Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Communicating molecules derived...
Communications between the liver...
Conclusions
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(7):3317-3330. doi:10.7150/thno.55795 This issue Cite
Review
Organ-organ communication: The liver's perspective
Fei Wang1, Kwok-Fai So2,3, Jia Xiao4 ![]() , Hua Wang5,6
, Hua Wang5,6 ![]()
1. Division of Gastroenterology, Seventh Affiliated Hospital of Sun Yat-sen University, Shenzhen, China.
2. GMH Institute of CNS Regeneration, Guangdong Medical Key Laboratory of Brain Function and Diseases, Jinan University, Guangzhou, China.
3. Laboratory of Neuroendocrinology, Fujian Key Laboratory of Developmental and Neurobiology, School of Life Sciences, Fujian Normal University, Fuzhou, China.
4. Clinical Medicine Research Institute and Department of Interventional Surgery, First Affiliated Hospital of Jinan University, Guangzhou, China.
5. Department of Oncology, the First Affiliated Hospital of Anhui Medical University, Anhui Medical University, Hefei, China.
6. Inflammation and Immune Mediated Disease Laboratory of Anhui Province; School of Pharmacy, Anhui Medical University, Hefei, China.
Received 2020-11-12; Accepted 2020-12-28; Published 2021-1-16
Abstract

Communication between organs participates in most physiological and pathological events. Owing to the importance of precise coordination among the liver and virtually all organs in the body for the maintenance of homeostasis, many hepatic disorders originate from impaired organ-organ communication, resulting in concomitant pathological phenotypes of distant organs. Hepatokines are proteins that are predominantly secreted from the liver, and many hepatokines and several signaling proteins have been linked to diseases of other organs, such as the heart, muscle, bone, and eyes. Although liver-centered interorgan communication has been proposed in both basic and clinical studies, to date, the regulatory mechanisms of hepatokine production, secretion, and reciprocation with signaling factors from other organs are obscure. Whether other hormones and cytokines are involved in such communication also warrants investigation. Herein, we summarize the current knowledge of organ-organ communication phenotypes in a variety of diseases and the possible involvement of hepatokines and/or other important signaling factors. This provides novel insight into the underlying roles and mechanisms of liver-originated signal transduction and, more importantly, the understanding of disease in an integrative view.
Keywords: Liver, Organ communication, Hepatokine, Cytokine, Disease mechanism.
Introduction
Over the past 30 years, we have witnessed advances in the understanding of how organs and tissues communicate under healthy and pathologic conditions by secreting proteins, lipids, metabolites, and small noncoding RNAs. Now, it is considered that most tissues are able to communicate with local and distant tissues/organs in an autocrine/paracrine/endocrine manner. Interorgan and intertissue communication is increasingly recognized as a critical way to maintain homeostasis and disease adaptation [1]. Furthermore, molecular biology has provided a toolkit to identify the functions of circulating proteins, such as hormones and cytokines, as communication messengers. Indeed, many new communication proteins are yet to be discovered that will widen our knowledge of organ-organ communication loops and provide potential novel strategies for treating some degenerative diseases [2].
The liver performs a number of essential functions related to detoxification, nutrient storage, digestion, metabolism, and immunity. Liver diseases, including viral hepatitis, nonalcoholic fatty liver disease (NAFLD), alcoholic liver disease (ALD), autoimmune hepatitis (AIH), fibrosis, and end-stage liver disease, account for approximately 2 million deaths per year worldwide [3]. Clinically, both acute and chronic liver diseases have evident extrahepatic complications, which in turn determine the disease progression and efficacy of therapy, forming 'organ-organ communication' [4, 5]. Thus, understanding the interorgan communicating mechanisms, especially the molecular mediators therein is important for the development of targeted therapies.
Communicating molecules derived from the liver
Although first identified in metabolic diseases (e.g., NAFLD and type 2 diabetes), signaling proteins exclusively or predominantly secreted from the liver termed 'hepatokines' are now known to directly affect other diseases. Hepatocytes secrete more than 560 types of hepatokines, many of which regulate metabolic and inflammatory diseases in the liver or at distant organs (e.g. the endocrine roles of hepatokines in bone and heart tissues) through circulation delivery [6]. Under extreme challenges, such as long-term starvation or overnutrition, the liver may secrete hepatokines to influence energy homeostasis and inflammation. Indeed, if the liver is unable to fulfill this process, the corresponding disease develops, such as steatosis from impaired hepatic insulin-sensitizing substance production [7].
Fibroblast growth factor-21
Fibroblast growth factor-21 (FGF-21) is a hormone predominantly secreted by the liver to regulate glucose metabolism in adipocytes in addition to insulin activity. It has been recognized as a therapeutic target of obesity and diabetes [8]. Injection of recombinant FGF-21 in obese mice successfully ameliorated hepatic steatosis, glucose intolerance, and insulin resistance through increased insulin production and secretion [9]. Recent studies also identified FGF-21 as a biomarker for a variety of human diseases, including metabolic disorders [10], drug-induced liver injury [11], hypertension [12], and mitochondrial disease [13]. Currently, the findings of high FGF-21 levels in those patients conflict with the beneficial effects of exogenous FGF-21 administration in patients with obesity and those with diabetes, which might be attributed to possible FGF-21 resistance, suggesting that high levels of FGF-21 are a compensation mechanism in response to metabolic stress in those patients.
Adropin
Adropin is a recently identified small peptide for the maintenance of energy homeostasis and insulin resistance. Its plasma level is positively correlated with nutritional status, that is, upregulated upon feeding and downregulated with fasting [14]. Patients with NAFLD, obesity, and cardiovascular disease are reported to have low levels of adropin, indicating that this peptide is associated with metabolic disorders [15]. In a NAFLD mouse model, overexpression of adropin significantly improved insulin resistance and glucose intolerance [16]. Moreover, muscle insulin signaling was also improved through the amelioration of mitochondrial dysfunction [17]. When adropin was knocked out, mice exhibited increased adiposity, dyslipidemia and whole-body insulin resistance [18]. A recent study unveiled the regulatory effects of adropin on fuel substrate utilization in the heart, probably via a novel G protein coupled receptor (GPCR)-mitogen-activated protein kinase (MAPK)-signaling pathway [19]. Adropin protein expression falls progressively with advancing age in the human peripheral vasculature. Restoration of adropin may facilitate the improvement in endothelial function via increased nitric oxide bioavailability [20].
Angiotensinogen
The renin-angiotensin system (RAS) is pivotal to the regulation of blood pressure and water/sodium metabolism. As a hepatokine, angiotensinogen (AGT) is the only precursor of all angiotensin peptides. Accumulating evidence shows that AGT is not only a passive substrate of the RAS, but also plays a critical role in the pathogenesis of obesity and atherosclerosis. Transgenic human AGT in mice showed augmented atherosclerosis when fed a high-fat diet for 14 weeks [21]. Hepatocyte-specific knockout of AGT in mice resulted in lowered body weight and ameliorated liver steatosis/atherosclerosis after high-fat diet feeding [22]. Indeed, deficiency of AGT could reduce basal plasma AGT levels and blood pressure and induce renal medial hyperplasia [23]. Since adipose tissue is the only extrahepatic source to have an impact on plasma AGT concentration, the sole role of adipocyte-derived AGT and the communication between the liver and adipose tissue during disease progression have attracted mass attention in recent years. Adipocyte-specific deficiency of AGT decreased systolic blood pressure and prevented obesity-induced hypertension in mice [24, 25].
Fetuins
Both fetuins-A and-B are important hepatokines in human metabolism regulation. Fetuin-A was first identified as a natural inhibitor of the insulin receptor tyrosine kinase in liver and skeletal muscle. Deficiency of fetuin-A in mice exhibited improved phenotypes of insulin signaling [26]. Human studies also confirmed that, independent of adiposity, circulating fetuin‑A levels positively correlated with high liver fat content, early atherosclerosis, and metabolic syndrome [27, 28]. In addition, fetuin-A induces insulin resistance and inflammation via multiple mechanisms, including the activation of the extracellular signal regulated-kinase-nuclear factor kappaB (ERK-NF-κB) pathway [29], promoting saturated fatty acid-induced activation of toll-like receptor 4 [30], inducing proinflammatory cytokine production in monocytes and adipocytes [31], and inhibiting insulin-sensitizing adiponectin production [32]. Recent articles also described the critical roles of fetuin-A in diseases of other organs, such as the heart, kidneys, and bone, which will be discussed in the following sections. Like fetuin-A, fetuin-B is also a liver-derived plasma protein. It is increased in patients with type 2 diabetes and impairs insulin sensitivity in myotubes and hepatocytes [33]. The current knowledge of the effects of liver-secreted cytokines and hepatokines on distant organs under physiological and pathological conditions is illustrated in Figure 1 and Table 1.
Effective communicating molecules, mostly hepatokines and cytokines, from the liver to other major distant organs under physiological and pathological conditions. AGT, angiotensinogen; AMBP, alpha-1-microglobulin; ANGPTL, angiopoietin-like protein; FGF, fibroblast growth factor; IGF, insulin growth factor; IGFBP, insulin growth factor binding protein (Created with BioRender.com).
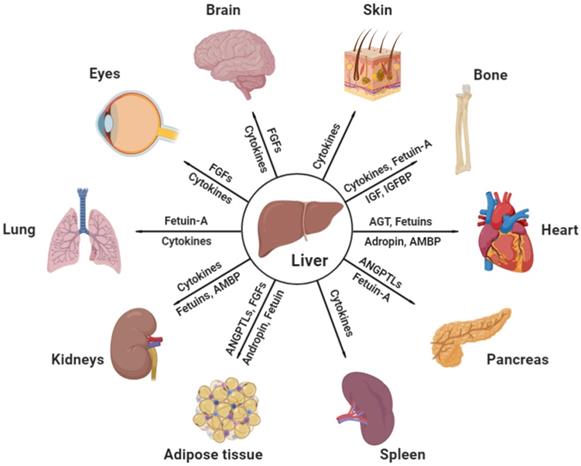
Identified key hepatokines linking hepatic and non-hepatic disorders
| Hepatokine | Organ sources | Key functions | Involved diseases | Refs |
|---|---|---|---|---|
| FGF-21 | Mostly liver, also thyroid and pancreas | Reverses hepatic steatosis and increases energy expenditure; Immuno-regulation; Blood pressure and hydration controls | Metabolic disease, DILI, cancers, mitochondrial disease | [8, 9] |
| Adropin | Brain, liver, prostate, testis, stomach | Improves insulin sensitivity and energy metabolism; Redox control; Cerebellum development | Metabolic disease, I/R injury, CVD, PCOS | [14-17] |
| Angiotensinogen | Mostly liver, also adipose tissue, brain, gall bladder, heart, and kidneys | Blood pressure and water/sodium metabolism regulations | Hypertension, atherosclerosis, metabolic disease, renal disease | [22-25] |
| ANGPTLs | ANGPTL1, 3, 4, 6, 8: Mostly liver, also adipose tissue and muscle. ANGPTL 2 and 5: Mostly heart, also adipose tissue; ANGPTL 7: eyes | Lipid and energy regulation; Angiogenesis; Inflammatory regulation; Carcinogenesis and inhibition | Metabolic disease, cardiovascular disease, cancer | [61, 62, 67] |
| Fetuin-A | Predominantly liver | Induces insulin resistance and inflammation | Metabolic disease, atherosclerosis, CVD, renal dysfunction, cancer, lung disease | [27-30, 167] |
| Fetuin-B | Mostly liver, also esophagus and pancreas | Causes glucose intolerance; Essential for fertilization | Metabolic disease, COPD, CVD, renal disease, osteoporosis | [33] |
| IGFBP-1 | Mostly liver, also placenta and endometrium | Metabolic regulation; Carcinogenesis, | Bone loss, cancer, metabolic disease | [106] |
| AMBP | Mostly liver, also gall bladder | Direct heme binding | CVD, renal disease | [115, 132] |
| Hepcidin | Mostly liver, also heart | Inhibits iron transport; Regulatory target of interleukins; Strong antimicrobial activity | CVD, renal disease | [116, 117, 133] |
| Tsukushi | Mostly liver | Controls energy expenditure via brown fat sympathetic innervation | Metabolic disease | [170, 175] |
AMBP, alpha-1-microglobulin; ANGPTL, angiopoietin-like protein; COPD, chronic obstructive pulmonary disease; CVD, cardiovascular disease; FGF, fibroblast growth factor; IGFBP-1, insulin-like growth factor binding protein 1; I/R injury, ischemia-reperfusion injury; PCOS, polycystic ovary syndrome.
