Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Theranostic implications
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(3):1377-1395. doi:10.7150/thno.52442 This issue Cite
Research Paper
Phosphoproteomics identify arachidonic-acid-regulated signal transduction pathways modulating macrophage functions with implications for ovarian cancer
Raimund Dietze1#, Mohamad K. Hammoud1#, María Gómez-Serrano1, Annika Unger1, Tim Bieringer1,6, Florian Finkernagel1, Anna M. Sokol2,3, Andrea Nist4, Thorsten Stiewe4, Silke Reinartz1, Viviane Ponath5, Christian Preußer5, Elke Pogge von Strandmann5, Sabine Müller-Brüsselbach1, Johannes Graumann2,3, Rolf Müller1 ![]()
1. Tranlational Oncology Group, Center for Tumor Biology and Immunology, Philipps University, Marburg, Germany.
2. Biomolecular Mass Spectrometry, Max-Planck-Institute for Heart and Lung Research, Bad Nauheim, Germany.
3. The German Centre for Cardiovascular Research (DZHK), Partner Site Rhine-Main, Max Planck Institute for Heart and Lung Research, Bad Nauheim, Germany.
4. Genomics Core Facility, Philipps University, Marburg, Germany.
5. Institute for Tumor Immunology, Philipps University, Marburg, Germany.
6. Present address: Hochschule Landshut, 84036 Landshut, Germany.
#These authors contributed equally to this work.
Received 2020-8-26; Accepted 2020-9-28; Published 2021-1-1
Abstract

Arachidonic acid (AA) is a polyunsaturated fatty acid present at high concentrations in the ovarian cancer (OC) microenvironment and associated with a poor clinical outcome. In the present study, we have unraveled a potential link between AA and macrophage functions.
Methods: AA-triggered signal transduction was studied in primary monocyte-derived macrophages (MDMs) by phosphoproteomics, transcriptional profiling, measurement of intracellular Ca2+ accumulation and reactive oxygen species production in conjunction with bioinformatic analyses. Functional effects were investigated by actin filament staining, quantification of macropinocytosis and analysis of extracellular vesicle release.
Results: We identified the ASK1 - p38δ/α (MAPK13/14) axis as a central constituent of signal transduction pathways triggered by non-metabolized AA. This pathway was induced by the Ca2+-triggered activation of calmodulin kinase II, and to a minor extent by ROS generation in a subset of donors. Activated p38 in turn was linked to a transcriptional stress response associated with a poor relapse-free survival. Consistent with the phosphorylation of the p38 substrate HSP27 and the (de)phosphorylation of multiple regulators of Rho family GTPases, AA impaired actin filament organization and inhibited actin-driven macropinocytosis. AA also affected the phosphorylation of proteins regulating vesicle biogenesis, and consistently, AA enhanced the release of tetraspanin-containing exosome-like vesicles. Finally, we identified phospholipase A2 group 2A (PLA2G2A) as the clinically most relevant enzyme producing extracellular AA, providing further potentially theranostic options.
Conclusion: Our results suggest that AA contributes to an unfavorable clinical outcome of OC by impacting the phenotype of tumor-associated macrophages. Besides critical AA-regulated signal transduction proteins identified in the present study, PLA2G2A might represent a potential prognostic tool and therapeutic target to interfere with OC progression.
Keywords: arachidonic acid, phosphoproteomics, macropinocytosis, ovarian cancer
Introduction
Cancer progression, metastatic spread and escape of tumor cells from immune surveillance are crucially dependent on the signaling network of the tumor microenvironment (TME) [1]. In ovarian cancer (OC), the peritoneal fluid (or ascites at advanced stages) is a crucial component of the peritoneal TME [2, 3]. Ascites contains large numbers of tumor spheroids and immune cells, in particular tumor-associated macrophages (TAMs), T cells and NK cells, which produce soluble factors and extracellular vesicles [4-6], collectively referred to as the tumor secretome. In spite of its key role in OC progression and therapy resistance, many crucial components of this secretome have not been identified, and the cellular origins and targets of individual mediators as well as their biological functions in the crosstalk between tumor and host cells remain obscure.
Cytokines and growth factors released into the TME are pivotal to all aspects of tumor growth, progression, dissemination and immune escape by affecting numerous biological processes, including cell proliferation, differentiation, survival and motility, immune cell attraction and activation as well as angiogenesis [1, 3]. Another essential class of soluble mediators with similar functions in the TME are bioactive lipids [7, 8]. These include breakdown products of phospholipids, i.e., lysophosphatidic acids (LPAs), polyunsaturated fatty acids (PUFAs) and their metabolites, in particular those derived from arachidonic acid (AA) by the cyclooxygenase and lipoxygenase pathways. The importance of lipid mediators for tumorigenesis is probably best understood for LPA in cancer cell invasion [9-11], and for prostaglandin E2 in immune suppression and angiogenesis [12]. Intriguingly, the ascites levels of several bioactive lipids, including several LPA species, AA and AA-derived eicosanoids (with median AA levels as high as 35 µM) are associated with a short relapse-free survival (RFS) of OC [13, 14]. As the ascites levels of these mediators do not correlate with one another, they are most likely independently associated with RFS.
While the pro-tumorigenic functions of LPA and AA metabolites are partly understood, it is unclear whether, and if so how, non-metabolized AA might be able to exert tumorigenesis-promoting functions. AA can interact with cellular proteins in various ways, which include the membrane-bound G-protein-coupled receptor (GPCR) free fatty acid receptor 4 (FFAR4) [15-17] and the nuclear receptor PPARβ/δ [18]. PPARβ/δ is indeed activated in TAMs by PUFAs present in OC ascites at high concentrations [19]. It is, however, unlikely that PPARβ/δ mediates the adverse effect of AA on RFS, because the major ascites-associated PUFA linoleic acid with strong agonistic effect on PPARβ/δ is not linked to survival [19].
AA has also been linked to the production of reactive oxygen species (ROS), partly via NADPH oxidase NOX-2 located in the plasma membrane [20-22], but the functional relevance of this mechanism remains unclear. Other targets of non-metabolized AA have been identified, including protein kinase C [23-27] and the MAP kinases p38 and JNK [28-30]. In monocytes and neutrophils, the AA-triggered induction of p38 phosphorylation was dependent on Ca2+, phospholipase C, and/or a pertussis toxin (PTX)-sensitive signaling compound, indicative of signaling via a GPCR [28, 29]. Whether FFAR4 is involved in this AA-regulated pathway is currently unknown. A role for GPCR(s) in AA-induced signaling is also suggested by the observation that AA-triggered superoxide generation [22] and extracellular signal-regulated kinase (ERK) activation are inhibited by PTX [31]. It remains, however, unclear how these events are interconnected and contribute to signaling transduction cascades, transcriptional programs and cancer-related biological functions. Non-metabolized AA can also exert direct effects on cellular membranes, leading to in altered mechanical properties of the bilayer, which can modulate the function of membrane channels [21] and transmembrane receptors [32].
TAMs represent one of the most prominent host cell types in ascites and promote tumor growth, metastasis, immunosuppression and chemoresistance. The pivotal role of TAMs in the TME has not only been shown in mouse models but is also evident in cancer patients, where the intratumoral macrophage density is associated with a poor clinical outcome [33], which also applies to OC [34]. These findings are consistent with the shortened RFS observed in OC patients with a high frequency of CD163high TAMs in ascites [35].
TAMs can be derived from both resident tissue macrophages and blood monocytes [36]. They are polarized by factors of the TME to adopt mixed-polarized phenotypes with components of both inflammatory and alternatively activated macrophages [35-39], but the underlying signaling mechanisms are only partly understood. In the present study, we have addressed the question whether AA modulates signal transduction pathways in macrophages and thereby interferes with their physiological functions. To this end we used monocyte-derived macrophages (MDMs) as an experimental system to mimic the infiltration of monocytes/macrophages into the tumor microenvironment with is high levels of AA.
Materials and Methods
Isolation and culture of monocyte-derived macrophages (MDMs) from healthy donors
Leukoreduction System (LRS) chambers with leucocytes from healthy adult volunteers were kindly provided by the Center for Transfusion Medicine and Hemotherapy at the University Hospital Gießen and Marburg. Mononuclear cells were isolated by Ficoll density gradient centrifugation. Subsequently, monocytes were purified by adherence selection and used for subsequent differentiation in RPMI1640 (Life Technologies, Darmstadt, Germany) supplemented with 5 % human AB serum (Sigma), 1 mM sodium pyruvate (Sigma Aldrich, Taufkirchen, Germany) at approximately 5 × 107 cells per 150 mm dish, 1.5 × 107 cells per 100 mm dish or 2.5 × 106, 1 × 106, 0.5 × 106 or 0.1 × 106 cells per well in 6-well, 12-well, 24-well or 96-well plates, respectively. The adherent cells were allowed to differentiate for at least 7 days and used within 3 days thereafter. Under these culture conditions, the macrophage-specific markers CD206 (MRC1) and HLA-DR were >95% as determined by flow cytometry. For the last 24 h prior to any experiment, the medium was replaced with serum-free medium for serum starvation.
Small-molecule compounds
Fatty acids, Latrunculin A, AH7614, BAPTA-AM and U73122 were obtained from Cayman chemicals (Hamburg, Germany), YM-254890 and BIRB796 (Doramapimod) from Biomol (Hamburg, Germany), pertussis toxin from Merck (Darmstadt, Germany), and SB203580 and Selonsertib from Biozol (Eching, Germany).
RNA Sequencing
MDMs were treated with 50 µm AA or solvent (ethanol) for 3 h and total RNA was isolated using the NucleoSpin RNA II kit (740955.250, Macherey-Nagel, Düren, Germany). RNA-Seq was performed and data were processed as described previously (Reinartz et al., 2016; Worzfeld et al., 2018) using Ensembl 96 [40]. RNA-Seq data were deposited at EBI ArrayExpress (accession numbers E-MTAB-8833). Another RNA-Seq dataset for MDMs and TAM was used for Fig. 3E and Table S7 [19, 41] (accession numbers E-MTAB-3114, E-MTAB-3398 and E-MTAB-3167). These datasets are not directly comparable because cDNA libraries were constructed by different methods (Illumina Truseq mRNA kit v2 for E-MTAB-8833, Epicentre Scriptseq human/mouse/rat low input Gold for E-MTAB-3114, E-MTAB-3398 and E-MTAB-3167). RNA expression data for solid tumor tissue from OC patients was retrieved from The Cancer Genome Atlas (TCGA) (https://www.cancer.gov/tcga).
