Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(10):4825-4838. doi:10.7150/thno.55330 This issue Cite
Research Paper
SIRT2 ablation inhibits glucose-stimulated insulin secretion through decreasing glycolytic flux
Feiye Zhou1*, Linlin Zhang1*, Kecheng Zhu1, Mengyao Bai1, Yuqing Zhang2, Qin Zhu1, Shushu Wang1, Chunxiang Sheng1, Miaomiao Yuan1, Yun Liu1, Jieli Lu1, Li Shao3 ![]() , Xiao Wang1
, Xiao Wang1 ![]() , Libin Zhou1
, Libin Zhou1 ![]()
1. Department of Endocrine and Metabolic Diseases/ Shanghai institute of Endocrine and Metabolic Diseases, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China.
2. Center for Reproductive Medicine, Shandong University, Jinan 250000, China.
3. Department of VIP Clinic, Shanghai East Hospital, Tongji University School of Medicine, Shanghai 200123, China.
* These authors contributed equally to this study
Received 2020-11-2; Accepted 2021-2-6; Published 2021-3-4
Abstract

Rationale: Sirtuins are NAD+-dependent protein deacylases known to have protective effects against age-related diseases such as diabetes, cancer, and neurodegenerative disease. SIRT2 is the only primarily cytoplasmic isoform and its overall role in glucose homeostasis remains uncertain.
Methods: SIRT2-knockout (KO) rats were constructed to evaluate the role of SIRT2 in glucose homeostasis. The effect of SIRT2 on β-cell function was detected by investigating the morphology, insulin secretion, and metabolomic state of islets. The deacetylation and stabilization of GKRP in β-cells by SIRT2 were determined by western blot, adenoviral infection, and immunoprecipitation.
Results: SIRT2-KO rats exhibited impaired glucose tolerance and glucose-stimulated insulin secretion (GSIS), without change in insulin sensitivity. SIRT2 deficiency or inhibition by AGK2 decreased GSIS in isolated rat islets, with lowered oxygen consumption rate. Adenovirus-mediated overexpression of SIRT2 enhanced insulin secretion from rat islets. Metabolomics analysis revealed a decrease in metabolites of glycolysis and tricarboxylic acid cycle in SIRT2-KO islets compared with control islets. Our study further demonstrated that glucokinase regulatory protein (GKRP), an endogenous inhibitor of glucokinase (GCK), was expressed in rat islets. SIRT2 overexpression deacetylated GKRP in INS-1 β-cells. SIRT2 knockout or inhibition elevated GKRP protein stability in islet β-cells, leading to an increase in the interaction of GKRP and GCK. On the contrary, SIRT2 inhibition promoted the protein degradation of ALDOA, a glycolytic enzyme.
Conclusions: SIRT2 ablation inhibits GSIS through blocking GKRP protein degradation and promoting ALDOA protein degradation, resulting in a decrease in glycolytic flux.
Keywords: SIRT2, islets, acetylation, glucokinase regulatory protein, glucose-stimulated insulin secretion, glycolysis.
Introduction
Protein lysine acetylation is a reversible post-translational modification that links acetyl-coenzyme A metabolism with cellular signaling [1]. Recent advances in proteomics and mass spectrometry technology reveal that both histones and non-histone proteins are highly acetylated and constitute a major portion of the acetylome in mammalian cells [2]. Indeed, acetylation is involved in key cellular processes, such as gene regulation, cell cycle, signal transduction, protein folding, autophagy, and metabolism [3]. Notably, the majority of identified acetylated proteins are enzymes involved in energy metabolism, including glycolysis, tricarboxylic acid (TCA) cycle, electron transport chain, and fatty acid β oxidation [2, 4]. This acetylation modification of metabolic proteins and enzymes occurs in response to altered nutrient status [5, 6], which regulates the balance between energy storage and expenditure [7]. Pancreatic β-cells, extremely sensitive to nutrients alterations, secrete insulin at appropriate rates to maintain blood glucose levels within a relatively narrow range. Our recent study provides a comprehensive picture of protein acetylation in rat islets [8]. But the direct effect of acetylation on islets function remains largely unknown.
Lysine acetyltransferases (KATs) and class I, II, III, and IV lysine deacetylases (KDACs) are responsible for reversible changes in protein acetylation status [9]. Class III KDACs, termed sirtuins (SIRT1-SIRT7), require NAD+ as a substrate and keep highly conserved from bacteria to humans [10]. Since the concentration of NAD+ is determined by the cellular nutritional state, sirtuins are considered to be metabolic and stress-sensor proteins that play important roles in energetic regulation. The deacetylase SIRT1, located in cell nuclei, is involved in carbohydrate and lipid metabolism by regulating key factors [11]. SIRT1 activation ameliorates hyperglycemia by promoting insulin secretion and β-cell expansion [12, 13]. SIRT3, SIRT4, and SIRT5 are mitochondrial sirtuins. SIRT3 targets key enzymes in fatty acid β oxidation pathway and regulates palmitate-stimulated insulin secretion [8, 14]. SIRT4 regulates amino acid catabolism and maintains insulin secretion and glucose homeostasis during aging [15, 16]. SIRT6 is also a predominantly nuclear isoform and has been implicated in a variety of metabolic processes [17]. Pancreatic β-cell-specific SIRT6 knockout mice showed a significant decrease in glucose-stimulated insulin secretion (GSIS) [18].
Although SIRT2 is expressed in a wide range of tissues and organs, SIRT2 is still one of the least understood sirtuins. SIRT2 possesses efficient deacetylation activity and is the only primarily cytoplasmic isoform, which can also be found in the nucleus and mitochondria [19, 20]. Previous studies for SIRT2 mainly focus on tumorigenesis and neurodegenerative diseases [21]. Recently, accumulating evidence implicates the involvement of SIRT2 in various metabolic processes, including adipocyte differentiation, gluconeogenesis, and insulin sensitivity as well as inflammation [22]. However, little is known about the role of SIRT2 in islet function.
In the present study, we generated SIRT2-knockout (KO) rats to determine its impact on glucose homeostasis. Impaired glucose tolerance and GSIS were observed in SIRT2-ablated rats. Metabolomics and oxygen consumption measurements revealed the involvement of glycolysis in SIRT2-regulated insulin secretion, in which SIRT2 inhibition decreased glucose-induced metabolic flux via stabilizing glucokinase regulatory protein (GKRP) and increasing its binding to glucokinase (GCK).
Methods
Experimental animals
Male Sprague-Dawley (SD) rats were purchased from Shanghai Laboratory Animal Company. All rats were group-housed in a barrier facility, receiving a regular chow diet ad libitum.
CRISPR/Cas9-mediated SIRT2 knockout rats
For SIRT2 targeting, two sgRNAs were designed to target a region upstream of exon4 and downstream of exon7. sgRNAs were screened for on-target activity using the UCA kit and were successfully transcribed in vitro. Then Cas9/sgRNA plasmid and donor vector construction were completed. Cas9 mRNA, sgRNAs, and donor vector were mixed at proper concentrations and co-injected into the cytoplasm of fertilized eggs at one-cell stage. Surviving zygotes were transferred into the oviducts of pseudopregnant SD female rats (Beijing Biocytogen). Offspring rats were maintained on a Sprague-Dawley background and normal chow.
Glucose and insulin tolerance tests
For glucose tolerance test (GTT), 8-week-old rats were injected with 2 g/kg body weight of glucose intraperitoneally after initial measurement of 16-h fasting blood glucose. Blood glucose concentrations were measured from the tail vein using a portable blood glucose meter (Lifescan, Johnson &Johnson) at 0, 15, 30, 60, and 120 min after the glucose injection. For insulin tolerance test (ITT), rats were injected with 0.75 IU/kg body weight of insulin intraperitoneally after initial measurement of 6-h fasting blood glucose. Blood glucose concentrations were measured at 0, 15, 30, 60, and 120 min after the insulin injection. Plasma was collected for insulin assay using ELISA kits (Mercodia). Total pancreatic insulin content was determined by radioimmunoassay, in which acidified ethanol extractions were performed on whole pancreases.
