Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Classical viewpoint
Autophagy-related molecules...
The estrogen /...
Ectopic endometrium autophagy on...
Clinical implications and future...
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(7):3512-3526. doi:10.7150/thno.55241 This issue Cite
Review
Ovarian hormones-autophagy-immunity axis in menstruation and endometriosis
Hui-Hui Shen1#, Tao Zhang2#, Hui-Li Yang1, Zhen-Zhen Lai1, Wen-Jie Zhou3, Jie Mei4, Jia-Wei Shi1, Rui Zhu5, Feng-Yuan Xu6, Da-Jin Li1, Jiang-Feng Ye7, Ming-Qing Li1,8 ![]()
1. Laboratory for Reproductive Immunology, NHC Key Laboratory of Reproduction Regulation (Shanghai Institute of Planned Parenthood Research), Hospital of Obstetrics and Gynecology, Shanghai Medical School, Fudan University, Shanghai 200080, People's Republic of China.
2. Assisted Reproductive Technology Unit, Department of Obstetrics and Gynecology, Faculty of Medicine, Chinese University of Hong Kong, Hong Kong, People's Republic of China.
3. Center of Reproductive Medicine of Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, People's Republic of China.
4. Reproductive Medicine Center, Department of Obstetrics and Gynecology, Nanjing Drum Tower Hospital, The Affiliated Hospital of Nanjing University Medicine School, Nanjing, 210000, People's Republic of China.
5. Center for Human Reproduction and Genetics, Suzhou Municipal Hospital, Suzhou 215008, People's Republic of China.
6. Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta 30332, Georgia, USA.
7. Division of Obstetrics and Gynecology, KK Women's and Children's Hospital, 229899, Singapore.
8. Shanghai Key Laboratory of Female Reproductive Endocrine Related Diseases, Shanghai, 200080, People's Republic of China.
#Co-first authors.
Received 2020-10-30; Accepted 2021-1-2; Published 2021-1-19
Abstract

Menstruation occurs in few species and involves a cyclic process of proliferation, breakdown and regeneration under the control of ovarian hormones. Knowledge of normal endometrial physiology, as it pertains to the regulation of menstruation, is essential to understand disorders of menstruation. Accumulating evidence indicates that autophagy in the endometrium, under the regulation of ovarian hormones, can result in the infiltration of immune cells, which plays an indispensable role in the endometrium shedding, tissue repair and prevention of infections during menstruation. In addition, abnormal autophagy levels, together with resulting dysregulated immune system function, are associated with the pathogenesis and progression of endometriosis. Considering its potential value of autophagy as a target for the treatment of menstrual-related and endometrium-related disorders, we review the activity and function of autophagy during menstrual cycles. The role of the estrogen/progesterone-autophagy-immunity axis in endometriosis are also discussed.
Keywords: autophagy, endometrium, estrogen, macrophage, menstruation, neutrophil, NK cell, progesterone
Introduction
Menstruation refers to cyclical shedding and regeneration of the endometrium occurring in very few species. It is regarded as an inflammatory event regulated by ovarian hormones [1, 2]. Menstruation occurs up to 400 times during a woman's reproductive lifespan. Endometriosis is prevalent in 10% of women of reproductive age worldwide, characterized by growth of endometrial tissue outside of the uterus, mainly on the ovaries and the pelvic peritoneum [3, 4]. Although the mechanisms of endometriosis remain unclear, retrograde menstruation is known as a key factor in its pathogenesis [5]. Likewise, how ovarian hormones orchestrate cyclic changes of endometrium during menstruation has not been fully clarified. Recently, a growing number of studies have reported the crucial role of the interaction between estrogen signaling and autophagy in menstruation as well as endometriosis.
Macroautophagy/autophagy is an intracellular self-digestion pathway for the maintenance of homeostasis. This can be found in all eukaryotic cells at basal level and can be further activated by a variety of stimuli, including deprivation of nutrients, infection, hormones, oxidative stress and endoplasmic reticulum stress [6]. It is characterized by the formation of autophagosomes with double-membrane structure, which fuse with lysosomes to degrade cytosolic proteins, damaged or excess organelles, protein aggregates, and invasive microbes [7].
It is well accepted that autophagy plays a critical role in cancers, embryo implantation and reproduction [8-11]. Interestingly, increasing evidence indicates autophagy is also involved in menstruation and endometriosis. In this review, we discuss the role of autophagy-immune regulation axis controlled by ovarian hormones in menstruation, its role in the pathogenesis of endometriosis and its potential values in the prevention and treatment of menstrual-related and endometrium-related diseases.
Classical viewpoint
According to the conventional viewpoint, by exposure to sequential ovarian estrogen and progesterone will cause the endometrium to undergo well-defined cycles of proliferation, differentiation, and shedding (menstruation) on a monthly basis [12, 13]. The molecular mechanisms underlying these events involves complex crosstalk between the endocrine and immune system [13, 14]. In the absence of pregnancy, progesterone withdrawal drives a local inflammatory response in the uterus involving infiltration of leukocytes, release of cytokines, activation of matrix metalloproteinases (MMPs) and disintegration of extracellular matrix (ECM) [12, 15]. The presence of progesterone inhibits neutrophils entry into the endometrium [14], NF-κB activation, and the expression of several stromal cell-derived, lytic enzymes, including urokinase-type plasminogen activator (uPA), MMP-1, and MMP-3 [16]. Thus, withdrawal of progesterone can be followed by a dramatic rise in the endometrial leukocyte population. Besides on the generation of more cytokines (e.g. interleukin (IL)-1, IL-6) and proteases (e.g. MMPs), these cells are also involved in ECM breakdown and debris removal.
Among the inflammatory mediators stimulated, IL-8 [a neutrophil chemotactic factor, also called C-X-C motif chemokine ligand 8 (CXCL8)], monocyte chemoattractant protein-1 [MCP-1, also called C-C motif chemokine ligand 2 (CCL2)] and cyclooxygenase-2 [COX-2, the inducible enzyme responsible for synthesis of prostaglandins (PGs)], are well-studied [17]. COX-2 upregulation mediated by nuclear factor kappa-B (NF-κB) leads to increased levels of PGs. PGF2α causes myometrial contractions and vasoconstriction of the endometrial spiral arterioles, resulting in a hypoxic environment in the endometrium [18].
Hypoxia during menstruation can directly upregulate vascular endothelial growth factor (VEGF) expression in endometrial stromal cells (ESCs) by modulating the NF-κB pathway [19]. VEGF is an important regulator of angiogenesis that stimulates the proliferation, migration, and proteolytic activity of endothelial cells. It can also induce MMP-1, -3 and -9 expression in vascular smooth muscle cells, which is required for tissue repair. Following endometrial breakdown, MMP activity can be inhibited by tissue inhibitors of metalloproteinases (TIMPs) or by the protease inhibitor α2-macroglobulin, which can ensure the limitation of tissue damage. During the proliferative phase, the functional layer of the endometrium experiences a rapid growth under the influence of estrogen. As a result, concomitant angiogenesis occurs to repair ruptured blood vessels and maintains perfusion of the new tissue.
Autophagy-related molecules during the menstruation cycle
Autophagy-related genes and molecules in the endometrium
Autophagy involves five steps, namely, nucleation, elongation, maturation, fusion and degradation by lysosomes. Double-membraned structures first engulf portions of the cytoplasm to form autophagosomes. Autophagosome is initiated by the unc-51-like kinase 1 (ULK1) complex (including ULK1, ULK2, Atg13, FIP200 and Atg101). This complex can be suppressed via mammalian target of rapamycin complex (mTOR). During autophagosomal vesicle elongation and maturation, autophagy-related (Atg) protein 7 and Atg10 promote Atg12 and Atg5 conjugation. Such that, Atg 12 and Atg5 could interact with Atg16L1 to form a dimeric complex (Atg12-Atg5-Atg16L1), which is required for formation of the covalent bond between LC3 and phosphatidylethanolamine (PE). Then LC3 precursor (pro-LC3) converts into LC3-Ι in the action of Atg4, which is activated by Atg7 and transferred to Atg3. The Atg12-5-16L1 complex facilitates the transfer of LC3-Ι from Atg3 to PE to generate microtubule-associated protein 1 light chain 3 (LC3‑II) [20], which is a standard marker of autophagic flux and localizes to both the inner and outer autophagosomal membranes.
LC3-Ι, a marker of autophagy, is increased during autophagy induction, both in endometrial glandular epithelial cells (EPCs) and ESCs throughout the menstrual cycle. LC3-I expression is very low in the proliferative phase, but it surges to peak level at the late secretory phase [21]. LC3-II expression in the late proliferative phase is higher than in the early proliferative phase [22]. Additionally, autophagosome numbers are higher in secretory ESCs, compared with the proliferative phase [23].
Role of ovarian hormones on endometrium autophagy
Effects of ovarian hormones on endometrial autophagy
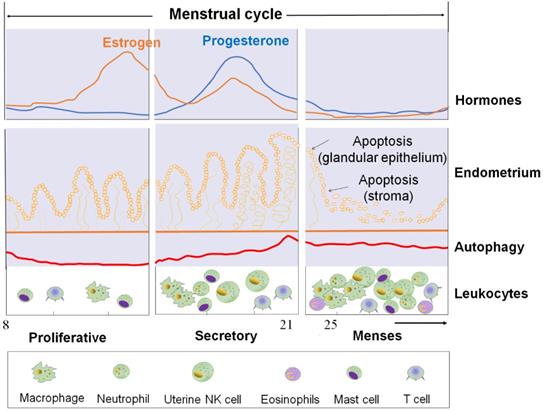
During the normal human secretory phase ESCs, estrogen is a potent suppressor of autophagy while progesterone can significantly stimulate the activities of autophagy [23, 24]. During menstruation cycle, autophagy levels remain low in the proliferative phase but significantly increase at the late secretory phase (Figure 1) [23, 24]. Choi et al. showed that the expression levels of LC3-II and cleaved caspase-3 increased significantly in normal ESCs cultured with estrogen and progesterone when compared with those cultured with estrogen alone [22]. In vitro experiment also reported the autophagy level in endometrial cells treated with estrogen alone (in vitro imitation of the proliferative phase) was low and was upregulated after the supplementation with progesterone (in vitro imitation of early-to-mid secretory phase) or following the removal of estrogen and progesterone (in vitro imitation of late secretory and menstrual phases) [25]. Specifically, progesterone significantly increased LC3-II and ATG5 expression in estrogen-treated normal ESCs, while AKT phosphorylation (which negatively regulate autophagy induction by activating mTOR) was significantly decreased [25]. This change could be suppressed by the addition of mifepristone (progesterone receptor modulator) [25]. Notably, the stimulatory effect of progesterone on ESC autophagy was also dependent on the CXCL12/CXCR4 axis [22].
