Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Summary
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(10):4672-4687. doi:10.7150/thno.54793 This issue Cite
Research Paper
Targeting lysosomal cysteine protease cathepsin S reveals immunomodulatory therapeutic strategy for oxaliplatin-induced peripheral neuropathy
Szu-Jung Chen1, Li-Hsien Chen1, Yu-Min Yeh1, Chou-Ching K Lin2, Peng-Chan Lin1, Han-Wei Huang2, Meng-Ru Shen3,4, Bo-Wen Lin5, Jeng-Chang Lee5, Cheng-Che Lee6, Yueh-Feng Lee1, Huai-Chueh Chiang1, Jang-Yang Chang1,7,8 ![]()
1. Department of Internal Medicine, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, Tainan, Taiwan.
2. Department of Neurology, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, Tainan, Taiwan.
3. Department of Obstetrics/Gynecology, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, Tainan, Taiwan.
4. Department of Pharmacology, College of Medicine, National Cheng Kung University, Tainan. Taiwan.
5. Department of Surgery, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, Tainan, Taiwan.
6. School of Medicine for International students, College of Medicine, I-SHOU University, Kaohsiung, Taiwan.
7. National Institute of Cancer Research, National Health Research Institutes, Zhunan, Taiwan.
8. Institute of Biotechnology and Pharmaceutical Research, National Health Research Institutes, Zhunan, Taiwan.
Received 2020-10-21; Accepted 2021-2-8; Published 2021-3-4
Abstract

Rationale: Oxaliplatin-induced peripheral neuropathy (OIPN) is a common adverse effect that causes delayed treatment and poor prognosis among colorectal cancer (CRC) patients. However, its mechanism remains elusive, and no effective treatment is available.
Methods: We employed a prospective cohort study of adult patients with pathologically confirmed stage III CRC receiving adjuvant chemotherapy with an oxaliplatin-based regimen for investigating OIPN. To further validate the clinical manifestations and identify a potential therapeutic strategy, animal models, and in vitro studies on the mechanism of OIPN were applied.
Results: Our work found that (1) consistent with clinical findings, OIPN was observed in animal models. Targeting the enzymatic activity of cathepsin S (CTSS) by pharmacological blockade and gene deficiency strategy alleviates the manifestations of OIPN. (2) Oxaliplatin treatment increases CTSS expression by enhancing cytosol translocation of interferon response factor 1 (IRF1), which then facilitates STIM-dependent store-operated Ca2+ entry homeostasis. (3) The cytokine array demonstrated an increase in anti-inflammatory cytokines and suppression of proinflammatory cytokines in mice treated with RJW-58. (4) Mechanistically, inhibiting CTSS facilitated olfactory receptors transcription factor 1 release from P300/CBP binding, which enhanced binding to the interleukin-10 (IL-10) promoter region, driving IL-10 downstream signaling pathway. (5) Serum CTSS expression is increased in CRC patients with oxaliplatin-induced neurotoxicity.
Conclusions: We highlighted the critical role of CTSS in OIPN, which provides a therapeutic strategy for the common adverse side effects of oxaliplatin.
Keywords: Oxaliplatin-induced peripheral neuropathy, Cathepsin S, olfactory receptor transcription factor 1, IL-10
Introduction
Oxaliplatin has exhibited significant efficacy in the treatment of human cancers, especially colorectal cancer (CRC). However, acute or chronic peripheral neuropathy and concurrent sensory and motor function deficiencies are a common adverse effect of oxaliplatin [1], resulting in chemotherapy dose reduction and early discontinuation [2]. Several mechanisms of oxaliplatin-induced peripheral neuropathy have been proposed, including direct damage, ROS generation, and dysfunction of voltage-gated ions channels in the dorsal root ganglia and axon [3, 4]. Several clinical trials were conducted to prevent or treat oxaliplatin-induced peripheral neuropathy based on the aforementioned mechanisms. However, these studies failed to demonstrate significant effectiveness in alleviating this side effect [5, 6]. Moreover, increasing evidence suggests that neuroinflammation also plays a role in the development of neurotoxicity [7]. In animal studies, inhibiting macrophage infiltration and proinflammatory cytokines in the dorsal root ganglion (DRG) suppressed the augmentation of paclitaxel-induced neuropathic nociception [7, 8]. Milligan et al. reported that intrathecal administration of anti-inflammatory cytokine Interleukin-10 (IL-10) briefly reversed chronic constriction injury-induced mechanical allodynia and thermal hyperalgesia [9, 10]. Furthermore, suppression of IL-20, a proinflammatory cytokine, with IL-20 neutralizing antibody alleviated paclitaxel-induced peripheral neuropathy in animal models [11]. Therefore, triggering anti-inflammatory cytokines and suppressing the proinflammatory response may be a therapeutic strategy for managing chemotherapy-induced peripheral neuropathy (CIPN).
Cathepsin S (CTSS), a lysosomal cysteine protease, is stably present in the cytoplasm of immune-related cells, such as antigen-presenting cells, B cells, and phagocytic cells (e.g., macrophages and microglial cells). Several lines of evidence have demonstrated that releasing CTSS from microglial cells causes neuropathic pain [12] that could be reversed by intraspinal injection of CTSS inhibitor, but the mechanisms remain unclear. Furthermore, CTSS is reportedly an autocrine factor that stimulates microglial cell activation and subsequently regulates the release of proinflammatory cytokines and chemokines [13]. Our studies have indicated that CTSS plays a vital role in mediating Ca2+ homeostasis by disturbing stromal interaction molecule 1 (STIM1) trafficking, which alters STIM1-Orai1 interaction and affects Ca2+ influx [14]. And, studies have reported that STIM1 regulates stored-operated Ca2+ entry (SOCE) by initiating Ca2+ influx and triggering immune activation in macrophages, chemotaxis, and neutrophil infiltration, which plays a role in the development of inflammatory pain [15].
IL-10, a “founding” member of IL-10 family cytokines, was initially described as a cytokine synthesis inhibitory factor produced by T helper cells with pleiotropic immunosuppressive functions [16]. Increasing evidence indicates that IL-10 is expressed by a wide variety of cell types, including macrophages, monocytes, dendritic cells, natural killer cells, and CD4+ and CD8+ T cells and B cells. The immunosuppressive activity of IL-10 is mediated by ligand binding with IL-10 receptor (IL-10R1, IL-10R2), which activates Janus tyrosine kinases (JAKs) and signal transducers and activators of transcription signaling pathways, which inhibit the expression profile of immunomodulatory genes [17] and release proinflammatory cytokines, such as IL-6, IL-1β, and TNF-α [18]. However, despite the fact that IL-10 acts as a prime drug target for autoimmune disease, tissue damage, and even cancer, effective therapies have yet to be developed.
Therefore, we investigated whether targeting CTSS can ameliorate oxaliplatin-induced peripheral neuropathy (OIPN) by upregulating IL-10 expression and suppressing proinflammatory cytokines. The detailed molecular mechanisms are described.
Materials and Methods
Clinical cohort
Patients with CRC enrolled in a clinical study investigating the CIPN (NCT02481336) were used for analysis. The details of this study are described elsewhere [19]. Briefly, patients with pathologically confirmed stage III or high-risk stage II CRC have been prospectively recruited from National Cheng Kung University Hospital (NCKUH) from January 2015 until the present. All patients received curative surgery followed by 6 months of oxaliplatin and infusional 5-fluorouracil and leucovorin (modified FOLFOX6) as adjuvant chemotherapy. Patients with poor performance status (ECOG performance score > 1), inadequate organ functions, prior treatment with neurotoxic chemotherapy (such as oxaliplatin, cisplatin, carboplatin, taxanes, or vinca alkaloids), preexisting peripheral neuropathy of any grade, or a family history of genetic or familial neuropathy were excluded. A complete neurological examination, patients-reported peripheral neuropathy CIPN20 questionnaire (QLQ-CIPN20), and quantitative examination of the peripheral nerve system (including nerve conduction velocity [NCV], quantitative sensory test [QST], nerve excitability test [NET], and total neuropathy score [TNS]) were assessed at baseline, the end of cycles 1 (week 2), 6 (week 12), and 12 (week 24) of chemotherapy, and 9 (week 36) and 12 months (week 48) after the beginning of chemotherapy. The CIPN20 questionnaire contained 20 items evaluating the severity of neurologic symptoms, including nine items for sensory, eight items for motor, and three items for autonomic symptoms. Scores from 1 to 4 were applied for each item. The scores of the items were added together for all 19 questions, and a final CIPN score, ranging from 19 to 76, was generated. The score of item 20, which rates men's sexual function, was not adopted in this study because the information is lacking for women and is often not provided for male patients either. In addition to collecting blood from all participants at baseline to identify predictive biomarkers of CIPN, blood samples at week 48 were also collected in 23 patients to monitor the changes in biomarkers. This study was approved by the institutional review board of NCKUH (A-ER-103-395). All participants provided written informed consent before being enrolled.
