like cell phenotypes with high tumorigenic potential under hypoxia. Oncotarget. 2017;8:28074-92
65. Souberan A, Cappai J, Chocry M, Nuccio C, Raujol J, Colin C. et al. Inhibitor of Apoptosis Proteins Determines Glioblastoma Stem-Like Cell Fate in an Oxygen-Dependent Manner. Stem Cells. 2019;37:731-42
66. Lehnus KS, Donovan LK, Huang X, Zhao N, Warr TJ, Pilkington GJ. et al. CD133 glycosylation is enhanced by hypoxia in cultured glioma stem cells. Int J Oncol. 2013;42:1011-7
67. Pistollato F, Chen HL, Rood BR, Zhang HZ, D'Avella D, Denaro L. et al. Hypoxia and HIF1alpha repress the differentiative effects of BMPs in high-grade glioma. Stem Cells. 2009;27:7-17
68. Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G. et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006;444:761-5
69. Man J, Yu X, Huang H, Zhou W, Xiang C, Huang H. et al. Hypoxic Induction of Vasorin Regulates Notch1 Turnover to Maintain Glioma Stem-like Cells. Cell Stem Cell. 2018;22(e6):104-18
70. Almiron Bonnin DA, Havrda MC, Lee MC, Liu H, Zhang Z, Nguyen LN. et al. Secretion-mediated STAT3 activation promotes self-renewal of glioma stem-like cells during hypoxia. Oncogene. 2018;37:1107-18
71. Evagelou SL, Bebenek O, Specker EJ, Uniacke J. DEAD Box Protein Family Member DDX28 Is a Negative Regulator of Hypoxia-Inducible Factor 2alpha- and Eukaryotic Initiation Factor 4E2-Directed Hypoxic Translation. Mol Cell Biol. 2020 40
72. Lee SB, Frattini V, Bansal M, Castano AM, Sherman D, Hutchinson K. et al. An ID2-dependent mechanism for VHL inactivation in cancer. Nature. 2016;529:172-7
73. Covello KL, Kehler J, Yu H, Gordan JD, Arsham AM, Hu CJ. et al. HIF-2alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006;20:557-70
74. Nusblat LM, Tanna S, Roth CM. Gene silencing of HIF-2alpha disrupts glioblastoma stem cell phenotype. Cancer Drug Resist. 2020;3:199-208
75. Lee G, Auffinger B, Guo D, Hasan T, Deheeger M, Tobias AL. et al. Dedifferentiation of Glioma Cells to Glioma Stem-like Cells By Therapeutic Stress-induced HIF Signaling in the Recurrent GBM Model. Mol Cancer Ther. 2016;15:3064-76
76. Johansson E, Grassi ES, Pantazopoulou V, Tong B, Lindgren D, Berg TJ. et al. CD44 Interacts with HIF-2alpha to Modulate the Hypoxic Phenotype of Perinecrotic and Perivascular Glioma Cells. Cell Rep. 2017;20:1641-53
77. Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res. 1989;49:6449-65
78. Damaghi M, Wojtkowiak JW, Gillies RJ. pH sensing and regulation in cancer. Front Physiol. 2013;4:370
79. Parks SK, Chiche J, Pouyssegur J. pH control mechanisms of tumor survival and growth. J Cell Physiol. 2011;226:299-308
80. Spugnini EP, Citro G, Fais S. Proton pump inhibitors as anti vacuolar-ATPases drugs: a novel anticancer strategy. J Exp Clin Cancer Res. 2010;29:44
81. Ibrahim Hashim A, Cornnell HH, Coelho Ribeiro Mde L, Abrahams D, Cunningham J, Lloyd M. et al. Reduction of metastasis using a non-volatile buffer. Clin Exp Metastasis. 2011;28:841-9
82. McIntyre A, Patiar S, Wigfield S, Li JL, Ledaki I, Turley H. et al. Carbonic anhydrase IX promotes tumor growth and necrosis in vivo and inhibition enhances anti-VEGF therapy. Clin Cancer Res. 2012;18:3100-11
83. Winum JY, Rami M, Scozzafava A, Montero JL, Supuran C. Carbonic anhydrase IX: a new druggable target for the design of antitumor agents. Med Res Rev. 2008;28:445-63
84. Gerweck LE, Seetharaman K. Cellular pH gradient in tumor versus normal tissue: potential exploitation for the treatment of cancer. Cancer Res. 1996;56:1194-8
85. Ribeiro MD, Silva AS, Bailey KM, Kumar NB, Sellers TA, Gatenby RA. et al. Buffer Therapy for Cancer. J Nutr Food Sci. 2012;2:6
86. Daniel C, Bell C, Burton C, Harguindey S, Reshkin SJ, Rauch C. The role of proton dynamics in the development and maintenance of multidrug resistance in cancer. Biochim Biophys Acta. 2013;1832:606-17
87. Fu Y, Wang D, Wang H, Cai M, Li C, Zhang X. et al. TSPO deficiency induces mitochondrial dysfunction, leading to hypoxia, angiogenesis and a growth-promoting metabolic shift towards glycolysis in glioblastoma. Neuro Oncol. 2019
88. Robey IF, Baggett BK, Kirkpatrick ND, Roe DJ, Dosescu J, Sloane BF. et al. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res. 2009;69:2260-8