Islet isolation and insulin secretion assay
Islets of Langerhans were isolated from 8- to 12-week-old SD male rats by collagenase digestion and density-gradient centrifugation as described previously [8]. Before insulin secretion assay, isolated islets were cultured in RPMI 1640 medium (GIBCO) containing 5% fatal bovine serum (FBS, GIBCO) and 5 mM glucose for 4-6 h at 37 °C. Then, these islets were preincubated in Krebs-Ringer Buffer (KRB) containing 0.25% bovine serum albumin (BSA) and 3.3 mM glucose for 30 min. Ten islets per assay in triplicate were incubated with KRB buffer containing either various concentrations of glucose or the indicated reagents for a further 1 h at 37 °C. Supernatants containing insulin were collected for analysis. Insulin secretion was normalized to insulin content extracted with acidified ethanol. Insulin levels of all samples were measured by ELISA kit (Mercodia).
Cell culture
INS-1 cells were cultured in RPMI 1640 medium containing 11.1 mM glucose, 10 mM HEPES, 10% FBS, 100 U/ml penicillin G sodium and 100 μg/ml streptomycin sulfate.
Immunofluorescence staining
Pancreases were dissected, weighed, fixed in 4% formalin overnight at 4 °C, washed with phosphate buffered saline (PBS), and embedded in paraffin. The pancreatic sections were stained as previously described [23] using SIRT2 (Abcam, ab67299), insulin (Abcam, ab7842), and glucagon (Abcam, ab10988) antibodies. For α/β cell number statistics, 7-10 5 μm sections of pancreas (paraffin embedded, n=3 per genotype), separated by at least 200 μm, were co-stained for insulin and glucagon. The number of α/β cells per islet on the section level was determined by counting the glucagon+ /insulin+ cells manually.
INS-1 cells were fixed for 20 min in 4% formalin, permeabilized in 0.1% Triton×100 for 5 min, washed with PBS, and blocked in 5% BSA for 1 h. Cells were then incubated with anti-GKRP antibody (1: 100) overnight at 4 °C and stained with FITC-labeled IgG (1: 200, Jackson ImmunoResearch Laboratories). DAPI was added to stain cell nuclei. The cellular localization of GKRP was photographed and analyzed using a fluorescence microscope (LSM 880 with Airyscan, ZEISS).
Adenovirus infection
For SIRT2/GKRP overexpression, vector adenovirus and adenovirus encoding wild-type rat SIRT2 or GKRP were transfected into islets or INS-1 cells for 48 h according to the manufacturer's instructions. The recombinant adenoviruses were generated by GeneChem (Shanghai, China).
Western blotting and immunoprecipitation
Islets or cells were homogenized in RIPA buffer (20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 1 mM Na3VO4) containing protease inhibitor and phosphatase inhibitor (MCE). Protein lysates were separated by SDS-PAGE on 10% polyacrylamide gels and transferred to PVDF membranes (Millipore). Primary antibodies were detected with horseradish peroxidase (HRP)-conjugated secondary antibodies. Blotted membrane was imaged with a LAS-4000 Super CCD Remote Control Science Imaging System (Fuji). Anti-Flag was from Sigma-Aldrich. Anti-tubulin was from Proteintech. Anti-ATP5A, anti-UQCRC2, anti-MTCO1, anti-SDHB, and anti-rabbit/mouse IgG conjugated with HRP were purchased from Cell Signaling Technology. Anti-GCK, anti-GKRP, and anti-ALDOA were from Santa Cruz biotechnology. Anti-phosphoenolpyruvate carboxykinase (PEPCK) was from Abcam. Anti-HSP90 was from Millipore. Anti-acetyllysine was from PTM Biolab. Immunoprecipitation was performed by incubating protein lysates with the indicated antibodies for 2 h and then with protein A/G-agarose beads (Santa Cruz biotechnology) overnight at 4 °C. The immunoprecipitates were extensively washed with lysis buffer and eluted with SDS loading buffer by boiling for 10 min. Then, standard western blotting was followed.
Real-time quantitative PCR
Total RNA was extracted from islets or cells by Trizol (Thermo Fisher). To quantify the transcript abundance of genes of interest, real-time quantitative PCR (RT-qPCR) was performed by using SYBR Green Premix Ex Taq (Takara) with an Applied Biosystems 7300 Real-Time PCR machine (Applied Biosystems).
Islet metabolomics
The untargeted metabolomics profiling was performed on XploreMET platform (Metabo-Profile, Shanghai, China). For samples preparation, isolated islets from SIRT2-WT and SIRT2-KO male rats were incubated in KRB buffer with 16.7 mM glucose for 1 h. Then, these islets were washed twice with PBS and stored at -80 °C freezer before metabolomics analysis. A total of 1,000 islets from one genotype were pooled to create a uniform sample. Untargeted metabolomics samples were collected from six independent biological replicates.
Oxygen consumption rate measurement
Islet oxygen consumption measurements were conducted on a Seahorse XFe24 analyser (Agilent Technologies). Islets were placed on a 24-well islet plate in XF Base Media (50 islets per well) with 3 mM glucose and equilibrated at 37 °C for 1 h in a CO2-free incubator. During the measurement of glucose-induced respiration, islets were treated with different concentrations of glucose and subsequently treated with carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP, 5 µM) at the indicated time point. For glucose-induced ATP-coupled OCR, islets were treated with 16.7 mM glucose and 5 µM oligomycin in succession. Each analysis was replicated five times.
Mitochondrial area and morphology analysis
INS-1 cells were seeded in a 96-well plate and subsequently treated with or without AGK2 for 4 h. Immunofluorescence dye Mito Tracker and hoechst were added into medium 15 min before the 96-well plate was moved into PerkinElmer Operetta CLS to obtain confocal images and the analysis of mitochondrial area and morphology.
Statistics
Data were expressed as mean ± SEM. Comparisons were performed using ANOVA for multiple groups or the Student's t-test for two groups. Statistical significance was established at P< 0.05.
Results
Deletion of SIRT2 impairs glucose tolerance and insulin secretion in rats
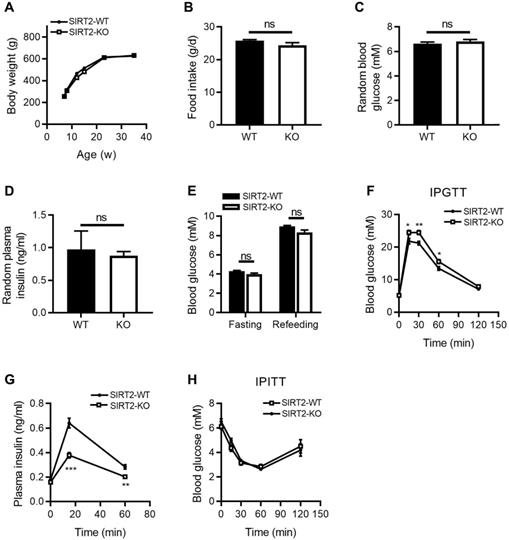
To investigate the metabolic role of SIRT2 in vivo, we characterized the metabolic phenotypes of SIRT2-KO rats, in which the gene encoding SIRT2 has been genetically disrupted by CRISPR/Cas9 system (Figure S1). The body weight (Figure 1A) and food intake (Figure 1B) were comparable between SIRT2-KO and wild-type (WT) male rats. No differences were observed in random blood glucose and insulin levels between two groups (Figure 1C-D), neither was fasting blood glucose level (Figure 1E). However, 2-month-old SIRT2-KO male rats exhibited glucose intolerance (Figure 1F) and decreased insulin secretion in response to glucose challenge (Figure 1G) compared with WT rats. Knockout of SIRT2 did not alter insulin sensitivity according to insulin tolerance test (Figure 1H). SIRT2-KO female rats showed a similar phenotype (Figure S2). SIRT2 has been demonstrated to regulate hepatic gluconeogenesis via destabilizing PEPCK protein [24]. However, our study revealed no significant differences in hepatic PEPCK mRNA and protein expressions at fasting and refeeding statuses between SIRT2-KO and WT male rats (Figure S3A-B), with comparable mRNA expressions of other gluconeogenic genes G6pase, Fbpase, and Foxo1 (Figure S3C). Consistent with the in-vivo result, SIRT2 inhibition by AGK2 had no impact on PEPCK protein level in primary mouse hepatocytes (Figure S3D).
