Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2021; 11(5):2080-2097. doi:10.7150/thno.50701 This issue Cite
Review
PDE inhibition in distinct cell types to reclaim the balance of synaptic plasticity
Ben Rombaut1,2, Sofie Kessels1, Melissa Schepers1,2, Assia Tiane1,2, Dean Paes1,2, Yevgeniya Solomina2,3, Elisabeth Piccart1,2, Daniel van den Hove2,4, Bert Brône1, Jos Prickaerts2, Tim Vanmierlo1,2 ![]()
1. Department of Neurosciences, European Graduate School of Neuroscience, Biomedical Research Institute, UHasselt, Hasselt University, Hasselt, Belgium.
2. Department Psychiatry and Neuropsychology, European Graduate School of Neuroscience, School for Mental Health and Neuroscience, Maastricht University, Maastricht, Netherlands.
3. Tomsk State University, Tomsk, Russia.
4. Department of Psychiatry, Psychosomatics, and Psychotherapy, University of Wuerzburg, Wuerzburg 97080, Germany.
Received 2020-7-15; Accepted 2020-11-17; Published 2021-1-1
Abstract

Synapses are the functional units of the brain. They form specific contact points that drive neuronal communication and are highly plastic in their strength, density, and shape. A carefully orchestrated balance between synaptogenesis and synaptic pruning, i.e., the elimination of weak or redundant synapses, ensures adequate synaptic density. An imbalance between these two processes lies at the basis of multiple neuropathologies. Recent evidence has highlighted the importance of glia-neuron interactions in the synaptic unit, emphasized by glial phagocytosis of synapses and local excretion of inflammatory mediators. These findings warrant a closer look into the molecular basis of cell-signaling pathways in the different brain cells that are related to synaptic plasticity. In neurons, intracellular second messengers, such as cyclic guanosine or adenosine monophosphate (cGMP and cAMP, respectively), are known mediators of synaptic homeostasis and plasticity. Increased levels of these second messengers in glial cells slow down inflammation and neurodegenerative processes. These multi-faceted effects provide the opportunity to counteract excessive synapse loss by targeting cGMP and cAMP pathways in multiple cell types. Phosphodiesterases (PDEs) are specialized degraders of these second messengers, rendering them attractive targets to combat the detrimental effects of neurological disorders. Cellular and subcellular compartmentalization of the specific isoforms of PDEs leads to divergent downstream effects for these enzymes in the various central nervous system resident cell types. This review provides a detailed overview on the role of PDEs and their inhibition in the context of glia-neuron interactions in different neuropathologies characterized by synapse loss. In doing so, it provides a framework to support future research towards finding combinational therapy for specific neuropathologies.
Keywords: neurodegeneration, synapses, phosphodiesterase, cell-signaling, glia-neuron
Introduction
The central nervous system (CNS) consists of different cell types with dedicated functions, i.e. neurons and glial cells, which together create and maintain the brain's circuitry. Synapses are contact points between neurons that allow them to communicate with each other and, hence, are vital to proper brain function. The amount of synapses is strictly and dynamically regulated throughout the human lifespan by constant turnover of synapses. Regular formation of synapses is offset by the elimination of synapses that are too weak or in excess [1]. In the aging or degenerating brain, this balance shifts towards loss of synapses, leading to a decline in neurological function. Synapse elimination can be influenced by physiological pathways within the neuron itself or by active removal of synapses by surrounding glial cells [2].
Synapse formation and elimination rely heavily on second messengers as effectors [3-5]. Among these second messengers, 3'5'-cyclic adenosine monophosphate (cAMP) and 3'5'-cyclic guanosine monophosphate (cGMP) show widespread expression, making them interesting targets in CNS resident cells. Compartmentalization of сAMP and cGMP is mainly determined by localization of macromolecular complexes formed by A-kinase anchor proteins (AKAPs) and G-kinase anchor proteins (GKAPs), respectively [6-8]. These anchoring proteins play an essential role by directing localization of cAMP and cGMP-dependent protein kinases (i.e. protein kinase A [PKA] and PKG, respectively) close to their substrates (cAMP and cGMP), allowing an adequate physiological response of the cell to a signal. Despite the signaling contribution of cGMP to neuroplasticity, GKAP/PKG signalosomes are not well characterized yet. This review will focus on AKAP/cAMP complexes. Extensive research has demonstrated that elevating cAMP and cGMP levels can drive the CNS towards a neuroprotective environment to prevent damage [9-17]. Achieving this elevation is possible through the anabolic pathway via guanylyl cyclase (GC) or adenylyl cyclase (AC), or by blocking phosphodiesterases (PDEs), which hydrolyze and break down cGMP and cAMP.
The physiology of PDEs carries a well-established diversity: 21 genes have been identified, which code for proteins that differ vastly in their regulatory properties and spatiotemporal organization. The 21 genes of the PDE superfamily can be distinguished by their catalytic unit and are grouped in 11 different families (PDE1-PDE11) based on protein structure and regulatory domains. These protein structures affect substrate selectivity (cGMP or cAMP, as shown in Table 1) and interactions with mediators of phosphorylation. The different genes (e.g., PDE4A-D) are unique in terms of their genomic organization, including the use of multiple promoters. Divergent isoforms (e.g., PDE4D1-9) exist owing to the existence of multiple promoters and the influence of alternative splicing. Isoforms of PDE4, for instance, can be classified into long, short, or supershort, based on the presence of upstream conserved regions (UCR's). Long isoforms have two of these UCR's, short isoforms have one, and the supershort has a truncated version of one. The presence of different PDE transcripts and their association with anchoring proteins in distinct pathways of cell signaling throughout the CNS leads to the widespread effects of PDE-mediated signaling pathways. These biochemical differences have also comprehensively been elucidated in research studies using C. elegans [18, 19]. Additionally, these studies have facilitated progress towards specialized drug targeting to influence specific pathways in neurobiology [20-24].
Summarizing different PDE genes with their substrate specificity and relative expression in different murine or human CNS cell types
| Gene family | Gene | Substrate | Cellular expression (murine) | Cellular expression (human) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Oligodendrocytes | Microglia/macrophages | Astrocytes | Neurons | Oligodendrocytes | Microglia/macrophages | Astrocytes | Neurons | |||
| PDE1 | PDE1A | Dual | -* | - | - | +++ | +++ | - | - | +++ |
| PDE1B | -* | + | +++* | +++ | - | + | - | +++ | ||
| PDE1C | +* | - | + | + | +++ | - | + | + | ||
| PDE2 | PDE2A | Dual | +* | +* | - | +++* | - | - | - | + |
| PDE3 | PDE3A | Dual | - | - | +++ | +* | - | - | +++ | +++ |
| PDE4 | PDE4A | cAMP | +* | +* | +* | +++* | - | - | - | + |
| PDE4B | +* | -* | +++* | -* | +++ | +++ | + | + | ||
| PDE4C | - | - | - | - | - | - | - | - | ||
| PDE4D | - | - | +++ | +++ | ||||||
| PDE5 | PDE5A | cGMP | - | - | - | - | ||||
| PDE6 | PDE6A | cGMP | - | - | - | - | - | - | - | - |
| PDE6B | -* | - | - | - | + | - | - | - | ||
| PDE6C | - | - | - | - | - | - | - | - | ||
| PDE7 | PDE7A | cAMP | +++* | -* | +++ | +++* | + | + | +++ | + |
| PDE7B | -* | - | + | -* | + | - | + | +++ | ||
| PDE8 | PDE8A | cAMP | +++ | -* | - | -* | +++ | + | - | + |
| PDE8B | +++* | +* | + | +++ | + | - | + | +++ | ||
| PDE9 | PDE9A | cGMP | +* | -* | +++* | +* | - | + | + | - |
| PDE10 | PDE10A | Dual | + | - | + | +++ | + | - | + | +++ |
| PDE11 | PDE11A | Dual | -* | - | - | - | + | - | - | - |
Expression per cell types is based on [214]. Differences in expression between murine and human cell types pose a challenge in the context of translational capacity of PDE inhibitors. *Murine expression different from human.
Synapses can differ according to the neurons with which they are made up. Among chemical synapses, two types can be distinguished, i.e. excitatory and inhibitory, of which the most prevalent in the mammalian brain are glutamatergic and GABAergic (relying on gamma-aminobutyric acid) synapses. Glutamatergic synapses rely on both metabotropic (mGluRs) and ionotropic glutamate receptors, such as kainite receptors, N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, of which the latter is responsible for baseline neurotransmission. Importantly, the synapse type dictates its structure. Inhibitory synapses (e.g. GABAergic) are mostly formed directly on dendritic shafts. Excitatory synapses (e.g. glutamatergic) are usually formed by using dendritic spines, which are small membrane protrusions from dendritic shafts, creating a postsynaptic partner for excitatory neurons [2, 25]. The importance of the two types of synapses is readily apparent in the complex interplay of neuronal excitation and inhibition, which dynamically regulates the neuronal circuitry. The tight coordination of these different inputs ensures a constant excitation/inhibition (E/I) ratio. This ratio has previously been described to be of crucial importance for efficient coding of information, while on the single-neuron level, it may safeguard efficient signal transduction and survival [26]. A plethora of neuropathologies, including autism, epilepsy, schizophrenia, and Alzheimer's disease (AD), have been linked to a disrupted E/I ratio [25, 27].
Apart from plasticity that is inherent to neuronal functioning, the effect of interaction of synapses with other, external, factors should not be overlooked. Glial cells, responsible for homeostatic balance and support of the neuronal circuit, play a major role in plasticity. Apart from indirectly changing the micro-environment, glial cells have emerged as direct regulators of synapse maintenance and elimination. Endfeet of astrocytes converge on neuronal contact sites to create the tripartite synapse [2]. On top of that, microglia interact dynamically with this tripartite synapse using their highly motile processes, leading to the emergence of the term 'quad-partite synapse' [28]. Both glial cell types contribute to synapse homeostasis by interacting with synapses directly, as well as indirectly by the release of soluble factors. The interplay between neurons, astrocytes, oligodendrocytes, and microglia involves a broad range of soluble factors, all contributing to the CNS environment and the dynamics of synapse formation or elimination. Moreover, mounting evidence describing crosstalk between microglia and astrocytes further highlights the importance of glial cells in synapse maintenance and plasticity [2, 29, 30].
The importance of glial cells in synaptic elimination has already been established [31]. Although the formation of synapses and strength of these connections is mainly attributable to neurons, the elimination of synapses that are too weak or in excess requires the involvement of glia. Mounting evidence underlines the importance of microglia [32, 33] and astrocytes [34] in synaptic elimination, highlighting neuron-glia signaling pathways crucial for synapse physiology. Astrocytes and microglia mediate this elimination by direct phagocytosis of synapses, as explained in detail below. Apart from clearing dead cells and debris, these glial cells thus actively participate in eliminating synapses [35]. As many neurodegenerative disorders include an inflammatory environment, the effects of inflammation on synaptic plasticity are also evaluated below. Based on the intricate cellular collaboration at the basis of synaptic plasticity, and the prominent role of PDE signaling therein [36, 37], the aim of our review is to highlight the present understanding of the beneficial effects of PDE inhibition. Additionally, deeper insights into PDE biology and the importance of isoforms were provided, emphasizing their potential as targets for personalized medicine in different neuropathologies that feature synapse loss. The present review summarizes the known effects of PDE inhibition on cellular mechanisms contributing to the elimination of synapses in the normal and pathological brain, throughout development and during aging (shown in Table 2 and Figure 1) [38].
Overview of selective PDE inhibition per cell type
| Cell type | Inhibitor | Target(s) | Specificity | Effect | In vitro/in vivo |
|---|---|---|---|---|---|
| Neuron | vinpocetine | PDE1 | dual | minimizes neuronal damage by oxidative stress [67] | In vivo |
| Bay 60-7550 | PDE2 | dual | increases hippocampal LTP, neuronal plasticity, and BDNF levels [56, 215, 216] | In vitro + In vivo | |
| Cilostazol | PDE3 | dual | stimulates CREB-mediated proliferation in the hippocampus [58] | In vivo | |
| Rolipram | PDE4 | cAMP | reduces neurotoxicity induced by amyloid β [12] | In vivo | |
| Roflumilast/BPN14770 Knockout/RNAi | increases CREB-mediated transcription and BDNF signaling [217-219] | In vitro + In vivo | |||
| Rolipram | enhances neuronal resilience and restorative capacity [70, 73, 75, 220-223] | In vitro + In vivo | |||
| ABI-4 | PDE4A-C | cAMP | diminishes the effect of LPS-induced neuronal inflammatory responses [13] | In vitro | |
| Transgenic model | PDE4B1 | cAMP | increased hippocampal CREB phosphorylation and LTP [65] | In vivo | |
| Ferulic acid | PDE4B2 | cAMP | downregulated Aβ-induced TNFα and IL-1β levels [224] | In vitro | |
| Sildenafil/vardenafil | PDE5 | cGMP | enhanced neuronal survival through BDNF and CREB signaling and increased membrane-bound AMPA receptors [4, 60] | In vivo | |
| BRL 50481/S14 | PDE7 | cAMP | displayed anti-inflammatory and neuroproductive effects [77, 78] | In vitro + In vivo | |
| BAY 73-6691 | PDE9 | cGMP | counteracts oxidative stress and reduced plasticity [61, 68] | In vitro + In vivo | |
| Transgenic model/TP10 | PDE10 | dual | neuroprotective effects in Huntington's model [222, 225] | In vivo | |
| Astrocyte | sildenafil | PDE5 | cGMP | prevents LPS-induced inflammation [176] | In vivo |
| ibudilast | PDE4/10 (+3/11) | cGMP | reduces astrogliosis during neuroinflammation and prevents astrocytic apoptosis [15, 194, 195] | In vitro + In vivo | |
| S14 | PDE7 | cAMP | stimulates Aβ degradation in a murine model for AD [79] | In vivo | |
| Oligodendrocyte | sildenafil | PDE5 | cGMP | improves BDNF signaling [100] | In vivo |
| rolipram | PDE4 | cAMP | improves OPC differentiation [208, 209] | In vitro + In vivo | |
| Microglia | amrinone | PDE3 | dual | skewing to an anti-inflammatory phenotype with decreased production of TNF-α, IL-1β, IL-12, and NO [94, 95] | In vitro |
| rolipram | PDE4 | cAMP | |||
| sildenafil | PDE5 | cGMP | |||
| upregulation of YM-1 [100] | In vivo | ||||
| pentoxifylline | Non-selective | dual | decrease LPS-dependent increase in TNF-α [97] | In vitro | |
| rolipram | PDE4 | cAMP | precognitive, neuroprotective and anti-inflammatory effects [98, 99] | In vitro + In vivo | |
| PDE4D-NAM | PDE4D | cAMP | |||
| ibudilast | PDE4/10 (+3/11) | multiple | antagonizing Toll-like receptor 4 [101] | In vivo | |
| cilostamide/amrinone | PDE3 | dual | regulate morphology and sensing mechanism (filopodia) of microglia [112] | In vitro | |
| IBMX | Non-selectuve | dual | |||
| inhibition of microglial phagocytosis [114, 141] | In vitro | ||||
| rolipram | PDE4 | cAMP | |||
| cilostamide | PDE3 | dual | |||
| Macrophage | shRNA | PDE1B2 | dual | increased phagocytic ability and augmented cell spreading [167] | In vitro |
| rolipram | PDE4 | cAMP | increased migratory capacity upon LPS challenge [154] | In vitro |
Type of inhibitor, the effect on the cell type in question, and modality of effects witnessed in the referred article is shown.
