Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Metabolic crosstalk: from TME to...
Metabolic crosstalk: from TAMs...
Conclusions and Perspectives
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(3):1016-1030. doi:10.7150/thno.51777 This issue Cite
Review
Metabolic regulatory crosstalk between tumor microenvironment and tumor-associated macrophages
Degao Chen1#, Xiaomei Zhang2#, Zhongjun Li2 ![]() , Bo Zhu1,3
, Bo Zhu1,3 ![]()
1. Institute of Cancer, Xinqiao Hospital, Third Military Medical University, Chongqing 400037, China
2. Department of Blood Transfusion, Lab of Radiation biology, Xinqiao Hospital, Third Military Medical University, Chongqing 400037, China
3. Chongqing Key Laboratory of Immunotherapy, Xinqiao Hospital, Third Military Medical University, Chongqing 400037, China
#These authors contributed equally: Degao Chen and Xiaomei Zhang.
Received 2020-8-10; Accepted 2020-10-18; Published 2021-1-1
Abstract

Macrophages phagocytize pathogens to initiate innate immunity and products from the tumor microenvironment (TME) to mediate tumor immunity. The loss of tumor-associated macrophage (TAM)-mediated immune responses results in immune suppression. To reverse this immune disorder, the regulatory mechanism of TAMs in the TME needs to be clarified. Immune molecules (cytokines and chemokines) from TAMs and the TME have been widely accepted as mutual mediators of signal transduction in the past few decades. Recently, researchers have tried to seek the intrinsic mechanism of TAM phenotypic and functional changes through metabolic connections. Numerous metabolites derived from the TME have been identified that induce the cell-cell crosstalk with TAMs. The bulk tumor cells, immune cells, and stromal cells produce metabolites in the TME that are involved in the metabolic regulation of TAMs. Meanwhile, some products from TAMs regulate the biological functions of the tumor as well. Here, we review the recent reports demonstrating the metabolic regulation between TME and TAMs.
Keywords: Tumor-associated macrophage, tumor microenvironment, metabolism, tumor immunotherapy
Introduction
Macrophages that reside within the tumor microenvironment (TME) are known as tumor-associated macrophages (TAMs). As the predominant infiltrated immune cells in the TME [1-3], TAMs have been extensively studied for their pro-tumoral activities, such as tumor initiation, angiogenesis, metastasis, drug-resistance, and antitumor immunosuppression [4]. To mirror the Th1/2 immune response, canonical concepts suggest that there are two distinct states of polarized activation in macrophages, namely classical (M1, induced by lipopolysaccharide and IFN-γ) and alternative (M2, induced by IL-4 or IL-13) [5]. M1 and M2 macrophages have different transcriptional profiles, such as cytokines, chemokines, metabolic pathways. Studies have indicated that TAMs predominantly have an M2-like phenotype, which manifests as an immunosuppressive state and pro-tumoral progression [6]. Hence, targeting M2-like TAMs and depleting them in the TME or reversing the M2-like TAMs into an M1-like phenotype, which directly boosts their cytotoxicity and indirectly stimulates cytotoxic T cells to eliminate tumor cells, is a potential strategy for antitumor immunotherapy [7, 8]. Numerous mechanisms underlying the role of TAMs in tumor immunosuppression have been elucidated, and novel therapeutic agents based on these new targets have been subjected to clinical trials in recent years [8, 9]. In addition, some recent reports focused on phagocytosis, the primordial function of macrophages, to reactivate antitumor immunity by targeting CD47/SIRPα [10], PD-1/PD-L1 [11], and CD24/Siglec-10 [12] pathways. All of these strategies have moved TAMs to the forefront of tumor immunotherapy.
In addition to the cytokine and chemokine regulatory mechanisms, metabolic regulation between the TME and TAMs has also been widely studied. All members of the TME rely on nutrients for their survival, maintenance, and proliferation. Meanwhile, the competition and symbiosis between TAMs and other members of the TME form a crosstalk mechanism linked by metabolites. Excessive metabolites uptake or secretion reprograms the phenotype and function of such components and interferes with tumor outcomes. Therefore, clarifying the metabolic regulatory mechanisms would help elucidate the landscape of metabolite connections between the TME and TAMs and promote metabolic immunotherapy targeting TAMs. Some excellent reviews have focused on TAM metabolism and the metabolic crosstalk between tumor cells and TAMs [13-15]. However, other than tumor cells, the metabolic regulation of immune and stromal cells in TME also needs to be concerned, especially the interaction with TAMs. Besides, the metabolism pattern of TAMs affects the outcome of cancer. This review discusses the metabolic crosstalk from TME to TAMs based on different cell types and from TAMs to TME by diverse oncobiological functions.
Metabolic crosstalk: from TME to TAMs
TAMs and other TME members make up the tumor ecosystem, suggesting that there are some interactions between them through cytokines, chemokines, and other factors [16]. Most often, all members of the TME consume oxygen and nutrients from the host for their phenotypic and functional performance [17, 18]. Thus, metabolites are accumulated in the TME and recycled from cell to cell. In particular, as messengers for cell-cell contact, the metabolites, which are derived from the TME (tumor cells, T cells, mast cells, cancer-associated fibroblasts, adipocytes, except TAMs), are ingested by TAMs to change their phenotype and function. In turn, TAMs promote tumor progression via metabolic reprogramming, which is triggered by the metabolites that are shuttled in the TME. Moreover, blockade of the metabolic pathways in the TME and TAMs is being used for drug discovery and tumor therapy [8, 19]. Next, we describe the recent findings regarding metabolic crosstalk between the TME and TAMs (Figure 1).
The metabolic crosstalk: from TME to TAMs. Tumor cell-derived lactic acid, succinate, α-ketoglutarate, ATP/Adenosine, PGE2 (prostaglandin E2), oleate, Retinoic acid (RA), BCKA (branched-chain ketoacids), kynurenine, POSTN (periostin), versican, sonic hedgehog and the extracellular acidosis promote the protumoral TAM formation to accelerate tumor progression. Artificially increasing HRG (histidine-rich glycoprotein) or stearic acid expression from tumor cell induce the antitumoral TAM. Metabolites from T cell to TAM are rarely identified, but activated T cell enhances IDO and SREBP1 (sterol regulatory element-binding protein 1) in TAM to facilitate tumor growth. CAF (cancer-associated fibroblast)-produced lactic acid, ADMA (asymmetric dimethyl arginine), BHB (beta-hydroxybutyrate), hyaluronan and STC1 (stanniocalcin-1) assist TAM to be protumoral. Mast cell-released LTB4 (leukotriene B4), chondroitin sulfate and tryptase/chymase guide TAM toward into tumor promoting. However, PGD2 (prostaglandin D2) from mast cell may bind DP (PGD2 receptors) on TAM and skews TAM to be antitumoral. Histamine triggers H4R (histamine H4-receptor) on macrophage into protumoral TAM, but H2R (histamine H2-receptor) into antitumoral TAM. Adipocyte-derived APN (adiponectin), leptin and A-FABP (adipose fatty acid binding protein) induce the protumoral TAM. Chemerin from adipocyte causing the phenotypic change of TAM is tumor context dependent. MCT1/4 (monocarboxylate transporter 1/4); SUCNR1 (succinate receptor1); GPCR (G protein-coupled receptors); FAO (fatty acid oxidation); P2XRs/P2YRs (P2 purinergic receptors); A2AR/A2BR (adenosine receptors); BCAA (branched-chain amino acid); HCA2 (hydroxycarboxylic acid receptor 2); LTB4R1/2 (leukotriene B4 receptor 1/2); AdipoR (adiponectin receptor); ChemR23 (chemokine-like receptor 1, CMKLR1); PIGF (placental growth factor); E-FABP (epidermal fatty acid binding protein); ICER (inducible cyclic AMP (cAMP) early repressor); CREB (cyclic AMP-responsive element binding); AHR (aryl hydrocarbon receptor); RAR (retinoid acid receptor); UCP2 (uncoupling protein 2).
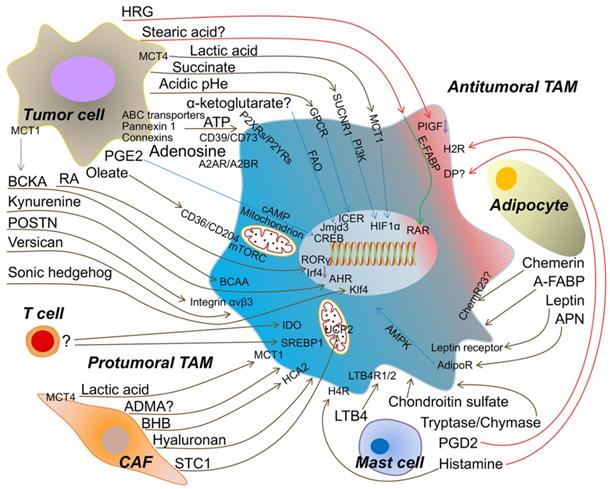
Tumor cells
The bulk of cells in the TME are tumor cells. Tumor cells possess the self-serving characteristic of educating the TME to provide pro-tumoral conditions. Numerous tumor cells deprive glucose from the TME for their progression [20]. Thereby, other cells, including TAMs, have no alternative but mainly rely on oxidative phosphorylation (OXPHOS) to produce energy for cellular processes [21]. However, most tumor cells do not fully transmit glucose into the mitochondrial tricarboxylic acid (TCA) cycle to generate ATP (adenosine triphosphate) efficiently, but instead use glucose in aerobic glycolysis, which is termed the “Warburg Effect” [22]. Thus, tumor cells produce a large amount of lactic acid through aerobic glycolysis and release the redundant lactic acid into the extracellular microenvironment via monocarboxylate transporter 4 (MCT4) [23].
Reports indicate that lactic acid-treatment suppresses TNF secretion in human monocytes through glycolysis inhibition [24]. Furthermore, lactic acid is reportedly the messenger between tumor cells and TAMs. In addition, tumor cell-derived lactic acid induces the expression of vascular endothelial growth factor (VEGF) and the M2-like polarization of TAMs, which is mediated by hypoxia-inducible factor 1α (HIF1α) [25]. Moreover, a recent study demonstrated that lactic acid induces M2-like gene activation in macrophages through histone lactylation [26]. Besides, extracellular acidosis is involved in tumor progression via the stimulation of autophagy and immunosuppression [23], which was also found to promote tumor progression by increasing tumor-promoting macrophages in prostate cancer [27]. Regarding the underlying mechanism, tumor acidosis is sensed by G protein-coupled receptors (GPCRs) in TAMs. It induces transcriptional repressor ICER (inducible cyclic AMP (cAMP) early repressor) expression to enhance pro-tumoral macrophage polarization, especially in tumors with a high glycolytic rate, such as melanoma [28]. It is noteworthy that tumor-derived lactic acid was found to be dispensable for the induction of ICER expression in TAMs in this study [28], which means that other organic acids and hydrogen ions together, with or without lactic acid, contribute to prime TAMs for tumor growth.