Reagents
Oxaliplatin was purchased from Sanofi Pharmaceutical Company (NY, USA). The procedure for synthesizing the CTSS inhibitor RJW-58 was described previously [14]. The following primary antibodies were purchased: Cathepsin S (Thermo Fisher catalog: #PA5-81369), EBF (Santa Cruz, #sc-137065), ATF-3 (Biossua, #BS-0519R), α-tubulin (Sigma, MO, USA), IRF-1 (Bio-Rad, #VPA00801), CBP (GeneTex, #GTX101249), Iba1 (Abcam, #ab-5076), NeuN (Novus, #NBP2-10491), IL-10 (Abcam, #ab189392), STIM1 (Cell signaling (D88E10), #5668S), CD11b (Abcam, #ab52478), CD45 (BD Biosciences, #553079), STAT3 (BD, #50402), P-STAT3 (Cell signaling, #9145S), NFκB (Cell signaling, #4727S), OR5B3 (Abcam, #ab186624), OR5M3 (LSBio, #LS-C200406), and β-actin (GeneTex, #GTX109639). Horseradish-peroxidase-conjugated secondary antibodies were purchased from GeneTex (#GTX213110, #GTX213111, #GTX224125, and #GTX232040).
Cell culture
DRG cell lines (ND 7/23) and mouse microglial cells (SIM-A9) were obtained from Sigma-Aldrich (#92090903, USA) and Kerafast (END001, USA), respectively. The ND 7/23 cells were grown in Dulbecco's Modified Eagle Medium (DMEM, #12100046; Gibco, USA) and the SIM-A9 cells were maintained in DMEM/F12 (#11320033; Gibco, USA) supplemented with 10% FBS, 100 U/ml of penicillin, and 100 μg/ml of streptomycin-glutamate (Thermo Scientific, Utah, USA). Both cell lines were maintained in a 5% CO2 incubator (humidified atmosphere) at 37 °C.
Animals
The 6-week-old C57BL/6JNarl mice were maintained in the pathogen-free facility of the Animal Laboratory of National Cheng Kung University (Institutional Animal Care and Use Committee [IACUC] number: 107078). These animals were housed in a temperature-controlled (25 ± 2 °C); humidity of approximately (60%-80%) and light-controlled environment (12 / 12 h light-dark cycle, lights on at 6:00 AM) with free access to food and water, following the approved guidelines by the IACUC of National Cheng Kung University. Before the experiments were performed, all animals had adapted to the housing environments for at least 7 days. Our preliminary study revealed that no body weight loss was observed in animals treated with oxaliplatin 3 mg/kg or RJW-58 25 mg/kg intraperitoneally (i.p.) for 5 days a week over two alternative weeks.
CTSS transgenic mice production
Ctss-/- mice were generated using CRISPR/Cas9-induced knockout in embryos obtained from the National Laboratory Animal Center. Mouse fertilized eggs were microinjected with CRISPR/Cas9-related point mutation CTSS sequence, TGACTA, including a stop code and restriction enzyme SpeI recognition cutting site to verify the mutations at exons 2. The point mutation was designed right after the CTSS transcription start site to inhibit CTSS RNA and protein expression. Recombinant embryonic stem cell lines were injected into 161 C57BL/6JNarl blastocysts, 122 of which were transferred into pseudo‐pregnant females, and 28 live pups were born in 2018. Only one Ctss+/- female strain was detected using polymerase chain reaction (PCR) genotyping. For recessive phenotypes, intercross between Ctss+/- female mice with male Ctss+/+ mice in the related C57BL/6JNarl background generated Ctss-/- mice. The litters were backcrossed three times to generate Ctss-/- mice. Ctss+/+ C57BL/6JNarl mice were used as controls. The following primers were used for Ctss-/- genotyping: forward 5′-GTCAGGCAGATTGCTACAAG-3′, reverse 5′-ACACTGCTCGGGTGGCAATC-3′, and digestion with restriction SpeI enzymes (Figure S1).
Mouse behavioral models of oxaliplatin-induced peripheral neuropathy
Following housing adaptions, 7-week-old C57BL/6JNarl mice (weight range: 18-20 g) were used to investigate oxaliplatin-induced neuropathy. Oxaliplatin (3 mg/kg, Sanofi, USA), vehicle (saline), or RJW-58 (25 mg/kg) was administered i.p. for 5 consecutive days every other week for 2 weeks. A von Frey filament test (Part #2390, IITC Inc., CA) for mechanical hyperalgesia, tail immersion test (water temperature: 48-49 °C or ice) for thermal sensitivity, and grip test (BIO-EVF3, Bioseb Inc., USA) for grip strength were used, following a previously described method [11, 20]. To quantify locomotor reactivity, we subjected each mouse to an open field chamber with photocell emitters and receptors equally spaced along the perimeter of the chamber. These photocell emitters and receptors created an x-y grid of invisible infrared beams and signals, recorded by computer software to calculate the quantity of mouse motor activity in 10 min. The baseline measurement of each behavior test was established before drug treatment, and five additional sessions were measured weekly.
Primary DRG isolation
Primary DRG tissues were removed from adult C57BL/6JNarl mice after the behavior tests, and the tissues were digested with 0.1% collagenase for 1 h, followed by 0.25% trypsin for 25 min for cell mixture. The cell mixtures were stained with NeuN (Novus, #NBP2-10491) antibodies to isolate primary spinal dorsal horn neurons for flow cytometry analysis to detect ATF3 expression. The cell mixtures were further stained with both CD11b (Abcam, #ab52478) and CD45 (BD Biosciences, #553079) antibodies to isolate the microglia for flow cytometry analysis to detect IL-10 expression.
Flow cytometry analysis
After treatment, the cells were fixed with 3.7% formaldehyde (dissolved in phosphate buffered saline [PBS]) for 15 min at room temperature and permeabilized with 0.5% Triton X-100 in 3.7% formaldehyde for another 15 min. The cells were washed three times with PBS and blocked with CAS-BlockTM solution (#008120; Life Technologies) for 1 h. The antibodies were prepared in the CAS-BlockTM solution (#008120; Life Technologies) and stained overnight. The cells were then washed three times with PBS and stained with secondary antibodies for 2 h. Finally, the cells were counterstained with DAPI for 2 min and then washed with 1-2 mL of BD FACSFlow buffer (LOT:1833202826) twice. After washing, the cells were resuspended with BD FACSFlow buffer. The data were acquired on a flow cytometer, following the manufacturer's recommendations.
Cytokine array analysis
Ctss+/+ mouse serums were collected after RJW-58 injection (week 4). Cytokine array analysis was performed following the protocol of the MILLIPLEXMAP mouse cytokine/chemokine magnetic bead panel (Millipore, MCYTOMAG-70K).
Gene expression profiling and statistical analysis of microarray data
Total RNA samples were extracted using TRIzol (Invitrogen) and purified using a RNeasy mini-kit (QIAGEN). Gene expression analysis was performed on a GeneChip platform (Human Genome U133A 2.0 Plus; Affymetrix) following the National Health Research Institutes Microarray Core facility technique support. Gene expression changes were calculated as the ratio of 1.2-fold significant alterations of the experimental groups (CTSS overexpression or si-CTSS treated) compared with each control group. Compared with the gene expression variance within each experimental group, most of the olfactory receptor genes detected were differentially expressed.
Ultrastructure of the sciatic nerve
After neurobehavioral tests at week 6 following oxaliplatin treatments, sciatic nerve samples were obtained from mice, fixed in 4% glutaraldehyde, and postfixed in 1% osmium tetroxide solution at 4 °C. These samples were then dehydrated in graded ethanol series and embedded in EMbed 812 (EMS-14120; Hatfield, PA, USA). The sciatic nerve sections (90 nm) were prepared by the Department of Pathology of NCKUH and observed and imaged using a transmission electron microscope (H7650, Hitachi, Japan).
Quantification of intraepidermal nerve fiber density (IENF) density
Mouse hind paws were isolated after neurobehavioral tests. Dissected tissues were fixed with 10% paraformaldehyde overnight and cryoprotected for 72 h with 30% sucrose in PBS at 4 °C. The samples were embedded in OCT (#CM 3050S; Leica) and sliced into 30 μm thick sections. The slides were washed with 0.05% Triton X-100 in PBS for 4 h and incubated with CAS-BlockTM solution (#008120; Life Technologies) at room temperature for 1 h. The primary antibodies used were rabbit anti-PGP9.5 (1:100; #ab108986, Abcam) and goat anti-Collagens VI (1:100, #AB769; Chemicon). For double staining, two antibodies from different species were mixed and incubated overnight with the sample sections at 4 °C. After washing away the primary antibodies with PBS, the tissues were incubated with secondary antibodies (Alexa-488, 594, or 647, 1:200; Invitrogen) and Hoechst 33342 (10 mg/mL, #H1399; Invitrogen) for 2 h at room temperature. Immunofluorescence images were acquired using a multiphoton laser scanning microscope with a 40× objective (FV1000MPE, Olympus).
Intracellular [Ca2+]i measurement
ND7/23 or SIM-A9 cells (passage numbers: 10-15) were prepared and cultured for 3 days to measure intracellular Ca2+ concentration ([Ca2+]i). The cells were incubated with 2 μM Fluo-4/acetoxymethyl ester (Fluo-4/AM, F14201, Molecular Probes, USA) in DMEM medium at 37 °C for 30 min and then washed three times with Ca2+ free HEPES solution (145 mM NaCl, 5 mM KCl, 0 mM CaCl2, 10 mM glucose, 1 mM MgCl2, and 5 mM HEPES, with an osmolarity of 297-300, and pH 7.4). Fluo-4 was excited alternatively at 488 nm and the fluorescence intensity was analyzed at 520 nm. The Image XpressMicro automated wide-field fluorescent microscope (Molecular Devices, Sunnyvale, CA, USA) was then used to calculate [Ca2+]i. For [Ca2+]i measurement, the cells were stimulated using 2 μM thapsigargin (TG, Cayman Chemical, USA), recorded for 7 min.
