Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(4):1918-1936. doi:10.7150/thno.48865 This issue Cite
Research Paper
Development of the phenylpyrazolo[3,4-d]pyrimidine-based, insulin-like growth factor receptor/Src/AXL-targeting small molecule kinase inhibitor
Ho Jin Lee1, Phuong Chi Pham2, Honglan Pei1, Bumhee Lim2, Seung Yeob Hyun1, Byungyeob Baek2, Byungjin Kim2, Yunha Kim2, Min-Hwan Kim2, Nae-Won Kang2, Hye-Young Min1,2, Dae-Duk Kim2, Jeeyeon Lee2 ![]() , Ho-Young Lee1,2
, Ho-Young Lee1,2 ![]()
1. Creative Research Initiative Center for concurrent control of emphysema and lung cancer, College of Pharmacy, Seoul National University, Seoul 08826, Republic of Korea.
2. College of Pharmacy and Research Institute of Pharmaceutical Sciences, Seoul National University, Seoul 08826, Republic of Korea.
Received 2020-5-30; Accepted 2020-11-4; Published 2021-1-1
Abstract

Rationale: The type I insulin-like growth factor receptor (IGF-1R) signaling pathway plays key roles in the development and progression of numerous types of human cancers, and Src and AXL have been found to confer resistance to anti-IGF-1R therapies. Hence, co-targeting Src and AXL may be an effective strategy to overcome resistance to anti-IGF-1R therapies. However, pharmacologic targeting of these three kinases may result in enhanced toxicity. Therefore, the development of novel multitarget anticancer drugs that block IGF-1R, Src, and AXL is urgently needed.
Methods: We synthesized a series of phenylpyrazolo[3,4-d]pyrimidine (PP)-based compounds, wherein the PP module was conjugated with 2,4-bis-arylamino-1,3-pyrimidines (I2) via a copper(I)-catalyzed alkyne-azide cycloaddition reaction. To develop IGF-1R/Src/AXL-targeting small molecule kinase inhibitors, we selected LL6 as an active compound and evaluated its antitumor and antimetastatic effects in vitro and in vivo using the MTT assay, colony formation assays, migration assay, flow cytometric analysis, a tumor xenograft model, the KrasG12D/+-driven spontaneous lung tumorigenesis model, and a spontaneous metastasis model using Lewis lung carcinoma (LLC) allografts. We also determined the toxicity of LL6 in vitro and in vivo.
Results: LL6 induced apoptosis and suppressed viability and colony-forming capacities of various non-small cell lung cancer (NSCLC) cell lines and their sublines with drug resistance. LL6 also suppressed the migration of NSCLC cells at nontoxic doses. Administration of LL6 in mice significantly suppressed the growth of NSCLC xenograft tumors and metastasis of LLC allograft tumors with outstanding toxicity profiles. Furthermore, the multiplicity, volume, and load of lung tumors in KrasG12D/+ transgenic mice were substantially reduced by the LL6 treatment.
Conclusions: Our results show the potential of LL6 as a novel IGF-1R/Src/AXL-targeting small molecule kinase inhibitor, providing a new avenue for anticancer therapies.
Keywords: small molecule kinase inhibitor, type I insulin-like growth factor receptor, Src, AXL
Introduction
Cancer is the leading cause of human deaths worldwide [1]. Despite extensive efforts to develop therapeutic interventions, the 5-year survival rate for certain types of cancers did not show much improvement [1]. Conventional chemotherapy is a standard therapeutic option [2]. However, resistance and toxicity of chemotherapy have led the focus of anticancer drug development to molecularly targeted therapy, in which anticancer drugs act on specific proteins, thereby reducing undesired side effects on normal cells [3]. However, drug resistance due to the cross-talk between signaling pathways and high mutation rates of oncogenes are the main drawbacks of single targeted therapy. Inhibitors targeting epidermal growth factor receptor (EGFR) are one of the most widely developed molecularly targeted therapeutic options and have shown efficacy in a subset of patients with specific genetic abnormalities [4]. However, most patients do not show a response to therapy because of primary resistance [4]. Furthermore, acquired resistance to therapy frequently develops in primarily susceptible patients [4-6].
Accumulating evidence supports the role of the type I insulin-like growth factor receptor (IGF-1R) signaling pathway in resistance to chemotherapy, molecularly targeted therapy, and recently developed immunotherapy [7-11]. Consequently, IGF-1R has been considered as an attractive therapeutic target, especially when combined with other anticancer treatments [10]. However, anti-IGF-1R therapies have shown marginal efficacy because of inherent or acquired resistance [10, 12-16]. Blockade of bypass mechanisms by combination therapies may circumvent drug resistance. However, potential severe side effects through drug-drug interactions, poor patient compliance, and complex pharmacokinetic and pharmacodynamic profiles are the main hurdles of combination therapies [17, 18]. Thus, there is an urgent need to develop novel IGF-1R-targeting drugs that overcome resistance.
Canonical IGF-1R signaling activity is controlled by ligands (IGF1, IGF2, and insulin) binding to IGF-1R, insulin receptor (IR), or hybrid receptors of IGF-1R/IR [19]. Additionally, Src, a non-receptor tyrosine kinase (non-RTK), has the ability to phosphorylate IGF-1R and IR at the same amino acid residues as the ligand-induced autophosphorylation sites [20]. Increased levels of Src expression and kinase activity have been reported in various human cancers [21, 22] and often induce resistance to various molecularly targeted anticancer drugs, including IGF-1R inhibitor-based therapies [12]. AXL, a RTK involved in various tumor activities [23-25], has also been implicated in the resistance to chemotherapy and anti-IGF-1R therapies [26, 27] and associated with poor prognosis in human cancers [28, 29].
A previous study revealed potent inhibition of AXL by bosutinib (SKI-606), which was originally developed as an Src and Abl kinase inhibitor [30]. Pyrazolo[3,4-d]pyrimidines (PP), a well-known scaffold for Src, functions as an AXL inhibitor [31]. Owing to its antitumor activity, bosutinib is currently under clinical evaluation as an AXL inhibitor [23]. In addition, several inhibitors targeting AXL, including UNC569 [32], UNC1062 [33], and eSM134 [31], or Src, including AD80 [34], KB SRC4 [35], PP2 [36], PP121 [37], and eCF506 [38], possess the PP fragment. These findings provide the rationale for the potential use of Src tyrosine kinase inhibitors (TKIs) to discover novel AXL inhibitors. Indeed, in the current study, our docking analysis revealed structural similarity between the ATP-binding pockets of AXL and Src. Hence, we have attempted to develop small molecule kinase inhibitors (SMKIs) that target IGF-1R, Src, and AXL by utilizing PP and 2,4-bis-arylamino-1,3-pyrimidines modules (I2) as scaffolds directed for Src and IGF-1R, respectively. Here, we report a novel multikinase inhibitor, designated as LL6, which has broad antitumor effects on non-small cell lung cancer (NSCLC) cells, including those carrying de novo or acquired resistance to chemotherapy or EGFR TKIs, both in vitro and in vivo, by concurrently targeting IGF-1R, Src, and AXL. These results suggest that LL6 is a useful multitarget SMKI in the treatment of cancer.
Materials and Methods
Cell culture
Human lung cancer cell lines (A549, H1299, H1993, H1944, H226B, H226Br, H460, H522, HCC15, and PC9), a diploid human lung fibroblast cell line (Wi38), and the Lewis lung carcinoma (LLC) cell line were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) or kindly provided by Dr. John V. Heymach (MD Anderson Cancer Center, Houston, TX, USA). Human retinal pigment epithelial (RPE) cells were kindly provided by Dr. Jeong Hun Kim (College of Medicine, Seoul National University, Seoul, Republic of Korea). HT-22 cells were provided by Dr. Dong Gyu Jo (College of Pharmacy, Sungkyunkwan University, Suwon, Republic of Korea). Lung cancer cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and antibiotics (Welgene, Gyeongsan-si, Republic of Korea). LLC, HT-22, and Wi38 cells were maintained in DMEM supplemented with 10% FBS and antibiotics. RPE cells were maintained in DMEM/F12 supplemented with 10% FBS and antibiotics. NSCLC cell lines with acquired resistance to chemotherapy (cisplatin-resistant H1299/CsR and pemetrexed-resistant H1299/PmR and H460/PmR) and molecular targeted therapy (erlotinib-resistant PC9/ER) were generated by continuous exposure to corresponding anticancer drugs for more than six months. Cells were maintained at 37 °C in a humidified atmosphere with 5% CO2. Cell lines were authenticated and validated using the AmpFLSTR identifier PCR Amplification Kit (Applied Biosystems, Foster City, CA, USA; cat. No. 4322288) in 2013 and 2016. Cells that had been passaged for < 6 months after receipt or resuscitation of validated cells were used in this study.
Reagents
Antibodies against AXL, pIGF-1R (Y1135/6), IGF-1R, pSrc (Y416), Src, pMet (Y1234/5), Met, tubulin, and cleaved caspase-3 were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies against cleaved PARP were purchased from BD Biosciences (San Jose, CA, USA). Antibodies against pAXL (Y702) were purchased from R&D systems (Minneapolis, MN, USA). Primary antibodies against IGF-1R and actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Primary antibodies against pIR/IGF-1R (Y1162/3) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Horseradish peroxidase-conjugated secondary antibodies were purchased from GeneTex (Irvine, CA, USA). Linsitinib, dasatinib, and bemcentinib (R428) were purchased from Selleckchem (Houston, TX, USA). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) and other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise specified. The detailed information on used primary and secondary antibodies, including vendor, catalogue number, application, and dilution ratio (or concentration) is listed in Table S1.
Molecular docking simulations
Molecular docking analysis was implemented using the Surflex-Dock module in Sybyl-X2.2.1 (Tripos Inc, St Louis, MO, USA) with the known crystal structure of AXL complexed with ligands (PDB ID: 5U6B). To prepare the protein, hydrogen was added and energy was minimized using Powell's method with the Tripos force field until the root-mean-square derivation (RMSD) values were < 0.05 Kcal/mol·Å. Initial optimization and termination of minimization were set as simplex and gradient, respectively. The new ligands were prepared using Chem3D (PerkinElmer, Waltham, MA, USA). Molecular docking simulations were conducted using the Surflex-Dock mode with the extraction of the original ligand. To generate the active site, a threshold of 0.5 Å and bloat of 0 Å were applied based on the original ligand in the crystal structure. Other parameters were used as default. The results of the docking simulation were validated by comparing the redocked structure to the original pose of the ligand. Molecular interactions between proteins and ligands were further analyzed using Discovery Studio 4.0 Visualizer (BIOVIA, San Diego, CA, USA).
MTT assay
Cells were seeded into 96-well plates at a density of 1-2 × 103 cells/well and incubated for 24 h. Cells were treated with vehicle or various concentrations of test compounds for two (PC9 and PC9/ER cells) or three (other cells) days. Cells were further incubated with MTT solution (final concentration of 500 μg/mL) for 4 h at 37 °C. The formazan products were dissolved in dimethyl sulfoxide (DMSO), and the absorbance was measured at 570 nm. The data are presented as a percentage of the control group.