The cyclical changes of ovarian steroid hormones, endometrium, and autophagy level in ESC and autophagy levels of ESC and the infiltration of immune cells during the menstrual cycle. First panel: Estrogen and progesterone levels throughout the menstrual cycle. Second panel: The morphological changes in the endometrium during the menstrual cycle. Third panel: Autophagy during the menstrual cycle. Bottom panel: Trafficking of Leukocytes into the endometrium during the menstrual cycle. The size of the cells represents the abundance. ESC: endometrial stromal cell.
In addition to ESCs, a large number of EPCs also reside in the endometrium, especially during the secretory phase of the menstrual cycle. The regulation of autophagy in normal EPCs by ovarian steroids has not been reported so far, which may be due to the fact that the primary normal EPCs are very difficult to obtain and culture. Nevertheless, some insights can still be obtained from studies on autophagy in uterine corpus endometrial carcinoma (UCEC) cells. Consistent with estrogen's inhibitory effect on the autophagy of ESCs, our previous studies also show that estrogen significantly restricts autophagy of UCEC cells in vitro and in vivo [26, 27]. Many drugs targeting autophagy-suppression are already in clinical use for endometrial hyperplasia [28]. Additionally, protopanaxadiol (PPD) and metformin alone or in combination exert their therapeutic roles by inducing autophagy of UCEC cell with higher levels of Beclin1 and LC3 II/LC3 I, and low level of p62 expression [27]. These reports demonstrate that estrogen has a strong inhibitor effect on the autophagy of ESCs and EPCs. Admittedly, there are some inconsistent results in other types of cells due to different local microenvironments [29-31].
Therefore, it may be hypothesized that, with estrogen and progesterone withdrawal, there should be higher levels of autophagy in ESC and EPC due to the absence of powerful estrogen-mediated autophagy inhibition (Figure 1).
How do ovarian hormones affect endometrial autophagy?
Ovarian hormones, that are the upstream of signaling mediators to regulate various aspects of uterine physiology, also serve as regulators of autophagy. Estrogen may mediate autophagy through its receptors (ER), namely ERα, ERβ and G-protein coupled estrogen receptor (GPER) [31, 32]. In both the human and nonhuman primate endometrium, ERα and ERβ are expressed in the nuclei and cytoplasm of glandular epithelial and stromal cells, which decline in the mid secretory phase in the functional layer [33]. Endometrial ERα levels are upregulated during the proliferative phase by estrogen, which is at its peak during the mid- to late proliferative phase of the menstrual cycle [34], and subsequently is downregulated in the secretory phase by progesterone [35, 36]. Estrogen suppresses autophagy through ERα in neurons, which contributes to less severity of iron-induced brain injury women compared with that in men [37]. Our previous research showed that estrogen could suppress autophagy through CXCL12/CXCR4 interaction and NF-κB signal activation in ESCs [23]. However, PPD downregulates the expression of ERα, upregulates the expression of progesterone receptors (PGRs), which restricts estrogen-mediated anti-autophagy effects in ESCs [38]. Moreover, fulvestrant is a selective ER antagonist that can abrogate the inhibitory effect of estrogen on autophagy in UCEC cells [26].
According to a number of recent studies, progesterone and progestins can be particularly important as inhibitors of mTOR in various cell types [25, 39], which can result in the activation of autophagy. These effects are mediated by PGR, consisting of PGRA and PGRB. Progesterone can activate PGRB and further induce autophagy via mediating nuclear translocation and activation of transcription factor EB (TFEB) [40]. These can then act as a master transcriptional regulator that would control the expression of autophagy and lysosomal genes. Therefore, TFEB upregulates the expression of autophagy and lysosomal genes to activate autophagy. Moreover, inhibition of progesterone receptor membrane component 1 (PGRMC1, also known as sigma-2 receptor) can increase the activation of adenosine 5'-monophosphate (AMP)-activated protein kinase (AMPK) and promotes the levels of tuberous sclerosis complex (TSC), which causing the inactivation of mTOR and the elevated autophagic flux [41]. Combined with progesterone, PGR can activate MAPK/ERK and the transcription factor cyclic AMP response-element binding protein (CREB), thereby modulating the expression of genes required for cell proliferation, inflammatory responses, differentiation, and apoptosis [42].
Hypoxia-inducible factor-1 (HIF-1), a transcription factor which regulates cellular response to hypoxia, is expressed exclusively during the secretory and menstrual phases, suggesting hypoxia would occur in the endometrium late in the menstrual cycle [43]. It has been reported that estrogen negatively correlatives with HIF-1 levels in other cells (e.g, macrophages and breast cancer cells) [44, 45]. Following treatment with follicle-stimulating hormone (FSH), autophagy signaling was activated in mouse granulosa cells via HIF-1α [46], contributing to follicle development and atresia. Estrogen withdrawal is linked to endometrial hypoxia during menstruation when spiral arterioles contract under the action of PGF2. Notably, overloaded reactive oxygen species (ROS) are generated due to the lack of oxygen, nutrient starvation, mitochondrial toxins and oxidative stress, resulting in organelle damage and the production of toxic substances [47, 48]. ROS elicits autophagy by reducing the ATP (adenosine triphosphate)/AMP (adenosine monophosphate) ratio. As a result, AMPK activates the TSC, leading to mTOR inactivation and initiation of autophagy [49]. Studies have shown that estrogen can inhibit ROS production, thus protecting cells from oxidative stress damage [50, 51]. Interesting, ERβ and GPER have also been reported to be involved in this process [50, 52]. Sugino et al. have reported that estrogen and progesterone withdrawal stimulated PGF2α production through ROS-induced NF-κB activation in human ESCs [53]. From these reports, it can be concluded that estrogen withdrawal might induce autophagy of endometrium during menstruation through the activation of the HIF-1/ROS/AMPK signaling pathway and further inactivation of mTOR signaling (Figure 2).
Signal transduction mechanisms activated by ovarian steroid hormones on endometrial autophagy. Estrogen (E2) inhibits reactive oxygen species (ROS) production, which further suppresses autophagy in the endometrium. ROS has been shown to suppress the mTOR signaling pathway and further induce autophagy through several mechanisms, including activation of endoplasmic reticulum (ER) stress. Additionally, ER stress induces autophagy via the PI3K/AKT pathway and the COX-2/prostaglandins (PGs) axis. Of note, progesterone (P4) suppresses ER stress by restricting the PGF2α/ROS axis. However, the inhibition of mTOR signaling aggregates ER stress. Under stimulation from estrogen or progesterone, autophagy is induced through the activation of the MAPK/ERK pathway, which regulates autophagy-related genes through the transcription factor CREB. Therefore, the withdrawal of estrogen and progesterone leads to the inactivation of mTOR signaling and high levels of endometrial autophagy during menstrual phase through the HIF-1/ROS/AMPK signaling pathway, ER stress and MAPK/ERK signaling pathway.
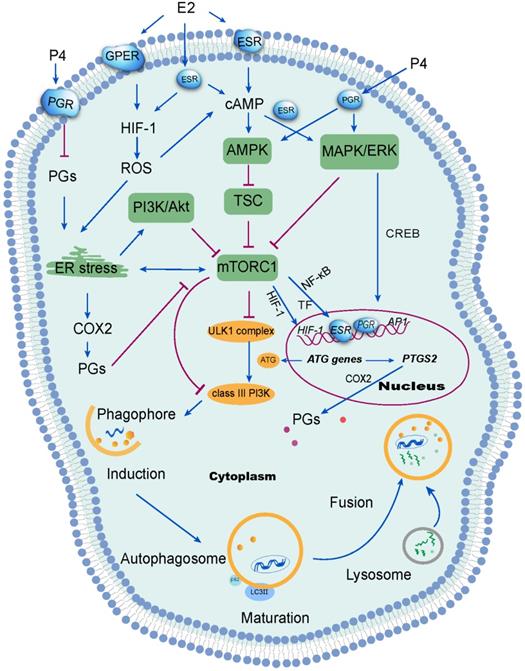
Several studies have identified the AMPK/TSC/mTOR signaling pathway as the classical signaling pathway regulating autophagy [54]. In mammals, various stimuli, including hypoxia, activate the AMPK or mitogen-activated protein kinases (MAPK)/ERK (extracellular signal-regulated kinases) signaling pathways can result in the suppression of mTOR and the activation ULK1 complex [55]. Alternatively, the inhibition of class I phosphatidylinositol 3-kinase (PI3K) and its downstream target AKT suppresses mTOR signaling. In turn, the inhibition of mTOR reduces the phosphorylation of ATG13 [56]. Thereafter, ATG13 forms a ULK1 complex with ULK1, focal adhesion kinase family interacting protein of 200 kDa (FIP200) and ATG101, to induce autophagy by phosphorylating Beclin 1 and activate class-III PI3K complex (hVps34-hVps15-Beclin1) [57]. Under nutrient-rich conditions, the mTOR complex 1 (mTORC1) mediates phosphorylation-dependent inactivation of the kinase activities of ATG13 and ULK1, leading to the inhibition of autophagy. In the event of starvation or treatment with rapamycin, mTORC1 will dissociate from this complex to decrease mTORC1-mediated phosphorylation of ATG13 and ULK1. During the initiation of autophagy, ATG7 (E1 ubiquitin-activating enzyme-like) activates the ubiquitin-like protein ATG12. Activated ATG12 subsequently is transferred to ATG10 (E2 ubiquitin-conjugating enzyme-like) and is ultimately linked covalently to ATG5 to form a complex (E3-like) with ATG16L1 [58]. This large complex is vital for the elongation of phagophores, but dissociates after autophagosome formation [59]. In addition, the LC3 precursor undergoes C-terminal cleavage by ATG4B to produce LC3-I. LC3-I is then conjugated to PE via interaction with Atg7 (E1-like), Atg3 (E2-like), ATG16L1 complex in sequence, generating LC3-II. Previous studies indicated that ovarian hormones (estrogen and progestogen) promoted breast tumorigenesis by enhancing insulin like growth factor receptor (IGFR) and Akt/mTOR signaling to inhibit apoptotic stimuli [60], suggesting that the suppression of autophagy induced by estrogen in breast cancer cells could be abolished by silencing ERα or knocking out ATG7 [37]. Akt is considered as a survival factor of endometrial cells in rodents [61]. Rapid activation of Akt by estrogen and progestogen has also been shown in human ESCs, resulting in proliferation and migration of ESCs [24, 61]. Moreover, phospho-Akt levels decreased in human ESCs when decidualization was induced triggered by high levels of progesterone [62]. These reports suggest that the Akt/mTOR signaling pathways could be involved in the induction of autophagy in the endometrium, due to estrogen and progesterone withdrawal during the menstrual phases (Figure 2). However, the specific mechanism has yet to be clarified.