Overview of the proven effective PDE inhibition on different cell types involved in synapse maintenance or loss in the physiological state (A) or neurodegeneration (B). Resident neuroglial cells have dual roles in the physiological state (A), highlighted by the presence of pro-(red) inflammatory and anti-(green) inflammatory cytokines. The positive effects on individual synapses are shown above the dotted line, while detrimental effects are shown below. In neurodegenerative pathologies (B), in which disruption of the blood-brain-barrier is common, the resident neuroglia become activated, and patrolling macrophages (pale green) extravasate into the central nervous system and intermingle with microglia (bright blue). Local upregulation of complement proteins (C1q and C3b) leads to targeted clearance of synapses by microglia and astrocytes (bright green). Inhibition of various PDEs (PDEXi) has been found to influence synaptic plasticity directly in neurons and indirectly via glial cells by skewing them toward an anti-inflammatory phenotype and promoting transcription factors to express for instance Brain-Derived Neurotrophic Factor (BDNF) from oligodendrocytes (purple). Images were modified from Reactome icon library and Servier Medical Art (http://smart.servier.com/), licensed under a Creative Common Attribution 3.0 Generic License [226].
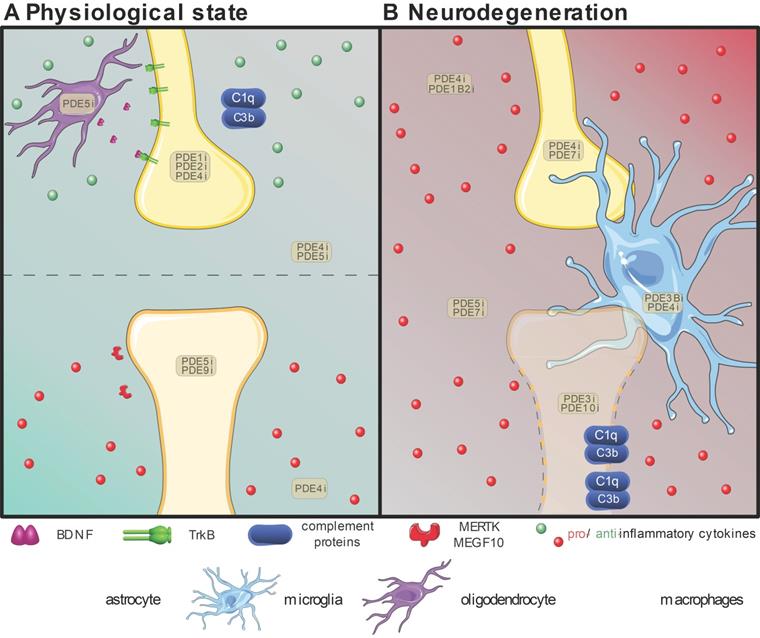
Modulating the synaptic unit at the core of synaptic plasticity in health and disease
Loss of spines and synapse elimination is critical both in the primary and secondary phase of human brain circuit formation, i.e. from early development up to childhood and during adolescence. Synaptic elimination during development is well-documented, as observed in the developing neuromuscular junction in rodents. Aside from the neuromuscular junction, experiments have highlighted that synapse elimination also occurs in sensory and executive circuits of the brain. As such, synaptic elimination represents a key developmental process in the cerebellum, cortex, thalamus, and the hippocampus [30, 39, 40].
As synapse elimination occurs in different stages of life, an important distinction has to be made between normal and pathological aging. Medical advances and public health efforts have led to a substantial increase in the percentage of older adults in the general population. Aging remains an underlying risk factor for many neurodegenerative diseases and is often accompanied by notably deteriorating cognitive capacities [41]. Physiological aging features neuroanatomical changes independent of pathological processes, such as a decrease in grey matter volume. This grey matter loss is not attributable to neuronal loss but rather the result of a gradual decline of dendritic arborization and synapse density. The main regions affected are the prefrontal cortex, medial temporal lobe, and hippocampus, associating with cognitive decline [42, 43]. In addition, a decline in energy utilization coincides with physiological neurodegeneration [44]. Loss of energy is attributable to the aging brain's hypometabolism, caused by hypoperfusion and loss of blood-brain barrier (BBB) integrity [45]. Notably, excitatory synapses have relatively high rates of energy consumption, rendering them vulnerable to diminished production of adenosine triphosphate (ATP) by neurons [46]. Apart from supporting neurons in maintaining baseline synaptic plasticity, glial cells also affect synaptic spine density. In these specialized signaling cascades, PDEs represent interesting players that control cellular and subcellular cAMP pools.
An important challenge of pharmacological inhibition of PDEs is the ability to translate preclinical work to therapeutics in humans. As illustrated below to a greater extent, the proven beneficial effects of PDE inhibition have been established in in vitro experiments or in in vivo animal model studies. However, to date, few PDE inhibitors have bridged the translational gap to become an approved therapy in the area of neurological disorders. Alzheimer's research, specifically, has looked into the potential of PDE inhibitors quite extensively [47, 48]. The effect of PDE inhibition on different CNS cell types is discussed below and summarized in table 2. These studies largely reach the same conclusion: blocking of a PDE protein derived from a gene does not result in the desired effects. For added value without unwanted side effects, the role of the different isoforms needs to be better understood. In addition, an in-depth analysis of the importance of the distinct isoforms in the different cell types involved in neurodegeneration is needed. This review aims to highlight and summarize decades of PDE inhibition research to argue for the development of isoform-specific PDE inhibitors.
Neurons - at the core of synaptic plasticity
The brain circuitry is known to undergo many neuronal changes throughout the human lifespan [46, 49]. On the micro-level, alterations include synaptic plasticity [50]. Neural activity alters the morphology and associated function of synapses [51]. The creation, modification, maintenance, or elimination of synapses results in the ever-changing brain environment to facilitate signal propagation and storage of information. Electrophysiological properties, mainly long-term potentiation (LTP) and long-term depression (LTD), are at the basis of these alterations [51]. Despite producing functionally opposite effects on synapse physiology, under physiological conditions, both LTP and LTD promote brain plasticity by changing synaptic strength. They can take place at the same synapse through subtle differences in NMDA receptor signaling. On the one hand, intense or persistent NMDA receptor activation can ultimately increase the presence of postsynaptic AMPA receptors, facilitating LTP. LTD, on the other hand, is induced by no or weak NMDA receptor activation, among other ways. The occurrence of LTD in the adult brain reduces synaptic strength, leading to the removal of postsynaptic AMPA receptors and, ultimately, when persistent, loss of spines [25, 30, 52].
Synapses are subject to activity-dependent competition. Active synaptic connections are retained, while poorly used synapses are prone to elimination. Furthermore, coincident activity of pre- and postsynaptic compartments leads to synapse strengthening. This dynamic nature of synapse strengthening or elimination enables a high degree of plasticity, which is crucial in learning and memory processes. Accordingly, synapse elimination would promote plasticity as long as it is appropriately targeted to rather inactive synapses. In neurodegenerative diseases, a decline in plasticity can be observed due to deficient strengthening and excessive elimination of synapses, even for adequately used synapses. Importantly, the increased inflammatory state in the aging and neurodegenerative brain, mediated by overactive astrocytes, microglia, and macrophages, poses a challenge for the maintenance and strengthening of these synapses. As such, excessive and non-specific synapse loss in neurodegeneration can be limited by promoting synaptic strengthening and dampening inflammatory signaling.
Activity-dependent synapse strengthening is tightly regulated by and dependent on cyclic nucleotide signaling cascades. Upon receptor activation on the postsynaptic neuron, calcium influx and G-protein coupled receptor stimulation promotes the synthesis of cGMP and cAMP. The second messengers cGMP and cAMP can then, either directly or via downstream effectors, modulate both transient and prolonged plasticity changes [5]. Transient plasticity changes consist of, for example, phosphorylation of AMPA receptors, inducing trafficking into the postsynaptic membrane [4]. Retrograde nitric oxide (NO) signaling enhances cGMP signaling presynaptically, a phenomenon that can promote presynaptic neurotransmitter release. More sustained plasticity effects involve mRNA translation and de novo protein synthesis (e.g., via cAMP response element-binding protein (CREB)-mediated transcription [53]) that produce more reliable postsynaptic densities (e.g., by increases in scaffolding proteins such as PSD95 [50]), synthesis of more receptive synapses (e.g., owing to more membrane-bound AMPA receptors [4]), or increased paracrine and autocrine signaling to stimulate neuronal survival (e.g., via brain-derived neurotrophic factor (BDNF) [54]). Pharmacological PDE2 inhibition increased hippocampal LTP, neuronal plasticity, and BDNF levels both in healthy animals and in disease models [55-57]. PDE3 inhibition showed restorative effects upon neuronal loss in the hippocampus through stimulation of CREB-mediated proliferation [58], while neurons were also protected by PDE10 inhibition in the context of Huntington's disease [59]. Modulation of neuronal cGMP specifically can be achieved by inhibition of mainly PDE5 and PDE9. Enhanced neuronal survival through BDNF and CREB signaling and increased membrane-bound AMPA receptors was observed upon PDE5 inhibition in Alzheimer's models [4, 60, 61]. Furthermore, inhibiting PDE4 and its subtypes and isoforms specifically increased CREB-mediated transcription and BDNF signaling to enhance neuroplasticity [62-64]. Additionally, it has been found that ablation of the activity of the PDE4B isoform, PDE4B1, increased hippocampal CREB phosphorylation, and LTP [65].
In addition to the modulation of neuroplasticity effects, cyclic nucleotides also affect neuroinflammatory processes [14, 66]. Through compartmentalization of signaling cascades, generic cAMP and cGMP can selectively influence different processes. This compartmentalization is partly established by the wide variety of PDE enzymes, enabling inhibition of certain PDEs to control inflammatory responses in neurons selectively. As cAMP and cGMP are exclusively degraded by these compartmentalized PDEs, PDEs are attractive pharmacological targets to, on the one hand, enhance neuronal plasticity and, on the other hand, reduce inflammatory responses. Moreover, in aging and neurodegenerative diseases, baseline cyclic nucleotide signaling is found to be altered, as extensively reviewed by Kelly et al. [3]. Accordingly, inhibition of several PDE families has proven useful to enhance both transient and prolonged plasticity and dampen inflammatory responses upon insults. Levels of both cAMP and cGMP can be elevated simultaneously via inhibition of dual-specific PDEs, leading to proven beneficial effects. For example, neuronal damage by oxidative stress was minimized by the PDE1 inhibitor vinpocetine [67]. In addition to PDE9 inhibition counteracting the oxidative stress and reduced plasticity in Alzheimer's disease [68], cAMP-elevating PDE4 inhibition was found to reduce neurotoxicity induced by amyloid-β [12]. Similarly, the effect of lipopolysacharide (LPS)-induced neuronal inflammatory responses on neurons was diminished by the PDE4D-sparing inhibitor ABI-4 [13]. PDE4 inhibition also enhanced neuronal resilience and restorative capacity in different disease models [69-75]. More specifically, PDE4B2 seems to be involved in mediating neuronal inflammation, as its inhibition decreased Aβ-induced TNFα and IL-1β levels [76]. Given the dual therapeutic action (i.e., enhancing plasticity and reducing inflammation) of PDE4 inhibition, this therapeutic strategy has been investigated in a variety of neurodegenerative disorders to protect neurons and their functional synapses [16]. Despite the therapeutic potential of PDE4 inhibitors, clinical use has been plagued by the occurrence of adverse side effects (e.g., headaches, diarrhea, and nausea) [77]. Consequently, more selective inhibition of PDE4 subtypes or isoforms is warranted. Alternatively, cAMP signaling can be elevated by other cAMP-selective PDEs to circumvent PDE4-mediated side effects; PDE7 inhibition also displayed anti-inflammatory and neuroprotective effects [78, 79]. Although PDE8 is expressed in the brain, and several PDE8 inhibitors do exist, its role in neuronal plasticity has not yet been investigated [80, 81].
cAMP compartmentalization plays a vital role in synaptic plasticity. The AKAP5-PDE4D5 complex, which is attached to the postsynaptic membrane, regulates cAMP signaling and plays a crucial role in learning and memory by facilitating activation of AMPA receptors in response to a signal [5]. In vitro and in vivo studies demonstrated that LRRK2 is responsible for the localization of cAMP/PKA signaling in dendritic spines and negatively regulates cAMP/PKA signaling during synaptogenesis. In particular, LRRK2 knock-out mice exhibit increased PKA phosphorylation of AMPA receptors, leading to abnormal dendrite morphology. Thus, LRRK2/cAMP complexes play a dual role, protecting neuronal cells from inflammation and balancing intracellular signaling for the healthy development of dendrites during synaptogenesis. Furthermore, research has shown that LRRK2 is associated with Parkinson's disease [82]. Yotiao, a specific splice variant of AKAP9, is found in neurons and neuromuscular junctions. This protein binds PKA and NMDA receptors, localizing cAMP signaling close to the receptor and modulating calcium permeability of the receptor [83]. Although Yotiao has not been shown to bind to PDEs, its longer form, AKAP9, anchors PDE4D3, restricting cAMP signaling in close vicinity to the centrosome, and participates in microtubule dynamics [84]. Two AKAP9 variants have been associated with AD; however, their contribution to neurodegeneration via cAMP/PKA signaling remains to be explored [85].