Competition binding assay
Kd values were obtained by KINOMEscan from DiscoverX (San Diego, CA, USA). To briefly summarize, Kd was measured by a competition binding assay that quantitatively measures the ability of a compound to compete with an immobilized ligand. The ligand-displayed affinity bead and DNA (T7 bacteriophage)-tagged kinases are combined with a test compound, and the amount of fusion protein bound to the solid support is quantified by qPCR. Dose response curves of 11 points are obtained for Kd values, which are measured in duplicate. See more details in the cited references [39-42] and see also at www.discoverx.com.
Anchorage-dependent colony formation assay
Cells were seeded into 6-well plates at a density of 300 cells/well and treated with LL6. The drug-containing medium was changed once or twice a week. After incubation for 10-14 days, colonies were fixed with 100% methanol, stained with 0.002% crystal violet solution, and washed with deionized water several times. The colonies were imaged and manually counted.
Anchorage-independent colony formation assays
Cells (2-3 × 103 cells/well) were mixed with sterile 1% agar solution (final concentration of 0.4%) and poured onto 1% base agar in 24-well plates. After solidification of the top agar, LL6 was diluted in complete medium and added to the agar. Cells embedded in the top agar were incubated for 10-14 days at 37 °C with 5% CO2. The LL6-containing medium was changed twice a week. After incubation, the colonies were stained with MTT solution, imaged, and counted using ImageJ software (National Institutes of Health, Bethesda, MA, USA).
Immunofluorescence staining
H1944 cells, seeded onto coverslip, were treated with increasing concentrations of LL6 (0, 1, 2.5 μM) for 24 h. Cells were fixed with 4% paraformaldehyde for 10 min at room temperature, washed with PBS, and then permeabilized with 0.3% Triton X-100 for 15 min at room temperature. After washing cells with PBS, the cells were incubated with blocking solution [3% bovine serum albumin (BSA) in Tris-buffered saline containing 0.1% Tween-20] for 1 h at room temperature. Cells were incubated with primary antibodies (1:200 dilution) at 4 ºC overnight. Cells were washed several times with PBS and incubated with fluorochrome-conjugated secondary antibodies (Thermo Fisher Scientific) for 1 h at room temperature. Cells were washed multiple times with PBS and counterstained with 4′,6-diamidino-2-phenylindole (DAPI). The coverslips were mounted with mounting solution (Dako, Glostrup, Denmark) and then observed under a fluorescence microscope (Zeiss Axio Observer Z1, Carl Zeiss AG, Oberkochen, Germany).
Western blot analysis
Cells were treated with LL6, linsitinib, or dasatinib for the indicated time intervals. Before harvesting, the cells were stimulated with 10% FBS for 20 min. Total cell lysates were prepared with modified radioimmunoprecipitation assay lysis buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 0.25% sodium deoxycholate, 1% Triton X-100, 100 mM NaF, 5 mM Na3VO4, 1 mM PMSF, 1 μg/mL aprotinin, 1 μg/mL leupeptin, and 1 μg/mL pepstatin). Equal amounts of protein (20 μg) were subjected to 8% SDS-PAGE and electrically transferred onto polyvinylidene difluoride membranes (Bio-Rad Laboratories, Hercules, CA, USA). Membranes were blocked with blocking buffer (3% BSA in Tris-buffered saline containing 0.1% Tween-20 [TBST]) for 1 h at room temperature. The membranes were incubated with primary antibodies diluted in 3% BSA in TBST (1:1,000) overnight at 4 °C, washed multiple times with TBST, and incubated with secondary antibodies diluted in 3% nonfat dry milk in TBST (1:5,000) for 1 h at room temperature. The membranes were washed multiple times with TBST and visualized using an enhanced chemiluminescence detection kit (Thermo Fisher Scientific). Densitometric analysis was performed using ImageJ software.
Targeted sequencing for analysis of EGFR mutation status
To extract genomic DNA from PC9 and PC9/ER cells, cells were treated with a lysis buffer (150 mM Tris-HCl [pH 8.5], 200 mM NaCl, 5 mM EDTA [pH 8.0], 0.2% SDS, and 300 μg/mL proteinase K) and subsequently incubated at 55 °C overnight. DNA was extracted using isopropanol as reported previously [43], further purified with PCI buffer (phenol : chloroform : isoamyl alcohol, 25:24:1), washed with 70% ethanol, and dissolved in TE buffer. For target capture, DNA was sheared into approximately 180 bp fragments, end-repaired, dA-tailed, and adapter-ligated using Illumina adapter pairs. Hybridization probes (Celemics, Seoul, Republic of Korea) were mixed with the target DNA and then separated by streptavidin beads. Target capture libraries were sequenced with the Nextseq500 platform (Illumina, San Diego, USA) using 2 × 150 bp paired-end run. The BWA aligner was used to map the sequence reads genome. Local alignment and duplication removal were performed with the Genome Analysis Tool Kit (GATK, Broad Institute, Cambridge, MA, USA) and the Picard software (Broad Institute). VarScan were used to call SNVs, and BWA aligner was performed with the Indel Detector in GATK. Mutation candidates at various loci were annotated with the ANNOVAR tool [44].
Animal experiments
All animal experiments were performed according to protocols approved by the Seoul National University Institutional Animal Care and Use Committee. Mice were fed standard mouse chow and water ad libitum and housed in temperature- and humidity-controlled facilities with a 12-h light/12-h dark cycle. For xenograft experiments, H1944 and A549 cells (1×106 cells/spot, diluted in equal amounts of PBS and Matrigel) were subcutaneously injected into the right flank of 6-week-old non-obese diabetic-severe combined immunodeficiency (NOD/SCID) mice. After the tumor volume reached 50-150 mm3, the mice were randomly grouped and treated with vehicle [10% DMSO in 60% polyethylene glycol (PEG) 400 solution] or LL6 (80 mg/kg) 6 days per week for 2 weeks. Tumor growth was determined by measuring the short and long diameters of the tumor with a caliper, and body weight was measured once or twice per week to monitor toxicity. Additionally, to evaluate the effect of LL6 on mutant Kras-driven lung tumorigenesis, 2-month-old KrasG12D/+ transgenic mice [48] were randomized and treated with vehicle or LL6 (80 mg/kg) for 8 weeks. The mice were euthanized, and tumor formation was evaluated and compared with that of the vehicle-treated control group. Microscopic evaluations of the H&E-stained lung tissue were also performed to measure mean tumor number (N) and volume (V) in a blinded fashion. The number and size of tumors were calculated in five sections uniformly distributed throughout each lung. In both animal experiments, the tumor volume and burden of each sample were calculated using the following formulas: Tumor volume (mm3) = (short diameter)2 × (long diameter) × 0.5; Tumor burden (mm3) = number of tumors × the average of tumor volume.
We used the IVIS-Spectrum microCT and Living Image (ver. 4.2) software (PerkinElmer, Alameda, CA, USA) for monitoring metastatic tumor formation in the lungs. The instrument was operated according to the manufacturer's instruction. To facilitate the detection of photons emitted from metastatic lung tumors, we performed ex vivo imaging analysis. In brief, mice were injected at 60 mg/kg with the 15 mg/mL stock of luciferin prior to anesthesia. After 10-15 min, mice were euthanized, and lung tissues were excised and placed into a 60 mm dish. Tissues were immediately imaged after exposure for 1 min. Regions of interest (ROIs) from displayed images were quantified as photons/second (ph/s) using the Living Image software (PerkinElmer).
Toxicity test
FVB mice were treated with vehicle, LL6 (20, 40, and 80 mg/kg, dissolved in 10% DMSO in 60% PEG 400 solution) or combination of linsitinib (25 mg/kg, dissolved in a 25 mM tartaric acid solution) and dasatinib (20 mg/kg, dissolved in an 80 mM citric acid solution) daily for 12 days. Blood was collected from euthanized mice under isoflurane-induced deep anesthesia by cardiac puncture. After allowing blood coagulation at 4 °C, serum was collected by centrifugation at 3,000 rpm for 10 min at 4 °C. Analysis of the serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) was performed using a veterinary hematology analyzer (Fuji DRI-Chem 3500s, Fujifilm, Tokyo, Japan) according to the manufacturer' s protocols.
Glucose tolerance test
Glucose tolerance tests were performed as described in the previously published literature [49].
Application to a Pharmacokinetic study in rats
The pharmacokinetic properties of LL6 were investigated in SD rats (n = 3). The left femoral artery and vein were catheterized with polyethylene tube (IntramedicTM PE-50; Becton-Dickinson Diagnostics, MD, USA) under Zoletil (Virbac, Carros, France) anesthesia (50 mg/kg, intramuscular injection). LL6 was dissolved in DMSO/PEG 400/normal saline mixture (35:35:30, v/v/v) and single dose solution (1 mg/kg) was administered to rat intravenously. Approximately 150 µL of blood samples were collected via femoral artery at predetermined time (1, 3, 5, 10, 15, 30, 60, 120 and 180 min) and equivalent volume of normal saline solution with 20 U/mL of heparin was replenished to prevent blood coagulation. After centrifugation of the sample at 16,000 × g for 5 min, 50 µL aliquot of plasma samples were stored at -20 °C until HPLC-MS/MS analysis. The Pharmacokinetic parameters, including terminal half-life (T½), area under the plasma concentration-time curve from time zero to time last (AUClast), area under the plasma concentration-time curve from time zero to time infinity (AUCinf), volume of distribution at steady-state (Vss) and mean residence time (MRT) were calculated using non-compartmental analysis (WinNonlin, version 3.1, NCA 201; Pharsight, Mountain View, CA, USA).
Immunohistochemistry
Sections derived from formalin-fixed and paraffin-embedded murine lung tissues were deparaffinized by incubation overnight at 65 °C, followed by rehydration in sequential xylene and ethanol rinses. After incubation with hydrogen peroxide, the slides were washed with PBS and then incubated with 0.3% Triton X-100. The sections were incubated with blocking solution (Dako Protein Block, Dako, Glostrup, Denmark) for 30 min at room temperature after washing with PBS. The sections were further incubated with primary antibodies (pAXL, pIGF-1R, and pSrc, diluted at 1:200) overnight at 4 °C, washed with PBS several times, incubated with the corresponding biotinylated secondary antibodies (diluted at 1:500), and then washed with PBS multiple times. After adding avidin-biotin complexes (Vector Laboratories, Burlingame, CA, USA), the sections were visualized using diaminobenzidine detection reagent (Enzo Life Sciences, Farmingdale, NY, USA) and mounted with a mounting solution (Vector Laboratories).
Statistical analysis
Data are presented as mean ± SD. All in vitro experiments were independently performed at least twice, and a representative result is presented. The data were calculated or analyzed using Microsoft Excel software (Microsoft Corp., Redmond, MA, USA). The IC50 values were determined by nonlinear regression analysis using GraphPad Prism 8 (GraphPad Software, Inc., La Jolla, CA, USA). Statistical significance was determined using a two-tailed Student's t-test or one-way analysis of variance (ANOVA) using GraphPad Prism 8. An F-test for equality of variances was performed to ensure the same variance of two test groups. The Shapiro-Wilk test was performed to determine whether the in vitro or in vivo data follows a normal distribution. A P-value < 0.05 was considered statistically significant.