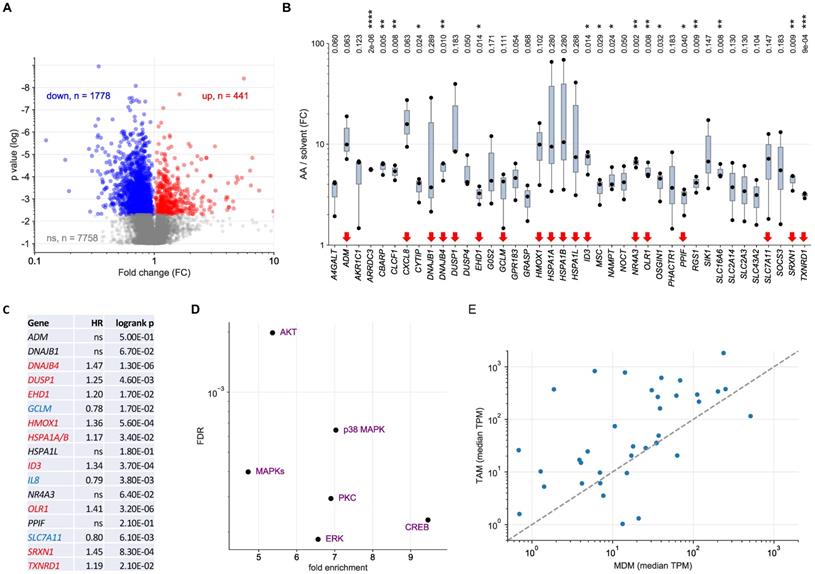
Transcriptional profiling of AA-treated MDMs and ex vivo TAMs. MDMs were treated with 50 µM AA or solvent for 3 h and RNA was analyzed by next-generation sequencing (RNA-Seq) for 3 independent donors. (A) Volcano plot depicting genes significantly (nominal p < 0.05) repressed by AA (blue), induced (red) by AA) or showing no significant change (grey). (B) RNA-Seq results for the top AA-induced genes (FC > 3; TPM > 3 in AA-treated cells). Red arrows: genes linked to stress response according to GO term enrichment analysis (GO:0006950 in Table S6; genes in Table S8). Box pots show medians (line), upper and lower quartiles (box) and range (whiskers). Significance was tested by paired t test: **** p < 0.0001; *** p < 0.001; ** p < 0.01; * p < 0.05 for AA versus solvent; p values were adjusted for multiple hypothesis testing by Benjamini Hochberg correction. (C) Association of the expression of stress-response-linked genes (red arrows in panel B) with relapse-free survival of serous ovarian cancer according to the Kaplan-Meier Plotter database (see Materials and Methods for details; HSPA1A and HSPA1B are combined in panel C; CXCL8 is referred to as IL8 in panel C). Red letters: positive hazard ratio (HR); blue: negative HR; ns: not significant logrank p value (≥0.05). (D) Ingenuity Upstream Regulator Analysis of the AA-induced genes identified in panel A. The plot shows the regulators with the lowest false discovery rate (FDR: < 0.05) and the strongest enrichment (> 3-fold). (E) Comparison of upregulated genes in MDMs and TAMs (TPM > 1 in TAMs) in an independent dataset (RNA-Seq; see Table S11 for details). The Pearson correlation coefficient for the data points shown is r = 0.32 (p = 0.052).
Proteomic and phosphoproteomics analyses
Differentiation of the cells was performed as described above. The cells were serum starved for 40 h and then treated with 50 µM AA or solvent for 15 min. Proteomic and phosphoproteomic analyses were performed in triplicate as described [42] with modifications. 4% SDS, 100 mM Tris, pH 7.6 supplemented with PhosStop and complete protease inhibitor cocktail (both Roche) was used as a lysis buffer, followed by 10 min boiling and sonication prior to centrifugational extract clearing performed at 12000 rpm for 15 min at room temperature. Post-trypsinization, samples were acidified bringing them to 0.75% TFA and cleared by centrifugation. Starting from 400 μg protein extract per condition and replicate, 150 μg of peptides each were TMT labeled (6plex; channels 126-129) and combined by replicate to 600 μg samples, of which 50 μg were subjected to high pH reverse phase separation and 550 μg to TiSH [43]. Following the batch enrichment steps of TiSH, ¾ of the product were HILIC fractionated and ¼ Measured unfractionated. Documentation of liquid chromatography/tandem mass spectrometry (LC/MS2) instrumentation and parametrization was extracted as well as summarized from raw data using MARMoSET [44] and is provided in the supplemental material (Table S1). The MaxQuant suite of algorithms [45, 46] (v. 1.6.8.0) together with the human canonical and isoforms UniProt database (downloaded on 2019/08/19; 173799 entries) and parametrized as included in the supplemental information (Table S2) were used for peptide/spectrum matching and quantitation. Phosphosite occupancies were normalized by dividing phosphosite intensities by the median protein group intensities associated with them using R. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository [47] with the dataset identifier PXD021038 (http://proteomecentral.proteomexchange.org). Reviewer PRIDE account details: Username 'reviewer33406@ebi.ac.uk', password 'IpfMUJwR'.
Immunoblotting and quantification
Immunoblots were performed according to standard Western blotting protocols using the following antibodies: pp38 T180/Y182 (4511, Cell Signaling, Frankfurt, Germany), pHSP27 S82 (SC-16669, Santa Cruz Biotechnology, Heidelberg, Germany), pCREB S133 (9198, Cell signaling), pCamKII T287 (PA-537833, Life Technologies Carlsbad, CA) p38 (9228, Cell Signaling), HSP27 (SC13132, Santa Cruz Biotechnology), CREB (9104, Cell Signaling), pan-CamKII (4436, Cell Signaling), β-Actin, (A5441,Sigma), α-rabbit IgG HRP-linked AB (27, Cell Signaling) and α-mouse IgG HRP-linked AB (32, Cell Signaling). Imaging and quantification were carried out using the ChemiDoc MP system and Image Lab software version 5 (Bio-Rad, Hercules, CA, USA). As phospho-p38 was frequently not detectable in solvent control samples, immunoblots were quantified relative to AA-treated samples for all immunoblots as indicated in the respective figure legends. In addition, the phosphoform signals were normalized against the respective protein signals to correct for unequal protein expression or loading.
Out of the four known CamKII isoforms, MDMs express mRNA for CamKIIβ, -γ and -δ, with CamKIIδ as the main form. The phosphoforms of CamKIIγ and -δ were readily detectable using the polyclonal phospho-CamKIIβ, -γ and -δ antibody (see above). The monoclonal pan-CamKII antibody used for CamKII detection, however, was unable to detect any CamKII isoform besides the δ variant, presumably because CamKIIδ is expressed at approximately 10-fold higher levels in MDMs compared to the other isoforms. For this reason, the upper pCaMKII band, representing pCamKIIγ, was normalized against β-actin.
Reactive oxygen species (ROS) assay
MDMs were prepared and differentiated in 12-well plates as described above. A total ROS/superoxide detection kit from Enzo Life Sciences was used according to the manufacturer's protocol. In brief, macrophages were pretreated with the ROS scavenger N-acetylcysteine (NAC) or the NOX2 inhibitor VAS3947 for 60 min prior to exposure to AA, ETYA or solvent. Cells were treated with AA and stained simultaneously with ROS/superoxide detection reagent for 30 min. Pyocyanin-treated samples served as positive controls. After the incubation period the cells were washed once with assay buffer, gently scraped off the plates and ROS levels were analyzed by flow cytometry (FACSCanto II BD Biosciences). Gating strategies and compensations were performed according to the assay user manual.
Measurement of intracellular free calcium
Calcium assays were carried out in 96-well microplate using a Fura-2 Calcium Flux Assay Kit (ab176766; abcam, Cambridge, UK) according to the instructions of the manufacturer with MDMs cultured as described above. Shortly, the MDMs were incubated with Fura-2 solution for 60 and 20 min at 37°C and room temperature, respectively. Kinetic measurements at 340 and 380 nm were carried out on a Victor Nivo Microplate Readers (PerkinElmer, Waltham, USA) for 0-105 sec after the addition of AA, ETYA or solvent to MDMs, and 340/380 nm excitation ratios were calculated. The 340/380 nm ratio before each treatment was used as reference for normalization.
Staining of actin filaments
MDMs were differentiated on glass coverslips as described above. For actin filament staining the cells were incubated with 50 µM AA for 24 h, fixed with 4 % paraformaldehyde for 10 min at room temperature and permeabilized with 0.3% Triton X100 for 5 min. Actin filament was stained with 1:1000 diluted Phalloidin-California Red Conjugate (AAT Bioquest, Sunnyvale, USA) for 30 min at RT. Glass coverslips were mounted onto the microscope slides using a drop of mounting medium with DAPI (VEC-H-1200, Vector, Burlingame, USA) and sealed with nail polish. Images were taken at 100× magnification and processed on a widefield microscope (Leica DM5500, Leica Microsystems, Wetzlar, Germany).
Macropinocytosis assay
MDMs cultured as described above were treated with AA 50 µM for 23 h, 200 nM of Latrunculin for 30 min A or solvent (ethanol) for 30 min. Macropinocytotic activity was evaluated the following day 60 minutes after addition of FITC-Dextran (70 kDa, 0.5 mg/ml). After gently scraping the cells of the culture plates, the samples were analyzed by flow cytometry. Gating was performed against cells preincubated for 60 min on ice to subtract FITC-Dextran binding from the uptake. Latrunculin is a powerful inhibitor of phagocytosis and macropinocytosis and was therefore used as an additional control.
Isolation and characterization of EVs
After 7 days of differentiation, culture medium was replaced by fresh serum-free RPMI1640 supplemented with 1% sodium pyruvate, and AA at a final concentration of 50 µM or solvent (ethanol) control. Supernatants were harvested after 0 and 24 hrs of incubation from MDMs at a maximum cell density of 80%. Conditioned media were sequentially centrifuged (at 4°C) at 300 × g for 10 min to remove cell debris, 2,000 × g for 15 min to remove organelles, and 10,000 × g for 1 hr to remove large particles. EV release was determined in 10,000 × g supernatants (0.5 ml per 6-well) by high-sensitivity flow cytometry (HSFC). Time-point zero sample media were used as blanks. The presence of protein aggregates or other supramolecular complexes was estimated by treating 10,000 × g supernatants with Triton X-100 at a final concentration of 0.05% to disrupt EVs, as previously described [48]. Further isolation of EVs was performed from culture supernatants (12 ml per 150 mm dish) by additional centrifugation at 100,000 × g for 90 min at 4°C in an OptimaTM XPN-80 ultracentrifuge equipped with an SW 41 Ti Swinging-bucket rotor (Beckman Coulter; Krefeld, Germany). After ultracentrifugation, EV pellets were re-suspended in 1 ml PBS and further centrifuged at 100,000 x g for 100 min in an OptimaTM MAX-XP ultracentrifuge (TLA-45 fixed-angle rotor, Beckman Coulter, Inc.). EV samples were re-suspended in 50 µl PBS and stored at -20°C until further analysis.
As recommended by the MISEV 2018 guidelines [49], three tetraspanin markers were assessed by HSFC in order to verify the EV nature of the samples. Immunostaining of at least 2 × 107 particles in a final volume of 100 µl PBS was performed over-night at 4°C with FITC-coupled antibodies against human CD9 (200 ng, Cat. 312104), CD63 (100 ng, Cat. 353006) and CD81 (200 ng, Cat. 349504; all from BioLegend, Koblenz, Germany). Isotype control antibodies were used as negative controls (200 ng, BioLegend). Removing of unlabeled antibody was performed by PBS washing and subsequent ultracentrifugation at 100,000 × g for 100 min (4°C).
Particle concentration of EV samples was determined by HSFC and NanoFCM Professional Version (V1.0) software analysis. Immunostaining data from HSFC (FCS 3.0 files) were analyzed using FlowJoTM (v10) software. Staining was considered positive when the percentage of positive events showed a minimum fold change of 3 relative to the matching isotype control.
