Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Background
Sources of ROS
Antioxidant defense mechanisms
ROS, a double-edged sword in...
The application of antioxidants...
The roles of pro-oxidants in...
Future perspectives
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(10):4839-4857. doi:10.7150/thno.56747 This issue Cite
Review
The double-edged roles of ROS in cancer prevention and therapy
Yawei Wang1,2, Huan Qi2, Yu Liu1,2, Chao Duan1,2, Xiaolong Liu2, Tian Xia2, Di Chen2, Hai-long Piao2,3 ![]() , Hong-Xu Liu1
, Hong-Xu Liu1 ![]()
1. Department of Thoracic Surgery, Cancer Hospital of China Medical University, Liaoning Cancer Hospital & Institute, Shenyang 110042, China.
2. CAS Key Laboratory of Separation Science for Analytical Chemistry, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China.
3. Department of Biochemistry & Molecular Biology, School of Life Sciences, China Medical University, Shenyang, 110122, China.
Received 2020-12-3; Accepted 2021-1-31; Published 2021-3-4
Abstract

Reactive oxygen species (ROS) serve as cell signaling molecules generated in oxidative metabolism and are associated with a number of human diseases. The reprogramming of redox metabolism induces abnormal accumulation of ROS in cancer cells. It has been widely accepted that ROS play opposite roles in tumor growth, metastasis and apoptosis according to their different distributions, concentrations and durations in specific subcellular structures. These double-edged roles in cancer progression include the ROS-dependent malignant transformation and the oxidative stress-induced cell death. In this review, we summarize the notable literatures on ROS generation and scavenging, and discuss the related signal transduction networks and corresponding anticancer therapies. There is no doubt that an improved understanding of the sophisticated mechanism of redox biology is imperative to conquer cancer.
Background
Reactive oxygen species (ROS) are two electron reduction products of oxygen, including superoxide anion, hydrogen peroxide, hydroxyl radical, lipid peroxides, protein peroxides and peroxides formed in nucleic acids [1]. They are maintained in a dynamic balance by a series of reduction-oxidation (redox) reactions in biological systems and act as signaling molecules to drive cellular regulatory pathways [2, 3]. Excessive oxidative stress derived from ROS accumulation deregulates the antioxidative defense system, which is closely associated with various diseases [4, 5], especially cancers [6]. Though emerging studies have demonstrated the primary ligand stimulants, the enzymatic generation mechanisms as well as the putative downstream targets [6, 7], the major mechanisms by which ROS participates in cancer development in concentration-dependent, spatially dependent and temporally dependent manners remain insufficiently understood.
During different stages of cancer formation, abnormal ROS levels play paradoxical roles in cell growth and death [8]. A physiological concentration of ROS that maintained in equilibrium is necessary for normal cell survival. Ectopic ROS accumulation promotes cell proliferation and consequently induces malignant transformation of normal cells by initiating pathological conversion of physiological signaling networks. Excessive ROS levels lead to cell death by damaging cellular components, including proteins, lipid bilayers, and chromosomes. Therefore, both scavenging abnormally elevated ROS to prevent early neoplasia and facilitating ROS production to specifically kill cancer cells are promising anticancer therapeutic strategies, in spite of their contradictoriness and complexity. Consequently, a better understanding of the sophisticated mechanism of ROS in tumorigenesis is critical to conquering cancer.
In this review, we not only discuss the double-edged roles and molecular regulatory mechanisms of ROS in cancer prevention and therapy, but also summarize the relevant small molecule interventions and envision the future perspectives of ROS-targeted cancer treatment.
Sources of ROS
Approximately 2.4-3.8 billion years ago, ROS most likely appeared on the earth along with the first oxygen molecule in the atmosphere, and they have existed with the aerobic life ever since [9, 10]. In human cells, biologically relevant ROS are derived from both the exogenous environment and endogenous metabolism.
Exogenous ROS generation
Exogenous ROS can be generated from exposure to air pollutants, tobacco, metals, asbestos or radiation. From the perspective of air pollutants, a recent study has shown that exposure to fine particulate matter with a diameter of less than 2.5 µm (PM 2.5) can lead to DNA damage in bronchial epithelial cells 16HBE by inducing oxidative stress [11]. Regarding tobacco, detrimental components generated from tobacco smoking can induce oxidative stress by decreasing the circulating concentrations of antioxidant micronutrients such as cryptoxanthin, α carotene, β carotene, and ascorbic acid [12]. As a class I carcinogenic heavy metal, arsenic can increase ROS levels via Fenton reaction, thus participating in the progressions of multiple malignancies [13, 14]. Inhaled asbestos fibers accumulate in lungs and induce the generation of ROS due to the presence of iron associated with the fibrous silicates, which promote the malignant transformation of mesothelial cells [15]. Ultraviolet radiation (UVR) increases oxidative stress not only by upregulating nitric oxide synthase (NOS) synthesis but also by impeding catalase (CAT) to scavenge hydrogen peroxide, thus leading to increased risk of sunburn, photoaging and skin cancer [16]. Ionizing radiation (IR) stimulates ROS generation by immediately inducing extracellular water radiolysis or causing intracellular mitochondrial metabolic disorder, thereby destroying cancer cells or, conversely, facilitating their survival and metastasis [17]. Besides, many carcinogens in the environment play oncogenic roles by inducing ROS accumulation.
Endogenous ROS generation
The two major sources of endogenous ROS are the mitochondrial respiratory chain, which generates ROS as a byproduct [18, 19], and active NADPH oxidases (NOXs), whose primary function is ROS production. In addition, peroxisomes and endoplasmic reticulum membranes have also been identified as cellular sites of ROS generation [20, 21]. Here, we mainly discuss research status on ROS production in mitochondria and by NOXs (Figure 1).