Communications between the liver and other major organs
Gut-liver-brain axis
The concept of the gut-liver-brain axis was first reported in the study of lipid metabolism and glucose synthesis by Wang, et al. in 2008 [34]. To date, this axis is the most well-studied interorgan pathway for the liver. Bile acids (BAs), the metabolites of cholesterol, have long been considered to play an essential role in lipid digestion and fat-soluble vitamin absorption. Two typical bile acid receptors, farnesoid X receptor (FXR) and Takeda G protein-coupled receptor 5 (TGR5), are abundantly expressed not only in the enterohepatic circulation but also in the brain [35, 36]. Thus, studies have focused on the central nervous system activities in which BAs are involved [37, 38]. A recent study discussed the relationship among bile acids, intestinal microbiota and Alzheimer's disease in which the interaction between bile acids and intestinal microbiota influenced the function of the brain [39]. Mechanistically, intestine-restricted agonism of FXR induced TGF5 activity, promoted secretion of FGF-15/21 to improve insulin resistance and promote white adipose tissue browning in mice [40]. This provides a theoretical basis for the direct or indirect risk factors of cognitive decline diseases caused by cholesterol metabolism disorder, including cholestatic liver disease, dyslipidemia, fatty liver disease, cardiovascular disease and diabetes.
Hepatic encephalopathy (HE) manifests as brain dysfunction caused by liver diseases, and portosystemic shunting is one of the late complications of cirrhosis [41]. HE occurs due to different pathogeneses in which hyperammonemia and gut-derived toxins (e.g. indoles, oxindoles and endotoxins) have been considered to be the major pathological causes. Ammonia and the proinflammatory environment are mainly derived from intestinal microbiota, especially Enterobacteriaceae and Streptococcaceae, and subsequently, ammonia is transported to the liver and metabolized [42, 43]. The critical involvement of Kupffer cells in HE pathogenesis was revealed in an ammonium acetate-challenged model where specific deletion of TLR9 in macrophages ameliorated brain edema and lymphocyte cytokine production [44]. Since the ammonia levels in chronic liver failure do not reliably correlate with HE severity and many HE cases are caused in an ammonia-independent manner, treating HE through microbiota modulation (e.g. lactulose and rifaximin) has up to now been considered a first line of intervention [45].
The gut-liver-brain axis is also involved in the regulation of glucose metabolism. Intestinal lipids activate the intestinal vagus nerve and transduce the signal to the brain, which then regulates the synthesis of glycogen [34]. Additionally, it was reported that cholecystokinin, protein kinase A, and protein kinase C activation were all involved in regulating the synthesis of glycogen through the gut-liver-brain axis [46-48]. In alcoholic liver disease, new evidence shows that coexistent depression and other psychiatric conditions can have a role in ALD progression, because only ~15-20% alcohol use disorder patients develop ALD [49]. Physiologically, a complex consisting of one of the FGF-21 receptors (FGFR1c, FGFR2c or FGFR3c) and a single-pass transmembrane protein, β-Klotho, renders FGF-21 signals able to regulate circadian behavior by acting on the hypothalamus and the dorsal vagal complex of the hindbrain [50]. FGF-21 also mediates endocrine control of simple sugar intake and sweet taste preference by the liver [51]. Physical exercise is known to improve both hepatic and brain health, which involves an increased secretion of FGF-21 and IGF-1 (insulin-like growth factor 1), leading to improved cognitive function and brain metabolism [52].
Liver-adipose tissue communication
Expandability of adipose tissue directly contributes to the progression of metabolic liver diseases, such as NAFLD and ALD [53]. When adipose tissue reaches the upper limit of lipid storage capacity, excessive lipids will be redirected to other organs, most notably the liver. This process warrants a precise modulation of extracellular matrix remodeling and neovascularization of adipocytes. Once deregulated, hypertrophic subcutaneous adipocytes induce lipolysis and to secrete more pro-inflammatory cytokines (e.g. TNF-α and IL-1β) which promote local and hepatic insulin resistance and inflammation [54, 55]. In opposite, a hyperplastic expansion, where the number of adipocytes increases, is thought to be linked to the maintenance of normal insulin sensitivity [56]. We recently reported that chronic-binge ethanol consumption significantly reduced both the mass and adipocyte size of white adipose tissues and increased the secretion of pro-lipolysis and inflammatory molecules (e.g. adipose triglyceride lipase, TNF-α, and IL-6), which aggravated the severity of ALD in a mice model [57].
FGF-21 exerts profound effects on energy expenditure and whole-body glucose metabolism largely in the adipose tissue via an insulin-independent manner. Adipose tissue production of adiponectin is increased in FGF21 transgenic mice and markedly inhibited in FGF21-null mice, possibly because of the impaired 'autocrine loop' between FGF21 and PPARγ [58]. Similarly, serum adropin level is higher in mice fed with HFD than mice with chow diet. Adropin knock-out mice exhibit a 50%-increase in increase in adiposity while injection of bioactive peptide adropin effectively improves insulin resistance and metabolic flexibility in HFD-fed mice [17, 18]. Fetuin-A mRNA expression is elevated in NASH compared with steatosis patients [59]. Interestingly, accumulated fetuin A protein (but no mRNA can be detected) is observed in subcutaneous adipose tissue of obese than that of non-obese human, implying that fetuin A potentially acts as a hepatokine taken up by adipose tissue directly from the liver or circulation rather than local production [60]. Angiopoietin-like protein-3 and -4 (ANGPTL3/4) are circulating hepatokines acting on lipoprotein lipase (LPL) inhibition to regulate plasma lipid levels [61]. ANGPTL8/betatrophin, or lipasin, is expressed primarily in liver and visceral adipose tissue and can promote hepatosteatosis and plasma triglyceride levels in humans. Its action on LPL activation needs the co-inhibition of ANGPTL3/4 to enhance triglyceride uptake [62]. Retinol-binding protein 4 (RBP4) is primarily secreted from the liver but also released from adipose tissue as an adipokine. Serum and hepatic RBP4 levels are associated with NAFLD to NASH progression in clinical trials [63, 64]. Adipocyte-derived RBP4 further contributes to insulin resistance and obesity [65]. Hepatocyte-specific knockdown of rbp4 (without influence in white adipose tissue) improved insulin sensitivity and glucose metabolism in diabetic mice [66]. Recent study found that increased serum concentrations of ANGPTL1, 2, 8, and FGF21, as well as decreased levels of ANGPTL4 and adiponectin were associated with NAFLD-associated HCC [67].
Liver-pancreas communication
Liver and pancreas play complementary roles in glucose metabolism and homeostasis. Interlobular and total pancreatic fat are both positively related to NAFLD activity score and total pancreatic fat is a significant predictor for the presence of NAFLD in patients [68]. Another study also indicates that fatty liver plays a prognostic role in acute pancreatitis [69]. Excessive alcohol ingestion often leads to evident pancreatic injury. Both a single occasion of binge drinking and chronic alcohol consumption can increase the risk of an acute attack of pancreatitis. In the US, approximately 1 in 3 cases of acute pancreatitis and 4 in 10 causes of chronic pancreatitis are caused by alcohol, respectively [70, 71]. Metabolites of alcohol (e.g. acetaldehyde) processed by the pancreas damage the acinar cells, promote premature intracellular digestive enzyme activation, and thereby induce the local autodigestive injury [72]. Importantly, acute pancreatitis patients with liver cirrhosis have higher inpatient mortality compared to non-cirrhotics, possibly due to complications of cirrhosis and portal hypertension itself during the decompensated stage [73].
In 2013, Yi et al., claimed that ANGPTL8 secreted from the liver controlled pancreatic beta cell proliferation and mass increase [74]. However, this observation was quickly challenged and proved to be incorrect by following studies which found that Angptl8 knockout mice exhibited no abnormalities in terms of glucose homeostasis and/or in beta cell expansion in response to HFD- or insulin receptor antagonist S961-induced insulin resistance [75], and liver-specific overexpression of Angptl8 did not increase beta cell proliferation in mice [76]. Fetuin-A induces chemokine (MCP-1 and IL-8) and cytokine (IL-6) production in human pancreatic pre-adipocytes and adipocytes. It also stimulates IL-1β expression in islet-infiltrating macrophages through TLR4, which reveals the role of fetuin-A-mediated metabolic crosstalk between fatty pancreas and fatty liver [77]. Hepatocyte-derived kisspeptin1, under the control of glucagon, will transport to pancreas and binds to abundantly expressed kisspeptin 1 receptor in the beta cells, to inhibit cyclic AMP production and insulin secretion [78]. It has been demonstrated that pancreatic FGF21 was a digestive enzyme secretagogue whose physiologic function was to maintain acinar cell proteostasis [79], and restoring pancreatic FGF21 reverses pancreatitis induced by cerulein, alcohol, or endoscopic retrograde cholangiopancreatography [80]. However, whether liver-secreted FGF21 participate such process remains largely unknown. Moreover, damaged pancreas may secrete pro-inflammatory cytokines (e.g. TNF-α) to directly attack the liver per se.