Metabolic phenotypes of SIRT2-KO male rats. (A) Body weight curves of SIRT2-WT and SIRT2-KO rats (n=8). (B) Food intake in SIRT2-WT and SIRT2-KO rats (n=6). (C) Random blood glucose of 10-week-old SIRT2-WT and SIRT2-KO rats (n=7). (D) Random plasma insulin of SIRT2-WT and SIRT2-KO rats (n=6). (E) Fasting blood glucose was measured after 16-h fasting (n=6). (F) Blood glucose excursions during a 2 g/kg i.p. glucose tolerance test (n=13). (G) Plasma insulin levels during glucose tolerance test (n=5). (H) Insulin tolerance test of SIRT2-WT and SIRT2-KO rats (n=11). Data are expressed as means ± SEM. *P< 0.05, **P< 0.01, ***P<0.001 vs WT rats.
Morphology of islets and pancreatic insulin content in SIRT2 knockout rats
Similar to other sirtuin family members, the tissue distribution of SIRT2 is ubiquitous. SIRT2 has been detected in a number of tissues and organs, including liver, brain, muscle, kidney, colon, and adipose tissue [25-29]. To explore the function of SIRT2 in islet, we firstly observed the abundance of SIRT2 in normal islet and found that its mRNA level was more than twice as much as SIRT1 (Figure 2A), which is the most extensively characterized family member. SIRT2 was mainly located in the cytoplasm of islet cells (Figure 2B) as in other tissue cells [22]. Elevated glucose did not lead to a significant decrease in SIRT2 expression (Figure S4).
Morphology of islet and pancreatic insulin content in SIRT2-KO rats. (A) The mRNA expression of sirtuin family in normal islets (n=3). (B) Representative examples of normal islets stained by immunofluorescence for SIRT2 (red), insulin (green), and nuclei (DAPI, blue) (bar=40 μm). (C) Immunoblotting of SIRT2 in islets isolated from SIRT2-WT and SIRT2-KO rats. (D) Total pancreatic insulin content normalized to total protein (n=4-5). (E) Pancreatic weight of SIRT2-WT and SIRT2-KO rats (n=4-5). (F) Histological analysis of pancreas isolated from SIRT2-WT and SIRT2-KO rats. Hematoxylin-eosin staining samples were photographed at 200 magnification (bar=0.5 mm). (G) Representative islet of SIRT2-WT and SIRT2-KO rats stained by immunofluorescence for insulin (green), glucagon (red), and nuclei (DAPI, blue) (bar=40 μm). (H-I) Average number of β cells and α cells per islet section (n=3). Data are expressed as means ± SEM.
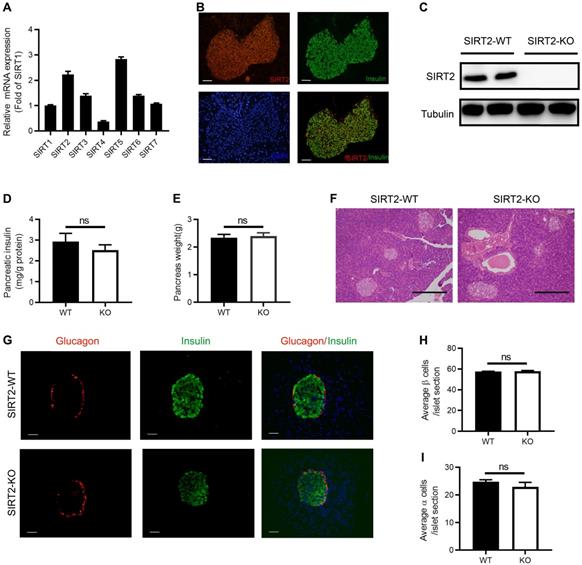
We further investigated the impact of SIRT2 knockout on pancreatic insulin content and islet morphology. Western blotting confirmed the knockout of SIRT2 gene in the islets isolated from SIRT2-KO male rats (Figure 2C). There was no significant difference in the total pancreatic insulin content (Figure 2D) or pancreas weight (Figure 2E) between SIRT2-KO and WT male rats. Islet morphology, assessed by hematoxylin and eosin (H&E) staining of pancreas sections (Figure 2F) and by immunofluorescence staining for insulin and glucagon (Figure 2G), was not appreciably altered in the SIRT2-KO rats compared with WT rats, without significant change in the average number of β-cells or α-cells per islet section (Figure 2H-I). Therefore, it is reasonable to suppose that the decreased GSIS is mainly attributed to the impaired islet secretory function.
Knockdown or inhibition of SIRT2 decreases glucose-stimulated insulin secretion
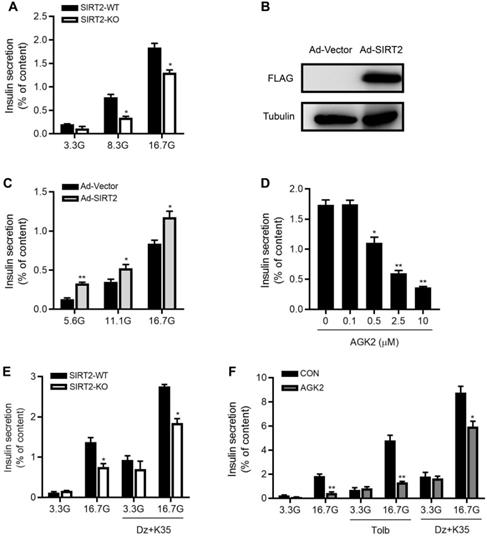
To directly evaluate the effect of SIRT2 knockout on islet function, we isolated male rat islets and performed static incubation assays. Compared with control islets, insulin secretion was decreased by 56% and 29% in SIRT2-KO islets in response to 8.3 and 16.7 mM glucose (Figure 3A). After SIRT2-overexpressing adenovirus (Ad-SIRT2) was transfected into isolated WT rat islets (Figure 3B), insulin secretion was enhanced at 5.6, 11.1 and 16.7 mM glucose (Figure 3C). The pharmacological inhibition of SIRT2 by AGK2 also led to reduced GSIS in a dose-dependent manner, with a significant action at the concentration of 0.5 μM (Figure 3D). It has been widely accepted that glucose stimulates insulin secretion via generating triggering and amplifying signals in β-cells [30]. We investigated whether the amplifying pathway was involved in SIRT2 deficiency-inhibited insulin secretion by treating islets with either 3.3 mM or 16.7 mM glucose in the presence or absence of KCl (35 mM) and the KATP channel activator diazoxide (250 μM). At 3.3 mM glucose, no difference was observed in insulin secretion between two groups. At 16.7 mM glucose, SIRT2-KO islets displayed a significant decrease in insulin secretion compared with control islets (Figure 3E). This was the case in AGK2-treated islets (Figure 3F), in which a high concentration of sulfonylurea tolbutamide (500 μM) was also used to explore the effect of SIRT2 on amplifying pathway. These results indicate that the amplifying pathway is involved in the regulation of SIRT2 on islet function.
SIRT2 promotes glucose-stimulated insulin secretion in rat islets. (A) Islets isolated from SIRT2-WT and SIRT2-KO rats were stimulated with 3.3, 8.3 or 16.7 mM glucose for 1 h, and insulin secretion was assayed (n=4). (B) Protein level of FLAG-SIRT2 in rat islets transfected with control vector (Ad-Vector) or SIRT2-overexpressing adenovirus (Ad-SIRT2). (C) After transfected with Ad-Vector or Ad-SIRT2 adenovirus, islets isolated from normal rats were stimulated with 5.6, 11.1 or 16.7 mM glucose for 1 h, and insulin secretion was assayed (n=4). (D) Islets isolated from normal rats were stimulated with various concentrations of AGK2 at 16.7 mM glucose for 1 h, and insulin secretion was assayed (n=4). (E) Islets isolated from SIRT2-WT and SIRT2-KO rats were stimulated with 3.3 or 16.7 mM glucose for 1 h. The amplifying pathway of GSIS was revealed by addition of 250 μM diazoxide (Dz) and 35 mM KCl (K35) to islets (n=4). (F) Islets isolated from normal rats were incubated with or without AGK2 (3 μM) in the presence of 3.3 or 16.7 mM glucose for 1 h. The amplifying pathway of GSIS was revealed by using 500 μM tolbutamide (Tolb) or 250 μM diazoxide plus 35 mM KCl (n=4). Data are expressed as means ± SEM. *P< 0.05, **P< 0.01 vs control group.