High-sensitivity flow cytometry (HSFC) fluorescence analysis of EVs
A Flow NanoAnalyzer fitted with a Blue Laser (488 nm) (NanoFCM, Inc.) was used for the multiparameter analysis of EV samples, allowing for the quantitative analysis of single particles down to 40 nm with high sensitivity. All experiments were performed in compliance with the NanoFCM system's recommendations (more information on http://www.nanofcm.com/). Briefly, dilutions of all samples were individually tested in order to record a total number of 3,000-12,500 events. PBS or RPMI1640 time-point zero samples were used for threshold setting and as blanks. Monodisperse silica nanoparticles cocktail (68-155 nm, Cat. S16M-Exo, NanoFCM, Inc.), and polystyrene 200 nm beads at a 1/100 dilution (QC Beads, Cat. S08210, NanoFCM, Inc.), were used as size and particle concentration reference, respectively. Standard beads and all samples were collected under the same sampling pressure (1 kPa) for 1 min. Light scattering was used for the measurement of nanoparticle size and size distributions. The EV size range was set between 40-300 nm. Fluorescence analyses were based on FITC-A. All applicable assay controls to HSFC described by MIFLowCyt-EV guidelines [50] (i.e. buffer controls, unstained samples or detergent treatment of stained samples) were evaluated. All samples were measured with at least two technical replicates.
Statistical analysis
Comparative data were statistically analyzed by unpaired or paired Student's t test (two-sided, equal variance) as indicated. Significance levels are indicated as ****, ***, ** and * for p < 0.0001, p < 0.001, p < 0.01 and p < 0.05, respectively. Box pots in Fig. 3 and S1 depicting medians (line), upper and lower quartiles (box), range (whiskers) and outliers/fliers (diamonds) were constructed using the Seaborn boxplot function with Python. Variance of expression in Table 1 was determined by means of the Python function pandas.DataFrame.var(). Progression-free survival data for serous OC patients were obtained from the Kaplan-Meier Plotter meta-analysis database (https://kmplot.com; version 06/2020 with data for 2.190 OC patients) [51]. Data associating gene expression with overall survival (OS) of OC were obtained from the PRECOG database (https://precog.stanford.edu) [52].
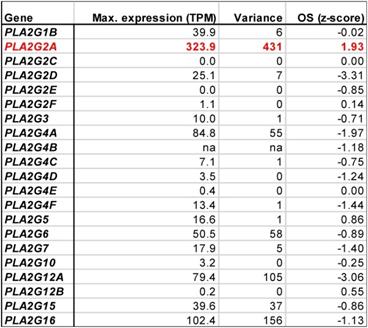
Expression of phospholipase A2 genes in in solid tumor tissue (TCGA RNA-Seq data) and association of expression with overall survival (OS, PRECOG data). A positive z-score indicates a hazard ratio > 1, a negative z-score a hazard ratio < 1; |z| = 1.96 corresponds to p = 0.05. The expression data show the maximum expression levels (TPM) and the variance of expression among samples (variance is the average squared deviation from the mean). na: not available
Results
Phosphoproteome analysis of AA-treated MDMs
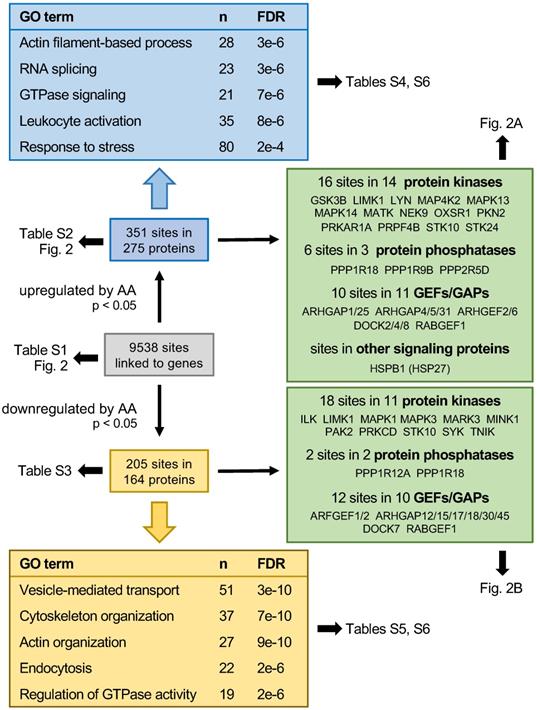
Mass-spectrometry-based phosphoproteomics identified 9538 phosphosites in MDMs treated with 50 µM AA or solvent for 15 min (see schematic summary in Fig. 1; Table S3; values normalized for total proteome signals). After normalization (see Materials and Methods), 351 of these sites sites in 275 proteins were significantly upregulated by AA (nominal p value < 0.05 by t test; Table S4), while 205 sites in 164 proteins were significantly downregulated (Table S5). These included 28 upregulated and 23 downregulated phosphosites, respectively, in signal-transducing protein kinases, phosphatases and G-protein regulators (Fig. 1). Gene ontology (GO) term enrichment analysis of upregulated phosphosites identified actin filament-based process, GTPase signaling, leukocyte activation and stress response as the most significant and enriched terms defining specific functions (Fig. 1; Table S6). The analogous analyses of downregulated phosphosites also yielded cytoskeleton/actin organization and regulation of GTPase activity as clearly enriched terms but also identified vesicle-mediated transport and endocytosis as highly significant hits (Fig. 1; Table S7). The AA-regulated phosphoproteins associated with these GO terms are listed in Table S8.
Summary of the phosphoproteome analysis of AA-treated MDMs. Blue boxes show the data for significantly upregulated phosphosites (nominal p value < 0.05; n = 3), yellow boxes the results downregulated phosphosites. Boxes at the top and bottom show for the most significantly (FDR) enriched terms associated with specific functions. n: number of proteins linked to the respective GO term. Green boxes: AA regulated phosphoproteins with functions in signal transduction mediated by protein kinases, phosphatases, G proteins (GTPases) or heat shock protein 27 (HSP27).
The AA-mediated phosphorylation of protein kinases linked to stress response (red arrows) or actin filament organization (black arrows) is illustrated in Figure 2A (upregulated phosphosites) and Fig. 2B (downregulated phosphosites). In line with current knowledge, the stress response proteins include two members of the p38 family, p38δ (HUGO name: MAPK13) and p38α (HUGO name: MAPK14) as well as their upstream regulator mitogen-activated protein kinase kinase kinase kinase 2 (MAP4K2) and serine/threonine kinase 24 (STK24) (Fig. 2A), suggesting a central role for p38 in AA-mediated signaling. Protein kinases of the second group (black arrows) mainly encompass components of Rho family GTPase-regulated signal transduction pathways regulating actin filament organization [53], i.e., LIM domain kinase 1 (LIMK1), serine/threonine kinase 10 (STK10) and p21Rac1 activated kinase 2 (PAK2), consistent with a role for AA-signaling in actin reorganization. Both, LIMK1 and STK10 contain phosphosites that are affected by AA in opposite ways, which may indicate opposite regulatory effects of these sites (inhibitory or stimulatory).
Phosphosites showing a significant response to AA treatment in MDMs. (A) Protein kinases with phosphosites upregulated by AA versus solvent (p < 0.05 by paired t test; FC > 1.1; n = 3 biological replicates) identified by phosphoproteome analysis. (B) Protein kinases with phosphosites downregulated by AA (p < 0.05; FC < 0.9; n = 3). Red arrows: protein kinases linked to stress response; black arrows: protein kinases associated with actin filament organization. (C) Phosphosites in proteins other than protein kinases strongly upregulated in response to AA treatment ( p < 0.05; FC > 1.7; n = 3). ID refers to the peptide identified by MS in Table S3. Fold change values were calculated by dividing normalized signals (see Materials and Methods). Data points represent individual samples.
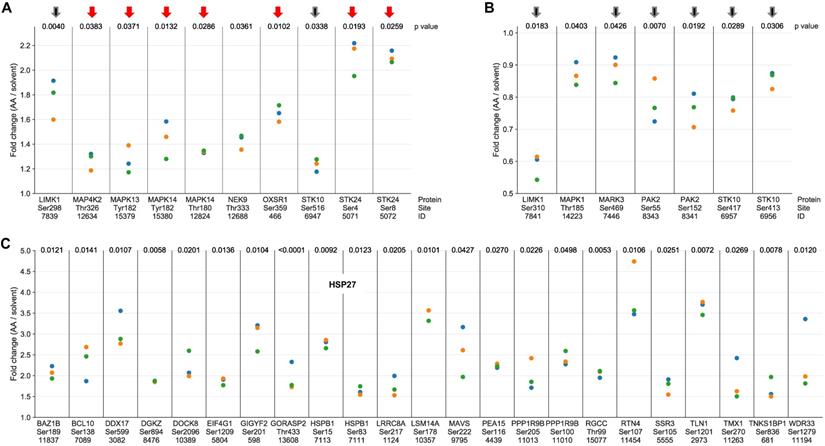
The notion that AA-signaling impacts actin reorganization is supported by AA-mediated effects on a large number of other phosphoproteins with known functions in actin filament organization, as exemplified for AA-upregulated phosphosites in Fig. 2C. Intriguingly, these include two sites in heat shock protein 27 (HSP27; HUGO name: HSPB1), which is a direct substrate of p38 and a regulator of actin filaments [54], thus suggesting a potential link of AA-regulated signal transduction and biological functions. This question is addressed further below.
Transcriptional profiling of AA-treated MDMs
To determine the AA-regulated transcriptome of MDMs we performed RNA-Seq analyses of 3 biological replicates of cells treated with 50 µM AA or solvent for 3 h (Table S9). As shown by the volcano plot in Fig. 3A, 441 genes were significantly upregulated and 1,778 genes significantly downregulated by AA compared to solvent (nominal p value < 0.05 by t test). The most robust group of AA-induced genes (n = 40; Fig. 3B), i.e., showing a minimum fold change (FC) of 3 and a minimum expression of 3 transcripts per million (TPM), were further analyzed by functional annotations. GO term enrichment analysis identified response to oxidative stress as the most significant and highly enriched specific category, followed by apoptosis-related terms (Table S10). Genes associated with response to stress comprised 45% (n = 18) of all genes analyzed (red arrows in Fig. 3B), supporting the relevance of these genes with respect to AA-induced signaling. Intriguingly, the expression of 9 of these 18 genes in serous OC is associated with a poor RFS (Fig. 3C) according to data in the Kaplan-Meier Plotter databases (https://kmplot.com) [51], pointing to a possible connection of the AA-induced stress response to tumor progression.
Ingenuity Pathway Analysis (IPA) of AA-induced genes identified MAP kinase signaling (including p38) as the most significant upstream regulators (Fig. 3D). Other enriched upstream regulators were cAMP Responsive Element Binding Protein (CREB), a known p38 target [55], as well as protein kinase C (PKC) and AKT pathways, which are also linked to MAPK signaling by various interconnections [56, 57]. Taken together, these results in conjunction with the results of the phosphorproteome analysis clearly identify a p38-mediated stress response, including a specific transcriptional program, as a major component of AA-induced signaling. On the other hand, transcriptional profiling did not reveal any evidence for actin-mediated effects, suggesting that Rho-family-GTPase-related signaling events via actin filaments are relevant primarily with regard to non-transcriptional mechanisms. We also identified 23 “robustly” downregulated genes (FC< 0.33; TPM > 3; Fig. S1), but functional annotation did not yield any significant conclusion.