Western blot
Cells were lysed with CelLytic MT Cell Lysis Reagent (Sigma) containing a protease inhibitor cocktail (Roche). The total cell lysates were separated with SDS-PAGE gel, transferred to polyvinylidene fluoride membranes (Amersham Life Science), blocked with 5% nonfat milk at room temperature for 1 h, washed three times with TBST (0.1% Tween-20), and then hybridized overnight with the primary antibody at 4 °C. The membranes were then washed three times with TBST (0.1% Tween-20) and hybridized with the appropriate peroxidase-conjugated secondary antibody (Santa Cruz) at room temperature for 1 h. Finally, the membranes were washed three times with TBST (0.1% Tween-20) and immunoreactivity was detected using Chemiluminescence (Millipore).
Nuclear extraction assay
Samples were lysed using 500 μL of buffer A (10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, and 0.05% NP-40; pH 7.9). After 10 min of centrifugation at 3,000 rpm at 4 °C, the supernatant was collected as a cytosolic fraction. The pellet was washed three times with a wash buffer (10 mM HEPES, 1.5 mM MgCl2, and 10 mM KCl; pH 7.9) and lysed using 450 μL of buffer B (5 mM HEPES, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 26% glycerol, and 300 mM NaCl; pH 7.9). The supernatant was collected after 20 min of centrifugation at 13,200 rpm at 4 °C as a nuclear fraction. Finally, 100 μg of the cytosolic and nuclear fractions were subjected to gel electrophoresis and analyzed using immunoblotting.
Coimmunoprecipitation and immunoblotting
Samples were lysed using an immunoprecipitation (IP) lysis buffer (0.5% NP-40 and 10% glycerol in PBS). A mixture of 2 mg of cell protein lysates, 5 μg of anti-STIM1 or anti-OLF1 antibodies, and 100 μL of protein-A/G sepharose beads were incubated overnight at 4 °C. The mixture was washed twice with wash buffer (0.5% NP-40, 0.1% Triton X-100 in PBS). The precipitated protein samples were processed for SDS-PAGE and detected using immunoblotting with the appropriate primary antibodies.
Gene reporter assay
The reporter gene plasmids of CTSS-, IL10-pGL4.17 were purchased from GENEWIZ (product numbers: BB9283-1/N332028). The plasmids were introduced into cells using the oligofectamine transfection reagent for 24 h and then processed using the Dual-Luciferase 1000 Assay system (Promega, Madison, USA, E1910), following the manufacturer protocol.
Chromatin immunoprecipitation PCR (ChIP)
The samples were crosslinked with 37% formaldehyde for 10 min at 37 °C. The cells were then harvested and processed for DNA fragmentation using sonication on ice. Following the manufacturer instructions, the resultant chromatin-DNA fractions were purified using a ChIP assay kit (Millipore, 2487694). The chromatin was immunoprecipitated at 4 °C using anti-IRF1, anti-OLF1, and normal IgG (negative control). The primer sequences used were as follows:
OLF1 on IL-10 promoter: forward 5′-CTGCCTTCATTATCAGTGTG-3′, reverse 5′-CAGCACAAAGCAAGATGTTG-3′; IRF1 on CTSS promoter: forward 5′-ATGCTCTATCTTTTCATCAGTCAG-3′, reverse 5′-GCCAACAAGAATCTCTCAAACAG-3′.
Quantitative reverse transcription PCR
The total RNA was extracted from samples using TRIzol reagent (Thermo Fisher, #74108). First-strand cDNA was synthesized using the SuperScript III first-strand synthesis system following the manufacturer instructions. The transcription level of CTSS, IL-10, TNF-α, IL-1β, IL-6, and MCP-1 were quantified using the Applied Biosystems 7500 Real-Time PCR System, with RPL13A as an internal control. The primer sequences used were as follows: CTSS: forward 5′-CCATTGGGATCTCTGGAAGAAAA-3′, reverse 5′-TCATGCCCACTTGGTAGGTAT-3′; IL-10: forward 5′-GCTCTTACTGACTGGCATGAG-3′, reverse 5′-CGCAGCTCTAGGAGCATGTG-3′; MCP-1 Forward: 5′-AGGTCCCTGTCATGCTTCTG-3′, Reverse: 5′-GCTGCTGGTGATCCTCTTGT-3′; TNF-α Forward: 5'-AGTGACAAGCCCGTAGCCC-3', Reverse: 5'-AGCCTTGTCCCTTGAAGAG-3'; IL-1β Forward: 5'-GCTGAAAGCTCTCCACCTC-3', Reverse: 5'-GAGGTGCTGATGTACCAGTT-3'; IL-6 Forward: 5'-CAGGAAATTTGCCTATTGAAAAT-3', Reverse: 5'-TTGGATGGTCTTGGTCCTTAG-3'.
Statistical analysis
At least seven mice were randomized to each group for all animal behavioral tests. The results are expressed as the mean ± SEM. Differences between groups were compared using a two-tailed Student's t-test or two-way analysis of variance, performed using Prism. Statistical significance was set as P < 0.05. Experimenters were blinded to all drug treatments.
Results
Evaluation of oxaliplatin-induced peripheral neuropathy in patients with colorectal cancer
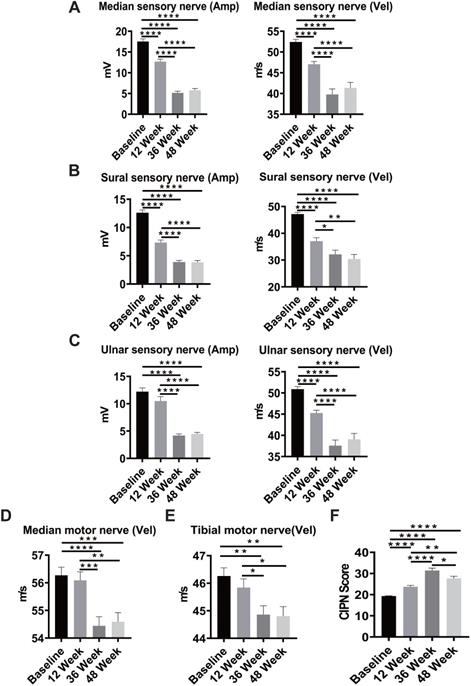
From January 2015 to July 2019, 87 patients were recruited into a clinical study investigating chemotherapy-induced peripheral neuropathies: 45 (51.7%) men and 42 (48.3%) women. By July 2019, 76 of 87 patients had completed the 6-month adjuvant chemotherapy with modified FOLFOX6, and 75 patients (86.2%) had completed the neurologic study scheduled at week 12, 67 (77%) at week 36, and 63 (71.3%) at week 48. A significant reduction in both amplitude and conduction velocity was observed in sensory nerves, including the median sensory nerve (Figure 1A), sural sensory nerve (Figure 1B), and ulnar sensory nerve (Figure 1C), after 6 cycles of chemotherapy (week 12) compared with the baseline results of nerve conduction (P < 0.0001). The slowing in conduction velocities and the reduced amplitude deteriorated further at week 36 and reached a plateau at week 48, which was 6 months after completion of the oxaliplatin-containing chemotherapy. Peripheral sensory neuropathy is a well-known toxicity of oxaliplatin. However, motor impairments have been less well characterized. Notably, we observed a similar pattern of reduction in conduction velocity in median motor nerves (P < 0.0001) and tibia motor nerves (P = 0.0015, P = 0.0018; Figure 1D-E). The CIPN20 questionnaire was used to assess patient-reported severity of peripheral neuropathy, and the CIPN scores increased significantly from baseline to week 12, suggesting an increase in neurologic symptoms after six cycles of chemotherapy (Figure 1F). The CIPN20 scores reached a peak at week 36 and declined significantly at week 48 (P < 0.0001). These results indicate that oxaliplatin-containing chemotherapy-induced subjective and objective sensory and motor neuropathies in CRC patients.
Sensory and motor nerve deficit in patients with colorectal cancer (CRC) after oxaliplatin treatment. A-C, Sequential measurement of the median, sural, and ulnar sensory nerve by NCV test before and after oxaliplatin treatment at baseline and weeks 12, 36, and 48 in CRC patients. D-E, Sequential measurement of median and tibial motor nerve before and after oxaliplatin treatment at weeks 12, 36, and 48 in CRC patients. F, The CIPN score at baseline and weeks 12, 36, and 48 in patients with colorectal cancer. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05 compared with baseline using a two-tailed Student's t-test. Each value represents the mean ± SEM from CRC patients.