The primary generation and elimination mechanisms of intracellular ROS. (Ⅰ) The two major sources of endogenous ROS are mitochondrial ETC and NOXs: oxygen gains electrons leaked from complex Ⅰ, Ⅱ and Ⅲ of ETC to generate superoxide; NOXs transfer electrons from NADPH in the cytosol to oxygen at extracellular to produce superoxide; superoxide is dismutated to H2O2 by SOD for subsequent transmembrane transportation. (Ⅱ) Intracellular ROS are scavenged by three enzymatic antioxidants: PRXs, GPXs and CAT. PRXs and GPXs use NADPH as reducing equivalent; CAT use non-NADPH hydrogen donors. (Ⅲ) NADPH is synthesized in cytoplasm and mitochondria: cytosolic NADPH is primarily generated from PPP pathway; in mitochondria, GLUD generate NADPH by converting glutamate to α-KG; IDHs and MEs are contributed to NADPH pools both in the cytosol and mitochondrial by respectively catalyzing the oxidative decarboxylation of isocitrate to α-KG and malate to pyruvate; one-carbon metabolism contributes to NADPH production both in cytosol and in mitochondria.
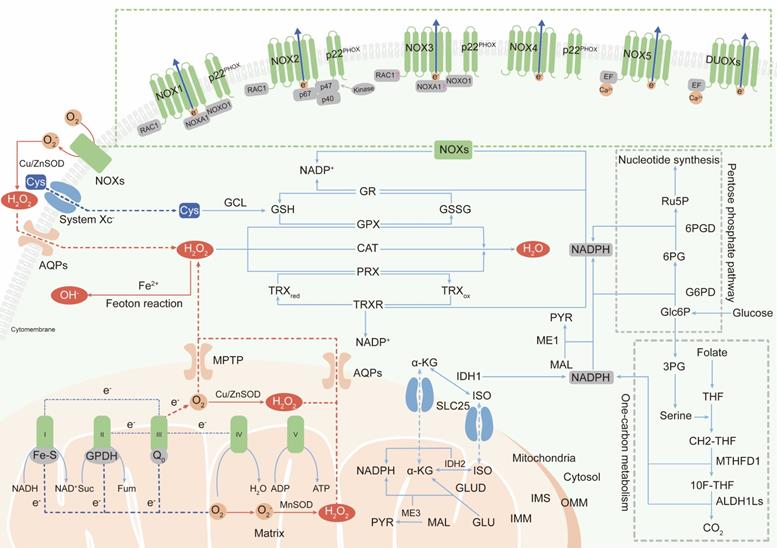
Mitochondria-derived ROS
In mammalian cells, the mitochondrial electron transport chain (ETC) is the main source of ATP [22]. However, during oxidative phosphorylation and energy transduction, approximately 1% of molecular oxygen gains electrons leaked from the ETC, yielding superoxide [23]. Some superoxide is released into the cytoplasm through the mitochondrial permeability transition pore (MPTP) located in the outer mitochondrial membrane (OMM) [24]. However, most superoxide is dismutated to H2O2 by superoxide dismutases (SODs) in the mitochondrial matrix or intermembrane space (IMS) [25]. H2O2 is highly diffusible and specifically carried into the cytoplasm by aquaporins (aquaporins 3 and 8) as a second messenger to regulate multiple signaling pathways [26, 27].
The mammalian mitochondrial respiratory chain mainly contains five complexes including NADH-ubiquinone oxidoreductase (complex I), succinate-ubiquinone oxidoreductase (complex II), cytochrome bc1 complex (complex III), cytochrome c oxidase (complex IV), and ATP synthase (complex V) [28]. It is believed that complexes I and III are the main sites of superoxide generation [29]. The iron-sulfur cluster (Fe-S) in the matrix-protruding hydrophilic arm in complex I is the potential site of electron leakage, and the generated superoxide is exclusively discharged into the mitochondrial matrix [30, 31]. The ubiquinol oxidation site (Qo site) is the locus of superoxide production in complex III and releases superoxide to both sides of the inner mitochondrial membrane (IMM) [32]. No more than 50% of the superoxide produced from electron leakage at complex III is excreted to the intermembrane space; the remaining 50% is released into the mitochondrial matrix [31]. In addition to complexes I and III, a fraction of the superoxide production in mitochondria is attributed to flavin-reducing site (IIF) within complex II [33].
Although the initial anionic form of superoxide generated via the ETC is too strongly charged to readily cross the inner mitochondrial membrane, instantaneously conversion into H2O2 by SODs permits the transportation of ROS from mitochondria to cytoplasm [34]. However, it has also been reported that a fraction of superoxide released by complex III in the intermembrane space can diffuse directly into the cytoplasm through the MPTP [35]. Metabolic equilibrium of mitochondrial ROS is important in processes promoting the normal function of cells, including Ca2+ homeostasis and ATP synthesis [36, 37]. Under aberrant physiopathological conditions, as a casualty, mitochondrial DNA damage and component dysfunction induced by ROS accumulation lead to malignant transformation [38].
NOX-derived ROS
The transmembrane NADPH oxidases are identified as the only enzyme family with the sole role of generating ROS and are first described to show bactericidal activity in phagocytes by yielding superoxide [39]. Later studies identified the other six cytochrome homolog subunits including NOX1, NOX3-5, dual oxidase 1 (DUOX1) and dual oxidase 2 (DUOX2) [40]. Together with the phagocyte NADPH oxidase (NOX2/gp91PHOX), these homologs are now referred to as the NOX family, and all of them have the ability to transport electrons across the plasma membrane and produce ROS [41]. All NOXs share similar structures including six transmembrane domains with two heme-binding regions and an NADPH-binding region in the intracellular C-terminus, but their organ distribution and regulatory mechanisms are different [42].