To assess the relevance of these findings in the context of the OC microenvironment we compared the expression of the upregulated genes in MDMs and TAMs from OC ascites (Table S11; TPM > 1 in TAMs). As illustrated in Fig. 3E, the vast majority of these genes (30/37; 81.1%) were expressed at higher levels in TAMs compared to MDMs (median TPM in TAMs > 1), suggesting that AA affects a similar set of genes in TAMs in vivo and MDMs in vitro.
Induction of ASK1-p38 signaling by non-metabolized AA
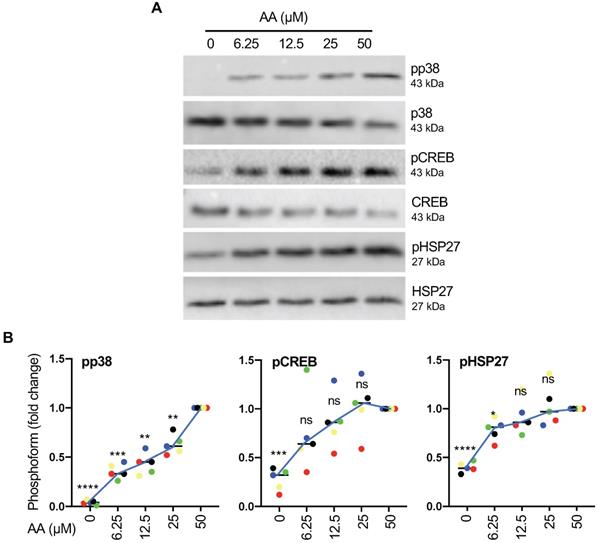
To understand the regulation by AA in more detail we next analyzed the connection of p38 phosphorylation to other components of the p38 signaling cascade. To this end, we first analyzed the dose dependence of p38 phosphorylation (Thr-180, Tyr-182) on the p38 substrates HSP27 (Ser-82) and CREB (Ser-133) by immunoblotting. Figs. 4A and B show that p38 phosphorylation increased nearly linearly within the tested concentration range of 6.25 - 50 µM of AA, while CREB and HSP27 phosphorylation reached saturation below 50 µM. This suggests that maximum levels of activated p38 (pp38) are not required for full phosphorylation of CREB and even less so for HSP27 phosphorylation. However, in view of maximum phosphorylation p38 being observed at 50 µM AA, which is close to the median level found in OC ascites [19], we chose this concentration for all subsequent experiments.
Signaling events induced by AA in MDMs. (A) Dose-dependent induction of p38, HSP27 and CREB phosphorylation by AA analyzed by immunoblot analysis. MDMs were incubated with different concentrations of AA for 30 min. Depicted are representative immunoblots. (B) Quantification of n = 5 biological replicates. Data points represent individual samples.The lines connect the median values depicted as horizontal lines. Statistical analysis was performed by paired t test and p values were adjusted by Benjamini-Hochberg correction. **** adjusted p < 0.0001; *** p < 0.001; ** p < 0.01; * p < 0.05 against samples treated with 50 µM AA. ns: not significant. Values represent the fold change relative to 50 µM.
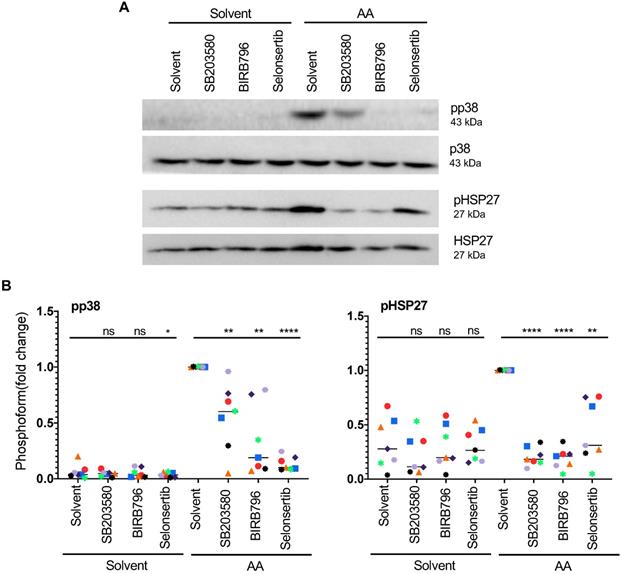
The phosphorylation of HSP27 in response to AA is of particular interest in view of a reported role for HSP27 in actin filament reorganization. To analyze the AA-induced pathway leading to HSP27 activation in more detail, we investigated the effect of pharmacological inhibitors. As shown in Fig. 5A and B, both p38 inhibitors tested, SB203580 and BIRB796 strongly diminished AA-induced p38 autophosphorylation and HSP27 phosphorylation. A well-known activating kinase upstream of p38 is apoptosis signal regulating kinase 1 (ASK1; HUGO name: MAP3K5). Selonsertib, a small-molecule clinically tested inhibitor of ASK1 [58, 59], also inhibited AA-induced p38 and HSP27 phosphorylation by 92% and 69%, respectively, thus confirming the AA-mediated induction of an ASK1 → p38 → HSP27 signaling pathway.
Effect of pharmacological inhibition of ASK1 or p38 on AA-induced HSP27 phosphorylation. (A) Immunoblot of MDMs pretreated with the selective ASK1 inhibitor Selonsertib (1 µM) or the p38 inhibitors BIRB796 or SB203580 (10 µM) for 1 h prior to treatment with AA for 30 min. (B) Quantification of n = 7 biological replicates. Data points represent individual samples, horizontal lines indicate the median. Statistical analysis was performed by paired t test. **** adjusted p < 0.0001; *** p < 0.001; ** p < 0.01; * p < 0.05 against samples treated with AA + solvent. Detection of pp38 was not reliably possible in solvent only samples. ns: not significant. Values represent the fold change relative to AA + solvent.
Next, we asked whether these signaling events are triggered by non-metabolized AA or an eicosanoid generated by the cyclooxygenase (COX) or lipoxygenase (LOX) pathways. Figs. 6A and B unequivocally show that the non-metabolizable AA-analogue and dual COX/LOX inhibitor 5,8,11,14-eicosatetraynoic acid (ETYA) [60] induced p38 and HSP27 phosphorylation to a very similar extent as AA, indicating that metabolizing AA is not required.
Signaling events induced by AA in MDMs compared to other PUFAs. (A) Immunoblot analysis of p38, HSP27 and CREB phosphorylation in response to 50 µM AA compared to 50 µM ETYA, LA, DHA or EPA (representative example). Details as in Fig. 4. (B) Quantification of n = 8 biological replicates. Data points represent individual samples, horizontal lines indicate the median. Statistical analysis was performed as in Fig. 4. Values represent the fold change relative to AA.
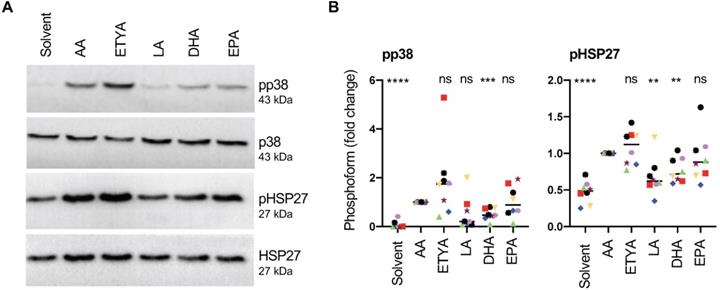
We also tested the effect of other structurally related PUFAs and found that linoleic acid (LA), eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) were also able to induce p38 phosphorylation. While the median effect of EPA approached that of AA and ETYA (89% versus AA), LA and DHA induced p38 phosphorylation to a considerably weaker extent (LA: 20.5%; DHA: 47%; Figs. 6A and B). HSP27 phosphorylation was also induced by all PUFAs, closely following the pattern observed for pp38 (Figs. 6A and B). The observed phosphorylation of p38 by PUFAs other than AA is consistent with the conclusion that this AA-mediated effect is triggered by AA itself rather than its metabolites.
Collectively, these observations indicate that induction of p38 → HSP27 signaling is selective, but not specific for AA, and is not dependent on the metabolization of AA.
Role of Ca2+-mediated signal transduction pathways in the AA-induced activation of p38
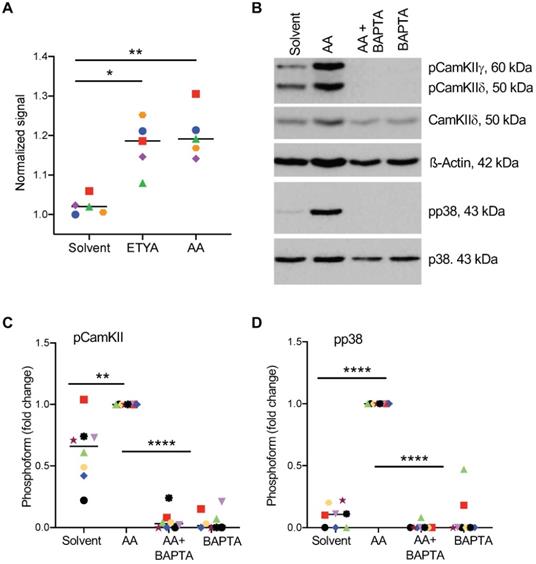
To elucidate the AA-triggered signaling event upstream of p38, we analyzed the role of Ca2+-mediated signaling previously implicated in conveying AA-triggered signals in rat neutrophils [29]. As shown in Figs. 7A and S2, both AA and ETYA stimulation of MDMs induced a rapid cytoplasmic influx of Ca2+ and phosphorylation of calcium/calmodulin-dependent protein kinase II (CAMKII; HUGO name: CAMK2; Fig. 7B), known as a direct activator of ASK1 [61, 62]. Consistent with these findings, sequestration of Ca2+ by the calcium chelator BAPTA-AM (1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid) [63] blocked the AA-triggered phosphorylation of p38 (Fig. 7B). A quantification of these data is shown in Fig. 7C and D.
Effect of Ca2+-dependent signaling on AA-mediated phosphorylation of p38. (A) Increase in intracellular Ca2+ levels 0-105 sec after stimulation with 50 µM AA or ETYA. Kinetic measurements of Fura-2 fluorescence were carried out as described in Materials and Methods. (B) AA-mediated phosphorylation of p38 and CAMK IIγ and δ, and its inhibition by the Ca2+ chelator BAPTA-AM. The cells were pretreated with 50 µM BAPTA-AM for 1 h prior to treatment with 50 µM AA for 30 min. (C, D). Quantification of 8 biological replicates as in panels A and B. Data points represents individual samples, horizontal lines indicate the median. Statistical analysis was performed by paired t test: **** adjusted p < 0.0001; *** p < 0.001; ** p < 0.01; * p < 0.05 against AA-treated samples.
To elucidate the potential origin of Ca2+ impinging on the CAMKII - ASK - p38 axis, we tested the effects of the FFAR4 antagonist AH7614, the PLCγ inhibitor U-73122, the Gi/0 inhibitor pertussis toxin and the Gq/11 inhibitor YM-254890. These compounds partially inhibited the AA-induced phosphorylation of p38 in MDMs from some donors, while no effect was detectable in others (Fig. S3). These findings suggest that an AA → GPCR/FFAR4 →PLCγ pathways triggers the release of Ca2+ from intracellular stores via inositol trisphosphate in a subset of donors to a minor extent, suggesting that a GPCR-/PLCγ-independent mechanism(s) plays a more prominent role in transducing AA-elicited signal to p38.