Metabolomic state of SIRT2-ablated islets
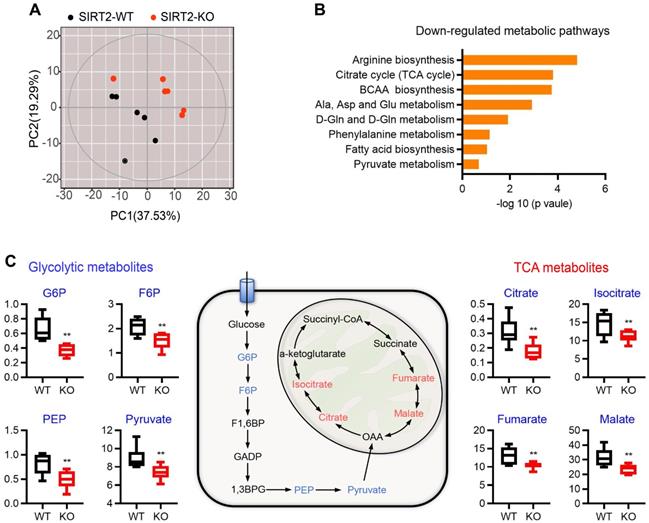
Pancreatic islets secrete insulin in response to nutrient metabolism. In addition to triggering signal, a number of glucose-derived metabolites influence insulin secretion through the metabolic amplifying pathway [31, 32]. Therefore, untargeted metabolomics analysis was performed on islets from WT and SIRT2-KO male rats to gain insight into the molecular pathways regulated by SIRT2. A total of 201 metabolites were detected and 125 of them were annotated with mammalian metabolite database JiaLibTM using a strict matching algorithm incorporated in XploreMET software that uses both retention times and fragmentation patterns in the mass spectrum. The annotated metabolites were distributed into several chemical classes, including amino acid, organic acids, carbohydrates, and fatty acids. The overview of global metabolic profiles, as revealed by PCA scores plots (Figure 4A), elucidated overall profile similarities and dissimilarities between the two genotypes. Visualization of differential metabolite profiles was shown in volcano-plot (Figure S5). A total of 46 metabolites (located in the left side of volcano-plot, VIP≥ 1, p< 0.1) were decreased in SIRT2-deleted islets, while only 12 metabolites were increased (located in the right side of volcano-plot, VIP≥ 1, p< 0.1). Pathway analysis revealed that many metabolic pathways were significantly downregulated in SIRT2-KO islets, including arginine biosynthesis, TCA cycle, branched-chain amino acid (BCAA) biosynthesis, alanine/aspartate/glutamate metabolism, and pyruvate metabolism (Figure 4B). We further analyzed metabolomics results and found that glucose 6-phosphate (G6P) in SIRT2-KO islets was significantly reduced compared with control islets. G6P is derived by the phosphorylation of glucose catalyzed by the rate-limiting enzyme GCK, which is the first step of glycolysis. Besides G6P, other glycolysis intermediates such as fructose 6-phosphate (F6P), phosphoenolpyruvate, and pyruvate were also decreased by SIRT2 ablation, without increased intermediates of glycolysis. In addition, many metabolic intermediates among TCA cycle are involved in the regulation of GSIS [33]. In our study, some TCA intermediates (citrate, isocitrate, fumarate, and malate) were reduced in SIRT2-KO islets while no annotated intermediates of TCA cycle were found to be significantly elevated. The change in these intermediates on a schematic of glycolysis and TCA cycle (Figure 4C) suggests that SIRT2 knockout lowers glucose-induced metabolic flux involved in GSIS.
Metabolomic change of islets isolated from SIRT2-KO rats. (A) Overview of metabolic profiles. Metabolomics data were obtained from islet extracts (n=6). (B) Main down-regulated metabolic pathways obtained from differential metabolites of SIRT2-KO islets compared with SIRT2-WT islets. (C) Schematic representation of intermediates in glycolysis and TCA cycle. The relative levels of indicated metabolites are shown on both sides. Data are expressed as means ± SEM. *P< 0.05, **P< 0.01 vs WT rats.
SIRT2 knockout or inhibition lowers glucose-stimulated oxygen consumption rate (OCR) in rat islets
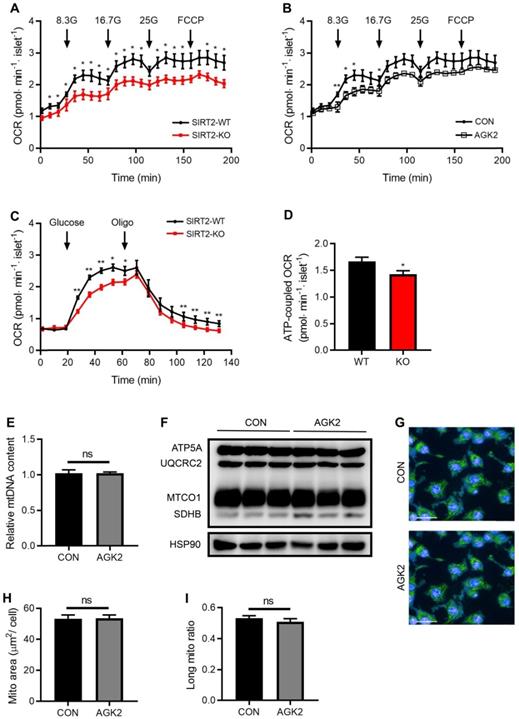
In consistent with decreased intermediates of glycolysis and TCA cycle by SIRT2 deletion, glucose supplementation-stimulated OCR was markedly decreased in SIRT2-KO islets (Figure 5A). SIRT2 inhibition by AGK2 treatment also led to a similar result in the oxygen consumption of rat islets (Figure 5B). In addition, glucose-induced mitochondrial ATP-coupled OCR was significantly reduced in SIRT2 deficient islets (Figure 5C-D). A critical role of the mitochondria is to generate ATP from ADP and organophosphate via oxidative phosphorylation. Previous study demonstrates that SIRT2 functions as a regulator of autophagy/mitophagy to maintain mitochondrial biology in mouse neurons and embryonic fibroblasts [20]. We further investigated whether SIRT2 exerted a similar action in β-cell mitochondria. mtDNA content and protein levels of TCA-related enzymes (ATP5A, UQCRC2, MTCO1, and SDHB) showed no changes in INS-1 cells after AGK2 treatment (Figure 5E-F). In addition, the mitochondrial morphology in INS-1 cells was observed using MitoTracker staining. AGK2 treatment had no significant impact on mitochondrial morphology as shown in Figure 5G. Further analysis demonstrated that the mean mitochondrial area and ratio of long mitochondria in INS-1 cells were comparable between the two groups (Figure 5H-I). Therefore, it is likely that SIRT2-regulated OCR on glucose stimulation is independent of mitochondrial function.
SIRT2 knockout or inhibition decreases glucose-stimulated OCR. (A) Oxygen consumption rate (OCR) in SIRT2-WT and SIRT2-KO islets stimulated with 8.3 mM, 16.7 mM, 25 mM glucose, and 5 µM FCCP (n=5). (B) OCR was measured in the presence of 8.3 mM, 16.7 mM, 25mM glucose or 5 µM FCCP after rat islets were pretreated with 3 μM AGK2 (n=5). (C) OCR in SIRT2-WT and SIRT2-KO islets supplemented with 16.7 mM glucose and 5 µM oligomycin (n=5). (D) ATP-coupled OCR was determined by the difference between the 16.7 mM glucose-stimulated OCR and the OCR after adding oligomycin in C (n=5). (E) mtDNA content in INS-1 cells stimulated with 3 μM AGK2 for 12 h (n=3). (F) Immunoblotting of mitochondrial genes including ATP5A, UQCRC2, MTCO1, and SDHB was detected in INS-1 cells. (G) INS-1 cells were stained with MitoTracker (green) as well as Hoechst (blue), and representative IFC images were shown (bar=20 μm). (H-I) Mitochondrial area and the ratio of long mitochondria in INS-1 cells treated with 3 μM AGK2 for 4 h. Data are expressed as means ± SEM. *P< 0.05, **P< 0.01 vs control group.