In response to progesterone, endometrial stromal fibroblasts acquire epithelioid characteristics, such as a highly developed endoplasmic reticulum required for greater secretory function, substantial lysosomes, abundant glycogen and lipids droplets in their cytoplasm [63]. In animal models, the expression of endoplasmic reticulum stress-related proteins and the autophagy marker LC3-II are increased in luteal cells treated with PGF2α [64]. Physiologically, elevated PGF2α in menstruation induces endoplasmic reticulum stress. Consequently, autophagy is induced by endoplasmic reticulum stress, with the activation of inositol-requiring protein-1α (IRE1α), protein kinase RNA (PKR)-like ER kinase (PERK), and activating transcription factor 6 (ATF6) [65, 66]. The activation of PERK inhibits general protein translation by inducing eIF2α phosphorylation, enabling translation of activating transcription factor 4 (ATF4) [67]. ATF4 can activate autophagy via the induction of several ATG genes [68]. For example, endoplasmic reticulum stress eIF2α-ATF4 pathway-mediated COX-2 overexpression contributes to kidney autophagy and injury [69]. It has been reported that progesterone has an inhibitory effect on PG secretion of pig endometrial glandular and stromal cells in vitro [70]. In addition, progesterone can alleviate ROS stress in UCEC cells [71]. Therefore, it can be speculated that progesterone prevents endometrial cells from endoplasmic reticulum stress by controlling the PGF2α/ROS signaling pathway. The withdrawal of progesterone may participate in the induction of high autophagy in endometrium and menstruation through the regulation of endoplasmic reticulum stress and these downstream metabolic signaling pathways. Further studies are needed to clarify the detailed mechanisms.
The estrogen / progesterone-autophagy-immunity axis in menstruation
Infiltration of immune cells in endometrium and immune defense
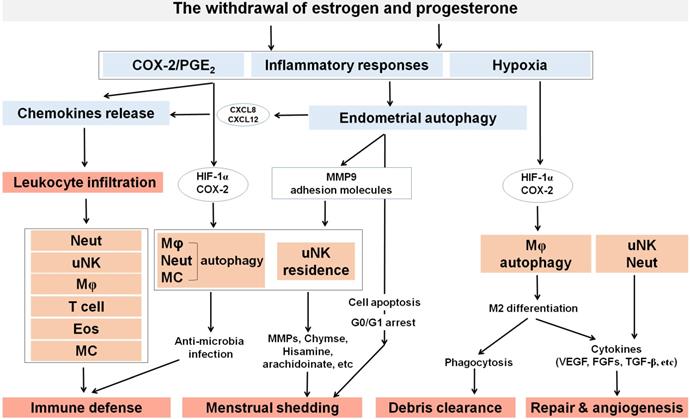
Under the cyclic regulation of estrogen and progesterone, leukocytes populations within the endometrium vary considerably across the menstrual cycle, and the mass infiltration of immune cells is closely associated with embryo implantation and menstruation [72]. During the mid-secretory phase of the human menstrual cycle, a large increase of specific subpopulations of immune cells [mainly uterine NK (uNK) cells and macrophages] infiltrate the endometrium to modulate uterine receptivity and embryo implantation [72]. In particular, many chemokines (e.g, CXCL12, CCL2, CCL4 and CX3CL1) are involved in these processes [23, 72]. With the withdrawal of estrogen and progesterone, however, the increase of COX-2 and PGF2α promotes uterine contractility and constricts the spiral arterioles in the late secretory phase endometrium. Subsequently, inflammatory responses and the formation of a hypoxic microenvironment further induce an influx of more immune cells (predominantly macrophages, uNK cells, neutrophils, eosinophils and mast cells) by the upregulation of chemokines and their receptors, including CXCL8 and CCL2, eotaxin, MMPs, and CX3CR1 (Figure 1) [72]. These immune cells play an important role on the immunity during the menstrual period.
It has been reported that adipose mTORC1 suppresses PGs signaling and beige adipogenesis via the CREB-regulated transcription coactivator 2 (CRTC2)-COX-2 pathway [73]. Such that, ATG7 regulates ultraviolet-induced cytokine expression and secretion, which can promote COX-2 expression in keratinocytes in a CREB1/CREB-dependent and IL1β-dependent manners [74]. In addition, autophagy promotes CCL2 transcription in epidermal keratinocytes through the AMPK-BRAF-MAPK1/3/ERK-activator protein 1 (AP1) pathway [75], and upregulates CXCL8 to trigger mesenchymal stem cell-mediated CD4+T cell migration and differentiation [76]. The presence of microbes in the uterine cavity has been widely demonstrated, which initiates immunologic reactions [77]. High levels of autophagy may participate in the regulation of endometrial infiltration of immune cells to play a protective role during the menstrual cycle, and the activation of COX-2/PGE2 and up-regulation of chemokines may be involved in these processes (Figure 2).
Macrophages are present throughout the menstrual cycle but display a substantial increase in number during the perimenstrual phase in response to progesterone withdrawal [78]. After macrophage infiltration, other immune cells, which are critical in cell-mediated immunity and in the resolution of inflammation will further be recruited [79]. Evidence shows that induction of autophagy is critical for the survival and differentiation of monocytes [80]. Under the stimulation of colony-stimulating factor 1 (CSF1) or granulocyte-macrophage colony-stimulating factor (GM-CSF), macrophages will be differentiated from monocytes. These processes are initiated dependently by the release of Beclin1 from Bcl2 from activated c-Jun N-terminal kinase (JNK) and blockage of ATG5 cleavage [80, 81]. Additionally, ROS production can be induced by CSF2, which is a potent stimulator of autophagy and persists in the uterus during the secretory phase [82]. In addition, estrogen can reduce the expression of HIF-1α in macrophages during the proliferative phase [45]. Therefore, autophagy level of macrophage should be elevated during menstrual phase. Adequate autophagy allows macrophages to maintain immune homeostasis and function, for pattern recognition, cytokine release, inflammasome activation, and LC3-associated phagocytosis [83, 84]. Hence, this making COX-2 an enhancer of the anti-microbial function of macrophages through the activation of autophagy [17, 85].
Typically, NK cell numbers increase dramatically from post-ovulation (around LH+3) to few days prior to menstruation [72]. As found, uNK cells (CD56brightCD16-), which is usually found near the endometrial glands and spiral arteries, increase in number from the proliferative phase and peak in the late-secretory phase [86]. These processes are mainly due to the recruitment of high levels of CXCL12 in the endometrium, which is modulated by estrogen and progesterone [23]. The cytotoxicity of uNK cells with high levels of the activation markers (CD69 and HLA-DR) is, comparable to peripheral NK cells in the late-proliferative phase. These characteristics of uNK cells are likely to prevent microbial infections [77]. Our recent studies demonstrated that the increase of autophagy of ESCs should promote the residence of NK cells in endometrium/decidua through upregulating MMP-9 and adhesion molecules expression [87]. Therefore, the increased numbers of resident uNK cell in the endometrium, triggered by high levels of autophagy during menstruation, supports immune defense.
During the early proliferative phase of the menstrual cycle, neutrophils and eosinophils are scarce in the human endometrium. However, their numbers significantly increase when reaching into premenstrual period [77]. Mast cells, located in close proximity to vessels, the neutrophil chemoattractants CXCL1 and CXCL2, which then works with macrophages [88] to recruit neutrophils [89]. LC3-II is mainly localized in the secretory granules of mast cells. Besides, autophagy is involved in the degranulation of mast cells, consolidating the antimicrobial capacity [90]. Such that, inhibition of autophagy or deficiency of either ATG5 or ATG7 was shown to reduce the degranulation of neutrophils in mouse models [91]. Importantly, the presence of autophagy in neutrophils is beneficial for bacteria clearance [91] and act as a modulator for multiple functions, including phagocytosis, degranulation, ROS production, formation of NET, and IL-1β production [92-94]. During menstruation, therefore, the increase in uterine contractility and local hypoxia should trigger autophagy in neutrophils and mast cells in the endometrium to act as a beneficiary for fighting various pathogens within uterus.
Removal debris and dead cells during menstruation
The spontaneous periodic apoptosis during the menstrual cycle is essential for maintaining the normal structure and function of the endometrium, while cell autophagy plays a critical role in the apoptosis of human endometrial cells at different phases of the menstrual cycle [95, 96]. High levels of autophagy in the endometrium during the mid- and late secretory stage can trigger autophagic cell death and initiate apoptosis to prepare for endometrial exfoliation during menses, this may then contribute cyclic remodeling of the endometrium [95, 97]. Coincident with changes in autophagic activity, cleaved caspase 3 expression levels in glandular epithelial cells increase significantly during the secretory phase, reaching a maximum during the late-secretory phase [95]. In addition, autophagy induces G0/G1 arrest and apoptosis in menstrual blood-derived endometrial stem cells [97]. Autophagy of ESC promotes the upregulation of MMP9 in a melanocyte inducing transcription factor (MITF)- herpesvirus entry mediator (HVEM)-dependent manner [87], resulting in rapid ECM degradation and endometrial exfoliation occur during menstruation. Remarkably, the intracellular composition of the ESC can be renewed through autophagy. This is brought upon by the rapid clearance of mRNA and new proteins synthesized by the gene expression program, as well as the recycling of damaged organelles [98].
Under the regulation of endometrial autophagy, a large number of infiltrating leukocytes accelerate endometrial exfoliation and become the main force for removing cellular debris, apoptotic and necrotic cells (Figure 3). Among these, macrophages and uNK cells secrete MMP-7 and MMP-9, resulting in the apoptosis of smooth muscle cells and endothelial cells [99]. The proinflammatory cytokines secreted by these immune cells, such as TNF-α, play a role in menstrual shedding, by upregulating the expression of Fas and facilitating Fas-mediated apoptosis [100]. Mast cells in endometrium express tryptase and chymase, likely contributing to menstruation. Chymase, in particular, can play an important role in establishing a cascade of MMPs activation [77]. In addition, mast cells produce histamine, arachidonate, heparin products and a large number of pleiotropic cytokines. They have important effects on endothelial cell function and local induction of edema. More importantly, CD68+ macrophages are abundant in the human endometrium during tissue breakdown and repair. As scavenger cells, macrophages can eliminate apoptotic cells in the uterus and endometrial cells in the peritoneal cavity following the retrograde flow of menstrual blood [101]. Certainly, this process is dependent on the bioavailability on autophagy of macrophages.
The roles of ovarian steroid hormones-autophagy-immunity axis in menstruation. With the withdrawal of estrogen and progesterone, local inflammatory responses are initiated in the endometrium together with the activation of endometrial autophagy. Subsequently, MMPs are released and synergize with other inflammatory factors to participate in menstrual shedding. The release of a large number of chemokines (e.g., CXCL8, CXCL12, CCL2, CCL4 and CX3CL1) results in leukocyte infiltrations (neutrophils, uterine NK cell, macrophage, T cell, eosinophils, and mast cells) that can contribute to immune defense during menstruation. Residence of uterine NK cell (uNK) activated by endometrial autophagy and mast cells accelerate menstrual shedding by MMP9, Chymase, and etc. Additionally, elevated levels of COX-2 and PGs not only generate a hypoxic environment, but also induce autophagy in macrophages, neutrophils and mast cells that protect the endometrium from infections. COX-2 is also produced as a result of hypoxia during late menstruation, facilitating M2 macrophages polarization which participates in debris clearance, endometrial repair and remodeling. Meanwhile, uNK and neutrophils also contribute to endometrium repair and remodeling by secreting various cytokines (e.g, VEGF, FGF, and TGF-β). Neut: neutrophils; Mφ: macrophage; EOS: eosinophils; MC: mast cell.