Results
Synthesis of LL6 as a novel SMKI targeting IGF-1R, Src, and AXL
We assessed the structural similarity between AXL and Src or IGF-1R by comparing the X-ray structures of Src, IGF-1R, and AXL (pdb: 2SRC, 3D94, and 5U6B, respectively). Intriguingly, AXL displayed a significant structural similarity with Src; that is, the microenvironments of ATP-binding pockets in AXL and Src were extremely similar and the RMSD value from AXL and Src alignment was markedly lower compared with that from AXL and IGF-1R alignment (2.304 vs 4.706, Figure 1A). In addition, ATP-binding pockets of AXL and Src are well overlaid, as depicted in the enlarged Figures. To assess the potential of PP to bind to AXL, we performed molecular docking simulations and examined the docking poses of the linker-attached PPs bound to AXL (Figure 1B). Results from in silico docking studies using various PP derivatives are summarized in Figure S1. Compound PP-5 with the extended linker possessing an ether group in addition to a triazole ring showed a significant increase in docking scores with proper orientation of the linker toward the surface side of AXL (pdb: 5U6B) [Figure 1B-(i)]. Docking with the Src protein (pdb: 2SRC) showed a similar trend (Figure S1C-D), suggesting that the PP module conjugated with the linker in PP-5 binds to AXL. In addition, our results suggested that the reported key interactions of the original ligand in the hinge region of the AXL shown in the X-ray crystal structure (pdb: 5U6B) were retained with PP-5. Particularly, hydrogen bonding interactions of PP-5 with Pro621 and Met623 in the hinge region of AXL [Figure 1B-(ii)] along with several hydrophobic interactions [Figure 1B-(iii)] in the binding pocket indicated that PP-5 may also bind to AXL. To discover new compounds that potently inhibit IGF-1R, Src, and AXL, we employed PP as a privileged scaffold directed to Src and AXL and I2 to IGF-1R. These two modules were conjugated via copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) (Figure 1C). Details of the synthesis and characterization of the synthesized compounds are given in Appendix of Supplementary Materials. Various functional groups (R1, R2, and R3) were introduced to PP and the phenyl ring of I2 to assess their anticancer activities. The key intermediates 1a-d were N-alkylated to afford compounds 3a-c, which possess a direct ether linkage with 6f. N-alkylation of 1a-d with 2-bromoethanol and mesylation, followed by the substitution reaction with sodium azide, provided 2a-d. CuAAC between alkyne-containing 6a-e and various azide-containing pyrazolopyrimidines 2a-d afforded 4a-l and 5a-h with a triazole linker at different positions on the phenyl group (R2 and R3) of the IGF-1R module (6a-e) (Figure 1C and Table 1).
Anti-proliferation effects of compounds 3-5 against A549 cancer cell line
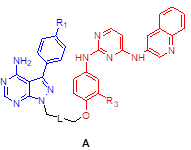 | 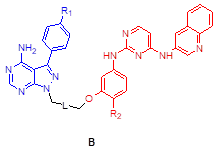 | ||||
|---|---|---|---|---|---|
| Compound | Structure | R1 | R2/R3 | L (linker) | % cells growth at 10 μM |
| 3a | A | Me | H | 98 | |
| 3b | H | H | 96 | ||
| 3c | Cl | H | 82 | ||
| 4a | A | Me | H | 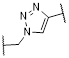 | 82 |
| 4b | H | H | 20 | ||
| 4c (LL6) | Cl | H | 6.4 | ||
| 4d | CF3 | H | 26 | ||
| 4e | Me | Cl | 101 | ||
| 4f | H | Cl | 103 | ||
| 4g | Cl | Cl | 99 | ||
| 4h | CF3 | Cl | 27 | ||
| 4i | Me | F | 88 | ||
| 4j | H | F | 96 | ||
| 4k | Cl | F | 94 | ||
| 4l | CF3 | F | 47 | ||
| 5a | B | Me | H | 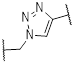 | 68 |
| 5b | H | H | 35 | ||
| 5c | Cl | H | 52 | ||
| 5d | CF3 | H | 76 | ||
| 5e | Me | OMe | 65 | ||
| 5f | H | OMe | 91 | ||
| 5g | Cl | OMe | 100 | ||
| 5h | CF3 | OMe | 106 | ||
| Bosutinib | 46 | ||||
| 1a | 98 | ||||
| 23a | 69 |
Synthesis of LL6 and determination of its cellular targets. (A) Superposed X-ray structures of AXL (magenta, PDB: 5U6B) and Src (yellow, PDB: 2SRC). Close-up views of overlaid binding pockets in cartoon (top) and surface (down) models are presented with the ligands shown in stick. (B) (i) Docking pose of PP-5 in the binding pocket of AXL. A close-up view of the binding orientation of PP-5 is shown in the box. See Figure S1 for further details. (ii) Suggested hydrogen bonding interactions of the critical binding site residues of AXL with PP-5; dashed green line and light green line represent conventional hydrogen bonds and carbon hydrogen bonds, respectively. (iii) Residues with hydrophobic interactions with PP-5 in the binding site of AXL. (C) Synthesis of compounds 3a-c, 4a-l, and 5a-h. Reagents and conditions: a) 6f, Cs2CO3, DMF, 80ºC, 3 h, 36-48%; b) 2-bromoethanol, Cs2CO3, DMF, 80ºC, 3 h, 50-63%; c) MsCl, pyridine, rt, 5 h, 45-76%; d) NaN3, DMF, 60°C, 8 h, 80-88%; e) 6a-e, CuSO4·5H2O, NaAsc, tBuOH/H2O/DMF, 80ºC, 3 h, 39-80%. See Table 1 for structural details. (D) Human kinome profile screened for LL6 at 10 µM. Red circles denote > 65% inhibition with a S-score of 0.12. The image was generated using TREEspot™ analysis from KINOMEscan (DiscoveRx). (E) Binding titrations for IGF-1R, Src, KIT, IR, AXL, and CSF1R were performed by competition binding assays from DiscoveRx. aDose dependent inhibition curve was not obtained with these three proteins (IR, KIT, and CSF1R).
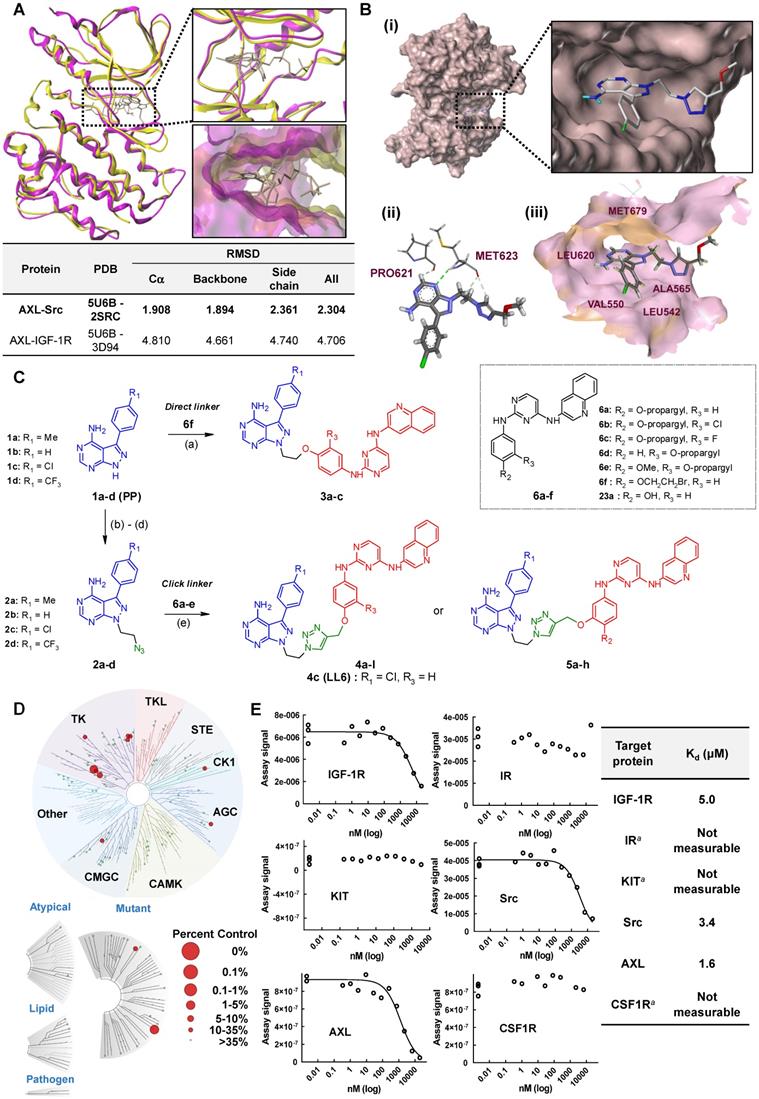
To assess the preliminary anticancer activity, the synthesized compounds were tested using the MTT assay for growth inhibition against human NSCLC (A549) cells (Table 1 and Figure S2). Bosutinib was also included in the assay as a control. To identify whether dual-targeting compounds would be better than a single targeting molecule, two key intermediates 1a and 23a for Src and IGF-1R, respectively, were also examined using the MTT assay (Table 1 and Figure S2) as control compounds. Several active compounds were selected to determine IC50 values against the viability of A549 cells (Table S2). Among the compounds, we selected 4c (LL6 hereafter) based on its highest potency in A549 cells (Table 1).
The structure-activity relationship (SAR) analyses based on our depicted data are as follows. Three compounds with a direct ether linker 3a-c did not show any inhibition on the viability of A549 cells at 10 μM (Table 1), indicating that the linker length between the two modules was extremely short. In contrast, several compounds with the triazole linker (4b-d, 4l, and 5b-c) displayed significant anticancer activity, with IC50 values shown in Table S2. Moreover, 4b-d with the linker attached at the para position showed greater potency in A549 cells than the derivatives linked at the meta position (5b-d), as indicated by the structural analysis on the previously reported X-ray structure of IGF-1R (pdb: 3QQU) shown in Figure S3A-B. Changes made at the aminophenol ring to include chlorine, fluorine, and methoxy groups (R2 or R3) in triazole-linked compounds (4e-l and 5e-h) resulted in significant loss of potency, implying that the large substituents adjacent to the linking position caused significant steric clashes upon binding to target proteins.