Ample research has detailed the active role of neurons in phagocytic signaling. For instance, neurophagy, the phagocytosis of live neurons and synapses by glia, is dependent on active intercellular signaling. The neuronal cell surface displays various “eat-me” signals to attract and activate phagocytic glia [86]. Conversely, TAR DNA Binding Protein 43 kDa (TDP43) and CD47 are surface molecules expressed by neurons to avoid phagocytic clearance, thus called “don't eat me” signals [87, 88]. Expression of CD47, in particular, is regulated by transcription factors signal transducer and activator of transcription 3 (STAT3) and nuclear factor κB (NF-κB). Consequently, these downstream effectors of cAMP signaling can benefit from PDE inhibition.
Microglia - the brain's residing overseers
Microglia develop from erythromyeloid progenitor cells. During early development, they travel via the bloodstream and infiltrate the brain [89]. Once they arrive, microglia populate the brain by proliferation and migration (reviewed by Smolders et al. [90]). Thereafter, the influx of circulating bone marrow-derived progenitors is redundant, as the self-renewal capacity of brain-resident microglia accounts for their maintenance and local expansion in the healthy adult brain [91]. As the resident macrophage population, microglia act as the first and foremost form of active innate immune defense in the brain. Micro-organisms, damaged cells, neurofibrillary tangles, or plaques activate microglia and consequently strengthen their capacity to proliferate, migrate, induce cell death, phagocytose, and present antigens [92].
Microglia also regulate the inflammatory response by producing cytokines, such as tumor necrosis factor alpha (TNF-α), interleukin-1 beta (IL-1β), chemokines, free radicals, and nitric oxide (NO) [93]. Pharmacological modulation of microglial activation and synaptic elimination has garnered considerable attention as a therapeutic approach to treat neurodegenerative diseases. Elevating microglial cAMP and cGMP levels has been shown to counteract inflammatory activation and promote neuroprotection. Inhibition of dual substrate PDE3, cAMP-specific PDE4 and cGMP-specific PDE5 using amrinone, rolipram, and sildenafil, respectively, drives cultured microglia towards an anti-inflammatory phenotype with decreased production of TNF-α, IL-1β, IL-12 and NO, suggesting a neuroprotective effect exerted via the microglial cGMP or cAMP signaling pathways [94-96]. The non-selective PDE inhibitor pentoxifylline decreases LPS-dependent increases in TNF-α [97].
PDE4 inhibitors (including rolipram) and a PDE4D-negative allosteric modulator (D158681) [98] have pro-cognitive, neuroprotective, and anti-inflammatory effects in vivo (reviewed in [99]). Of the PDE4 subtypes that are known to modulate inflammation in the CNS, the high expression of PDE4B in microglia seems to underline its importance. Notably, PDE4B is highly expressed in activated microglia found in traumatic brain and spinal cord injury [99]. However, inhibition of PDE4 subtype expression is not microglia-specific; its effects cannot be directly attributed to microglia only in in vivo studies. Microglial cAMP and cGMP have also been shown to upregulate Arginase (Arg-1) in microglia, while sildenafil has been found to upregulate chitinase 3-like 3 (YM-1), skewing them towards an anti-inflammatory phenotype [96, 100]. In inflammatory reactions, Toll-like receptors (TLRs) amplify the release of pro-inflammatory cytokines and chemokines. The non-selective PDE inhibitor Ibudilast, targeting PDE4, -10 and to a lesser extent, PDE3 and -11, has been shown to antagonize TLR4 [101]. Interestingly, cAMP/PKA signaling is anchored and regulated by the leucine-rich repeat kinase 2 (LRRK2)-PDE4 complex. Although LRRK2 is not microglia-specific, research has shown that its knock-out decreased the LPS-dependent pro-inflammatory response of microglia by modulation of cAMP/PKA activity in vivo [102]. It has also been shown that LRRK2 restricts cAMP signaling mainly to intercellular endosomal-lysosomal compartments and mitochondria, affecting vesicle trafficking and autophagy [103]. These findings support the fact that PDEs associated with cAMP/AKAP complexes and their localization have to be taken into account when designing drugs to improve neuroprotection and synaptogenesis.
In addition to their protective functions, microglia are critical cells in overall brain maintenance. Both during development and homeostasis, microglia actively survey and shape the brain parenchyma by physically monitoring neurons and synapses [49, 104, 105]. This contact is dependent on neuronal activity: the frequency of these contacts decreases with decreasing neuronal activity and the duration of contact increases in pathological circumstances [106, 107]. During development, an excess of newly born neurons is generated, and microglia quickly clear dead and dying cells through phagocytosis to prevent the spillover of pro-inflammatory and neurotoxic molecules [108-110]. Similarly, microglia are vital regulators of the balance between synaptogenesis and synaptic elimination: synaptic connections between neurons are formed in abundance during postnatal brain development, and microglia eliminate weak synaptic connections and strengthen the remaining synapses [111]. In vivo pre- and postnatal microglial depletion or perturbation models reveals the essential role for microglia in synapse formation, refinement, and elimination (16, 24, 62-74). Microglial cAMP and cGMP have also been shown to be critical for microglial surveillance of the brain parenchyma and, therefore, rapid and subtle manipulation of cAMP and cGMP levels can dramatically affect their role in synaptic elimination. The addition of the non-selective, also called pan-PDE blocker, IBMX, prevents the breakdown of cAMP and triggers a fast retraction of microglial processes in situ, leading to reduced elimination. Specific blocking of PDE3B, whose expression is highly enriched in microglia, has a similar effect, suggesting that intracellular cAMP and cGMP levels regulate the morphology and sensing mechanism of microglia [112]. General microglial phagocytosis is also regulated through cAMP and cGMP signaling. In addition, PDE4B inhibition has been shown to alter microglial phagocytosis and reactivity, as inflammation regulates PDE4 expression (reviewed in [113]). Treating microglia with IBMX (all PDEs), rolipram (PDE4), or cilostamide (PDE3) resulted in inhibition of microglial phagocytosis [114], suggesting a potential mechanism to counteract phagocytosing of synapses [115]. It is clear that the structural and functional consequences of impaired microglial surveillance of the neuronal network require further investigation.
An important point to raise is that ex vivo live-imaging has not found evidence of active phagocytosis of complete synapses by microglia [116]. The same study did confirm direct engulfment and subsequent elimination of pre-synaptic material. The distinction is that active phagocytosis (which was not witnessed) entails direct phagocytosis of spines as a functional unit on neurons, constituting direct breakdown of functional spines by microglia. Conversely, elimination of synaptic material encompasses the clearance of synaptic debris following disruption of the synapse or targeting of the pre-synaptic compartment. More specifically, the authors observed trogocytosis -or nibbling- of presynaptic boutons, while contacts at postsynaptic sites were shown to elicit transient filopodia from mature spines. A microglia depletion study suggests that microglial support in synapse formation is maintained throughout the lifespan, while microglial-mediated synapse elimination declines from adolescence [117]. However, synaptic elimination might be turned back on aberrantly in the aged and diseased brain [118].
Synaptic elimination involves many cues that promote and inhibit the detection and removal of specific synapses (reviewed in [119-121]). To date, the best-described mediator of synaptic elimination is the classic complement cascade. The soluble complement protein C1q targets synapses that need to be eliminated in an activity-dependent and transforming growth factor beta (TGF-β)-induced manner, after which a sequence of proteolytic reactions leads to the covalent association of C3b proteins to the same synapses. The complement receptor CR3 (alternatively CD11b/CD18), which is expressed on the surface of microglia, binds C3b and promotes phagocytosis of the opsonized synapses [122]. The importance of functional cell-cell communication is again highlighted in this process. TGF-β is produced by astrocytes, and its binding to its specific receptor on neurons triggers these neurons to produce C1q, which functions as an 'eat me' signal for microglia [123]. Conversely, TGF-β is needed by neurons for conversion from early phase LTP to late phase LTP and correlates with an increased expression of the phosphorylated transcription factor CREB, downstream of cAMP signaling [124].
The classic complement cascade ensures the removal of overproduced synapses during postnatal development, and the expression of its components decreases in adulthood [125]. Tight regulation is a necessity, as dysfunctional synaptic elimination leads to different neuropathologies depending on age and the affected brain region. Complement-dependent synaptic elimination is implicated in, among others, AD [118], frontotemporal lobar dementia [126], West Nile virus-associated neurodegeneration [127], schizophrenia [128, 129], epilepsy [130], and multiple sclerosis [131]. In contrast, microglia and the complement system were initially seen as neuroprotective in AD, as the complement system counteracts amyloid plaque deposition by inducing microglial phagocytosis of Aβ [132]. In a later stage, by encapsulating and thereby segregating the amyloid plaques, microglia appear neuroprotective. Thus, microglial functioning has been extensively described as being a double-edged sword in several neuropathological disorders. However, studies investigating microglial ablation found no difference in plaque burden, but showed considerable cognitive improvement, which suggests an alternate, neurodegenerative process mediated by microglia in AD [29, 133]. All in all, disturbances in the complement cascade may represent a shared etiological pathway in neurological diseases that needs to be further explored.
Age-dependent synapse loss is characterized by a decline in cognitive functions and is provoked by a combination of increased synaptic elimination, loss of cortico-cortical connections, and neuronal apoptosis [134]. Aging mouse and human brains show increased levels of components of the complement system, such as C1q [135]. During adulthood and in normal aging, microglia produce the majority of C1q, which is a harbinger for increased synaptic elimination [136]. Aging microglia also upregulate the expression of CR3, which implicates a microglial contribution in synaptic loss during normal aging that utilizes the complement pathway [137]. Increased expression and presence of CR3 is one of the possible mechanisms explaining phagocytic clearance of focal apoptotic synaptic compartments by microglia, the other being increased Ca2+ levels in dendritic spines, triggering 'eat me' signals for microglia [138]. Aging microglia have also been shown to be activated, accompanied by elevated levels of chemokines and cytokines such as TNF-α and IL-1β [139], and an altered expression of various membrane receptors like triggering receptor expressed on myeloid cells 2 (TREM2), scavenger receptor class A (SR-A/Scara-1), and CD33 [140]. This increased activation state can result in excessive synaptic elimination. Interestingly, many pathways found in studies aimed at elucidating the mechanisms behind microglial phagocytosis share cAMP as an upstream effector [141, 142]. Administering PDE inhibitors, like IBMX and rolipram, or reagents that alter cAMP levels has been shown to inhibit phagocytosis in macrophages [115], but also phagocytosis of myelin mediated by CR3 and SR-A [141]. The mechanisms underlying age-dependent changes in microglial function remain elusive, but are thought to be similar, at least in part, to those in neurodegenerative diseases.
Macrophages - the reinforcements for hire
The central myeloid cell population of the CNS is composed of parenchymal microglia and non-parenchymal macrophages, including perivascular macrophages, meningeal macrophages, macrophages of the choroid plexus and blood-borne monocytes [143-145]. Non-parenchymal macrophages in the CNS are known to be crucial for the preservation of capillary stability and BBB integrity [146]. In the past few years, CNS macrophages have gained interest for their involvement in synaptic maintenance and associated CNS disorders. Even though the process of synapse refinement has been mainly attributed to microglia, it is likely that non-parenchymal macrophages also interact with neurons, as perivascular and meningeal macrophages even show increased phagocytic activity when compared to microglia [147]. Blood-borne monocytes, once in the brain, polarize into monocyte-derived macrophages, which subsequently share similar characteristics with parenchymal microglia (e.g., the expression of complement receptors, signal regulatory protein alpha (SIRPα), chemokine receptors, and TREM2) [148, 149]. Studies using in vitro co-cultures of monocyte-derived macrophages and neurons have further emphasized the possible involvement of these macrophages with regards to synapse refinements, as synapse material was found present within these macrophages [148, 149].