Targeting cathepsin S alleviates oxaliplatin-induced peripheral neuropathy in vitro and in vivo
We had previously successfully established an integrated image-based high-content screening platform to identify neuroprotective compounds [11, 20]. By applied the high-content drug screening platform, several compounds were tested for protective effects against chemotherapy-induced neurotoxicity, including cathepsin S inhibitors. Furthermore, our studies revealed that inhibition of cathepsin S by RJW-58 could retard tumor progression and metastasis [14]. Importantly, similarly with previous findings, elevated levels of CTSS are associated with increased risk in the development of neuropathic pain [21, 22]. And, blocking CTSS activity could relieve inflammatory hyperalgesia [23, 24]. Above evidence suggested that cathepsin S inhibitors, as potential compounds, prevent chemotherapy-induced neurotoxicity. Therefore, we investigated whether cathepsin S inhibitor (RJW-58) could prevent oxaliplatin-induced neurotoxicity in vitro. Our results indicated that RJW-58 suppressed oxaliplatin-induced ATF3 expression in ND 7/23 cells (Figure 2A). ATF3 is a marker of damaged neurons [25].
The neuropathic effects of oxaliplatin in the Ctss+/+ mouse model. A, Flow analysis of ATF3 expression in ND 7/23 cell treated with control, oxaliplatin (1 µM), RJW-58 (5 µM), or a combination of oxaliplatin (1 µM) and RJW-58 (5 µM), respectively. B, The protocol of drug administration and behavioral tests in the mouse model. The basal levels of each behavioral assay were obtained before treatment. In the first week, PBS control (100 µL), oxaliplatin (3 mg/kg, 100 µL), and RJW-58 (25 mg/kg, 100 µL) were injected intraperitoneally five times per week for 2 weeks. Behavioral tests were performed weekly. C, Von Frey filament test to detect the paw withdrawal threshold to mechanical stimulus. The Y-axis is the normalized pressure from touch to paw withdrawal. D-E, Tail immersion test to assess thermal sensation. The Y-axis is the normalized latency from tail immersion to tail withdrawal. F, Locomotor activity test to detect allodynia. The Y-axis is the normalized moved distance baseline. G, Grip test to detect neuromuscular function. The Y-axis is the normalized moved distance baseline. The black arrow indicates drug infusion. H, Flow analysis of ATF3 expression of primary DRG cells at week 4 for NeuN+-ATF3+ double staining. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05 compared with baseline using a two-tailed Student's t-test. Each value is represented as mean ± SEM from at least seven mice in each group.
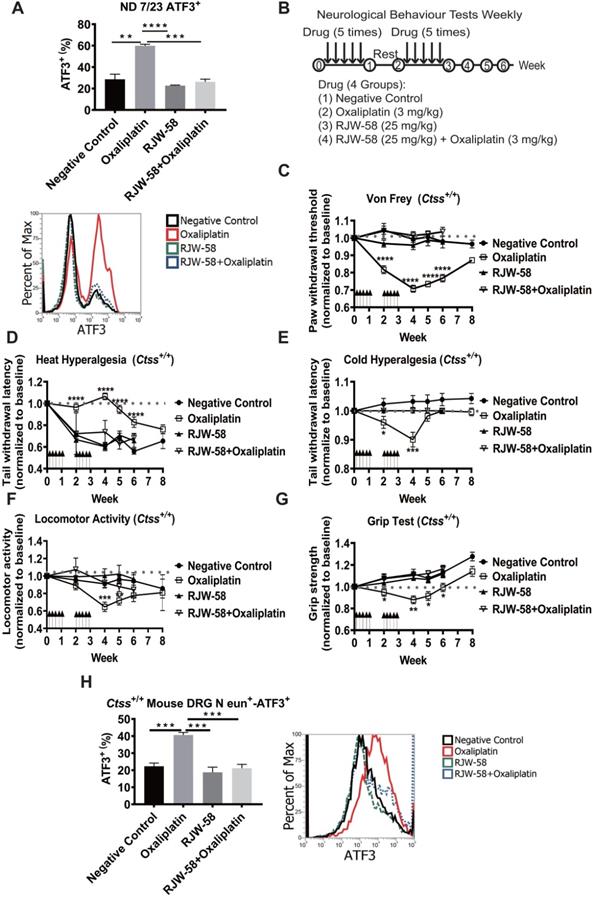
To validate the neuroprotective effects of RJW-58 in vivo, C57BL/6JNarl-Ctss+/+ mice were treated with either oxaliplatin, RJW-58, or both (Figure 2B). Mouse behavioral tests, including tail immersion, von Frey filament, locomotor activity, and grip tests, were then performed weekly to assess the thermal threshold, mechanical allodynia, motor function, and fine motor skills, respectively. The oxaliplatin-treated mice exhibited a dramatic reduction in mechanical thresholds (Figure 2C) and an increase in thermal thresholds (Figure 2D), which was consistent with the clinical neurological findings for CRC patients treated with oxaliplatin (Figure 1). Notably, cold hyperalgesia recovered gradually, 2 weeks after the cession of oxaliplatin treatment, and returned to baseline at week 6 (Figure 2E). Furthermore, the locomotor activity and grip strength were also dramatically reduced by oxaliplatin treatment (Figure 2F-G), but recovered gradually to baseline at week 6. The post-hoc analysis data obtained at post-treatment week 6 revealed a significant protective effect of RJW-58 on oxaliplatin-induced sensory and motor deficits. Moreover, we isolated the primary DRG cells from Ctss+/+ mouse spinal cord before and after drug treatment. Consistent with the results from ND 7/23 cell lines, oxaliplatin treatment enhanced ATF3 expression, and RJW-58 reversed the effect of oxaliplatin on mouse DRG cells (Figure 2H).
Ctss-/- mice were generated to confirm that CTSS plays a critical role in protecting neurons from oxaliplatin damage (Figure S1A). Western blot analysis confirmed no expression of CTSS in Ctss-/- mouse (Figure S1B). Mice were then treated with the same dose and schedule of oxaliplatin, as illustrated in Figure 2B, and neurobehavioral tests were then performed. No differences were observed in sensory and motor function test results between Ctss-/- mice treated with or without oxaliplatin (Figure S2A-E). Together, these data indicate that CTSS plays a pivotal role in the development of OIPN.
Histology of sciatic nerve and quantification of intraepidermal nerve fiber density
Two methods were used to examine histopathological differences before and after oxaliplatin treatment. First, we inspected the ultrastructure of sciatic nerves using a transmission electron microscope at week 6 after drug treatment. Our data demonstrated that oxaliplatin damaged both myelinated and nonmyelinated nerve fibers. These fibers exhibited varying degrees of axon death (black arrow), axons with detached compact myelin, and accumulated myelin debris. In contrast, RJW-58 cotreatment reduced oxaliplatin-induced nerve damage (Figure 3A). Furthermore, we assessed the integrity of the myelinated nerve by calculating the ratio of inner axon circumference to outer myelin circumference (G-ratio). As depicted in Figure 3B, oxaliplatin treatment significantly reduced the G-ratio of both large myelinated (axon diameter > 5 μm; P = 0.01) and small myelinated fibers (< 5 μm; P = 0.008) compared with the control, whereas RJW-58 significantly reduced the axonal damage (P = 0.003).
RJW-58 protects peripheral nerves from oxaliplatin-induced damage. A, Transmission electron micrographs of the sciatic nerve from Ctss+/+ mice treated with control, oxaliplatin, RJW-58, or the combination of oxaliplatin and RJW-58. Bottom: G-ratio's measurement represents the ratio of axon diameter (yellow line) to myelin (red line). N: nucleus of Schwann cell. Scale bar: 10 µm (top), 500 nm (middle and bottom). B, The myelinated fiber density of sciatic nerves from mice in different treatment groups was measured at week 6 (n = 5 mice/group). The G-ratio was calculated and compared between different treatment groups. C-D, Representative immunofluorescence images and quantitative analysis of intraepidermal nerve fibers of mice treated with control, oxaliplatin, RJW-58, and a combination of oxaliplatin and RJW-58. Scale bar: 25 µm. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05 compared with baseline using a two-tailed Student's t-test. Each value represents the mean ± SEM from at least 7 mice in each group.
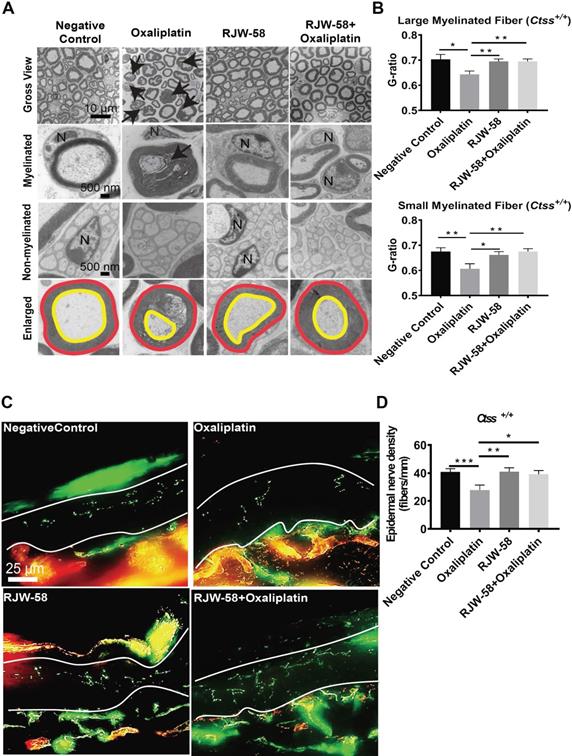
Second, morphologic changes in skin innervations are associated with clinical measures of small-fiber neuropathy and have proposed that oxaliplatin could induce intraepidermal nerve fiber (IENF) loss [26]. Therefore, IENF density in the mouse hind-paw interplanar skin was calculated before and after oxaliplatin treatment with or without RJW-58. As illustrated in Figure 3C, oxaliplatin treatment caused 68% IENF loss in the interplanar epidermis (Figure 3D; P = 0.003). However, no differences in IENF density were observed between oxaliplatin-treated mice co-treated with RJW-58 and control mice.