Next, we assessed the kinase selectivity of LL6, the most potent among the synthesized compounds, by kinome-wide inhibition profiling. Among the 97 tested kinases (DiscoveRx), several kinases showed > 75% inhibition by LL6 treatment (Table S3). In the TREEspot visualization map of the kinome selectivity profile, the target proteins of LL6 are shown in red circles and mainly distributed in the TK family of the phylogenetic tree (Figure 1D). To date, most kinases targeted by clinically approved drugs are located in the TK family [45, 46]. KIT, Src, IGF-1R, AXL, IR, CSF1R, and PDGFRB belong to the TK family and show > 65% inhibition with LL6 (Figure 1D and Table S3). Based on the hierarchical clustering analysis of the human kinome dataset, PDGFR, KIT, and CSF1R are found to be promiscuous kinases [47]. Indeed, further validation of Kd values using an equilibrium binding titration method ruled out the possibility of LL6 binding to KIT, IR, and CSF1R, as they did not show dose-dependent inhibition curves and were found to be false positives in the assay (Figure 1E). These results are in accordance with the notion that some kinases are promiscuous. LL6 exhibited binding affinity to IGF-1R (Kd = 5 μM), Src (Kd = 3.4 μM), and AXL (Kd = 1.6 μM). It is worth noting that LL6 possessed weak binding affinity to IR (Kd : not accurately measurable as indicated in Figure 1E), conferring selectivity toward IGF-1R, although both proteins are structurally related. An opposite orientation of the I2 ligand was suggested in the docking study with IR (Figure S3C-D). Note that the measured Kd values are comparable to IC50 values obtained from cellular assays shown in Table S2.
LL6 inhibits the viability and colony-forming ability of NSCLC cells by inducing apoptosis
We evaluated the in vitro antitumor effects of LL6 against various NSCLC cell lines. LL6 displayed dose-dependent inhibitory effects on the viability of NSCLC cells with various genetic backgrounds and histologies with IC50 values of approximately 2.5 μM (Figure 2A and Table S4), whereas LL6 exhibited significantly less cytotoxicity in Wi38 cells, human normal diploid lung fibroblasts, than in the NSCLC cell lines used; the percentage of cell viability at the concentration of 10 μM was above 60%. The concentration of 10 μM was less than the theoretical initial plasma concentration of LL6 [22.7 μg/mL (33.3 μM)] determined by a pharmacokinetic study in rats after intravenous administration of 1 mg/mL LL6 (Table S5 and Figure S4), indicating the effectiveness of LL6 even at the concentration lower than predicted plasma concentration.
Effects of LL6 on the viability, colony formation, and migration of several NSCLC cells. (A, D, E-G) Dose-dependent inhibitory effects of LL6 on the viability of naïve (A), erlotinib-resistant (D), and chemoresistant (E) NSCLC cells and NSCLC colony formation grown in anchorage-dependent (F) and anchorage-independent (G) culture conditions evaluated using the MTT assay (A, D, E), anchorage-dependent colony formation assay (F), and soft agar colony formation assay (G), respectively. For cell viability assay, cells were treated with various concentrations of LL6 diluted in 10% FBS- (A, E) or 5% FBS (D)-containing media for three (A, E) or two (D) days. (B, C) Western blot analysis showing basal expression of total and phosphorylated forms of AXL, IGF-1R, and Src in various NSCLC cell lines (B), PC9 (C), and erlotinib-resistant PC9 (PC9/ER) cells (C). (H) The effect of LL6 on the migration of NSCLC cells evaluated using Transwell migration assay. (I) The effect of LL6 on the viability of NSCLC cells under the same experimental conditions used for cell migration determined using the MTT assay. Bars represent mean ± SD. P < 0.05, **P < 0.01 and ***P < 0.001, as determined using the two-tailed Student's t-test compared with the vehicle-treated control.
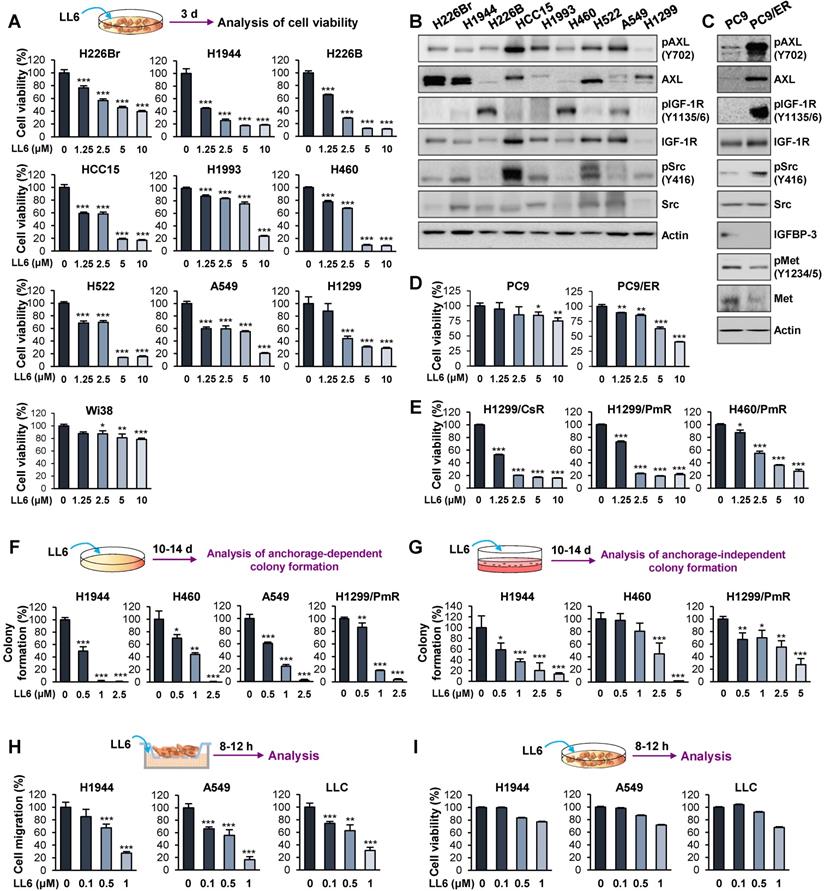
We then examined the correlation between the IC50 values of LL6 and the expression/activity of cellular targets of LL6, IGF-1R, Src, and AXL. Western blot analysis showed the basal levels of total and phosphorylated forms of IGF-1R, Src, and AXL expression in the indicated NSCLC cells (Figure 2B). We observed no obvious correlation between the IC50 value of LL6 and the basal expression/activation levels of IGF-1R, Src, and AXL in each NSCLC cell line. These results might be due to the heterogenous activation status of IGF-1R, Src, and AXL at basal levels; only one to two kinases among IGF-1R, Src, and AXL were activated in the NSCLC cell lines used in this study. Of note, the cellular target of LL6, IGF-1R, Src, and AXL can be activated through acquired mechanism of drug resistance. Studies have shown that various cancer cells acquire resistance to IGF-1R inhibitor-based therapies, through activation of Src and AXL [12, 26]. These points may explain the lack of correlation between the IC50 value of LL6 and the basal expression/activation levels of targets in NSCLC cells.
We next assessed the effects of LL6 on EGFR TKI (erlotinib)-resistant subline (PC9/ER) [48]. Targeted next-generation sequencing analysis to determine the mutation status of EGFR in PC9/ER showed that the PC9/ER subline has various mutations in the tyrosine kinase domain (exon 18-21) of EGFR, including T790M (Table S6). In line with the findings, PC9/ER cells showed resistance to the corresponding EGFR TKI after a prolonged culture in the absence of drug for more than a month (Figure S5). PC9/ER and PC9 cells were further analyzed for activation status of several RTKs [MET, IGF-1R, and AXL], a nonreceptor tyrosine kinase Src, and IGFBP-3, all of which have been implicated in resistance to EGFR TKIs [49, 50]. We observed greater activation of IGF-1R, Src, and AXL and weaker expression of IGFBP-3 in PC9/ER than in PC9 cells (Figure 2C). In contrast, Met was not activated in PC9/ER cells (Figure 2C). More importantly, LL6 exhibited greater inhibitory effects on viability of PC9/ER than that of PC9 cells (Figure 2D). These results suggested that IGF-1R, Src, and AXL might act as drivers of resistance to EGFR TKIs and inhibition of these kinases by using a multikinases inhibitor such as LL6 efficiently suppresses the viability of EGFR TKI-resistant cells.
We also found that the inhibitory effects of LL6 on the chemoresistant sublines (H1299/CsR, H1299/PmR and H460/PmR) [51] were comparable with those on the corresponding parental cells (H1299 and H460) (Figure 2E). These findings collectively suggest that LL6 is applicable to both naïve and anticancer drug-resistant lung cancer cells. We further observed that LL6 significantly suppressed colony formation in a subset of NSCLC cells in anchorage-dependent (Figure 2F) and anchorage-independent (Figure 2G) culture conditions, which indicates cell survival and tumorigenicity, respectively [52, 53]. LL6 effectively suppressed the migration of NSCLC cells in a concentration-dependent manner 8-12 h after treatment (Figure 2H) when the cytotoxic activity of the drug was minimal (Figure 2I).
Superior in vitro anticancer effects of LL6 compared with the pharmacological blockade of IGF-1R, Src, and AXL in combination
We validated the inhibitory effect of LL6 on the activation of IGF-1R, Src, and AXL by Western blot analysis (Figure 3A) and immunofluorescence staining (Figure 3B). Because IGF-1R, Src, and AXL are potential cellular targets of LL6 in NSCLC cells, we assessed whether LL6 has comparable or superior antitumor activities compared with the concurrent blockade of IGF-1R, Src, and AXL by combined treatment with small molecular TKIs against IGF-1R (linsitinib), Src-family kinase (SFK, dasatinib), and AXL (bemcentinib). We first determined the appropriate doses of each drug that suppress their corresponding targets. Linsitinib (2 μM), dasatinib (50 nM), and bemcentinib (100 nM) as monotherapy suppressed phosphorylation of IGF-1R, Src, and AXL, respectively, in H1944 and A549 NSCLC cell lines (Figure S6). LL6 efficiently suppressed activation (phosphorylation) of IGF-1R, Src, and AXL at the concentration of 2.5 μM in the two cell lines (Figure 3A). Because the IGF-1R, Src, and AXL signaling pathways have been implicated in tumor growth and metastasis [22, 23, 54], we compared the effects of single treatment of LL6 and linsitinib (L), dasatinib (D), and bemcentinib (B) combination (LDB) on proliferation and metastatic activities of NSCLC cells in vitro. Compared to the LDB combination, LL6 treatment resulted in significantly greater inhibitory effects on viability (Figure 3C), colony-forming capacity (Figure 3D), and migration (Figure 3E) of two representative H1944 and A549 NSCLC cells.