During aging, blood-borne monocytes infiltrate the brain parenchyma, where they differentiate into microglia-like monocyte-derived macrophages [148]. The monocyte-derived macrophage population subsequently becomes intermingled with tissue-resident microglia and either facilitates or counteracts neurological signaling [143, 145]. In the aged brain, peripheral monocytes infiltrate to a greater degree and consequently alter the CNS microenvironment, skewing it to a more inflammatory environment. The infiltrated monocytes negatively influence synaptic plasticity, as they decrease the LTP response in hippocampal brain slices, suggestive of a detrimental effect of infiltrated macrophages [150, 151]. In vivo studies further confirmed the destructive nature of the infiltrated macrophages, since the level of macrophage infiltration in aged rats correlated with reduced synaptic functioning [152]. This age-related pro-inflammatory environment of the CNS has negative consequences. Research has shown that aged animals are less protected against acute traumatic CNS insults, as they exhibited an increase in macrophage infiltration leading to exacerbated synapse loss and accelerated cognitive decline [153]. Rolipram-mediated PDE4 inhibition increased the migratory capacity of a murine macrophage cell line when challenged with LPS, an outcome that could lead to a more efficient pathogen clearance and subsequent wound repair [154]. This migratory phenotype was primarily mediated by activating the cAMP-Epac signaling cascade [154].
Even though the exact role of non-parenchymal macrophages remains inconclusive, several current studies have aimed at skewing specific macrophage-mediated responses to resolve neurodegenerative disease. One strategy to regulate macrophage functions is to alter intracellular levels of second messengers by inhibiting specific PDEs. To understand which type of PDE inhibition could be beneficial in particular processes, differences in PDE expression profiles have been mapped. Interestingly, when peripheral monocytes differentiate towards macrophages in vitro, dramatic changes can be observed with regards to PDE expression [155]. For instance, while monocytes are characterized by profound PDE4 activity, its activity rapidly declines during differentiation [155]. A detailed PDE4 isoform profiling showed a selective downregulation of PDE4D3 and PDE4D5. Conversely, PDE4A10 and PDE4B2 protein levels increased, shifting the primary source of PDE4 activity from long PDE4D isoforms in monocytes to short PDE4B isoforms in macrophages [156]. While the total PDE4 activity reduced during monocyte to macrophage differentiation, PDE1 and PDE3 activity increased [155].
The importance of synaptic maintenance and preservation is especially highlighted in schizophrenia. In schizophrenic patients, an increase in the absolute number of circulating peripheral blood monocytes was found, whereas an increase of macrophages was observed in the cerebrospinal fluid of schizophrenia patients during an acute psychotic episode [157, 158]. However, when differentiating peripheral monocytes from schizophrenia patients or healthy controls into macrophages in vitro, only a minor decrease in purinergic receptor P2X7 gene expression was seen in schizophrenia material upon pro-inflammatory LPS stimulation [159]. No other functional irregularities were present in response to alternative pro- or anti-inflammatory stimuli, questioning the active involvement of non-parenchymal macrophages in alleviating or exacerbating schizophrenic disease progression.
Owing to their phagocytic capacity, macrophages have been extensively investigated for their possible neuroprotective potential in AD, as they may regulate Aβ clearance [160]. In both AD patients and animal models for AD, macrophages have been shown to accumulate around senile plaques [161, 162]. When stimulating perivascular macrophage turnover, a reduced cerebral amyloid angiopathy load was observed in TgCRND8 mice, an outcome that was found to occur independent of microglia-mediated Aβ clearance [163]. The role of not only perivascular but also monocyte-derived macrophages has been studied in relationship to AD. Both in vitro and in vivo studies have showed an increased capacity of these monocyte-derived macrophages to remove Aβ fibrils, a process mediated by intracellular CD36/early endosomal antigen-1 (EEA1) early endosomal proteolysis [164-166]. Interestingly, not only fibrils, but also Aβ42 oligomers, were shown to be cleared by activated macrophages in vivo [166]. In contrast to fibrils, oligomer clearance was primarily mediated via extracellular/matrix metallopeptidase 9 (MMP-9) enzymatic degradation, leading to macrophage-mediated vesicular glutamate transporter 1/postsynaptic density protein 95 (VGlut1/PSD95) synapse preservation through the prevention of oligomer build-up and subsequent synaptotoxicity [166].
Understanding isoform-specific PDE expression changes can offer insights into PDE inhibitor development as a therapeutic strategy to alter macrophage-mediated responses in the CNS. As such, PDE inhibitors can be beneficial in multiple CNS disorders, either by changing the phagocytic capacity of macrophages or by downregulating their pro-inflammatory markers and therefore resolving neuroinflammation. Besides PDE4, PDE1B2 suppression has been studied for its effect on macrophages. When PDE1B2 was specifically suppressed, macrophages showed an increased phagocytic ability and enhanced cell spreading [167]. Finally, the pro-inflammatory microenvironment in the aged and diseased brain has been shown to increase PDE4B2 expression in macrophages, specifically (138). Therefore, inhibiting the PDE4B family or specific isoforms is an interesting strategy for controlling the age-related pro-inflammatory environment in the brain.
There is little information available about the compartmentalization of second messengers within CNS macrophages. Evidence has been found to show that AKAP10 (AKAP-D) and AKAP95 regulate secretion of anti-inflammatory cytokines IL-10, IL-6, and TNF-α through cAMP/PKA signaling in alveolar macrophages [168]. AKAP95, observed in nuclear fractions, formed complexes with nuclear PDE4 and PDE3, where the latter serves as a barrier to prevent the accumulation of cAMP [169].
Astrocytes - from support structure to clean-up crew
Astrocytes are the most abundant cells within the CNS. Their supportive and protective role renders them indispensable in normal brain functioning. As such, astrocytes are involved in extracellular matrix (ECM) homeostasis, neurotransmitter recycling, neuroplasticity, and many other processes. Their particular contribution to synaptic formation, maintenance, and elimination has become more appreciated over the past decades. Astrocytes secrete synaptogenic factors, such as thrombospondins, which are required during synaptic formation and act to strengthen functional synapses [170]. Furthermore, through direct physical contact with astrocytes, synaptogenesis happens considerably faster (as evidenced in vitro [171]), emphasizing the importance of these glial cells during synaptogenesis [172]. As part of the so-called 'tripartite synapse', along with pre- and postsynaptic neurons, astrocytes exert a wide range of regulatory functions to maintain synaptic stability. However, here, we mainly focus on their role in synaptic elimination and E/I balance maintenance during neurodegeneration.
In addition to microglia, astrocytes have been shown to be involved in synaptic elimination. A study by Chung et al. showed that astrocytes express phagocytic receptors, such as MER Proto-Oncogene, Tyrosine Kinase (MERTK) and multiple epidermal growth factor like domains 10 (MEGF10), which are responsible for synapse targeting and elimination [170, 173]. Interestingly, the phagocytosis of synapses by astrocytes seemed to be independent of the complement protein C1q, suggesting a different phagocytic pathway than employed by microglia. Additionally, the study revealed that the phagocytic functions of astrocytes are strongly dependent on neural activity, indicating that astrocytes actively prune active synapses rather than just clean up synaptic debris. Specifically, blocking spontaneous retinal waves in both eyes significantly reduced astrocyte-mediated phagocytosis of bilateral synaptic inputs, whereas selective blocking of activity in only one eye induced preferential engulfment of the silenced synapses by astrocytes [173]. Reactive astrocytes have also been shown to ?? phagocytose synapses in hippocampal regions of amyloid precursor protein/presenilin 1 (APP/PS1) mice as well as in post-mortem AD brains. Dystrophic presynaptic vGlut1-positive terminals seem to be targeted and degraded by astrocytic endfeet, yet it remains unclear whether these mechanisms contribute to disease pathology or become impaired during disease progression [174]. The involvement of PDEs in astrocytic-mediated synaptic control remains an unstudied topic. PDE inhibition or cAMP elevation within astrocytes seems to regulate distinct neuroinflammatory pathways and could, therefore, influence synaptic integrity in an indirect manner [66]. Treatment of astrocytes with 8-Br-cAMP, a cAMP analog, has been shown to decrease the levels of the pro-inflammatory cytokine TNFα [11]. TNFα stimulates C3 expression, facilitating complement-mediated synapse elimination, suggesting that astrocytic cAMP might attenuate inflammatory induced synaptic elimination [175, 176].
Reactive astrocytes have also been shown to be closely associated with amyloid plaques in AD brains and were found to internalize Aβ both in vitro and in vivo [177-179]. Subsequent astrocytic Aβ-induced release of pro-inflammatory factors leads to synaptic disturbances and neuronal death in models for AD [180, 181]. A genotype-specific phagocytic ability of astrocytes has been shown within the context of AD. Astrocytes of apolipoprotein E 4 (APOE4) knock-in (KI) mice exhibit a defective phagocytic capacity, while astrocytes from APOE2 KI mice have an enhanced rate of synaptic elimination [182]. Interestingly, induced pluripotent stem cell-derived astrocytes from APOE4 AD patients that had been genetically converted with CRISPR-Cas9 to an APOE3 genotype showed a rescued overall phagocytic capacity, confirming the APOE allele-dependent phagocytic capacity of astrocytes in AD pathology [183].
Astrocytes target both excitatory and inhibitory synapses, yet the balance varies during different developmental stages. In the first phase of postnatal development, more excitatory synapses are engulfed, likely owing to the higher rate of synaptogenesis in this phase. This observation shows that astrocytes play a pivotal role in maintaining the E/I equilibrium [173]. One of the significant functions of astrocytes in the vicinity of excitatory synapses is the reuptake of excessive glutamate via the excitatory amino acid transporters (EAAT)1, and EAAT3, to prevent excitotoxicity [184]. Both amyotrophic lateral sclerosis (ALS) and Huntington mutant mouse models, i.e., superoxide dismutase 1 (SOD1)- and huntingtin (htt)-mediated, respectively, show defective astrocytic glutamate transporters, eventually leading to extracellular glutamate accumulation and neuronal death through excitotoxicity [185, 186]. Similarly disrupted glutamate receptors have been proposed within the context of Parkinson's disease (PD), resulting from α-synuclein accumulation within astrocytes [187]. Furthermore, astrocytes are capable of producing and releasing GABA as a gliotransmitter, potentially activating GABA receptors on adjacent neurons [188]. However, reactive astrocytes produce markedly less glutamine. As a precursor for GABA, glutamine starvation leads to a reduction in inhibitory currents as a result of GABA depletion, while excitatory currents remain unaltered [189]. In contrast, overreactive astrocytes in AD have been shown to disrupt synaptic function through excessive production and release of GABA by the GABA-producing enzyme monoamine oxidase B (MAO-B). The released GABA strongly inhibits neuronal synaptic release, leading to impairment in LTP and learning and memory [190]. In line with this, it has been shown that Aβ increases astrocytic GABA synthesis, suggesting a crucial role of astrocytes in the E/I imbalance in AD [191]. The opposite has been shown in a mouse model for Huntington's disease (HD), where astrocytic GABA release through GABA transporter 3 (GAT-3) is reduced, resulting in reduced tonic inhibition in striatal output neurons [192]. It has been shown that PDE5 inhibition through sildenafil treatment has a preventive and restorative effect on LPS-induced inflammation in astrocytes in vivo [193]. Accordingly, ibudilast administration has been proven to reduce astrogliosis during neuroinflammation and seems to prevent astrocytic apoptosis via cGMP signaling [15, 194, 195]. Inhibition of PDE7 by the inhibitor S14 has been shown to stimulate astrocyte-mediated Aβ degradation in a murine model for AD [79]. Taken together, these data highlight the therapeutic potential of PDE modulation in astrocytes within the context of neuroinflammation. However, further research is necessary to find whether PDE inhibition has a direct effect on astrocytic-mediated synapse maintenance. Since astrocytes are crucial mediators during synaptic elimination and in preserving the E/I balance, future research should focus on these mechanisms to elucidate the potential benefits of PDE inhibitors as therapeutic strategies for neurodegenerative diseases.
Oligodendrocytes - silent modulators of the synaptic unit
Oligodendrocytes are indispensable in CNS functioning. In the case of synapse loss, however, their role is not yet fully understood. The supportive, seemingly static role of oligodendrocytes reaches well beyond their prominent role as myelinating cells of the CNS; they are active participants in cell-signaling pathways. The oligodendrocytic processes are in intimate contact with neurons at both pre- and postsynaptic structures, facilitating nutritional and structural support for synapses [100, 196]. Therefore, oligodendrocyte cell signaling pathways should not be overlooked in the context of synaptogenesis. Cyclic nucleotide signaling has proven to be vital in oligodendrocyte metabolism, implicating them as interesting effector cells [66]. For instance, inhibition of the cGMP-specific PDE5 by sildenafil improves BDNF signaling of oligodendrocytes and thus synaptogenesis [100]. The role of compartmentalized cAMP/PKA and cGMP/PKG signaling, to the best of our knowledge, remains to be elucidated in oligodendrocyte functioning pathways.
Oligodendrocytes are the myelinating glia of the brain. Though proven to be indispensable for brain functioning, to the best of our knowledge, they have not been studied extensively in the context of synapse development, maintenance, or plasticity. As previously illustrated, glial-secreted factors are of undeniable importance for synaptic maturation and maintenance. Notably, in the developing brain, oligodendrocytes actively participate in synaptic plasticity through BDNF signaling, promoting neuronal survival and plasticity by inducing LTP [197, 198]. BDNF plays a fundamental role in age-related synaptic loss, preventing cerebral atrophy and cognitive decline [199, 200]. Oligodendrocyte-astrocyte crosstalk has been proven to be of importance in the formation and maintenance of the CNS. Importantly, these interactions are not only involved in activity dependent adaptive myelination but also provide metabolic support to axons [201-203].