Interestingly, the restricted OXPHOS in tumor cells is accompanied by glutamine consumption to fuel the TCA cycle. Glutamine is converted to α-ketoglutarate by glutamate dehydrogenase or aminotransferases [29]. One study has reported that intracellular α-ketoglutarate leads to the M2-like activation of macrophages via FAO (fatty acid oxidation) and Jmjd3-dependent epigenetic reprogramming of M2 genes [30]. However, more research is needed to elucidate the mechanism of α-ketoglutarate shuttling between tumor cells and TAMs. Succinate, the downstream product of α-ketoglutarate, is an intermediate of the TCA cycle in mitochondria, and its levels are mainly controlled by succinate dehydrogenase (SDH), which is regarded as a tumor suppressor [31]. Inherited or somatic mutations and the inhibition of SDH result in tumor formation, which importantly causes the accumulation of succinate in tumor cells [32]. Nevertheless, the excessive uptake of glutamine by tumor cells induces glutamine-based anaplerosis, and the γ-aminobutyric acid shunting pathway also increases succinate levels [33]. The tumor-derived succinate is taken up through succinate receptor (SUCNR1) and results in the polarization of TAMs into a pro-tumoral form via the SUCNR1/PI3K/HIF1α signaling pathway to enhance tumor metastasis, which can also be mediated by autocrine succinate [34]. These studies suggest that serum succinate could be a potential clinical biomarker.
ATP has long been known as the energy molecule. Hypoxia and chemotherapy lead to the accumulation of abundant ATP in the TME [35], which is primarily derived from tumor cells via ATP-binding cassette (ABC) transporters [36], pannexin 1 [37], or connexins [38]. The extracellular ATP triggers the P2 purinergic receptors (P2XRs and P2YRs) in tumor cells for its growth or inhibition [39]. In most cases, ATP is unstable in biological fluids and is rapidly degraded to adenosine, which is abundantly accumulated in extracellular fluids of solid tumors for tumor immunosuppression [40]. This biological process is mediated through the ectonucleotidases CD39, which converts ATP into ADP/AMP, and CD73, degrading AMP into adenosine [41]. One study reported that adenosine induces TAMs to express CD39 and CD73 for tumor immune escape [42]. Moreover, adenosine receptors (A1R, A2AR, A2BR, and A3R) have been found to sense adenosine for tumor growth, survival, metastasis, and immunosuppression [38]. Furthermore, reports suggest that CD39/CD73 and adenosine receptors are mainly induced and regulated by HIF1α [43-45]. Furthermore, A2AR and A2BR were reportedly to be expressed on macrophages and stimulated by adenosine, inducing M2-like TAM polarization [38, 41]. Additionally, A2AR depletion on myeloid cells, especially macrophages, slows melanoma growth and reduces metastasis by relieving T and NK cell suppression [46]. Studies have recently revealed that ectonucleotidases, adenosine receptors, and the adenosinergic pathway are potential targets to enhance antitumor immunity [38, 40], which would accelerate drug discovery in this field.
Prostaglandin E2 (PGE2), a prostanoid lipid, is associated with tumor progression [47] and is synthesized by tumor cyclooxygenase to mediate tumor immune escape [48]. Reports suggested that PGE2 induces M2 polarization in macrophages through the cyclic AMP-responsive element binding (CREB) pathway [49, 50]. Furthermore, PD-L1 expression in TAMs can be regulated by PGE2 levels [51]. Recently, one study clarified that PGE2 modulates mitochondrial membrane potential to regulate M2-like gene expression in the nucleus [52]. Thereby, PGE2 from tumor cells polarizes TAMs through cAMP and mitochondrial signaling.
Fatty acids are reportedly enriched in the TME and mainly tumor cell-derived [53]. A recent paper indicated that long-chain fatty acids, exemplified by oleate, are scavenged by CD36/204, facilitate mitochondrial respiration in TAMs, and polarize immune-suppressive TAMs through mTORC signaling [54]. Thus, tumor lipid metabolites orchestrate a TAM phenotype, and this might explain why a lipid droplet-rich TME is associated with more infiltrating TAMs. Nevertheless, researchers revealed that free fatty acids, particularly stearic acid, facilitate the inflammatory functions of CD11c+ macrophages via activation of the nuclear retinoic acid receptor and the cytosolic expression of epidermal fatty acid-binding proteins (E-FABP) in obesity models [55]. Therefore, whether various categories of fatty acids have different effects on TAM polarization needs more clarification.
Retinoic acid (RA) is a metabolite of vitamin A that has an established role in organ development [56], cancer therapy [57], and immune homeostasis [58]. In vitro, RA-treated head and neck squamous cell carcinoma cells fail to activate macrophages by decreasing VEGF and IL-8 secretion [59]. In addition, the clinical use of available all-trans-retinoic acid (ATRA) abrogates the pro-tumorigenic phenotype of TAMs in prostate cancer by suppressing the activation of NF-κB p50 [60] and reduces osteosarcoma metastasis by downregulating MMP12 (matrix metalloproteinase 12) secretion from M2-type TAMs [61]. Further, ATRA could prevent osteosarcoma initiation and stemness by inhibiting M2-like TAMs [62]. However, retinoic-acid-related orphan receptor (RORC1/RORγ), one of the retinoid nuclear receptors, has been found to prevent myeloid-derived suppressor cells (MDSCs) from undergoing apoptosis, and to promote M2-like TAM differentiation in the TME [63]. Moreover, a recent study indicated that tumor cell-derived RA promotes TAMs and suppresses immunostimulatory dendritic cell differentiation from monocytes [64]. Combined with the RA mechanism in treating acute promyelocytic leukemia, the concept of differentiation therapy, and hence, RA from tumor cells, seems more like an immunosuppressant to mediate TAM formation in the TME.
Nevertheless, the effects of other tumor cell-derived metabolites on TAMs have been investigated. Branched-chain ketoacids, which are excreted from glioblastoma cells via monocarboxylate transporter 1 (MCT1), are taken up by TAMs and re-purposed to form branched-chain amino acids, which reduces phagocytosis by TAMs and mediates tumor immunosuppression [65]. Moreover, a recent study revealed that glioblastoma cell-derived kynurenine activates AHR (aryl hydrocarbon receptor) in TAMs to modulate TAM recruitment and T cell dysfunction [66]. With respect to glioblastoma, researchers found that periostin (POSTN) from glioma stem cells (GSCs) recruits monocyte-derived macrophages from the peripheral blood through integrin αvβ3 to induce an M2 phenotype, and POSTN knockdown in GSCs inhibits tumor growth [67]. One report revealed that bladder tumor cell-derived versican drives lung metastasis in a TAM dependent manner [68]; however, the mechanism underlying CCL2 expression in TAMs via versican warrants additional investigation. Another recent study reported that tumor cell-derived sonic hedgehog triggers hedgehog signaling in TAMs for M2 polarization and impedes CD8+ T cell recruitment by hindering CXCL9 and CXCL10 production by TAMs, which is mediated by Kruppel-like factor 4 (Klf4) [69]. Meanwhile, some tumor metabolites can re-educate TAMs to induce antitumor activity. HRG (histidine-rich glycoprotein) levels are decreased in the TME, and researchers have increased tumor levels by using a genetic gain-of-function strategy to find that HRG derived from these tumor cells inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through the downregulation of PIGF (placental growth factor), suggesting that HRG contributes to the skewing of TAMs to an antitumor polarization phenotype [70].
T cells
T cell-based immune-checkpoint inhibitors have resulted in tremendous achievements [71]. Given that T cells are the cytotoxic cells of the antitumor immune response, their persistent survival, and the production of toxicity factors are critical to eliminate tumor cells and surveil tumor initiation and recurrence. All of these functions are based on metabolic support [72]. The naïve T cells mainly use the OXPHOS pathway to produce ATP [73]. When activated by immunological signals, the metabolic model switches to be glycolysis-dependent [74]. In the TME, glucose, oxygen, and other essential metabolites are deprived by tumor cells. T cells must thus adapt to the hypoxic and nutrient-deficient conditions, which becomes tumor immunosuppressive. Thus, the TME reprograms T cell metabolism into weak glycolysis and OXPHOS, accompanying the decrease in IFN-γ and perforin/granzyme B secretion [72]. However, the impact of T cell-derived metabolites on TAMs has not received much research attention with respect to the TME. One report indicated that tumor-infiltrating CD69+-activated T cells increase IDO (indoleamine 2,3-dioxygenase) expression in TAMs, but the metabolic mediator has not been identified [75]. Another group revealed that CD8 T cell-derived IFN-γ could block sterol regulatory element-binding protein 1 (SREBP1)-mediated fatty acid metabolism in TAMs [76]. In summary, cytokine/chemokine crosstalk is common between T cells and TAMs, but metabolites are also important.
Mast cells
Mast cells are derived from bone marrow precursors and reside in peripheral tissues after maturation; further, they play the role of sentinels to recruit an immune army during infection, especially in allergic responses [77]. To date, the beneficial effects of mast cells on both the tumor and the host have been reported [78, 79]. A recent report found that IL-33-mediated mast cell activation in gastric cancer enhances TAM infiltration and tumorigenesis through CSF2, CCL3, and IL-6 secretion [80]. Other than cytokines and chemokines, mast cells also secrete distinct biogenic amines, eicosanoids, and proteoglycans when they are activated, which are either pro- or anti-tumoral. Theoharis C. Theoharides proposed M1- and M2-type mast cells according to their different effects on the tumor [78]. Moreover, some of these molecules have been studied on TAMs.
Histamine, one of the biogenic amines from mast cells, combined with IL-2, increases Th1 immune responses in patients with stage IV melanoma [81]. Later clinical data suggested that histamine with IL-2 does not change the number of intratumoral macrophages [82]. With respect to the mechanism, histamine can interact with the H2-receptor on macrophages to inhibit ROS production, which relieves NK and T cell inhibition to favor tumor inhibition [83]. Further, histamine deficiency causes a high incidence of colon and skin carcinogenesis via abnormal myeloid differentiation [84]. Moreover, macrophages also express other histamine receptors, including H1/4-receptor, and the H4-receptor is required to induce histamine-mediated chemotaxis and phagocytic activity [85]. Further, H4-receptor knockout mice show suppressed mammary tumor growth and metastasis through a reduction in CD4 T and Treg cells and an increase in NK cells in tumor-draining lymph nodes; besides, stimulation of the H4-receptor increases Treg numbers, enhances IL-10 expression, and decreases IFN-γ [86]. Moreover, the same group recently found that histamine has an antitumor effect, with increased cytotoxic lymphocyte infiltration, in H4-receptor knockout mice [87]. Thus, the specific histamine receptor that induces the antitumor immune response is still unknown. Histamine is generated by histidine decarboxylase (HDC), and HDC-knockout mice fail to form M1-like macrophages [88]. To date, although no systematic study has revealed the phenotypic and functional effect of histamine on TAMs, we speculate that histamine could polarize M1-like TAMs, based on these data combined with results of a previous report [89].