Improved efficacy of LL6 compared with the concurrent blockade of IGF-1R, Src, and AXL by treatment with a combination of linsitinib, dasatinib, and bemcentinib. (A) Inhibition of the phosphorylation of AXL, IGF-1R, and Src by treatment with LL6 was determined using Western blot analysis. (B) Inhibitory effects of LL6 on the phosphorylation of AXL, Src, and IGF-1R were determined using immunofluorescence analysis. (C-E) Inhibitory effects of LL6 (2.5 μM) on the viability (C), anchorage-dependent colony formation (D), and migration (E) evaluated in comparison with the treatment with a combination of linsitinib (L, 2 µM), dasatinib (D, 50 nM), or bemcentinib (B, 100 nM) (LD or LDB). (F-H) Proapoptotic effects of LL6 (2.5 µM), as determined by induction of PARP cleavage (F), chromatin condensation (G), and accumulation of the cell population in the sub-G1 phase (H), evaluated in comparison with the treatment with a combination of linsitinib (L, 2 µM), dasatinib (D, 50 nM), or bemcentinib (B, 100 nM) (LD or LDB). Bars represent mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001, as determined using the two-tailed Student's t-test compared with the vehicle-treated control (A, B) and one-way ANOVA with Dunette's post-hot test (C-H). Scale bar: 20 µm.
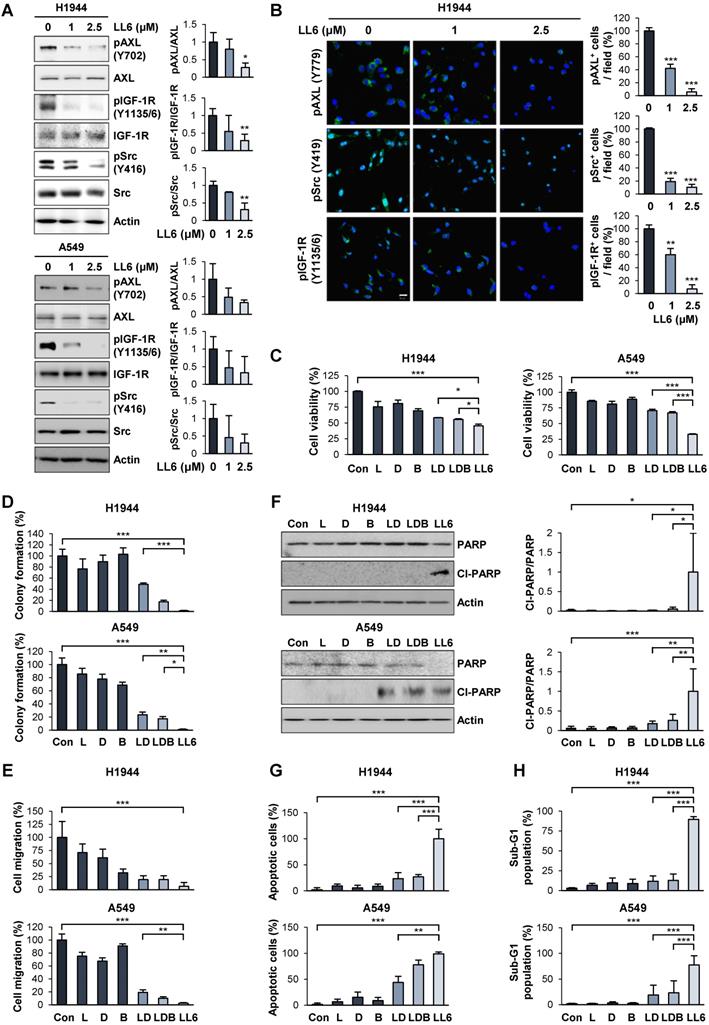
Characteristics of apoptotic cells include membrane blebbing, chromatin condensation, and DNA fragmentation [55, 56]. During apoptosis, activated caspase-3 cleaves the inhibitory proteins of caspase-activated DNases, leading to upregulation of DNase enzymatic activity and subsequent induction of internucleosomal DNA fragmentation [55]. Flow cytometry analysis recognized apoptotic cells with fractional DNA contents as the cells with deficit in DNA contents (sub-G1 phase, sub-G1 peak, or hypodiploid peak) [57]. Poly (ADP-ribose) polymerase (PARP) is another substrate for caspases, and the ability of PARP to repair damaged DNA is inactivated by caspase-mediated cleavage [58]. Previous studies have also shown that DNA fragmentation by caspase-activated DNase and apoptosis-inducing factor induce chromatin condensation [59-61]. Therefore, sub-G1 phase, PARP cleavage, and chromatin condensation can be regarded as caspase-mediated events during apoptosis execution. As sub-G1 phase is a flow cytometric indicator of apoptotic cells and PARP cleavage is an indicator of caspase activation, the level of the cells in the sub-G1 phase is positively correlated with the level of PARP cleavage. The level of the cells in the sub-G1 phase is also believed to be positively correlated with the level of chromatin condensation, a morphological phenotype of apoptotic cells. Therefore, we also determined whether the proapoptotic effects of LL6 might be superior to those of the LDB combination in terms of increases in PARP cleavage, chromatin condensation, and accumulation of cell population in the sub-G1 phase. Compared with the minimal proapoptotic effects of LDB, LL6 notably induced apoptosis, as measured by PARP cleavage (Figure 3F), a number of cells carried condensed chromatin in the nucleus (Figure 3G), and cell population accumulated in the sub-G1 phase of the cell cycle (Figure 3H). These results collectively suggest that the LL6-mediated combined blockade of IGF-1R, Src, and AXL may have greater improved cytotoxic, antimigratory, and proapoptotic efficacies compared with IGF-1R, Src, and AXL blockade by treatment with a combination of linsitinib, dasatinib, and bemcentinib.
Minimal toxicity of LL6 in vitro and in vivo
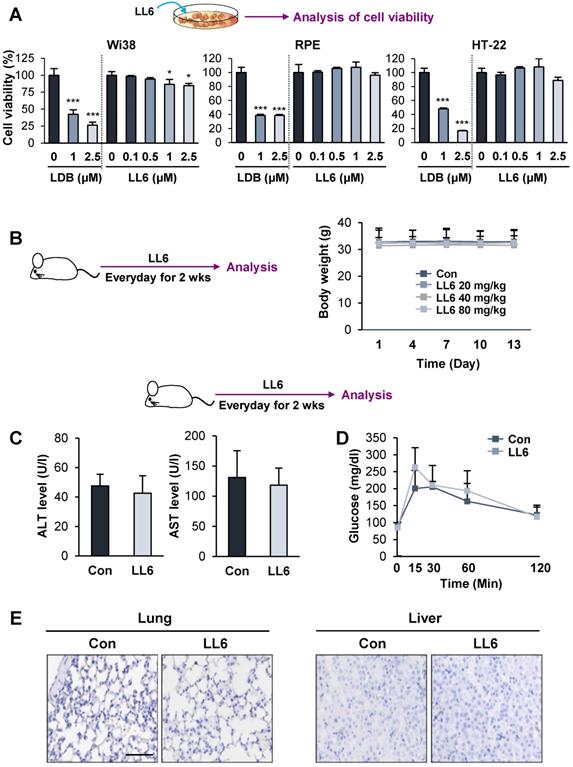
We then examined the toxicity profile of LL6 by comparing the effects of LL6 and LDB combination on the viability of diploid lung fibroblasts (Wi38) and two normal epithelial cells (RPE human retinal pigment epithelial cells and HT-22 mouse hippocampus cells). We observed that LL6 exhibited markedly less cytotoxicity in these normal cells compared with the LDB combination (Figure 4A). We further evaluated the toxicity profiles of LL6 in vivo. To this end, mice were treated with LL6 (20, 40, and 80 mg/kg) daily for 2 weeks, and changes in body weight were monitored. Our pharmacokinetic study suggested poor oral bioavailability of LL6 due to its poor water solubility and rapid elimination in plasma (Table S5 and Figure S4). Previous reports showed that drug administration by oral gavage required more than 10-fold higher dose compared to that by intravenous administration to achieve similar plasma concentration [62-64]. Given the differences in the route of administration (intravenous injection vs oral gavage), the species (rat vs mouse), and poor oral bioavailability of LL6 as predicted by the pharmacokinetic study, we decided the dose of LL6 (80 mg/kg) for oral administration in mice. As shown in Figure 4B, the body weight of mice treated with LL6 up to 80 mg/kg remained unchanged. Compared with the vehicle-treated control mice, the levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) (markers of liver function) in the LL6-treated mice remained unchanged (Figure 4C). The blockade of IR by linsitinib treatment markedly delayed glucose clearance in mice [54]. In line with weak binding affinity of LL6 on IR (Figure 1E), the LL6-administered mice displayed minimal difference in glucose clearance compared to vehicle-treated mice (Figure 4D). We also observed that LL6 treatment did not cause detectable histological alterations in major organs, including the lung and liver (Figure 4E). These results suggested that co-targeting IGF-1R, Src, and AXL by LL6 may have greater antitumor activities with less cytotoxicity compared with that LDB-mediated IGF-1R/Src/AXL blockade. These results indicated minimal toxicity of LL6 in vitro and in vivo.
Minimal toxicity of LL6 in vitro and in vivo. (A) The effects of combined treatment with linsitinib, dasatinib, and bemcentinib (LDB) or treatment with LL6 on the viability of Wi38, RPE, and HT-22 cells were determined using the MTT assay. (B) Body weight changes in mice treated with vehicle or LL6 (20, 40, and 80 mg/kg). (C, D) Changes in ALT and AST levels in the serum (C) and blood glucose (D) of the vehicle- (Con) or LL6-treated mice. (E) Changes in the histological features of lung and liver in the vehicle- or LL6-treated mice were determined using analysis of H&E-stained tissues. Bars represent mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001, as determined using the two-tailed Student's t-test compared with the vehicle-treated control. Scale bar: 50 µm.