Endometrial tissue remodeling during late menstruation
A high level of autophagy during late menstruation facilitates M2 macrophages polarization to participate in efficient repair and remodeling (Figure 3) [102, 103]. It is well established that classically activated “M1” macrophages produce inflammatory cytokines while the activated “M2” macrophages (CD68+/CD163+) promote tissue remodeling and anti-inflammatory reactions due to reduced proinflammatory cytokine secretion and increased endocytic clearance capacity [104, 105]. Endometrial macrophages constantly change their phenotype to adapt to their microenvironment [78]. Macrophages at wound site express transforming growth factor (TGF) α, fibroblast growth factors (FGFs), platelet-derived growth factor (PDGF), VEGF and TGFβ, leading to rapid removal of cell debris and replacement by granulation tissue containing inflammatory cells and blood vessels [106]. As reported, the depletion of macrophages at other sites of injury results delayed and defective repair [106]. High levels of VEGF induced by hypoxia during menstruation promote M2 macrophage recruitment, migration and polarization by the VEGFR1/VEGF axis [107]. Besides, MMP-27 is mainly expressed by M2 macrophages in normal endometrium throughout the menstrual cycle and in endometriotic lesions. This plays a critical role in the regeneration of the endometrial epithelial lining and angiogenesis [108-110]. Wang et al. demonstrated that the hypoxia-autophagy axis induced VEGFA expression in peritoneal mesothelial cells [111], which suggests that the hypoxia-autophagy axis could be involved in endometrial tissue remodeling by triggering VEGFA-mediated M2 macrophage differentiation and vascular remodeling.
Additionally, the function of neutrophils and uNK cells cannot be ignored. It has been reported that neutrophil depletion with the antibody RB6-8C5 not only affected endometrial breakdown, but also can remarkably delayed endometrial repair [112], highlighting their importance in the destruction of endometrial tissue and concomitant repair. Further, neutrophils promote cyclical endometrial vascular proliferation by expressing VEGF but do not express ERα nor PGR [113]. uNK cells may initiate arterial remodeling and maintain vascular stability as they are a rich source of many different cytokines and growth factors including TNF-α, IL-10, GM-CSF, IL-1β, TGF-β1, CSF-1, leukemia inhibitory factor (LIF) and IFN-γ [114]. Moreover, Xu et al. have reported that the LKB1/p53/TIGAR/autophagy-dependent VEGF expression contributes to pulmonary inflammatory responses [115]. Additionally, autophagy has been implicated to cause tumor resistance to anti-angiogenic therapy [116], which is probably dependent on the dual roles of the AMPK/autophagy in angiogenesis [117]. Collectively, autophagy affects the function of neutrophils and NK cells, indicating their participation in tissue remodeling.
Ectopic endometrium autophagy on endometriosis
Menstruation and endometriosis
Endometriosis is characterized by implantation and growth of endometrial tissue outside the uterine cavity, affecting 10% of women in their reproductive life [3]. It has been proposed that endometrial fragments reaching the pelvis via retrograde menstruation, as well as their subsequent adhesion and implantation onto the peritoneum and abdominal organs, resulting in chronic pelvic pain and infertility. However, the pathogenesis and etiology of endometriosis remains unclear. There is increasing evidence that menstruation is linked with this condition. Firstly, this only occurs in women and menstruating primates. Secondly, there are various risk factors for this disease, including early menarche and nulliparity, and sexual activity leading to orgasm during menstruation, which further emphasizes that menstruation may play a key role toward the origin of endometriosis [118, 119]. Furthermore, patients with endometriosis have shorter menstrual cycle intervals, heavier menstrual effluent [120] and increased duration of menstrual flow than those without [121]. In addition, evidence from animal models showed that intra-peritoneal inoculation with menstrual in baboons resulted in the development of endometriosis [122]. Thus, it suggests that retrograde-menstruation is closely linked with the occurrence of endometriosis.
The link between autophagy in menstruation and endometriosis
Endometriosis is characterized by estrogen dependence and progesterone resistance [123]. With the upregulation of 17β-hydroxysteroid dehydrogenase-1 and aromatase genes, the level of estradiol in endometriotic lesions is higher than normal endometrium [124].This increased estradiol would bind and further activate ERs in endometriotic lesions, specifically ERβ [125]. A previous report also identified an epigenetic defect involving hypomethylation of a CpG island occupying ERβ promoter, which was found to cause the enhanced expression of ERβ [126]. Pathologically high ERβ levels repress ERα and PGR by negatively on regions on their promoter [33]. In turn, the reduced PGR activity further promotes estrogen synthesis. Of note, ERβ may upregulate PGE2 in endometriotic cells via the induction of COX2 expression [33]. Increased PGE2 induces the production of estradiol [127, 128]. As ERα mainly promotes angiogenesis, ERβ has a predominant role in anti-apoptosis, activation of the inflammasome and invasion of ectopic lesions [129, 130]. Therefore, excessive estrogen and PGE2 entail inflammation, immune responses, angiogenesis, and promotes the survival of endometriotic lesions [131, 132].
Far beyond that, high estrogen levels and progesterone resistance are also considered to be the regulators on the decreased autophagy in patients with endometriosis [133]. When compared with normal ESCs [134], the secretory ectopic and eutopic ESCs in endometriosis patients exhibited decreased expression patterns of autophagy-related genes, mRNA and proteins [135]. The difference is even greater when endometriotic tissues are compared with eutopic endometrium [136]. Our previous work has identified autophagy-related genes, including SNCA, RGS19, IGF1, ATG9B, ATG12, ATG10, IFNG, PIK3CG, and DAPK1, with lower expression levels in eutopic and ectopic secretory phase ESCs than normal ESCs. Moreover, the genes involved in autophagy initiation and regulation, such as CXCR4, ESR1, and mTOR, are up-regulated, while ATG16L1, IRGM and ULK1 are further weakened in ectopic ESCs [23]. Choi et al. demonstrated that progesterone had no significant effects on AKT, mTOR activity, autophagy or apoptosis in endometriotic cyst stromal cells. While for endometriotic tissues, they are noticed to have constant autophagy and apoptosis throughout the menstrual cycle [25]. These phenomena suggest that the dysregulation of autophagy levels in response to progesterone may play a role in the pathogenesis of endometriosis. For epithelial cells from endometriotic tissues, it has also been shown that both CXCR4 and CXCL12 are higher when compared with normal uterine endometrium [137]. Thus, high concentrations of estrogen may contribute to lower autophagy levels of ectopic ESCs and epithelial cells through the activation of the CXCL12-CXCR4 axis [23]. In addition, elevated levels of mTOR and the decreased ratio of LC3-II/LC3-I ratio in the eutopic ESCs have been observed [138, 139]. It is widely accepted that endometrial cell autophagy exerts an active role in the regulation of apoptosis [140]. Reduced autophagy induction via AKT/mTOR signaling and decreased apoptosis were observed during the late secretory phase of the menstrual cycle in endometriotic tissues [25].
By contrast, treatment with PPD markedly reverses the inhibitory effect of estrogen on ectopic ESC autophagy by upregulating PGR and downregulating ER. Moreover, the cytotoxic activity of NK cells in ectopic ESCs was found to be enhanced [38]. Taken together, the autophagy levels in patients with endometriosis are strongly reduced due to overexpression of estrogen through complex signaling pathway compared with normal endometrium.
Yet, the reduced autophagy and abnormal immune responses in endometriosis are not conducive to the removal of endometrial debris due to impaired immune functions. As previously mentioned, NK cells are involved in the removal of menstrual debris and endometrial fragments that are likely to reach the peritoneal cavity by retrograde menses. Differing from uNK cells in normal endometrium increased remarkedly from the proliferative phase with highest levels in the late secretory phase, uNK cells in ectopic lesions remained significantly low throughout the cycle [86], and allowing refluxed uterine cells to survive in ectopic lesions. Many cells secrete IL-15, including epithelial cells, monocytes, macrophages, and ESCs, that can function to mediate NK cells activation, expansion, function and survival [141]. Differentially, IL-15 plays a major role in recruiting unique CD16-NK cells with low cytotoxicity in the human endometrium. This is partly via selective extravasation of peripheral blood counterparts from the local microvascular circulation [142]. Decreased autophagy levels in ESCs promotes cell viability and invasion, restricts cell apoptosis, and suppresses the activation of NK cells (low CD16, Granzyme B and IFN-γ) by upregulating IL-15Rα and IL-2Rβ, thus contributing to the growth and immunosurveillance of ectopic lesions [143]. Importantly, impaired autophagy of ectopic ESCs mediated by estrogen dependence and progesterone resistance can lead to high levels of IL-8 and IL-23 secretion in a STAT3-HCK (hematopoietic cellular kinase)-dependent manner, which further promotes immunosurveillance of ectopic lesions by inducing the differentiation of COX-2highCD16- NK cells with low cytotoxicity and high levels of IL-10 and TGF-β (Figure 4) [144]. Additionally, IL-8 protects ectopic cells against death by apoptosis, and promotes cell adhesion and implantation in the pelvic cavity [145]. Interestingly, treatment with rapamycin, an inhibitor of the mTOR pathway, decreases the levels of IL-15 receptors in ESCs by downregulating IL-15-mediated ESC growth and invasion [143], and restricts endometriosis development by inhibiting COX-2highCD16- NK cell differentiation [144]. These reports highlight the potential value of rapamycin in the treatment of endometriosis [143, 144, 146].
The involvement of the ovarian steroid hormones-autophagy-immunity axis in the pathogenesis of endometriosis. In physiological situation, under the periodic regulation of estrogen and progesterone, autophagy levels in the endometrium also change in a CXCL12/CXCR4 dependent manner. During the menstrual period, the withdrawal of estrogen and progesterone leads to high levels of endometrial autophagy, contributing to endometrial exfoliation, immune defense, menstrual debris and endometrial fragment clearance by CD16+Granzyme B (Grzm)+INF-γ+ NK cells. In the presence of high concentrations of estrogen and progesterone resistance, however, autophagy-related genes (e.g., SNCA, RGS19, IGF1 ATGs, CXCR4, ESR1, and mTOR) are altered, leading to decreased endometrial autophagy. The suppression of endometrium autophagy directly accelerates the implantation, growth and angiogenesis of endometriotic lesions, whilst promoting the immune escape of endometriotic lesions through IL-8 and IL-23-mediated COX-2+CD16-NK cell differentiation. IL-15 is also involved in this process. Additionally, IL-8 inhibits activated T cells, and COX-2-induced PGE2 release stimulates M2 macrophage differentiation, which can facilitate the immune escape, remodeling of ECM and neovascularization of endometriotic lesions. Therefore, the effects of aberrant ovarian steroid hormones-autophagy-immunity axis contribute to the occurrence and development of endometriosis.