Furthermore, Ctss-/- mice were also used to examine the neuroprotective role of CTSS in oxaliplatin-induced nerve damage. Our results indicated that no differences in ultrastructure changes in the sciatic nerve or the number of IENF between oxaliplatin-treated and control mice (Figure S2F-I). Furthermore, no differences were revealed in the expression level of ATF3 in microglial cells isolated from DRG in mice treated with or without oxaliplatin (Figure S2J). These findings further confirm the neuroprotective effect of CTSS inhibition in OIPN.
Oxaliplatin treatment increases CTSS expression and binding to STIM1
DNA damage caused by drugs, such as cisplatin and doxorubicin, results in the upregulation of interferon regulatory factor 1 (IRF1) [27]. A study revealed that CTSS expression is regulated by transcriptional factor IRF1 [28]. Therefore, we determined whether oxaliplatin could enhance CTSS expression by regulating IRF1 in SIM-A9 cells. Our data demonstrated that oxaliplatin treatment increased cytosol translocation of IRF1 to the nucleus (Figure 4A) and enhanced IRF1 binding to the CTSS promoter region, which augmented the transcriptional level of CTSS in vitro and in vivo (Figure 4B-E).
Effects of oxaliplatin-induced remodeling of calcium homeostasis on CTSS expression via IRF1. A, The effect of oxaliplatin on the nuclear localization of IRF1. Cells were treated with or without oxaliplatin (1 µM) for 24 h, and the cytosolic and nuclear fractions were then separated, as described in the Materials and Methods section. B, The effect of oxaliplatin treatment for 24 h on IRF1 binding to the CTSS promoter regions in SIM-A9 cells was analyzed using ChIP. C, The effect of oxaliplatin treatment for 24 h on the transcriptional activity of CTSS promoters was determined using the promoter assay. D, The effect of oxaliplatin treatment for 24 h on the transcription levels of CTSS genes in SIM-A9 was determined using RT-qPCR. E, The effect of oxaliplatin on the transcription levels analysis of CTSS genes in mouse primary DRG at week 4 was determined using RT-qPCR. F, The time-dependent mouse serum CTSS level (n = 10). G, Patients with colorectal cancer' serum CTSS level at week 48 compared to baseline (n = 23). ****P < 0.0001, Two-way ANOVA analysis. H, For the drug treatment, the cells were pretreated with DMSO or RJW-58 (5 µM). The treatment time for ND 7/23 and SIM-A9 was 24 h. The calcium influx from the ER and SOCE in CTSS overexpression (n = 50 for each condition) cell was assessed using single-cell intracellular Ca2+ measurement. I, CTSS was disassociated from STIM1 following RJW-58 treatment. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05. The statistical significance was determined using a two-tailed Student's t-test.
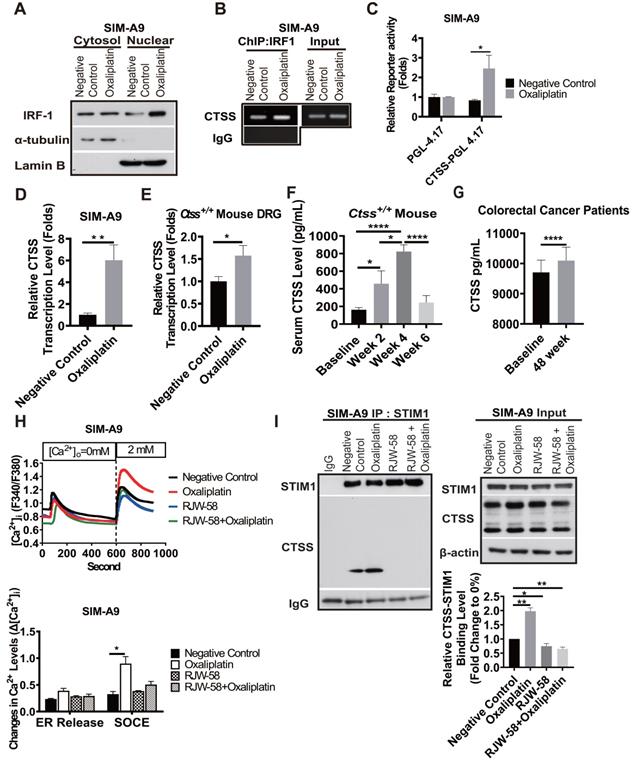
We further investigated whether serum CTSS level could serve as a predictive biomarker of oxaliplatin-induced peripheral neuropathy. Our data revealed that the serum level of CTSS in mice increased at week 2 (P = 0.049), peaked at week 4 (P < 0.0001), and returned to baseline level at week 6 after oxaliplatin treatment (Figure 4F). These changes were consistent with findings that sensory and motor function started to decline at week 2, reached their nadir at week 4, and recovered at week 6 after oxaliplatin treatment, as illustrated in Figure 2. Furthermore, we retrospectively verified serum CTSS level in 23 patients at week 48 (Figure 4G) compared for the nerve conduction velocity test. Consistent with animal models' findings, motor function recovers more rapidly than sensory function (Figure 2).
Disruption of store-operated calcium entry (SOCE) through STIM1 has been reported that also underlies chemotherapy-induced neuropathy [11]. Our previous study demonstrated that CTSS plays a crucial role in mediating Ca2+ homeostasis by binding to STIM1, thus interfering with Orail1 [14]. Therefore, we investigated whether oxaliplatin augments CTSS expression results in the dysregulation of Ca2+ homeostasis and also examined whether inhibiting CTSS enzyme activity may restore the SOCE disruption. Compared to control group, oxaliplatin significantly increased SOCE Ca2+ level after thapsigargin-induced endoplasmic reticulum (ER) store depletion (Figure 4H, P = 0.04). RJW-58 treatment alone did not affect the SOCE activation. However, co-treated with oxaliplatin, blocking CTSS by RJW-58 prevented oxaliplatin-induced SOCE dysregulation. Our previous study had reported that CTSS inhibition reduced Ca2+ influx through SOCE, but did not affect STIM1 and Orai 1 expression [14]. Hence, immunoprecipitation assays were performed to confirm whether oxaliplatin evoked SOCE disruption via the augmentation of CTSS-STIM1 binding affinity. Our results demonstrated that CTSS bound to STIM1, which was enhanced by oxaliplatin treatment in SIM-A9 cells and reduced by cotreated with RJW-58 (Figure 4I).
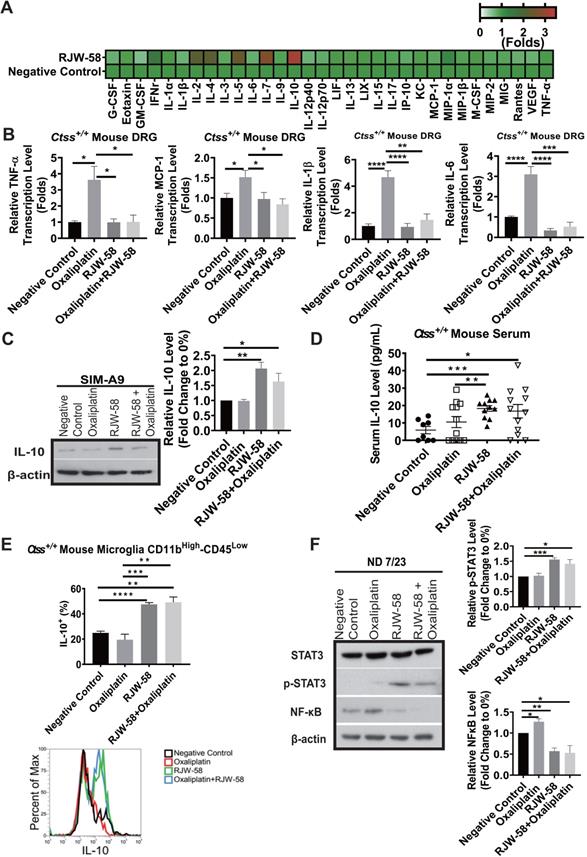
RJW-58 treatment induces anti-inflammatory cytokine production and suppresses proinflammatory cytokine production
Neuroinflammation is a pathological feature of CIPN, and consequently, therapeutics targeting the suppression of immune response has become an amenable strategy for CIPN. Furthermore, our data indicated that inhibition of CTSS reduced intracellular Ca2+ influx, which was reported to initiate macrophage activation [15, 29]. Therefore, investigating cytokine profiles before and after CTSS inhibition is reasonable. We then collected mouse serums at week 4 after RJW-58 i.p. injection and analyzed the cytokine profiles using a cytokine array. Our findings indicated that RJW-58 dramatically increased anti-inflammatory cytokine expression, with a predominant IL-10, IL-5, and IL-4 expression and a concomitant reduction in proinflammatory cytokine expression, including TNF-α, IL-6, IL-1β, and MCP-1 (Figure 5A). The transcriptional levels of TNF-α, MCP-1, IL-1β, and IL-6 in mouse DRG were increased significantly after oxaliplatin treatment, which was reversed by RJW-58 (Figure 5B). We focused on CTSS and IL-10 because of the pleiotropic immunosuppressive function of IL-10. We determined that translational levels of IL-10 increased significantly in SIM-A9 cells treated with either RJW-58 alone or cotreated with oxaliplatin (Figure 5C). Furthermore, the serum level of IL-10 in RJW-58-treated Ctss+/+ mice was significantly higher than the animals with vehicle or oxaliplatin alone (Figure 5D; P = 0.0002 and P = 0.0042, respectively). Moreover, the serum level of IL-10 in Ctss-/- mice was significantly higher than in Ctss+/+ mice (Figure S2K, P < 0.0001). Furthermore, a similar result was obtained in microglial (CD11b+-CD45-) cells isolated from mice (Figure 5E). IL-10 was reported to exert its function by activating the IL-10/STAT3 anti-inflammatory pathway cascade. In contrast to increasing NF-κB levels after treatment with oxaliplatin in ND 7/23 cells, RJW-58 treatment alone or cotreatment with oxaliplatin increased p-STAT3 and reduced NF-κB expression levels (Figure 5F). Our finding supported that CTSS inhibition results in an anti-inflammatory response in oxaliplatin-induced neuropathy, through the upregulation of IL-10.