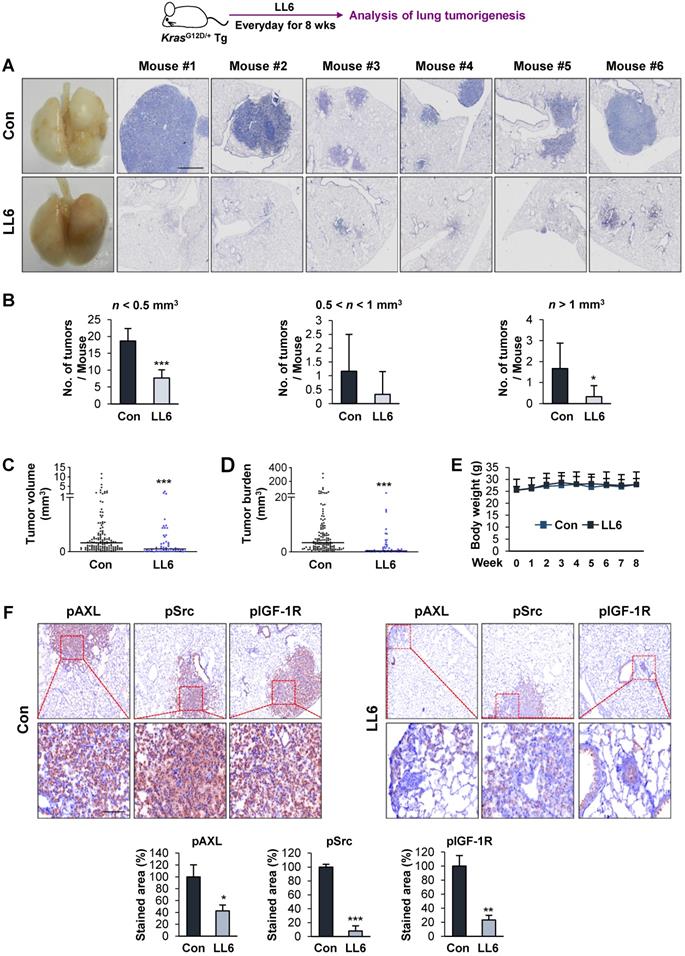
Inhibition of mutant Kras-driven lung tumorigenesis by administration of LL6
We further evaluated the antitumor and antimetastatic effects of LL6 in vivo. To this end, we first explored the effects of LL6 on the growth of lung tumors in transgenic mice carrying mutant Kras (KrasG12D/+) that spontaneously develops lung tumors with a 100% incidence [65] (Figure 5A). Mice were randomly grouped and received LL6 (80 mg/kg) for 8 weeks. Postmortem analysis of the mice showed that LL6-treated mice had significantly decreased lung tumor growth (Figure 5A). Microscopic analysis of hematoxylin and eosin (H&E)-stained lung tissues further showed that the LL6-administered mice had significantly decreased lung tumor nodules, especially those > 1 mm3, compared with vehicle-treated mice (Figure 5B). Substantial decreases in tumor volume (Figure 5C) and burden (Figure 5D) were also found in LL6-treated mice. These data collectively indicate the inhibitory effect of LL6 on mutant Kras-mediated lung tumorigenesis in mice. Notably, body weight of vehicle- and LL6-treated mice showed no detectable difference (Figure 5E). We also confirmed significantly decreased levels of pIGF-1R, pSrc, and pAXL in lung tumors derived from LL6-treated mice compared with those from vehicle-treated mice (Figure 5F). These results suggested that targeting AXL, Src, and IGF-1R by LL6 may result in a significant suppression in mutant Kras-driven lung tumor growth.
Inhibitory effect of LL6 on KrasG12D/+-driven murine lung tumorigenesis. (A) Representative images showing inhibition of lung tumor formation in the lungs of LL6 (80 mg/kg)-treated mice. Left. Representative photographs of the lungs from vehicle- or LL6-treated mice. Right. Representative photographs of H&E-stained lung sections derived from vehicle- or LL6-treated mice. (B-D) Microscopic analysis of the H&E-stained lung sections for lung tumor multiplicity (B), tumor volume (C), and tumor burden (D). (E) Changes in body weight of vehicle- or LL6-treated mice. (F) The tumoral expression of phosphorylated AXL, Src, and IGF-1R (pAXL, pSrc, and pIGF-1R) in the lungs of vehicle- or LL6-treated mice was analyzed using immunohistochemistry. Bottom. Quantification of the positive cells for each marker per field of view. Bars represent mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001, as determined using the two-tailed Student's t-test compared with the vehicle-treated control. Scale bars: 50 µm.
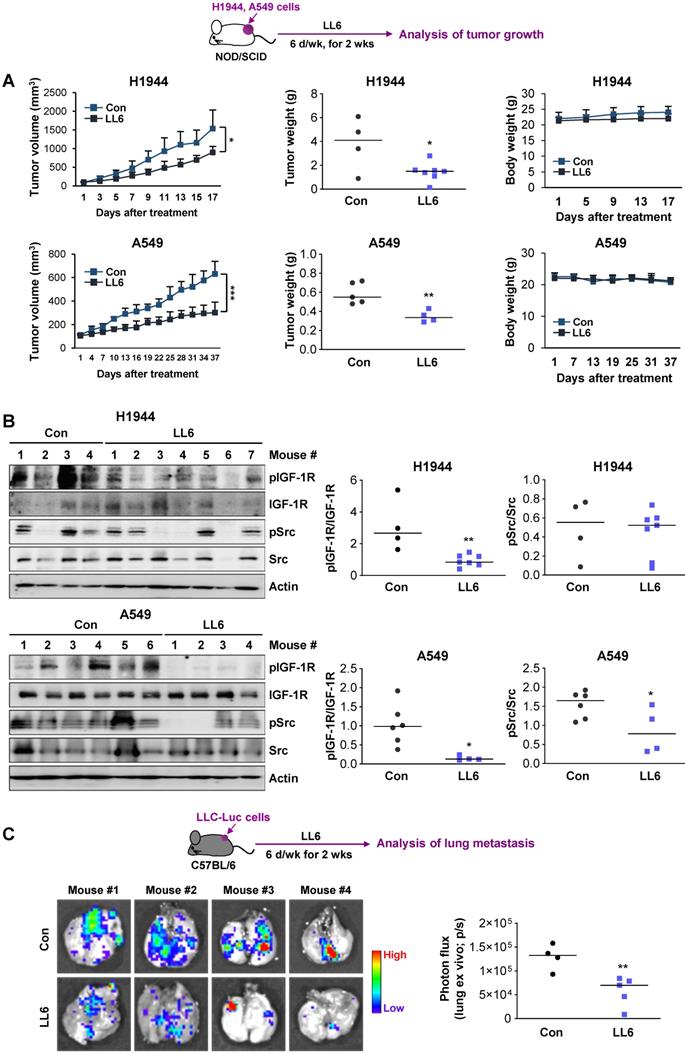
Effects of LL6 on the growth and metastatic activities of NSCLC cells with limited toxicity in vivo
We evaluated the effects of LL6 on the tumor growth using xenograft tumors of human NSCLC cell lines (H1944 and A549). Compared with vehicle-treated mice, the LL6-administered mice showed significantly reduced tumor growth and weight with minimal changes in body weight (Figure 6A). Western blot analysis of the tumors further showed that the expression of phosphorylated IGF-1R and Src was substantially suppressed in tumors derived from the LL6-treated tumors (Figure 6B). LL6 showed quite variable impact on IGF-1R and Src activation in xenograft tumor models, which might be due to the tumor heterogeneity. A subpopulation of slow-cycling cancer cells, characterized by lack of the proliferation marker Ki67 expression and chemoresistance [51, 66, 67], has been found even in rapidly growing tumors and cancer cell lines. Hence, growth rate of subpopulations in the tumors in different mice might have impacted on drug sensitivity. To examine the effects of LL6 on the metastasis of lung cancer cells, C57BL/6 mice bearing subcutaneous LLC allografts [68] were orally treated with vehicle or LL6 daily for 2 weeks (Figure 6C). Compared with control lungs from vehicle-treated mice, lungs from LL6-treated mice showed significantly decreased metastatic tumor nodules (Figure 6C). Taken together, these results suggest the antitumor and antimetastatic activities of LL6 without overt toxicity in vivo.
Inhibition of the growth of xenograft tumors and spontaneous lung metastasis of LLC allografts by administration of LL6 in vivo. (A) Inhibition of tumor growth (left) and tumor weight at endpoint (middle) by treatment with LL6 (80 mg/kg) with negligible changes in body weight (right). (B) Suppression of the activation of IGF-1R and Src in tumors from mice treated with LL6. Right. Quantification of the level of phosphorylated IGF-1R or Src expression versus corresponding total protein expression by densitometric analysis using ImageJ software. (C) Significant reduction in metastatic tumor formation in the lungs after treatment with LL6 (80 mg/kg). Bioluminescence imaging shows a decrease in tumor formation in the lungs of LL6-treated mice. Bars represent mean ± SD. P < 0.05 and P < 0.01, as determined using the two-tailed Student's t-test compared with the vehicle-treated control.
Discussion
Numerous studies have demonstrated the plasticity of cancer cells, which mediate anticancer drug resistance through activation of compensatory pathways once specific pathways are blocked. Hence, a paradigm in drug discovery is shifting from one drug-one target to one drug-multiple target model [69, 70]. Based on previous studies supporting the function of Src and AXL in resistance to IGF-1R inhibitors [12, 14, 26], we attempted to develop efficacious SMKIs targeting IGF-1R, Src, and AXL. Herein, we report a novel multitarget SMKI, LL6, which effectively inactivates these kinases and has potent anticancer activities with outstanding safety profiles both in vitro and in vivo. These results provide preclinical evidence for the use of LL6 as a novel anticancer drug targeting IGF-1R, Src, and AXL without overt toxicity.
Molecularly targeted anticancer therapy has been utilized as the first-line therapy either as a single treatment or in combination with other anticancer therapeutics [11, 71]. However, drug resistance through mutations in cellular targets and activation of bypass signaling pathways hamper their clinical utility [72]. Hence, selection of appropriate targets after full understanding of complex signaling networks in cancer cells and development of SMKIs that simultaneously act on targets involved in bypass signaling pathways have emerged as a novel strategy for anticancer drug discovery [73-75]. IGF-1R signaling has shown crucial roles in the development and progression of several types of cancer and resistance to various anticancer therapies [54]. However, currently available IGF-1R-targeted therapies have shown minimal efficacy in advanced clinical trials owing to drug resistance through activation of bypass signaling mechanisms. Previous studies have shown the implication of Src and AXL signaling pathways in resistance to IGF-1R-targeted anticancer drugs [12, 14, 26]. Src and AXL are frequently overexpressed in various types of human cancers, including NSCLC, and serve as poor prognosis markers in cancer patients [21-23, 76, 77]. As several Src- and AXL-targeting anticancer drugs have been evaluated in clinical studies [22, 23], concurrent inhibition of IGF-1R, Src, and AXL by combinatorial treatment with each SMKI would be an effective therapeutic strategy. However, as with many other anticancer regimens, various toxicities have limited the use of Src and AXL inhibitors in the clinic [22, 23, 78]. Therefore, we attempted to develop potent but safe, multitarget SMKIs that concurrently target IGF-1R, Src, and AXL as a single entity.
In this regard, our observation of the structural similarity between the ATP-binding pocket of the Src kinase and that of AXL kinase and previous studies showing the blockade of AXL activity by an Src inhibitor provided a starting point for the development of such a candidate. In fact, a recent study employed a ligand-based drug design as an approach to develop PP-based focused library to target AXL and FLT3 [31]. Our investigation of the syntheses of multitarget SMKIs using the PP-I2 hybrids offered structural implications for the development of such SMKIs, and further SAR analysis and in silico studies provided one lead candidate, LL6. Our findings suggested the following: 1) linker size and attachment position for conjugating two modules with different orientations played an important role in the design of potent anticancer agents, and 2) substituents on the meta position (R3) of the aminophenol ring of I2 are detrimental to anticancer activity because of steric clashes. The attachment of the linker on the methoxy group in the meta position resulted in loss of potency in 5a-h, indicating that the conformation of the IGF-1R ligand (2,4-bis-arylamino-1,3-pyrimidines, I2) with the U shape, shown in the X-ray structure (pdb: 3QQU) bound to IGF-1R (Figure S3) [79], is retained in LL6. Although the experimentally measured binding affinities of LL6 to the target kinases have low micromolar ranges (< 5 μM), the kinome-wide selectivity of LL6 was observed. Of note, LL6 showed distinct binding affinities for IR and IGF-1R. It was previously observed that the binding pocket of IR TK is wider than that of IGF-1R [80]. Our docking simulations of LL6 using the X-ray structure of IR TK (pdb: 5E1S) suggest that the aminophenol group attached to the triazole linker is buried in the hydrophobic pocket of the IR TK, displaying opposite orientations of the original ligand bound to IGF-1R. Meanwhile, removing all water molecules from the X-ray structure resulted in similar poses. Therefore, potential steric clashes when the PP module is attached to the triazole linker-attached IGF-1R ligand module (I2) might cause the observed abrogated binding affinity of LL6 to IR TK.