Studies looking into the involvement of oligodendrocyte precursor cells (OPCs) have found evidence of them receiving signals from axons for myelination in an activity-dependent manner via glutamatergic signaling. The abundantly present AMPA- and NMDA-type glutamate receptors mediate OPC proliferation and migration or myelination, respectively [204, 205]. An essential functional overlap in synaptic strengthening and myelination by oligodendrocytes has been noted in the process of learning. Both processes are enhanced in animals living in an enriched environment, prompting the conclusion that oligodendrocytes are involved in cell-cell communication processes that take place during learning. Proliferation and differentiation of OPCs have been shown in animals subjected to an enriched environment that involves learning exercises [206, 207]. As PDE4 inhibition, mainly by rolipram [208, 209], has proven to significantly enhance OPC differentiation, this process may aid in creating a positive environment for synaptic plasticity. OPCs receive synaptic input from pyramidal neurons and interneurons in the hippocampus and axons in the corpus callosum [210]. How exactly this communication between neurons and oligodendrocytes takes place remains to be elucidated, and understanding this process should contribute to efforts to counteract synapse loss. In addition, cAMP and PDE remain important to oligodendrocyte signaling. By modulating these signaling mechanisms, and skewing them towards a differentiated state, trophic support can be used as a driving factor in synaptic plasticity. The associated activity-induced myelination is a byproduct of PDE inhibition, which contributes to efficient support of the neuronal network.
Concluding remarks
PDEs have been at the center of many investigative strategies aimed at understanding various diseases. Increased understanding of signaling cascades surrounding specific PDE isoforms has uncovered their potential as therapeutic targets. As non-selective inhibition often proves ineffective or is accompanied by adverse effects, only a small number of PDE inhibitors have translated to the clinic, and many inhibitors still require validation for clinical neuromodulation [48, 211].
Elevation of cyclic nucleotide levels in astrocytes ameliorates neuromodulatory processes, contributing to a suitable environment for synaptic plasticity. In addition to astrocytes, microglia are the other neuroglial cells directly interacting with synapses. They influence not only elimination, but also the formation and protection of synapses. This influence is exerted during development and learning, as well as in aging and neurodegeneration. In the latter case, when neuroinflammation is widespread, PDE inhibition has proven useful in counteracting the activation of microglia, limiting the effect of inflammatory signaling. Additionally, the phagocytic capacity of astrocytes and microglia contributes to the elimination of synapses via its focal clearance. Comparison of astrocytic and microglial phagocytosis addresses an interesting knowledge gap, as it remains unclear if and, if so, how neural activity controls the rate of astrocyte-mediated synapse phagocytosis, whether astrocytes and microglia phagocytose different synapse types or circuits, and which different 'eat me signals' are involved [173]. If different receptors with their respective signaling molecules are at the basis of phagocytosis by these two cell types, this fact presents an opportunity for differential PDE inhibition interventions. The inhibition of PDEs can counteract inflammatory pruning by microglia and astrocytes.
Notably, the effects of PDE inhibition have a direct modulatory effect on neurons, contributing to synaptic plasticity and neuroprotection. Both transient and lasting changes on plasticity can be achieved by targeting neurons, ultimately strengthening the synaptic network. The direct effects of the inflammatory environment on neurons can be affected by PDE inhibition. Previous work digging deeper into targeting isoforms of PDEs has focused largely on neurons, yet an exciting avenue might be to pursue glial cells. The effects of isoform compartmentalization are readily apparent in various forms of synaptic plasticity in neurons, so finding out to what extent varying isoforms alter glial cell functioning might prove valuable to understanding neurodegeneration. An interesting avenue that has been the focus of many research labs is the neuronal expression of intercellular signaling molecules that can activate surrounding glial cells. Many of these signals have second messengers lying downstream or upstream to PDE-directed pathways, rendering them vulnerable to PDE signaling [212, 213].
In closing, the role of glial cells and glia-neuron communications has gained increasing interest as a driving factor behind many diseases. Enhancement of both cAMP and cGMP signaling can successfully promote synapse strengthening and diminish inflammatory signaling, as is evident by the use of a variety of PDE inhibitors. As such, PDE inhibition has potential as a therapeutic approach to make neurons and their synapses more resilient to (disease-induced) glia-mediated elimination. The usefulness of PDE inhibitors is not limited to protecting neurons, however. By actively altering cell signaling in glial cells, a more direct approach can also be pursued. Recent advances in understanding different neuropathologies have opened up interesting avenues towards personalized medicine. As summarized in this review, the specific expression pattern of PDEs in different cell types allows targeted treatment strategies. Expanding on this notion, the targeting of isoform-specific PDE signatures per cell type may prevent neuroinflammation, while promoting synaptic plasticity in divergent pathologies. Because of this, the holistic impact of PDE inhibition can be beneficial to the entire synaptic unit. Consequently, combination therapy consisting of inhibitors targeting multiple PDEs, over multiple cell types, paves the way towards personalized medicine. This underlying potential solidifies that PDE inhibitors remain a viable strategy, yet require additional research to reach their full potential. Although the use of PDE inhibitors can lead to adverse side effects, targeting of specific isoforms holds promise to circumvent these unwanted effects. By determining the predominant isoforms of PDEs in fundamental pathways of neurodegenerative diseases, a deeper understanding of their causative mechanisms can be achieved. The additive effect of combining multiple isoform-specific drugs into pathology-specific therapy holds promise to finally unlock the full potential of PDE inhibitors.
Abbreviations
Aβ: amyloid beta; AD: Alzheimer's disease; AKAP: A-kinase-anchoring protein; ALS: amyotrophic lateral sclerosis; AMPA: α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; Arg-1: arginase; APOE: apolipoprotein E; APP/PS1: amyloid precursor protein/presenilin 1; ATP: adenosine triphosphate; BBB: blood-brain-barrier; BDNF: brain derived neurotrophic factor; cAMP: 3',5'-cyclic adenosine monophosphate; CNS: central nervous system; CR: complement receptor; CREB: cAMP response element-binding protein; cGMP: 3',5'-cyclic guanosine monophosphate; CRISPR: Clustered Regularly Interspaced Short Palindromic Repeats; EAAT: excitatory amino acid transporter; ECM: extracellular matrix; EEA1+: early endosomal antigen-1; E/I: excitation/inhibition; GABA: gamma-aminobutyric acid; GAT-3: GABA transporter 3; GKAP: G-kinase anchoring protein; HD: Huntington's disease; htt: huntingtin; IBMX: 3-isobutyl-1-methylxanthine; IL1β: interleukin 1 beta; KI: knock-in; LPS: lipopolysacharide; LRRK2: leucine-rich repeat kinase 2; LTD: long term depression; LTP: long term potentiation; MAO-B: monoamine oxidase B; MEGF10: multiple epidermal growth factor like domains 10; MERTK: MER Proto-Oncogene, Tyrosine Kinase; MMP-9: matrix metallopeptidase 9; NMDA: N-methyl-D-aspartate; NO: nitric oxice; OPC: oligodendrocyte precursor cell; PD: Parkinson's disease; PDE: phosphodiesterase; PKA: protein kinase A; PKG: protein kinase G; PSD95: postsynaptic density protein 95; RNA: ribonucleic acid; SIRP: signal regulatory protein alpha; SOD1: superoxide dismutase 1; SR-A: scavenger receptor class A; TGF: transforming growth factor beta; TLR4: Toll-like receptor 4; TNFα: tumor necrosis factor α; TREM2: triggering receptor expressed on myeloid cells 2; UCR: upstream conserved region; vGlut1: vesicular glutamate transporter 1; YM-1: chitinase 3-like 3.
Acknowledgements
The authors sincerely thank prof. dr. David M. Wilson III for proofreading the paper.
Competing Interests
TV and JP have a proprietary interest in selective PDE4D inhibitors for the treatment of demyelinating disorders. JP has a proprietary interest in the PDE4 inhibitor roflumilast for the treatment of cognitive impairment.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Rakic P, Bourgeois JP, Eckenhoff MF, Zecevic N, Goldman-Rakic PS. Concurrent overproduction of synapses in diverse regions of the primate cerebral cortex. Science. 1986;232:232-5
2. Henstridge CM, Tzioras M, Paolicelli RC. Glial Contribution to Excitatory and Inhibitory Synapse Loss in Neurodegeneration. Front Cell Neurosci. 2019 13
3. Kelly MP. Cyclic nucleotide signaling changes associated with normal aging and age-related diseases of the brain. Cell Signal. 2018;42:281-91
4. Argyrousi EK, Heckman PR, van Hagen BT, Muysers H, van Goethem NP, Prickaerts J. Pro-cognitive effect of upregulating cyclic guanosine monophosphate signalling during memory acquisition or early consolidation is mediated by increased AMPA receptor trafficking. J Psychopharmacol. 2020;34:103-14
5. Argyrousi EK, Heckman PRA, Prickaerts J. Role of cyclic nucleotides and their downstream signaling cascades in memory function: Being at the right time at the right spot. Neurosci Biobehav Rev. 2020;113:12-38
6. Kritzer MD, Li J, Dodge-Kafka K, Kapiloff MS. AKAPs: the architectural underpinnings of local cAMP signaling. J Mol Cell Cardiol. 2012;52:351-8
7. Arora K, Sinha C, Zhang W, Ren A, Moon CS, Yarlagadda S. et al. Compartmentalization of cyclic nucleotide signaling: a question of when, where, and why? Pflugers Arch. 2013;465:1397-407
8. Arora K, Sinha C, Zhang W, Ren A, Moon CS, Yarlagadda S. et al. Compartmentalization of cyclic nucleotide signaling: a question of when, where, and why? Pflugers Arch. 2013;465:1397-407
9. Ghosh M, Xu Y, Pearse DD. Cyclic AMP is a key regulator of M1 to M2a phenotypic conversion of microglia in the presence of Th2 cytokines. J neuroinflammation. 2016;13:9
10. Li J, Liu C-N, Wei N, Li X-D, Liu Y-Y, Yang R. et al. Protective effects of BAY 73-6691, a selective inhibitor of phosphodiesterase 9, on amyloid-β peptides-induced oxidative stress in in-vivo and in-vitro models of Alzheimer's disease. Brain Res. 2016;1642:327-35
11. Paco S, Hummel M, Pla V, Sumoy L, Aguado F. Cyclic AMP signaling restricts activation and promotes maturation and antioxidant defenses in astrocytes. BMC genomics. 2016;17:304
12. Cameron RT, Whiteley E, Day JP, Parachikova AI, Baillie GS. Selective inhibition of phosphodiesterases 4, 5 and 9 induces HSP20 phosphorylation and attenuates amyloid beta 1-42-mediated cytotoxicity. FEBS open bio. 2017;7:64-73
13. Hedde JR, Hanks AN, Schmidt CJ, Hughes ZA. The isozyme selective phosphodiesterase-4 inhibitor, ABI-4, attenuates the effects of lipopolysaccharide in human cells and rodent models of peripheral and CNS inflammation. Brain Behav Immun. 2017;64:285-95
14. Knott EP, Assi M, Rao SN, Ghosh M, Pearse DD. Phosphodiesterase Inhibitors as a Therapeutic Approach to Neuroprotection and Repair. Int J Mol Sci. 2017 18
15. Schwenkgrub J, Zaremba M, Joniec-Maciejak I, Cudna A, Mirowska-Guzel D, Kurkowska-Jastrzębska I. The phosphodiesterase inhibitor, ibudilast, attenuates neuroinflammation in the MPTP model of Parkinson's disease. PLOS ONE. 2017;12:e0182019
16. Blokland A, Heckman P, Vanmierlo T, Schreiber R, Paes D, Prickaerts J. Phosphodiesterase Type 4 Inhibition in CNS Diseases. Trends Pharmacol Sci. 2019;40:971-85
17. Schepers M, Tiane A, Paes D, Sanchez S, Rombaut B, Piccart E. et al. Targeting phosphodiesterases (PDE)-towards a tailor-made approach in multiple sclerosis treatment. Front Immunol. 2019;10:1727
18. Schuster KD, Mohammadi M, Cahill KB, Matte SL, Maillet AD, Vashisth H. et al. Pharmacological and molecular dynamics analyses of differences in inhibitor binding to human and nematode PDE4: Implications for management of parasitic nematodes. PLoS One. 2019;14:e0214554
19. Wang D, O'Halloran D, Goodman MB. GCY-8, PDE-2, and NCS-1 are critical elements of the cGMP-dependent thermotransduction cascade in the AFD neurons responsible for C. elegans thermotaxis. J Gen Physiol. 2013;142:437-49
20. Yu B, Wang X, Wei S, Fu T, Dzakah EE, Waqas A. et al. Convergent Transcriptional Programs Regulate cAMP Levels in C. elegans GABAergic Motor Neurons. Dev Cell. 2017;43:212-26.e7
21. Rojo Romanos T, Petersen JG, Pocock R. Control of Neuropeptide Expression by Parallel Activity-dependent Pathways in Caenorhabditis elegans. Sci Rep. 2017;7:38734
22. Charlie NK, Thomure AM, Schade MA, Miller KG. The Dunce cAMP phosphodiesterase PDE-4 negatively regulates G alpha(s)-dependent and G alpha(s)-independent cAMP pools in the Caenorhabditis elegans synaptic signaling network. Genetics. 2006;173:111-30
23. Fujiwara M, Hino T, Miyamoto R, Inada H, Mori I, Koga M. et al. The Importance of cGMP Signaling in Sensory Cilia for Body Size Regulation in Caenorhabditis elegans. Genetics. 2015;201:1497-510
24. Beets I, Zhang G, Fenk LA, Chen C, Nelson GM, Félix M-A. et al. Natural Variation in a Dendritic Scaffold Protein Remodels Experience-Dependent Plasticity by Altering Neuropeptide Expression. Neuron. 2020;105:106-21.e10
25. Yu W, Lu B. Synapses and Dendritic Spines as Pathogenic Targets in Alzheimer's Disease. Neural Plast. 2012;2012:8
26. He H-Y, Cline HT. What Is Excitation/Inhibition and How Is It Regulated? A Case of the Elephant and the Wisemen. J Exp Neurosci. 2019;13:1179069519859371 -
27. Morelli G, Avila A, Ravanidis S, Aourz N, Neve RL, Smolders I. et al. Cerebral Cortical Circuitry Formation Requires Functional Glycine Receptors. Cereb Cortex. 2016;27:1863-77
28. Schafer DP, Lehrman EK, Stevens B. The "quad-partite" synapse: microglia-synapse interactions in the developing and mature CNS. Glia. 2013;61:24-36
29. Hong S, Dissing-Olesen L, Stevens B. New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol. 2016;36:128-34
30. Neniskyte U, Gross CT. Errant gardeners: glial-cell-dependent synaptic pruning and neurodevelopmental disorders. Nat Rev Neurosci. 2017;18:658-70
31. Berbel P, Innocenti GM. The development of the corpus callosum in cats: a light- and electron-microscopic study. J Comp Neurol. 1988;276:132-56
32. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P. et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456-8
33. Schafer DP, Stevens B. Microglia Function in Central Nervous System Development and Plasticity. Cold Spring Harb Perspect Biol. 2015;7:a020545
34. Clarke LE, Barres BA. Emerging roles of astrocytes in neural circuit development. Nat Rev Neurosci. 2013;14:311-21
35. Lee E, Chung W-S. Glial Control of Synapse Number in Healthy and Diseased Brain. Front Cell Neurosci. 2019;13:42 -
36. Bolger G. The PDE4 cAMP-Specific Phosphodiesterases: Targets for Drugs with Antidepressant and Memory-Enhancing Action. 2017; p: 63-102.