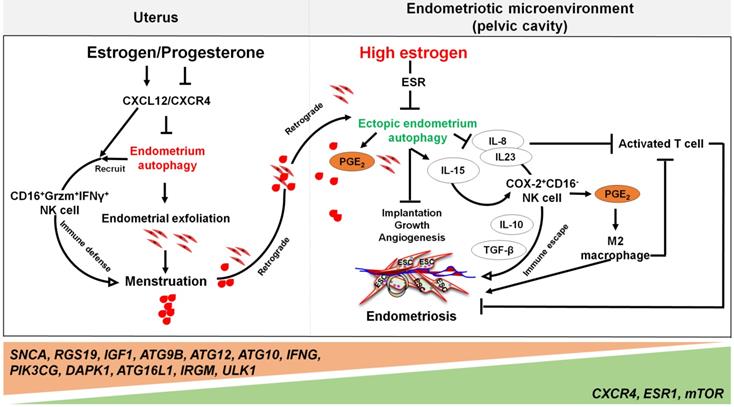
Notably, the elevated levels of peritoneal IL-8 due to defect autophagy enhance the FasL-induced apoptosis in activated T lymphocytes, contributing to the immune-privileged environment around the endometriotic implants and supporting their survival [145]. Additionally, IL-8 exerts chemotactic activities primarily on neutrophils to further accelerate the development of endometriosis [147]. A number of studies have shown that high levels of COX-2 are present in ectopic lesions and large amounts of PGE2 in PF [17]. Considering the effects of COX-2 and PGE2 on promoting cell autophagy [69, 148], and the key roles of autophagy in M2 macrophage differentiation, it can be hypothesized that elevated M2 macrophages could be associated with COX-2/PGE2/autophagy signaling. These M2 macrophages can facilitate the remodeling of ECM and neovascularization of lesions [149, 150].
Programmed cell death 4 (PDCD4), a well-documented tumor suppressor, could inhibit tumor cell proliferation, migration and invasion at the transcriptional and translational levels [151]. It has been shown that their expression was downregulated in the eutopic and ectopic endometrium from patients with endometriosis compared with normal endometrium. But its expression in the proliferative phase was comparable to that in the secretory phase. While in normal endometrium, their expression was significantly decreased in the progesterone-predominated secretory ESCs compared with that in estrogen-predominated proliferative phase in the proliferative phase was comparable to that in the secretory phase [152]. PDCD4 inhibits the proliferation and colony-forming ability of endometrial cells via the NF-κB/MMP2/MMP9 signaling pathway, resulting in a reduction in autophagy-dependent degradation of proteins and programmed cell death, as well as promoting the survival of ectopic lesions [152]. However, owing to the limited number of substantial clinical data, the role of autophagy in the pathogenesis of endometriosis remains controversial [132, 153]. Based on the reports discussed above, we believe that the role of the estrogen/progesterone-autophagy-immunity axis in endometriosis should be better emphasized. Additionally, the molecular mechanisms involving these pathways require further study.
Clinical implications and future perspectives
As discussed previously, the estrogen/ progesterone-autophagy-immunity axis is involved in the shedding of the endometrium, tissue regeneration and the defense mechanism during menstruation. Aberrant levels of autophagy are responsible for menstrual-related and endometrium-related diseases [154]. Under the regulation of high levels of estrogen and progesterone resistance, reduced autophagy in ESCs can be combined with dysregulated immune responses to accelerate the development of endometriosis. Theoretically, we postulate the potential risk of endometriosis by detecting autophagy levels, subsets and phenotypes of immune cells in exfoliated endometrium in menstrual tissues, especially in patients with abnormal bleeding, extended menstrual periods, or abnormal endometrial thickness. Herein, we may possibly establish an immunological classification and evaluate its potential value in predicting the recurrence of endometriosis and other endometrial-related diseases (e.g., UCEC) by analyzing the autophagy levels and the phenotype subsets of immune cell in endometrial lesions and/or PF, as well as their genetic characteristics. Besides endometriosis, the dysfunctional autophagy can also contribute to a variety of other diseases, including cardiovascular disease [155], diabetes [156] and hepatocellular carcinoma [157]). Several synthetic autophagy modulators have been identified as promising candidates for the treatment of cancers or benign lesions [158]. Notably, rapamycin has been shown to protect spontaneous miscarriage by inducing decidual stromal cell autophagy-mediated decidual NK cell residence [87]. Similarly, PPD also has therapeutic potential in endometriosis and UCEC by inducing cell autophagy [27, 38]. Therefore, prospective studies will be needed to assess their potential values in early warning, diagnosis, recurrence risk prediction and personalized therapy for endometriosis and other endometrial diseases.
Given that autophagy is a dynamic process, static measurements could lead to misinterpretation of the result. Additionally, studies on the mechanism of menstruation are limited, due to the lack of suitable animal models (menstruation does not take place in rodents) and in vivo interventions in humans is not plausible. Thus, further well-designed studies would be required to fully understand the mechanisms underlying the pleiotropic roles of this axis in order to enrich our knowledge of physiological mechanisms associated with menstruation and pathological events such as menstrual dysfunction, endometriosis, and miscarriage.
Abbreviations
AA: arachidonic acid; AMP: adenosine monophosphate; AMPK: adenosine 5'-monophosphate (AMP)-activated protein kinase; AP1: activator protein 1; Atg: autophagy-related; ATP: adenosine triphosphate; COX2: cyclooxygenase-2; CREB: cAMP responsive element binding protein; CRTC2: CREB-regulated transcription coactivator 2; CSF1: colony-stimulating factor 1; CXCL8: neutrophil chemotactic factor; ECM: extracellular matrix; EPC: endometrial glandular epithelial cells; ER: estrogen receptor; ESC: endometrial stromal cells; FIP200: focal adhesion kinase family interacting protein of 200kDA; FGF: fibroblast growth factors; FSH: follicle-stimulating hormone; GM-CSF: granulocyte-macrophage colony- stimulating factor; GPER: G-protein coupled estrogen receptor; HIF-1: hypoxia-inducible factor-1; HVEM: herpesvirus entry mediator; IGFR: insulin like growth factor receptor; IL-8: interleukin-8; JNK: c-Jun N-terminal kinase; LC3‑II: microtubule‑associated protein 1 light chain 3; LIF: leukemia inhibitory factor; MCP-1: monocyte chemoattractant protein-1; MITF: melanocyte inducing transcription factor; MMP: matrix metalloproteinases; mTOR: mammalian target of rapamycin complex; mTORC1: mTOR complex 1; NF-κB: nuclear factor kappa-B; PE: phosphatidylethanolamine; PDCD4: Programmed cell death 4; PDGF: platelet-derived growth factor; PF: peritoneal fluid; PGs: prostaglandins; PGE2: prostaglandin E2; PGF2α: prostaglandin F2α; PI3K: class I phosphatidylinositol 3-kinase; PPD: protopanaxadiol; PGR: progesterone receptors; PGRMC1: progesterone receptor membrane component 1; proLC3: LC3 precursor; ROS: reactive oxygen species; TFEB: transcription factor EB; TGF: transforming growth factor; TIMPS: tissue inhibitors of metalloproteinases; TSC: tuberous sclerosis complex; UCEC: uterine corpus endometrial carcinoma cells; ULK1: unc-51-like kinase 1; uPA: urokinase-type plasminogen activator; uNK: uterine NK cells; VEGF: vascular endothelial growth factor.
Acknowledgements
We thank Prof. Asgi Fazleabas in Department of Obstetrics, Gynecology & Reproductive Biology at Michigan State University and Dr. Gene Chi Wai Man in Department of Obstetrics and Gynecology at The Chinese University of Hong Kong for their generous help in reviewing and revising the manuscript. This study was supported by the National Natural Science Foundation of China (NSFC) (No. 92057119, 31970798, 31671200, 81901563, 81671460 and 82071646), the National Key Research and Development Program of China (2017YFC1001404), the Shanghai Sailing Program (19YF1438500), the Innovation-oriented Science and Technology Grant from NPFPC Key Laboratory of Reproduction Regulation (CX2017-2) and the Program for Zhuoxue of Fudan University (JIF157602).