89. Ibrahim-Hashim A, Cornnell HH, Abrahams D, Lloyd M, Bui M, Gillies RJ. et al. Systemic buffers inhibit carcinogenesis in TRAMP mice. J Urol. 2012;188:624-31
90. Lock FE, McDonald PC, Lou Y, Serrano I, Chafe SC, Ostlund C. et al. Targeting carbonic anhydrase IX depletes breast cancer stem cells within the hypoxic niche. Oncogene. 2013;32:5210-9
91. Chafe SC, McDonald PC, Saberi S, Nemirovsky O, Venkateswaran G, Burugu S. et al. Targeting Hypoxia-Induced Carbonic Anhydrase IX Enhances Immune-Checkpoint Blockade Locally and Systemically. Cancer Immunol Res. 2019;7:1064-78
92. Xu L, Fukumura D, Jain RK. Acidic extracellular pH induces vascular endothelial growth factor (VEGF) in human glioblastoma cells via ERK1/2 MAPK signaling pathway: mechanism of low pH-induced VEGF. J Biol Chem. 2002;277:11368-74
93. Hjelmeland AB, Wu Q, Heddleston JM, Choudhary GS, MacSwords J, Lathia JD. et al. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ. 2011;18:829-40
94. Bokstein F, Shpigel S, Blumenthal DT. Treatment with bevacizumab and irinotecan for recurrent high-grade glial tumors. Cancer. 2008;112:2267-73
95. Buie LW, Valgus J. Bevacizumab: a treatment option for recurrent glioblastoma multiforme. The Annals of pharmacotherapy. 2008;42:1486-90
96. Kang TY, Jin T, Elinzano H, Peereboom D. Irinotecan and bevacizumab in progressive primary brain tumors, an evaluation of efficacy and safety. Journal of neuro-oncology. 2008;89:113-8
97. Vredenburgh JJ, Desjardins A, Herndon JE 2nd, Marcello J, Reardon DA, Quinn JA. et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol. 2007;25:4722-9
98. Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA. et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370:699-708
99. Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R. et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014;370:709-22
100. Hu P, Li S, Tian N, Wu F, Hu Y, Li D. et al. Acidosis enhances the self-renewal and mitochondrial respiration of stem cell-like glioma cells through CYP24A1-mediated reduction of vitamin D. Cell Death Dis. 2019;10:25
101. Boyd NH, Walker K, Ayokanmbi A, Gordon ER, Whetsel J, Smith CM. et al. Chromodomain Helicase DNA-Binding Protein 7 Is Suppressed in the Perinecrotic/Ischemic Microenvironment and Is a Novel Regulator of Glioblastoma Angiogenesis. Stem Cells. 2019;37:453-62
102. Chiche J, Ilc K, Laferriere J, Trottier E, Dayan F, Mazure NM. et al. Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res. 2009;69:358-68
103. Kovacs GG, Zsembery A, Anderson SJ, Komlosi P, Gillespie GY, Bell PD. et al. Changes in intracellular Ca2+ and pH in response to thapsigargin in human glioblastoma cells and normal astrocytes. Am J Physiol Cell Physiol. 2005;289:C361-71
104. Ihnatko R, Kubes M, Takacova M, Sedlakova O, Sedlak J, Pastorek J. et al. Extracellular acidosis elevates carbonic anhydrase IX in human glioblastoma cells via transcriptional modulation that does not depend on hypoxia. Int J Oncol. 2006;29:1025-33
105. Sathornsumetee S, Cao Y, Marcello JE, Herndon JE 2nd, McLendon RE, Desjardins A. et al. Tumor angiogenic and hypoxic profiles predict radiographic response and survival in malignant astrocytoma patients treated with bevacizumab and irinotecan. J Clin Oncol. 2008;26:271-8
106. Proescholdt MA, Merrill MJ, Stoerr EM, Lohmeier A, Pohl F, Brawanski A. Function of carbonic anhydrase IX in glioblastoma multiforme. Neuro Oncol. 2012;14:1357-66
107. Boyd NH, Walker K, Fried J, Hackney JR, McDonald PC, Benavides GA. et al. Addition of carbonic anhydrase 9 inhibitor SLC-0111 to temozolomide treatment delays glioblastoma growth in vivo. JCI Insight. 2017 2
108. Xu X, Wang Z, Liu N, Cheng Y, Jin W, Zhang P. et al. Association between SOX9 and CA9 in glioma, and its effects on chemosensitivity to TMZ. Int J Oncol. 2018;53:189-202
109. Salaroglio IC, Mujumdar P, Annovazzi L, Kopecka J, Mellai M, Schiffer D. et al. Carbonic Anhydrase XII Inhibitors Overcome P-Glycoprotein-Mediated Resistance to Temozolomide in Glioblastoma. Mol Cancer Ther. 2018;17:2598-609
110. Hannen R, Selmansberger M, Hauswald M, Pagenstecher A, Nist A, Stiewe T. et al. Comparative Transcriptomic Analysis of Temozolomide Resistant Primary GBM Stem-Like Cells and Recurrent GBM Identifies Up-Regulation of the Carbonic Anhydrase CA2 Gene as Resistance Factor. Cancers (Basel). 2019 11