Based on this data AA-triggered signal transduction is initiated by multiple, donor-dependent events (including GPCR(s) and PLCγ), which merge on a common Ca2+-regulated pathway: Ca2+ → CAMK II → ASK1 → (MKK3/6) → p38 → HSP27/CREB (parentheses: not analyzed in the present study; see scheme in Fig. 8).
Model of AA-regulated signal transduction pathways. An AA-triggered Ca2+- ASK1 - p38 pathway induces a transcriptional stress response and mediates HSP27 phosphorylation, thereby contributing to actin filament reorganization in concert with AA-regulated RhoGEFs and RhoGAPs. Other protein kinases phosphorylated in response to AA, such MAP4K2, may also impinge on this pathway. ROS produced by different mechanisms may contribute to p38 phosphorylation to a minor extent in a subset of donors. Orange: phosphosites identified by MS and confirmed by immunoblotting; yellow: phosphosites identified by MS. EX: extracellular space; PM: plasma membrane; CYT: cytosol; NUC: nucleus. AA is most likely derived from extracellular phospholipids with PLA2G2A possibly playing a crucial role (see text and Table 1).
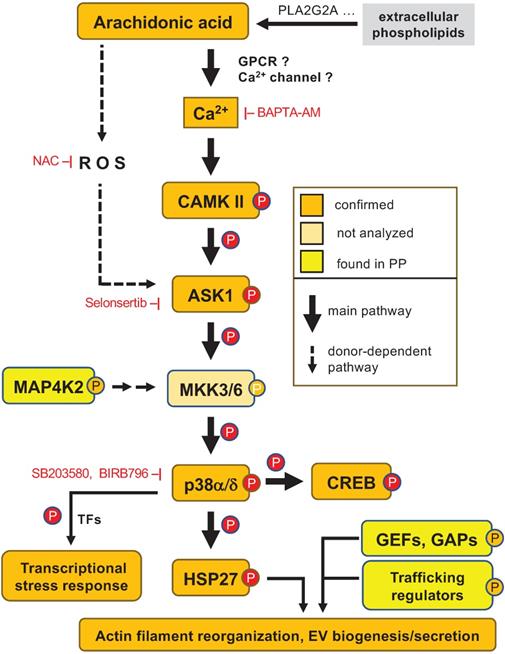
Role of ROS in AA-triggered p38 phosphorylation
Next, we addressed the potential role of ROS in light of previously published findings suggesting the generation of intracellular ROS in response to AA [20, 21]. Fig. S4 shows an example of AA-mediated induction of total ROS and superoxide generation in one MDM sample. However, AA-induced ROS production varied strongly among individual MDM samples and, despite a similar AA-induced p38 phosphorylation across all samples was even undetectable in most cases (Fig. S5). Furthermore, quenching of ROS by N-acetylcysteine (NAC) or the NOX inhibitor VAS3947 only partially affected p38 phosphorylation, if at all (Fig. S5). We conclude from these observations that ROS production, if any, plays a secondary role as a mediator of AA-induced p38 activation in a subset of donors (Fig. 8).
Interference of AA with actin filament-dependent processes
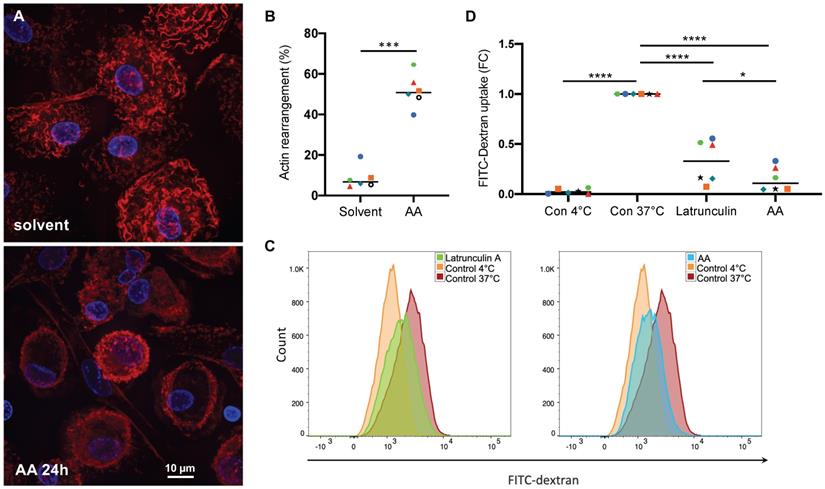
As shown above, AA affects the phosphorylation status of the pp38 substrate HSP27 as well as multiple regulators of the Rho family GTPases (Figs. 1 and 2), pointing to an impact on actin filament organization. To test this hypothesis we stained MDMs with fluorescently labeled phalloidin (California red conjugate) as an actin-binding probe [64]. Figs. 9A shows actin fibers evenly distributed in solvent-treated cells, which was altered in 40-65% of cells (Fig. 9B) after a 24-hr incubation with 50 µM AA, with actin staining largely restricted to the cell edges and/or reduced to a low intensity, suggesting that AA prevents the assembly or promotes the disassembly of actin filaments. In view of these findings, we studied the effect of AA on macropinocytosis as an actin-driven process [65]. As shown in Figs. 9C and D AA inhibited the uptake of FITC-labelled dextran by MDMs by 91% after 24 hrs incubation. This inhibitory effect was even stronger than that of Latrunculin A, which sequesters monomeric G-actin and prevents actin filament assembly [66], thereby blocking endocytotic uptake by pinocytosis or phagocytosis [67, 68]. Taken together, these findings confirm that AA-induced signaling interferes with endocytotic uptake functions of MDMs by interfering with actin filament organization.
Effect of AA on actin filament organization and macropinocytosis. (A) Phalloidin staining of actin filaments in MDMs treated with 50 µM AA or solvent (control) for 24 hrs (representative experiment). (B) Quantification of actin filament rearrangement in MDMs from n = 6 donors after treatment as in panel A (each donor is represented by a specific symbol). Cells were counted as positive for actin rearrangement, if the typically wavy actin fibers were undetectable and/or actin filaments were accumulated at the cell edges. For each sample, on average 70 cells were evaluated. (C, D) Flow cytometric analysis of FITC-Dextran pinocytosis by MDMs treated with 50 µM AA for 24 h. Latrunculin A was included as a known pinocytosis inhibitor [67]. Untreated MDMs incubated on ice to invoke a complete inhibition of pinocytosis were used as negative control. Significance was tested by paired t test: ****p < 0.0001, ***p < 0.001, * p < 0.05. The gating strategy is shown in Fig. S6.
Impact of AA on the release of EVs
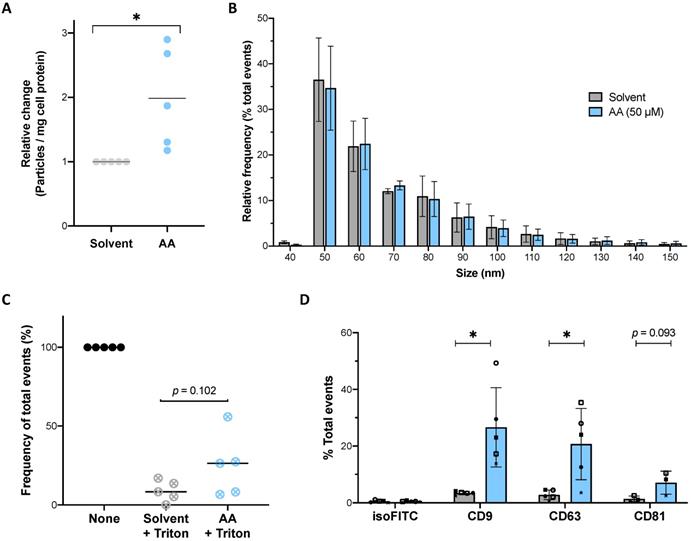
The functional annotation analysis (Fig. 1 and Table S7) identified a strong association of AA-downregulated phosphosites with vesicle-mediated transport. We therefore analyzed the effect of AA on the release of extracellular vesicles (EVs) by MDMs. High-sensitivity flow cytometry (HSFC) analysis of conditioned medium from MDMs cultured in the presence of 50 µM AA under serum-free conditions revealed a significant approximately 2-fold increase in the frequency of released particles (Fig. 10A) within a size range characteristic of EVs [49, 69] (~90% ranging from 50 to 100 nm; Fig. 10B). These particles were highly sensitive to detergent treatment, consistent with their classification as EVs (Fig. 10C). We also quantified EVs isolated by differential ultracentrifugation from conditioned media, which confirmed a higher release from AA-treated MDMs (average 2.94 × 109 particles, n = 9 donors) compared to solvent controls (average 1.61 × 109 particles) (relative change illustrated in Fig. S7). To further characterize these samples, we applied flow cytometry to analyze the expression of the tetraspanins CD9, CD63 and CD81 as classical exosomal markers [70, 71]. This analysis showed positive events for all markers in both AA-treated and solvent controls with a 5- to 8-fold increase in samples from AA-induced MDMs (Figs. 10D and S8). In conjunction with the results of the size analyses (Fig. 10B), these findings confirm the released particles as bona fide EVs with exosomal features. These observations also lend strong support to the hypothesis that AA-mediated signaling events affect proteins that regulate the biogenesis and/or release of tetraspanin-containing EVs as suggested by our phosphoproteomics results.
Effect of AA on the release of EVs. (A) Relative number of particles released by MDMs into the culture medium after 24 h of treatment with AA (blue dots) or solvent (grey dots) determined by HSFC. Values were calculated per mg total cell protein and normalized to 1 for solvent-treated cells. (B) Size distribution of particles released by MDMs shown in panel A. Histogram of particle size with a bin width of 10 nm for AA (blue bars) and solvent control (grey bars) samples. Data were normalized to the total number of acquired events separately for solvent and AA-treated samples. The plot shows the mean (± SEM) of the relative frequency of total events detected by HSFC in conditioned media from n = 5 donors. (C) Relative decrease of particle count in conditioned media after treatment with Triton X-100 (final concentration 0.05%). Values obtained before addition of detergent were set to 100%. (D) Immunostaining results on EV pellets obtained by differential centrifugation. The presence of tetraspanin markers (CD9, CD63 and CD81) was evaluated on particles obtained from n = 3-5 donors. A histogram of particle size of positive events is shown in Figure S8 for a representative sample. Bars represent mean (± SD) of total % of positive events. Symbols represent the values obtained with independent donors. Significance was tested by paired t test: * p < 0.05 for AA versus solvent.
Expression and survival association of AA-generating phospholipase A2 isoforms
The association of both AA [13] and AA-induced genes (Fig. 3C) with a poor clinical outcome suggests that decreased AA levels could be a therapeutically beneficial achievement. AA is generated by the phospholipase A2-mediated cleavage of both extra- and intracellular phospholipids [72-74]. Phospholipase A2 enzymes are encoded by 21 different PLA2 genes, whose contribution to OC progression is poorly understood. To identify potential therapeutic targets we analyzed the expression of PLA2 genes in solid tumor samples from OC patients (which contain both tumor and tumor-associated host cells as possible cellular sources of PLA2) and their association with the clinical outcome. Intriguingly, analysis of the data in the PRECOG database revealed that within this gene family, only PLA2G2A was associated with the overall survival (OS) of OC with near-significance (Table 1; z-score = 1.93). Consistently, PL2G2A was also associated with a shorter progression-free survival of high-grade serous OC bases of the data in the Kaplan Meier Plotter meta-analysis database (Fig. S9). In agreement with these findings, PLA2G2A showed the highest variance of expression in solid OC tumor samples (Table 1). Furthermore, PLA2G2A exhibited the highest maximum expression among all PLA2 family members (Table 1). These findings suggest that the PLA2G2A (PLA2 Group 2A) isoform plays a crucial role in the generation of clinically relevant AA, and thus represents an interesting target for pharmacological intervention with OC progression.