Liver-eye communication
Modern clinical investigations suggest that noticing common manifestations in the eyes can help people prevent or address an issue with liver well-being, such as scleral icterus reflecting jaundice and xanthelasma palpebra for possible hepatic steatosis. Supplementation with lutein, a well-recognized eye health-promoting carotenoid, improved oxidative stress and inflammation in the liver and eyes of guinea pigs fed a hypercholesterolemic diet [81]. A recent study even found that retinitis pigmentosa-induced oxidative stress in the retina might influence soluble macromolecules exiting the retina or significantly impair the melanopsin system, resulting in chronic circadian desynchronization and weakened systemic antioxidant defense, which ultimately caused hepatic oxidative stress [82]. Another interesting study found that bright-light therapy towards the eyes could clinically ameliorate pruritus of cholestasis since this chronic liver disease-related skin disorder was centrally mediated by endogenous opioid peptides that induce disrupted circadian rhythm [83]. In a mice HE model, we identified that thioacetamide-challenged liver could release proinflammatory cytokines (TNF-α and IL-6) to cause impairments in the brain and eyes. Amelioration of liver injury significantly improved eyesight and cognition in mice [84]. This is in line with a previous clinical report in which a patient with severe acute liver failure had uncontrolled intracranial hypertension and needed a hepatectomy that resulted in stabilization of the systemic and cerebral hemodynamics. The removal of the liver was associated with a sharp and sustained reduction in the circulating proinflammatory cytokine concentration [85]. A recent study revealed that FGF-21 administration suppressed retinal neovessel growth and local TNF-α expression in a mouse model, partly through an adiponectin-dependent manner [86]. What and how liver-derived hepatokines and cytokines are delivered to the eyes are the most urgent questions to be answered.
Liver-bone communication
The prevalence of osteopenia/osteoporosis/fractures in chronic liver disease is between 12% and 55%, which is much higher in cholestatic diseases [87]. Hepatic osteodystrophy commonly manifests with osteoporosis and osteopenia, while osteomalacia is rare [88]. The pathogenic mechanisms for hepatic osteodystrophy are not well defined and are considered to be multifactorial. Impaired IGF-1 secretion from the liver might be one of the causative factors of hepatic osteodystrophy because it is a bone collagen and osteoblast stimulator [89]. In addition, increased unconjugated bilirubin can inhibit osteoblast differentiation and proliferation [90]. The association between viral hepatitis and bone dysfunction has been established in recent years. For example, untreated hepatitis C virus (HCV) infection is found to have a higher fracture risk [91]. Interestingly, except in the African-American population, untreated hepatitis B viral (HBV) infection had no significant difference in the incidence of hip fracture when compared with uninfected controls [92]. Low body weight, malnutrition, smoking habits, alcohol consumption, opioids or antidepressive drug use, and hypogonadism will also contribute to viral hepatitis-associated bone disease [93]. Similarly, several recent cross-sectional and case-control studies have shown that NAFLD patients have a greater prevalence of decreased BMD than age-, sex-, and body mass index-matched healthy controls [94, 95]. Proven links between NAFLD and decreased bone mass include vitamin D insufficiency [96], the growth hormone/IGF-I axis [97], TNF-α [98], the receptor activator of NF-κB (RANK)/osteoprotegerin pathway [99], fetuin-A [100], osteopontin [101], and adiponectin [102]. In addition, chronic inflammation and irisin from osteoblasts have been proposed as mediators of mutual interactions among the skeleton, fatty tissue, and liver [103]. Patients with ALD often have accompanying decreased vitamin D levels, which is primarily attributed to reduced hepatic 25-hydroxylase activity, malabsorption, irregular feeding habits and lack of sun exposure [104]. Zinc deficiency is also described among chronic alcoholics. Since zinc stimulates osteoblastic bone formation and mineralization, its deficiency decreases bone mass and is associated with an increased risk of fracture [105]. Blockade of liver-derived IGFBP-1 inhibits bone loss mediated by FGF-21 without inhibiting insulin sensitivity. In vivo administration of IGFBP-1 promotes osteoclast differentiation and bone resorption [106].
Liver-heart communication
Manifestations from the liver are very common in cardiac dysfunction or congestive hepatopathy. The pathological features of chronic cardiogenic liver disease include liver congestion, sinusoidal dilatation, necrosis and fibrosis in the central lobule of the liver [107]. Patients with acute heart failure and/or hypovolemia often develop hypoxic hepatitis, which has similar clinical manifestations as acute viral hepatitis. In addition, cirrhosis can further lead to the redistribution of circulating blood volume, as well as abnormal cardiac contractility and electrophysiology, which in turn leads to cirrhotic cardiomyopathy [108]. Half of the patients with cirrhosis had prolonged QT (Q wave to T wave) intervals, which might be an important factor to evaluate patient survival and prognosis. In addition, the plasma membrane fluidity, calcium channel disorders, and many factors such as cytokines, nitric oxide and endocannabinoids, also participate in the pathophysiological processes of cirrhotic cardiomyopathy [109].
Several systematic conditions affect the liver and heart simultaneously. NAFLD and cardiovascular diseases are usually related to metabolic disorders, such as obesity and diabetes. A multicenter community-based longitudinal cohort study has shown that NAFLD is an independent risk factor for subclinical myocardial remodeling and dysfunction after the assessment of body mass index (BMI) and ventricular activation time (VAT). Patients with NAFLD demonstrated cardiac diastolic dysfunction and had higher left ventricular (LV) mass, LV end-diastolic volume, and LV relative wall thickness than non-NAFLD participants [110]. A NAFLD risk allele TM6SF2 (transmembrane 6 superfamily member 2; rs58542926 c.449 C>T, p.Glu167Lys) was found to be associated with dyslipidemia and cardiovascular risk, primarily due to impaired lipidation and reduced secretion of VLDL (very-low-density lipoprotein) particles from the liver [111]. The heart and liver are also common targets of alcohol abuse, which has been proved to damage the heart structure to a greater extent than to the liver [112]. The liver-heart inflammatory axis has a pivotal pathological role in the development of hepatic cardiomyopathy. Cannabinoid-2 receptor activation markedly improved hepatic/myocardial inflammation, decreased serum TNF-α level, and cardiac dysfunction, underlining the importance of inflammatory mediators in the pathology of this disease [113]. Hepatocyte-specific knockout of IL-6 was sufficient to block aging-induced cardiac arrhythmia in a fruit fly model [114]. In addition to the characterized linking function of adropin [19] and fetuin [33], a recent study reported that alpha-1-microglobulin (AMBP) exacerbated inflammation and disturbed hepatic fibrotic repair after myocardial infarction through activating Akt, NF-κB, and ERK signaling and promoting macrophage migration and polarization [115]. Another hepatokine, hepcidin, has been proven to regulate macrophage inflammation and arterial stiffness, thus promoting the development of atherosclerosis [116, 117]. Upregulated fetuin B aggravates myocardial ischemia/reperfusion injury through inhibiting insulin signaling in diabetic mice [118]. Gut microbial metabolism of choline results in hepatic production of trimethylamine-N-oxide (TMAO), which exacerbates atherosclerosis via promoting forward cholesterol transport [119, 120]. In an acute myocardial infarction mice model, it was revealed that supplementation of IL-22 markedly prevented left ventricular dysfunction and heart failure via liver-derived STAT3-FGF21 production, in which hepatocyte-specific knockout of STAT3 or FGF21 blocked such alleviation of IL-22 [121].
Liver-kidney communication
Hepatorenal syndrome is a serious complication that can lead to death. It is caused by severe liver diseases (e.g., acute liver failure and cirrhosis) and induce progressive kidney failure [122]. Apart from that, a number of liver disorders may also involve renal damage, such as viral hepatitis, ALD, NAFLD, and Wilson disease [123]. Two major renal diseases associated with chronic HBV are membranous nephropathy and polyarteritis nodosa. Membranoproliferative glomerulopathy, IgA nephropathy, and amyloidosis may also occur [124]. In a recent retrospective cohort analysis involving 56,448 HCV patients in the United States, these patients had a 27% increased risk of chronic kidney diseases (e.g., membranoproliferative glomerulonephritis, and cryoglobulinemia) compared with patients without HCV in which the viral protein deposits may mediate the interorgan pathogenesis [125]. Patients with ALD have a tendency to exhibit impaired sodium and fluid handling, as well as severe alterations in the body's acid-base balance, which directly influence the homeostasis of the kidneys [126]. Intuitively, alcoholics should have a higher risk of chronic kidney disease. However, one meta-analysis comprising 20 studies with a total of 292,431 patients found a decreased risk or no risk of kidney disease in heavy alcohol consumers [127]. This result was confirmed by other similar studies showing that the incidence of kidney disease is comparable or even lower in heavier drinkers (>210 g/wk of ethanol) than in moderate drinkers (70-210 g/wk of ethanol) [128, 129]. Emerging evidence suggests that NAFLD aggravates insulin resistance and atherogenic dyslipidemia, promoting the release of proinflammatory, prooxidant, profibrogenic, and procoagulant mediators from the liver or kidneys to induce renal injury [130]. Interestingly, although vitamin D deficiency has been linked to the pathogenesis and severity of NAFLD and renal diseases, benefits of vitamin D supplementation remain controversial, which may require higher dose treatment or vitamin D receptor agonists (e.g., paricalcitol) to be overcome [131]. Although the detailed mechanism remains controversial, the application of a recombinant hepatokine protein, AMBP, has been proven to be effective for ischemic acute kidney injury and chronic renal disorder therapies [132]. In an angiotensin II-induced vascular dysfunction mouse model, replenishment of FGF-21 directly regulates the multi-organ crosstalk between liver, adipose tissue, kidney, and blood vessels to improve hypertension and renal injury [12]. As a key regulator of the entry of iron into the circulation, hepcidin is also believed to contribute to increased iron sequestration and subsequent anemia in patients with chronic kidney disease [133]. A very recent study found that IL-6-mediated hepatocyte production was the primary source of plasma and urine neutrophil gelatinase-associated lipocalin during acute kidney injury [134].
Liver-skin communication
The notion of 'skin reflects liver health' have been recognized for a long time. For example, jaundice is usually recognizable when serum bilirubin levels are high. Whether the excessive bilirubin is conjugated or unconjugated implies whether the cause is prehepatic, intrahepatic, or posthepatic [135]. When the neutralizing function of the liver is disrupted, deposition of toxins and filtered bile salts in the skin causes irritation and itching [136]. Another well-known skin sign of severe liver disorder is spider angiomas induced by elevated estrogen levels, such as in cirrhosis [137]. Recently, interorgan communication between the liver and the skin under pathological conditions has received much attention. Acyl-CoA-binding protein (ACBP) knockout mice display delayed metabolic adaptation to weaning. Further mechanistic studies found that this phenomenon, including hepatic lipid adaptation, was caused by ACBP deficiency in the skin rather than in the liver [138]. The association between liver disease and psoriasis has been reported by several studies. For example, studies suggested that psoriasis may be more severe in patients with NAFLD/NASH [139, 140]. Mice fed a high-fat diet developed steatohepatitis reminiscent of human NASH. Cotreatment with imiquimod, a commonly used agent for psoriasis-like systemic inflammation models, exacerbated psoriatic phenotypes, including scale formation and acanthosis, through an IL-17A-dependent mechanism [141]. Conversely, another study found that a 9-week topical treatment with imiquimod induced typical psoriasiform dermatitis and, surprisingly, moderate portal/periportal hepatitis and liver fibrosis in mice. Livers from psoriatic mice were enriched for macrophages, polymorphonuclear neutrophils, and T cells [142]. AIH is often concomitant with extrahepatic autoimmune diseases. The positive association between AIH (particularly type 2 AIH) and vitiligo is well documented. Probable associations are also identified with alopecia areata, psoriasis, and pyoderma gangrenosum, although they were probably underdiagnosed and underreported in patients with AIH [143]. IGF-1 released from the liver contributes to skin wound healing. Deficiency of IGF-1 receptor keratinocytes disrupts epidermal homeostasis and stem cell maintenance [144]. Of note, all of the organs from liver specific IL-22 transgenic mice had a normal histology except for slightly thicker epidermis and minor inflammation in the skin compared with WT mouse skin [145]. However, the exact communicating mechanisms are not well characterized.