111. Wu L, Bernal GM, Cahill KE, Pytel P, Fitzpatrick CA, Mashek H. et al. BCL3 expression promotes resistance to alkylating chemotherapy in gliomas. Sci Transl Med. 2018 10
112. Cui J, Zhang Q, Song Q, Wang H, Dmitriev P, Sun MY. et al. Targeting hypoxia downstream signaling protein, CAIX, for CAR T-cell therapy against glioblastoma. Neuro Oncol. 2019;21:1436-46
113. Stephanou A, Ballesta A. pH as a potential therapeutic target to improve temozolomide antitumor efficacy: A mechanistic modeling study. Pharmacol Res Perspect. 2019;7:e00454
114. Reichert M, Steinbach JP, Supra P, Weller M. Modulation of growth and radiochemosensitivity of human malignant glioma cells by acidosis. Cancer. 2002;95:1113-9
115. Ohtsubo T, Wang X, Takahashi A, Ohnishi K, Saito H, Song CW. et al. p53-dependent induction of WAF1 by a low-pH culture condition in human glioblastoma cells. Cancer Res. 1997;57:3910-3
116. Libby CJ, McConathy J, Darley-Usmar V, Hjelmeland AB. The Role of Metabolic Plasticity in Blood and Brain Stem Cell Pathophysiology. Cancer Res. 2020;80:5-16
117. Vlashi E, Lagadec C, Vergnes L, Matsutani T, Masui K, Poulou M. et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc Natl Acad Sci U S A. 2011;108:16062-7
118. Shibao S, Minami N, Koike N, Fukui N, Yoshida K, Saya H. et al. Metabolic heterogeneity and plasticity of glioma stem cells in a mouse glioblastoma model. Neuro Oncol. 2018;20:343-54
119. Mendoza EE, Pocceschi MG, Kong X, Leeper DB, Caro J, Limesand KH. et al. Control of Glycolytic Flux by AMP-Activated Protein Kinase in Tumor Cells Adapted to Low pH. Transl Oncol. 2012;5:208-16
120. Stadlbauer A, Zimmermann M, Doerfler A, Oberndorfer S, Buchfelder M, Coras R. et al. Intratumoral heterogeneity of oxygen metabolism and neovascularization uncovers 2 survival-relevant subgroups of IDH1 wild-type glioblastoma. Neuro Oncol. 2018;20:1536-46
121. Bao B, Azmi AS, Ali S, Ahmad A, Li Y, Banerjee S. et al. The biological kinship of hypoxia with CSC and EMT and their relationship with deregulated expression of miRNAs and tumor aggressiveness. Biochimica et biophysica acta. 2012;1826:272-96
122. Folkins C, Shaked Y, Man S, Tang T, Lee CR, Zhu Z. et al. Glioma tumor stem-like cells promote tumor angiogenesis and vasculogenesis via vascular endothelial growth factor and stromal-derived factor 1. Cancer Res. 2009;69:7243-51
123. Baisiwala S, Auffinger B, Caragher SP, Shireman JM, Ahsan R, Lee G. et al. Chemotherapeutic Stress Induces Transdifferentiation of Glioblastoma Cells to Endothelial Cells and Promotes Vascular Mimicry. Stem Cells Int. 2019;2019:6107456
124. Rocha R, Torres A, Ojeda K, Uribe D, Rocha D, Erices J. et al. The Adenosine A(3) Receptor Regulates Differentiation of Glioblastoma Stem-Like Cells to Endothelial Cells under Hypoxia. Int J Mol Sci. 2018 19
125. Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T. et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 2010;468:824-8
126. Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A. et al. Glioblastoma stem-like cells give rise to tumour endothelium. Nature. 2010;468:829-33
127. Takahashi H, Inoue A, Kawabe Y, Hosokawa Y, Iwata S, Sugimoto K. et al. Oct-3/4 promotes tumor angiogenesis through VEGF production in glioblastoma. Brain tumor pathology. 2015;32:31-40
128. Treps L, Perret R, Edmond S, Ricard D, Gavard J. Glioblastoma stem-like cells secrete the pro-angiogenic VEGF-A factor in extracellular vesicles. J Extracell Vesicles. 2017;6:1359479
129. Zhao C, Wang H, Xiong C, Liu Y. Hypoxic glioblastoma release exosomal VEGF-A induce the permeability of blood-brain barrier. Biochem Biophys Res Commun. 2018;502:324-31
130. Sun X, Ma X, Wang J, Zhao Y, Wang Y, Bihl JC. et al. Glioma stem cells-derived exosomes promote the angiogenic ability of endothelial cells through miR-21/VEGF signal. Oncotarget. 2017;8:36137-48
131. Hamerlik P, Lathia JD, Rasmussen R, Wu Q, Bartkova J, Lee M. et al. Autocrine VEGF-VEGFR2-Neuropilin-1 signaling promotes glioma stem-like cell viability and tumor growth. J Exp Med. 2012;209:507-20
132. Zhao D, Pan C, Sun J, Gilbert C, Drews-Elger K, Azzam DJ. et al. VEGF drives cancer-initiating stem cells through VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene. 2015;34:3107-19