NOX2 is a prototypical NOX family member and first identified as cytochrome b558, which is absent in the bactericidal-deficient leukocytes of chronic granulomatous disease patients [43, 44]. NOX2 is widely distributed in various tissues and is composed of the protein scaffold gp91PHOX, the membrane partner p22PHOX, the GTP-binding protein RAC1 and three cytosolic subunits (p67PHOX, p47PHOX and p40PHOX) [45]. Phosphorylated p47PHOX acts as a “launching switch” to interact with p22PHOX and organizes the translocation of the catalytic subunits (p67PHOX, p40PHOX) and the energy-providing subunit (RAC1) to the complex. Once assembled, NOX2 is activated and transfers electrons from NADPH in cytosol to oxygen in extracellular space to produce superoxide [46].
NOX1 is the first discovered isoform of NOX2 and is highly expressed in colonic epithelium [47, 48]. NOX organizer 1 (NOXO1, a homolog of p47PHOX) and NOX activator 1 (NOXA1, a homolog of p67PHOX) are novel cytosolic subunits of NOX1. Besides, NOX1 activation also requires the membrane partner p22PHOX and the GTPase Rac1 [49].
NOX3 is defined by two seminal contributions which reveal that NOX3 mutations give rise to vestibular defects in head tilt mice [50, 51]. p22PHOX and NOXO1 are essential partners for NOX3 activation [52, 53]. Whether the other cytosolic subunits, such as NOXA1, p47PHOX, p67PHOX and RAC1, are also involved in NOX3 assembly is still controversial.
NOX4 is initially identified as a NADPH oxidase in the kidney [54, 55]. In contrast to NOX1-3 activation, NOX4 activation is independent of the cytosolic subunit [56], and the subunit required for NOX4-derived ROS production is p22PHOX [57]. A question about NOX4 is its uncertain subcellular localization. As a transmembrane protein, NOX4 has been found to be localized at the cytomembrane and endoplasmic reticulum (ER), but NOX4 expression has also been observed in the nucleus [58, 59], which is difficult to understand how a membrane-spanning protein can be located in a presumably membrane-free space.
NOX5 is a unique and unusual NADPH oxidase. Its structure is distinguished from the NOX1-4 homologs by the presence of a long intracellular NH2 terminus containing a Ca2+-binding EF-hand domain and it does not require membrane partner, cytosolic subunit or GTPase for activation [60]. The only activator of superoxide generation by NOX5 is the intracellular Ca2+ concentration, which is documented by its inactivated function of ROS production under Ca2+-free conditions [61]. To date, only the NOX5 crystal structure has been discovered (in 2017; the first and only NOX crystal structure to be solved) [62].
DUOX proteins, also known as thyroid oxidases, were first defined as NADPH oxidases in thyroid epithelial cells [63, 64]. DUOXs can be directly activated by Ca2+ and do not require cytosolic activator or organizer subunits, suggesting that their EF-hand Ca2+-binding domains are functional [65]. Although DUOXs are homologous with peroxidases, it is not clear whether they can function as peroxidases [66].
The superoxide produced by NOXs in extracellular is transported into the cytoplasm in the form of H2O2 [67], which acts as a secondary messenger to maintain cell survival and proliferation or, paradoxically, to induce cell death [68, 69]. Abundant researches have shown that ROS accumulation induced by over-activation of NOXs contribute to both the initiation and the progression of malignancies. For example, NOX-2 derived superoxide drives mitochondrial to transfer from bone marrow stromal cells to leukemic blasts[70]. NOX1-generated ROS promotes the self-renewal activity of CD133+ thyroid cancer cells through activation of the Akt signaling [71]. NOX4-driven ROS formation regulates proliferation and apoptosis of gastric cancer cells through the GLI1 pathway [72]. Therefore, a deep understanding of the precise roles of NOXs will pave the way for their validation as anticancer therapeutic targets.
Antioxidant defense mechanisms
Antioxidant defense systems sustain the balance between the generation and neutralization of ROS to maintain redox equilibrium and protect macromolecules from indiscriminate destruction inflicted by oxidative stress (Figure 1).
Intracellular enzymatic antioxidants
Superoxide dismutases (SODs), categorized as cytosolic SOD1, mitochondrial SOD2, and extracellular SOD3, are widely distributed in various cellular compartments and can rapidly dismutate superoxide into H2O2 [73]. Then, peroxiredoxin (PRX), glutathione peroxidase (GPX) and catalase (CAT) convert H2O2 into water (H2O). The primary sites in PRXs, GPXs and CAT that react with H2O2 are cysteine, selenocysteine and heme, respectively [74-76]. Among these enzymes, PRXs are thought to be ideal H2O2 scavengers, given their high-affinity binding sites for H2O2 and their abundant expressions and broad distributions in subcellular compartments [77]. Oxidized PRXs are reduced by thioredoxin (TRX). TRX is then restored to their reduced form by thioredoxin reductase (TRXR) and the reducing equivalent provided by NADPH [77]. GPXs convert H2O2 to H2O by oxidizing reduced glutathione (GSH) to glutathione disulfide (GSSG), and glutathione reductase (GR) reduces oxidized GSSG back to GSH using NADPH as an electron donor [78]. Unlike PRXs and GPXs, CAT converts H2O2 to H2O without the participation of any cofactors. CAT participates in two different antioxidant reactions according to the H2O2 concentration. At high H2O2 levels, CAT exhibits catalytic activity by converting H2O2 into H2O and O2. At low H2O2 levels, CAT exhibits peroxidatic activity to reduce one H2O2 molecule into two H2O molecules by consuming two reducing equivalents from non-NADPH hydrogen donors, such as alcohols, phenols, hormones and metals [79].