Our results from several biological analyses shown in the current study demonstrate that LL6 offers reasonable antitumor effects by inducing apoptosis. Clonogenicity under adherent conditions is associated with survival capacity without cell-to-cell interactions [52]; thus, it is likely that LL6 suppresses disseminated cancer cells. In support of this notion, LL6 exhibited significant inhibitory effects on cancer cell migration. Considering the observed feature of LL6 to suppress IGF-1R, Src, and AXL, LL6 offered the expected results, that is, more efficacious antitumor activities compared with concurrent pharmacologic suppression of IGF-1R, Src, and AXL. Combinatorial therapies can be challenged by various drawbacks, especially enhanced side effects and toxicity. Of note, LL6 minimally affected the viability of normal cells derived from various organs in vitro and displayed no obvious toxicities in vivo, such as diabetes-like symptoms, hepatotoxicity, and histological alterations in major organs. Moreover, LL6 significantly suppressed mutant Kras-driven lung tumorigenesis and growth and metastatic tumor formation in a syngeneic mouse model. Although additional investigations on the efficacy and toxicity profiles of LL6 are required, these results collectively support its clinical utility as an anticancer drug.
Multifunctional therapeutic strategies using multitarget-directed ligands that act simultaneously on various biological targets have emerged as a new principle for drug discovery [73-75], especially for the treatment of cancers with heterogeneous natures. Although monoclonal antibody has a defined specificity, a dual-targeting approach with antibodies requires substantial costs for manufacturing. Moreover, bispecific antibodies cope with issues related to stability and pharmacokinetic properties [81]. Compared with antibody engineering, concurrent targeting of several oncogenic pathways by multitargeting SMKIs would have a number of potential advantages: synergistic antitumor activities, reduced drug resistance, and limited toxicity may lead to a potent efficacy. Indeed, enormous efforts have been dedicated to developing rationally designed multitarget SMKIs since the FDA approval of sunitinib in 2006 [82]. In fact, several multitarget SMKIs have been approved for clinical use [83]. Most FDA-approved SMKIs have been found to interact with more than one target [46].
Therefore, our study presents LL6 as a novel multitarget SMKI that concurrently targets IGF-1R, Src, and AXL with great potency but limited toxicity. Our strategy could be a starting point in understanding polypharmacological networks for the development of drug candidates with improved therapeutic effects. Considering the antitumor efficacy and safety of LL6, LL6 could provide a new design principle to develop novel multitarget SMKI candidates. Our study also provides the rationale to discover novel AXL inhibitors by repositioning the chemical library of potential Src inhibitors. Additional studies are warranted to validate our ligand-based target profiling approach to evaluate the efficacy, pharmacokinetic and pharmacodynamic profiles, and toxicity of LL6 in advanced preclinical and clinical settings.
Abbreviations
ALT: alanine aminotransferase; AST: aspartate aminotransferase; CuAAC: copper(I)-catalyzed alkyne-azide cycloaddition; EGFR: epidermal growth factor receptor; IGF-1R: type I insulin-like growth factor receptor; IR: insulin receptor; NSCLC: non-small cell lung cancer; NOD/SCID: non-obese diabetic-severe combined immunodeficiency; PARP: poly (ADP-ribose) polymerase; RMSD: root-mean-square derivation; ROI: region of interest; SAR: structure-activity relationship; SMKI: small molecule kinase inhibitor; TK: tyrosine kinase; TKI: tyrosine kinase inhibitor.
Supplementary Material
Supplementary figures.
Acknowledgements
This study was supported by the grants from the National Research Foundation of Korea (NRF), the Ministry of Science and ICT (MSIT), Republic of Korea (No. NRF-2016R1A3B1908631 to H.-Y. L. and NRF-2018R1A2B2005535 to J. L.).
Contributions
H.J.L. carried out in vitro and in vivo experiments. P.C.P designed and synthesized the compounds. H.P. and S.Y.H. contributed to in vitro and in vivo experiments. B.L. carried out in silico docking study. B.L., B.B., and Y.K. synthesized the compounds. B.K. carried out the initial in vitro screening. M.H.K., N.W.K., and D.D.K performed or contributed to a pharmacokinetic study. H.Y.M. wrote the initial draft of the manuscript. J.L designed the compounds, conceived and supervised the study, and wrote the manuscript. H.Y.L. designed in vitro and in vivo studies, conceived and supervised the study, and wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394-424
2. Palumbo MO, Kavan P, Miller WH Jr, Panasci L, Assouline S, Johnson N. et al. Systemic cancer therapy: achievements and challenges that lie ahead. Front Pharmacol. 2013;4:57
3. Dobbelstein M, Moll U. Targeting tumour-supportive cellular machineries in anticancer drug development. Nature reviews Drug discovery. 2014;13:179-96
4. Chong CR, Janne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med. 2013;19:1389-400
5. Ma P, Fu Y, Chen M, Jing Y, Wu J, Li K. et al. Adaptive and Acquired Resistance to EGFR Inhibitors Converge on the MAPK Pathway. Theranostics. 2016;6:1232-43
6. Queiroz AL, Vakifahmetoglu-Norberg H, Norberg E. Resistant to Targeted Therapy - Aim for Metabolic Liabilities. Theranostics. 2018;8:2061-3
7. Yuan J, Yin Z, Tao K, Wang G, Gao J. Function of insulin-like growth factor 1 receptor in cancer resistance to chemotherapy. Oncol Lett. 2018;15:41-7
8. Denduluri SK, Idowu O, Wang Z, Liao Z, Yan Z, Mohammed MK. et al. Insulin-like growth factor (IGF) signaling in tumorigenesis and the development of cancer drug resistance. Genes Dis. 2015;2:13-25
9. Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT. et al. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science. 2016;352:aad3018
10. Simpson A, Petnga W, Macaulay VM, Weyer-Czernilofsky U, Bogenrieder T. Insulin-Like Growth Factor (IGF) Pathway Targeting in Cancer: Role of the IGF Axis and Opportunities for Future Combination Studies. Target Oncol. 2017;12:571-97
11. Schrank Z, Chhabra G, Lin L, Iderzorig T, Osude C, Khan N. et al. Current Molecular-Targeted Therapies in NSCLC and Their Mechanism of Resistance. Cancers (Basel). 2018 10
12. Shin DH, Lee HJ, Min HY, Choi SP, Lee MS, Lee JW. et al. Combating resistance to anti-IGFR antibody by targeting the integrin beta3-Src pathway. J Natl Cancer Inst. 2013;105:1558-70
13. Lee JS, Kang JH, Boo HJ, Hwang SJ, Hong S, Lee SC. et al. STAT3-mediated IGF-2 secretion in the tumour microenvironment elicits innate resistance to anti-IGF-1R antibody. Nat Commun. 2015;6:8499
14. Min HY, Yun HJ, Lee JS, Lee HJ, Cho J, Jang HJ. et al. Targeting the insulin-like growth factor receptor and Src signaling network for the treatment of non-small cell lung cancer. Mol Cancer. 2015;14:113
15. Heske CM, Yeung C, Mendoza A, Baumgart JT, Edessa LD, Wan X. et al. The Role of PDGFR-beta Activation in Acquired Resistance to IGF-1R Blockade in Preclinical Models of Rhabdomyosarcoma. Transl Oncol. 2016;9:540-7
16. Wan X, Yeung C, Heske C, Mendoza A, Helman LJ. IGF-1R Inhibition Activates a YES/SFK Bypass Resistance Pathway: Rational Basis for Co-Targeting IGF-1R and Yes/SFK Kinase in Rhabdomyosarcoma. Neoplasia. 2015;17:358-66
17. McLeod HL. Clinically relevant drug-drug interactions in oncology. Br J Clin Pharmacol. 1998;45:539-44
18. Mouzon A, Kerger J, D'Hondt L, Spinewine A. Potential interactions with anticancer agents: a cross-sectional study. Chemotherapy. 2013;59:85-92
19. Dziadziuszko R, Camidge DR, Hirsch FR. The insulin-like growth factor pathway in lung cancer. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer. 2008;3:815-8
20. Peterson JE, Kulik G, Jelinek T, Reuter CW, Shannon JA, Weber MJ. Src phosphorylates the insulin-like growth factor type I receptor on the autophosphorylation sites. Requirement for transformation by src. J Biol Chem. 1996;271:31562-71
21. Irby RB, Yeatman TJ. Role of Src expression and activation in human cancer. Oncogene. 2000;19:5636-42
22. Wheeler DL, Iida M, Dunn EF. The role of Src in solid tumors. Oncologist. 2009;14:667-78
23. Zhu C, Wei Y, Wei X. AXL receptor tyrosine kinase as a promising anti-cancer approach: functions, molecular mechanisms and clinical applications. Mol Cancer. 2019;18:153
24. Taniguchi H, Yamada T, Wang R, Tanimura K, Adachi Y, Nishiyama A. et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat Commun. 2019;10:259
25. Meyer AS, Miller MA, Gertler FB, Lauffenburger DA. The receptor AXL diversifies EGFR signaling and limits the response to EGFR-targeted inhibitors in triple-negative breast cancer cells. Sci Signal. 2013;6:ra66
26. Huang F, Hurlburt W, Greer A, Reeves KA, Hillerman S, Chang H. et al. Differential mechanisms of acquired resistance to insulin-like growth factor-i receptor antibody therapy or to a small-molecule inhibitor, BMS-754807, in a human rhabdomyosarcoma model. Cancer Res. 2010;70:7221-31
27. Wang C, Jin H, Wang N, Fan S, Wang Y, Zhang Y. et al. Gas6/Axl Axis Contributes to Chemoresistance and Metastasis in Breast Cancer through Akt/GSK-3beta/beta-catenin Signaling. Theranostics. 2016;6:1205-19
28. Hutterer M, Knyazev P, Abate A, Reschke M, Maier H, Stefanova N. et al. Axl and growth arrest-specific gene 6 are frequently overexpressed in human gliomas and predict poor prognosis in patients with glioblastoma multiforme. Clin Cancer Res. 2008;14:130-8
29. Ben-Batalla I, Schultze A, Wroblewski M, Erdmann R, Heuser M, Waizenegger JS. et al. Axl, a prognostic and therapeutic target in acute myeloid leukemia mediates paracrine crosstalk of leukemia cells with bone marrow stroma. Blood. 2013;122:2443-52