Liver-spleen communication
Liver cirrhosis often leads to splenomegaly and frequently accompanying hypersplenism, which is considered the main cause of cytopenia and thrombocytopenia in those patients [146]. Portal vein blockade is widely considered the initial cause of splenomegaly during liver cirrhosis. Conversely, since the spleen is the body's largest immune organ and plays an important role in the production of antigen-specific T cells and numerous cytokines, a recent study demonstrated the effects of splenic immunity on liver fibrosis and reported that after liver injury, T helper (Th)1 cells suppressed liver fibrosis, while Th2-dominant splenic lymphocytes migrated into the liver and promoted liver fibrosis by shifting the cytokine balance towards Th2 dominance [147]. Splenectomy also accelerates hepatic regeneration in cirrhotic animals and patients with cirrhosis and inhibits the formation of hepatic fibrosis [148, 149]. Potential activation of splenic macrophages and subsequent transforming growth factor (TGF)-β1 production might be the major mechanisms. After splenectomy, the capacity of liver regeneration is improved by the decrease in TGF-β1 and the increase in hepatocyte growth factor. Splenectomy significantly increased the hepatic accumulation of Ly-6C(lo) monocytes or macrophages in a thioacetamide-induced murine model of liver cirrhosis with hypersplenism, implicating a role for the splenic control of hepatic monocyte or macrophage phenotypes [150]. Moreover, splenectomy suppressed regulatory T cell and hepatic fibrogenesis levels, implicating a role for the splenic modulation of the liver via alterations in T cell subsets [151]. Splenectomy improved liver function in patients with liver cirrhosis, especially with large spleen and lower alanine aminotransferase (ALT) levels, and stem cell therapy efficacy [152].
Several reports have discussed the messengers linking the liver and the spleen. An early interesting study found that the modification of inflammatory mediator generation by splenectomy or inhibition of Kupffer cell function may be beneficial for the prevention of endotoxin-induced liver injury after partial hepatectomy [153]. Another study demonstrated that cytokine expression in the spleen affects the progression of liver cirrhosis through liver-spleen crosstalk. Expression of TGF-β1 and cytokines such as IL-6 produced by macrophages could affect the progression of liver fibrosis and regeneration in patients with liver cirrhosis [154]. Foxp3-expressing CD25+CD4+ Treg cells in the spleen lead to tolerance of immunity after liver transplantation [155]. Enlargement of spleen is commonly seen in patients with of NAFLD, which is caused by disrupted iron metabolism, low vitamin D status, and increased cytokine communication (e.g. IL-6). Moreover, inflamed the visceral adipose tissue during NAFLD may also induce splenic injury via adipokines and cytokines [156].
Liver-lung communication
From a physiological point of view, the lung and the liver are closely coordinated. When liver function is perturbed, the inactivation of enterogenous pulmonary vasodilators by hepatocytes is lost, contributing to the accumulation of vasodilators [157]. At the same time, the increase in nonenteric vasodilators leads to the abnormal expansion of pulmonary vascular and gas exchange disorders, which further triggers hypoxemia and a series of other pathophysiological changes and clinical symptoms. These symptoms are clinically known as hepatopulmonary syndrome, which is common in patients with cirrhosis [158]. Tuberculosis (TB) is a chronic respiratory infection caused by Mycobacterium tuberculosis, which invades multiple organs and systems, with lung infections being the most common. Hepatic manifestation is seen in up to 50-80% of TB cases. Coinfection with HCV greatly promotes the occurrence of liver damage in patients with pulmonary tuberculosis, possibly because of decreased erythrocytes and T lymphocytes [159]. It should be noted that anti-TB drugs, including the three major first-line drugs isoniazid, rifampicin, and pyrazinamide, are the most common adverse events necessitating therapy interruption and causes of DILI, which has a general incidence of 5.3% [160]. Acute lung injury/acute respiratory distress syndrome (ALI/ARDS) is an inflammatory response regulated by multiple inflammatory mediators and cytokines. Activation of the liver X receptor alpha (LXRα) pathway has been shown to alleviate LPS-induced ALI by the natural product platycodin D [161]. Decreased fetuin A predicts increased disease activity in obstructive lung disease, Crohn's disease, and ulcerative colitis [162]. Serum fetuin A promotes Lewis lung carcinoma in a calcium-dependent fashion [163]. By using omics methods, it was identified that circulating fetuin B and andropin levels could be used as biomarkers for lung function evaluation in COPD (chronic obstructive pulmonary disease) and myeloperoxidase anti-neutrophil cytoplasm autoantibody-associated lung injury patients, respectively [164, 165]. Other communicating proteins are yet to be discovered for the liver-lung communication.
Conclusions
Keeping a stable internal environment requires precise regulation of whole-body homeostasis in which organ-organ communication plays critical roles (Figure 2 and Table 2). Since the liver is responsible for a variety of physiological processes, disruptions of homeostatic balance warrant a rapid and well-regulated response from it, in most cases, via paracrine and endocrine signaling molecules, to rebalance itself and distant organs. Importantly, new study has revealed that NAFLD had higher overall risk of incident cancers than obesity without NAFLD, possibly because hepatic ectopic fat act as a paracrine source for cancer development in the liver, gastrointestinal tract and uterus [166]. The past decade has witnessed the discovery of a set of hepatokines in many physiological and pathological conditions, in particular, metabolic liver diseases [167]. Indeed, other common and rare liver diseases also require communication from the liver to other organs to alleviate pathological events such as inflammation and cell death and to promote local tissue regeneration. Although the functions of several hepatokines and cytokines in organ-organ communications have been preliminarily analyzed, most studies only described the phenotypic relationships between the liver and other organs under different conditions, with the mystery of the direct communicating mediators unanswered. Hence, a future challenge will be to focus on the signals and to elucidate the modulatory mechanisms of liver-generated molecules in disease progression and communication. Specifically, (1) although substantial progress has been made in understanding disease-controlled production of hepatokines, little is known about the inductive mechanism of transcriptional reprogramming, protein translation, modification, and secretion of hepatokines, particularly through the ER and Golgi, by pathological processes; (2) feedback signaling from distant organs that are induced by hepatokines and the reciprocation between hepatokines and other 'organokines' are poorly recognized. Several proteins (e.g. FGF21 and soluble epidermal growth factor receptor) can be secreted from the liver and other organs, which act as hepatokine, adipokine, and/or myokine under different circumstances [168, 169]. How these signals are spatio-temporally regulated to maintain functional interorgan communication is largely unknown, which needs cutting-edge techniques, such as specific cell type-transgenic method and single-cell sequencing, to answer; (3) whether different liver diseases (e.g., viral hepatitis and metabolic liver diseases) share similar hepatokine secretion profiles needs further investigation. That is, from a translational perspective, searching a single or set of hepatokines as biomarkers for certain diseases is critical for clinical applications. For metabolic liver diseases, one of the central problems is that they rarely induce specific symptoms and diagnosis is frequently incidental. Recent studies using multiomics approach identified that hepatokine tsukushi was a potential blood biomarker and drug treatment target of NAFLD and subsequent atherosclerosis [170, 171]. Indeed, such findings need to be confirmed in large cohorts of corresponding patients (e.g. the pilot clinical findings of FGF21 and LECT2 in diabetic patients) [172, 173]; (4) Through the use of advanced mass spectrometry "omics" approaches, we now recognized that liver functions as an endocrine organ by secreting a wide array of hormones (e.g. IGF-1) and noncoding RNAs (miR-9 and -375 for pancreatic insulin regulation), in addition to hepatokines and cytokines, that exert powerful effects on metabolic processes both in the liver and in peripheral tissues [174]. Future studies that interrogate the hepatocyte or liver secretomes using multiomics approaches are well positioned to identify novel communicating 'messengers'. Addressing these challenges will be of major interest for the understanding of liver-centered organ-organ communication, as well as the development of rational therapeutic strategies for certain diseases.
Distant communicating mechanisms, including neuronal, hormonal, metabolic, and other factors, between major organs under pathological conditions. 5-HT, 5-hydroxytryptamine; AIH, auto-immune hepatitis; ANGPTL, angiopoietin-like protein; DILI, drug-induced liver injury; FFA, free fatty acids; GI, gastrointestinal; IgA, immunoglobulin A; I/R injury, ischemia/reperfusion injury; PBA, primary bile acids; RAS, renin-angiotensin system (Created with BioRender.com).
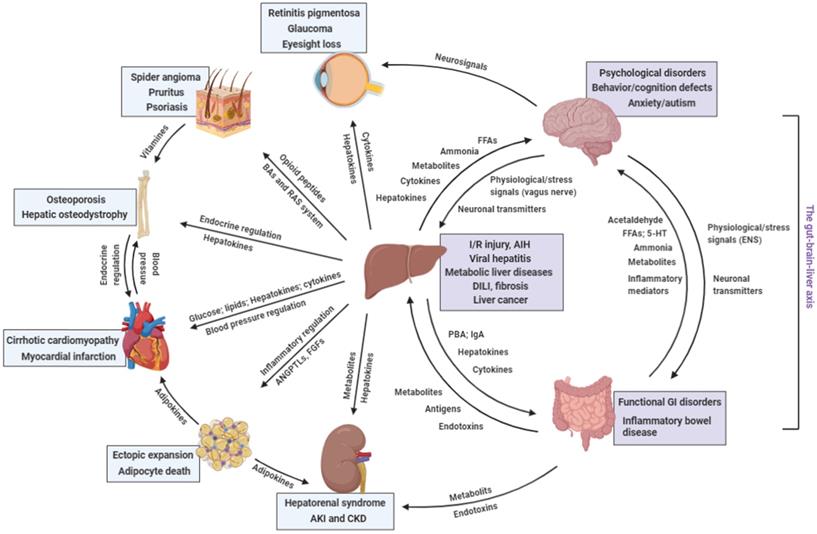
Involvement of the liver in other organ's disorders
| Organ | Disorder | Hepatic manifestation | Possible linking mechanisms | Refs |
|---|---|---|---|---|
| Brain | Cognitive disorder (e.g. Alzheimer's disease) | Cholestatic liver disease and fatty liver disease | Bile acids | [39] |
| Hepatic encephalopathy | Cirrhosis and liver failure | Ammonia, cytokines, and hepatokines | [42, 43, 176, 177] | |
| Circadian behavior disorder | Lipid dysregulation | FGF-21 | [50, 51] | |
| Adipose tissue | Ectopic expansion and adipocyte death | NAFLD and ALD | FGF-21, ANGPTLs, adropin, fetuin-A | [17, 57-59, 67] |
| Pancreas | Acute and chronic pancreatitis | NAFLD and ALD | ANGPTLs, fetuin-A, kispeptin | [75, 77, 78] |
| Eyes | Retina injury | NAFLD and ALD | Oxidative stress and cytokines | [81, 178] |
| Retinitis pigmentosa | Liver injury | Oxidative stress | [82] | |
| Bone | Hepatic osteodystrophy | PBC and PSC | IGF-1, bilirubin, | [88, 90] |
| Fracture | Viral hepatitis, NAFLD, and ALD | Hepatokines, hormones, and vitamins | [91, 92, 95, 96, 98, 100, 101, 106] | |
| Heart | Heart failure and cardiomyopathy | Liver fibrosis/cirrhosis | Cytokines, calcium, NO, and endocannabinoids | [107-109] |
| Diastolic dysfunction | NAFLD | Free fatty acids (?) | [110] | |
| Myocardial infarction | Hepatic inflammation and disorders | Hepatokines | [115-117] | |
| Kidneys | Hepatorenal syndrome | severe liver diseases (e.g. alcoholic cirrhosis, acute liver failure, and cirrhosis) | Cytokines and hepatokines | [122] |
| Chronic kidney diseases | Viral hepatitis, NAFLD, ALD | Viral-protein deposits, cytokines, hepatokines, ion level, excessive IgA loads and ROS | [125, 126] | |
| Skin | Spider angiomas | Severe liver diseases | Elevated estrogen level | [137] |
| Psoriasis | NAFLD, ALD, and AIH | Cytokines and hepatokines (?) | [137, 139, 141-143] | |
| Spleen | Splenomegaly and splenic disorders | Cirrhosis | Portal vein blockade, cytokines and TGF signaling | [41, 147, 150] |
| Lung | Hepatopulmonary syndrome | Cirrhosis and acute liver failure | Vasodilator secretion (e.g. NO) | [158] |
| Tuberculosis | Viral hepatitis, DILI, | Cytokines | [160] | |
| ALI/ARDS | Liver injury | Hepatokines and cytokines | [161, 179] |
AIH, auto-immune hepatitis; ALD, alcoholic liver disease; ALI/ARDS, acute lung injury/acute respiratory distress syndrome; ANGPTL, angiopoietin-like protein; DILI, drug-induced liver injury; FGF, fibroblast growth factor; IGF, insulin growth factor; NAFLD, non-alcoholic liver disease; NO, nitric oxide; PBC, primary biliary cirrhosis; PSC, primary sclerosing cholangitis; ROS, reactive oxygen species; TGF, transforming growth factor.