Activated mast cells can produce three eicosanoids, prostaglandin D2 (PGD2), leukotriene B4 (LTB4), and LTC4 [90]. These three eicosanoids are produced de novo from arachidonic acid derived from nuclear membrane phospholipids via cytosolic phospholipase A2. Prostaglandin H2 (PGH2) is generated from arachidonic acid by prostaglandin H synthase 1/2 (PGHS1/2, also called cyclooxygenase 1/2) and then is converted to PGD2 by hematopoietic prostaglandin D2 synthase (H-PGDS, in leukocytes) or lipocalin prostaglandin D2 synthase (L-PGDS, in the central nervous system). PGD2 has been found to induce leukocyte chemotaxis via PGD2 receptors (DP) in Th2 cells [91]. Enhancing the PGD2/DP pathway results in a suppressive effect on tumor growth by modulating tumor hyperpermeability and angiogenesis [92]. Moreover, decreasing PGD2 through H-PGDS deficiency increases tumor growth, and mast cell-derived PGD2 reduces tumor expansion [93]. These findings suggest that the PGD2/DP pathway favors the inhibition of tumor progression. Although many studies mentioned PGD2 in the TME [94], DP expression and the regulation of PGD2 biosynthesis in TAMs are yet to be identified. Mast cell-derived leukotrienes are potent pro-inflammatory lipid mediators implicated in cancers, which require 5-lipoxygenase (5-LOX) for their production. Generally, 5-LOX plays a pro-carcinogenic role [95], whereas its repression in TAMs also promotes tumor progression [96]. Thus, the effect of the 5-LOX pathway on tumors is diverse and might depend on the metabolites from 5-LOX. LTB4, one of several leukotrienes derived from mast cells, binds to two GPCR LTB4Rs (LTB4R1 with high affinity and LTB4R2 with low affinity [47]) and favors murine melanoma growth [97]. Elevated TAMs can be attracted to the TME inducing immunosuppression via LTB4 [98]. The addition of LTC4 has no remarkably increasing effect on the tumoricidal function of TAMs [99]. Leukotrienes and the expression of their receptors, especially LTB4, are increased in some human cancers, including colon, prostate, and pancreatic cancer [47]. Except with respect to chemotaxis functions, other mechanisms of leukotriene-mediated TAM polarization are comparatively less well known.
For proteoglycans, mast cell-derived heparin has been reported to increase angiogenesis [100]. However, the interaction between heparin and TAMs has not been elucidated. Chondroitin sulfate, another proteoglycan from mast cells, is associated with decreased T cells and increases in TAM infiltration in the TME [68, 101]. However, the use of chondroitinase gene therapy to digest chondroitin sulfate increases CD206 (a marker of M2-like macrophages) expression after spinal cord contusion injury [102]. These data suggest that chondroitin sulfate deficiency might favor wound healing. Thus, further clarification of the mechanism underlying the effect of mast cell-derived proteoglycans on TAMs might explain the different functional changes. Mast cells are typically characterized by protease storage and secretion, including tryptase and chymase [103]. The expression of these two enzymes correlates with tumor progression [104]. These enzymes' primary function is to degrade the extracellular matrix, which might indirectly mediate the immune response in the TME. However, fewer studies have mentioned the impact on TAMs.
In general, mast cells were known to produce chemotactic materials. These chemoattractants induce immune responses in the TME, and especially, TAMs have been overlooked. Increasing research findings indicate that the number of immune cells in the TME is not the crucial factor with respect to tumor elimination, and increasing the tumoricidal immune cells by regulating these metabolites might be the future task, for instance, by reversing the M2-like phenotype of TAMs to generate antitumor M1-like macrophages with these mast cell-derived metabolites.
Cancer-associated fibroblasts
The tumor stroma constructs the framework of a solid tumor. Cancer-associated fibroblasts (CAFs), which can be derived from endothelial cells, pericytes, adipocytes, and mesenchymal stem cells [105], are the dominant component of the tumor stroma and have been suggested to boost tumor progression and metastasis [106, 107]. A variety of makers have been used to identify CAFs, such as α-SMA (smooth muscle actin), FSP1 (fibroblast specific protein 1), NG2 (neuron-glial antigen-2), and CD90, among others [105]. To date, CAFs cannot be identified with a single marker, even in the same tumor. In line with this, CD10+GPR77+ CAFs have been found to promote tumor formation and chemoresistance by sustaining tumor stemness [108]. Studies have indicated that fibroblast activation protein and microRNAs can reprogram normal fibroblasts into CAFs [109]. Moreover, CAFs and TAMs are often used together to assess tumor progression [110].
In recent years, some discoveries in CAF metabolism have been made. Michael P. Lisanti's group has presented many studies on metabolism in CAFs, and they termed this new paradigm “The Reverse Warburg Effect” [111], which is characterized by increased lactate and pyruvate secretion from CAFs to fuel tumor cells [112]. As in the tumor cells, they found that lactate is released from CAFs via MCT4 [113, 114]. Even though no study has determined whether lactate from CAFs differentially affects TAMs than that from tumor cells, we believe that CAF-derived lactate polarizes M2-like TAMs, as previously mentioned herein. Moreover, they found that the loss of caveolin-1 (Cav-1) in CAFs is associated with tumor progression [115]. Asymmetric dimethyl arginine (ADMA) and beta-hydroxybutyrate (BHB), which are markers of oxidative stress and mitochondrial dysfunction, are profoundly upregulated in Cav-1-knockout CAFs [116]. Interestingly, endogenous ADMA has been demonstrated to inhibit nitric oxide synthase activity, probably inducing immune-suppressive TAMs in the TME [117]; however, more evidence is needed to indicate if ADMA can be taken up into TAMs. Furthermore, BHB, a ketone body produced by β-oxidation, also induces anti-inflammatory macrophages through HCA2 (hydroxycarboxylic acid receptor 2) [118]. Therefore, ADMA and BHB from CAFs might be conducive to TAM polarization, but the two metabolites' primary source needs to be fully elucidated.
Additionally, other CAF-specific metabolites have been found to affect TAMs. It has been found a high level of hyaluronan in the TME predicts poor prognosis for tumor patients [119]. Nobutaka Kobayashi and co-workers found that TAMs preferentially traffic to hyaluronan-enriched areas, and genetic disruption of the hyaluronan synthase 2 (Has2) gene in fibroblasts impairs TAM recruitment and reduces tumor angiogenesis and lymphangiogenesis, which indicates that CAF secretion of hyaluronan results in TAM pro-tumoral activities [120]. Besides, stanniocalcin-1 (STC1), which is secreted by CAFs, is recognized as a mediator of colorectal cancer growth and metastasis [121]. The previous studies suggested that STC1 is an anti-inflammatory protein, especially for macrophages, which is mediated by inducing mitochondrial UCP2 (uncoupling protein 2) to suppress superoxide generation [122]. Thus, STC1 might inhibit the antitumor chemokine expression in TAMs to impede antitumor T cell infiltration. In summary, very few studies have been performed to assess CAF metabolites' effect on TAMs, except for some specific metabolites.
Adipocytes
Besides CAFs, as another type of stromal cell, adipocytes, also called cancer-associated adipocytes (CAAs), are through to promote tumor growth and metastasis by secreting inflammatory factors and metabolites in the TME [123], especially in breast cancer [124]. CAAs have been reported to increase tumor growth and vascularization through TAMs [125]. Meanwhile, the impact of metabolites from CAAs on TAMs has been studied. For example, adiponectin (APN), one such adipokine, is produced by adipocytes and has antitumor activities [126] and pro-tumoral activities [127]. In response to APN, macrophages are mainly characterized by their anti‐inflammatory effect [128] and M2-like phenotype, specifically, negatively regulating the growth of macrophage progenitors [129], inhibiting the production of CXCR3 ligands to reduce T cell migration [130], and decreasing the production of pro-inflammatory cytokines by suppressing IκB, JNK, p38, and STAT3 phosphorylation [131, 132]. Likewise, full-length APN was reported to induce M2 macrophage polarization via AMPK and shift macrophage metabolism to OXPHOS [133]. Moreover, in the TME, APN deficiency induces TAM polarization to an M1-like phenotype to favor an antitumor immune response for tumor inhibition [134]. In mechanistic terms, APN might trigger downstream signaling of adiponectin receptors (AdipoRs) to polarize M2-like TAMs, such as AdipoR1 [135, 136], AdipoR2 [136, 137], C1qRp [129], and calreticulin receptor [138], among others. AdipoR1/2 probably mediates this on TAMs, which is suggested by the observation that AdipoR1/2 expression on dendritic cells mediates T cell anergy and tolerance in breast cancer [139].
Leptin, another adipokine, was also found to shape the TME and contribute to tumor development [140]. In macrophages, leptin induces some inflammatory mediators' production, including TNF-α, IL-6, and leukotriene B4 [141]. Moreover, leptin modulates cellular lipid metabolism and storage by enhancing ADRP (adipose differentiation-related protein) accumulation within macrophages via the PI3K/mTOR pathway [141]. Although the phenotypic changes in TAMs mediated by leptin have not been revealed, leptin-mediated macrophage chemotaxis and activation were demonstrated through the leptin receptor, which was found to require JAK/STAT, MAPK, and PI3K pathways [142]. This indicates that leptin might mediate the link between CAAs and M2-like macrophages in high-fat diet-induced obesity-associated tumor metastasis [143].
Chemerin, a novel adipokine, regulates adipogenesis and adipocyte metabolism in an autocrine and paracrine manner [144] and was found to be a potent chemoattractant for macrophages that functions in a ChemR23-dependent manner [145]. ChemR23, also known as chemokine-like receptor 1 (CMKLR1), is downregulated in mouse M1-like macrophages but upregulated in mouse M2-like macrophages [146]. However, subsequent research suggested that human M1-like macrophages express ChemR23 but human M2-like macrophages do not [147]. In DSS-induced colitis, chemerin aggravates mucosal damage and increases pro-inflammatory cytokines by suppressing M2-like macrophage polarization [148]. This suggests that the mechanism of chemerin-induced inflammation is intricate. In the TME, chemerin has been reported to inhibit tumor growth by increasing NK cell infiltration and reducing MDSC accumulation [149]. Furthermore, chemerin-deficient (Rarres2-/-) mice show significantly increased TAM numbers in the hepatocellular carcinoma TME [150]. Moreover, tumor cells favor CMKLR1 expression in macrophages, and chemerin enhances pro-inflammatory gene expression in TAMs [151]. This reveals that chemerin might skew TAMs to have an antitumor effect. However, chemerin is downregulated in most tumor tissues, including melanoma, acute myeloid leukemia, breast cancer, and adrenocortical carcinoma [152], indicating that chemerin downregulation can induce tumor immune escape. Thus, the re-introduction of chemerin into the TME might represent a therapeutic strategy [153]. Meanwhile, chemerin is elevated in colorectal cancer, gastric cancer, glioblastoma, hepatocellular carcinoma, and others [152]. Therefore, the effect of treating tumors with chemerin could be tumor context-dependent, and further study is warranted. In addition, recent research indicated that circulating adipose fatty acid binding protein (A-FABP) is mainly derived from CAAs, and elevated circulating A-FABP is associated with obesity-associated tumor development [154]. However, the impact of A-FABP on TAMs is still elusive, since CD11b+F4/80+MHCII-Ly6C- TAMs produce A-FABP as well [155]. Together, the metabolite-mediated interaction between CAAs and TAMs is complicated, and further studies are urgently needed.