37. Sanderson TM, Sher E. The role of phosphodiesterases in hippocampal synaptic plasticity. Neuropharmacology. 2013;74:86-95
38. Conti M, Beavo J. Biochemistry and Physiology of Cyclic Nucleotide Phosphodiesterases: Essential Components in Cyclic Nucleotide Signaling. Annu Rev Biochem. 2007;76:481-511
39. Tapia JC, Wylie JD, Kasthuri N, Hayworth KJ, Schalek R, Berger DR. et al. Pervasive synaptic branch removal in the mammalian neuromuscular system at birth. Neuron. 2012;74:816-29
40. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P. et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456-8
41. Xia X, Jiang Q, McDermott J, Han J-DJ. Aging and Alzheimer's disease: Comparison and associations from molecular to system level. Aging cell. 2018;17:e12802-e
42. Sigurdsson T, Duvarci S. Hippocampal-Prefrontal Interactions in Cognition, Behavior and Psychiatric Disease. Front Syst Neurosci. 2016 9
43. Samson RD, Barnes CA. Impact of aging brain circuits on cognition. Eur J Neurosci. 2013;37:1903-15
44. Zilberter Y, Zilberter M. The vicious circle of hypometabolism in neurodegenerative diseases: Ways and mechanisms of metabolic correction. J Neurosci Res. 2017;95:2217-35
45. Noe CR, Noe-Letschnig M, Handschuh P, Noe CA, Lanzenberger R. Dysfunction of the Blood-Brain Barrier—A Key Step in Neurodegeneration and Dementia. Front Aging Neurosci. 2020 12
46. Camandola S, Mattson MP. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017;36:1474-92
47. Heckman P, Wouters C, Prickaerts J. Phosphodiesterase inhibitors as a target for cognition enhancement in aging and Alzheimer's disease: a translational overview. Curr Pharm Des. 2015;21:317-31
48. Prickaerts J, Heckman PRA, Blokland A. Investigational phosphodiesterase inhibitors in phase I and phase II clinical trials for Alzheimer's disease. Expert Opin Investig Drugs. 2017;26:1033-48
49. Mosser CA, Baptista S, Arnoux I, Audinat E. Microglia in CNS development: Shaping the brain for the future. Prog Neurobiol. 2017;149-150:1-20
50. Meyer D, Bonhoeffer T, Scheuss V. Balance and stability of synaptic structures during synaptic plasticity. Neuron. 2014;82:430-43
51. Feldman DE. Developmental Synaptic Plasticity: LTP, LTD, and Synapse Formation and Elimination. In: Squire LR, editor. Encyclopedia of Neuroscience. Oxford: Academic Press. 2009 p: 495-501
52. Piochon C, Kano M, Hansel C. LTD-like molecular pathways in developmental synaptic pruning. Nat Neurosci. 2016;19:1299-310
53. Kandel ER. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Molecular brain. 2012;5:14
54. Kowianski P, Lietzau G, Czuba E, Waskow M, Steliga A, Morys J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell Mol Neurobiol. 2018;38:579-93
55. Boess FG, Hendrix M, van der Staay FJ, Erb C, Schreiber R, van Staveren W. et al. Inhibition of phosphodiesterase 2 increases neuronal cGMP, synaptic plasticity and memory performance. Neuropharmacology. 2004;47:1081-92
56. Soares LM, Meyer E, Milani H, Steinbusch HWM, Prickaerts J, de Oliveira RMW. The phosphodiesterase type 2 inhibitor BAY 60-7550 reverses functional impairments induced by brain ischemia by decreasing hippocampal neurodegeneration and enhancing hippocampal neuronal plasticity. Eur J Neurosci. 2017;45:510-20
57. Xu Y, Pan J, Sun J, Ding L, Ruan L, Reed M. et al. Inhibition of phosphodiesterase 2 reverses impaired cognition and neuronal remodeling caused by chronic stress. Neurobiol Aging. 2015;36:955-70
58. Yoneyama M, Tanaka M, Hasebe S, Yamaguchi T, Shiba T, Ogita K. Beneficial effect of cilostazol-mediated neuronal repair following trimethyltin-induced neuronal loss in the dentate gyrus. J Neurosci Res. 2015;93:56-66
59. Beaumont V, Zhong S, Lin H, Xu W, Bradaia A, Steidl E. et al. Phosphodiesterase 10A Inhibition Improves Cortico-Basal Ganglia Function in Huntington's Disease Models. Neuron. 2016;92:1220-37
60. Puerta E, Hervias I, Barros-Miñones L, Jordan J, Ricobaraza A, Cuadrado-Tejedor M. et al. Sildenafil protects against 3-nitropropionic acid neurotoxicity through the modulation of calpain, CREB, and BDNF. Neurobiol Dis. 2010;38:237-45
61. Li J, Liu C-N, Wei N, Li X-D, Liu Y-Y, Yang R. et al. Protective effects of BAY 73-6691, a selective inhibitor of phosphodiesterase 9, on amyloid-β peptides-induced oxidative stress in in-vivo and in-vitro models of Alzheimer's disease. Brain Res. 2016;1642:327-35
62. Feng H, Wang C, He W, Wu X, Li S, Zeng Z. et al. Roflumilast ameliorates cognitive impairment in APP/PS1 mice via cAMP/CREB/BDNF signaling and anti-neuroinflammatory effects. Metab Brain Dis. 2019;34:583-91
63. Zhang C, Xu Y, Chowdhary A, Fox D, Gurney ME, Zhang H-T. et al. Memory enhancing effects of BPN14770, an allosteric inhibitor of phosphodiesterase-4D, in wild-type and humanized mice. Neuropsychopharmacology. 2018;43:2299-309
64. Li Y-F, Cheng Y-F, Huang Y, Conti M, Wilson SP, O'Donnell JM. et al. Phosphodiesterase-4D Knock-Out and RNA Interference-Mediated Knock-Down Enhance Memory and Increase Hippocampal Neurogenesis via Increased cAMP Signaling. J Neurosci. 2011;31:172-83
65. Campbell SL, van Groen T, Kadish I, Smoot LHM, Bolger GB. Altered phosphorylation, electrophysiology, and behavior on attenuation of PDE4B action in hippocampus. BMC Neurosci. 2017;18:77
66. Schepers M, Tiane A, Paes D, Sanchez S, Rombaut B, Piccart E. et al. Targeting phosphodiesterases (PDE)-towards a tailor-made approach in multiple sclerosis treatment. Front Immunol. 2019;10:1727
67. Gupta S, Singh P, Sharma BM, Sharma B. Neuroprotective Effects of Agomelatine and Vinpocetine Against Chronic Cerebral Hypoperfusion Induced Vascular Dementia. Curr Neurovasc Res. 2015;12:240-52
68. Kroker KS, Mathis C, Marti A, Cassel J-C, Rosenbrock H, Dorner-Ciossek C. PDE9A inhibition rescues amyloid beta-induced deficits in synaptic plasticity and cognition. Neurobiol Aging. 2014;35:2072-8
69. Schaal SM, Garg MS, Ghosh M, Lovera L, Lopez M, Patel M. et al. The therapeutic profile of rolipram, PDE target and mechanism of action as a neuroprotectant following spinal cord injury. PLoS One. 2012 7
70. Nikulina E, Tidwell JL, Dai HN, Bregman BS, Filbin MT. The phosphodiesterase inhibitor rolipram delivered after a spinal cord lesion promotes axonal regeneration and functional recovery. Proc Natl Acad Sci U S A. 2004;101:8786-90
71. Pearse DD, Pereira FC, Marcillo AE, Bates ML, Berrocal YA, Filbin MT. et al. cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat Med. 2004;10:610-6
72. Hulley P, Hartikka J, Abdel'Al S, Engels P, Buerki HR, Wiederhold KH. et al. Inhibitors of type IV phosphodiesterases reduce the toxicity of MPTP in substantia nigra neurons in vivo. Eur J Neurosci. 1995;7:2431-40
73. DeMarch Z, Giampà C, Patassini S, Bernardi G, Fusco FR. Beneficial effects of rolipram in the R6/2 mouse model of Huntington's disease. Neurobiol Dis. 2008;30:375-87
74. Giampà C, Patassini S, Borreca A, Laurenti D, Marullo F, Bernardi G. et al. Phosphodiesterase 10 inhibition reduces striatal excitotoxicity in the quinolinic acid model of Huntington's disease. Neurobiol Dis. 2009;34:450-6
75. Yang L, Calingasan NY, Lorenzo BJ, Beal MF. Attenuation of MPTP neurotoxicity by rolipram, a specific inhibitor of phosphodiesterase IV. Exp Neurol. 2008;211:311-4
76. Huang H, Ma Z-C, Wang Y-G, Hong Q, Tan H-L, Xiao C-R. et al. Ferulic acid alleviates Aβ25-35-and lipopolysaccharide-induced PC12 cellular damage: a potential role in Alzheimer's disease by PDE inhibition. Int J Clin Pharmacol Ther. 2015;53:828-37
77. Giembycz MA. Life after PDE4: overcoming adverse events with dual-specificity phosphodiesterase inhibitors. Curr Opin Pharmacol. 2005;5:238-44
78. Morales-Garcia JA, Redondo M, Alonso-Gil S, Gil C, Perez C, Martinez A. et al. Phosphodiesterase 7 inhibition preserves dopaminergic neurons in cellular and rodent models of Parkinson disease. PloS one. 2011 6
79. Perez-Gonzalez R, Pascual C, Antequera D, Bolos M, Redondo M, Perez DI. et al. Phosphodiesterase 7 inhibitor reduced cognitive impairment and pathological hallmarks in a mouse model of Alzheimer's disease. Neurobiol Aging. 2013;34:2133-45
80. Martinez A, Gil C. cAMP-specific phosphodiesterase inhibitors: promising drugs for inflammatory and neurological diseases. Expert Opin Ther Pat. 2014;24:1311-21
81. Tsai LC, Chan GK, Nangle SN, Shimizu-Albergine M, Jones G, Storm DR. et al. Inactivation of Pde8b enhances memory, motor performance, and protects against age-induced motor coordination decay. Genes Brain Behav. 2012;11:837-47
82. Parisiadou L, Yu J, Sgobio C, Xie C, Liu G, Sun L. et al. LRRK2 regulates synaptogenesis and dopamine receptor activation through modulation of PKA activity. Nat Neurosci. 2014;17:367-76
83. Skeberdis VA, Chevaleyre V, Lau CG, Goldberg JH, Pettit DL, Suadicani SO. et al. Protein kinase A regulates calcium permeability of NMDA receptors. Nat Neurosci. 2006;9:501-10
84. Terrin A, Monterisi S, Stangherlin A, Zoccarato A, Koschinski A, Surdo NC. et al. PKA and PDE4D3 anchoring to AKAP9 provides distinct regulation of cAMP signals at the centrosome. J Cell Biol. 2012;198:607-21
85. Logue MW, Schu M, Vardarajan BN, Farrell J, Bennett DA, Buxbaum JD. et al. Two rare AKAP9 variants are associated with Alzheimer's disease in African Americans. Alzheimers Dement. 2014;10:609-18.e11
86. Vilalta A, Brown GC. Neurophagy, the phagocytosis of live neurons and synapses by glia, contributes to brain development and disease. FEBS J. 2018;285:3566-75
87. Huang C-Y, Ye Z-H, Huang M-Y, Lu J-J. Regulation of CD47 expression in cancer cells. Transl Oncol. 2020;13:100862
88. Lehrman EK, Wilton DK, Litvina EY, Welsh CA, Chang ST, Frouin A. et al. CD47 Protects Synapses from Excess Microglia-Mediated Pruning during Development. Neuron. 2018;100:120-34.e6
89. Ginhoux F, Prinz M. Origin of microglia: current concepts and past controversies. Cold Spring Harb Perspect Biol. 2015;7:a020537
90. Smolders SM, Kessels S, Vangansewinkel T, Rigo JM, Legendre P, Brone B. Microglia: Brain cells on the move. Prog Neurobiol. 2019;178:101612
91. Ginhoux F, Guilliams M. Editorial for Cellular Immunology special issue on "Tissue Macrophages". Cell Immunol. 2018;330:1-4
92. Filiano AJ, Gadani SP, Kipnis J. Interactions of innate and adaptive immunity in brain development and function. Brain Res. 2015;1617:18-27
93. Chew LJ, Takanohashi A, Bell M. Microglia and inflammation: impact on developmental brain injuries. Ment Retard Dev Disabil Res Rev. 2006;12:105-12
94. Zhao W, Xie W, Xiao Q, Beers DR, Appel SH. Protective effects of an anti-inflammatory cytokine, interleukin-4, on motoneuron toxicity induced by activated microglia. J Neurochem. 2006;99:1176-87
95. Haskó G, Szabó C, Németh ZH, Salzman AL, Vizi ES. Suppression of IL-12 production by phosphodiesterase inhibition in murine endotoxemia is IL-10 independent. Eur J Immunol. 1998;28:468-72
96. Santiago A, Soares LM, Schepers M, Milani H, Vanmierlo T, Prickaerts J. et al. Roflumilast promotes memory recovery and attenuates white matter injury in aged rats subjected to chronic cerebral hypoperfusion. Neuropharmacology. 2018;138:360-70
97. Wood PL. Differential regulation of IL-1α and TNFα release from immortalized murine microglia (BV-2). Life Sci. 1994;55:661-8