Abbreviations
ACBP: Acyl-CoA-binding protein; AGT: angiotensinogen; AIH: autoimmune hepatitis; AKI: acute kidney injury; ALD: alcoholic liver disease; ALT: alanine aminotransferase; BA: bile acid; CKD: chronic kidney disease; ERK: extracellular signal regulated kinase; FGF: fibroblast growth factor; FXR: farnesoid X receptor; GPCR: G protein coupled receptor; HBV: hepatitis B virus; HCV: hepatitis C virus; HE: hepatic encephalopathy; LPL: lipoprotein lipase; LV: left ventricule; LXR: liver X receptor; MAPK: mitogen-activated protein kinase; NAFLD: nonalcoholic fatty liver disease; NF-κB: nuclear factor-kappa B; RANK: receptor activator of NF-κB; RAS: renin-angiotensin system; RBP: retinol-binding protein; TB: tuberculosis; TGF: transforming growth factor; TNF: tumor necrosis factor.
Acknowledgements
This study has been graciously supported by grants from the National Natural Science Foundation of China (81970515; 81873573) and Guangdong Natural Science Funds for Distinguished Young Scholar (2019B151502013).
Authors' contributions
All authors contributed to the draft of the text. FW and JX created the figures and tables. KFS and HW made critical revisions.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Karsenty G, Olson EN. Bone and Muscle Endocrine Functions: Unexpected Paradigms of Inter-organ Communication. Cell. 2016;164:1248-56
2. Leitner LM, Wilson RJ, Yan Z, Godecke A. Reactive Oxygen Species/Nitric Oxide Mediated Inter-Organ Communication in Skeletal Muscle Wasting Diseases. Antioxid Redox Signal. 2017;26:700-17
3. Xiao J, Wang F, Wong NK, He J, Zhang R, Sun R. et al. Global liver disease burdens and research trends: Analysis from a Chinese perspective. J Hepatol. 2019;71:212-21
4. Rahimi RS, Rockey DC. End-stage liver disease complications. Curr Opin Gastroenterol. 2013;29:257-63
5. Armstrong MJ, Adams LA, Canbay A, Syn WK. Extrahepatic complications of nonalcoholic fatty liver disease. Hepatology. 2014;59:1174-97
6. Meex RCR, Watt MJ. Hepatokines: linking nonalcoholic fatty liver disease and insulin resistance. Nat Rev Endocrinol. 2017;13:509-20
7. Petersen MC, Shulman GI. Mechanisms of Insulin Action and Insulin Resistance. Physiol Rev. 2018;98:2133-223
8. Degirolamo C, Sabba C, Moschetta A. Therapeutic potential of the endocrine fibroblast growth factors FGF19, FGF21 and FGF23. Nat Rev Drug Discov. 2016;15:51-69
9. Xu J, Lloyd DJ, Hale C, Stanislaus S, Chen M, Sivits G. et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes. 2009;58:250-9
10. Keipert S, Lutter D, Schroeder BO, Brandt D, Stahlman M, Schwarzmayr T. et al. Endogenous FGF21-signaling controls paradoxical obesity resistance of UCP1-deficient mice. Nat Commun. 2020;11:624
11. Ye D, Wang Y, Li H, Jia W, Man K, Lo CM. et al. Fibroblast growth factor 21 protects against acetaminophen-induced hepatotoxicity by potentiating peroxisome proliferator-activated receptor coactivator protein-1alpha-mediated antioxidant capacity in mice. Hepatology. 2014;60:977-89
12. Pan X, Shao Y, Wu F, Wang Y, Xiong R, Zheng J. et al. FGF21 Prevents Angiotensin II-Induced Hypertension and Vascular Dysfunction by Activation of ACE2/Angiotensin-(1-7) Axis in Mice. Cell Metab. 2018;27:1323-37.e5
13. Morovat A, Weerasinghe G, Nesbitt V, Hofer M, Agnew T, Quaghebeur G. et al. Use of FGF-21 as a Biomarker of Mitochondrial Disease in Clinical Practice. J Clin Med. 2017 6
14. Gao S, McMillan RP, Jacas J, Zhu Q, Li X, Kumar GK. et al. Regulation of substrate oxidation preferences in muscle by the peptide hormone adropin. Diabetes. 2014;63:3242-52
15. Aydin S. Three new players in energy regulation: preptin, adropin and irisin. Peptides. 2014;56:94-110
16. Gao S, Ghoshal S, Zhang L, Stevens JR, McCommis KS, Finck BN. et al. The peptide hormone adropin regulates signal transduction pathways controlling hepatic glucose metabolism in a mouse model of diet-induced obesity. J Biol Chem. 2019;294:13366-77
17. Gao S, McMillan RP, Zhu Q, Lopaschuk GD, Hulver MW, Butler AA. Therapeutic effects of adropin on glucose tolerance and substrate utilization in diet-induced obese mice with insulin resistance. Mol Metab. 2015;4:310-24
18. Ganesh Kumar K, Zhang J, Gao S, Rossi J, McGuinness OP, Halem HH. et al. Adropin deficiency is associated with increased adiposity and insulin resistance. Obesity (Silver Spring). 2012;20:1394-402
19. Thapa D, Stoner MW, Zhang M, Xie B, Manning JR, Guimaraes D. et al. Adropin regulates pyruvate dehydrogenase in cardiac cells via a novel GPCR-MAPK-PDK4 signaling pathway. Redox Biol. 2018;18:25-32
20. Kwon OS, Andtbacka RHI, Hyngstrom JR, Richardson RS. Vasodilatory function in human skeletal muscle feed arteries with advancing age: the role of adropin. J Physiol. 2019;597:1791-804
21. Sugiyama F, Haraoka S, Watanabe T, Shiota N, Taniguchi K, Ueno Y. et al. Acceleration of atherosclerotic lesions in transgenic mice with hypertension by the activated renin-angiotensin system. Lab Invest. 1997;76:835-42
22. Yiannikouris F, Wang Y, Shoemaker R, Larian N, Thompson J, English VL. et al. Deficiency of angiotensinogen in hepatocytes markedly decreases blood pressure in lean and obese male mice. Hypertension. 2015;66:836-42
23. Matsusaka T, Niimura F, Shimizu A, Pastan I, Saito A, Kobori H. et al. Liver angiotensinogen is the primary source of renal angiotensin II. J Am Soc Nephrol. 2012;23:1181-9
24. Yiannikouris F, Karounos M, Charnigo R, English VL, Rateri DL, Daugherty A. et al. Adipocyte-specific deficiency of angiotensinogen decreases plasma angiotensinogen concentration and systolic blood pressure in mice. Am J Physiol Regul Integr Comp Physiol. 2012;302:R244-51
25. Yiannikouris F, Gupte M, Putnam K, Thatcher S, Charnigo R, Rateri DL. et al. Adipocyte deficiency of angiotensinogen prevents obesity-induced hypertension in male mice. Hypertension. 2012;60:1524-30
26. Mathews ST, Singh GP, Ranalletta M, Cintron VJ, Qiang X, Goustin AS. et al. Improved insulin sensitivity and resistance to weight gain in mice null for the Ahsg gene. Diabetes. 2002;51:2450-8
27. Rittig K, Thamer C, Haupt A, Machann J, Peter A, Balletshofer B. et al. High plasma fetuin-A is associated with increased carotid intima-media thickness in a middle-aged population. Atherosclerosis. 2009;207:341-2
28. Ix JH, Wassel CL, Kanaya AM, Vittinghoff E, Johnson KC, Koster A. et al. Fetuin-A and incident diabetes mellitus in older persons. Jama. 2008;300:182-8
29. Takata H, Ikeda Y, Suehiro T, Ishibashi A, Inoue M, Kumon Y. et al. High glucose induces transactivation of the alpha2-HS glycoprotein gene through the ERK1/2 signaling pathway. J Atheroscler Thromb. 2009;16:448-56
30. Pal D, Dasgupta S, Kundu R, Maitra S, Das G, Mukhopadhyay S. et al. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat Med. 2012;18:1279-85
31. Dasgupta S, Bhattacharya S, Biswas A, Majumdar SS, Mukhopadhyay S, Ray S. et al. NF-kappaB mediates lipid-induced fetuin-A expression in hepatocytes that impairs adipocyte function effecting insulin resistance. Biochem J. 2010;429:451-62
32. Chattopadhyay M, Mukherjee S, Chatterjee SK, Chattopadhyay D, Das S, Majumdar SS. et al. Impairment of energy sensors, SIRT1 and AMPK, in lipid induced inflamed adipocyte is regulated by Fetuin A. Cell Signal. 2018;42:67-76
33. Meex RC, Hoy AJ, Morris A, Brown RD, Lo JC, Burke M. et al. Fetuin B Is a Secreted Hepatocyte Factor Linking Steatosis to Impaired Glucose Metabolism. Cell Metab. 2015;22:1078-89
34. Wang PY, Caspi L, Lam CK, Chari M, Li X, Light PE. et al. Upper intestinal lipids trigger a gut-brain-liver axis to regulate glucose production. Nature. 2008;452:1012-6
35. McMillin M, Frampton G, Quinn M, Ashfaq S, de los Santos M 3rd, Grant S. et al. Bile Acid Signaling Is Involved in the Neurological Decline in a Murine Model of Acute Liver Failure. Am J Pathol. 2016;186:312-23
36. Yanguas-Casas N, Barreda-Manso MA, Nieto-Sampedro M, Romero-Ramirez L. TUDCA: An Agonist of the Bile Acid Receptor GPBAR1/TGR5 With Anti-Inflammatory Effects in Microglial Cells. J Cell Physiol. 2017;232:2231-45
37. Mertens KL, Kalsbeek A, Soeters MR, Eggink HM. Bile Acid Signaling Pathways from the Enterohepatic Circulation to the Central Nervous System. Front Neurosci. 2017;11:617
38. Ackerman HD, Gerhard GS. Bile Acids in Neurodegenerative Disorders. Front Aging Neurosci. 2016;8:263
39. MahmoudianDehkordi S, Arnold M, Nho K, Ahmad S, Jia W, Xie G. et al. Altered bile acid profile associates with cognitive impairment in Alzheimer's disease-An emerging role for gut microbiome. Alzheimers Dement. 2019;15:76-92