Although cellular metabolism in the TME is different from that in normal host cells, all cells share the same metabolic pathways [156]. It is difficult to find some specific metabolites from one component of the TME, but it is feasible to analyze the quantity of various metabolites based on corresponding key metabolic enzymes' expression. In general, the quantity of metabolites in early tumor patients is relatively small. Thereby, the diagnostic validity of metabolic tumor markers is limited to advanced and metabolically hyperactive tumors. TAMs are surrounded by the bulk of tumor cells and other components in the TME in either early- or late-stage tumors. Even slight changes in TME metabolic secretions could shape the immune phenotype of TAMs. However, it is a technical challenge to detect changes in local TME metabolites directly.
Metabolic crosstalk: from TAMs to TME
Metabolites from the TME affect the polarization of TAMs to induce tumor progression and inhibition. In turn, TAMs regulate the TME as well, such as via known cytokines and vesicles. As mentioned previously herein, tumor tissues are hypoxic, and tumor cells mainly use aerobic glycolysis to generate energy. TAMs have a direct impact on tumor cell metabolism. A recent study reported that TAMs make non-small cell lung cancer (NSCLC) cells more glycolytic through TNF-α secretion and promote enhanced hypoxia through AMP-activated protein kinase and PGC-1α activation. Moreover, the depletion of TAMs increases T cell infiltration and PD-L1 expression in tumor cells, which is beneficial for the PD-L1 blockade in NSCLC [157]. Another recent study suggested that enhanced aerobic glycolysis and decreased apoptosis in breast cancer cells are mediated by TAM-derived extracellular vesicle-packaged HIF-1α-stabilizing long noncoding RNA, which is upregulated by lactate in the TME [158]. An additional recent report focused on clear cell renal cell carcinoma, which highly relies on glutamine, and found that glutamine-deprived TAMs secrete IL-23 to decrease survival and enhance the immunosuppressive function of Tregs [159]. In addition to the altered tumor cell metabolism mediated by TAM-derived cytokines and vesicles, metabolites from TAMs extensively regulate the tumor immune microenvironment, metastasis, angiogenesis, and drug-resistance (Figure 2).
The metabolic crosstalk: from TAMs to TME. TAM-derived cholesterol, itaconic acid, creatine and A-FABP switch its own phenotype into protumoral TAM. E-FABP (epidermal fatty acid binding protein) from TAM induces the antitumoral TAM formation. TAM-caused arginine consumption, eicosanoid and kynurenine accumulation result in immunosuppressive T cell. These effects reflect the immune microenvironment changes induced by metabolites from TAM. TAM-produced osteopontin, collagen and MMP9 (matrix metalloproteinase 9) promote tumor metastasis. Lower vitamin D-caused decreased cathelicidin in TAM and TAM-released deoxycytidine induce drug resistance. REDD1(regulated in development and DNA damage response 1)-dependent Sema4D (semaphorin 4D) secretion from TAM accelerates tumor angiogenesis. ABHD5 (abhydrolase domain-containing 5); 15-LOX2 (15-lipoxygenase-2); Irg1 (immune-responsive gene 1).
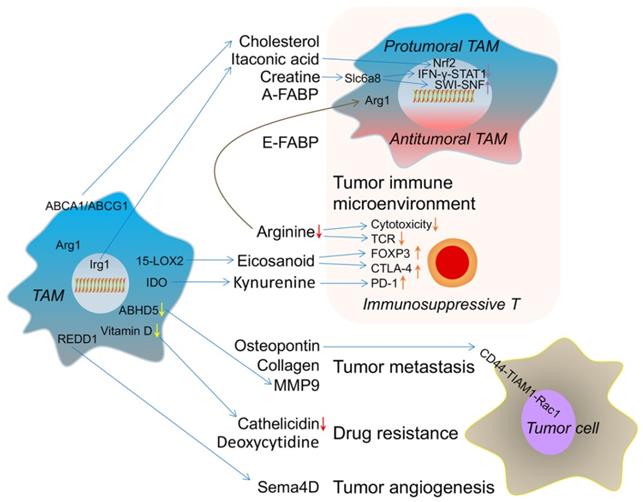
First, some secreted molecules from TAMs can shape their immune phenotype. Reducing circulating cholesterol levels has been considered a useful strategy to treat breast cancer [160]. One study recently suggested that membrane cholesterol efflux in TAMs is increased by ovarian cancer cell-derived hyaluronic acid. On the one hand, these TAM-derived cholesterols promote tumor growth, whereas, on the other hand, high cholesterol efflux disrupts lipid rafts in TAMs and results in TAM reprogramming toward a tumor-promoting phenotype [161]. The cholesterol efflux in TAMs has a positive correlation with tumor burden. The genetic deletion of ABCA1 and/or ABCG1 revert the tumor-promoting functions of TAMs and reduces tumor growth. Activated macrophages could produce itaconic acid [162], which is regulated by immune-responsive gene 1 (Irg1) and an essential regulator of macrophage metabolism and inflammation [163]. Interestingly, itaconic acid acts as a metabolic brake in activated macrophages to avoid hyperinflammatory reactions. In mechanistic terms, itaconic acid inhibits succinate dehydrogenase to reprogram immune responses in macrophages [164] and activates the anti-inflammatory transcription factor Nrf2 via KEAP1 alkylation [165]. Importantly, itaconic acid, a key anti-inflammatory metabolite in macrophages, is the most highly upregulated metabolite of TAMs in a peritoneal tumor model and mediates tumor growth. Correspondingly, the genetic downregulation of Irg1 remarkably reduces peritoneal tumors [166]. FABPs are known as the carrier of free fatty acids that regulate lipid metabolism and inflammation in the host cells. The tissue distributions and ligand-binding specificities of a series of FABPs have been elucidated [167]. In tumors, one report found that F4/80+CD11b+MHCII+CD11c+ TAMs highly express E-FABP, which promotes the recruitment of T and NK cells to mediate antitumor effects through IFN-β production [168]. However, the same group later found that F4/80+CD11b+MHCII-CD11c- TAMs express adipocyte/macrophage FABP (A-FABP) and promote tumor progression through NF-κB-miR-29b-regulated IL-6 production [155]. Therefore, TAMs expressing FABPs represent a double-edged sword for tumor immune surveillance. Moreover, the marker of M2-like TAMs is arginase 1 (Arg1), which depletes arginine to form urea and ornithine[169]. Another result of L-arginine metabolism is the generation of creatine. In addition to ATP, earlier reports indicated that creatine, another direct energy molecule, is likely an antitumor molecule [170]. However, a recent study suggested that creatine uptake via Slc6a8 skews the alternative macrophage polarization by suppressing IFN-γ-STAT1 signaling and promoting ATP-dependent SWI-SNF-mediated chromatin remodeling [171]. Creatine is highly needed in muscle, liver, and brain tissues [172]. Thus, it is assumed that the TME, especially tumors initiated from these organs, maintains high creatine amounts. However, the crosstalk between creatine in the TME and TAMs is not understood. Meanwhile, arginine-generated ornithine is the precursor of polyamines and proline, TAM-derived ornithine might be beneficial for tumor cells and their proliferation [173]. Additionally, T cell immune responses are affected by TAM metabolites [174]. Early research reported that arginine consumption via Arg1 suppresses T cell receptor expression and T cell cytotoxicity [175, 176]. Later, a study found that arginine is critical for T cell survival and antitumor T cell immunity [177]. This is one of the metabolic reasons why increased TAM accumulation in tumor tissues hinders the antitumor T cell immune response. Furthermore, the 15-lipoxygenase-2 (15-LOX2) pathway enhances eicosanoid production in renal cell carcinoma TAMs, which increases the expression of FOXP3 and CTLA-4 in T lymphocytes to mediate immunosuppression [178]. Tumor-repopulating cell-derived kynurenine, the downstream metabolite of tryptophan via IDO catalysis, upregulates PD-1 expression in CD8 T cells by activating the transcriptional factor AhR [179]. This study indicated that metabolites from IDO signaling modulate T cell immunosuppression. It should be noted that IDO is also overexpressed in TAMs [180]. In addition, TAM-derived kynurenine might similarly induce immunosuppressive T cells.
Metabolites from TAMs contribute to tumor invasion and metastasis. TAM-secreted microvesicle-packaged microRNAs have been found to promote breast cancer cell invasion [181]. Osteopontin from TAMs induces bladder cancer metastasis through the osteopontin-CD44-TIAM1 (T cell lymphoma invasion and metastasis 1)-Rac1 pathway [182]. The blockade of this pathway disrupts the initial steps in bladder cancer metastasis. Furthermore, monocyte-derived TAMs upregulate collagenous extracellular matrix, such as collagen types I, VI, and XIV, for tumor invasion [183]. MMP9 has been involved in tumor lung-specific metastasis [184]. Recently, researchers found that ABHD5 (abhydrolase domain-containing 5) deficiency in TAMs induces the NF-κB p65-dependent production of MMP, which indicates that low-level triglyceride hydrolysis-mediated MMP is one of the mediators of tumor metastasis from TAMs [185].
Moreover, TAMs influence tumor angiogenesis. An earlier study found that high TAM numbers are associated with human breast tumor angiogenesis [186]. In terms of the mechanism, cytokines and chemokines are the predominant pro-angiogenic factors produced by TAMs [187]. Besides, MMPs [188] and cathepsins [189] from TAMs have been suggested to support tumor angiogenesis through extracellular matrix degradation. Semaphorin 4D, a ligand of plexin B1 on endothelial cells, is mainly derived from TAMs in the TME and promotes tumor angiogenesis and vessel maturation [190]. Subsequent research also found that TAMs enhance the abnormal vessel formation that occurs through REDD1 (regulated in development and DNA damage response 1)-mediated mTOR inhibition, and REDD1-deficient TAMs deprive glucose from endothelial cells, with consequent formation of an organized tumor vasculature to prevent metastasis [191]. However, the metabolic derivatives of REDD1-deficient TAMs are still unknown.
In principle, macrophages can kill tumor cells via antibody-dependent cellular cytotoxicity (ADCC). For example, the M1-like macrophages target proliferating high-grade B cell lymphoma cells by releasing cathelicidin in a vitamin D-dependent manner. Instead, tumor-educated M2-like TAMs downregulate vitamin D metabolism and produce less cathelicidin resulting in failed cytotoxicity [192]. This phenomenon suggests that reduced cathelicidin from TAMs mediates ADCC resistance. Another recent report found that TAMs release deoxycytidine, based on liquid chromatography coupled tandem mass spectrometry metabolomics, which induces gemcitabine resistance by competitively inhibiting gemcitabine uptake in pancreatic ductal adenocarcinoma [193]. Using the colony-stimulating factor receptor 1 (CSFR1) inhibitor AZD7507, depleting TAMs prolongs survival in an autochthonous pancreatic cancer model. In summary, metabolites from TAMs affect multiple biological functions of the TME, especially remodeling the tumor immune microenvironment. With the clinical application of immunotherapy, an evaluation of metabolic flux in TAMs might guide tumor immunotherapy and elucidate the mechanism underlying drug-resistance and tumor immunotherapy progression.