98. Burgin AB, Magnusson OT, Singh J, Witte P, Staker BL, Bjornsson JM. et al. Design of phosphodiesterase 4D (PDE4D) allosteric modulators for enhancing cognition with improved safety. Nat Biotechnol. 2010;28:63-70
99. Song GJ, Suk K. Pharmacological Modulation of Functional Phenotypes of Microglia in Neurodegenerative Diseases. Front Aging Neurosci. 2017;9:139 -
100. Pifarré P, Gutierrez-Mecinas M, Prado J, Usero L, Roura-Mir C, Giralt M. et al. Phosphodiesterase 5 inhibition at disease onset prevents experimental autoimmune encephalomyelitis progression through immunoregulatory and neuroprotective actions. Exp Neurol. 2014;251:58-71
101. Anwar MA, Shah M, Kim J, Choi S. Recent clinical trends in Toll-like receptor targeting therapeutics. Med Res Rev. 2019;39:1053-90
102. Russo I, Di Benedetto G, Kaganovich A, Ding J, Mercatelli D, Morari M. et al. Leucine-rich repeat kinase 2 controls protein kinase A activation state through phosphodiesterase 4. J Neuroinflammation. 2018;15:297
103. Singh A, Zhi L, Zhang H. LRRK2 and mitochondria: Recent advances and current views. Brain Res. 2019;1702:96-104
104. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314-8
105. Augusto-Oliveira M, Arrifano GP, Lopes-Araujo A, Santos-Sacramento L, Takeda PY, Anthony DC. et al. What Do Microglia Really Do in Healthy Adult Brain? Cells. 2019 8
106. Li Y, Du XF, Liu CS, Wen ZL, Du JL. Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Dev Cell. 2012;23:1189-202
107. Fontainhas AM, Wang M, Liang KJ, Chen S, Mettu P, Damani M. et al. Microglial morphology and dynamic behavior is regulated by ionotropic glutamatergic and GABAergic neurotransmission. PLoS One. 2011;6:e15973
108. Wolf SA, Boddeke HW, Kettenmann H. Microglia in Physiology and Disease. Annu Rev Physiol. 2017;79:619-43
109. Sierra A, Abiega O, Shahraz A, Neumann H. Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Front Cell Neurosci. 2013;7:6
110. Marquez-Ropero M, Benito E, Plaza-Zabala A, Sierra A. Microglial Corpse Clearance: Lessons From Macrophages. Front Immunol. 2020;11:506
111. Low D, Ginhoux F. Recent advances in the understanding of microglial development and homeostasis. Cell Immunol. 2018;330:68-78
112. Bernier LP, Bohlen CJ, York EM, Choi HB, Kamyabi A, Dissing-Olesen L. et al. Nanoscale Surveillance of the Brain by Microglia via cAMP-Regulated Filopodia. Cell Rep. 2019;27:2895-908 e4
113. Pearse DD, Hughes ZA. PDE4B as a microglia target to reduce neuroinflammation. Glia. 2016;64:1698-709
114. Schmidt C, Schneble N, Müller JP, Bauer R, Perino A, Marone R. et al. Phosphoinositide 3-kinase γ mediates microglial phagocytosis via lipid kinase-independent control of cAMP. Neuroscience. 2013;233:44-53
115. Rossi AG, McCutcheon JC, Roy N, Chilvers ER, Haslett C, Dransfield I. Regulation of macrophage phagocytosis of apoptotic cells by cAMP. J Immunol (Baltimore, Md: 1950). 1998;160:3562-8
116. Weinhard L, di Bartolomei G, Bolasco G, Machado P, Schieber NL, Neniskyte U. et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat Commun. 2018;9:1228 -
117. Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ. et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. 2013;155:1596-609
118. Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S. et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352:712-6
119. Wu Y, Dissing-Olesen L, MacVicar BA, Stevens B. Microglia: Dynamic Mediators of Synapse Development and Plasticity. Trends Immunol. 2015;36:605-13
120. Whitelaw BS. Microglia-mediated synaptic elimination in neuronal development and disease. J Neurophysiol. 2018;119:1-4
121. Wilton DK, Dissing-Olesen L, Stevens B. Neuron-Glia Signaling in Synapse Elimination. Annu Rev Neurosci. 2019;42:107-27
122. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N. et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164-78
123. Bialas AR, Stevens B. TGF-β signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat Neurosci. 2013;16:1773-82
124. Caraci F, Gulisano W, Guida CA, Impellizzeri AAR, Drago F, Puzzo D. et al. A key role for TGF-β1 in hippocampal synaptic plasticity and memory. Sci Rep. 2015;5:11252
125. Presumey J, Bialas AR, Carroll MC. Complement System in Neural Synapse Elimination in Development and Disease. Adv Immunol. 2017;135:53-79
126. Lui H, Zhang J, Makinson SR, Cahill MK, Kelley KW, Huang HY. et al. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell. 2016;165:921-35
127. Vasek MJ, Garber C, Dorsey D, Durrant DM, Bollman B, Soung A. et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature. 2016;534:538-43
128. Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N. et al. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530:177-83
129. Druart M, Le Magueresse C. Emerging Roles of Complement in Psychiatric Disorders. Front Psychiatry. 2019;10:573
130. Chu Y, Jin X, Parada I, Pesic A, Stevens B, Barres B. et al. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc Natl Acad Sci U S A. 2010;107:7975-80
131. Hammond JW, Bellizzi MJ, Ware C, Qiu WQ, Saminathan P, Li H. et al. Complement-dependent synapse loss and microgliosis in a mouse model of multiple sclerosis. Brain Behav Immun. 2020
132. Czirr E, Castello NA, Mosher KI, Castellano JM, Hinkson IV, Lucin KM. et al. Microglial complement receptor 3 regulates brain Abeta levels through secreted proteolytic activity. J Exp Med. 2017;214:1081-92
133. Dagher NN, Najafi AR, Kayala KMN, Elmore MRP, White TE, Medeiros R. et al. Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg-AD mice. J Neuroinflammation. 2015;12:139 -
134. Anderson B, Rutledge V. Age and hemisphere effects on dendritic structure. Brain. 1996;119:1983-90
135. Stephan AH, Madison DV, Mateos JM, Fraser DA, Lovelett EA, Coutellier L. et al. A dramatic increase of C1q protein in the CNS during normal aging. J Neurosci. 2013;33:13460-74
136. Luchena C, Zuazo-Ibarra J, Alberdi E, Matute C, Capetillo-Zarate E. Corrigendum to "Contribution of Neurons and Glial Cells to Complement-Mediated Synapse Removal during Development, Aging and in Alzheimer's Disease". Mediators Inflamm. 2019;2019:7539620
137. VanGuilder HD, Bixler GV, Brucklacher RM, Farley JA, Yan H, Warrington JP. et al. Concurrent hippocampal induction of MHC II pathway components and glial activation with advanced aging is not correlated with cognitive impairment. J Neuroinflammation. 2011;8:138
138. Nonaka S, Nakanishi H. Microglial clearance of focal apoptotic synapses. Neurosci Lett. 2019;707:134317
139. Sierra A, Gottfried-Blackmore AC, McEwen BS, Bulloch K. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia. 2007;55:412-24
140. Yuan C, Aierken A, Xie Z, Li N, Zhao J, Qing H. The age-related microglial transformation in Alzheimer's disease pathogenesis. Neurobiol Aging. 2020;92:82-91
141. Makranz C, Cohen G, Reichert F, Kodama T, Rotshenker S. cAMP cascade (PKA, Epac, adenylyl cyclase, Gi, and phosphodiesterases) regulates myelin phagocytosis mediated by complement receptor-3 and scavenger receptor-AI/II in microglia and macrophages. Glia. 2006;53:441-8
142. Serezani CH, Ballinger MN, Aronoff DM, Peters-Golden M. Cyclic AMP: master regulator of innate immune cell function. Am J Respir Cell Mol Biol. 2008;39:127-32
143. Prinz M, Priller J, Sisodia SS, Ransohoff RM. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat Neurosci. 2011;14:1227-35
144. Goldmann T, Wieghofer P, Jordao MJ, Prutek F, Hagemeyer N, Frenzel K. et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol. 2016;17:797-805
145. Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. 2014;15:300-12
146. Yamamoto S, Muramatsu M, Azuma E, Ikutani M, Nagai Y, Sagara H. et al. A subset of cerebrovascular pericytes originates from mature macrophages in the very early phase of vascular development in CNS. Sci Rep. 2017;7:3855
147. Zhang GX, Li J, Ventura E, Rostami A. Parenchymal microglia of naive adult C57BL/6J mice express high levels of B7.1, B7.2, and MHC class II. Exp Mol Pathol. 2002;73:35-45
148. Wohleb ES, Delpech JC. Dynamic cross-talk between microglia and peripheral monocytes underlies stress-induced neuroinflammation and behavioral consequences. Prog Neuropsychopharmacol Biol Psychiatry. 2017;79:40-8
149. Linnartz-Gerlach B, Schuy C, Shahraz A, Tenner AJ, Neumann H. Sialylation of neurites inhibits complement-mediated macrophage removal in a human macrophage-neuron Co-Culture System. Glia. 2016;64:35-47
150. Costello DA, Keenan K, McManus RM, Falvey A, Lynch MA. The age-related neuroinflammatory environment promotes macrophage activation, which negatively impacts synaptic function. Neurobiol Aging. 2016;43:140-8
151. Girard S, Brough D, Lopez-Castejon G, Giles J, Rothwell NJ, Allan SM. Microglia and macrophages differentially modulate cell death after brain injury caused by oxygen-glucose deprivation in organotypic brain slices. Glia. 2013;61:813-24
152. Blau CW, Cowley TR, O'Sullivan J, Grehan B, Browne TC, Kelly L. et al. The age-related deficit in LTP is associated with changes in perfusion and blood-brain barrier permeability. Neurobiol Aging. 2012;33:1005 e23-35
153. Krukowski K, Chou A, Feng X, Tiret B, Paladini MS, Riparip LK. et al. Traumatic Brain Injury in Aged Mice Induces Chronic Microglia Activation, Synapse Loss, and Complement-Dependent Memory Deficits. Int J Mol Sci. 2018 19
154. Yang JX, Hsiung TC, Weng FC, Ding SL, Wu CP, Conti M. et al. Synergistic effect of phosphodiesterase 4 inhibitor and serum on migration of endotoxin-stimulated macrophages. Innate Immun. 2018;24:501-12
155. Gantner F, Kupferschmidt R, Schudt C, Wendel A, Hatzelmann A. In vitro differentiation of human monocytes to macrophages: change of PDE profile and its relationship to suppression of tumour necrosis factor-alpha release by PDE inhibitors. Br J Pharmacol. 1997;121:221-31
156. Shepherd MC, Baillie GS, Stirling DI, Houslay MD. Remodelling of the PDE4 cAMP phosphodiesterase isoform profile upon monocyte-macrophage differentiation of human U937 cells. Br J Pharmacol. 2004;142:339-51
157. Theodoropoulou S, Spanakos G, Baxevanis CN, Economou M, Gritzapis AD, Papamichail MP. et al. Cytokine serum levels, autologous mixed lymphocyte reaction and surface marker analysis in never medicated and chronically medicated schizophrenic patients. Schizophr Res. 2001;47:13-25
158. Nikkila HV, Muller K, Ahokas A, Rimon R, Andersson LC. Increased frequency of activated lymphocytes in the cerebrospinal fluid of patients with acute schizophrenia. Schizophr Res. 2001;49:99-105
159. Ormel PR, van Mierlo HC, Litjens M, Strien MEV, Hol EM, Kahn RS. et al. Characterization of macrophages from schizophrenia patients. NPJ Schizophr. 2017;3:41
160. Schwartz M, Shechter R. Systemic inflammatory cells fight off neurodegenerative disease. Nat Rev Neurol. 2010;6:405-10
161. Solito E, Sastre M. Microglia function in Alzheimer's disease. Front Pharmacol. 2012;3:14
162. Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489-502
163. Hawkes CA, McLaurin J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc Natl Acad Sci U S A. 2009;106:1261-6
164. Koronyo Y, Salumbides BC, Sheyn J, Pelissier L, Li S, Ljubimov V. et al. Therapeutic effects of glatiramer acetate and grafted CD115(+) monocytes in a mouse model of Alzheimer's disease. Brain. 2015;138:2399-422