SIRT2 inhibition increases GKRP protein level and its interaction with GCK
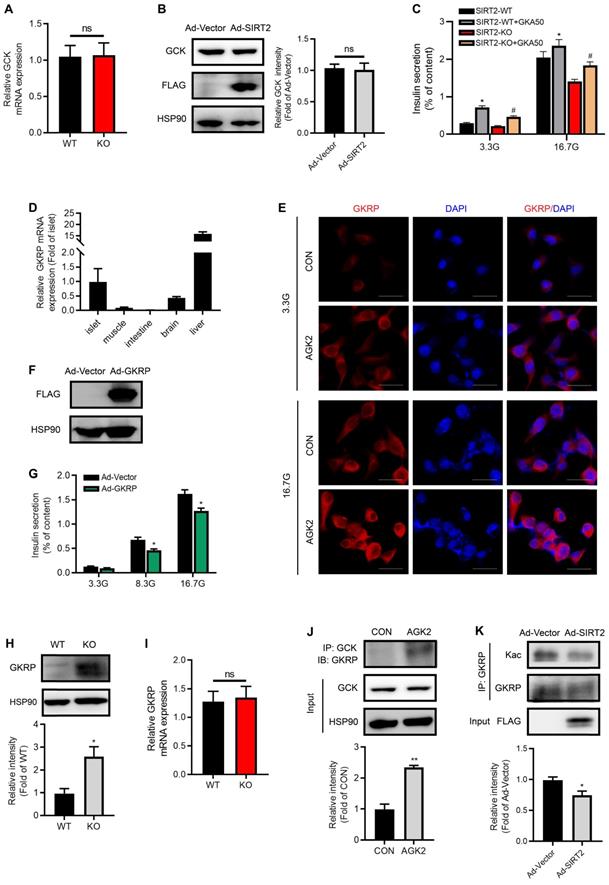
We further sought to explore SIRT2 effectors in the process of glycolysis. In spite of low G6P level observed in SIRT2-KO islets, no significant changes for GCK mRNA and protein expressions were observed after SIRT2 ablation or overexpression (Figure 6A-B). These results drew our attention to the activity of GCK. As shown in Figure 6C, the glucokinase activator GKA50 antagonized SIRT2 deficiency-impaired GSIS, suggesting that SIRT2 may regulate GCK activity so as to control the first step in glycolysis after glucose enters β-cells. GKRP has been demonstrated to inhibit the GCK activity competitively with the substrate glucose [34, 35]. Previous study reported that GKRP was expressed only in liver [36]. Our study showed that GKRP mRNA level in islet was much less than that in liver, but more than twice as much as that in brain (Figure 6D), where the expression of GKRP was previously proved [37]. GKRP expression was hardly detectable in muscle and intestine (Figure 6D). In INS-1 β-cell line, GKRP protein was mainly located in the cytoplasm at both low and high glucose concentrations as shown by immunofluorescence staining (Figure 6E), inconsistent with its distribution in hepatocytes, where GKRP binds GCK and sequesters the enzyme to the nucleus at low glucose. Elevated glucose concentration leads to the disruption of GKRP-GCK complex, enabling the migration of GCK and GKRP to the cytoplasm [38]. AGK2 increased the abundance of GKRP protein in INS-1 cells at both 3.3 and 16.7 mM glucose (Figure 6E). Insulin secretory response to glucose stimulation was significantly reduced by GKRP overexpression (Figure 6F-G). Similarly, GKRP protein expression in SIRT2-KO islets was much higher than that in control islets (Figure 6H). However, the change in GKRP mRNA expression level was not observed (Figure 6I), indicating a possibility that SIRT2 regulates GKRP protein expression at posttranslational level. Similar to the result in hepatocytes previously described [39], the interaction of GCK with GKRP was enhanced by the treatment of AGK2 (Figure 6J). GKRP has been reported to be a direct target of SIRT2 in mice liver [39]. We wondered whether its acetylation was also regulated by SIRT2 in islet β-cells. Adenovirus-mediated SIRT2 overexpression significantly decreased GKRP acetylation level in INS-1 cells (Figure 6K). Collectively, SIRT2 knockout or inhibition markedly enhances GKPR protein expression and its interaction with GCK in islets, leading to a decrease in GCK activity.
SIRT2 inhibition stabilizes GKRP protein and enhances its binding to GCK. (A) mRNA levels of GCK in the islets isolated from SIRT2-WT and SIRT2-KO rats (n=3). (B) Protein expression of GCK in INS-1 cells transfected with Ad-SIRT2 or Ad-Vector adenovirus for 36 h. (C) Islets isolated from SIRT2-WT and SIRT2-KO rats were incubated with 3 µM GKA50 in the presence of 3.3 or 16.7 mM glucose for 1 h, and insulin secretion was assayed (n=4). (D) mRNA levels of GKRP in different tissues (n=6). (E) INS-1 cells treated with 3 μM AGK2 for 6 h in the presence of 3.3 or 16.7 mM glucose were stained by immunofluorescence for GKRP (red) and nuclei (DAPI, blue) (bar=20 μm). (F) Protein level of GKRP in rat islets transfected with Ad-Vector or GKRP-overexpressing adenovirus (Ad-GKRP). (G) Rat islets transfected with Ad-Vector or Ad-GKRP adenovirus were stimulated with 3.3, 8.3 or 16.7 mM glucose for 1 h, and insulin secretion was assayed (n=4). (H-I) Protein and mRNA expressions of GKRP in the islets isolated from SIRT2-WT and SIRT2-KO rats (n=3). (J) The interaction between GCK and GKRP was detected in INS-1 cells treated with 3 μM AGK2 for 6 h. (K) After INS-1 cells were transfected with Ad-Vector or Ad-SIRT2 adenovirus, cell lysates were immunoprecipitated with GKRP antibody and subjected to Western blot with anti-acetyllysine antibody. Data are expressed as means ± SEM. *P< 0.05, **P< 0.01 vs control group. #P< 0.05 vs SIRT2-KO group.
SIRT2 inhibition promotes GKRP protein stability and ALDO protein degradation
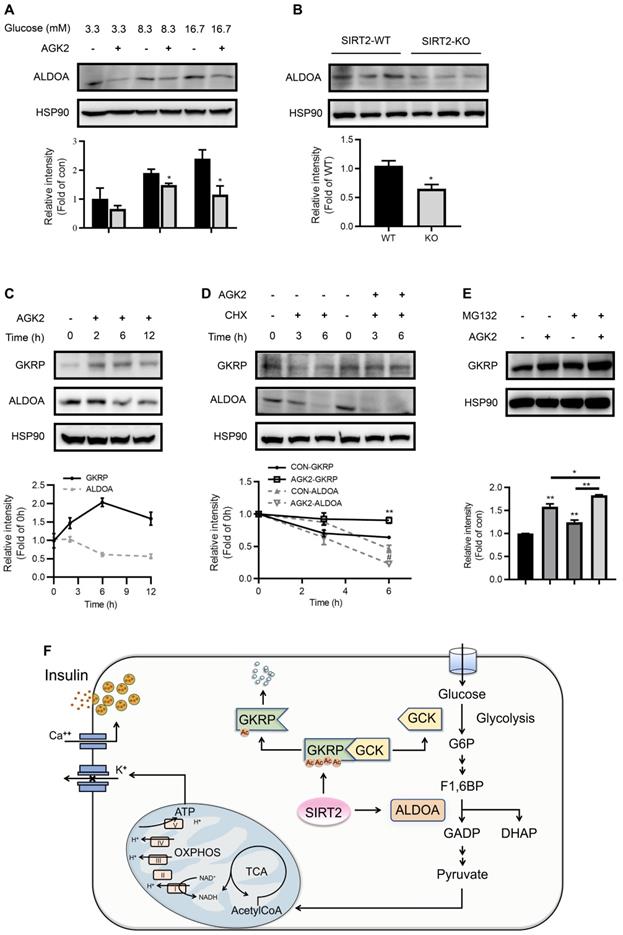
Aldolase is a crucial enzyme in glycolysis responsible for the reversible aldol reaction of fructose-1,6-bisphosphate (FBP) to glyceraldehyde-3-phosphate and dihydroxyacetone phosphate. Aldolase A (ALDOA) revealed a significantly higher expression in rat islets compared with other two aldolase isozymes (B and C) (Figure S6). In contrast to GKRP, the protein expression of ALDOA was suppressed by AGK2 (Figure 7A). Islets isolated from SIRT2-KO rats also exhibited a significant decrease in ALDOA protein level compared with control islets (Figure 7B). The inverse changes in GKRP and ALDOA mediated by AGK2 proved to be a rapid progress, which occurred in INS-1 cells treated with AGK2 for just 6 h. The increase of GKRP protein level could even be observed just after 2-h treatment of AGK2 (Figure 7C). Moreover, SIRT2 pharmacological inhibition by AGK2 significantly prevented the degradation of GKRP protein in INS-1 cells in the presence of protein synthesis inhibitor cycloheximide (CHX). On the other hand, AGK2 dramatically shortened the half-life of ALDOA in INS-1 cells (Figure 7D). Combined treatment of AGK2 and the proteasome inhibitor MG132 exerted a synergistic effect on the prevention of GKRP protein degradation in INS-1 cells (Figure 7E). Taken together, these results indicate that knockout or inhibition of SIRT2 lessens glycolytic flux via promoting GKRP protein stability and ALDOA protein degradation.