Discussion
The present study is based on our original discovery that the concentration of AA in OC ascites is associated with short RFS [13], for which the underlying molecular mechanisms remeined unknown. TAMs represent the major host cell population in ascites, and in view of their link to a poor prognosis are of particular interest in this context. In the present study, we therefore asked the question, which signal transduction pathways and biological functions are regulated by AA in macrophages. We addressed this problem by a combination of omics, biochemical and functional analyses, which revealed a comprehensive picture of AA-regulated signal transduction pathways in MDMs and identified AA-affected biological functions potentially relevant in the TME.
The AA-triggered p38 signaling pathway
Our phosphoproteomics, transcriptomics and complementary immunoblotting analyses revealed the stress-response-associated p38 MAP kinase pathway as a central target of AA signaling in MDMs (Figs. 1-4). Consistent with this finding we identified the stress-response kinase ASK1 [75-77] as the upstream regulator of p38 (Fig. 5), presumably via MKK3/6 [78]. In agreement with this finding, the known p38 substrates CREB and HSP27 [55] were also phosphorylated in response to AA in an ASK1-dependent manner (Fig. 5). Importantly, these results were confirmed with the non-metabolizable AA analog and COX/LOX inhibitor ETYA [60] (Fig. 6), indicating that the observed effects were caused by AA itself rather than its metabolites. This is consistent with the observation that other PUFAs triggered the effects as well, albeit to a reduced extent (Fig. 6).
Even though redox signaling has been reported as the main activator of ASK1 in other experimental systems [79-81], this was not the case in AA-stimulated MDMs. AA-induced ROS production was observed in MDMs only from a subset of donors, and even in positive cases its contribution to p38 phosphorylation was low (Figs. S4 and S5). We therefore analyzed CAMK II as another known direct activator of ASK1 [61, 62]. Our data clearly show that CAMK II phosphorylation was induced by AA in all MDM samples analyzed. Consistent with this observation, AA induced a rapid accumulation of intracellular Ca2+, and the Ca2+ chelator BAPTA-AM [63] prevented CAMK II and p38 phosphorylation (Figs. 7A-C).
Others have reported that GPCR-mediated signaling and PLC are instrumental in AA-triggered signaling in rat neutrophils, but we were unable to confirm this for MDMs. The pharmacological inhibition of PLC, FFAR4 or GPCR-associated G proteins reduced p38 phosphorylation only in a subset of samples, and if so, only to a low extent (< 50%; Fig. S3). This suggests that GPCR-mediated PLC activation only plays a minor role in the intracellular accumulation of Ca2+ and the ensuing activation of CAMK II in AA-stimulated MDMs. It is possible that lipid membrane structures associated with Ca2+ channels are affected by AA in MDMs, as reported for other experimental systems [82-86], potentially resulting in increased intracellular Ca2+ levels in AA-treated cells. Taken together, these findings indicate the following pathway to play a major and donor-independent role in AA-stimulated MDMs: AA → multiple mediators → Ca2+ → CAMK II → ASK1 → MKK3/6 → p38 → HSP27 / CREB.
AA signaling impinges on actin-dependent processes
GO term enrichment analyses of our phosphoproteomics data identified several potentially interesting functions affected by AA apart from “response to stress”, one of which is “actin cytoskeletal organization” (Fig. 1; Tables S4 and S5). In agreement with this finding, an enrichment was also found for the term “GTPase signaling”, representing a group of G protein regulators [guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs), Table S8] impacting Rho/Rac pathways and thereby actin cytoskeletal structures [53]. At present, little is known about the precise function of most of these proteins, in particular with respect to the functional consequences of their phosphorylation, complicating the interpretation of these findings. This applies in particular to the observation that several proteins, including LIMK1 and STK10, contain phosphosites that are upregulated by AA, while other sites in the same protein are downregulated. It may be speculated that such modifications exert opposite regulatory effects, but this question may only be clarified once the functions of these proteins and their regulation by phosphorylation are known.
Another interesting aspect in view of the observed AA-induced morphological changes is a potential involvement of signaling pathways upstream of myosin II. While the myosin light chain kinase MLCK was not detected in the MDM proteome (Table S3), two other proteins phosphorylated in response to AA may be of interest in this context, i.e., MYH9 and MYO9B (Table S4). MYO9B is an unconventional myosin involved in intracellular movements. It interacts with actin, is inhibited by Ca2+, figures as a RhoA GTPase activator and plays a role in cell migration [87, 88]. Furthermore, the AA-induced phosphorylation of MYO9B at S1354 found in the present study was also observed in platelets, where MYO9B phosphorylation mediated by PKC and PKD increased its RhoA GTPase activity [89].
MYH9 is a non-muscle myosin IIA heavy chain, which is involved in the generation of intracellular mechanical force and translocation of the actin cytoskeleton [90]. MYH9 is phosphorylated by AA in MDMs at S1943 (Table S4), a site described to regulate cellular protrusions and invasion [91]. Taken together, these observations raise the possibility that the observed AA-induced phosphorylation of MYH9 and MYO9B play a role in mediating the observed morphological changes.
In agreement with these findings and the predictions of the GO term enrichment analysis, we found microscopic evidence for an impact of AA on actin organization, indicating an interference with the assembly of actin filaments and the formation of an actin ring at the cellular edges (Figs. 9A and B). This observation is consistent with previous reports describing AA-induced actin ring formation in osteoclasts [92], and actin clustering at the cell membrane of MDMs to be regulated by external signals impacting their polarization [93]. AA had no cytotoxic effects on CD14+ monocytes after 48 hrs of exposure (tested for up to 80 µM) [92], and cytotoxicity affected only few cells in our own experiments (as indicated by normal nuclear morphology and a low fraction of detached cells after AA treatment), ruling out a major role for cell-death-triggering events in AA-mediated actin reorganization.
AA also strongly inhibited macropinocytosis (Figs. 9C and D), a process known to be dependent on actin filament organization [65]. Macropinocytosis is functionally similar to phagocytosis, suggesting that the observed effect of AA may be relevant in the context of the tumor microenvironment, since the inability to phagocytize tumor cells is a characteristic trait of TAMs [94]. Apart from the G protein regulators alluded to above, another interesting protein in this context is HSP27, which has been described as a regulator of actin filaments and macropinocytosis [54] and is phosphorylated in response to AA at two different sites (Fig. 2C).
AA signaling regulates extracellular vesicle release
Another intriguing AA-regulated mechanism suggested by GO term enrichment analysis of our phosphoproteomics data is “vesicle-mediated transport” (Fig. 1; Table S7). The 51 proteins linked this GO term (Table S8) encompass G-protein modulators and actin filament modulators (e.g., ARFGEFs, ARHGAP Rho GTPase-activating proteins, RAB family regulators; LIMK1, MAPK1/3 and STK10 protein kinases) as wells as many numerous regulators of membrane trafficking and secretion, including exocyst complex component 7, intersectin-2, the SNARE component SNAP29, synaptogyrin-2 and vacuolar protein sorting-associated proteins, consistent with the established role of Rho family GTPases in vesicle trafficking [95].
EVs comprise a heterogeneous population of membrane vesicles of different cellular origins. Their size varies and encompasses two main classes: exosomes (50-150 nm), which are of endosomal origin, and microvesicles (50-500 nm), which directly bud from the plasma membrane [69]. Their overlapping size spectrum and similar compositions, hamper their distinction after isolation from culture supernatants.
In the present study, we were able to confirm the prediction of the GO term enrichment analysis experimentally by demonstrating an increased release of particles upon AA treatment within a size range expected for EVs (Fig. 10A and B). These particles were sensitive to detergent (Fig. 10C) confirming their lipid nature and were endowed with membrane organizers, i.e., the tetraspanins CD9, CD63 and CD81 (Fig. 10D). These tetraspanins are classical exosomal markers [70], indicating that AA induces the release of exosome-like vesicles from MDMs. In agreement with these results, it has been reported by others that M2-like polarization of human macrophages increases the release of EVs [96]. Interestingly, our data also showed that AA treatment led to a higher proportion of tetraspanin-positive particles relative to solvent controls (Fig. 10D). While CD9 and CD81 have been associated with both exosomes and microvesicles, CD63 has been shown to be enriched in exosome populations in different experimental systems [69]. Taken together, these data suggest that AA boosts the release of EVs, and in particular exosome-like particles.
As discussed above, our data demonstrate that AA treatment triggers actin-filament rearrangement and Ca2+ release in MDMs. Interestingly, both events have been previously linked to the EV releasing process [69], lending further support to our conclusions. In view of the prominent role of EVs in the microenvironment of OC [97-100], it is tantalizing to speculate that AA might promote tumor progression via the increased release of vesicles carrying pro-tumorigenic and/or immune suppressive proteins, lipids and/or nucleic acids from macrophages. In line with this hypothesis are the previously reported tumor tropism of macrophage-derived EVs [101] and the tumor-promoting role of EVs in cancer [6]. It will therefore be of great interest to determine the cargo of these vesicles by a combined omics approach in future studies.
Theranostic implications
Our work has uncovered two levels within the AA-governed signaling network that are associated with the clinical outcome of AA and may thus provide prognostic and/or therapeutic options. The first level is extracellular AA itself. As previously shown, the ascites concentration of AA associated with a short RFS [13]. AA is generated either intracellularly or extracellularly from phospholipids by cytosolic or secretory PLA2 isoforms (cPLA2 and sPLA2), constituting a family of 21 proteins [72, 74]. As reported in the present study, PLA2G2A showed the highest variance of expression in tumor tissue from different patients and reached the highest expression level among all PLA2 isoforms in a subset of patients. Intriguingly, the latter patients are characterized by a short OS and a short RFS, while no association with a short OS was observed for others PLA2 isoforms. sPLA2s are well known as proinflammatory enzymes but their role in human cancer is controversial [73], which may also be reflected by the observation that three PLA2 isoforms (PLA2D, PL4a, PLA12D) are linked to a longer OS of OC (Table 1). Our data suggest that pharmacological inhibition of PLA2G2A might be therapeutically beneficial in OC, although direct experimental evidence is lacking at this point. Furthermore, PLA2G2A might be a useful prognostic marker, as suggested for breast cancer [102], a question to be addressed in future biomarker studies.
We have previously reported that a high ascites levels of PLA2G7 protein is associated with a short RFS [13], which is not the case for PLA2G7 RNA levels in solid tumor tissue (Table 1). A possible explanation for this apparent discrepancy is the high expression of PLA2G7 in ascites-associated macrophages [13], which in turn represent the major population of ascites cells in the majority of OC patients, thereby increasing the potential functional relevance of PLA2G7. This would suggest that both tumor-associated PLA2G2A as well as ascites-associated PLA2G7 may be useful theranostic candidates.