Conclusions and Perspectives
Outstanding achievements have been made with respect to tumor immunotherapy; however, we still cannot completely overcome tumors. Chimeric antigen receptor (CAR)-T therapy effectively saves patients from refractory B cell malignancies, especially acute lymphoblastic leukemia (ALL), but the macrophage-involved cytokine-release syndrome (CRS) and neurotoxicity after CAR-T injection are associated with life-threatening consequences [194, 195]. Some cytokines from macrophages have been identified in CAR-T-mediated CRS and neurotoxicity, such as IL-1 and IL-6. However, no metabolites have been clarified in this field. Meanwhile, even after receiving immune-checkpoint inhibitors, a so-called hyperprogressive phenomenon is observed in the clinic [196]. The mechanism of hyperprogression in immunotherapy has puzzled clinical immunologists. We might consider that the phenotype of TAMs becomes tumoricidal after immunotherapy, but little is known about the metabolic changes in these cells. An analysis of unique shuttling metabolites between TAMs and the TME might help us understand hyperprogression in immunotherapy.
Even though the M1/2 classification is useful to analyze macrophages in vitro, this oversimplification cannot convey the heterogeneity of TAMs in vivo. Recent studies have indicated that TAM subsets cannot be explained by the M1/2-associated genes [197-199]. Using single-cell RNA sequencing and/or mass cytometry, the immune landscape of the TME has been revealed in human clear cell renal cell carcinoma [200], breast cancer [201], hepatocellular carcinoma [199], brain cancer [202], and colon cancer [203]. Thus, combined with metabolomics, the subtype classification of TAMs will more abundantly reflect the heterogeneity of TAMs in the TME, and novel metabolic targets might be discovered.
Abbreviations
TME: tumor microenvironment; TAM: tumor-associated macrophage; IFN-γ: interforn-γ; SIRPα: signal regulatory proteinα; OXPHOS: oxidative phosphorylation; TCA: tricarboxylic acid; ATP: adenosine triphosphate; MCT1: monocarboxylate transporter 1; MCT4: monocarboxylate transporter 4; TNF: tumor necrosis factor; VEGF: vascular endothelial growth factor; HIF1α: hypoxia-inducible factor 1α; GPCRs: G protein-coupled receptors; ICER: inducible cyclic AMP (cAMP) early repressor; FAO: fatty acid oxidation; SDH: succinate dehydrogenase; SUCNR1: succinate receptor; P2XRs and P2YRs: P2 purinergic receptors; PGE2: prostaglandin E2; CREB: cyclic AMP-responsive element binding; mTOR: mammalian target of rapamycin; RA: retinoic acid; ATRA: all trans-retinoic acid; MMP12: matrix metalloproteinase 12; RORC1/RORγ: retinoic-acid-related orphan receptor; MDSCs: myeloid-derived suppressor cells; AHR: aryl hydrocarbon receptor; POSTN: periostin; GSCs: glioma stem cells; Klf4: Kruppel-like factor 4; HRG: histidine-rich glycoprotein; PIGF: placental growth factor; IDO: indoleamine 2,3-dioxygenase; SREBP1: sterol regulatory element-binding protein 1; HDC: histidine decarboxylase; PGD2: prostaglandin D2; LTB4: leukotriene B4; LTC4: leukotriene C4; PGH2: prostaglandin H2; H-PGDS: hematopoietic prostaglandin D2 synthase; L-PGDS: lipocalin prostaglandin D2 synthase; 5-LOX: 5-lipoxygenase; CAFs: cancer-associated fibroblasts; α-SMA: smooth muscle actin; FSP1: fibroblast specific protein 1; NG2: neuron-glial antigen-2; Cav-1: caveolin-1; ADMA: asymmetric dimethyl arginine; BHB: beta-hydroxybutyrate; HCA2: hydroxycarboxylic acid receptor 2; STC1: stanniocalcin-1; UCP2: uncoupling protein 2; CAAs: cancer-associated adipocytes; APN: adiponectin; AdipoRs: adiponectin receptors; ADRP: adipose differentiation-related protein; CMKLR1: chemokine-like receptor 1; A-FABP: adipose fatty acid binding protein; NSCLC: non-small cell lung cancer; Arg1: arginase 1; TIAM1: T cell lymphoma invasion and metastasis 1; ABHD5: abhydrolase domain-containing 5; REDD1: regulated in development and DNA damage response 1; ADCC: antibody-dependent cellular cytotoxicity; CSFR1: colony stimulating factor receptor 1; CAR: chimeric antigen receptor; ALL: acute lymphoblastic leukemia; CRS: cytokine-release syndrome.
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 81620108023; 81821003; 81925030; 82003018), China Postdoctoral Science Foundation (No. 2018M643861), and Talent Training Program of Army Military Medical University (No. 2018XLC2015).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Noy R, Pollard JW. Tumor-associated macrophages: From mechanisms to therapy. Immunity. 2014;41:49-61
2. Mantovani A, Allavena P. The interaction of anticancer therapies with tumor-associated macrophages. J Exp Med. 2015;212:435-45
3. Sun X, Gao D, Gao L, Zhang C, Yu X, Jia B. et al. Molecular imaging of tumor-infiltrating macrophages in a preclinical mouse model of breast cancer. Theranostics. 2015;5:597-608
4. Cassetta L, Pollard JW. Targeting macrophages: Therapeutic approaches in cancer. Nat Rev Drug Discov. 2018;17:887-904
5. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat Immunol. 2010;11:889-96
6. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized m2 mononuclear phagocytes. Trends Immunol. 2002;23:549-55
7. Chen D, Xie J, Fiskesund R, Dong W, Liang X, Lv J. et al. Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward m1 phenotype. Nat Commun. 2018;9:873
8. Pathria P, Louis TL, Varner JA. Targeting tumor-associated macrophages in cancer. Trends Immunol. 2019;40:310-27
9. Miao L, Qi J, Zhao Q, Wu QN, Wei DL, Wei XL. et al. Targeting the sting pathway in tumor-associated macrophages regulates innate immune sensing of gastric cancer cells. Theranostics. 2020;10:498-515
10. Logtenberg MEW, Scheeren FA, Schumacher TN. The cd47-sirpalpha immune checkpoint. Immunity. 2020;52:742-52
11. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN. et al. Pd-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495-9
12. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW. et al. Cd24 signalling through macrophage siglec-10 is a target for cancer immunotherapy. Nature. 2019;572:392-6
13. Mehla K, Singh PK. Metabolic regulation of macrophage polarization in cancer. Trends Cancer. 2019;5:822-34
14. Dehne N, Mora J, Namgaladze D, Weigert A, Brune B. Cancer cell and macrophage cross-talk in the tumor microenvironment. Curr Opin Pharmacol. 2017;35:12-9
15. Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and metabolism in the tumor microenvironment. Cell Metab. 2019;30:36-50
16. Cassetta L, Fragkogianni S, Sims AH, Swierczak A, Forrester LM, Zhang H. et al. Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell. 2019;35:588-602
17. Henze AT, Mazzone M. The impact of hypoxia on tumor-associated macrophages. J Clin Invest. 2016;126:3672-9
18. Muir A, Vander Heiden MG. The nutrient environment affects therapy. Science. 2018;360:962-3
19. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14:399-416
20. Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD. et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162:1229-41
21. Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and metabolism in the tumor microenvironment. Cell metabolism. 2019;30:36-50
22. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science. 2009;324:1029-33
23. Corbet C, Feron O. Tumour acidosis: From the passenger to the driver's seat. Nat Rev Cancer. 2017;17:577-93
24. Dietl K, Renner K, Dettmer K, Timischl B, Eberhart K, Dorn C. et al. Lactic acid and acidification inhibit tnf secretion and glycolysis of human monocytes. J Immunol. 2010;184:1200-9
25. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V. et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559-63
26. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y. et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574:575-80
27. El-Kenawi A, Gatenbee C, Robertson-Tessi M, Bravo R, Dhillon J, Balagurunathan Y. et al. Acidity promotes tumour progression by altering macrophage phenotype in prostate cancer. Br J Cancer. 2019;121:556-66
28. Bohn T, Rapp S, Luther N, Klein M, Bruehl TJ, Kojima N. et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat Immunol. 2018;19:1319-29
29. Altman BJ, Stine ZE, Dang CV. From krebs to clinic: Glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16:749
30. Sun Y, Zheng Z, Zhang H, Yu Y, Ma J, Tang K. et al. Chemotherapeutic tumor microparticles combining low-dose irradiation reprogram tumor-promoting macrophages through a tumor-repopulating cell-curtailing pathway. Oncoimmunology. 2017;6:e1309487
31. Mills E, O'Neill LA. Succinate: A metabolic signal in inflammation. Trends Cell Biol. 2014;24:313-20
32. Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD. et al. Succinate links tca cycle dysfunction to oncogenesis by inhibiting hif-alpha prolyl hydroxylase. Cancer Cell. 2005;7:77-85
33. Grimolizzi F, Arranz L. Multiple faces of succinate beyond metabolism in blood. Haematologica. 2018;103:1586-92
34. Wu JY, Huang TW, Hsieh YT, Wang YF, Yen CC, Lee GL. et al. Cancer-derived succinate promotes macrophage polarization and cancer metastasis via succinate receptor. Mol Cell. 2020;77:213-27 e5 doi:10.1016/j.molcel.2019.10.023
35. Di Virgilio F, Sarti AC, Falzoni S, De Marchi E, Adinolfi E. Extracellular atp and p2 purinergic signalling in the tumour microenvironment. Nat Rev Cancer. 2018;18:601-18
36. Locher KP. Mechanistic diversity in atp-binding cassette (abc) transporters. Nat Struct Mol Biol. 2016;23:487-93
37. Sandilos JK, Chiu YH, Chekeni FB, Armstrong AJ, Walk SF, Ravichandran KS. et al. Pannexin 1, an atp release channel, is activated by caspase cleavage of its pore-associated c-terminal autoinhibitory region. J Biol Chem. 2012;287:11303-11
38. Vijayan D, Young A, Teng MWL, Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer. 2017;17:709-24
39. Di Virgilio F, Adinolfi E. Extracellular purines, purinergic receptors and tumor growth. Oncogene. 2017;36:293-303
40. Allard B, Beavis PA, Darcy PK, Stagg J. Immunosuppressive activities of adenosine in cancer. Curr Opin Pharmacol. 2016;29:7-16