Contributions
M.Q.L. and H.H.S. conceived and designed the review; H.H.S., T. Z., M.Q.L., W.J.Z., J.M., J.W.S., R.Z., D.J.L., and J.F.Y. drafted and revised the manuscript. F.Y.X edited the manuscript. H.H.S. and M.Q.L. generated the figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jarrell J. The significance and evolution of menstruation. Best Pract Res Clin Obstet Gynaecol. 2018;50:18-26
2. Azlan A, Salamonsen LA, Hutchison J, Evans J. Endometrial inflammasome activation accompanies menstruation and may have implications for systemic inflammatory events of the menstrual cycle. Hum Reprod. 2020;35:1363-76
3. Bulun SE, Yilmaz BD, Sison C, Miyazaki K, Bernardi L, Liu S. et al. Endometriosis. Endocr Rev. 2019;40:1048-79
4. Macer ML, Taylor HS. Endometriosis and infertility: a review of the pathogenesis and treatment of endometriosis-associated infertility. Obstet Gynecol Clin North Am. 2012;39:535-49
5. Critchley HOD, Babayev E, Bulun SE, Clark S, Garcia-Grau I, Gregersen PK. et al. Menstruation: science and society. Am J Obstet Gynecol. 2020;223:624-64
6. Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3-12
7. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1-222
8. Su Y, Zhang J, He J, Liu X, Chen X, Ding Y. et al. Endometrial autophagy is essential for embryo implantation during early pregnancy. J Mol Med (Berl). 2020;98:555-67
9. Tan H, Yang S, Li M, Wang H. Autophagy suppression of trophoblast cells induces pregnancy loss by activating decidual NK cytotoxicity and inhibiting trophoblast invasion. Cell Commun Signal. 2020;18:73
10. Oestreich AK, Chadchan SB, Medvedeva A, Lydon JP, Jungheim ES, Moley KH. et al. The autophagy protein, FIP200 (RB1CC1) mediates progesterone responses governing uterine receptivity and decidualization. Biol Reprod. 2020;102:843-51
11. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27-42
12. Critchley HO, Kelly RW, Brenner RM, Baird DT. The endocrinology of menstruation - a role for the immune system. Clin Endocrinol (Oxf). 2001;55:701-10
13. Critchley HOD, Maybin JA, Armstrong GM, Williams ARW. Physiology of the Endometrium and Regulation of Menstruation. Physiol Rev. 2020;100:1149-79
14. Jabbour HN, Kelly RW, Fraser HM, Critchley HO. Endocrine regulation of menstruation. Endocr Rev. 2006;27:17-46
15. Salamonsen LA, Lathbury LJ. Endometrial leukocytes and menstruation. Hum Reprod Update. 2000;6:16-27
16. Itoh H, Kishore AH, Lindqvist A, Rogers DE, Word RA. Transforming growth factor beta1 (TGFbeta1) and progesterone regulate matrix metalloproteinases (MMP) in human endometrial stromal cells. J Clin Endocrinol Metab. 2012;97:E888-97
17. Lai ZZ, Yang HL, Ha SY, Chang KK, Mei J, Zhou WJ. et al. Cyclooxygenase-2 in Endometriosis. Int J Biol Sci. 2019;15:2783-97
18. Milne SA, Jabbour HN. Prostaglandin (PG) F(2alpha) receptor expression and signaling in human endometrium: role of PGF(2alpha) in epithelial cell proliferation. J Clin Endocrinol Metab. 2003;88:1825-32
19. Popovici RM, Irwin JC, Giaccia AJ, Giudice LC. Hypoxia and cAMP stimulate vascular endothelial growth factor (VEGF) in human endometrial stromal cells: potential relevance to menstruation and endometrial regeneration. J Clin Endocrinol Metab. 1999;84:2245-8
20. Walczak M, Martens S. Dissecting the role of the Atg12-Atg5-Atg16 complex during autophagosome formation. Autophagy. 2013;9:424-5
21. Choi JY, Jo MW, Lee EY, Yoon BK, Choi DS. The role of autophagy in follicular development and atresia in rat granulosa cells. Fertil Steril. 2010;93:2532-7
22. Choi J, Jo M, Lee E, Kim HJ, Choi D. Differential induction of autophagy by mTOR is associated with abnormal apoptosis in ovarian endometriotic cysts. Mol Hum Reprod. 2014;20:309-17
23. Mei J, Zhu XY, Jin LP, Duan ZL, Li DJ, Li MQ. Estrogen promotes the survival of human secretory phase endometrial stromal cells via CXCL12/CXCR4 up-regulation-mediated autophagy inhibition. Hum Reprod. 2015;30:1677-89
24. Choi J, Jo M, Lee E, Lee DY, Choi D. Dienogest enhances autophagy induction in endometriotic cells by impairing activation of AKT, ERK1/2, and mTOR. Fertil Steril. 2015;104:655-64 e1
25. Choi J, Jo M, Lee E, Hwang S, Choi D. Aberrant PTEN expression in response to progesterone reduces endometriotic stromal cell apoptosis. Reproduction. 2017;153:11-21
26. Zhou WJ, Zhang J, Yang HL, Wu K, Xie F, Wu JN. et al. Estrogen inhibits autophagy and promotes growth of endometrial cancer by promoting glutamine metabolism. Cell Commun Signal. 2019;17:99
27. Gu CJ, Cheng J, Zhang B, Yang SL, Xie F, Sun JS. et al. Protopanaxadiol and metformin synergistically inhibit estrogen-mediated proliferation and anti-autophagy effects in endometrial cancer cells. Am J Transl Res. 2017;9:4071-82
28. Wang Q, Guo X, Li L, Gao Z, Ji M. Treatment with metformin and sorafenib alleviates endometrial hyperplasia in polycystic ovary syndrome by promoting apoptosis via synergically regulating autophagy. J Cell Physiol. 2020;235:1339-48
29. Wei Y, Zhou J, Wu J, Huang J. ERbeta promotes Abeta degradation via the modulation of autophagy. Cell Death Dis. 2019;10:565
30. Fan D, Liu SY, van Hasselt CA, Vlantis AC, Ng EK, Zhang H. et al. Estrogen receptor alpha induces prosurvival autophagy in papillary thyroid cancer via stimulating reactive oxygen species and extracellular signal regulated kinases. J Clin Endocrinol Metab. 2015;100:E561-71
31. Xiang J, Liu X, Ren J, Chen K, Wang HL, Miao YY. et al. How does estrogen work on autophagy? Autophagy. 2019;15:197-211
32. Zhang L, Zhao Y, Guo L. 17beta-estradiol protects INS-1 insulinoma cells from mitophagy via G protein-coupled estrogen receptors and the PI3K/Akt signaling pathway. Int J Mol Med. 2018;41:2839-46
33. Bulun SE, Monsavais D, Pavone ME, Dyson M, Xue Q, Attar E. et al. Role of estrogen receptor-beta in endometriosis. Semin Reprod Med. 2012;30:39-45
34. Lessey BA, Killam AP, Metzger DA, Haney AF, Greene GL, McCarty KS Jr. Immunohistochemical analysis of human uterine estrogen and progesterone receptors throughout the menstrual cycle. J Clin Endocrinol Metab. 1988;67:334-40
35. Snijders MP, de Goeij AF, Koudstaal J, Thunnissen EB, de Haan J, Bosman FT. Oestrogen and progesterone receptor immunocytochemistry in human hyperplastic and neoplastic endometrium. J Pathol. 1992;166:171-7
36. Wang H, Critchley HO, Kelly RW, Shen D, Baird DT. Progesterone receptor subtype B is differentially regulated in human endometrial stroma. Mol Hum Reprod. 1998;4:407-12
37. Chen CW, Chen TY, Tsai KL, Lin CL, Yokoyama KK, Lee WS. et al. Inhibition of autophagy as a therapeutic strategy of iron-induced brain injury after hemorrhage. Autophagy. 2012;8:1510-20
38. Zhang B, Zhou WJ, Gu CJ, Wu K, Yang HL, Mei J. et al. The ginsenoside PPD exerts anti-endometriosis effects by suppressing estrogen receptor-mediated inhibition of endometrial stromal cell autophagy and NK cell cytotoxicity. Cell Death Dis. 2018;9:574
39. Hong Y, Liu Y, Zhang G, Wu H, Hou Y. Progesterone suppresses Aβ(42)-induced neuroinflammation by enhancing autophagy in astrocytes. Int Immunopharmacol. 2018;54:336-43
40. Tan S, Bajalovic N, Wong ESP, Lin VCL. Ligand-activated progesterone receptor B activates transcription factor EB to promote autophagy in human breast cancer cells. Exp Cell Res. 2019;382:111433
41. Mir SU, Schwarze SR, Jin L, Zhang J, Friend W, Miriyala S. et al. Progesterone receptor membrane component 1/Sigma-2 receptor associates with MAP1LC3B and promotes autophagy. Autophagy. 2013;9:1566-78
42. Kahnamouyi S, Nouri M, Farzadi L, Darabi M, Hosseini V, Mehdizadeh A. The role of mitogen-activated protein kinase-extracellular receptor kinase pathway in female fertility outcomes: a focus on pituitary gonadotropins regulation. Ther Adv Endocrinol Metab. 2018;9:209-15
43. Critchley HO, Osei J, Henderson TA, Boswell L, Sales KJ, Jabbour HN. et al. Hypoxia-inducible factor-1alpha expression in human endometrium and its regulation by prostaglandin E-series prostanoid receptor 2 (EP2). Endocrinology. 2006;147:744-53
44. Chu PY, Wang SM, Chen PM, Tang FY, Chiang EI. Expression of MTDH and IL-10 Is an Independent Predictor of Worse Prognosis in ER-Negative or PR-Negative Breast Cancer Patients. J Clin Med. 2020;9:3153
45. Bajbouj K, Shafarin J, Muhammad JS, Ali A, Unnikannan H, Suleiman B. et al. Estrogen signaling differentially alters iron metabolism in monocytes in an Interleukin 6-dependent manner. Immunobiology. 2020;225:151995
46. Zhou J, Yao W, Li C, Wu W, Li Q, Liu H. Administration of follicle-stimulating hormone induces autophagy via upregulation of HIF-1alpha in mouse granulosa cells. Cell Death Dis. 2017;8:e3001
47. Azad MB, Chen Y, Gibson SB. Regulation of autophagy by reactive oxygen species (ROS): implications for cancer progression and treatment. Antioxid Redox Signal. 2009;11:777-90
48. Scherz-Shouval R, Elazar Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007;17:422-7
49. Poillet-Perez L, Despouy G, Delage-Mourroux R, Boyer-Guittaut M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 2015;4:184-92
50. Liao TL, Lee YC, Tzeng CR, Wang YP, Chang HY, Lin YF. et al. Mitochondrial translocation of estrogen receptor beta affords resistance to oxidative insult-induced apoptosis and contributes to the pathogenesis of endometriosis. Free Radic Biol Med. 2019;134:359-73
51. Xu Z, Mu S, Liao X, Fan R, Gao W, Sun Y. et al. Estrogen protects against liver damage in sepsis through inhibiting oxidative stress mediated activation of pyroptosis signaling pathway. PLoS One. 2020;15:e0239659
52. Ogola BO, Zimmerman MA, Sure VN, Gentry KM, Duong JL, Clark GL. et al. G Protein-Coupled Estrogen Receptor Protects From Angiotensin II-Induced Increases in Pulse Pressure and Oxidative Stress. Front Endocrinol (Lausanne). 2019;10:586
53. Sugino N, Karube-Harada A, Taketani T, Sakata A, Nakamura Y. Withdrawal of ovarian steroids stimulates prostaglandin F2alpha production through nuclear factor-kappaB activation via oxygen radicals in human endometrial stromal cells: potential relevance to menstruation. J Reprod Dev. 2004;50:215-25
54. Ghayad SE, Cohen PA. Inhibitors of the PI3K/Akt/mTOR pathway: new hope for breast cancer patients. Recent Pat Anticancer Drug Discov. 2010;5:29-57
55. Nuñez-Olvera SI, Gallardo-Rincón D, Puente-Rivera J, Salinas-Vera YM, Marchat LA, Morales-Villegas R. et al. Autophagy Machinery as a Promising Therapeutic Target in Endometrial Cancer. Front Oncol. 2019;9:1326
56. Papandreou I, Lim A, Laderoute K, Denko N. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ. 2008;15:1572-81
57. Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3'-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem. 2000;275:992-8
58. Devis-Jauregui L, Eritja N, Davis M, Matias-Guiu X, Llobet-Navàs D. Autophagy in the physiological endometrium and cancer. Autophagy. 2020 p: 1-19
59. Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T. et al. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci. 2003;116:1679-88
60. Arumugam A, Parada J, Rajkumar L. Mammary cancer promotion by ovarian hormones involves IGFR/AKT/mTOR signaling. Steroids. 2012;77:791-7
61. Lee II, Kim JJ. Influence of AKT on progesterone action in endometrial diseases. Biol Reprod. 2014;91:63
62. Yin X, Pavone ME, Lu Z, Wei J, Kim JJ. Increased activation of the PI3K/AKT pathway compromises decidualization of stromal cells from endometriosis. J Clin Endocrinol Metab. 2012;97:E35-43
63. Lane B, Oxberry W, Mazella J, Tseng L. Decidualization of human endometrial stromal cells in vitro: effects of progestin and relaxin on the ultrastructure and production of decidual secretory proteins. Hum Reprod. 1994;9:259-66
64. Wen X, Liu L, Li S, Lin P, Chen H, Zhou D. et al. Prostaglandin F2alpha Induces Goat Corpus Luteum Regression via Endoplasmic Reticulum Stress and Autophagy. Front Physiol. 2020;11:868
65. Song S, Tan J, Miao Y, Zhang Q. Crosstalk of ER stress-mediated autophagy and ER-phagy: Involvement of UPR and the core autophagy machinery. J Cell Physiol. 2018;233:3867-74
66. Wu P, Tian T, Zhao J, Song Q, Wu X, Guo Y. et al. IRE1alpha-JNK pathway-mediated autophagy promotes cell survival in response to endoplasmic reticulum stress during the initial phase of hepatic steatosis. Life Sci. 2020: 118668.
67. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89-102
68. B'Chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y. et al. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013;41:7683-99
69. Luo B, Lin Y, Jiang S, Huang L, Yao H, Zhuang Q. et al. Endoplasmic reticulum stress eIF2alpha-ATF4 pathway-mediated cyclooxygenase-2 induction regulates cadmium-induced autophagy in kidney. Cell Death Dis. 2016;7:e2251
70. Zhang Z, Davis DL. Prostaglandin E and E2 alpha secretion by glandular and stromal cells of the pig endometrium in vitro: effects of estradiol-17 beta, progesterone, and day of pregnancy. Prostaglandins. 1991;42:151-62
71. Nguyen H, Syed V. Progesterone inhibits growth and induces apoptosis in cancer cells through modulation of reactive oxygen species. Gynecol Endocrinol. 2011;27:830-6
72. Hannan NJ, Evans J, Salamonsen LA. Alternate roles for immune regulators: establishing endometrial receptivity for implantation. Expert Rev Clin Immunol. 2011;7:789-802
73. Zhang X, Luo Y, Wang C, Ding X, Yang X, Wu D. et al. Adipose mTORC1 Suppresses Prostaglandin Signaling and Beige Adipogenesis via the CRTC2-COX-2 Pathway. Cell Rep. 2018;24:3180-93
74. Qiang L, Sample A, Shea CR, Soltani K, Macleod KF, He YY. Autophagy gene ATG7 regulates ultraviolet radiation-induced inflammation and skin tumorigenesis. Autophagy. 2017;13:2086-103
75. Qiang L, Yang S, Cui YH, He YY. Keratinocyte autophagy enables the activation of keratinocytes and fibroblasts and facilitates wound healing. Autophagy. 2020:1-16
76. Cen S, Wang P, Xie Z, Yang R, Li J, Liu Z. et al. Autophagy enhances mesenchymal stem cell-mediated CD4(+) T cell migration and differentiation through CXCL8 and TGF-beta1. Stem Cell Res Ther. 2019;10:265
77. Agostinis C, Mangogna A, Bossi F, Ricci G, Kishore U, Bulla R. Uterine Immunity and Microbiota: A Shifting Paradigm. Front Immunol. 2019;10:2387
78. Thiruchelvam U, Dransfield I, Saunders PT, Critchley HO. The importance of the macrophage within the human endometrium. J Leukoc Biol. 2013;93:217-25
79. Hunter M, Wang Y, Eubank T, Baran C, Nana-Sinkam P, Marsh C. Survival of monocytes and macrophages and their role in health and disease. Front Biosci (Landmark Ed). 2009;14:4079-102
80. Zhang Y, Morgan MJ, Chen K, Choksi S, Liu ZG. Induction of autophagy is essential for monocyte-macrophage differentiation. Blood. 2012;119:2895-905
81. Obba S, Hizir Z, Boyer L, Selimoglu-Buet D, Pfeifer A, Michel G. et al. The PRKAA1/AMPKalpha1 pathway triggers autophagy during CSF1-induced human monocyte differentiation and is a potential target in CMML. Autophagy. 2015;11:1114-29
82. Del Prete A, Zaccagnino P, Di Paola M, Saltarella M, Oliveros Celis C, Nico B. et al. Role of mitochondria and reactive oxygen species in dendritic cell differentiation and functions. Free Radic Biol Med. 2008;44:1443-51
83. Clarke AJ, Simon AK. Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat Rev Immunol. 2019;19:170-83
84. Wu MY, Lu JH. Autophagy and Macrophage Functions: Inflammatory Response and Phagocytosis. Cells. 2019;9:70
85. Ren H, Chen X, Jiang F, Li G. Cyclooxygenase-2 Inhibition Reduces Autophagy of Macrophages Enhancing Extraintestinal Pathogenic Escherichia coli Infection. Front Microbiol. 2020;11:708
86. Drury JA, Parkin KL, Coyne L, Giuliani E, Fazleabas AT, Hapangama DK. The dynamic changes in the number of uterine natural killer cells are specific to the eutopic but not to the ectopic endometrium in women and in a baboon model of endometriosis. Reprod Biol Endocrinol. 2018;16:67
87. Lu H, Yang HL, Zhou WJ, Lai ZZ, Qiu XM, Fu Q. et al. Rapamycin prevents spontaneous abortion by triggering decidual stromal cell autophagy-mediated NK cell residence. Autophagy. 2020 p: 1-17
88. Abtin A, Jain R, Mitchell AJ, Roediger B, Brzoska AJ, Tikoo S. et al. Perivascular macrophages mediate neutrophil recruitment during bacterial skin infection. Nat Immunol. 2014;15:45-53
89. De Filippo K, Dudeck A, Hasenberg M, Nye E, van Rooijen N, Hartmann K. et al. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood. 2013;121:4930-7
90. Ushio H, Ueno T, Kojima Y, Komatsu M, Tanaka S, Yamamoto A. et al. Crucial role for autophagy in degranulation of mast cells. J Allergy Clin Immunol. 2011;127:1267-76 e6
91. Bhattacharya A, Wei Q, Shin JN, Abdel Fattah E, Bonilla DL, Xiang Q. et al. Autophagy Is Required for Neutrophil-Mediated Inflammation. Cell Rep. 2015;12:1731-9
92. Ullah I, Ritchie ND, Evans TJ. The interrelationship between phagocytosis, autophagy and formation of neutrophil extracellular traps following infection of human neutrophils by Streptococcus pneumoniae. Innate Immun. 2017;23:413-23
93. Jin L, Batra S, Jeyaseelan S. Deletion of Nlrp3 Augments Survival during Polymicrobial Sepsis by Decreasing Autophagy and Enhancing Phagocytosis. J Immunol. 2017;198:1253-62
94. Iula L, Keitelman IA, Sabbione F, Fuentes F, Guzman M, Galletti JG. et al. Autophagy Mediates Interleukin-1beta Secretion in Human Neutrophils. Front Immunol. 2018;9:269
95. Choi J, Jo M, Lee E, Oh YK, Choi D. The role of autophagy in human endometrium. Biol Reprod. 2012;86:70
96. Choi J, Jo M, Lee E, Choi D. The role of autophagy in corpus luteum regression in the rat. Biol Reprod. 2011;85:465-72
97. Du J, Zhu X, Guo R, Xu Z, Cheng FF, Liu Q. et al. Autophagy induces G0/G1 arrest and apoptosis in menstrual blood-derived endometrial stem cells via GSK3-beta/beta-catenin pathway. Stem Cell Res Ther. 2018;9:330
98. Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280-93
99. Smith S, Dunk C, Aplin J, Harris L, Jones R. Evidence for immune cell involvement in decidual spiral arteriole remodeling in early human pregnancy. The American journal of pathology. 2009;174:1959-71
100. Gogacz M, Gałczyński K, Wojtaś M, Winkler I, Adamiak A, Romanek-Piva K. et al. Fas-Related Apoptosis of Peritoneal Fluid Macrophages in Endometriosis Patients: Understanding the Disease. Journal of immunology research. 2017;2017:3175394
101. Ottnad E, Parthasarathy S, Sambrano GR, Ramprasad MP, Quehenberger O, Kondratenko N. et al. A macrophage receptor for oxidized low density lipoprotein distinct from the receptor for acetyl low density lipoprotein: partial purification and role in recognition of oxidatively damaged cells. Proc Natl Acad Sci U S A. 1995;92:1391-5
102. Zhang Q, Li Y, Miao C, Wang Y, Xu Y, Dong R. et al. Anti-angiogenesis effect of Neferine via regulating autophagy and polarization of tumor-associated macrophages in high-grade serous ovarian carcinoma. Cancer Lett. 2018;432:144-55
103. Liu T, Wang L, Liang P, Wang X, Liu Y, Cai J. et al. USP19 suppresses inflammation and promotes M2-like macrophage polarization by manipulating NLRP3 function via autophagy. Cell Mol Immunol. 2020
104. Gu S, Dai H, Zhao X, Gui C, Gui J. AKT3 deficiency in M2 macrophages impairs cutaneous wound healing by disrupting tissue remodeling. Aging (Albany N Y). 2020;12:6928-46
105. Zhang S, Liu Y, Zhang X, Zhu D, Qi X, Cao X. et al. Prostaglandin E2 hydrogel improves cutaneous wound healing via M2 macrophages polarization. Theranostics. 2018;8:5348-61
106. van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH, van Luyn MJ. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol. 2007;170:818-29
107. Wheeler K, Jena M, Pradhan B, Nayak N, Das S, Hsu C. et al. VEGF may contribute to macrophage recruitment and M2 polarization in the decidua. PLoS One. 2018;13:e0191040
108. Jetten N, Verbruggen S, Gijbels MJ, Post MJ, De Winther MP, Donners MM. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis. 2014;17:109-18
109. Adamson R. Role of macrophages in normal wound healing: an overview. J Wound Care. 2009;18:349-51
110. Bacci M, Capobianco A, Monno A, Cottone L, Di Puppo F, Camisa B. et al. Macrophages are alternatively activated in patients with endometriosis and required for growth and vascularization of lesions in a mouse model of disease. Am J Pathol. 2009;175:547-56
111. Wang X, Che X, Yu Y, Cheng Y, Bai M, Yang Z. et al. Hypoxia-autophagy axis induces VEGFA by peritoneal mesothelial cells to promote gastric cancer peritoneal metastasis through an integrin alpha5-fibronectin pathway. J Exp Clin Cancer Res. 2020;39:221
112. Kaitu'u-Lino TJ, Morison NB, Salamonsen LA. Neutrophil depletion retards endometrial repair in a mouse model. Cell Tissue Res. 2007;328:197-206
113. Mueller MD, Lebovic DI, Garrett E, Taylor RN. Neutrophils infiltrating the endometrium express vascular endothelial growth factor: potential role in endometrial angiogenesis. Fertil Steril. 2000;74:107-12
114. Jokhi PP, King A, Sharkey AM, Smith SK, Loke YW. Screening for cytokine messenger ribonucleic acids in purified human decidual lymphocyte populations by the reverse-transcriptase polymerase chain reaction. J Immunol. 1994;153:4427-35
115. Xu H, Xu X, Wang H, Qimuge A, Liu S, Chen Y. et al. LKB1/p53/TIGAR/autophagy-dependent VEGF expression contributes to PM2.5-induced pulmonary inflammatory responses. Sci Rep. 2019;9:16600