The second level is represented by the signal transduction pathways triggered by AA. In this context, signaling components at the crossroads with other pro-tumorigenic pathways are of particular interest. These include CAMKII [103], ASK1 [75], p38 [104], HSP27 [105] and Rho GTPase regulators [106]. Modulating the activity of any of these signaling molecules might not only dampen the pro-tumorigenic effect of TAMs but might also reduce the growth, survival and metastatic potential of cancer cells. It is possible that these signal molecules also have prognostic value in OC, but this option can only be assessed once data directly addressing this issue are available.
Acknowledgements
We are grateful to Margitta Alt and Sylvia Jeratsch for expert technical assistance.
Funding source
This work was supported by a grant from the Wilhelm Sander Stiftung to S.M.B and S.R. (2016.123.1), a Postgraduate Scholarship for Foreign Studies (Fundación Ramón Areces, XXXI Edition) to MGS and a Scholarship from the German Academic Exchange Service (DAAD) to M.H. R.M. holds an Anneliese Pohl Senior Research Professorship.
Author contributions
RD, MGS, SR, EPvS, SMB, JG and RM designed the study and supervised the project. RD, MKH, MGS, FF, JG and RM analyzed the raw data. RM and FF carried out bioinformatic analyses. RD and MKH performed immunoblotting experiments. MKH analyzed actin filaments and micropinocytosis, and MGS, VP and CP EVs. AU and TB performed initial experiments providing the basis for the present study. AMS performed phosphoproteomics. TB, AN and TS carried out RNA-Seq analyses. RM wrote the manuscript.
Supplementary Material
Supplementary figures.
Supplementary tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Roma-Rodrigues C, Mendes R, Baptista PV, Fernandes AR. Targeting Tumor Microenvironment for Cancer Therapy. Int J Mol Sci. 2019 20
2. Ahmed N, Stenvers KL. Getting to Know Ovarian Cancer Ascites: Opportunities for Targeted Therapy-Based Translational Research. Front Oncol. 2013;3:256
3. Worzfeld T, Pogge von Strandmann E, Huber M, Adhikary T, Wagner U, Reinartz S. et al. The Unique Molecular and Cellular Microenvironment of Ovarian Cancer. Front Oncol. 2017;7:24
4. Finkernagel F, Reinartz S, Schuldner M, Malz A, Jansen JM, Wagner U. et al. Dual-platform affinity proteomics identifies links between the recurrence of ovarian carcinoma and proteins released into the tumor microenvironment. Theranostics. 2019;9:6601-17
5. Graumann J, Finkernagel F, Reinartz S, Stief T, Brodje D, Renz H. et al. Multi-platform Affinity Proteomics Identify Proteins Linked to Metastasis and Immune Suppression in Ovarian Cancer Plasma. Front Oncol. 2019;9:1150
6. Hoshino A, Kim HS, Bojmar L, Gyan KE, Cioffi M, Hernandez J. et al. Extracellular Vesicle and Particle Biomarkers Define Multiple Human Cancers. Cell. 2020;182:1044-61 e18
7. Wymann MP, Schneiter R. Lipid signalling in disease. Nat Rev Mol Cell Biol. 2008;9:162-76
8. Luo X, Zhao X, Cheng C, Li N, Liu Y, Cao Y. The implications of signaling lipids in cancer metastasis. Exp Mol Med. 2018;50:127
9. Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer. 2003;3:582-91
10. Tsujiuchi T, Araki M, Hirane M, Dong Y, Fukushima N. Lysophosphatidic acid receptors in cancer pathobiology. Histol Histopathol. 2013;29:313-21
11. Brindley DN, Lin FT, Tigyi GJ. Role of the autotaxin-lysophosphatidate axis in cancer resistance to chemotherapy and radiotherapy. Biochim Biophys Acta. 2013;1831:74-85
12. Kalinski P. Regulation of immune responses by prostaglandin E2. J Immunol. 2012;188:21-8
13. Reinartz S, Finkernagel F, Adhikary T, Rohnalter V, Schumann T, Schober Y. et al. A transcriptome-based global map of signaling pathways in the ovarian cancer microenvironment associated with clinical outcome. Genome Biol. 2016;17:108
14. Reinartz S, Lieber S, Pesek J, Brandt DT, Asafova A, Finkernagel F. et al. Cell-type-selective pathways and clinical associations of lysophosphatidic acid biosynthesis and signaling in the ovarian cancer microenvironment. Mol Oncol. 2019;23:185-201
15. Villegas-Comonfort S, Takei Y, Tsujimoto G, Hirasawa A, Garcia-Sainz JA. Effects of arachidonic acid on FFA4 receptor: Signaling, phosphorylation and internalization. Prostaglandins Leukot Essent Fatty Acids. 2017;117:1-10
16. Milligan G, Alvarez-Curto E, Hudson BD, Prihandoko R, Tobin AB. FFA4/GPR120: Pharmacology and Therapeutic Opportunities. Trends Pharmacol Sci. 2017;38:809-21
17. Mobraten K, Haug TM, Kleiveland CR, Lea T. Omega-3 and omega-6 PUFAs induce the same GPR120-mediated signalling events, but with different kinetics and intensity in Caco-2 cells. Lipids Health Dis. 2013;12:101
18. Rieck M, Meissner W, Ries S, Müller-Brüsselbach S, Müller R. Ligand-mediated regulation of peroxisome proliferator-activated receptor (PPAR) beta/delta: a comparative analysis of PPAR-selective agonists and all-trans retinoic acid. Mol Pharmacol. 2008;74:1269-77
19. Schumann T, Adhikary T, Wortmann A, Finkernagel F, Lieber S, Schnitzer E. et al. Deregulation of PPARβ/δ target genes in tumor-associated macrophages by fatty acid ligands in the ovarian cancer microenvironment. Oncotarget. 2015;6:13416-33
20. Matono R, Miyano K, Kiyohara T, Sumimoto H. Arachidonic acid induces direct interaction of the p67(phox)-Rac complex with the phagocyte oxidase Nox2, leading to superoxide production. J Biol Chem. 2014;289:24874-84
21. Pompeia C, Cury-Boaventura MF, Curi R. Arachidonic acid triggers an oxidative burst in leukocytes. Braz J Med Biol Res. 2003;36:1549-60
22. Abramson SB, Leszczynska-Piziak J, Weissmann G. Arachidonic acid as a second messenger. Interactions with a GTP-binding protein of human neutrophils. J Immunol. 1991;147:231-6
23. Bordin L, Priante G, Musacchio E, Giunco S, Tibaldi E, Clari G. et al. Arachidonic acid-induced IL-6 expression is mediated by PKC alpha activation in osteoblastic cells. Biochemistry. 2003;42:4485-91
24. O'Flaherty JT, Chadwell BA, Kearns MW, Sergeant S, Daniel LW. Protein kinases C translocation responses to low concentrations of arachidonic acid. J Biol Chem. 2001;276:24743-50
25. Huang XP, Pi Y, Lokuta AJ, Greaser ML, Walker JW. Arachidonic acid stimulates protein kinase C-epsilon redistribution in heart cells. J Cell Sci. 1997;110( Pt 14):1625-34
26. Schaechter JD, Benowitz LI. Activation of protein kinase C by arachidonic acid selectively enhances the phosphorylation of GAP-43 in nerve terminal membranes. J Neurosci. 1993;13:4361-71
27. Khan WA, Blobe GC, Hannun YA. Arachidonic acid and free fatty acids as second messengers and the role of protein kinase C. Cell Signal. 1995;7:171-84
28. Guijas C, Perez-Chacon G, Astudillo AM, Rubio JM, Gil-de-Gomez L, Balboa MA. et al. Simultaneous activation of p38 and JNK by arachidonic acid stimulates the cytosolic phospholipase A2-dependent synthesis of lipid droplets in human monocytes. J Lipid Res. 2012;53:2343-54
29. Chang LC, Wang JP. The upstream regulation of p38 mitogen-activated protein kinase phosphorylation by arachidonic acid in rat neutrophils. J Pharm Pharmacol. 2000;52:539-46
30. Rizzo MT, Carlo-Stella C. Arachidonic acid mediates interleukin-1 and tumor necrosis factor-alpha-induced activation of the c-jun amino-terminal kinases in stromal cells. Blood. 1996;88:3792-800
31. Capodici C, Pillinger MH, Han G, Philips MR, Weissmann G. Integrin-dependent homotypic adhesion of neutrophils. Arachidonic acid activates Raf-1/Mek/Erk via a 5-lipoxygenase- dependent pathway. J Clin Invest. 1998;102:165-75
32. Norris PC, Dennis EA. Omega-3 fatty acids cause dramatic changes in TLR4 and purinergic eicosanoid signaling. Proc Natl Acad Sci U S A. 2012;109:8517-22
33. Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263-6
34. Kawamura K, Komohara Y, Takaishi K, Katabuchi H, Takeya M. Detection of M2 macrophages and colony-stimulating factor 1 expression in serous and mucinous ovarian epithelial tumors. Pathol Int. 2009;59:300-5
35. Reinartz S, Schumann T, Finkernagel F, Wortmann A, Jansen JM, Meissner W. et al. Mixed-polarization phenotype of ascites-associated macrophages in human ovarian carcinoma: Correlation of CD163 expression, cytokine levels and early relapse. Int J Cancer. 2014;134:32-42
36. Laviron M, Boissonnas A. Ontogeny of Tumor-Associated Macrophages. Front Immunol. 2019;10:1799
37. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39-51
38. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253-68
39. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787-95
40. Yates AD, Achuthan P, Akanni W, Allen J, Allen J, Alvarez-Jarreta J. et al. Ensembl 2020. Nucleic Acids Res. 2020;48:D682-D8
41. Finkernagel F, Reinartz S, Lieber S, Adhikary T, Wortmann A, Hoffmann N. et al. The transcriptional signature of human ovarian carcinoma macrophages is associated with extracellular matrix reorganization. Oncotarget. 2016;17:75339-52
42. Iring A, Jin YJ, Albarran-Juarez J, Siragusa M, Wang S, Dancs PT. et al. Shear stress-induced endothelial adrenomedullin signaling regulates vascular tone and blood pressure. J Clin Invest. 2019;129:2775-91
43. Engholm-Keller K, Larsen MR. Improving the Phosphoproteome Coverage for Limited Sample Amounts Using TiO2-SIMAC-HILIC (TiSH) Phosphopeptide Enrichment and Fractionation. Methods Mol Biol. 2016;1355:161-77
44. Kiweler M, Looso M, Graumann J. MARMoSET - Extracting Publication-ready Mass Spectrometry Metadata from RAW Files. Mol Cell Proteomics. 2019;18:1700-2
45. Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367-72
46. Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res. 2011;10:1794-805
47. Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ. et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47:D442-D50
48. Gyorgy B, Modos K, Pallinger E, Paloczi K, Pasztoi M, Misjak P. et al. Detection and isolation of cell-derived microparticles are compromised by protein complexes resulting from shared biophysical parameters. Blood. 2011;117:e39-48