41. Young A, Mittal D, Stagg J, Smyth MJ. Targeting cancer-derived adenosine: New therapeutic approaches. Cancer Discov. 2014;4:879-88
42. Montalban Del Barrio I, Penski C, Schlahsa L, Stein RG, Diessner J, Wockel A. et al. Adenosine-generating ovarian cancer cells attract myeloid cells which differentiate into adenosine-generating tumor associated macrophages - a self-amplifying, cd39- and cd73-dependent mechanism for tumor immune escape. J Immunother Cancer. 2016;4:49
43. Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK. et al. Ecto-5'-nucleotidase (cd73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. 2002;110:993-1002
44. Li J, Wang L, Chen X, Li L, Li Y, Ping Y. et al. Cd39/cd73 upregulation on myeloid-derived suppressor cells via tgf-beta-mtor-hif-1 signaling in patients with non-small cell lung cancer. Oncoimmunology. 2017;6:e1320011
45. Kong T, Westerman KA, Faigle M, Eltzschig HK, Colgan SP. Hif-dependent induction of adenosine a2b receptor in hypoxia. Faseb j. 2006;20:2242-50
46. Cekic C, Day YJ, Sag D, Linden J. Myeloid expression of adenosine a2a receptor suppresses t and nk cell responses in the solid tumor microenvironment. Cancer Res. 2014;74:7250-9
47. Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. 2010;10:181-93
48. Zelenay S, van der Veen AG, Bottcher JP, Snelgrove KJ, Rogers N, Acton SE. et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell. 2015;162:1257-70
49. Luan B, Yoon YS, Le Lay J, Kaestner KH, Hedrick S, Montminy M. Creb pathway links pge2 signaling with macrophage polarization. Proc Natl Acad Sci U S A. 2015;112:15642-7
50. Na YR, Jung D, Yoon BR, Lee WW, Seok SH. Endogenous prostaglandin e2 potentiates anti-inflammatory phenotype of macrophage through the creb-c/ebp-beta cascade. Eur J Immunol. 2015;45:2661-71
51. Prima V, Kaliberova LN, Kaliberov S, Curiel DT, Kusmartsev S. Cox2/mpges1/pge2 pathway regulates pd-l1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc Natl Acad Sci U S A. 2017;114:1117-22
52. Sanin DE, Matsushita M, Klein Geltink RI, Grzes KM, van Teijlingen Bakker N, Corrado M. et al. Mitochondrial membrane potential regulates nuclear gene expression in macrophages exposed to prostaglandin e2. Immunity. 2018;49:1021-33
53. Spector AA. The importance of free fatty acid in tumor nutrition. Cancer Res. 1967;27:1580-6
54. Wu H, Han Y, Rodriguez Sillke Y, Deng H, Siddiqui S, Treese C. et al. Lipid droplet-dependent fatty acid metabolism controls the immune suppressive phenotype of tumor-associated macrophages. EMBO Mol Med. 2019;11:e10698
55. Zeng J, Zhang Y, Hao J, Sun Y, Liu S, Bernlohr DA. et al. Stearic acid induces cd11c expression in proinflammatory macrophages via epidermal fatty acid binding protein. J Immunol. 2018;200:3407-19
56. Cunningham TJ, Duester G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat Rev Mol Cell Biol. 2015;16:110-23
57. de The H. Differentiation therapy revisited. Nat Rev Cancer. 2018;18:117-27
58. Erkelens MN, Mebius RE. Retinoic acid and immune homeostasis: A balancing act. Trends Immunol. 2017;38:168-80
59. Liss C, Fekete MJ, Hasina R, Lingen MW. Retinoic acid modulates the ability of macrophages to participate in the induction of the angiogenic phenotype in head and neck squamous cell carcinoma. Int J Cancer. 2002;100:283-9
60. Tsagozis P, Augsten M, Pisa P. All trans-retinoic acid abrogates the pro-tumorigenic phenotype of prostate cancer tumor-associated macrophages. Int Immunopharmacol. 2014;23:8-13
61. Zhou Q, Xian M, Xiang S, Xiang D, Shao X, Wang J. et al. All-trans retinoic acid prevents osteosarcoma metastasis by inhibiting m2 polarization of tumor-associated macrophages. Cancer Immunol Res. 2017;5:547-59
62. Shao XJ, Xiang SF, Chen YQ, Zhang N, Cao J, Zhu H. et al. Inhibition of m2-like macrophages by all-trans retinoic acid prevents cancer initiation and stemness in osteosarcoma cells. Acta Pharmacol Sin. 2019;40:1343-50
63. Strauss L, Sangaletti S, Consonni FM, Szebeni G, Morlacchi S, Totaro MG. et al. Rorc1 regulates tumor-promoting "emergency" granulo-monocytopoiesis. Cancer Cell. 2015;28:253-69
64. Devalaraja S, To TKJ, Folkert IW, Natesan R, Alam MZ, Li M. et al. Tumor-derived retinoic acid regulates intratumoral monocyte differentiation to promote immune suppression. Cell. 2020;180:1098-114 e16
65. Silva LS, Poschet G, Nonnenmacher Y, Becker HM, Sapcariu S, Gaupel AC. et al. Branched-chain ketoacids secreted by glioblastoma cells via mct1 modulate macrophage phenotype. EMBO Rep. 2017;18:2172-85
66. Takenaka MC, Gabriely G, Rothhammer V, Mascanfroni ID, Wheeler MA, Chao CC. et al. Control of tumor-associated macrophages and t cells in glioblastoma via ahr and cd39. Nat Neurosci. 2019;22:729-40
67. Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J. et al. Periostin secreted by glioblastoma stem cells recruits m2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol. 2015;17:170-82
68. Said N, Sanchez-Carbayo M, Smith SC, Theodorescu D. Rhogdi2 suppresses lung metastasis in mice by reducing tumor versican expression and macrophage infiltration. J Clin Invest. 2012;122:1503-18
69. Petty AJ, Li A, Wang X, Dai R, Heyman B, Hsu D. et al. Hedgehog signaling promotes tumor-associated macrophage polarization to suppress intratumoral cd8+ t cell recruitment. J Clin Invest. 2019;129:5151-62
70. Rolny C, Mazzone M, Tugues S, Laoui D, Johansson I, Coulon C. et al. Hrg inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of plgf. Cancer Cell. 2011;19:31-44
71. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252-64
72. Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic instruction of immunity. Cell. 2017;169:570-86
73. MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of t lymphocytes. Annu Rev Immunol. 2013;31:259-83
74. Chapman NM, Boothby MR, Chi H. Metabolic coordination of t cell quiescence and activation. Nat Rev Immunol. 2020;20:55-70
75. Zhao Q, Kuang DM, Wu Y, Xiao X, Li XF, Li TJ. et al. Activated cd69+ t cells foster immune privilege by regulating ido expression in tumor-associated macrophages. J Immunol. 2012;188:1117-24
76. Liu C, Chikina M, Deshpande R, Menk AV, Wang T, Tabib T. et al. Treg cells promote the srebp1-dependent metabolic fitness of tumor-promoting macrophages via repression of cd8(+) t cell-derived interferon-gamma. Immunity. 2019;51:381-97.e6
77. Abraham SN, St John AL. Mast cell-orchestrated immunity to pathogens. Nat Rev Immunol. 2010;10:440-52
78. Theoharides TC, Conti P. Mast cells: The jekyll and hyde of tumor growth. Trends Immunol. 2004;25:235-41
79. Ribatti D, Crivellato E. Mast cells, angiogenesis, and tumour growth. Biochim Biophys Acta. 2012;1822:2-8
80. Eissmann MF, Dijkstra C, Jarnicki A, Phesse T. Il-33-mediated mast cell activation promotes gastric cancer through macrophage mobilization. Nat Commun. 2019;10:2735
81. Asemissen AM, Scheibenbogen C, Letsch A, Hellstrand K, Thoren F, Gehlsen K. et al. Addition of histamine to interleukin 2 treatment augments type 1 t-cell responses in patients with melanoma in vivo: Immunologic results from a randomized clinical trial of interleukin 2 with or without histamine (mp 104). Clin Cancer Res. 2005;11:290-7
82. Donskov F, Hokland M, Marcussen N, Torp Madsen HH, von der Maase H. Monocytes and neutrophils as 'bad guys' for the outcome of interleukin-2 with and without histamine in metastatic renal cell carcinoma-results from a randomised phase ii trial. Br J Cancer. 2006;94:218-26
83. Hellstrand K, Brune M, Naredi P, Mellqvist UH, Hansson M, Gehlsen KR. et al. Histamine: A novel approach to cancer immunotherapy. Cancer Invest. 2000;18:347-55
84. Yang XD, Ai W, Asfaha S, Bhagat G, Friedman RA, Jin G. et al. Histamine deficiency promotes inflammation-associated carcinogenesis through reduced myeloid maturation and accumulation of cd11b+ly6g+ immature myeloid cells. Nat Med. 2011;17:87-95
85. Czerner CP, Klos A, Seifert R, Neumann D. Histamine induces chemotaxis and phagocytosis in murine bone marrow-derived macrophages and raw 264.7 macrophage-like cells via histamine h 4-receptor. Inflammation Research. 2014;63:239-47
86. Sterle HA, Nicoud MB, Massari NA, Delgado MAT, Ducloux MVH, Cremaschi GA. et al. Immunomodulatory role of histamine h4 receptor in breast cancer. British journal of cancer. 2019;120:128-38
87. Nicoud MB, Sterle HA, Massari NA, Delgado MAT, Formoso K, Ducloux MVH. et al. Study of the antitumour effects and the modulation of immune response by histamine in breast cancer. British journal of cancer. 2020;122:348-60
88. Xu L, Cheng D, Huang Z, Ding S, Zhang W, Tan H. et al. Histamine promotes the differentiation of macrophages from cd11b+ myeloid cells and formation of foam cells through a stat6-dependent pathway. Atherosclerosis. 2017;263:42-52
89. Naredi P. Histamine as an adjunct to immunotherapy. Semin Oncol. 2002;29:31-4
90. Boyce JA. Mast cells and eicosanoid mediators: A system of reciprocal paracrine and autocrine regulation. Immunol Rev. 2007;217:168-85
91. Xue L, Gyles SL, Wettey FR, Gazi L, Townsend E, Hunter MG. et al. Prostaglandin d2 causes preferential induction of proinflammatory th2 cytokine production through an action on chemoattractant receptor-like molecule expressed on th2 cells. J Immunol. 2005;175:6531-6
92. Murata T, Lin MI, Aritake K, Matsumoto S, Narumiya S, Ozaki H. et al. Role of prostaglandin d2 receptor dp as a suppressor of tumor hyperpermeability and angiogenesis in vivo. Proc Natl Acad Sci U S A. 2008;105:20009-14
93. Murata T, Aritake K, Matsumoto S, Kamauchi S, Nakagawa T, Hori M. et al. Prostagladin d2 is a mast cell-derived antiangiogenic factor in lung carcinoma. Proc Natl Acad Sci U S A. 2011;108:19802-7
94. Kobayashi K, Omori K, Murata T. Role of prostaglandins in tumor microenvironment. Cancer Metastasis Rev. 2018;37:347-54
95. Pidgeon GP, Lysaght J, Krishnamoorthy S, Reynolds JV, O'Byrne K, Nie D. et al. Lipoxygenase metabolism: Roles in tumor progression and survival. Cancer Metastasis Rev. 2007;26:503-24