133. Cheng L, Huang Z, Zhou W, Wu Q, Donnola S, Liu JK. et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell. 2013;153:139-52
134. Yao X, Ping Y, Liu Y, Chen K, Yoshimura T, Liu M. et al. Vascular endothelial growth factor receptor 2 (VEGFR-2) plays a key role in vasculogenic mimicry formation, neovascularization and tumor initiation by Glioma stem-like cells. PloS one. 2013;8:e57188
135. Ping YF, Yao XH, Jiang JY, Zhao LT, Yu SC, Jiang T. et al. The chemokine CXCL12 and its receptor CXCR4 promote glioma stem cell-mediated VEGF production and tumour angiogenesis via PI3K/AKT signalling. The Journal of pathology. 2011;224:344-54
136. Wurth R, Bajetto A, Harrison JK, Barbieri F, Florio T. CXCL12 modulation of CXCR4 and CXCR7 activity in human glioblastoma stem-like cells and regulation of the tumor microenvironment. Frontiers in cellular neuroscience. 2014;8:144
137. Calinescu AA, Yadav VN, Carballo E, Kadiyala P, Tran D, Zamler DB. et al. Survival and Proliferation of Neural Progenitor-Derived Glioblastomas Under Hypoxic Stress is Controlled by a CXCL12/CXCR4 Autocrine-Positive Feedback Mechanism. Clinical cancer research: an official journal of the American Association for Cancer Research. 2017;23:1250-62
138. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741-51
139. Cheng L, Wu Q, Guryanova OA, Huang Z, Huang Q, Rich JN. et al. Elevated invasive potential of glioblastoma stem cells. Biochem Biophys Res Commun. 2011;406:643-8
140. Joseph JV, Conroy S, Pavlov K, Sontakke P, Tomar T, Eggens-Meijer E. et al. Hypoxia enhances migration and invasion in glioblastoma by promoting a mesenchymal shift mediated by the HIF1alpha-ZEB1 axis. Cancer Lett. 2015;359:107-16
141. Torres A, Erices JI, Sanchez F, Ehrenfeld P, Turchi L, Virolle T. et al. Extracellular adenosine promotes cell migration/invasion of Glioblastoma Stem-like Cells through A3 Adenosine Receptor activation under hypoxia. Cancer letters. 2019;446:112-22
142. Chen JE, Lumibao J, Blazek A, Gaskins HR, Harley B. Hypoxia activates enhanced invasive potential and endogenous hyaluronic acid production by glioblastoma cells. Biomater Sci. 2018;6:854-62
143. Delpech B, Maingonnat C, Girard N, Chauzy C, Maunoury R, Olivier A. et al. Hyaluronan and hyaluronectin in the extracellular matrix of human brain tumour stroma. Eur J Cancer. 1993;29A:1012-7
144. Pietras A, Katz AM, Ekstrom EJ, Wee B, Halliday JJ, Pitter KL. et al. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell. 2014;14:357-69
145. Anido J, Saez-Borderias A, Gonzalez-Junca A, Rodon L, Folch G, Carmona MA. et al. TGF-beta Receptor Inhibitors Target the CD44(high)/Id1(high) Glioma-Initiating Cell Population in Human Glioblastoma. Cancer Cell. 2010;18:655-68
146. Gilg AG, Tye SL, Tolliver LB, Wheeler WG, Visconti RP, Duncan JD. et al. Targeting hyaluronan interactions in malignant gliomas and their drug-resistant multipotent progenitors. Clin Cancer Res. 2008;14:1804-13
147. Maciaczyk D, Picard D, Zhao L, Koch K, Herrera-Rios D, Li G. et al. CBF1 is clinically prognostic and serves as a target to block cellular invasion and chemoresistance of EMT-like glioblastoma cells. British journal of cancer. 2017;117:102-12
148. Fujimura A, Michiue H, Cheng Y, Uneda A, Tani Y, Nishiki T. et al. Cyclin G2 promotes hypoxia-driven local invasion of glioblastoma by orchestrating cytoskeletal dynamics. Neoplasia. 2013;15:1272-81
149. Xie Q, Wu Q, Horbinski CM, Flavahan WA, Yang K, Zhou W. et al. Mitochondrial control by DRP1 in brain tumor initiating cells. Nat Neurosci. 2015;18:501-10
150. Heddleston JM, Wu Q, Rivera M, Minhas S, Lathia JD, Sloan AE. et al. Hypoxia-induced mixed-lineage leukemia 1 regulates glioma stem cell tumorigenic potential. Cell Death Differ. 2012;19:428-39
151. Prasad P, Mittal SA, Chongtham J, Mohanty S, Srivastava T. Hypoxia-Mediated Epigenetic Regulation of Stemness in Brain Tumor Cells. Stem Cells. 2017;35:1468-78
152. Liau BB, Sievers C, Donohue LK, Gillespie SM, Flavahan WA, Miller TE. et al. Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell. 2017;20:233-46 e7
153. Sherry-Lynes MM, Sengupta S, Kulkarni S, Cochran BH. Regulation of the JMJD3 (KDM6B) histone demethylase in glioblastoma stem cells by STAT3. PloS one. 2017;12:e0174775