49. Thery C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R. et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7:1535750
50. Welsh JA, Van Der Pol E, Arkesteijn GJA, Bremer M, Brisson A, Coumans F. et al. MIFlowCyt-EV: a framework for standardized reporting of extracellular vesicle flow cytometry experiments. J Extracell Vesicles. 2020;9:1713526
51. Gyorffy B, Lanczky A, Szallasi Z. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr Relat Cancer. 2012;19:197-208
52. Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D. et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21:938-45
53. Sit ST, Manser E. Rho GTPases and their role in organizing the actin cytoskeleton. J Cell Sci. 2011;124:679-83
54. Lavoie JN, Hickey E, Weber LA, Landry J. Modulation of actin microfilament dynamics and fluid phase pinocytosis by phosphorylation of heat shock protein 27. J Biol Chem. 1993;268:24210-4
55. Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochem J. 2010;429:403-17
56. Tanaka Y, Gavrielides MV, Mitsuuchi Y, Fujii T, Kazanietz MG. Protein kinase C promotes apoptosis in LNCaP prostate cancer cells through activation of p38 MAPK and inhibition of the Akt survival pathway. J Biol Chem. 2003;278:33753-62
57. McGuire VA, Gray A, Monk CE, Santos SG, Lee K, Aubareda A. et al. Cross talk between the Akt and p38alpha pathways in macrophages downstream of Toll-like receptor signaling. Mol Cell Biol. 2013;33:4152-65
58. Lanier M, Pickens J, Bigi SV, Bradshaw-Pierce EL, Chambers A, Cheruvallath ZS. et al. Structure-Based Design of ASK1 Inhibitors as Potential Agents for Heart Failure. ACS Med Chem Lett. 2017;8:316-20
59. Nelson CH, Etchevers K, Yi S, Breckenridge D, Hepner M, Patel U. et al. Pharmacokinetics, Safety, and Tolerability of Selonsertib, an Apoptosis Signal-Regulating Kinase 1 (ASK1) Inhibitor, Following First-in-Human Single and Multiple Ascending Doses in Healthy Subjects. Clin Pharmacokinet. 2020
60. Tobias LD, Hamilton JG. The effect of 5,8,11,14-eicosatetraynoic acid on lipid metabolism. Lipids. 1979;14:181-93
61. Takeda K, Matsuzawa A, Nishitoh H, Tobiume K, Kishida S, Ninomiya-Tsuji J. et al. Involvement of ASK1 in Ca2+-induced p38 MAP kinase activation. EMBO Rep. 2004;5:161-6
62. Kashiwase K, Higuchi Y, Hirotani S, Yamaguchi O, Hikoso S, Takeda T. et al. CaMKII activates ASK1 and NF-kappaB to induce cardiomyocyte hypertrophy. Biochem Biophys Res Commun. 2005;327:136-42
63. Lincoln TM, Cornwell TL. Intracellular cyclic GMP receptor proteins. FASEB J. 1993;7:328-38
64. Huang ZJ, Haugland RP, You WM, Haugland RP. Phallotoxin and actin binding assay by fluorescence enhancement. Anal Biochem. 1992;200:199-204
65. Canton J. Macropinocytosis: New Insights Into Its Underappreciated Role in Innate Immune Cell Surveillance. Front Immunol. 2018;9:2286
66. Coue M, Brenner SL, Spector I, Korn ED. Inhibition of actin polymerization by latrunculin A. FEBS Lett. 1987;213:316-8
67. Racoosin EL, Swanson JA. Macrophage colony-stimulating factor (rM-CSF) stimulates pinocytosis in bone marrow-derived macrophages. J Exp Med. 1989;170:1635-48
68. Oliveira CA, Kashman Y, Mantovani B. Effects of latrunculin A on immunological phagocytosis and macrophage spreading-associated changes in the F-actin/G-actin content of the cells. Chem Biol Interact. 1996;100:141-53
69. van Niel G, D'Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19:213-28
70. Jeppesen DK, Fenix AM, Franklin JL, Higginbotham JN, Zhang Q, Zimmerman LJ. et al. Reassessment of Exosome Composition. Cell. 2019;177:428-45 e18
71. Panagopoulou MS, Wark AW, Birch DJS, Gregory CD. Phenotypic analysis of extracellular vesicles: a review on the applications of fluorescence. J Extracell Vesicles. 2020;9:1710020
72. Murakami M, Nakatani Y, Atsumi GI, Inoue K, Kudo I. Regulatory Functions of Phospholipase A2. Crit Rev Immunol. 2017;37:121-79
73. Yarla NS, Bishayee A, Vadlakonda L, Chintala R, Duddukuri GR, Reddanna P. et al. Phospholipase A2 Isoforms as Novel Targets for Prevention and Treatment of Inflammatory and Oncologic Diseases. Curr Drug Targets. 2016;17:1940-62
74. Balsinde J, Winstead MV, Dennis EA. Phospholipase A(2) regulation of arachidonic acid mobilization. FEBS Lett. 2002;531:2-6
75. Ogier JM, Nayagam BA, Lockhart PJ. ASK1 inhibition: a therapeutic strategy with multi-system benefits. J Mol Med (Berl). 2020;98:335-48
76. Shiizaki S, Naguro I, Ichijo H. Activation mechanisms of ASK1 in response to various stresses and its significance in intracellular signaling. Adv Biol Regul. 2013;53:135-44
77. Hattori K, Naguro I, Runchel C, Ichijo H. The roles of ASK family proteins in stress responses and diseases. Cell Commun Signal. 2009;7:9
78. Barros-Minones L, Orejana L, Goni-Allo B, Suquia V, Hervias I, Aguirre N. et al. Modulation of the ASK1-MKK3/6-p38/MAPK signalling pathway mediates sildenafil protection against chemical hypoxia caused by malonate. Br J Pharmacol. 2013;168:1820-34
79. Jarvis RM, Hughes SM, Ledgerwood EC. Peroxiredoxin 1 functions as a signal peroxidase to receive, transduce, and transmit peroxide signals in mammalian cells. Free Radic Biol Med. 2012;53:1522-30
80. Nadeau PJ, Charette SJ, Landry J. REDOX reaction at ASK1-Cys250 is essential for activation of JNK and induction of apoptosis. Mol Biol Cell. 2009;20:3628-37
81. Kekulandara DN, Nagi S, Seo H, Chow CS, Ahn YH. Redox-Inactive Peptide Disrupting Trx1-Ask1 Interaction for Selective Activation of Stress Signaling. Biochemistry. 2018;57:772-80
82. Striggow F, Ehrlich BE. Regulation of intracellular calcium release channel function by arachidonic acid and leukotriene B4. Biochem Biophys Res Commun. 1997;237:413-8
83. Meves H. Arachidonic acid and ion channels: an update. Br J Pharmacol. 2008;155:4-16
84. Shuttleworth TJ. Arachidonic acid activates the noncapacitative entry of Ca2+ during [Ca2+]i oscillations. J Biol Chem. 1996;271:21720-5
85. Muslikhov ER, Sukhanova IF, Avdonin PV. Arachidonic acid activates release of calcium ions from reticulum via ryanodine receptor channels in C2C12 skeletal myotubes. Biochemistry (Mosc). 2014;79:435-9
86. Richieri GV, Mescher MF, Kleinfeld AM. Short term exposure to cis unsaturated free fatty acids inhibits degranulation of cytotoxic T lymphocytes. J Immunol. 1990;144:671-7
87. Post PL, Bokoch GM, Mooseker MS. Human myosin-IXb is a mechanochemically active motor and a GAP for rho. J Cell Sci. 1998;111( Pt 7):941-50
88. Kong R, Yi F, Wen P, Liu J, Chen X, Ren J. et al. Myo9b is a key player in SLIT/ROBO-mediated lung tumor suppression. J Clin Invest. 2015;125:4407-20
89. Comer S, Nagy Z, Bolado A, von Kriegsheim A, Gambaryan S, Walter U. et al. The RhoA regulators Myo9b and GEF-H1 are targets of cyclic nucleotide-dependent kinases in platelets. J Thromb Haemost. 2020
90. Pecci A, Ma X, Savoia A, Adelstein RS. MYH9: Structure, functions and role of non-muscle myosin IIA in human disease. Gene. 2018;664:152-67
91. Rai V, Thomas DG, Beach JR, Egelhoff TT. Myosin IIA Heavy Chain Phosphorylation Mediates Adhesion Maturation and Protrusion in Three Dimensions. J Biol Chem. 2017;292:3099-111
92. Kasonga AE, Deepak V, Kruger MC, Coetzee M. Arachidonic acid and docosahexaenoic acid suppress osteoclast formation and activity in human CD14+ monocytes, in vitro. PLoS One. 2015;10:e0125145
93. Rostam HM, Reynolds PM, Alexander MR, Gadegaard N, Ghaemmaghami AM. Image based Machine Learning for identification of macrophage subsets. Sci Rep. 2017;7:3521
94. Zhou J, Tang Z, Gao S, Li C, Feng Y, Zhou X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front Oncol. 2020;10:188
95. Chi X, Wang S, Huang Y, Stamnes M, Chen JL. Roles of rho GTPases in intracellular transport and cellular transformation. Int J Mol Sci. 2013;14:7089-108
96. Hohensinner PJ, Mayer J, Kichbacher J, Kral-Pointner J, Thaler B, Kaun C. et al. Alternative activation of human macrophages enhances tissue factor expression and production of extracellular vesicles. Haematologica. 2020
97. Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020 367
98. Pogge von Strandmann E, Müller R. Shipping Drug Resistance: Extracellular Vesicles in Ovarian Cancer. Trends Mol Med. 2016;22:741-3
99. Tang MK, Wong AS. Exosomes: Emerging biomarkers and targets for ovarian cancer. Cancer Lett. 2015;367:26-33
100. Beach A, Zhang HG, Ratajczak MZ, Kakar SS. Exosomes: an overview of biogenesis, composition and role in ovarian cancer. J Ovarian Res. 2014;7:14
101. Guo L, Zhang Y, Wei R, Zhang X, Wang C, Feng M. Proinflammatory macrophage-derived microvesicles exhibit tumor tropism dependent on CCL2/CCR2 signaling axis and promote drug delivery via SNARE-mediated membrane fusion. Theranostics. 2020;10:6581-98
102. Qu J, Zhao X, Wang J, Liu C, Sun Y, Cai H. et al. Plasma phospholipase A2 activity may serve as a novel diagnostic biomarker for the diagnosis of breast cancer. Oncol Lett. 2018;15:5236-42
103. Wang YY, Zhao R, Zhe H. The emerging role of CaMKII in cancer. Oncotarget. 2015;6:11725-34
104. Martinez-Limon A, Joaquin M, Caballero M, Posas F, de Nadal E. The p38 Pathway: From Biology to Cancer Therapy. Int J Mol Sci. 2020 21
105. Acunzo J, Andrieu C, Baylot V, So A, Rocchi P. Hsp27 as a therapeutic target in cancers. Curr Drug Targets. 2014;15:423-31
106. Clayton NS, Ridley AJ. Targeting Rho GTPase Signaling Networks in Cancer. Front Cell Dev Biol. 2020;8:222
Author contact
![]() Corresponding author: Rolf Müller, Center for Tumor Biology and Immunology (ZTI), Philipps University, Hans-Meerwein-Strasse 3, 35043 Marburg, Germany. E-mail: rolf.muellerde. Phone: +49 6421 2866236.
Corresponding author: Rolf Müller, Center for Tumor Biology and Immunology (ZTI), Philipps University, Hans-Meerwein-Strasse 3, 35043 Marburg, Germany. E-mail: rolf.muellerde. Phone: +49 6421 2866236.