96. Ringleb J, Strack E. Apoptotic cancer cells suppress 5-lipoxygenase in tumor-associated macrophages. J Immunol. 2018;200:857-68
97. Bachi AL, Kim FJ, Nonogaki S, Carneiro CR, Lopes JD, Jasiulionis MG. et al. Leukotriene b4 creates a favorable microenvironment for murine melanoma growth. Mol Cancer Res. 2009;7:1417-24
98. Wen Z, Liu H, Li M, Li B, Gao W, Shao Q. et al. Increased metabolites of 5-lipoxygenase from hypoxic ovarian cancer cells promote tumor-associated macrophage infiltration. Oncogene. 2015;34:1241-52
99. Braun DP, Ahn MC, Harris JE, Chu E, Casey L, Wilbanks G. et al. Sensitivity of tumoricidal function in macrophages from different anatomical sites of cancer patients to modulation of arachidonic acid metabolism. Cancer Res. 1993;53:3362-8
100. Azizkhan RG, Azizkhan JC, Zetter BR, Folkman J. Mast cell heparin stimulates migration of capillary endothelial cells in vitro. J Exp Med. 1980;152:931-44
101. Gorter A, Zijlmans HJ, van Gent H, Trimbos JB, Fleuren GJ, Jordanova ES. Versican expression is associated with tumor-infiltrating cd8-positive t cells and infiltration depth in cervical cancer. Mod Pathol. 2010;23:1605-15
102. Bartus K, James ND, Didangelos A, Bosch KD, Verhaagen J, Yanez-Munoz RJ. et al. Large-scale chondroitin sulfate proteoglycan digestion with chondroitinase gene therapy leads to reduced pathology and modulates macrophage phenotype following spinal cord contusion injury. J Neurosci. 2014;34:4822-36
103. Metcalfe DD, Baram D, Mekori YA. Mast cells. Physiol Rev. 1997;77:1033-79
104. de Souza DA Jr, Toso VD, Campos MR, Lara VS, Oliver C, Jamur MC. Expression of mast cell proteases correlates with mast cell maturation and angiogenesis during tumor progression. PLoS One. 2012;7:e40790
105. Harper J, Sainson RC. Regulation of the anti-tumour immune response by cancer-associated fibroblasts. Semin Cancer Biol. 2014;25:69-77
106. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16:582-98
107. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392-401
108. Su S, Chen J, Yao H, Liu J, Yu S, Lao L. et al. Cd10(+)gpr77(+) cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell. 2018;172:841-56.e16
109. Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W. et al. Fap promotes immunosuppression by cancer-associated fibroblasts in the tumor microenvironment via stat3-ccl2 signaling. Cancer Res. 2016;76:4124-35
110. Comito G, Giannoni E, Segura CP, Barcellos-de-Souza P, Raspollini MR, Baroni G. et al. Cancer-associated fibroblasts and m2-polarized macrophages synergize during prostate carcinoma progression. Oncogene. 2014;33:2423-31
111. Sotgia F, Martinez-Outschoorn UE, Howell A, Pestell RG, Pavlides S, Lisanti MP. Caveolin-1 and cancer metabolism in the tumor microenvironment: Markers, models, and mechanisms. Annu Rev Pathol. 2012;7:423-67
112. Balliet RM, Capparelli C, Guido C, Pestell TG, Martinez-Outschoorn UE, Lin Z. et al. Mitochondrial oxidative stress in cancer-associated fibroblasts drives lactate production, promoting breast cancer tumor growth: Understanding the aging and cancer connection. Cell Cycle. 2011;10:4065-73
113. Whitaker-Menezes D, Martinez-Outschoorn UE, Lin Z, Ertel A, Flomenberg N, Witkiewicz AK. et al. Evidence for a stromal-epithelial "lactate shuttle" in human tumors: Mct4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle. 2011;10:1772-83
114. Martinez-Outschoorn UE, Lisanti MP, Sotgia F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin Cancer Biol. 2014;25:47-60
115. Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC. et al. Loss of stromal caveolin-1 leads to oxidative stress, mimics hypoxia and drives inflammation in the tumor microenvironment, conferring the "reverse warburg effect": A transcriptional informatics analysis with validation. Cell Cycle. 2010;9:2201-19
116. Pavlides S, Tsirigos A, Migneco G, Whitaker-Menezes D, Chiavarina B, Flomenberg N. et al. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle. 2010;9:3485-505
117. Rodriguez PC, Ochoa AC, Al-Khami AA. Arginine metabolism in myeloid cells shapes innate and adaptive immunity. Front Immunol. 2017;8:93
118. Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D. et al. The ketone metabolite beta-hydroxybutyrate blocks nlrp3 inflammasome-mediated inflammatory disease. Nat Med. 2015;21:263-9
119. Tammi RH, Kultti A, Kosma VM, Pirinen R, Auvinen P, Tammi MI. Hyaluronan in human tumors: Pathobiological and prognostic messages from cell-associated and stromal hyaluronan. Semin Cancer Biol. 2008;18:288-95
120. Kobayashi N, Miyoshi S, Mikami T, Koyama H, Kitazawa M, Takeoka M. et al. Hyaluronan deficiency in tumor stroma impairs macrophage trafficking and tumor neovascularization. Cancer Res. 2010;70:7073-83
121. Pena C, Cespedes MV, Lindh MB, Kiflemariam S, Mezheyeuski A, Edqvist PH. et al. Stc1 expression by cancer-associated fibroblasts drives metastasis of colorectal cancer. Cancer Res. 2013;73:1287-97
122. Wang Y, Huang L, Abdelrahim M, Cai Q, Truong A, Bick R. et al. Stanniocalcin-1 suppresses superoxide generation in macrophages through induction of mitochondrial ucp2. J Leukoc Biol. 2009;86:981-8
123. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR. et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011;17:1498-503
124. Wu Q, Li B, Li Z, Li J, Sun S, Sun S. Cancer-associated adipocytes: Key players in breast cancer progression. J Hematol Oncol. 2019;12:95
125. Wagner M, Steinskog ES, Wiig H. Blockade of lymphangiogenesis shapes tumor-promoting adipose tissue inflammation. Am J Pathol. 2019;189:2102-14
126. Perrier S, Jarde T. Adiponectin, an anti-carcinogenic hormone? A systematic review on breast, colorectal, liver and prostate cancer. Curr Med Chem. 2012;19:5501-12
127. Nakajima TE, Yamada Y, Hamano T, Furuta K, Matsuda T, Fujita S. et al. Adipocytokines as new promising markers of colorectal tumors: Adiponectin for colorectal adenoma, and resistin and visfatin for colorectal cancer. Cancer Sci. 2010;101:1286-91
128. Ohashi K, Parker JL, Ouchi N, Higuchi A, Vita JA, Gokce N. et al. Adiponectin promotes macrophage polarization toward an anti-inflammatory phenotype. J Biol Chem. 2010;285:6153-60
129. Yokota T, Oritani K, Takahashi I, Ishikawa J, Matsuyama A, Ouchi N. et al. Adiponectin, a new member of the family of soluble defense collagens, negatively regulates the growth of myelomonocytic progenitors and the functions of macrophages. Blood. 2000;96:1723-32
130. Okamoto Y, Folco EJ, Minami M, Wara AK, Feinberg MW, Sukhova GK. et al. Adiponectin inhibits the production of cxc receptor 3 chemokine ligands in macrophages and reduces t-lymphocyte recruitment in atherogenesis. Circ Res. 2008;102:218-25
131. Folco EJ, Rocha VZ, Lopez-Ilasaca M, Libby P. Adiponectin inhibits pro-inflammatory signaling in human macrophages independent of interleukin-10. J Biol Chem. 2009;284:25569-75
132. Lovren F, Pan Y, Quan A, Szmitko PE, Singh KK, Shukla PC. et al. Adiponectin primes human monocytes into alternative anti-inflammatory m2 macrophages. Am J Physiol Heart Circ Physiol. 2010;299:H656-63
133. Mandal P, Pratt BT, Barnes M, McMullen MR, Nagy LE. Molecular mechanism for adiponectin-dependent m2 macrophage polarization: Link between the metabolic and innate immune activity of full-length adiponectin. J Biol Chem. 2011;286:13460-9
134. Peng J, Tsang JY, Ho DH, Zhang R, Xiao H, Li D. et al. Modulatory effects of adiponectin on the polarization of tumor-associated macrophages. Int J Cancer. 2015;137:848-58
135. Luo N, Chung BH, Wang X, Klein RL, Tang CK, Garvey WT. et al. Enhanced adiponectin actions by overexpression of adiponectin receptor 1 in macrophages. Atherosclerosis. 2013;228:124-35
136. Parker-Duffen JL, Nakamura K, Silver M, Zuriaga MA, MacLauchlan S, Aprahamian TR. et al. Divergent roles for adiponectin receptor 1 (adipor1) and adipor2 in mediating revascularization and metabolic dysfunction in vivo. J Biol Chem. 2014;289:16200-13
137. Chinetti G, Zawadski C, Fruchart JC, Staels B. Expression of adiponectin receptors in human macrophages and regulation by agonists of the nuclear receptors pparalpha, ppargamma, and lxr. Biochem Biophys Res Commun. 2004;314:151-8
138. Takemura Y, Ouchi N, Shibata R, Aprahamian T, Kirber MT, Summer RS. et al. Adiponectin modulates inflammatory reactions via calreticulin receptor-dependent clearance of early apoptotic bodies. J Clin Invest. 2007;117:375-86
139. Tan PH, Tyrrell HE, Gao L, Xu D, Quan J, Gill D. et al. Adiponectin receptor signaling on dendritic cells blunts antitumor immunity. Cancer Res. 2014;74:5711-22
140. Ando S, Catalano S. The multifactorial role of leptin in driving the breast cancer microenvironment. Nat Rev Endocrinol. 2011;8:263-75
141. Maya-Monteiro CM, Almeida PE, D'Avila H, Martins AS, Rezende AP, Castro-Faria-Neto H. et al. Leptin induces macrophage lipid body formation by a phosphatidylinositol 3-kinase- and mammalian target of rapamycin-dependent mechanism. J Biol Chem. 2008;283:2203-10
142. Gruen ML, Hao M, Piston DW, Hasty AH. Leptin requires canonical migratory signaling pathways for induction of monocyte and macrophage chemotaxis. Am J Physiol Cell Physiol. 2007;293:C1481-8
143. Jung JI, Cho HJ, Jung YJ, Kwon SH, Her S, Choi SS. et al. High-fat diet-induced obesity increases lymphangiogenesis and lymph node metastasis in the b16f10 melanoma allograft model: Roles of adipocytes and m2-macrophages. Int J Cancer. 2015;136:258-70
144. Goralski KB, McCarthy TC, Hanniman EA, Zabel BA, Butcher EC, Parlee SD. et al. Chemerin, a novel adipokine that regulates adipogenesis and adipocyte metabolism. J Biol Chem. 2007;282:28175-88
145. Wittamer V, Franssen JD, Vulcano M, Mirjolet JF, Le Poul E, Migeotte I. et al. Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J Exp Med. 2003;198:977-85
146. Zabel BA, Ohyama T, Zuniga L, Kim JY, Johnston B, Allen SJ. et al. Chemokine-like receptor 1 expression by macrophages in vivo: Regulation by tgf-beta and tlr ligands. Exp Hematol. 2006;34:1106-14