40. Pathak P, Xie C, Nichols RG, Ferrell JM, Boehme S, Krausz KW. et al. Intestine farnesoid X receptor agonist and the gut microbiota activate G-protein bile acid receptor-1 signaling to improve metabolism. Hepatology. 2018;68:1574-88
41. Hadjihambi A, Arias N, Sheikh M, Jalan R. Hepatic encephalopathy: a critical current review. Hepatol Int. 2018;12:135-47
42. Zhang Z, Zhai H, Geng J, Yu R, Ren H, Fan H. et al. Large-scale survey of gut microbiota associated with MHE Via 16S rRNA-based pyrosequencing. Am J Gastroenterol. 2013;108:1601-11
43. Shawcross DL. Is it time to target gut dysbiosis and immune dysfunction in the therapy of hepatic encephalopathy? Expert Rev Gastroenterol Hepatol. 2015;9:539-42
44. Manakkat Vijay GK, Hu C, Peng J, Garcia-Martinez I, Hoque R, Verghis RM. et al. Ammonia-Induced Brain Edema Requires Macrophage and T Cell Expression of Toll-Like Receptor 9. Cell Mol Gastroenterol Hepatol. 2019;8:609-23
45. Mancini A, Campagna F, Amodio P, Tuohy KM. Gut: liver: brain axis: the microbial challenge in the hepatic encephalopathy. Food Funct. 2018;9:1373-88
46. Cheung GW, Kokorovic A, Lam CK, Chari M, Lam TK. Intestinal cholecystokinin controls glucose production through a neuronal network. Cell Metab. 2009;10:99-109
47. Kokorovic A, Cheung GW, Breen DM, Chari M, Lam CK, Lam TK. Duodenal mucosal protein kinase C-delta regulates glucose production in rats. Gastroenterology. 2011;141:1720-7
48. Rasmussen BA, Breen DM, Luo P, Cheung GW, Yang CS, Sun B. et al. Duodenal activation of cAMP-dependent protein kinase induces vagal afferent firing and lowers glucose production in rats. Gastroenterology. 2012;142:834-43.e3
49. de Timary P, Leclercq S, Starkel P, Delzenne N. A dysbiotic subpopulation of alcohol-dependent subjects. Gut Microbes. 2015;6:388-91
50. Bookout AL, de Groot MH, Owen BM, Lee S, Gautron L, Lawrence HL. et al. FGF21 regulates metabolism and circadian behavior by acting on the nervous system. Nat Med. 2013;19:1147-52
51. von Holstein-Rathlou S, BonDurant LD, Peltekian L, Naber MC, Yin TC, Claflin KE. et al. FGF21 Mediates Endocrine Control of Simple Sugar Intake and Sweet Taste Preference by the Liver. Cell Metab. 2016;23:335-43
52. Pedersen BK. Physical activity and muscle-brain crosstalk. Nat Rev Endocrinol. 2019;15:383-92
53. Virtue S, Vidal-Puig A. Adipose tissue expandability, lipotoxicity and the Metabolic Syndrome-an allostatic perspective. Biochim Biophys Acta. 2010;1801:338-49
54. Wree A, Schlattjan M, Bechmann LP, Claudel T, Sowa JP, Stojakovic T. et al. Adipocyte cell size, free fatty acids and apolipoproteins are associated with non-alcoholic liver injury progression in severely obese patients. Metabolism. 2014;63:1542-52
55. Marcelin G, Silveira ALM, Martins LB, Ferreira AV, Clément K. Deciphering the cellular interplays underlying obesity-induced adipose tissue fibrosis. J Clin Invest. 2019;129:4032-40
56. Ghaben AL, Scherer PE. Adipogenesis and metabolic health. Nat Rev Mol Cell Biol. 2019;20:242-58
57. Luo P, Zheng M, Zhang R, Zhang H, Liu Y, Li W. et al. S-allylmercaptocysteine improves alcoholic liver disease partly through a direct modulation of insulin receptor signaling. Acta Pharmaceutica Sinica B. 2020
58. Lin Z, Tian H, Lam KS, Lin S, Hoo RC, Konishi M. et al. Adiponectin mediates the metabolic effects of FGF21 on glucose homeostasis and insulin sensitivity in mice. Cell Metab. 2013;17:779-89
59. Kahraman A, Sowa JP, Schlattjan M, Sydor S, Pronadl M, Wree A. et al. Fetuin-A mRNA expression is elevated in NASH compared with NAFL patients. Clin Sci (Lond). 2013;125:391-400
60. Khadir A, Kavalakatt S, Madhu D, Hammad M, Devarajan S, Tuomilehto J. et al. Fetuin-A levels are increased in the adipose tissue of diabetic obese humans but not in circulation. Lipids Health Dis. 2018;17:291
61. Carbone C, Piro G, Merz V, Simionato F, Santoro R, Zecchetto C. et al. Angiopoietin-Like Proteins in Angiogenesis, Inflammation and Cancer. Int J Mol Sci. 2018 19
62. Altun Ö, Dikker O, Arman Y, Ugurlukisi B, Kutlu O, Ozgun Cil E. et al. Serum Angiopoietin-like peptide 4 levels in patients with hepatic steatosis. Cytokine. 2018;111:496-9
63. Wang X, Chen X, Zhang H, Pang J, Lin J, Xu X. et al. Circulating retinol-binding protein 4 is associated with the development and regression of non-alcoholic fatty liver disease. Diabetes Metab. 2020;46:119-28
64. Petta S, Tripodo C, Grimaudo S, Cabibi D, Cammà C, Di Cristina A. et al. High liver RBP4 protein content is associated with histological features in patients with genotype 1 chronic hepatitis C and with nonalcoholic steatohepatitis. Dig Liver Dis. 2011;43:404-10
65. Graham TE, Yang Q, Blüher M, Hammarstedt A, Ciaraldi TP, Henry RR. et al. Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. N Engl J Med. 2006;354:2552-63
66. Ma X, Zhou Z, Chen Y, Wu Y, Liu Y. RBP4 functions as a hepatokine in the regulation of glucose metabolism by the circadian clock in mice. Diabetologia. 2016;59:354-62
67. Kucukoglu O, Sowa JP, Mazzolini GD, Syn WK, Canbay A. Hepatokines and adipokines in NASH-related hepatocellular carcinoma. J Hepatol. 2020
68. van Geenen EJ, Smits MM, Schreuder TC, van der Peet DL, Bloemena E, Mulder CJ. Nonalcoholic fatty liver disease is related to nonalcoholic fatty pancreas disease. Pancreas. 2010;39:1185-90
69. Mikolasevic I, Orlic L, Poropat G, Jakopcic I, Stimac D, Klanac A. et al. Nonalcoholic fatty liver and the severity of acute pancreatitis. Eur J Intern Med. 2017;38:73-8
70. Sadr Azodi O, Orsini N, Andrén-Sandberg Å, Wolk A. Effect of type of alcoholic beverage in causing acute pancreatitis. Br J Surg. 2011;98:1609-16
71. Coté GA, Yadav D, Slivka A, Hawes RH, Anderson MA, Burton FR. et al. Alcohol and smoking as risk factors in an epidemiology study of patients with chronic pancreatitis. Clin Gastroenterol Hepatol. 2011;9:266-73 quiz e27
72. Apte MV, Pirola RC, Wilson JS. Mechanisms of alcoholic pancreatitis. J Gastroenterol Hepatol. 2010;25:1816-26
73. Simons-Linares CR, Romero-Marrero C, Jang S, Bhatt A, Lopez R, Vargo J. et al. Clinical outcomes of acute pancreatitis in patients with cirrhosis. Pancreatology. 2020;20:44-50
74. Yi P, Park JS, Melton DA. Betatrophin: a hormone that controls pancreatic β cell proliferation. Cell. 2013;153:747-58
75. Gusarova V, Alexa CA, Na E, Stevis PE, Xin Y, Bonner-Weir S. et al. ANGPTL8/betatrophin does not control pancreatic beta cell expansion. Cell. 2014;159:691-6
76. Cox AR, Lam CJ, Bonnyman CW, Chavez J, Rios JS, Kushner JA. Angiopoietin-like protein 8 (ANGPTL8)/betatrophin overexpression does not increase beta cell proliferation in mice. Diabetologia. 2015;58:1523-31
77. Gerst F, Wagner R, Kaiser G, Panse M, Heni M, Machann J. et al. Metabolic crosstalk between fatty pancreas and fatty liver: effects on local inflammation and insulin secretion. Diabetologia. 2017;60:2240-51
78. Song WJ, Mondal P, Wolfe A, Alonso LC, Stamateris R, Ong BW. et al. Glucagon regulates hepatic kisspeptin to impair insulin secretion. Cell Metab. 2014;19:667-81
79. Coate KC, Hernandez G, Thorne CA, Sun S, Le TDV, Vale K. et al. FGF21 Is an Exocrine Pancreas Secretagogue. Cell Metab. 2017;25:472-80
80. Hernandez G, Luo T, Javed TA, Wen L, Kalwat MA, Vale K. et al. Pancreatitis is an FGF21-deficient state that is corrected by replacement therapy. Sci Transl Med. 2020 12
81. Kim JE, Clark RM, Park Y, Lee J, Fernandez ML. Lutein decreases oxidative stress and inflammation in liver and eyes of guinea pigs fed a hypercholesterolemic diet. Nutr Res Pract. 2012;6:113-9
82. Perdices L, Fuentes-Broto L, Segura F, Ben Gdara N, Sanchez-Cano AI, Insa G. et al. Hepatic oxidative stress in pigmented P23H rhodopsin transgenic rats with progressive retinal degeneration. Free Radic Biol Med. 2018;124:550-7
83. Bergasa NV, Link MJ, Keogh M, Yaroslavsky G, Rosenthal RN, McGee M. Pilot study of bright-light therapy reflected toward the eyes for the pruritus of chronic liver disease. Am J Gastroenterol. 2001;96:1563-70
84. Sun X, Lv Y, Huang L, Gao H, Ren C, Li J. et al. Pro-inflammatory cytokines serve as communicating molecules between the liver and brain for hepatic encephalopathy pathogenesis and Lycium barbarum polysaccharides protection. J Ethnopharmacol. 2020;248:112357
85. Jalan R, Pollok A, Shah SH, Madhavan K, Simpson KJ. Liver derived pro-inflammatory cytokines may be important in producing intracranial hypertension in acute liver failure. J Hepatol. 2002;37:536-8
86. Fu Z, Gong Y, Liegl R, Wang Z, Liu CH, Meng SS. et al. FGF21 Administration Suppresses Retinal and Choroidal Neovascularization in Mice. Cell Rep. 2017;18:1606-13
87. Gatta A, Verardo A, Di Pascoli M, Giannini S, Bolognesi M. Hepatic osteodystrophy. Clin Cases Miner Bone Metab. 2014;11:185-91