Effects of RJW-58 in IL-10 expression in vitro and in vivo. A, Heat map of cytokine array clustering analysis. RJW-58 increased IL-10 secretion in B6 mouse serum at week 4. B, The effect of RJW-58 (5 µM) in primary DRG cells on the transcription levels of TNF-α, IL-6, IL-1β, and MCP-1 genes at week 4 was determined using RT-qPCR. C, Western blotting of IL-10 expression level in SIM-A9 after RJW-58 for 24 h treatment. D, The mouse serum IL-10 level after RJW-58 treatment at week 4. E, Flow analysis of CD11b+-CD45- B6 primary microglial cells at week 4 for IL-10 expression. F, Western blotting of the NF-κB/STAT3 signaling pathway under treatment with RJW-58 for 24 h. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05. The statistical significance was determined using a two-tailed Student's t-test. Data are presented as the mean ± SEM.
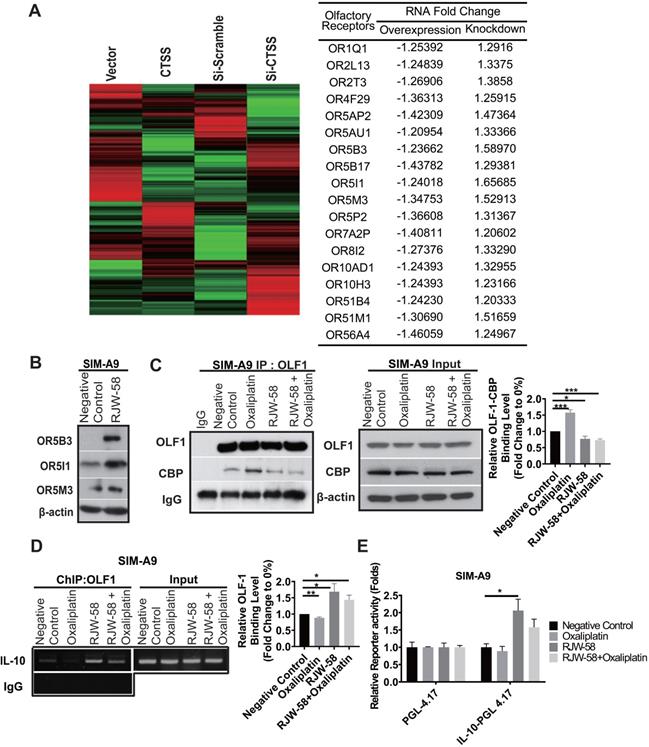
CTSS is involved in the regulations of the olfactory transcription factor, Olf-1
Previously, we investigated genes that may be regulated by the expression of CTSS. The siRNA knockdown and overexpression by transient transfection with CTSS in cancer cell lines was performed and applied to gene expression profiling. Notably, microarray results revealed that CTSS knockdown upregulated almost all olfactory receptor family genes, and vice versa, in cell overexpressing CTSS (Figure 6A). Western blot analysis confirmed the increase of the expression level of olfactory receptor 5B3, 5I1, and 5M3 proteins when SIM-A9 cells were treated with RJW-58 (Figure 6B). Evidence has indicated that olfactory receptors are regulated by an olfactory receptor transcription factor, Olf-1, which was reported that CBP competes with Olf-1 to bind DNA and activate target genes [30]. Above finding suggested that inhibition of CTSS might affect the interaction between CBP and Olf-1, which results in the release of Olf-1 and binding to the target genes. Oxaliplatin treatment increased the recruitment of p300/CBP and Olf-1, which was reduced when cells were treated with RJW-58 alone or combined with oxaliplatin (Figure 6C). Next, the ChIP experiment demonstrated that incubation with oxaliplatin decreased Olf-1 transcription factor binding to IL-10 promoter. RJW-58 alone or co-treatment with oxaliplatin promoted released Olf-1 from p300/CBP for binding to the Olf-1-binding sites on the IL-10 promoter regions (Figure 6D). We further performed a luciferase functional quantitative assay to assess the effects of RJW-58 on IL-10 promoter activity. Comparison of the PGL-4.17 vector, RJW-58 promotes a two-fold increase in IL-10 transcriptional activity (Figure 6E, P = 0.04). These findings indicate that oxaliplatin treatment facilitated the assembling of p300/CBP and Olf-1. By contrast, inhibiting CTSS by RJW-58 decreased the recruitment of CBP and Olf-1. These released Olf-1 transcription factors further bind to Olf-1-binding sites on the IL-10 promoter regions (Figure 6D), which results in an increased IL-10 transcriptional activity (Figure 6E).
Effects of RJW-58 in IL-10 expression via OLF-1. A, The cluster heat map reveals the results of microarray analysis. CTSS knockdown increases the mRNA levels of the olfactory receptor family. B, Western blot analysis of OR5B3, OR5I1, and OR5M3 proteins when SIM-A9 cells were treated RJW-58 (5 µM) for 24 h. C, OLF-1 disassociated with CBP by RJW-58 for 24 h treatment in SIM-A9 cells. D, The effect of RJW-58 for 24 h on OLF-1 binding to the IL-10 promoter region in SIM-A9 cells was analyzed using ChIP. E, RJW-58 treatment facilitated IL-10 promoter activity in SIM-A9. IL-10 promoters were identified using the promoter assay. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05. The statistical significance was determined using a two-tailed Student's t-test. Data are presented as the mean ± SEM.
Discussion
Oxaliplatin, a platinum derivative, is widely used as first-line adjuvant therapy in advanced CRC [31]. Oxaliplatin can cause acute pain syndromes, which usually subside within days after cessation of treatment, and chronic distal neuropathy, which occurs after several cycles of treatment [32]. However, less is known regarding oxaliplatin-induced motor impairments from clinical studies or animal models. Our clinical study demonstrated that oxaliplatin could cause both sensory and motor impairments in patients receiving a standard dose and course of adjuvant chemotherapy with an oxaliplatin-containing regimen after the definitive operation (Figure 1). Furthermore, despite numerous animal models having been established to understand the mechanisms and explore the therapeutic strategies to treat or prevent oxaliplatin-induced peripheral neuropathy, most of these models were not fully relevant to clinical findings. Therefore, establishing an appropriate animal model is warranted. By administering oxaliplatin according to the treatment protocol illustrated in Figure 2B, we demonstrated that oxaliplatin treatment caused both sensory and motor neuropathies without reducing body weight in the mice model. Furthermore, oxaliplatin treatment significantly reduced the G-ratio of both large myelinated and small myelinated fibers, compared with the control, assessed using electron microscopy (Figure 3). These animal data further support that oxaliplatin can cause both sensory and motor impairments, which is consistent with clinical findings (Figures 1 and 2). Discrepancies between the findings in this study and other studies are mainly caused by different dosing schedules and cumulative doses of oxaliplatin [33, 34]. We demonstrated, using this animal model, that CTSS inhibition could protect animals from oxaliplatin-induced neuropathy, which was supported by the results from mouse behavioral tests, EM studies of sciatic nerves, and quantification of IENF density (Figures 2 and 3). The pivotal role of CTSS in oxaliplatin-induced neuropathy was further validated in the Ctss-/- knockout mouse model (Figure S1).