165. Bernstein KE, Gonzalez-Villalobos RA, Giani JF, Shah K, Bernstein E, Janjulia T. et al. Angiotensin-converting enzyme overexpression in myelocytes enhances the immune response. Biol Chem. 2014;395:1173-8
166. Li S, Hayden EY, Garcia VJ, Fuchs DT, Sheyn J, Daley DA. et al. Activated Bone Marrow-Derived Macrophages Eradicate Alzheimer's-Related Abeta42 Oligomers and Protect Synapses. Front Immunol. 2020;11:49
167. Hertz AL, Beavo JA. Cyclic nucleotides and phosphodiesterases in monocytic differentiation. Handb Exp Pharmacol. 2011:365-90
168. Kim SH, Serezani CH, Okunishi K, Zaslona Z, Aronoff DM, Peters-Golden M. Distinct protein kinase A anchoring proteins direct prostaglandin E2 modulation of Toll-like receptor signaling in alveolar macrophages. J Biol Chem. 2011;286:8875-83
169. Clister T, Greenwald EC, Baillie GS, Zhang J. AKAP95 Organizes a Nuclear Microdomain to Control Local cAMP for Regulating Nuclear PKA. Cell Chem Biol. 2019;26:885-91.e4
170. Chung W-S, Allen NJ, Eroglu C. Astrocytes Control Synapse Formation, Function, and Elimination. Cold Spring Harb Perspect Biol. 2015;7:a020370-a
171. Kuijlaars J, Oyelami T, Diels A, Rohrbacher J, Versweyveld S, Meneghello G. et al. Sustained synchronized neuronal network activity in a human astrocyte co-culture system. Sci Rep. 2016;6:36529
172. Barker AJ, Koch SM, Reed J, Barres BA, Ullian EM. Developmental Control of Synaptic Receptivity. Journal Neurosci. 2008;28:8150-60
173. Chung W-S, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C. et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature. 2013;504:394-400
174. Gomez-Arboledas A, Davila JC, Sanchez-Mejias E, Navarro V, Nunez-Diaz C, Sanchez-Varo R. et al. Phagocytic clearance of presynaptic dystrophies by reactive astrocytes in Alzheimer's disease. Glia. 2018;66:637-53
175. Sheerin NS, Zhou W, Adler S, Sacks SH. TNF-<em>α</em> regulation of C3 gene expression and protein biosynthesis in rat glomerular endothelial cells. Kidney Int. 1997;51:703-10
176. Zhou Z, Ikegaya Y, Koyama R. The Astrocytic cAMP Pathway in Health and Disease. Int J Mol Sci. 2019;20:779
177. Olabarria M, Noristani HN, Verkhratsky A, Rodriguez JJ. Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer's disease. Glia. 2010;58:831-8
178. Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F. et al. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med. 2003;9:453-7
179. Pihlaja R, Koistinaho J, Malm T, Sikkila H, Vainio S, Koistinaho M. Transplanted astrocytes internalize deposited beta-amyloid peptides in a transgenic mouse model of Alzheimer's disease. Glia. 2008;56:154-63
180. Garwood CJ, Pooler AM, Atherton J, Hanger DP, Noble W. Astrocytes are important mediators of Abeta-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis. 2011;2:e167
181. Iram T, Trudler D, Kain D, Kanner S, Galron R, Vassar R. et al. Astrocytes from old Alzheimer's disease mice are impaired in Abeta uptake and in neuroprotection. Neurobiol Dis. 2016;96:84-94
182. Chung WS, Verghese PB, Chakraborty C, Joung J, Hyman BT, Ulrich JD. et al. Novel allele-dependent role for APOE in controlling the rate of synapse pruning by astrocytes. Proc Natl Acad Sci U S A. 2016;113:10186-91
183. Lin Y-T, Seo J, Gao F, Feldman HM, Wen H-L, Penney J. et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer's Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron. 2018;98:1141-54.e7
184. Zagami CJ, O'Shea RD, Lau CL, Cheema SS, Beart PM. Regulation of glutamate transporters in astrocytes: evidence for a relationship between transporter expression and astrocytic phenotype. Neurotox Res. 2005;7:143-9
185. Shin JY, Fang ZH, Yu ZX, Wang CE, Li SH, Li XJ. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol. 2005;171:1001-12
186. Faideau M, Kim J, Cormier K, Gilmore R, Welch M, Auregan G. et al. In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with Huntington's disease subjects. Hum Mol Genet. 2010;19:3053-67
187. Gu XL, Long CX, Sun L, Xie C, Lin X, Cai H. Astrocytic expression of Parkinson's disease-related A53T alpha-synuclein causes neurodegeneration in mice. Mol Brain. 2010;3:12
188. Yoon B-E, Lee CJ. GABA as a rising gliotransmitter. Front Neural Circuits. 2014 8
189. Ortinski PI, Dong J, Mungenast A, Yue C, Takano H, Watson DJ. et al. Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat Neurosci. 2010;13:584-91
190. Jo S, Yarishkin O, Hwang YJ, Chun YE, Park M, Woo DH. et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer's disease. Nat Med. 2014;20:886-96
191. Mitew S, Kirkcaldie MTK, Dickson TC, Vickers JC. Altered synapses and gliotransmission in Alzheimer's disease and AD model mice. Neurobiol Aging. 2013;34:2341-51
192. Wojtowicz AM, Dvorzhak A, Semtner M, Grantyn R. Reduced tonic inhibition in striatal output neurons from Huntington mice due to loss of astrocytic GABA release through GAT-3. Front Neural Circuits. 2013;7:188
193. de Santana Nunes AK, Raposo C, Bjorklund U, da Cruz-Hofling MA, Peixoto CA, Hansson E. Sildenafil (Viagra((R))) prevents and restores LPS-induced inflammation in astrocytes. Neurosci Lett. 2016;630:59-65
194. Cueva Vargas JL, Belforte N, Di Polo A. The glial cell modulator ibudilast attenuates neuroinflammation and enhances retinal ganglion cell viability in glaucoma through protein kinase A signaling. Neurobiol Dis. 2016;93:156-71
195. Takuma K, Lee E, Enomoto R, Mori K, Baba A, Matsuda T. Ibudilast attenuates astrocyte apoptosis via cyclic GMP signalling pathway in an in vitro reperfusion model. Br J Pharmacol. 2001;133:841-8
196. Gallo V, Ghiani CA. Glutamate receptors in glia: new cells, new inputs and new functions. Trends Pharmacol Sci. 2000;21:252-8
197. Jang M, Gould E, Xu J, Kim EJ, Kim JH. Oligodendrocytes regulate presynaptic properties and neurotransmission through BDNF signaling in the mouse brainstem. Elife. 2019;8:e42156
198. Rosenblum K. 4.13 - Conditioned Taste Aversion and Taste Learning: Molecular Mechanisms. In: Byrne JH, editor. Learning and Memory: A Comprehensive Reference. Oxford: Academic Press; 2008. p. 217-34
199. Molinari C, Morsanuto V, Ruga S, Notte F, Farghali M, Galla R. et al. The Role of BDNF on Aging-Modulation Markers. Brain Sci. 2020;10:285
200. Wang S, Yao H, Xu Y, Hao R, Zhang W, Liu H. et al. Therapeutic potential of a TrkB agonistic antibody for Alzheimer's disease. Theranostics. 2020;10:6854-74
201. Nutma E, van Gent D, Amor S, Peferoen LAN. Astrocyte and Oligodendrocyte Cross-Talk in the Central Nervous System. Cells. 2020 9
202. Hirrlinger J, Nave KA. Adapting brain metabolism to myelination and long-range signal transduction. Glia. 2014;62:1749-61
203. Mot AI, Depp C, Nave KA. An emerging role of dysfunctional axon-oligodendrocyte coupling in neurodegenerative diseases. Dialogues Clin Neurosci. 2018;20:283-92
204. Andrea R, Ilaria V, José Julio Rodríguez A, Arthur B. Decreased Regenerative Capacity of Oligodendrocyte Progenitor Cells (NG2-Glia) in the Ageing Brain: A Vicious Cycle of Synaptic Dysfunction, Myelin Loss and Neuronal Disruption? Curr Alzheimer Res. 2016;13:413-8
205. Bergles DE, Roberts JD, Somogyi P, Jahr CE. Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature. 2000;405:187-91
206. Okuda H, Tatsumi K, Makinodan M, Yamauchi T, Kishimoto T, Wanaka A. Environmental enrichment stimulates progenitor cell proliferation in the amygdala. J Neurosci Res. 2009;87:3546-53
207. Fields RD. A new mechanism of nervous system plasticity: activity-dependent myelination. Nat Rev Neurosci. 2015;16:756-67
208. Sun X, Liu Y, Liu B, Xiao Z, Zhang L. Rolipram promotes remyelination possibly via MEK-ERK signal pathway in cuprizone-induced demyelination mouse. Exp Neurol. 2012;237:304-11
209. Syed YA, Baer A, Hofer MP, González GA, Rundle J, Myrta S. et al. Inhibition of phosphodiesterase-4 promotes oligodendrocyte precursor cell differentiation and enhances CNS remyelination. EMBO Mol Med. 2013;5:1918-34
210. Hughes AN, Appel B. Oligodendrocytes express synaptic proteins that modulate myelin sheath formation. Nat Commun. 2019;10:4125
211. Baillie GS, Tejeda GS, Kelly MP. Therapeutic targeting of 3′,5′-cyclic nucleotide phosphodiesterases: inhibition and beyond. Nat Rev Drug Discov. 2019;18:770-96
212. Biber K, Neumann H, Inoue K, Boddeke HWGM. Neuronal "On" and "Off" signals control microglia. Trends Neurosci. 2007;30:596-602
213. Qin L, Bouchard R, Pugazhenthi S. Regulation of cyclic AMP response element-binding protein during neuroglial interactions. J Neurochem. 2016;136:918-30
214. BA B. Brain RNA-Seq
215. Boess FG, Hendrix M, van der Staay FJ, Erb C, Schreiber R, van Staveren W. et al. Inhibition of phosphodiesterase 2 increases neuronal cGMP, synaptic plasticity and memory performance. Neuropharmacology. 2004;47:1081-92
216. Xu Y, Pan J, Sun J, Ding L, Ruan L, Reed M. et al. Inhibition of phosphodiesterase 2 reverses impaired cognition and neuronal remodeling caused by chronic stress. Neurobiol Aging. 2015;36:955-70
217. Li Y-F, Cheng Y-F, Huang Y, Conti M, Wilson SP, O'Donnell JM. et al. Phosphodiesterase-4D Knock-Out and RNA Interference-Mediated Knock-Down Enhance Memory and Increase Hippocampal Neurogenesis via Increased cAMP Signaling. Journal Neurosci. 2011;31:172-83
218. Zhang C, Xu Y, Chowdhary A, Fox D, Gurney ME, Zhang H-T. et al. Memory enhancing effects of BPN14770, an allosteric inhibitor of phosphodiesterase-4D, in wild-type and humanized mice. Neuropsychopharmacology. 2018;43:2299-309
219. Feng H, Wang C, He W, Wu X, Li S, Zeng Z. et al. Roflumilast ameliorates cognitive impairment in APP/PS1 mice via cAMP/CREB/BDNF signaling and anti-neuroinflammatory effects. Metab Brain Dis. 2019;34:583-91
220. Hulley P, Hartikka J, Abdel'Al S, Engels P, Buerki HR, Wiederhold KH. et al. Inhibitors of type IV phosphodiesterases reduce the toxicity of MPTP in substantia nigra neurons in vivo. Eur J Neurosci. 1995;7:2431-40
221. Pearse DD, Pereira FC, Marcillo AE, Bates ML, Berrocal YA, Filbin MT. et al. cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat Med. 2004;10:610-6
222. Giampà C, Patassini S, Borreca A, Laurenti D, Marullo F, Bernardi G. et al. Phosphodiesterase 10 inhibition reduces striatal excitotoxicity in the quinolinic acid model of Huntington's disease. Neurobiol Dis. 2009;34:450-6
223. Schaal SM, Garg MS, Ghosh M, Lovera L, Lopez M, Patel M. et al. The therapeutic profile of rolipram, PDE target and mechanism of action as a neuroprotectant following spinal cord injury. PLoS One. 2012 7
224. Huang H, Ma Z-C, Wang Y-G, Hong Q, Tan H-L, Xiao C-R. et al. Ferulic acid alleviates Aβ25-35-and lipopolysaccharide-induced PC12 cellular damage: a potential role in Alzheimer's disease by PDE inhibition. Int J Clin Pharmacol Ther. 2015;53:828-37
225. Beaumont V, Zhong S, Lin H, Xu W, Bradaia A, Steidl E. et al. Phosphodiesterase 10A Inhibition Improves Cortico-Basal Ganglia Function in Huntington's Disease Models. Neuron. 2016;92:1220-37
226. Sidiropoulos K, Viteri G, Sevilla C, Jupe S, Webber M, Orlic-Milacic M. et al. Reactome enhanced pathway visualization. Bioinformatics. 2017;33:3461-7
Author contact
![]() Corresponding author: Tim Vanmierlo, E-mail: tim.vanmierlobe; t.vanmierlonl.
Corresponding author: Tim Vanmierlo, E-mail: tim.vanmierlobe; t.vanmierlonl.