SIRT2 inhibition promotes GKRP protein stability and ALDOA protein degradation. (A) ALDOA protein level in INS-1 cells treated with 3 μM AGK2 in the presence of 3.3, 8.3 or 16.7 mM glucose. (B) Protein expression of ALDOA in the islets isolated from SIRT2-WT and SIRT2-KO rats. (C) GKRP and ALDOA protein expressions in INS-1 cells incubated with 3 μM AGK2 for the indicated time points. (D) GKRP and ALDOA protein expressions in INS-1 cells exposed to 3 μM AGK2 and 10 μg/ml cycloheximide (CHX) for 3 and 6 h. (E) GKRP protein expression in INS-1 cells treated with or without 3 μM AGK2 and 10 μM MG132 for 6 h. Signal intensity was quantified by image J software for statistical comparison. (F) The schematic illustration summarizes the role of SIRT2 in glucose-stimulated insulin secretion. GCK acts as a glucose sensor in the β-cells by converting glucose to G6P, which eventually yields ATP through glycolysis, TCA cycle, and oxidative phosphorylation for triggering insulin secretion. GKRP binds and inactivates GCK. SIRT2 inhibits ALDOA protein degradation and decreases GKRP protein stability by decreasing its acetylation level, thereby disrupting the GKRP-GCK complex and promoting glycolytic flux and insulin secretion. Data are expressed as means ± SEM. *P< 0.05, **P< 0.01 vs control group. #P< 0.05 vs CON-ALDOA group.
Discussion
Abnormalities in islet function are critical in defining the risk and development of type 2 diabetes [40]. SIRT2 is a key regulator of cytoplasmic protein acetylation status [41], but its biological function has not been examined in pancreatic β-cells. The present study demonstrated that genetic deletion or pharmacological inhibition of SIRT2 led to impaired insulin secretion from rat islets, in which the metabolic amplifying pathway was involved. SIRT2 ablation significantly reduced glucose-induced glycolytic influx in β-cells via stabilizing GKRP protein and promoting its binding to GCK. Therefore, glucose intolerance in SIRT2-KO rats is mainly attributed to impaired GSIS due to the deacceleration of glucose metabolism in islets.
As a fuel-sensing molecule, SIRT2 expression is induced during low-energy status and repressed under energy excess condition [42]. In contrast, SIRT2 expression displayed no change in islets exposed to high glucose in our study. SIRT2 protein expressions in visceral WAT from human obese subjects and high-fat diet-fed mice were decreased compared with lean controls [43]. It has been demonstrated that SIRT2 inhibits adipocyte differentiation through deacetylating Foxo1 and inhibiting PPARγ [44]. SIRT2 deacetylates and destabilizes ATP-citrate lyase (ACLY), a key enzyme for de novo fatty acid synthesis [45]. Overexpression of SIRT2 promotes fatty acid oxidation via deacetylating PGC-1α [43]. These results suggest that enhancing SIRT2 activity is beneficial for obesity-related disease, such as type 2 diabetes mellitus, nonalcoholic fatty liver, and metabolic syndrome. However, our study exhibited a comparable body weight between SIRT2-KO and WT rats.
Genetic polymorphism of SIRT2 has been reported to associate with diabetes development [46]. However, previous studies displayed contradictory results about the role of SIRT2 in maintaining glucose homeostasis. Watanabe et al. [39] reported that hepatic SIRT2 knockdown by tail vein injection of adenovirus expressing SIRT2 siRNA resulted in impaired glucose tolerance by suppressing hepatic glucose uptake. Another study showed a contrary result, in which SIRT2 silence decreased blood glucose level in mice by promoting PEPCK acetylation and degradation [24]. However, our study showed that neither genetic knockout nor pharmacological inhibition of SIRT2 altered the expression levels of hepatic gluconeogenic genes. It was reported that glucose intolerance was markedly improved in SIRT2-KO mice [47]. But in another study, SIRT2-KO did not alter glucose disposal in mice fed with chow diet [48]. In this current study, the deletion of SIRT2 led to glucose intolerance in chow diet-fed rats. In addition, there also exist conflicting results regarding the function of SIRT2 on insulin sensitivity. It has been demonstrated that SIRT2 overexpression improves insulin sensitivity in insulin-resistant hepatocytes [49] and enhances insulin-activated Akt signaling in HeLa cells and 3T3-L1 preadipocytes [50]. Lantier et al. [48] showed that SIRT2 deletion alone was sufficient to impair muscle insulin sensitivity in chow diet-fed mice and high fat diet-induced liver insulin resistance was worsened by SIRT2 KO. But in C2C12 muscle cells, SIRT2 negatively regulates insulin sensitivity and glucose uptake [51]. In SIRT2-KO mice reported by Belman et al. [47], insulin-stimulated whole body glucose uptake was greatly enhanced. In our study, the insulin sensitivity was comparable between SIRT2-KO and WT rats, but blood insulin level after glucose loading was markedly decreased in SIRT2-KO rats, which may explain glucose intolerance.
Among the Sirtuin family, SIRT1 and SIRT6 are the positive regulators of insulin secretion [12, 13, 18] while two mitochondrial deacetylases SIRT3 and SIRT4 negatively regulate insulin secretion [8, 16]. However, the role of SIRT2 in islet function still remains enigmatic. In the present study, the islets isolated from SIRT2-KO rats secreted less insulin in response to glucose challenge and the overexpression of SIRT2 in rat islets enhanced GSIS, indicating that SIRT2 is a direct regulator of islet function. The triggering pathway is essential for glucose-induced insulin secretion, which has been well characterized [30]. Glucose-stimulated amplitude of insulin secretion largely depends on the amplifying pathway that does not raise intracellular Ca2+ concentration further, but metabolic molecules augment the action of triggering Ca2+ on exocytosis [52]. Our study showed that genetic knockout or pharmacological inhibition of SIRT2 disrupted the metabolic amplifying pathway of GSIS, suggesting the involvement of metabolites in SIRT2-modulated insulin secretion.
The pancreatic β-cells sense circulating levels of nutrients to secrete insulin. Glucose is the primary stimulus for insulin secretion, whose metabolism in β-cells is achieved by tightly linking glycolysis with mitochondrial metabolism [53]. In this current study, decreased glucose-stimulated OCR and universal reduction of glycolytic and TCA intermediates with unaffected mitochondria were detected in SIRT2-KO islets, implicating the influx of insufficient substrate. Unlike most other tissues where metabolism primarily responds to nerve or hormonal stimulation and energy expenditures, glucose metabolism in β-cells is governed by substrate availability [54]. This unique feature is possible because: (1) glucose transport into β-cells is not limiting [55]. (2) glycolysis is controlled by GCK, lacking any form of feedback control [56]. (3) The use of G6P by other pathways (e.g., pentose phosphate pathway and glycogen synthesis) is very limited in islets [57]. Thereby the cellular levels of glucose as well as the expression and activity of GCK primarily determine glucose usage and regulate insulin secretion in islets. SIRT2 ablation lowered the concentrations of G6P and subsequent intermediates in islet without changing GCK expression, indicating a possibility that SIRT2 plays a regulatory role in GCK activity. The GCK activator did rescue SIRT2 knockout-impaired GSIS, suggesting that SIRT2 facilitates GSIS by promoting the activity of GCK.