Lines of evidence have demonstrated that the ubiquitous second messenger Ca2+ plays a crucial role in fundamental physiological functions [35], including cell motility, apoptosis, cell cycle control, and gene expression. Under inflammations, dysregulation of intracellular Ca2+ homeostasis is a vital mechanism for regulating neurite outgrowth and subsequent neurite damage [36]. Our previous studies have revealed that SOCE components participate in modulating calcium homeostasis in response to inflammation and partly contribute to paclitaxel-induced neuropathies, which were also determined in the present study [11, 20]. A study demonstrated that oxaliplatin was degraded to oxalate and platinum inside the cell. Oxalate by chelating intracellular Ca2+ disrupts neuronal membrane potentials and further dysregulates the activity of voltage-gated ion channels. However, these disruptions only caused cold allodynia in mice [37, 38]. Furthermore, oxaliplatin treatment was reported to induce intracellular Ca2+ influx, but the mechanism remains unclear [39]. Our previous study has shown CTSS inhibition reduced Ca2+ influx through affecting SOCE activation and interfering STIM1 trafficking, which leads to the suppression of tumor cell progression. Furthermore, immunofluorescence data further demonstrated that CTSS could bind to STIM1, which was reversed by CTSS inhibitor. This effect was caused by accumulating STIM1 puncta in the ER, thereby reducing the interaction between active STIM1 and EB1 [14]. In the present study, we demonstrated that oxaliplatin treatment significantly increased the SOCE Ca2+ level after thapsigargin-induced ER store depletion, which was reversed by CTSS inhibition with RJW-58 (Figure 4H). Notably, we determined that oxaliplatin treatment increased CTSS expression by promoting IRF1 translocation from the cytosol to the nucleus, thereby enhancing IRF1 binding to the CTSS promoter region, which amplified the transcriptional level of CTSS (Figure 4A-E). Furthermore, chemotherapeutics stimuli may cause alterations of lysosomal membrane permeability and leakage of cathepsins into the cytoplasm [28]. These data explain why oxaliplatin could increase CTSS expression, causing an increase in binding to STIM1 (Figure 4I), indicating that the regulation of CTSS expression contributes to the effect of oxaliplatin on intracellular Ca2+ homeostasis.
Neuroinflammation is a hallmark of the pathogenesis of CIPN. Increasing evidence demonstrates that excessive inflammation in the damaged nervous tissue after chemotherapy contributes to the initiation and maintenance of peripheral neuropathies [36]. Oxaliplatin treatment increased the production and release of TNF and IL-1β through the upregulation of P2X purinoreceptor 7 in human neuroblastoma cell lines [40]. Furthermore, in vivo study revealed that oxaliplatin treatment causes an increased number of activated satellite glial cells in the spinal cord [41]. CTSS is reportedly an autocrine factor that stimulates microglial cell activation and subsequently regulates the release of proinflammatory cytokines and chemokines [13]. In the present study, we demonstrated that CTSS inhibition with RJW-58 increases the serum levels of anti-inflammatory cytokines (predominantly IL-10, IL-4, and IL-7) and suppresses proinflammatory cytokines (Figure 5A). This effect was further validated in an animal model, which demonstrates that oxaliplatin treatment results in the upregulation of TNF-α, IL-6, IL-1β, and MCP-1 in mice DRG cells, which is suppressed by cotreated with RJW-58 (Figure 5B). Studies have reported that exogenous administration of the anti-inflammatory cytokine IL-10 suppresses allodynia in surgery [9, 10] and chemotherapy-induced neuropathy [42]. Furthermore, increasing evidence suggests that STIM-dependent Ca2+ influx is required for IL-10 production because dysregulation of Ca2+ homeostasis impairs IL-10 secretion [43].
In this study, we determined that RJW-58 reversed oxaliplatin-induced dysregulation of Ca2+ homeostasis and increased endogenous IL-10 by activating the transcription factor Olf-1. We discovered that Olf-1 and transcriptional coactivators CBP coregulated the induction of IL-10 genes. Studies have indicated that the HAT activity of p300/CBP is involved in regulating transcription by remodeling chromatin, which is inhibited by binding with Olf-1 [30, 44]. In the ChIP experiment, oxaliplatin treatment facilitated the assembling of p300/CBP and Olf-1, whereas RJW-58 reversed this assembling and further drove IL-10 gene transcription by activating Olf-1. Furthermore, the elevation of endogenous IL-10 after RJW-58 treatment sequentially triggers the IL-10/STAT3 cascade. IL-10 is also involved in raising feedback inhibition, which limits the inflammation cascade. Therefore, IL-10 decreases NF-κB expression, which causes a reduction in the induction of inflammation in mouse DRG neurons. These findings suggest that RJW-58 reverses oxaliplatin-induces CTSS production, activates the anti-inflammatory IL-10/STAT3 cascade, and reduces proinflammatory cytokines expression, which results in the alleviation of CIPN.
Summary
This study highlights the novel role of CTSS in oxaliplatin-induced neurotoxicity, as evidenced by in vitro cell-line studies, in vivo OIPN mouse models, and clinical sample analysis. Blocking the enzymatic activity of lysosomal cathepsin S alleviates OIPN by regulating the STIM-dependent store-operated Ca2+ entry homeostasis. Furthermore, RJW-58, CTSS inhibitor, reverses oxaliplatin-induced CTSS production, activates the anti-inflammatory IL-10/STAT3 cascade, and reduces proinflammatory cytokines expression, which results in the alleviation of OIPN. RJW-58 might represent a potential therapeutic strategy for cancer patients with oxaliplatin-induced neuropathy.
Conclusions
We demonstrated that CTSS plays a critical role in the development of oxaliplatin-induced neuropathy. Targeting CTSS may reveal a therapeutic strategy to protect neuron cells from oxaliplatin-induced damage. Furthermore, the serum level of CTSS may serve as a biomarker for the risk of oxaliplatin-induced peripheral neuropathies. A proof-of-concept large cohort prospective clinical study is ongoing.
Abbreviations
ANOVA: analysis of variance; APCs: antigen-presenting cells; ATF3: activating transcription factor 3; BSA: bovine serum albumin; CTSS: cathepsin S; cDNA: complementary DNA; CRISPR/Cas9: clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9; CRC: colorectal cancer; CIPN: chemotherapy-induced peripheral neuropathy; ChIP: chromatin immunoprecipitation PCR; DMEM: Dulbecco's modified Eagle's medium; DNA: deoxyribonucleic acid; DRG: dorsal root ganglion; EDTA: ethylenediaminetetraacetic acid; ELISA: enzyme-linked immunosorbent assay; ER: endoplasmic reticulum; FACS: BD fluorescence-activated cell sorter; FBS: fetal bovine serum; FITC: fluorescein isothiocyanate; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; GFP: green fluorescent protein; h: hour; HBSS: Hanks' balanced salt solution; HRP: horseradish peroxidase; HEPES: 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid; IRF1: interferon response factor 1; IL-10: interleukin-10; IL-1β: interleukin-1β; IL-6: interleukin-6; IL-7: interleukin-7; i.p.: intraperitoneal(ly); IENF: intraepidermal nerve fiber; JAKs: Janus tyrosine kinases; m: meter; M: molar; mRNA: messenger RNA; MCP1: monocyte chemoattractant protein 1; NF-κB: nuclear factor-κB; NP-40: Nonidet P-40; NCV: nerve conduction velocity; NET: nerve excitability test; OIPN: oxaliplatin-induced peripheral neuropathy; Olf-1: olfactory neuronal transcription factor 1; PAGE: polyacrylamide gel electrophoresis; PBS: phosphate-buffered saline; PCR: polymerase chain reaction; P300/CBP: p300 and CREB-binding protein; QST: quantitative sensory test; R: receptor; RNA: ribonucleic acid; RT: reverse transcription; ROS: reactive oxygen species; s: second; SDS: sodium dodecyl sulfate; SEM: standard error of the mean; siRNA: small interfering RNA; STAT3: signal transducer and activator of transcription 3; STIM1: stromal interaction molecule 1; SOCE: stored-operated Ca2+ entry; TNF-α: tumor necrosis factor-α; TNS: total neuropathy score.
Supplementary Material
Supplementary figures.
Acknowledgements
The authors would like to thank the Bio-image Core Facility of the National Core Facility Program for Biotechnology, Ministry of Science and Technology, Taiwan, for technical services. We also thank the NHRI Microarray Core facility for their help with technique. This study was supported by the following grants: the National Health Research Institutes (CA-107-PP-22), the Ministry of Science and Technology (MOST 106-2314-B-006-076-MY3), the Ministry of Health and Welfare (MOHW 109-TDU-B-211-134018, MOHW110-TDU-B-211-144018), Higher Education Sprout Project, Ministry of Education to the Headquarters of University Advancement at National Cheng Kung University (NCKU).