NADPH, the primary intracellular hydrogen donor
NADPH, as the primary hydrogen donor and component of the reducing equivalent pool, maintains moderate disulfide reduction of most proteins through NADPH-dependent TRXR and GR redox systems [80]. Maintaining the dynamic equilibrium of the compartmentalized NADP+/NADPH pools is essential for cellular redox homeostasis and modulation of thiol-disulfide transformation signaling [81].
NADPH is generated via various metabolic processes in the cytosol or mitochondria. Cytosolic NADPH is primarily generated via the pentose phosphate pathway (PPP) by glucose-6-phosphate dehydrogenase (G6PD) and 6-phosphogluconate dehydrogenase (6PGD) [82]. In addition, isocitrate dehydrogenases (IDHs) and malic enzymes (MEs) also contribute to the NADPH pools in the cytosol and mitochondria. IDHs and MEs catalyze the oxidative decarboxylation of isocitrate and malate to α-ketoglutarate (α-KG) and pyruvate, respectively [83, 84]. In mitochondria, glutamate dehydrogenases (GLUDs) can generate NADPH by converting glutamate (GLU) to α-KG [85]. In addition to the conventional knowledge described above, serine-driven one-carbon metabolism can also contribute to the NADPH production according to recent studies, in which the oxidation of methylene tetrahydrofolate (CH2-THF) to 10-formyl-tetrahydrofolate (10F-THF) by MTHFD1 and the conversion of 10F-THF to CO2 by ALDH1Ls (ALDH1L1 in cytosol and ALDH1L2 in mitochondria, respectively) are both accompanied by the reduction of NADP+ to NADPH [86, 87].
Because the IMM is impermeable to NADPH, NADPH communication between the cytosol and mitochondria is conducted via the isocitrate-a-KG shuttle through the mitochondrial transporter SLC25 [88]. In mitochondria, IDH2 converts α-KG to isocitrate using NADPH. Then, isocitrate turns into citrate and is transported to cytosol via SLC25. The cytosolic enzyme IDH1, finally, catalyzes the oxidative decarboxylation of isocitrate to α-ketoglutarate and produces NADPH [89].
Superoxide and hydrogen peroxide are the partially reduced forms of oxygen, as it is fully reduced to H2O with the addition of four electrons. NADPH, as the electron carrier, is not only a producer (incomplete reduction) but also a scavenger (complete reduction) of ROS. Paradoxically, inadequate NADPH leads to ROS accumulation, and excessive NADPH leads to reductive stress, in particular, being utilized by NOXs to produce ROS. Therefore, only if the NADP+/NADPH level remains in an equilibrium state, NADPH can perform its antioxidant defensive roles.
NRF2, the master regulator of the antioxidant defense system
Nuclear factor E2-related factor 2 (NRF2), an intracellular transcription factor, controls the expression of antioxidant genes and protects cells from oxidative and electrophilic stress [90]. NRF2 is a leucine zipper member of the cap 'n' collar (CNC) family and is composed of six highly conserved Nrf2-ECH (Neh) domains [91]. Neh family proteins (Neh1-Neh6) contain a CNC-type leucine zipper domain, which is crucial for DNA binding and dimerization with other transcription factors, such as CREB-binding protein (CBP) and brahma-related gene 1 (BRG1) [92]. Kelch-like ECH-associated protein 1 (KEAP1) triggers proteasomal degradation of Nrf2 via CUL3-dependent E3-ubiquitin ligase-mediated ubiquitination [93, 94]. When intracellular ROS are accumulated abnormally, the cysteine residues in KEAP1 are oxidized, thus blocking NRF2 interaction and the subsequent degradation [95]. Then, the stabilized NRF2 protein is translocated into the nucleus to bind with antioxidant response elements (AREs) and activate the transcription of enzymatic antioxidants such as CAT, PRX and GPX, as well as enzymes involved in GSH metabolism [96]. Furthermore, it has also been demonstrated that NRF2 activation increases the production of NADPH by transcriptionally activating the key enzymes in the PPP and one-carbon metabolism pathways [97, 98].
ROS, a double-edged sword in cancer progression
The double-edged roles of ROS in cancer progression are complicated. We hypothesize this contradiction is rooted in the fact that ROS do not operate as a single biochemical entity but play diverse roles as secondary messengers in a manner defined by their variable concentrations, distributions and durations.
The carcinogenic effects of ROS
Beginning in the 1990s, studies set the stage for the concept that ROS are a driving factor for tumorigenesis [99]. One important characteristic of cancer cells is their increased ROS levels compared to those in their counterpart cells and the subsequently elevated levels of antioxidants to detoxify the accumulated ROS to reinstitute a redox balance. ROS are thought to play oncogenic roles by contributing to activation of proto-oncogenes and inactivation of tumor suppressor genes and by acting as signaling molecules to induce abnormal cell growth and metastasis.
ROS-related carcinogenic genetic alterations
Excessive ROS generated by environmental carcinogens, mitochondrial ETC or NADPH oxidases induce DNA damage, including depurination and depyrimidination, single- and double-stranded DNA breaks, base modifications and DNA-protein crosslinks [100]. Moreover, ROS not only delay the identification of damaged regions by affecting sensor kinases (ATM and ATR) and downstream transducer kinases (CHK1 and CHK2) [101, 102], but also impair the activation of DNA repair enzyme OGG1 by oxidation of critical cysteine residues [103]. Accumulation of DNA lesions affects the interpretation and transmission of genetic information, which leads to permanent changes in genetic material and is one of the vital steps involved in carcinogenic mutagenesis and tumor transformation [104]. 8-Hydroxy-2 deoxyguanosine (8-oxo-dG), an oxidizing adduct generated by ROS-associated DNA damage, is commonly utilized to test intracellular oxidative stress levels and is highly expressed in a variety of malignant tumor tissues than in the matched normal ones [105, 106].