30. Zhang YX, Knyazev PG, Cheburkin YV, Sharma K, Knyazev YP, Orfi L. et al. AXL is a potential target for therapeutic intervention in breast cancer progression. Cancer Res. 2008;68:1905-15
31. Myers SH, Temps C, Houston DR, Brunton VG, Unciti-Broceta A. Development of Potent Inhibitors of Receptor Tyrosine Kinases by Ligand-Based Drug Design and Target-Biased Phenotypic Screening. J Med Chem. 2018;61:2104-10
32. Liu J, Yang C, Simpson C, Deryckere D, Van Deusen A, Miley MJ. et al. Discovery of Novel Small Molecule Mer Kinase Inhibitors for the Treatment of Pediatric Acute Lymphoblastic Leukemia. ACS Med Chem Lett. 2012;3:129-34
33. Liu J, Zhang W, Stashko MA, Deryckere D, Cummings CT, Hunter D. et al. UNC1062, a new and potent Mer inhibitor. Eur J Med Chem. 2013;65:83-93
34. Dar AC, Das TK, Shokat KM, Cagan RL. Chemical genetic discovery of targets and anti-targets for cancer polypharmacology. Nature. 2012;486:80-U101
35. Brandvold KR, Steffey ME, Fox CC, Soellner MB. Development of a Highly Selective c-Src Kinase Inhibitor. ACS Chemical Biology. 2012;7:1393-8
36. Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ. et al. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271:695-701
37. Schenone S, Radi M, Musumeci F, Brullo C, Botta M. Biologically driven synthesis of pyrazolo[3,4-d]pyrimidines as protein kinase inhibitors: an old scaffold as a new tool for medicinal chemistry and chemical biology studies. Chem Rev. 2014;114:7189-238
38. Fraser C, Dawson JC, Dowling R, Houston DR, Weiss JT, Munro AF. et al. Rapid Discovery and Structure-Activity Relationships of Pyrazolopyrimidines That Potently Suppress Breast Cancer Cell Growth via SRC Kinase Inhibition with Exceptional Selectivity over ABL Kinase. J Med Chem. 2016;59:4697-710
39. Fabian MA, Biggs WH, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG. et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23:329-36
40. Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT. et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127-32
41. Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G. et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046-51
42. Wodicka LM, Ciceri P, Davis MI, Hunt JP, Floyd M, Salerno S. et al. Activation State-Dependent Binding of Small Molecule Kinase Inhibitors: Structural Insights from Biochemistry. Chem Biol. 2010;17:1241-9
43. Laird PW, Zijderveld A, Linders K, Rudnicki MA, Jaenisch R, Berns A. Simplified mammalian DNA isolation procedure. Nucleic Acids Res. 1991;19:4293
44. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164
45. Eid S, Turk S, Volkamer A, Rippmann F, Fulle S. KinMap: a web-based tool for interactive navigation through human kinome data. BMC Bioinformatics. 2017 18
46. Wu P, Nielsen TE, Clausen MH. Small-molecule kinase inhibitors: an analysis of FDA-approved drugs. Drug Discov Today. 2016;21:5-10
47. Hanson SM, Georghiou G, Thakur MK, Miller WT, Rest JS, Chodera JD. et al. What Makes a Kinase Promiscuous for Inhibitors? Cell Chem Biol. 2019;26:390-99
48. Min HY, Jang HJ, Park KH, Hyun SY, Park SJ, Kim JH. et al. The natural compound gracillin exerts potent antitumor activity by targeting mitochondrial complex II. Cell Death Dis. 2019;10:810
49. Liu Q, Yu S, Zhao W, Qin S, Chu Q, Wu K. EGFR-TKIs resistance via EGFR-independent signaling pathways. Mol Cancer. 2018;17:53
50. Belli S, Esposito D, Servetto A, Pesapane A, Formisano L, Bianco R. c-Src and EGFR Inhibition in Molecular Cancer Therapy: What Else Can We Improve? Cancers (Basel). 2020 12
51. Cho J, Lee HJ, Hwang SJ, Min HY, Kang HN, Park AY. et al. The Interplay between Slow-Cycling, Chemoresistant Cancer Cells and Fibroblasts Creates a Proinflammatory Niche for Tumor Progression. Cancer Res. 2020;80:2257-72
52. Munshi A, Hobbs M, Meyn RE. Clonogenic cell survival assay. Methods Mol Med. 2005;110:21-8
53. Levenbook IS, Petricciani JC, Qi Y, Elisberg BL, Rogers JL, Jackson LB. et al. Tumorigenicity testing of primate cell lines in nude mice, muscle organ culture and for colony formation in soft agarose. J Biol Stand. 1985;13:135-41
54. Yee D. A tale of two receptors: insulin and insulin-like growth factor signaling in cancer. Clin Cancer Res. 2015;21:667-9
55. Saraste A, Pulkki K. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc Res. 2000;45:528-37
56. Li J, Yuan J. Caspases in apoptosis and beyond. Oncogene. 2008;27:6194-206
57. Kajstura M, Halicka HD, Pryjma J, Darzynkiewicz Z. Discontinuous fragmentation of nuclear DNA during apoptosis revealed by discrete "sub-G1" peaks on DNA content histograms. Cytometry A. 2007;71:125-31
58. Lyakhovich A, Surralles J. Constitutive activation of caspase-3 and Poly ADP ribose polymerase cleavage in fanconi anemia cells. Mol Cancer Res. 2010;8:46-56
59. Lu Z, Zhang C, Zhai Z. Nucleoplasmin regulates chromatin condensation during apoptosis. Proc Natl Acad Sci U S A. 2005;102:2778-83
60. Liu X, Li P, Widlak P, Zou H, Luo X, Garrard WT. et al. The 40-kDa subunit of DNA fragmentation factor induces DNA fragmentation and chromatin condensation during apoptosis. Proc Natl Acad Sci U S A. 1998;95:8461-6
61. Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM. et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441-6
62. Chen Y, Amantana A, Tyavanagimatt SR, Zima D, Yan XS, Kasi G. et al. Comparison of the safety and pharmacokinetics of ST-246(R) after i.v. infusion or oral administration in mice, rabbits and monkeys. PLoS One. 2011;6:e23237
63. Mus LM, Denecker G, Speleman F, Roman BI. Vehicle development, pharmacokinetics and toxicity of the anti-invasive agent 4-fluoro-3',4',5'-trimethoxychalcone in rodents. PLoS One. 2018;13:e0192548
64. Tung NT, Park CW, Oh TO, Kim JY, Ha JM, Rhee YS. et al. Formulation of solid dispersion of rebamipide evaluated in a rat model for improved bioavailability and efficacy. J Pharm Pharmacol. 2011;63:1539-47
65. Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA. et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111-6
66. Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer. 2007;7:834-46
67. Cho J, Min HY, Pei H, Wei X, Sim JY, Park SH. et al. The ATF6-EGF Pathway Mediates the Awakening of Slow-Cycling Chemoresistant Cells and Tumor Recurrence by Stimulating Tumor Angiogenesis. Cancers (Basel). 2020 12
68. Gomez-Cuadrado L, Tracey N, Ma R, Qian B, Brunton VG. Mouse models of metastasis: progress and prospects. Dis Model Mech. 2017;10:1061-74
69. Medina-Franco JL, Giulianotti MA, Welmaker GS, Houghten RA. Shifting from the single to the multitarget paradigm in drug discovery. Drug Discov Today. 2013;18:495-501
70. Morphy R, Rankovic Z. Designed multiple ligands. An emerging drug discovery paradigm. J Med Chem. 2005;48:6523-43
71. Lee YT, Tan YJ, Oon CE. Molecular targeted therapy: Treating cancer with specificity. Eur J Pharmacol. 2018;834:188-96
72. Keefe DMK, Bateman EH. Potential Successes and Challenges of Targeted Cancer Therapies. J Natl Cancer Inst Monogr. 2019. 2019
73. Silva MD, Dias KST, Gontijo VS, Ortiz CJC, Viegas C. Multi-Target Directed Drugs as a Modern Approach for Drug Design Towards Alzheimer's Disease: An Update. Curr Med Chem. 2018;25:3491-525
74. Bansal Y, Silakari O. Multifunctional compounds: smart molecules for multifactorial diseases. Eur J Med Chem. 2014;76:31-42
75. Costantino L, Barlocco D. Designed Multiple Ligands: Basic Research vs Clinical Outcomes. Curr Med Chem. 2012;19:3353-87
76. Giaccone G, Zucali PA. Src as a potential therapeutic target in non-small-cell lung cancer. Ann Oncol. 2008;19:1219-23
77. Wu X, Ma W, Zhou Q, Yan H, Lim ZF, Huang M. et al. AXL-GAS6 expression can predict for adverse prognosis in non-small cell lung cancer with brain metastases. J Cancer Res Clin Oncol. 2017;143:1947-57
78. Kopetz S, Shah AN, Gallick GE. Src continues aging: current and future clinical directions. Clin Cancer Res. 2007;13:7232-6
79. Buchanan JL, Newcomb JR, Carney DP, Chaffee SC, Chai L, Cupples R. et al. Discovery of 2,4-bis-arylamino-1,3-pyrimidines as insulin-like growth factor-1 receptor (IGF-1R) inhibitors. Bioorg Med Chem Lett. 2011;21:2394-9
80. Sanderson MP, Apgar J, Garin-Chesa P, Hofmann MH, Kessler D, Quant J. et al. BI 885578, a Novel IGF1R/INSR Tyrosine Kinase Inhibitor with Pharmacokinetic Properties That Dissociate Antitumor Efficacy and Perturbation of Glucose Homeostasis. Mol Cancer Ther. 2015;14:2762-72
81. Kontermann RE. Dual targeting strategies with bispecific antibodies. MAbs. 2012;4:182-97
82. Chen Z, Han L, Xu MH, Xu YF, Qian XH. Rationally Designed Multitarget Anticancer Agents. Curr Med Chem. 2013;20:1694-714
83. Broekman F, Giovannetti E, Peters GJ. Tyrosine kinase inhibitors: Multi-targeted or single-targeted? World J Clin Oncol. 2011;2:80-93
Author contact
![]() Corresponding authors: Ho-Young Lee, Ph.D., Creative Research Initiative Center for concurrent control of emphysema and lung cancer, College of Pharmacy, Seoul National University, Seoul 08826, Republic of Korea. Phone: +82-2-880-9277; Fax: +82-2-6280-5327; E-mail: hylee135ac.kr; Jeeyeon Lee, Ph.D., College of Pharmacy and Research Institute of Pharmaceutical Sciences, Seoul National University, Seoul 08826, Republic of Korea; Tel: +82-2-880-2471; Fax: +82-2-884-8334; E-mail: jyleeutac.kr.
Corresponding authors: Ho-Young Lee, Ph.D., Creative Research Initiative Center for concurrent control of emphysema and lung cancer, College of Pharmacy, Seoul National University, Seoul 08826, Republic of Korea. Phone: +82-2-880-9277; Fax: +82-2-6280-5327; E-mail: hylee135ac.kr; Jeeyeon Lee, Ph.D., College of Pharmacy and Research Institute of Pharmaceutical Sciences, Seoul National University, Seoul 08826, Republic of Korea; Tel: +82-2-880-2471; Fax: +82-2-884-8334; E-mail: jyleeutac.kr.