GKRP functions as an allosteric inhibitor of glucokinase and as a metabolic sensor [58]. GCK is bound to GKRP at low glucose status, resulting in its inactivation. Glucose induces GCK-GKRP dissociation and GCK activation [34]. GKRP was reported to be only expressed in liver, but one study described the existence of a brain GKRP and its functional interaction with GCK [37]. In our study, GKRP protein expression was detectable in islet β-cells, which was significantly induced by SIRT2 inhibition. Park J et al. [59] and Watanabe H et al. [39] implicate that SIRT2-dependent GKRP deacetylation facilitates the dissociation of GKRP from GCK, thereby promoting GCK activity in the liver. Our study exhibited a similar result that SIRT2 overexpression deacetylated GKRP in INS-1 cells and SIRT2 inhibition by AGK2 enhanced the interaction of GKRP and GCK. However, unlike the result in liver that SIRT2 failed to affect GKRP expression [39], SIRT2 directly regulated the protein stability of GKRP in islets. Thus, it is possible that SIRT2 inhibition-mediated GKRP protein stability is linked to its acetylation.
A recent study showed that knockdown of SIRT2 in fibroblasts considerably increased the acetylation levels of four glycolytic enzymes (ALDOA, GAPDH, PGK1, and ENO1) and enhanced their enzymatic activities, without affecting the total amount of enzymes [60]. In contrast to the result, we found that SIRT2 knockout or inhibition decreased the protein level of ALDOA via destabilizing its protein, which acted in concert with GKRP to reduce the glycolytic flux for insulin secretion. The effect of SIRT2 on glycolysis in islets and liver seems contrast to that in fibroblasts. After all, both islets and liver are the most metabolically relevant tissues, in which nutrients metabolism is quite different from that in other tissues concerning their vital roles in maintaining energy homeostasis. The opposing role of SIRT2 in different tissues needs further investigation.
In summary, this study demonstrates a positive role of SIRT2 played in islet function. Increased protein stability of GKRP followed by reduced glycolytic flux is responsible for the impaired GSIS in SIRT2-KO islets (Figure 7F). Since GKRP disruptors have been expected as a potential new class of glucose-lowering drugs [61], the action of GKRP in islets should be taken into account for these agents in treating type 2 diabetes mellitus.
Abbreviations
SIRT2: NAD-dependent deacetylase sirtuin 2; GSIS: glucose-stimulated insulin secretion; GKRP: glucokinase regulatory protein; GCK: glucokinase; ALDOA: aldolase A; TCA: tricarboxylic acid; KATs: lysine acetyltransferases; KDACs: lysine deacetylases; H&E: hematoxylin and eosin; G6P: glucose 6-phosphate; F6P: fructose 6-phosphate; FBP: fructose-1,6-bisphosphate; CHX: cycloheximide; FOXO1: forkhead box protein O1; PPARγ: peroxisome proliferator-activated receptor γ; ACLY: ATP-citrate lyase; PGC-1α: peroxisome proliferator-activated receptor gamma coactivator 1-α; PEPCK: phosphoenolpyruvate carboxykinase; G6pase: glucose 6-phosphatase; Fbpase: fructose 1,6-bisphosphatase.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was funded by grants from the National Key R&D Program of China (2018YFC2000700), the National Natural Science Foundation of China (81670795, 81770767, 81870526, and 81970691), Shanghai Municipal Education Commission-Gaofeng Clinical Medicine Grant (20191801).
Ethics approval and consent to participate
All animal protocols were reviewed and approved by the Animal Care Committee of Ruijin Hospital, Shanghai Jiaotong University School of Medicine.
Author Contributions
X.W., L.Z., L.S., and F.Z. conceived and designed the research. F.Z., L.Z., Y. Z., M.B., Q.Z., S.W., and K.Z. performed most experiments. M.Y., C.S. and Y.L. performed animal experiments. Data were analyzed by X.W., L.Z. F.Z. and L.Z. The paper was written by X.W., L.Z., J.L., and F.Z.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Verdin E, Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat Rev Mol Cell Biol. 2015;16:258-64
2. Zhao SM, Xu W, Jiang WQ, Yu W, Lin Y, Zhang TF. et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000-4
3. Narita T, Weinert BT, Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol. 2019;20:156-74
4. Wang QJ, Zhang YK, Yang C, Xiong H, Lin Y, Yao J. et al. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science. 2010;327:1004-7
5. Schwer B, Eckersdorff M, Li Y, Silva JC, Fermin D, Kurtev MV. et al. Calorie restriction alters mitochondrial protein acetylation. Aging cell. 2009;8:604-6
6. Kendrick AA, Choudhury M, Rahman SM, McCurdy CE, Friederich M, Van Hove JL. et al. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem J. 2011;433:505-14
7. Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol. 2014;15:536-50
8. Zhang YQ, Zhou FY, Bai MY, Liu Y, Zhang LL, Zhu Q. et al. The pivotal role of protein acetylation in linking glucose and fatty acid metabolism to beta-cell function. Cell Death Dis. 2019;10:66
9. Aka JA, Kim GW, Yang XJ. K-acetylation and its enzymes: overview and new developments. Handb Exp Pharmacol. 2011;206:1-12
10. Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587-91
11. Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, Li X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009;9:327-38
12. Luu L, Dai FF, Prentice KJ, Huang X, Hardy AB, Hansen JB. et al. The loss of Sirt1 in mouse pancreatic beta cells impairs insulin secretion by disrupting glucose sensing. Diabetologia. 2013;56:2010-20
13. Moynihan KA, Grimm AA, Plueger MM, Bernal-Mizrachi E, Ford E, Cras-Meneur C. et al. Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2005;2:105-17
14. Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB. et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464:121-5
15. Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ. et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell. 2006;126:941-54
16. Anderson KA, Huynh FK, Fisher-Wellman K, Stuart JD, Peterson BS, Douros JD. et al. SIRT4 is a lysine deacylase that controls leucine metabolism and insulin secretion. Cell Metab. 2017;25:838-55
17. Kim HS, Xiao C, Wang RH, Lahusen T, Xu X, Vassilopoulos A. et al. Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab. 2010;12:224-36
18. Xiong XW, Wang GH, Tao RY, Wu PF, Kono T, Li K. et al. Sirtuin 6 regulates glucose-stimulated insulin secretion in mouse pancreatic beta cells. Diabetologia. 2016;59:151-60
19. Vaquero A, Scher MB, Lee DH, Sutton A, Cheng HL, Alt FW. et al. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 2006;20:1256-61
20. Liu G, Park SH, Imbesi M, Nathan WJ, Zou X, Zhu Y. et al. Loss of NAD-dependent protein deacetylase Sirtuin-2 alters mitochondrial protein acetylation and dysregulates mitophagy. Antioxid Redox Signal. 2017;26:849-63
21. Wang Y, Yang JQ, Hong TT, Chen XJ, Cui LL. SIRT2: Controversy and multiple roles in disease and physiology. Ageing Res Rev. 2019;55:100961
22. Gomes P, Outeiro TF, Cavadas C. Emerging role of Sirtuin 2 in the regulation of mammalian metabolism. Trends Pharmacol Sci. 2015;36:756-68
23. Yang X, Zhang YQ, Xu W, Deng RY, Liu Y, Li FY. et al. Potential role of Hsp90 in rat islet function under the condition of high glucose. Acta Diabetol. 2016;53:621-8