Besides the direct carcinogenic effects of DNA damage and chromosomal instability, ROS also act as signaling molecules regulated by oncogene activation or anti-oncogene inactivation. Ectopic expression of the proto-oncogene p21RAS in NIH3T3 fibroblast cells produces large amounts of superoxide via RAC1 activation in NOX complexes and leads to enhanced mitogenic activity, which can be reversed by the chemical antioxidant N-acetyl-L-cysteine (NAC) [107]. Trp53-knockout mice, which succumb to neoplasia at the age of 6 months, exhibit elevated ROS levels and karyotypic abnormalities in different organs. Strikingly, prepartum administration of NAC-supplemented water and continuation of this treatment throughout the life of Trp53-/- progeny can extend the lifespan and suppress lymphoma formation [108]. The breast cancer susceptibility gene, BRCA1, can protect the cells under oxidative stress. Embryonic fibroblasts with genetic ablation of BRCA1 show higher ROS levels than those from wild-type mice [109]. In contrast, BRCA1 overexpression in breast cancer cells stimulates ARE-driven transcriptional activity and upregulates phase II antioxidant enzymes, including glutathione S-transferase and glutathione peroxidase, by enhancing the activity of NRF2 [110].
ROS driven cellular proliferation
In recent decades, researches have been focused on ROS-dependent stimulation of cellular proliferation. Among them, the seminal achievements are the identification of ROS as second messengers participating in growth factor activation via the PI3K/AKT/mTOR and MAPK/ERK mitogenic signaling cascades (Figure 2).
The carcinogenic effects of ROS. (Ⅰ) ROS drive proliferation by activating of PI3K/AKT/mTOR and MAPK mitogenic signaling cascades: the devitalized oxidation of PTEN and PTP1B impair their inhibition on PI3K and cause the hyper-activation of AKT and mTOR; ROS accumulation can respectively activate ASK1, PKG and JNK to further stimulate the downstream MAPKK and MAPK mitosis cascades. (Ⅱ) ROS participate in cancer cell EMT: RAC1 not only directly affects cytoskeleton rearrangement but also up-regulates FAK or inhibits RhoA expression through ROS generation to promote cytoskeleton rearrangement; ROS pile up increases MMP expression by activating NF-κB phosphorylation to enhance ECM degradation; ROS suppress HIF ubiquitin degradation and promote its interaction with p300 to induce angiogenesis.
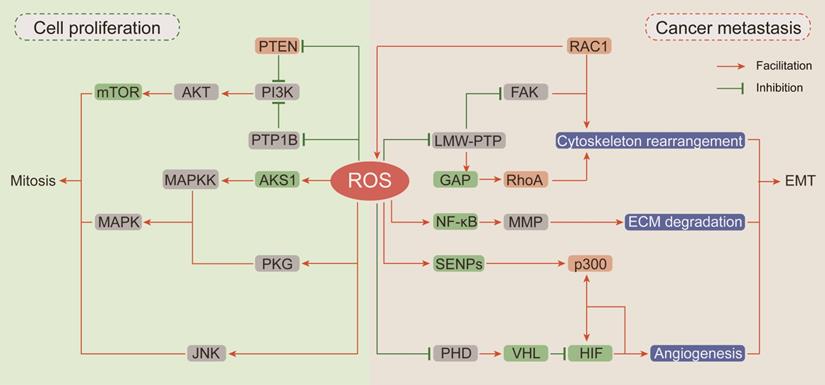
Thiol-disulfide transformation upon H2O2 treatment leads to reversible inactivation of phosphatase and tensin homolog (PTEN) [111], which prompted researchers to investigate the mechanism of ROS in neoplastic progression. Then, oxidation-dependent conversion of cysteine into sulfenyl amide in the catalytic subunit of protein tyrosine phosphatase (PTP) 1B was described, and this conversion is consequently accompanied by a conformational change and inhibition of substrate binding [112]. Both PTEN and PTP1B are negative regulators of phosphoinositide 3-kinase (PI3K) and protein kinase B (AKT) [113]. Given the importance of PI3K/AKT in mitogenic signaling cascades, hyperactivation of this pathway by upstream devitalized oxidation of PTEN/PTP1B is a hallmark of malignancies [114, 115]. In neuroblastoma cells, excessive ROS generated by NOXs after insulin stimulation causes oxidative inactivation of PTEN and phosphorylation activation of PI3K/AKT. In neuroblastoma cells pretreated with the NOX inhibitor diphenyleneiodonium (DPI) before insulin stimulation, insulin-induced phosphorylation of PI3K/Akt is markedly reduced [116]. In breast cancer cells, transient H2O2 accumulation via chemokine CXCL12-activated NOX2 results in oxidation of PTEN and PTP1B followed by activation of PI3K/AKT, but the accumulated H2O2 can be neutralized by DPI and CAT [113].
The mitogen-activated protein kinases (MAPKs) comprise four homologs including extracellular signal-related kinases (Erk1/2), c-Jun N-terminal kinase (JNK), p38 kinase (p38), and big MAP kinase 1 (BMK1/Erk5), and they are activated by a three-rung kinase tier: MAPK kinase kinases (MAPKKKs), MAPK kinases (MAPKKs) and MAPKs [117]. Apoptosis signal-regulated kinase 1 (ASK1), an MAPKKK, is inactivated by the reduced form of thioredoxin (TRX) through inhibition of Thr-838 phosphorylation in the activation loop [118]. ROS accumulation or antioxidant deficiency induces the oxidation of TRX, resulting in its dissociation from ASK1 and allowing subsequent restoration of ASK1 kinase activity [119]. cGMP-dependent protein kinase (PKG), a redox sensor activated by H2O2-dependent oxidation of Cys-42, is involved in MAPK activation [120, 121]. In addition to influencing the upstream of MAPKs, ROS can also activate MAPKs by directly inhibiting MAPK phosphatases. JNK-inactivating phosphatases have been shown to be inhibited by ROS through reversible oxidation of a catalytic site cysteine to sulfenic acid, thus sustaining JNK activation [122].