116. Chandra A, Rick J, Yagnik G, Aghi MK. Autophagy as a mechanism for anti-angiogenic therapy resistance. Semin Cancer Biol. 2020;66:75-88
117. Li Y, Sun R, Zou J, Ying Y, Luo Z. Dual Roles of the AMP-Activated Protein Kinase Pathway in Angiogenesis. Cells. 2019;8:752
118. Parazzini F, Esposito G, Tozzi L, Noli S, Bianchi S. Epidemiology of endometriosis and its comorbidities. Eur J Obstet Gynecol Reprod Biol. 2017;209:3-7
119. Mollazadeh S, Sadeghzadeh Oskouei B, Kamalifard M, Mirghafourvand M, Aminisani N, Jafari Shobeiri M. Association between Sexual Activity during Menstruation and Endometriosis: A Case-Control Study. Int J Fertil Steril. 2019;13:230-5
120. Matalliotakis IM, Cakmak H, Fragouli YG, Goumenou AG, Mahutte NG, Arici A. Epidemiological characteristics in women with and without endometriosis in the Yale series. Arch Gynecol Obstet. 2008;277:389-93
121. Parazzini F, Cipriani S, Bianchi S, Gotsch F, Zanconato G, Fedele L. Risk factors for deep endometriosis: a comparison with pelvic and ovarian endometriosis. Fertil Steril. 2008;90:174-9
122. D'Hooghe TM, Bambra CS, Raeymaekers BM, Koninckx PR. Increased prevalence and recurrence of retrograde menstruation in baboons with spontaneous endometriosis. Hum Reprod. 1996;11:2022-5
123. Patel BG, Rudnicki M, Yu J, Shu Y, Taylor RN. Progesterone resistance in endometriosis: origins, consequences and interventions. Acta Obstet Gynecol Scand. 2017;96:623-32
124. Delvoux B, Groothuis P, D'Hooghe T, Kyama C, Dunselman G, Romano A. Increased production of 17beta-estradiol in endometriosis lesions is the result of impaired metabolism. J Clin Endocrinol Metab. 2009;94:876-83
125. Han SJ, Jung SY, Wu SP, Hawkins SM, Park MJ, Kyo S. et al. Estrogen Receptor beta Modulates Apoptosis Complexes and the Inflammasome to Drive the Pathogenesis of Endometriosis. Cell. 2015;163:960-74
126. Dyson MT, Roqueiro D, Monsivais D, Ercan CM, Pavone ME, Brooks DC. et al. Genome-wide DNA methylation analysis predicts an epigenetic switch for GATA factor expression in endometriosis. PLoS Genet. 2014;10:e1004158
127. Mori T, Ito F, Koshiba A, Kataoka H, Takaoka O, Okimura H. et al. Local estrogen formation and its regulation in endometriosis. Reprod Med Biol. 2019;18:305-11
128. Bulun SE, Lin Z, Imir G, Amin S, Demura M, Yilmaz B. et al. Regulation of aromatase expression in estrogen-responsive breast and uterine disease: from bench to treatment. Pharmacol Rev. 2005;57:359-83
129. Wang Y, Nicholes K, Shih IM. The Origin and Pathogenesis of Endometriosis. Annu Rev Pathol. 2020;15:71-95
130. Han SJ, Jung SY, Wu SP, Hawkins SM, Park MJ, Kyo S. et al. Estrogen Receptor β Modulates Apoptosis Complexes and the Inflammasome to Drive the Pathogenesis of Endometriosis. Cell. 2015;163:960-74
131. Vercellini P, Vigano P, Somigliana E, Fedele L. Endometriosis: pathogenesis and treatment. Nat Rev Endocrinol. 2014;10:261-75
132. Zhan L, Yao S, Sun S, Su Q, Li J, Wei B. NLRC5 and autophagy combined as possible predictors in patients with endometriosis. Fertil Steril. 2018;110:949-56
133. Yang HL, Mei J, Chang KK, Zhou WJ, Huang LQ, Li MQ. Autophagy in endometriosis. Am J Transl Res. 2017;9:4707-25
134. Ruiz A, Rockfield S, Taran N, Haller E, Engelman RW, Flores I. et al. Effect of hydroxychloroquine and characterization of autophagy in a mouse model of endometriosis. Cell Death Dis. 2016;7:e2059
135. Sui X, Li Y, Sun Y, Li C, Li X, Zhang G. Expression and significance of autophagy genes LC3, Beclin1 and MMP-2 in endometriosis. Exp Ther Med. 2018;16:1958-62
136. Li M, Lu M, Liu M, Deng S, Tang X, Han C. et al. An Observation of the Role of Autophagy in Patients with Endometriosis of Different Stages during Secretory Phase and Proliferative Phase. Curr Gene Ther. 2018;18:286-95
137. Leconte M, Chouzenoux S, Nicco C, Chereau C, Arkwright S, Santulli P. et al. Role of the CXCL12-CXCR4 axis in the development of deep rectal endometriosis. J Reprod Immunol. 2014;103:45-52
138. Pei T, Huang X, Long Y, Duan C, Liu T, Li Y. et al. Increased expression of YAP is associated with decreased cell autophagy in the eutopic endometrial stromal cells of endometriosis. Mol Cell Endocrinol. 2019;491:110432
139. Jamali N, Zal F, Mostafavi-Pour Z, Samare-Najaf M, Poordast T, Dehghanian A. Ameliorative Effects of Quercetin and Metformin and Their Combination Against Experimental Endometriosis in Rats. Reprod Sci. 2020
140. Wang W, Zhang Y, Liu W, Zhang X, Xiao H, Zhao M. et al. CXCR4 induces cell autophagy and maintains EBV latent infection in EBVaGC. Theranostics. 2020;10:11549-61
141. Oka N, Markova T, Tsuzuki K, Li W, El-Darawish Y, Pencheva-Demireva M. et al. IL-12 regulates the expansion, phenotype, and function of murine NK cells activated by IL-15 and IL-18. Cancer Immunol Immunother. 2020;69:1699-712
142. Kitaya K, Yasuo T. Regulatory role of membrane-bound form interleukin-15 on human uterine microvascular endothelial cells in circulating CD16(-) natural killer cell extravasation into human endometrium. Biol Reprod. 2013;89:70
143. Yu JJ, Sun HT, Zhang ZF, Shi RX, Liu LB, Shang WQ. et al. IL15 promotes growth and invasion of endometrial stromal cells and inhibits killing activity of NK cells in endometriosis. Reproduction. 2016;152:151-60
144. Mei J, Zhou WJ, Zhu XY, Lu H, Wu K, Yang HL. et al. Suppression of autophagy and HCK signaling promotes PTGS2(high) FCGR3(-) NK cell differentiation triggered by ectopic endometrial stromal cells. Autophagy. 2018;14:1376-97
145. Sikora J, Smycz-Kubanska M, Mielczarek-Palacz A, Kondera-Anasz Z. Abnormal peritoneal regulation of chemokine activation-The role of IL-8 in pathogenesis of endometriosis. Am J Reprod Immunol. 2017 77
146. Leconte M, Nicco C, Ngo C, Chereau C, Chouzenoux S, Marut W. et al. The mTOR/AKT inhibitor temsirolimus prevents deep infiltrating endometriosis in mice. Am J Pathol. 2011;179:880-9
147. Selam B, Kayisli UA, Garcia-Velasco JA, Akbas GE, Arici A. Regulation of fas ligand expression by IL-8 in human endometrium. J Clin Endocrinol Metab. 2002;87:3921-7
148. Xiong W, Wen Q, Du X, Wang J, He W, Wang R. et al. Novel Function of Cyclooxygenase-2: Suppressing Mycobacteria by Promoting Autophagy via the Protein Kinase B/Mammalian Target of Rapamycin Pathway. J Infect Dis. 2018;217:1267-79
149. Yuan M, Li D, An M, Li Q, Zhang L, Wang G. Rediscovering peritoneal macrophages in a murine endometriosis model. Hum Reprod. 2017;32:94-102
150. Wynn TA, Vannella KM. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity. 2016;44:450-62
151. Wang X, Li Y, Wan L, Liu Y, Sun Y, Liu Y. et al. Downregulation of PDCD4 induced by progesterone is mediated by the PI3K/AKT signaling pathway in human endometrial cancer cells. Oncol Rep. 2019;42:849-56
152. Li Y, Wang X, Wang X, Wan L, Liu Y, Shi Y. et al. PDCD4 suppresses proliferation, migration, and invasion of endometrial cells by inhibiting autophagy and NF-kappaB/MMP2/MMP9 signal pathway. Biol Reprod. 2018;99:360-72
153. Zhan L, Li J, Wei B. Autophagy in endometriosis: Friend or foe? Biochem Biophys Res Commun. 2018;495:60-3
154. Yang S, Wang H, Li D, Li M. Role of Endometrial Autophagy in Physiological and Pathophysiological Processes. J Cancer. 2019;10:3459-71
155. Saliba-Gustafsson P, Pedrelli M, Gertow K, Werngren O, Janas V, Pourteymour S. et al. Subclinical atherosclerosis and its progression are modulated by PLIN2 through a feed-forward loop between LXR and autophagy. J Intern Med. 2019;286:660-75
156. Abad-Jimenez Z, Lopez-Domenech S, Diaz-Rua R, Iannantuoni F, Gomez-Abril SA, Perianez-Gomez D. et al. Systemic Oxidative Stress and Visceral Adipose Tissue Mediators of NLRP3 Inflammasome and Autophagy Are Reduced in Obese Type 2 Diabetic Patients Treated with Metformin. Antioxidants (Basel). 2020;9:892
157. Xiao Q, Liu H, Wang HS, Cao MT, Meng XJ, Xiang YL. et al. Histone deacetylase inhibitors promote epithelial-mesenchymal transition in Hepatocellular Carcinoma via AMPK-FOXO1-ULK1 signaling axis-mediated autophagy. Theranostics. 2020;10:10245-61
158. Marinkovic M, Sprung M, Buljubasic M, Novak I. Autophagy Modulation in Cancer: Current Knowledge on Action and Therapy. Oxid Med Cell Longev. 2018;2018:8023821
Author contact
![]() Corresponding author: Ming-Qing Li, MD, PhD, Laboratory for Reproductive Immunology, NHC Key Lab of Reproduction Regulation (Shanghai Institute of Planned Parenthood Research), Hospital of Obstetrics and Gynecology, Shanghai Medical School, Fudan University, Shanghai 200080, China. Telephone: +86-21-33189900-8286; E-mail address: mqliedu.cn.
Corresponding author: Ming-Qing Li, MD, PhD, Laboratory for Reproductive Immunology, NHC Key Lab of Reproduction Regulation (Shanghai Institute of Planned Parenthood Research), Hospital of Obstetrics and Gynecology, Shanghai Medical School, Fudan University, Shanghai 200080, China. Telephone: +86-21-33189900-8286; E-mail address: mqliedu.cn.