154. Sui A, Xu Y, Li Y, Hu Q, Wang Z, Zhang H. et al. The pharmacological role of histone demethylase JMJD3 inhibitor GSK-J4 on glioma cells. Oncotarget. 2017;8:68591-8
155. Wang B, Fan X, Ma C, Lei H, Long Q, Chai Y. Downregulation of KDM4A Suppresses the Survival of Glioma Cells by Promoting Autophagy. J Mol Neurosci. 2016;60:137-44
156. Hall JA, Georgel PT. CHD proteins: a diverse family with strong ties. Biochem Cell Biol. 2007;85:463-76
157. Marfella CG, Imbalzano AN. The Chd family of chromatin remodelers. Mutat Res. 2007;618:30-40
158. Schnetz MP, Bartels CF, Shastri K, Balasubramanian D, Zentner GE, Balaji R. et al. Genomic distribution of CHD7 on chromatin tracks H3K4 methylation patterns. Genome Res. 2009;19:590-601
159. Schnetz MP, Handoko L, Akhtar-Zaidi B, Bartels CF, Pereira CF, Fisher AG. et al. CHD7 targets active gene enhancer elements to modulate ES cell-specific gene expression. PLoS Genet. 2010;6:e1001023
160. Bergman JE, Janssen N, Hoefsloot LH, Jongmans MC, Hofstra RM, van Ravenswaaij-Arts CM. CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. Journal of medical genetics. 2011;48:334-42
161. Hsu P, Ma A, Wilson M, Williams G, Curotta J, Munns CF. et al. CHARGE syndrome: a review. J Paediatr Child Health. 2014;50:504-11
162. Colbert LE, Petrova AV, Fisher SB, Pantazides BG, Madden MZ, Hardy CW. et al. CHD7 expression predicts survival outcomes in patients with resected pancreatic cancer. Cancer Res. 2014;74:2677-87
163. Kim MS, Chung NG, Kang MR, Yoo NJ, Lee SH. Genetic and expressional alterations of CHD genes in gastric and colorectal cancers. Histopathology. 2011;58:660-8
164. Tahara T, Yamamoto E, Madireddi P, Suzuki H, Maruyama R, Chung W. et al. Colorectal carcinomas with CpG island methylator phenotype 1 frequently contain mutations in chromatin regulators. Gastroenterology. 2014;146:530-38 e5
165. Engelen E, Akinci U, Bryne JC, Hou J, Gontan C, Moen M. et al. Sox2 cooperates with Chd7 to regulate genes that are mutated in human syndromes. Nat Genet. 2011;43:607-11
166. Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood. 2004;104:2224-34
167. Kumar V, Gabrilovich DI. Hypoxia-inducible factors in regulation of immune responses in tumour microenvironment. Immunology. 2014;143:512-9
168. Wu A, Wei J, Kong LY, Wang Y, Priebe W, Qiao W. et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010;12:1113-25
169. Yi L, Xiao H, Xu M, Ye X, Hu J, Li F. et al. Glioma-initiating cells: a predominant role in microglia/macrophages tropism to glioma. J Neuroimmunol. 2011;232:75-82
170. Guo X, Xue H, Shao Q, Wang J, Guo X, Chen X. et al. Hypoxia promotes glioma-associated macrophage infiltration via periostin and subsequent M2 polarization by upregulating TGF-beta and M-CSFR. Oncotarget. 2016;7:80521-42
171. Bacher M, Schrader J, Thompson N, Kuschela K, Gemsa D, Waeber G. et al. Up-regulation of macrophage migration inhibitory factor gene and protein expression in glial tumor cells during hypoxic and hypoglycemic stress indicates a critical role for angiogenesis in glioblastoma multiforme. Am J Pathol. 2003;162:11-7
172. Polajeva J, Bergstrom T, Edqvist PH, Lundequist A, Sjosten A, Nilsson G. et al. Glioma-derived macrophage migration inhibitory factor (MIF) promotes mast cell recruitment in a STAT5-dependent manner. Mol Oncol. 2014;8:50-8
173. Otvos B, Silver DJ, Mulkearns-Hubert EE, Alvarado AG, Turaga SM, Sorensen MD. et al. Cancer Stem Cell-Secreted Macrophage Migration Inhibitory Factor Stimulates Myeloid Derived Suppressor Cell Function and Facilitates Glioblastoma Immune Evasion. Stem Cells. 2016;34:2026-39
174. Raychaudhuri B, Rayman P, Ireland J, Ko J, Rini B, Borden EC. et al. Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro Oncol. 2011;13:591-9
175. Zeiner PS, Preusse C, Blank AE, Zachskorn C, Baumgarten P, Caspary L. et al. MIF Receptor CD74 is Restricted to Microglia/Macrophages, Associated with a M1-Polarized Immune Milieu and Prolonged Patient Survival in Gliomas. Brain Pathol. 2015;25:491-504
176. Chen Q, Han B, Meng X, Duan C, Yang C, Wu Z. et al. Immunogenomic analysis reveals LGALS1 contributes to the immune heterogeneity and immunosuppression in glioma. International journal of cancer Journal international du cancer. 2019;145:517-30
177. Cai J, Chen Q, Cui Y, Dong J, Chen M, Wu P. et al. Immune heterogeneity and clinicopathologic characterization of IGFBP2 in 2447 glioma samples. Oncoimmunology. 2018;7:e1426516