Author Contributions
J.Y. Chang conceived and supervised this study. S.J. Chen, L.H. Chen, M.R. Shen, and J.Y. Chang designed the research. S.J. Chen, L.H. Chen, H.C. Chiang, C.C. Lee, Y.F. Lee developed methodology and performed the experiments. Y.M. Yeh, C.C. K Lin, P.C. Lin, H.W. Huang, M.R. Shen, B.W. Lin, J.C. Lee provided administrative or technical support. S.J. Chen, L.H. Chen, Y.M. Yeh and J.Y. Chang wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hill JM, Speer RJ. Organo-platinum complexes as antitumor agents (review). Anticancer Res. 1982;2:173-86
2. Izycki D, Niezgoda AA, Kazmierczak M, Piorunek T, Izycka N, Karaszewska B. et al. Chemotherapy-induced peripheral neuropathy - diagnosis, evolution and treatment. Ginekol Pol. 2016;87:516-21
3. Saif MW, Reardon J. Management of oxaliplatin-induced peripheral neuropathy. Ther Clin Risk Manag. 2005;1:249-58
4. Aromolaran KA, Goldstein PA. Ion channels and neuronal hyperexcitability in chemotherapy-induced peripheral neuropathy; cause and effect? Mol Pain. 2017;13:1744806917714693
5. Alberti P, Canta A, Chiorazzi A, Fumagalli G, Meregalli C, Monza L. et al. Topiramate prevents oxaliplatin-related axonal hyperexcitability and oxaliplatin induced peripheral neurotoxicity. Neuropharmacology. 2020;164:107905
6. Zareba P, Gryzlo B, Malawska K, Salat K, Hofner GC, Nowaczyk A. et al. Novel mouse GABA uptake inhibitors with enhanced inhibitory activity toward mGAT3/4 and their effect on pain threshold in mice. Eur J Med Chem. 2020;188:111920
7. Lees JG, Makker PG, Tonkin RS, Abdulla M, Park SB, Goldstein D. et al. Immune-mediated processes implicated in chemotherapy-induced peripheral neuropathy. Eur J Cancer. 2017;73:22-9
8. Zhang H, Li Y, de Carvalho-Barbosa M, Kavelaars A, Heijnen CJ, Albrecht PJ. et al. Dorsal Root Ganglion Infiltration by Macrophages Contributes to Paclitaxel Chemotherapy-Induced Peripheral Neuropathy. J Pain. 2016;17:775-86
9. Milligan ED, Langer SJ, Sloane EM, He L, Wieseler-Frank J, O'Connor K. et al. Controlling pathological pain by adenovirally driven spinal production of the anti-inflammatory cytokine, interleukin-10. Eur J Neurosci. 2005;21:2136-48
10. Shen KF, Zhu HQ, Wei XH, Wang J, Li YY, Pang RP. et al. Interleukin-10 down-regulates voltage gated sodium channels in rat dorsal root ganglion neurons. Exp Neurol. 2013;247:466-75
11. Chen LH, Yeh YM, Chen YF, Hsu YH, Wang HH, Lin PC. et al. Targeting interleukin-20 alleviates paclitaxel-induced peripheral neuropathy. Pain. 2020;161:1237-1254
12. Clark AK, Malcangio M. Microglial signalling mechanisms: Cathepsin S and Fractalkine. Exp Neurol. 2012;234:283-92
13. Barclay J, Clark AK, Ganju P, Gentry C, Patel S, Wotherspoon G. et al. Role of the cysteine protease cathepsin S in neuropathic hyperalgesia. Pain. 2007;130:225-34
14. Lin HH, Chen SJ, Shen MR, Huang YT, Hsieh HP, Lin SY. et al. Lysosomal cysteine protease cathepsin S is involved in cancer cell motility by regulating store-operated Ca(2+) entry. Biochim Biophys Acta Mol Cell Res. 2019;1866:118517
15. Nascimento Da Conceicao V, Sun Y, Zboril EK, De la Chapa JJ, Singh BB. Loss of Ca(2+) entry via Orai-TRPC1 induces ER stress, initiating immune activation in macrophages. J Cell Sci. 2019 133; jcs237610
16. Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science. 2017;356:513-9
17. Hutchins AP, Diez D, Miranda-Saavedra D. The IL-10/STAT3-mediated anti-inflammatory response: recent developments and future challenges. Brief Funct Genomics. 2013;12:489-98
18. Titos E, Rius B, Lopez-Vicario C, Alcaraz-Quiles J, Garcia-Alonso V, Lopategi A. et al. Signaling and Immunoresolving Actions of Resolvin D1 in Inflamed Human Visceral Adipose Tissue. J Immunol. 2016;197:3360-70
19. Huang HW, Wu PY, Su PF, Li CI, Yeh YM, Lin PC. et al. A Simplified Diagnostic Classification Scheme of Chemotherapy-Induced Peripheral Neuropathy. Dis Markers. 2020;2020:3402108
20. Chen YF, Chen LH, Yeh YM, Wu PY, Chen YF, Chang LY. et al. Minoxidil is a potential neuroprotective drug for paclitaxel-induced peripheral neuropathy. Sci Rep. 2017;7:45366
21. Inoue K, Tsuda M. Microglia in neuropathic pain: cellular and molecular mechanisms and therapeutic potential. Nat Rev Neurosci. 2018;19:138-52
22. Nieto FR, Clark AK, Grist J, Hathway GJ, Chapman V, Malcangio M. Neuron-immune mechanisms contribute to pain in early stages of arthritis. J Neuroinflammation. 2016;13:96
23. Clark AK, Yip PK, Grist J, Gentry C, Staniland AA, Marchand F. et al. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc Natl Acad Sci U S A. 2007;104:10655-60
24. Zhao P, Lieu T, Barlow N, Metcalf M, Veldhuis NA, Jensen DD. et al. Cathepsin S causes inflammatory pain via biased agonism of PAR2 and TRPV4. J Biol Chem. 2014;289:27215-34
25. Makker PG, Duffy SS, Lees JG, Perera CJ, Tonkin RS, Butovsky O. et al. Characterisation of Immune and Neuroinflammatory Changes Associated with Chemotherapy-Induced Peripheral Neuropathy. PLoS One. 2017;12:e0170814
26. Lauria G, Dacci P, Lombardi R, Cazzato D, Porretta-Serapiglia C, Taiana M. et al. Side and time variability of intraepidermal nerve fiber density. Neurology. 2015;84:2368-71
27. Pavan S, Olivero M, Cora D, Di Renzo MF. IRF-1 expression is induced by cisplatin in ovarian cancer cells and limits drug effectiveness. Eur J Cancer. 2013;49:964-73
28. Kramer L, Turk D, Turk B. The Future of Cysteine Cathepsins in Disease Management. Trends Pharmacol Sci. 2017;38:873-98
29. Zumerle S, Cali B, Munari F, Angioni R, Di Virgilio F, Molon B. et al. Intercellular Calcium Signaling Induced by ATP Potentiates Macrophage Phagocytosis. Cell Rep. 2019;27:1-10 e4
30. Zhao F, McCarrick-Walmsley R, Akerblad P, Sigvardsson M, Kadesch T. Inhibition of p300/CBP by early B-cell factor. Mol Cell Biol. 2003;23:3837-46
31. Alcindor T, Beauger N. Oxaliplatin: a review in the era of molecularly targeted therapy. Curr Oncol. 2011;18:18-25
32. Cassidy J, Misset JL. Oxaliplatin-related side effects: characteristics and management. Semin Oncol. 2002;29:11-20
33. Xiao WH, Zheng H, Bennett GJ. Characterization of oxaliplatin-induced chronic painful peripheral neuropathy in the rat and comparison with the neuropathy induced by paclitaxel. Neuroscience. 2012;203:194-206
34. Gadgil S, Ergun M, van den Heuvel SA, van der Wal SE, Scheffer GJ, Hooijmans CR. A systematic summary and comparison of animal models for chemotherapy induced (peripheral) neuropathy (CIPN). PLoS One. 2019;14:e0221787
35. Chen YF, Chen YT, Chiu WT, Shen MR. Remodeling of calcium signaling in tumor progression. J Biomed Sci. 2013;20:23
36. Sisignano M, Baron R, Scholich K, Geisslinger G. Mechanism-based treatment for chemotherapy-induced peripheral neuropathic pain. Nat Rev Neurol. 2014;10:694-707
37. Gamelin L, Capitain O, Morel A, Dumont A, Traore S, Anne le B. et al. Predictive factors of oxaliplatin neurotoxicity: the involvement of the oxalate outcome pathway. Clin Cancer Res. 2007;13:6359-68
38. Sakurai M, Egashira N, Kawashiri T, Yano T, Ikesue H, Oishi R. Oxaliplatin-induced neuropathy in the rat: involvement of oxalate in cold hyperalgesia but not mechanical allodynia. Pain. 2009;147:165-74
39. Schulze C, McGowan M, Jordt SE, Ehrlich BE. Prolonged oxaliplatin exposure alters intracellular calcium signaling: a new mechanism to explain oxaliplatin-associated peripheral neuropathy. Clin Colorectal Cancer. 2011;10:126-33
40. Massicot F, Hache G, David L, Chen D, Leuxe C, Garnier-Legrand L. et al. P2X7 Cell Death Receptor Activation and Mitochondrial Impairment in Oxaliplatin-Induced Apoptosis and Neuronal Injury: Cellular Mechanisms and In vivo Approach. PLoS One. 2013;8:e66830
41. LaPointe NE, Morfini G, Brady ST, Feinstein SC, Wilson L, Jordan MA. Effects of eribulin, vincristine, paclitaxel and ixabepilone on fast axonal transport and kinesin-1 driven microtubule gliding: implications for chemotherapy-induced peripheral neuropathy. Neurotoxicology. 2013;37:231-9
42. Binshtok AM, Wang H, Zimmermann K, Amaya F, Vardeh D, Shi L. et al. Nociceptors are interleukin-1beta sensors. J Neurosci. 2008;28:14062-73
43. Matsumoto M, Fujii Y, Baba A, Hikida M, Kurosaki T, Baba Y. The calcium sensors STIM1 and STIM2 control B cell regulatory function through interleukin-10 production. Immunity. 2011;34:703-14
44. Chan HM, La Thangue NB. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci. 2001;114:2363-73
Author contact
![]() Corresponding author: Dr. Jang-Yang Chang, Department of Internal Medicine, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, Tainan, Taiwan. E-mail: jychangedu.tw.
Corresponding author: Dr. Jang-Yang Chang, Department of Internal Medicine, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, Tainan, Taiwan. E-mail: jychangedu.tw.