88. Nakchbandi IA, van der Merwe SW. Current understanding of osteoporosis associated with liver disease. Nat Rev Gastroenterol Hepatol. 2009;6:660-70
89. Yan J, Herzog JW, Tsang K, Brennan CA, Bower MA, Garrett WS. et al. Gut microbiota induce IGF-1 and promote bone formation and growth. Proc Natl Acad Sci U S A. 2016;113:E7554-e63
90. Weinreb M, Pollak RD, Ackerman Z. Experimental cholestatic liver disease through bile-duct ligation in rats results in skeletal fragility and impaired osteoblastogenesis. J Hepatol. 2004;40:385-90
91. Hansen AB, Omland LH, Krarup H, Obel N. Fracture risk in hepatitis C virus infected persons: results from the DANVIR cohort study. J Hepatol. 2014;61:15-21
92. Byrne DD, Newcomb CW, Carbonari DM, Nezamzadeh MS, Leidl KB, Herlim M. et al. Risk of hip fracture associated with untreated and treated chronic hepatitis B virus infection. J Hepatol. 2014;61:210-8
93. Jeong HM, Kim DJ. Bone Diseases in Patients with Chronic Liver Disease. Int J Mol Sci. 2019 20
94. Pirgon O, Bilgin H, Tolu I, Odabas D. Correlation of insulin sensitivity with bone mineral status in obese adolescents with nonalcoholic fatty liver disease. Clin Endocrinol (Oxf). 2011;75:189-95
95. Pardee PE, Dunn W, Schwimmer JB. Non-alcoholic fatty liver disease is associated with low bone mineral density in obese children. Aliment Pharmacol Ther. 2012;35:248-54
96. Wang X, Li W, Zhang Y, Yang Y, Qin G. Association between vitamin D and non-alcoholic fatty liver disease/non-alcoholic steatohepatitis: results from a meta-analysis. Int J Clin Exp Med. 2015;8:17221-34
97. Perrini S, Laviola L, Carreira MC, Cignarelli A, Natalicchio A, Giorgino F. The GH/IGF1 axis and signaling pathways in the muscle and bone: mechanisms underlying age-related skeletal muscle wasting and osteoporosis. J Endocrinol. 2010;205:201-10
98. Yilmaz Y. Review article: non-alcoholic fatty liver disease and osteoporosis-clinical and molecular crosstalk. Aliment Pharmacol Ther. 2012;36:345-52
99. Indridason OS, Franzson L, Sigurdsson G. Serum osteoprotegerin and its relationship with bone mineral density and markers of bone turnover. Osteoporos Int. 2005;16:417-23
100. Brylka L, Jahnen-Dechent W. The role of fetuin-A in physiological and pathological mineralization. Calcif Tissue Int. 2013;93:355-64
101. Rosen CJ, Bouxsein ML. Mechanisms of disease: is osteoporosis the obesity of bone? Nat Clin Pract Rheumatol. 2006;2:35-43
102. Fuggle NR, Westbury LD, Syddall HE, Duggal NA, Shaw SC, Maslin K. et al. Relationships between markers of inflammation and bone density: findings from the Hertfordshire Cohort Study. Osteoporos Int. 2018;29:1581-9
103. Filip R, Radzki RP, Bienko M. Novel insights into the relationship between nonalcoholic fatty liver disease and osteoporosis. Clin Interv Aging. 2018;13:1879-91
104. Wijnia JW, Wielders JP, Lips P, van de Wiel A, Mulder CL, Nieuwenhuis KG. Is vitamin D deficiency a confounder in alcoholic skeletal muscle myopathy? Alcohol Clin Exp Res. 2013;37(Suppl 1):E209-15
105. Gonzalez-Reimers E, Duran-Castellon MC, Martin-Olivera R, Lopez-Lirola A, Santolaria-Fernandez F, De la Vega-Prieto MJ. et al. Effect of zinc supplementation on ethanol-mediated bone alterations. Food Chem Toxicol. 2005;43:1497-505
106. Wang X, Wei W, Krzeszinski JY, Wang Y, Wan Y. A Liver-Bone Endocrine Relay by IGFBP1 Promotes Osteoclastogenesis and Mediates FGF21-Induced Bone Resorption. Cell Metab. 2015;22:811-24
107. Koehne de Gonzalez AK, Lefkowitch JH. Heart Disease and the Liver: Pathologic Evaluation. Gastroenterol Clin North Am. 2017;46:421-35
108. Zardi EM, Abbate A, Zardi DM, Dobrina A, Margiotta D, Van Tassell BW. et al. Cirrhotic cardiomyopathy. J Am Coll Cardiol. 2010;56:539-49
109. Serste T, Melot C, Francoz C, Durand F, Rautou PE, Valla D. et al. Deleterious effects of beta-blockers on survival in patients with cirrhosis and refractory ascites. Hepatology. 2010;52:1017-22
110. VanWagner LB, Wilcox JE, Colangelo LA, Lloyd-Jones DM, Carr JJ, Lima JA. et al. Association of nonalcoholic fatty liver disease with subclinical myocardial remodeling and dysfunction: A population-based study. Hepatology. 2015;62:773-83
111. Holmen OL, Zhang H, Fan Y, Hovelson DH, Schmidt EM, Zhou W. et al. Systematic evaluation of coding variation identifies a candidate causal variant in TM6SF2 influencing total cholesterol and myocardial infarction risk. Nat Genet. 2014;46:345-51
112. Hankey G. Alcohol use and burden for 195 countries and territories, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2018;392:1015-35
113. Matyas C, Erdelyi K, Trojnar E, Zhao S, Varga ZV, Paloczi J. et al. Interplay of Liver-Heart Inflammatory Axis and Cannabinoid 2 Receptor Signaling in an Experimental Model of Hepatic Cardiomyopathy. Hepatology. 2020;71:1391-407
114. Huang K, Miao T, Chang K, Kim J, Kang P, Jiang Q. et al. Impaired peroxisomal import in Drosophila oenocytes causes cardiac dysfunction by inducing upd3 as a peroxikine. Nat Commun. 2020;11:2943
115. Hakuno D, Kimura M, Ito S, Satoh J, Nakashima Y, Horie T. et al. Hepatokine alpha1-Microglobulin Signaling Exacerbates Inflammation and Disturbs Fibrotic Repair in Mouse Myocardial Infarction. Sci Rep. 2018;8:16749
116. Wang X, Sheng L, Ye P, Cao R, Yang X, Xiao W. et al. The association between Hepcidin and arterial stiffness in a community-dwelling population. Lipids Health Dis. 2018;17:244
117. Malhotra R, Wunderer F, Barnes HJ, Bagchi A, Buswell MD, O'Rourke CD. et al. Hepcidin Deficiency Protects Against Atherosclerosis. Arterioscler Thromb Vasc Biol. 2019;39:178-87
118. Xing W, Tan Y, Li K, Tian P, Tian F, Zhang H. Upregulated hepatokine fetuin B aggravates myocardial ischemia/reperfusion injury through inhibiting insulin signaling in diabetic mice. J Mol Cell Cardiol. 2020
119. Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B. et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57-63
120. Trøseid M, Ueland T, Hov JR, Svardal A, Gregersen I, Dahl CP. et al. Microbiota-dependent metabolite trimethylamine-N-oxide is associated with disease severity and survival of patients with chronic heart failure. J Intern Med. 2015;277:717-26
121. Tang TT, Li YY, Li JJ, Wang K, Han Y, Dong WY. et al. Liver-heart crosstalk controls IL-22 activity in cardiac protection after myocardial infarction. Theranostics. 2018;8:4552-62
122. Gines P, Sola E, Angeli P, Wong F, Nadim MK, Kamath PS. Hepatorenal syndrome. Nat Rev Dis Primers. 2018;4:23
123. Noble J, Jouve T, Malvezzi P, Rostaing L. Renal complications of liver diseases. Expert Rev Gastroenterol Hepatol. 2018;12:1135-42
124. Shah AS, Amarapurkar DN. Spectrum of hepatitis B and renal involvement. Liver Int. 2018;38:23-32
125. Park H, Chen C, Wang W, Henry L, Cook RL, Nelson DR. Chronic hepatitis C virus (HCV) increases the risk of chronic kidney disease (CKD) while effective HCV treatment decreases the incidence of CKD. Hepatology. 2018;67:492-504
126. Varga ZV, Matyas C, Paloczi J, Pacher P. Alcohol Misuse and Kidney Injury: Epidemiological Evidence and Potential Mechanisms. Alcohol Res. 2017;38:283-8
127. Cheungpasitporn W, Thongprayoon C, Kittanamongkolchai W, Brabec BA, O'Corragain OA, Edmonds PJ. et al. High alcohol consumption and the risk of renal damage: a systematic review and meta-analysis. Qjm. 2015;108:539-48
128. Sato KK, Hayashi T, Uehara S, Kinuhata S, Oue K, Endo G. et al. Drinking pattern and risk of chronic kidney disease: the kansai healthcare study. Am J Nephrol. 2014;40:516-22
129. Koning SH, Gansevoort RT, Mukamal KJ, Rimm EB, Bakker SJ, Joosten MM. Alcohol consumption is inversely associated with the risk of developing chronic kidney disease. Kidney Int. 2015;87:1009-16
130. Targher G, Byrne CD. Non-alcoholic fatty liver disease: an emerging driving force in chronic kidney disease. Nat Rev Nephrol. 2017;13:297-310
131. Musso G, Cassader M, Cohney S, De Michieli F, Pinach S, Saba F. et al. Fatty Liver and Chronic Kidney Disease: Novel Mechanistic Insights and Therapeutic Opportunities. Diabetes Care. 2016;39:1830-45
132. Zager RA. Alpha 1 Microglobulin: A Potentially Paradoxical Anti-Oxidant Agent. Adv Tech Biol Med. 2017 5
133. Sheetz M, Barrington P, Callies S, Berg PH, McColm J, Marbury T. et al. Targeting the hepcidin-ferroportin pathway in anaemia of chronic kidney disease. Br J Clin Pharmacol. 2019;85:935-48
134. Skrypnyk NI, Gist KM, Okamura K, Montford JR, You Z, Yang H. et al. IL-6-mediated hepatocyte production is the primary source of plasma and urine neutrophil gelatinase-associated lipocalin during acute kidney injury. Kidney Int. 2020;97:966-79
135. Roche SP, Kobos R. Jaundice in the adult patient. Am Fam Physician. 2004;69:299-304