147. Herova M, Schmid M, Gemperle C, Hersberger M. Chemr23, the receptor for chemerin and resolvin e1, is expressed and functional on m1 but not on m2 macrophages. J Immunol. 2015;194:2330-7
148. Lin Y, Yang X, Yue W, Xu X, Li B, Zou L. et al. Chemerin aggravates dss-induced colitis by suppressing m2 macrophage polarization. Cell Mol Immunol. 2014;11:355-66
149. Pachynski RK, Zabel BA, Kohrt HE, Tejeda NM, Monnier J, Swanson CD. et al. The chemoattractant chemerin suppresses melanoma by recruiting natural killer cell antitumor defenses. J Exp Med. 2012;209:1427-35
150. Lin Y, Yang X, Liu W, Li B, Yin W, Shi Y. et al. Chemerin has a protective role in hepatocellular carcinoma by inhibiting the expression of il-6 and gm-csf and mdsc accumulation. Oncogene. 2017;36:3599-608
151. Rama D, Esendagli G, Guc D. Expression of chemokine-like receptor 1 (cmklr1) on j744a.1 macrophages co-cultured with fibroblast and/or tumor cells: Modeling the influence of microenvironment. Cell Immunol. 2011;271:134-40
152. Shin WJ, Zabel BA, Pachynski RK. Mechanisms and functions of chemerin in cancer: Potential roles in therapeutic intervention. Front Immunol. 2018;9:2772
153. Shin WJ, Pachynski RK. Chemerin modulation of tumor growth: Potential clinical applications in cancer. Discov Med. 2018;26:31-7
154. Hao J, Zhang Y, Yan X, Yan F, Sun Y, Zeng J. et al. Circulating adipose fatty acid binding protein is a new link underlying obesity-associated breast/mammary tumor development. Cell Metab. 2018;28:689-705.e5
155. Hao J, Yan F, Zhang Y, Triplett A, Zhang Y, Schultz DA. et al. Expression of adipocyte/macrophage fatty acid-binding protein in tumor-associated macrophages promotes breast cancer progression. Cancer Res. 2018;78:2343-55
156. Ghesquiere B, Wong BW, Kuchnio A, Carmeliet P. Metabolism of stromal and immune cells in health and disease. Nature. 2014;511:167-76
157. Jeong H, Kim S. Tumor-associated macrophages enhance tumor hypoxia and aerobic glycolysis. Cancer Res. 2019;79:795-806
158. Chen F, Chen J, Yang L, Liu J, Zhang X, Zhang Y. et al. Extracellular vesicle-packaged hif-1alpha-stabilizing lncrna from tumour-associated macrophages regulates aerobic glycolysis of breast cancer cells. Nat Cell Biol. 2019;21:498-510
159. Fu Q, Xu L, Wang Y, Jiang Q, Liu Z, Zhang J. et al. Tumor-associated macrophage-derived interleukin-23 interlinks kidney cancer glutamine addiction with immune evasion. Eur Urol. 2019;75:752-63
160. Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK. et al. 27-hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science. 2013;342:1094-8
161. Goossens P, Rodriguez-Vita J, Etzerodt A, Masse M, Rastoin O, Gouirand V. et al. Membrane cholesterol efflux drives tumor-associated macrophage reprogramming and tumor progression. Cell Metab. 2019;29:1376-89.e4
162. Strelko CL, Lu W, Dufort FJ, Seyfried TN, Chiles TC, Rabinowitz JD. et al. Itaconic acid is a mammalian metabolite induced during macrophage activation. J Am Chem Soc. 2011;133:16386-9
163. Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O. et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci U S A. 2013;110:7820-5
164. Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E. et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. 2016;24:158-66
165. Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z. et al. Itaconate is an anti-inflammatory metabolite that activates nrf2 via alkylation of keap1. Nature. 2018;556:113-7
166. Weiss JM, Davies LC, Karwan M, Ileva L, Ozaki MK, Cheng RY. et al. Itaconic acid mediates crosstalk between macrophage metabolism and peritoneal tumors. J Clin Invest. 2018;128:3794-805
167. Storch J, Thumser AE. The fatty acid transport function of fatty acid-binding proteins. Biochim Biophys Acta. 2000;1486:28-44
168. Zhang Y, Sun Y, Rao E, Yan F, Li Q, Zhang Y. et al. Fatty acid-binding protein e-fabp restricts tumor growth by promoting ifn-beta responses in tumor-associated macrophages. Cancer Res. 2014;74:2986-98
169. Arlauckas SP, Garren SB, Garris CS, Kohler RH, Oh J, Pittet MJ. et al. Arg1 expression defines immunosuppressive subsets of tumor-associated macrophages. Theranostics. 2018;8:5842-54
170. Miller EE, Evans AE, Cohn M. Inhibition of rate of tumor growth by creatine and cyclocreatine. Proc Natl Acad Sci U S A. 1993;90:3304-8
171. Ji L, Zhao X, Zhang B, Kang L, Song W, Zhao B. et al. Slc6a8-mediated creatine uptake and accumulation reprogram macrophage polarization via regulating cytokine responses. Immunity. 2019;51:272-84 e7
172. Wyss M, Kaddurah-Daouk R. Creatine and creatinine metabolism. Physiol Rev. 2000;80:1107-213
173. Popovic PJ, Zeh HJ 3rd, Ochoa JB. Arginine and immunity. J Nutr. 2007;137:1681s-6s
174. Ugel S, De Sanctis F, Mandruzzato S, Bronte V. Tumor-induced myeloid deviation: When myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest. 2015;125:3365-76
175. Rodriguez PC, Zea AH, Culotta KS, Zabaleta J, Ochoa JB, Ochoa AC. Regulation of t cell receptor cd3zeta chain expression by l-arginine. J Biol Chem. 2002;277:21123-9
176. Bronte V, Serafini P, Mazzoni A, Segal DM, Zanovello P. L-arginine metabolism in myeloid cells controls t-lymphocyte functions. Trends Immunol. 2003;24:302-6
177. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T. et al. L-arginine modulates t cell metabolism and enhances survival and anti-tumor activity. Cell. 2016;167:829-42.e13
178. Daurkin I, Eruslanov E, Stoffs T, Perrin GQ, Algood C, Gilbert SM. et al. Tumor-associated macrophages mediate immunosuppression in the renal cancer microenvironment by activating the 15-lipoxygenase-2 pathway. Cancer Res. 2011;71:6400-9
179. Liu Y, Liang X, Dong W, Fang Y, Lv J, Zhang T. et al. Tumor-repopulating cells induce pd-1 expression in cd8(+) t cells by transferring kynurenine and ahr activation. Cancer Cell. 2018;33:480-94 e7
180. Munn DH, Mellor AL. Ido in the tumor microenvironment: Inflammation, counter-regulation, and tolerance. Trends Immunol. 2016;37:193-207
181. Yang M, Chen J, Su F, Yu B, Su F, Lin L. et al. Microvesicles secreted by macrophages shuttle invasion-potentiating micrornas into breast cancer cells. Mol Cancer. 2011;10:117
182. Ahmed M, Sottnik JL, Dancik GM, Sahu D, Hansel DE, Theodorescu D. et al. An osteopontin/cd44 axis in rhogdi2-mediated metastasis suppression. Cancer Cell. 2016;30:432-43
183. Afik R, Zigmond E, Vugman M. Tumor macrophages are pivotal constructors of tumor collagenous matrix. J Exp Med. 2016;213:2315-31
184. Hiratsuka S, Nakamura K, Iwai S, Murakami M, Itoh T, Kijima H. et al. Mmp9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell. 2002;2:289-300
185. Shang S, Ji X, Zhang L, Chen J, Li C, Shi R. et al. Macrophage abhd5 suppresses nfκb-dependent matrix metalloproteinase expression and cancer metastasis. Cancer research. 2019;79:5513-26
186. Leek RD, Lewis CE, Whitehouse R, Greenall M, Clarke J, Harris AL. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. 1996;56:4625-9
187. De Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer. 2017;17:457-74
188. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell. 2010;141:52-67
189. Olson OC, Joyce JA. Cysteine cathepsin proteases: Regulators of cancer progression and therapeutic response. Nat Rev Cancer. 2015;15:712-29
190. Sierra JR, Corso S, Caione L, Cepero V, Conrotto P, Cignetti A. et al. Tumor angiogenesis and progression are enhanced by sema4d produced by tumor-associated macrophages. J Exp Med. 2008;205:1673-85
191. Wenes M, Shang M, Di Matteo M, Goveia J, Martin-Perez R, Serneels J. et al. Macrophage metabolism controls tumor blood vessel morphogenesis and metastasis. Cell Metab. 2016;24:701-15
192. Bruns H, Buttner M, Fabri M, Mougiakakos D, Bittenbring JT, Hoffmann MH. et al. Vitamin d-dependent induction of cathelicidin in human macrophages results in cytotoxicity against high-grade b cell lymphoma. Sci Transl Med. 2015;7:282ra47
193. Halbrook CJ, Pontious C, Kovalenko I, Lapienyte L, Dreyer S, Lee HJ. et al. Macrophage-released pyrimidines inhibit gemcitabine therapy in pancreatic cancer. Cell Metab. 2019;29:1390-9.e6
194. Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. Car t cell-induced cytokine release syndrome is mediated by macrophages and abated by il-1 blockade. Nat Med. 2018;24:731-8
195. Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M. et al. Monocyte-derived il-1 and il-6 are differentially required for cytokine-release syndrome and neurotoxicity due to car t cells. Nat Med. 2018;24:739-48
196. Champiat S, Ferrara R, Massard C, Besse B, Marabelle A, Soria JC. et al. Hyperprogressive disease: Recognizing a novel pattern to improve patient management. Nat Rev Clin Oncol. 2018;15:748-62
197. Muller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A. et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017;18:234
198. Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S. et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell. 2018;174:1293-308 e36
199. Zhang Q, He Y, Luo N, Patel SJ, Han Y, Gao R. et al. Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell. 2019;179:829-45 e20
200. Chevrier S, Levine JH, Zanotelli VRT, Silina K, Schulz D, Bacac M. et al. An immune atlas of clear cell renal cell carcinoma. Cell. 2017;169:736-49 e18
201. Wagner J, Rapsomaniki MA, Chevrier S, Anzeneder T, Langwieder C, Dykgers A. et al. A single-cell atlas of the tumor and immune ecosystem of human breast cancer. Cell. 2019;177:1330-45 e18
202. Friebel E, Kapolou K, Unger S, Nunez NG, Utz S, Rushing EJ. et al. Single-cell mapping of human brain cancer reveals tumor-specific instruction of tissue-invading leukocytes. Cell. 2020;181:1626-42 e20
203. Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O'Brien SA. et al. Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell. 2020;181:442-59 e29
Author contact
![]() Corresponding authors: Z.L. (Email: johnneyusccom) or B.Z. (Email: bo.zhuedu.cn). Tel: 86-23-68774622; Fax: 86-23-68755626
Corresponding authors: Z.L. (Email: johnneyusccom) or B.Z. (Email: bo.zhuedu.cn). Tel: 86-23-68774622; Fax: 86-23-68755626