ROS promote EMT
Epithelial-to-mesenchymal transition (EMT) has been defined as an early event in cancer metastasis, which is linked with loss of cell-to-cell adhesion, loss of interaction with the extracellular matrix (ECM) and migration towards blood and lymphatic vessels [123]. ROS are involved in these processes by inducing Rho family GTPase-dependent cytoskeletal rearrangement, promoting matrix metalloprotease (MMP)-dependent ECM protein degradation and accelerating hypoxia-inducible factor (HIF)-dependent angiogenesis [124, 125] (Figure 2).
EMT is initiated by loss of cell polarity and detachment from surrounding cells via a complex cascade of cytoskeletal rearrangement brought about by the coordinated action of small Rho family GTPases [126]. Among Rho family GTPases, RAC1 promotes membrane protrusion into lamellipodia and filopodia and the establishment of focal contacts at the leading edge [127]. An intriguing and obvious question is whether a potential relationship exists between the effects of RAC1 on cytoskeletal dynamics and its capacity for ROS generation by participating in NOX complex assembly. Along this line of investigation, Moldovan et al. (1999) showed that actin cytoskeleton reorganization induced by RAC1 in human endothelial cells required the production of superoxide [128]. Furthermore, the transient increase in ROS levels induced by RAC1 activation causes fibroblast cell adhesion and spreading onto fibronectin by upregulating focal adhesion kinase (FAK) expression through reversible oxidation of low molecular weight PTP (LMW-PTP) [129]. RAC1 and RhoA (Rho family GTPase A) appear to orchestrate two different and mutually exclusive motility programs in invading malignant cells. In an attempt to clarify the mechanism by which RAC1 regulates RhoA signaling, Nimnual et al. (2003) found that ROS derived from RAC1 inactivated LMW-PTP, causing hyperphosphorylation of p190Rho-GTPase activating protein (GAP), thereby inhibiting RhoA expression [130]. Collectively, these observations clearly indicate that RAC1 is an important node in the crosstalk between oxidative stress and cytoskeletal rearrangement.
The critical stage in EMT is scavenging the surrounding physical obstacles. MMPs compose a family of endopeptidases transactivated by nuclear factor-κB (NF-κB), which can cleave almost every protein component of the ECM [131]. It has been reported that RAC1-dependent ROS production regulates the expressions of MMPs. In human articular chondrocytes, stimulation of integrin-α5β1 by fragments of the ECM protein fibronectin increases intracellular levels of ROS and leads to increased MMP-13 expression by activating NF-κB phosphorylation [132]. The antioxidant agent NAC can completely block this regulatory cascade. Epidermal growth factor (EGF) enhances the invasive capacity of PANC-1 human pancreatic cancer cells by inducing secretion of the collagenase MMP-2. The signaling events downstream of the EGF receptor involve RAC1-generated ROS, which are responsible for NF-κB activation and MMP-2 secretion [133]. Taken together, these evidences show that MMPs can be regulated by integrin-ECM-interacting signaling pathways that are susceptible to modification by ROS.
Emerging lines of evidences indicate that hypoxia-induced factors (HIFs) are frequently active key transcriptional regulators which orchestrate signal transduction cascades to induce angiogenesis [134]. Major HIF isoforms are accumulated in hypoxic condition and are rapidly degraded in the presence of oxygen due to prolyl hydroxylation by prolyl hydroxylases (PHDs) and subsequent ubiquitin-mediated degradation by Von Hippel Lindau (VHL) [135]. Compelling lines of evidences indicate that HIFs are involved in redox regulation via multiple mechanisms. Mild oxidative stress promotes p300 de-SUMOylation by retarding the degradation of Sentrin/SUMO-specific proteases (SENPs) and subsequently enhances the binding of p300 to HIF-1α [136]. In addition, hydrogen peroxide inhibits prolyl hydroxylation to promote HIF stabilization by oxidizing catalytic ferrous iron in PHD and inhibiting its activity, and this effect can be reversed by the antioxidant agent vitamin C [137]. Moreover, it has been recently reported that NAC exerts antitumoral effects in tumorigenic mouse models mainly by decreasing HIF-1α expression [138].
The tumor-suppressive roles of ROS
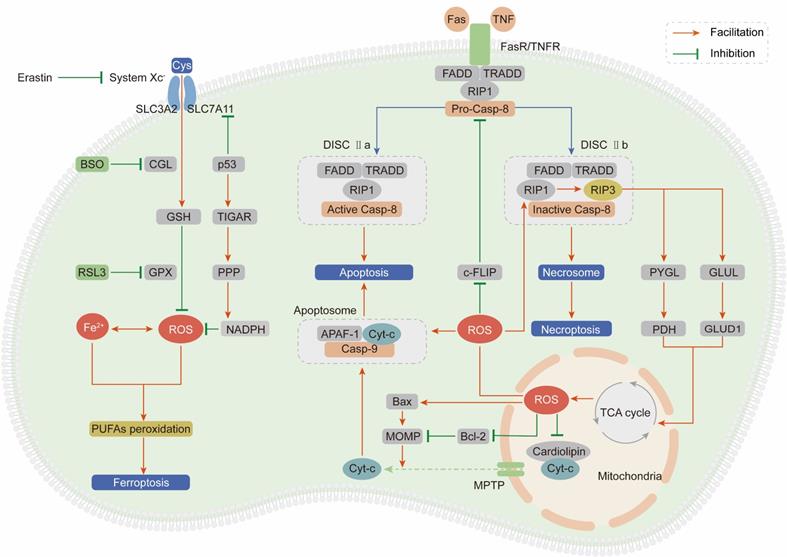
As outlined above, ROS comprise multiple signaling entities with opposite effects and diverse spatiotemporal functions in cancer progression. When the ROS accumulation exceeds the tipping point, their carcinogenic roles in proliferation and invasion are shifted to antitumor effects via the induction of regulated cell death (RCD) programs, mainly including apoptosis, necroptosis and ferroptosis (Figure 3).