178. Shen F, Song C, Liu Y, Zhang J, Wei Song S. IGFBP2 promotes neural stem cell maintenance and proliferation differentially associated with glioblastoma subtypes. Brain Res. 2019;1704:174-86
179. Hsieh D, Hsieh A, Stea B, Ellsworth R. IGFBP2 promotes glioma tumor stem cell expansion and survival. Biochem Biophys Res Commun. 2010;397:367-72
180. Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH. et al. The Immune Landscape of Cancer. Immunity. 2018;48:812-30 e14
181. Miska J, Lee-Chang C, Rashidi A, Muroski ME, Chang AL, Lopez-Rosas A. et al. HIF-1alpha Is a Metabolic Switch between Glycolytic-Driven Migration and Oxidative Phosphorylation-Driven Immunosuppression of Tregs in Glioblastoma. Cell Rep. 2019;27:226-37 e4
182. Kahlert UD, Maciaczyk D, Dai F, Claus R, Firat E, Doostkam S. et al. Resistance to hypoxia-induced, BNIP3-mediated cell death contributes to an increase in a CD133-positive cell population in human glioblastomas in vitro. Journal of neuropathology and experimental neurology. 2012;71:1086-99
183. Wang P, Wan W, Xiong S, Wang J, Zou D, Lan C. et al. HIF1alpha regulates glioma chemosensitivity through the transformation between differentiation and dedifferentiation in various oxygen levels. Sci Rep. 2017;7:7965
184. Yuan J, Narayanan L, Rockwell S, Glazer PM. Diminished DNA repair and elevated mutagenesis in mammalian cells exposed to hypoxia and low pH. Cancer Res. 2000;60:4372-6
185. Ramirez YP, Weatherbee JL, Wheelhouse RT, Ross AH. Glioblastoma multiforme therapy and mechanisms of resistance. Pharmaceuticals. 2013;6:1475-506
186. Scanlon SE, Glazer PM. Multifaceted control of DNA repair pathways by the hypoxic tumor microenvironment. DNA repair. 2015;32:180-9
187. Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11:393-410
188. Sullivan R, Graham CH. Hypoxia prevents etoposide-induced DNA damage in cancer cells through a mechanism involving hypoxia-inducible factor 1. Molecular cancer therapeutics. 2009;8:1702-13
189. King HO, Brend T, Payne HL, Wright A, Ward TA, Patel K. et al. RAD51 Is a Selective DNA Repair Target to Radiosensitize Glioma Stem Cells. Stem Cell Reports. 2017;8:125-39
190. Ong DST, Hu B, Ho YW, Sauve CG, Bristow CA, Wang Q. et al. PAF promotes stemness and radioresistance of glioma stem cells. Proc Natl Acad Sci U S A. 2017;114:E9086-E95
191. Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG. et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522-6
192. Chou CW, Wang CC, Wu CP, Lin YJ, Lee YC, Cheng YW. et al. Tumor cycling hypoxia induces chemoresistance in glioblastoma multiforme by upregulating the expression and function of ABCB1. Neuro Oncol. 2012;14:1227-38
193. Hosokawa Y, Takahashi H, Inoue A, Kawabe Y, Funahashi Y, Kameda K. et al. Oct-3/4 modulates the drug-resistant phenotype of glioblastoma cells through expression of ATP binding cassette transporter G2. Biochimica et biophysica acta. 2015;1850:1197-205
194. Lee CAA, Banerjee P, Wilson BJ, Wu S, Guo Q, Berg G. et al. Targeting the ABC transporter ABCB5 sensitizes glioblastoma to temozolomide-induced apoptosis through a cell-cycle checkpoint regulation mechanism. J Biol Chem. 2020;295:7774-88
195. Saggar JK, Yu M, Tan Q, Tannock IF. The tumor microenvironment and strategies to improve drug distribution. Frontiers in oncology. 2013;3:154
196. Primeau AJ, Rendon A, Hedley D, Lilge L, Tannock IF. The distribution of the anticancer drug Doxorubicin in relation to blood vessels in solid tumors. Clinical cancer research: an official journal of the American Association for Cancer Research. 2005;11:8782-8
197. Kyle AH, Huxham LA, Yeoman DM, Minchinton AI. Limited tissue penetration of taxanes: a mechanism for resistance in solid tumors. Clinical cancer research: an official journal of the American Association for Cancer Research. 2007;13:2804-10
198. Patel KJ, Tredan O, Tannock IF. Distribution of the anticancer drugs doxorubicin, mitoxantrone and topotecan in tumors and normal tissues. Cancer chemotherapy and pharmacology. 2013;72:127-38
199. Beig N, Patel J, Prasanna P, Hill V, Gupta A, Correa R. et al. Radiogenomic analysis of hypoxia pathway is predictive of overall survival in Glioblastoma. Sci Rep. 2018;8:7