24. Jiang WQ, Wang SW, Xiao MT, Lin Y, Zhou LH, Lei QY. et al. Acetylation regulates gluconeogenesis by promoting PEPCK1 degradation via recruiting the UBR5 ubiquitin ligase. Mol Cell. 2011;43:33-44
25. Wang J, Koh HW, Zhou L, Bae UJ, Lee HS, Bang IH. et al. Sirtuin 2 aggravates postischemic liver injury by deacetylating mitogen-activated protein kinase phosphatase-1. Hepatology. 2017;65:225-36
26. Donmez G, Outeiro TF. SIRT1 and SIRT2: emerging targets in neurodegeneration. EMBO Mol Med. 2013;5:344-52
27. Wang Q, Zhou Y, Weiss HL, Evers BM. Sirt2 protein is required for activation of mTOR signaling in colorectal cancer cells. Cancer Res. 2016 76
28. Jung YJ, Lee AS, Tung NT, Kim D, Kang KP, Lee S. et al. SIRT2 regulates LPS-induced renal tubular CXCL2 and CCL2 expression. J Am Soc Nephrol. 2015;26:1549-1560
29. Jing E, Gesta S, Kahn CR. SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab. 2007;6:105-14
30. Henquin JC. The dual control of insulin secretion by glucose involves triggering and amplifying pathways in beta-cells. Diabetes Res Clin Pract. 2011 93S: S27-31
31. Henquin JC. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes. 2000;49:1751-60
32. Henquin JC. Regulation of insulin secretion: a matter of phase control and amplitude modulation. Diabetologia. 2009;52:739-51
33. Prentki M, Matschinsky FM, Madiraju SR. Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 2013;18(2):162-85
34. Choi JM, Seo MH, Kyeong HH, Kim E, Kim HS. Molecular basis for the role of glucokinase regulatory protein as the allosteric switch for glucokinase. Proc Natl Acad Sci U S A. 2013;110:10171-6
35. Slosberg ED, Desai UJ, Fanelli B, St Denny I, Connelly S, Kaleko M. et al. Treatment of type 2 diabetes by adenoviral-mediated overexpression of the glucokinase regulatory protein. Diabetes. 2001;50:1813-20
36. Hayward BE, Dunlop N, Intody S, Leek JP, Markham AF, Warner JP. et al. Organization of the human glucokinase regulator gene GCKR. Genomics. 1998;49(1):137-42
37. Alvarez E, Ronce ro I, Chowen JA, Vazquez P, Blazquez E. Evidence that glucokinase regulatory protein is expressed and interacts with glucokinase in rat brain. J Neurochem. 2002;80(1):45-53
38. Brouwers M, Jacobs C, Bast A, Stehouwer CDA, Schaper NC. Modulation of glucokinase regulatory protein: A double-edged sword? Trends Mol Med. 2015;21(10):583-94
39. Watanabe H, Inaba Y, Kimura K, Matsumoto M, Kaneko S, Kasuga M. et al. Sirt2 facilitates hepatic glucose uptake by deacetylating glucokinase regulatory protein. Nat Commun. 2018;9:30
40. Weir GC, Bonner-Weir S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes. 2004 53S: S16-21
41. North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell. 2003;11:437-44
42. Wang F, Nguyen M, Qin XF, Tong Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell. 2007;6:505-14
43. Krishnan J, Danzer C, Simka T, Ukropec J, Walter KM, Kumpf S. et al. Dietary obesity-associated Hif1 alpha activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system. Genes Dev. 2012;26:259-70
44. Wang F, Tong Q. SIRT2 suppresses adipocyte differentiation by deacetylating FOXO1 and enhancing FOXO1's repressive interaction with PPAR gamma. Mol Biol Cell. 2009;20:801-8
45. Lin RT, Tao R, Gao X, Li TT, Zhou X, Guan KL. et al. Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Mol Cell. 2013;51:506-18
46. Liu TT, Yang WT, Pang SC, Yu SP, Yan B. Functional genetic variants within the SIRT2 gene promoter in type 2 diabetes mellitus. Diabetes Res Clin Pract. 2018;137:200-7
47. Belman JP, Bian RR, Habtemichael EN, Li DT, Jurczak MJ, Alcazar-Roman A. et al. Acetylation of TUG protein promotes the accumulation of GLUT4 glucose transporters in an insulin-responsive intracellular compartment. J Biol Chem. 2015;290:4447-63
48. Lantier L, Williams AS, Hughey CC, Bracy DP, James FD, Ansari MA. et al. SIRT2 knockout exacerbates insulin resistance in high fat-fed mice. PLoS One. 2014;13(12):e0208634
49. Lemos V, de Oliveira RM, Naia L, Szego E, Ramos E, Pinho S. et al. The NAD+-dependent deacetylase SIRT2 attenuates oxidative stress and mitochondrial dysfunction and improves insulin sensitivity in hepatocytes. Hum Mol Genet. 2017;26:4105-17
50. Ramakrishnan G, Davaakhuu G, Kaplun L, Chung WC, Rana A, Atfi A. et al. Sirt2 deacetylase is a novel AKT binding partner critical for AKT activation by insulin. J Biol Chem. 2014;289:6054-66
51. Arora A, Dey CS. SIRT2 negatively regulates insulin resistance in C2C12 skeletal muscle cells. Biochim Biophys Acta. 2014;1842:1372-8
52. Kalwat M, Cobb M. Mechanisms of the amplifying pathway of insulin secretion in the β cell. Pharmacol Ther. 2017;179:17-30
53. Newsholme P, Gaudel C, McClenaghan NH. Nutrient regulation of insulin secretion and beta-cell functional integrity. Adv Exp Med Biol. 2010;654:91-114
54. Matschinsky FM. Banting Lecture 1995. A lesson in metabolic regulation inspired by the glucokinase glucose sensor paradigm. Diabetes. 1996;45:223-41
55. Thorens B, Mueckler M. Glucose transporters in the 21st Century. Am J Physiol Endocrinol Metab. 2010;298:E141-5
56. Meglasson MD, Matschinsky FM. New perspectives on pancreatic islet glucokinase. Am J Physiol. 1984;246:E1-13
57. Schuit F, De Vos A, Farfari S, Moens K, Pipeleers D, Brun T. et al. Metabolic fate of glucose in purified islet cells. Glucose-regulated anaplerosis in beta cells. J Biol Chem. 1997;272:18572-9
58. Van Schaftingen E, Vandercammen A, Detheux M, Davies DR. The regulatory protein of liver glucokinase. Adv Enzyme Regul. 1992;32:133-48
59. Park JM, Kim TH, Jo SH, Kim MY, Ahn YH. Acetylation of glucokinase regulatory protein decreases glucose metabolism by suppressing glucokinase activity. Sci Rep. 2015;5:17395
60. Cha Y, Han MJ, Cha HJ, Zoldan J, Burkart A, Jung JH. et al. Metabolic control of primed human pluripotent stem cell fate and function by the miR-200c-SIRT2 axis. Nat Cell Biol. 2017;19(5):445-56
61. Lloyd DJ, St Jean DJJ, Kurzeja RJ, Wahl RC, Michelsen K, Cupples R. et al. Antidiabetic effects of glucokinase regulatory protein small-molecule disruptors. Nature. 2013;504:437-40
Author contact
![]() Corresponding authors: Libin Zhou and Xiao Wang, Department of Endocrine and Metabolic Diseases, Shanghai institute of Endocrine and Metabolic Diseases, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, 197 Ruijin Road Ⅱ, Shanghai 200025, China. Phone: 86-21-64315587. Fax: 86-21-64673639. E-mail address: libinzhou99com or wangxiao1976com; Li Shao, Department of VIP Clinic, Shanghai East Hospital, Tongji University School of Medicine, No. 1800 Yuntai Road Pudong District, Shanghai, 200123, China. E-mail address: dongfangshaolicom.
Corresponding authors: Libin Zhou and Xiao Wang, Department of Endocrine and Metabolic Diseases, Shanghai institute of Endocrine and Metabolic Diseases, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, 197 Ruijin Road Ⅱ, Shanghai 200025, China. Phone: 86-21-64315587. Fax: 86-21-64673639. E-mail address: libinzhou99com or wangxiao1976com; Li Shao, Department of VIP Clinic, Shanghai East Hospital, Tongji University School of Medicine, No. 1800 Yuntai Road Pudong District, Shanghai, 200123, China. E-mail address: dongfangshaolicom.