ROS induce cell death, mainly including apoptosis, necroptosis and ferroptosis. (Ⅰ) Exceeding ROS promote both the extrinsic and intrinsic apoptosis pathway: ROS activate extrinsic apoptosis pathway by accelerating the ubiquitin degradation of c-FLIP, then enhancing the binding between the adaptor protein and pro-caspase-8; ROS induce intrinsic apoptosis by facilitating the release of Cyt-c from mitochondria to cytoplasm to form apoptosome with casp-9 and APAF-1. (Ⅱ) ROS and necroptosis form a positive feedback loop: ROS stabilize RIP3 protein to lead to the formation of DISC Ⅱb (necrosome); in turn, RIP3 can facilitate the TCA cycle and aerobic respiration in mitochondria to induce ROS generation. (Ⅲ) Ferroptosis is a ROS-dependent form of RCD: the basic of ferroptosis is GSH anabolism disorder leads to the lethal accumulation of PUFAs peroxidation; p53 plays opposite roles on ROS and ferroptosis by inhibiting SLC7A11 expression or increasing NADPH production.
Apoptosis
Apoptosis, also known as type I programmed cell death, is mediated by extrinsic and intrinsic pathways, which are executed by specific cysteine proteases known as caspases [139].
The extrinsic apoptotic pathway is driven by the ligand-receptor interaction between death-inducing ligands such as Fas ligand (FasL) and tumor necrosis factor (TNF) and their respective receptors Fas receptor (FasR) and TNF receptor (TNFR) [140, 141]. After the ligand-receptor interaction, an adaptor protein (FADD for FasR and TRADD for TNFR), receptor-interacting protein kinase 1 (RIP1) and procaspase-8 are recruited to form the death-inducing signaling complex (DISC) and subsequently induce apoptosis [142]. ROS stimulate the extrinsic apoptotic pathway by accelerating the ubiquitin-mediated proteasomal degradation of the antiapoptotic factor cellular FLICE-inhibitory protein (c-FLIP), which impedes formation of the DISC through competitive binding with procaspase-8 for the adaptor protein [143, 144]. Pretreatment with NAC can effectively stabilize the c-FLIP protein and facilitate apoptosis, demonstrating that ROS are apoptosis-inducing factors.
The intrinsic apoptotic pathway is activated in a mitochondria-dependent manner by release of the proapoptotic factor cytochrome-c (Cyt-c) from mitochondria through the mitochondrial permeability transition pore (MPTP) [145]. In mitochondria, Cyt-c is immobilized by reduced cardiolipin. After cardiolipin is oxidized by ROS derived from ETC, its affinity for Cyt-c is attenuated, leading to Cyt-c penetration and release into the cytosol [146]. Then, Cyt-c interacts with apoptotic protease activating factor 1 (APAF-1) and procaspase-9 to form the apoptosome, subsequently activating the caspase-9 signaling cascade and inducing apoptosis [147]. Enoksson et al. (2005) showed that overexpression of glutaredoxin 2 (GRX2) in HeLa cells specifically inhibited Cyt-c release and caspase activation by preventing cardiolipin oxidation in mitochondria [148]. In addition to Cyt-c, caspase-9 is a direct target of ROS. Zuo et al. (2009) demonstrated that oxidative modification of Cys-403 in caspase-9 promoted a disulfide-mediated interaction with APAF-1 and facilitated autocleavage-mediated activation of caspase-9 [149]. The last but not the least, ROS accumulation enhances mitochondrial outer membrane permeabilization (MOMP) via regulating B-cell lymphoma-2 (Bcl-2) family and subsequently induces the release of Cyt-c [150, 151]. The ratio between two hostile proteins of BCL-2 family (pro-apoptotic Bax vs anti-apoptotic Bcl-2) is modulated by ROS via following mechanisms: direct oxidation of Bcl-2 at Cys-158 and Cys-229 [152], decreasing ubiquitination of Bax and increasing ubiquitination of Bcl-2 [153, 154].
Necroptosis
Necroptosis, termed type III programmed cell death, is initiated in a manner similar to the extrinsic apoptotic pathway: the ligand-receptor interaction between TNFR or FasR and their respective ligands [155]. However, necroptosis is caspase-independent and involves receptor-interacting protein kinase 3 (RIP3) to form DISC IIb, distinct from DISC IIa in apoptosis [156]. Accumulating evidences suggest that necroptosis plays a vital role in cancer biology and influences the prognosis of patients who receive antineoplastic chemotherapy or radiotherapy [157]. Inducing necroptosis has emerged as a novel approach for bypassing apoptosis-resistance and a new target for cancer therapy.
Based on a series of studies, we draw the conclusion that ROS and necroptosis can form a positive feedback loop. Excessive ROS oxidize the Cys-257, Cys-268 and Cys-586 residues of RIP1 and subsequently activate RIP1 through Ser-161 autophosphorylation, which protects the RIP3 protein from cleavage by caspase-8 and leads to the formation of the DISC IIb [158]. In turn, RIP3 can facilitate the TCA cycle and aerobic respiration in mitochondria to induce ROS generation through two distinct metabolic signaling pathways: (1) upregulation of glycogen phosphorylase (PYGL) and pyruvate dehydrogenase (PDH) expression [159, 160]; (2) elevation of glutamate-ammonia ligase (GLUL) and glutamate dehydrogenase 1 (GLUD1) expression to increase glutaminolysis [161].