200. Harris RJ, Cloughesy TF, Liau LM, Prins RM, Antonios JP, Li D. et al. pH-weighted molecular imaging of gliomas using amine chemical exchange saturation transfer MRI. Neuro-oncology. 2015;17:1514-24
201. Harris RJ, Cloughesy TF, Liau LM, Nghiemphu PL, Lai A, Pope WB. et al. Simulation, phantom validation, and clinical evaluation of fast pH-weighted molecular imaging using amine chemical exchange saturation transfer echo planar imaging (CEST-EPI) in glioma at 3 T. NMR Biomed. 2016;29:1563-76
202. Schure JR, Shrestha M, Breuer S, Deichmann R, Hattingen E, Wagner M. et al. The pH sensitivity of APT-CEST using phosphorus spectroscopy as a reference method. NMR Biomed. 2019;32:e4125
203. Rao JU, Coman D, Walsh JJ, Ali MM, Huang Y, Hyder F. Temozolomide arrests glioma growth and normalizes intratumoral extracellular pH. Sci Rep. 2017;7:7865
204. Brenner A, Zuniga R, Sun JD, Floyd J, Hart CP, Kroll S. et al. Hypoxia-activated evofosfamide for treatment of recurrent bevacizumab-refractory glioblastoma: a phase I surgical study. Neuro Oncol. 2018;20:1231-9
205. Stroka DM, Burkhardt T, Desbaillets I, Wenger RH, Neil DA, Bauer C. et al. HIF-1 is expressed in normoxic tissue and displays an organ-specific regulation under systemic hypoxia. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2001;15:2445-53
206. Ziello JE, Jovin IS, Huang Y. Hypoxia-Inducible Factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. The Yale journal of biology and medicine. 2007;80:51-60
207. Mimeault M, Batra SK. Hypoxia-inducing factors as master regulators of stemness properties and altered metabolism of cancer- and metastasis-initiating cells. Journal of cellular and molecular medicine. 2013;17:30-54
208. Zhu LL, Wu LY, Yew DT, Fan M. Effects of hypoxia on the proliferation and differentiation of NSCs. Mol Neurobiol. 2005;31:231-42
209. Lee DH, Cheul Oh S, Giles AJ, Jung J, Gilbert MR, Park DM. Cardiac glycosides suppress the maintenance of stemness and malignancy via inhibiting HIF-1alpha in human glioma stem cells. Oncotarget. 2017;8:40233-45
210. Chen W, Hill H, Christie A, Kim MS, Holloman E, Pavia-Jimenez A. et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature. 2016;539:112-7
211. Wallace EM, Rizzi JP, Han G, Wehn PM, Cao Z, Du X. et al. A Small-Molecule Antagonist of HIF2alpha Is Efficacious in Preclinical Models of Renal Cell Carcinoma. Cancer Res. 2016;76:5491-500
212. Bernstock JD, Ye D, Gessler FA, Lee YJ, Peruzzotti-Jametti L, Baumgarten P. et al. Topotecan is a potent inhibitor of SUMOylation in glioblastoma multiforme and alters both cellular replication and metabolic programming. Sci Rep. 2017;7:7425
213. Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D. et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiological reviews. 2011;91:1071-121
214. Rubenstein JL, Kim J, Ozawa T, Zhang M, Westphal M, Deen DF. et al. Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia. 2000;2:306-14
215. de Groot JF, Fuller G, Kumar AJ, Piao Y, Eterovic K, Ji Y. et al. Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro-oncology. 2010;12:233-42
216. Tamura R, Tanaka T, Miyake K, Yoshida K, Sasaki H. Bevacizumab for malignant gliomas: current indications, mechanisms of action and resistance, and markers of response. Brain tumor pathology. 2017;34:62-77
217. Jiang X, Wang C, Fitch S, Yang F. Targeting Tumor Hypoxia Using Nanoparticle-engineered CXCR4-overexpressing Adipose-derived Stem Cells. Theranostics. 2018;8:1350-60
218. Gagner JP, Sarfraz Y, Ortenzi V, Alotaibi FM, Chiriboga LA, Tayyib AT. et al. Multifaceted C-X-C Chemokine Receptor 4 (CXCR4) Inhibition Interferes with Anti-Vascular Endothelial Growth Factor Therapy-Induced Glioma Dissemination. The American journal of pathology. 2017;187:2080-94
219. Hua L, Wang Z, Zhao L, Mao H, Wang G, Zhang K. et al. Hypoxia-responsive lipid-poly-(hypoxic radiosensitized polyprodrug) nanoparticles for glioma chemo- and radiotherapy. Theranostics. 2018;8:5088-105
220. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature cell biology. 2011;13:132-41
221. Kahn J, Hayman TJ, Jamal M, Rath BH, Kramp T, Camphausen K. et al. The mTORC1/mTORC2 inhibitor AZD2014 enhances the radiosensitivity of glioblastoma stem-like cells. Neuro-oncology. 2014;16:29-37
222. Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279-90
223. Chapnick DA, Warner L, Bernet J, Rao T, Liu X. Partners in crime: the TGFbeta and MAPK pathways in cancer progression. Cell & bioscience. 2011;1:42
224. Mao H, Lebrun DG, Yang J, Zhu VF, Li M. Deregulated signaling pathways in glioblastoma multiforme: molecular mechanisms and therapeutic targets. Cancer investigation. 2012;30:48-56
225. Breunig JJ, Levy R, Antonuk CD, Molina J, Dutra-Clarke M, Park H. et al. Ets Factors Regulate Neural Stem Cell Depletion and Gliogenesis in Ras Pathway Glioma. Cell Rep. 2015;12:258-71
226. Franovic A, Gunaratnam L, Smith K, Robert I, Patten D, Lee S. Translational up-regulation of the EGFR by tumor hypoxia provides a nonmutational explanation for its overexpression in human cancer. Proc Natl Acad Sci U S A. 2007;104:13092-7
227. Yoshida D, Kim K, Noha M, Teramoto A. Hypoxia inducible factor 1-alpha regulates of platelet derived growth factor-B in human glioblastoma cells. J Neurooncol. 2006;76:13-21
228. Papa S, Choy PM, Bubici C. The ERK and JNK pathways in the regulation of metabolic reprogramming. Oncogene. 2019;38:2223-40
229. Chhipa RR, Fan Q, Anderson J, Muraleedharan R, Huang Y, Ciraolo G. et al. AMP kinase promotes glioblastoma bioenergetics and tumour growth. Nature cell biology. 2018;20:823-35
230. Guise CP, Mowday AM, Ashoorzadeh A, Yuan R, Lin WH, Wu DH. et al. Bioreductive prodrugs as cancer therapeutics: targeting tumor hypoxia. Chin J Cancer. 2014;33:80-6
231. Rauth AM, Melo T, Misra V. Bioreductive therapies: an overview of drugs and their mechanisms of action. International journal of radiation oncology, biology, physics. 1998;42:755-62
232. Patterson LH, McKeown SR. AQ4N: a new approach to hypoxia-activated cancer chemotherapy. British journal of cancer. 2000;83:1589-93
233. Albertella MR, Loadman PM, Jones PH, Phillips RM, Rampling R, Burnet N. et al. Hypoxia-selective targeting by the bioreductive prodrug AQ4N in patients with solid tumors: results of a phase I study. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008;14:1096-104
234. Zeman EM, Brown JM, Lemmon MJ, Hirst VK, Lee WW. SR-4233: a new bioreductive agent with high selective toxicity for hypoxic mammalian cells. International journal of radiation oncology, biology, physics. 1986;12:1239-42
235. Del Rowe J, Scott C, Werner-Wasik M, Bahary JP, Curran WJ, Urtasun RC. et al. Single-arm, open-label phase II study of intravenously administered tirapazamine and radiation therapy for glioblastoma multiforme. J Clin Oncol. 2000;18:1254-9
236. Hicks KO, Siim BG, Jaiswal JK, Pruijn FB, Fraser AM, Patel R. et al. Pharmacokinetic/pharmacodynamic modeling identifies SN30000 and SN29751 as tirapazamine analogues with improved tissue penetration and hypoxic cell killing in tumors. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16:4946-57
237. Gu Y, Chang TT, Wang J, Jaiswal JK, Edwards D, Downes NJ. et al. Reductive Metabolism Influences the Toxicity and Pharmacokinetics of the Hypoxia-Targeted Benzotriazine Di-Oxide Anticancer Agent SN30000 in Mice. Front Pharmacol. 2017;8:531
238. Kennedy KA, Rockwell S, Sartorelli AC. Preferential activation of mitomycin C to cytotoxic metabolites by hypoxic tumor cells. Cancer Res. 1980;40:2356-60
239. Ma H, Zhang Y, Wang H, Han C, Lei R, Zhang L. et al. Effect and mechanism of Mitomycin C combined with recombinant adeno-associated virus type II against glioma. Int J Mol Sci. 2013;15:1-14
240. Gu B, DeAngelis LM. Enhanced cytotoxicity of bioreductive antitumor agents with dimethyl fumarate in human glioblastoma cells. Anticancer Drugs. 2005;16:167-74
241. Okamura T, Kurisu K, Yamamoto W, Takano H, Nishiyama M. NADPH/quinone oxidoreductase is a priority target of glioblastoma chemotherapy. International journal of oncology. 2000;16:295-303
Author contact
![]() Corresponding author: Anita B. Hjelmeland, Ph.D. Associate Professor. Department of Cell, Developmental and Integrative Biology, University of Alabama at Birmingham, Birmingham, AL 35294. E-mail: hjelmeaedu; Phone: 205-996-4596.
Corresponding author: Anita B. Hjelmeland, Ph.D. Associate Professor. Department of Cell, Developmental and Integrative Biology, University of Alabama at Birmingham, Birmingham, AL 35294. E-mail: hjelmeaedu; Phone: 205-996-4596.