136. Mayo MJ, Handem I, Saldana S, Jacobe H, Getachew Y, Rush AJ. Sertraline as a first-line treatment for cholestatic pruritus. Hepatology. 2007;45:666-74
137. Dogra S, Jindal R. Cutaneous manifestations of common liver diseases. J Clin Exp Hepatol. 2011;1:177-84
138. Neess D, Bek S, Bloksgaard M, Marcher AB, Faergeman NJ, Mandrup S. Delayed hepatic adaptation to weaning in ACBP-/- mice is caused by disruption of the epidermal barrier. Cell Rep. 2013;5:1403-12
139. Miele L, Vallone S, Cefalo C, La Torre G, Di Stasi C, Vecchio FM. et al. Prevalence, characteristics and severity of non-alcoholic fatty liver disease in patients with chronic plaque psoriasis. J Hepatol. 2009;51:778-86
140. Madanagobalane S, Anandan S. The increased prevalence of non-alcoholic fatty liver disease in psoriatic patients: a study from South India. Australas J Dermatol. 2012;53:190-7
141. Vasseur P, Serres L, Jegou JF, Pohin M, Delwail A, Petit-Paris I. et al. High-Fat Diet-Induced IL-17A Exacerbates Psoriasiform Dermatitis in a Mouse Model of Steatohepatitis. Am J Pathol. 2016;186:2292-301
142. Vasseur P, Pohin M, Jegou JF, Favot L, Venisse N, McHeik J. et al. Liver fibrosis is associated with cutaneous inflammation in the imiquimod-induced murine model of psoriasiform dermatitis. Br J Dermatol. 2018;179:101-9
143. Terziroli Beretta-Piccoli B, Invernizzi P, Gershwin ME, Mainetti C. Skin Manifestations Associated with Autoimmune Liver Diseases: a Systematic Review. Clin Rev Allergy Immunol. 2017;53:394-412
144. Muraguchi T, Nanba D, Nishimura EK, Tashiro T. IGF-1R deficiency in human keratinocytes disrupts epidermal homeostasis and stem cell maintenance. J Dermatol Sci. 2019;94:298-305
145. Park O, Wang H, Weng H, Feigenbaum L, Li H, Yin S. et al. In vivo consequences of liver-specific interleukin-22 expression in mice: Implications for human liver disease progression. Hepatology. 2011;54:252-61
146. Lok AS, Zoulim F, Dusheiko G, Ghany MG. Hepatitis B cure: From discovery to regulatory approval. J Hepatol. 2017;67:847-61
147. Tanabe K, Taura K, Koyama Y, Yamamoto G, Nishio T, Okuda Y. et al. Migration of splenic lymphocytes promotes liver fibrosis through modification of T helper cytokine balance in mice. J Gastroenterol. 2015;50:1054-68
148. Lee SC, Jeong HJ, Choi BJ, Kim SJ. Role of the spleen in liver regeneration in relation to transforming growth factor-beta1 and hepatocyte growth factor. J Surg Res. 2015;196:270-7
149. Yamada S, Morine Y, Imura S, Ikemoto T, Arakawa Y, Iwahashi S. et al. Liver regeneration after splenectomy in patients with liver cirrhosis. Hepatol Res. 2016;46:443-9
150. Yada A, Iimuro Y, Uyama N, Uda Y, Okada T, Fujimoto J. Splenectomy attenuates murine liver fibrosis with hypersplenism stimulating hepatic accumulation of Ly-6C(lo) macrophages. J Hepatol. 2015;63:905-16
151. Romano A, Hou X, Sertorio M, Dessein H, Cabantous S, Oliveira P. et al. FOXP3+ Regulatory T Cells in Hepatic Fibrosis and Splenomegaly Caused by Schistosoma japonicum: The Spleen May Be a Major Source of Tregs in Subjects with Splenomegaly. PLoS Negl Trop Dis. 2016;10:e0004306
152. Tang WP, Akahoshi T, Piao JS, Narahara S, Murata M, Kawano T. et al. Splenectomy enhances the therapeutic effect of adipose tissue-derived mesenchymal stem cell infusion on cirrhosis rats. Liver Int. 2016;36:1151-9
153. Suzuki S, Nakamura S, Serizawa A, Sakaguchi T, Konno H, Muro H. et al. Role of Kupffer cells and the spleen in modulation of endotoxin-induced liver injury after partial hepatectomy. Hepatology. 1996;24:219-25
154. Asanoma M, Ikemoto T, Mori H, Utsunomiya T, Imura S, Morine Y. et al. Cytokine expression in spleen affects progression of liver cirrhosis through liver-spleen cross-talk. Hepatol Res. 2014;44:1217-23
155. Li W, Kuhr CS, Zheng XX, Carper K, Thomson AW, Reyes JD. et al. New insights into mechanisms of spontaneous liver transplant tolerance: the role of Foxp3-expressing CD25+CD4+ regulatory T cells. Am J Transplant. 2008;8:1639-51
156. Barrea L, Di Somma C, Muscogiuri G, Tarantino G, Tenore GC, Orio F. et al. Nutrition, inflammation and liver-spleen axis. Crit Rev Food Sci Nutr. 2018;58:3141-58
157. Rodriguez-Roisin R, Krowka MJ. Hepatopulmonary syndrome-a liver-induced lung vascular disorder. N Engl J Med. 2008;358:2378-87
158. Fuhrmann V, Krowka M. Hepatopulmonary syndrome. J Hepatol. 2018;69:744-5
159. Pai M, Behr MA, Dowdy D, Dheda K, Divangahi M, Boehme CC. et al. Tuberculosis. Nat Rev Dis Primers. 2016;2:16076
160. Teleman MD, Chee CB, Earnest A, Wang YT. Hepatotoxicity of tuberculosis chemotherapy under general programme conditions in Singapore. Int J Tuberc Lung Dis. 2002;6:699-705
161. Hu X, Fu Y, Lu X, Zhang Z, Zhang W, Cao Y. et al. Protective Effects of Platycodin D on Lipopolysaccharide-Induced Acute Lung Injury by Activating LXRalpha-ABCA1 Signaling Pathway. Front Immunol. 2016;7:644
162. Mukhopadhyay S, Mondal SA, Kumar M, Dutta D. Proinflammatory and antiinflammatory attributes of fetuin-a: a novel hepatokine modulating cardiovascular and glycemic outcomes in metabolic syndrome. Endocr Pract. 2014;20:1345-51
163. Kundranda MN, Henderson M, Carter KJ, Gorden L, Binhazim A, Ray S. et al. The serum glycoprotein fetuin-A promotes Lewis lung carcinoma tumorigenesis via adhesive-dependent and adhesive-independent mechanisms. Cancer Res. 2005;65:499-506
164. Diao WQ, Shen N, Du YP, Liu BB, Sun XY, Xu M. et al. Fetuin-B (FETUB): a Plasma Biomarker Candidate Related to the Severity of Lung Function in COPD. Sci Rep. 2016;6:30045
165. Gao F, Fang J, Chen F, Wang C, Chen S, Zhang S. et al. Enho Mutations Causing Low Adropin: A Possible Pathomechanism of MPO-ANCA Associated Lung Injury. EBioMedicine. 2016;9:324-35
166. Allen AM, Hicks SB, Mara KC, Larson JJ, Therneau TM. The risk of incident extrahepatic cancers is higher in non-alcoholic fatty liver disease than obesity - A longitudinal cohort study. J Hepatol. 2019;71:1229-36
167. Stefan N, Haring HU. The role of hepatokines in metabolism. Nat Rev Endocrinol. 2013;9:144-52
168. Itoh N. FGF21 as a Hepatokine, Adipokine, and Myokine in Metabolism and Diseases. Front Endocrinol (Lausanne). 2014;5:107
169. Kyohara M, Shirakawa J, Okuyama T, Togashi Y, Inoue R, Li J. et al. Soluble EGFR, a hepatokine, and adipsin, an adipokine, are biomarkers correlated with distinct aspects of insulin resistance in type 2 diabetes subjects. Diabetol Metab Syndr. 2020;12:83
170. Mouchiroud M, Camire E, Aldow M, Caron A, Jubinville E, Turcotte L. et al. The hepatokine Tsukushi is released in response to NAFLD and impacts cholesterol homeostasis. JCI Insight. 2019 4
171. Veyel D, Wenger K, Broermann A, Bretschneider T, Luippold AH, Krawczyk B. et al. Biomarker discovery for chronic liver diseases by multi-omics - a preclinical case study. Sci Rep. 2020;10:1314
172. Yu H, Xia F, Lam KS, Wang Y, Bao Y, Zhang J. et al. Circadian rhythm of circulating fibroblast growth factor 21 is related to diurnal changes in fatty acids in humans. Clin Chem. 2011;57:691-700
173. Okumura A, Unoki-Kubota H, Matsushita Y, Shiga T, Moriyoshi Y, Yamagoe S. et al. Increased serum leukocyte cell-derived chemotaxin 2 (LECT2) levels in obesity and fatty liver. Biosci Trends. 2013;7:276-83
174. Watt MJ, Miotto PM, De Nardo W, Montgomery MK. The Liver as an Endocrine Organ-Linking NAFLD and Insulin Resistance. Endocr Rev. 2019;40:1367-93
175. Wang Q, Sharma VP, Shen H, Xiao Y, Zhu Q, Xiong X. et al. The hepatokine Tsukushi gates energy expenditure via brown fat sympathetic innervation. Nat Metab. 2019;1:251-60
176. Oria RB, Murray-Kolb LE, Scharf RJ, Pendergast LL, Lang DR, Kolling GL. et al. Early-life enteric infections: relation between chronic systemic inflammation and poor cognition in children. Nutr Rev. 2016;74:374-86
177. Romero-Gomez M, Ramos-Guerrero R, Grande L, de Teran LC, Corpas R, Camacho I. et al. Intestinal glutaminase activity is increased in liver cirrhosis and correlates with minimal hepatic encephalopathy. J Hepatol. 2004;41:49-54
178. Sancho-Tello M, Muriach M, Barcia J, Bosch-Morell F, Genoves JM, Johnsen-Soriano S. et al. Chronic alcohol feeding induces biochemical, histological, and functional alterations in rat retina. Alcohol Alcohol. 2008;43:254-60
179. Birrell MA, Catley MC, Hardaker E, Wong S, Willson TM, McCluskie K. et al. Novel role for the liver X nuclear receptor in the suppression of lung inflammatory responses. J Biol Chem. 2007;282:31882-90
Author contact
![]() Corresponding authors: Hua Wang, MD, PhD. E-mail: wanghuaedu.cn; or Jia Xiao, PhD. E-mail: edwinsiuhku.hk
Corresponding authors: Hua Wang, MD, PhD. E-mail: wanghuaedu.cn; or Jia Xiao, PhD. E-mail: edwinsiuhku.hk