Ferroptosis
Ferroptosis is an iron- and ROS-dependent form of regulated cell death (RCD), and is morphologically, biochemically, and genetically distinct from apoptosis and necroptosis [162]. The oxidative stress burden caused by an excessive intracellular iron level and inadequate GSH leads to lethal accumulation of peroxidated polyunsaturated fatty acids (PUFAs), which is the fundamental characteristic of ferroptosis [163]. Recently, growing researches suggest that small molecules-induced ferroptosis has a strong inhibition of tumor growth and enhances the sensitivity of chemotherapeutic drugs, especially in the condition of drug resistance [164]. These evidences have highlighted the importance of ferroptosis in cancer therapy.
In 2012, Dixon et al. first proposed the concept of ferroptosis by using the oncogenic RAS-selective lethal (RSL) small molecule erastin to trigger an iron- and ROS-dependent form of cell death. This group unequivocally proved that erastin can inhibit cystine uptake through the cystine/glutamate antiporter (system XC-) and impair the GSH-dependent GPX antioxidant system, ultimately leading to ferroptosis [165]. Cotreatment with the iron chelator deferoxamine or the lipid ROS scavenger ferrostatin-1 can significantly reverse the lethal effect of erastin. In addition to erastin, RAS-selective lethal compound 3 (RSL3) can also lead to ferroptosis, not by blocking system XC- but by inhibiting GPX4, which can be counteracted by GPX4 overexpression [166]. Moreover, buthionine sulfoximine (BSO), a GSH deletion agent, has been proven to initiate ferroptosis by inhibiting the activity of glutamate-cysteine ligase (GCL), which is the rate-limiting enzyme in GSH synthesis [167].
System XC- is a dimeric channel protein that is structurally composed of SLC7A11 and SLC3A2 and is responsible for maintaining redox homeostasis by transporting cystine to synthesize GSH [168]. Recently, it was demonstrated that p53 can inhibit cystine uptake by repressing SLC7A11 expression and inducing ferroptosis [169]. Combining these findings with the abovementioned ROS-dependent carcinogenic roles in Trp53-knockout mice, we summarize strong evidences to support the idea that p53 plays paradoxical roles to dynamically maintain ROS balance. On one hand, p53 induces TIGAR expression to redirect glucose towards metabolism through the PPP and increase cytosolic NADPH production [170]. On the other hand, p53 impedes cystine uptake through system XC- to weaken the antioxidant defense system [169].
The application of antioxidants in cancer prevention
ROS accumulation in normal cells is one of the initiating factors in the early stage of the neoplastic process. Therefore, an appropriate application of antioxidants can decrease the oxidative stress burden, consequently preventing normal cells from sliding into the abyss of malignant transformation. Numerous epidemiologic data and preclinical/clinical studies have suggested that keeping an antioxidative dietary or pharmaceutical application of antioxidative phytochemicals can effectively prevent tumorigenesis.
An antioxidative dietary reduces cancer incidence
It is widely accepted that fruit and vegetables rich in antioxidant nutrients are important components of a healthy diet and can reduce the incidence of numerous malignancies. In a study with 77,446 participants, Han et al. (2013) measured the uptake of different antioxidants from the diet in relation to pancreatic cancer risk. They observed that dietary selenium intake was negatively associated with the incidence of pancreatic cancer [171]. In another cohort study, Wright et al. (2004) constructed a dietary antioxidant index that analyzed the comprehensive intake of individual selenium, flavonoids, vitamin C and carotenoids to predict the risk of lung cancer [172]. This group proved that integration of dietary antioxidants can significantly reduce lung cancer incidence in male smokers. In animal models, administration of tomato powder, which is rich in the antioxidant carotenoid lycopene, reduces steatosis and inflammatory foci and abolishes preneoplastic foci in liver tissues of mice injected with diethylnitrosamine (DEN) [173]. Cocoa, with abundant antioxidant properties, can decrease malondialdehyde (MDA) levels, which were elevated in a mouse model of azoxymethane (AOM)/dextran sulfate sodium (DSS)-induced colitis-associated colorectal cancer. Further investigations showed that cocoa treatment could upregulate cellular enzymatic antioxidants, such as SOD, CAT, GPX and GR, by activating the NRF2 signaling cascade [174]. In addition, it has been shown that consumption of ≥150 g of tea per month can effectively protect women against esophageal carcinoma [175].
Application of antioxidative phytochemicals prevents tumorigenesis
Phytochemicals, secondary plant metabolites with antioxidant properties, play important roles in cancer chemoprevention by reversing oxidative stress-induced malignant transformation. Detoxification of ROS via positively regulating phase II antioxidant enzymes is a main factor contributing to the chemopreventive potential of phytochemicals [176].
Curcumin, contained in turmeric has been demonstrated to have preventive effects against multiple types of chemical-induced spontaneous neoplasms in animal models by inducing the expression of phase II antioxidant enzymes through activating the KEAP1/NRF2/AME pathway [177, 178]. Epigallocatechin gallate (EGCG), a key active catechin in green tea known to possess antioxidant capacity, has been shown to slow the formation of aberrant colon crypt foci induced by 2-amino-3-methylimidazo [4,5-f] quinoline via activation of the NRF2 signaling pathway [179]. Moreover, in a one-year proof-of-principle clinical study, Bettuzzi et al. (2006) proved that green tea catechins significantly decreased total prostate-specific antigen levels and delayed the emergence of prostate cancer in volunteers with high-grade prostate intraepithelial neoplasia [180].
In recent years, great efforts have been made to investigate the roles of phytochemicals in cancer prevention. Owing to space constraints, these relevant outstanding achievements are summarized in Table 1.
Antioxidative phytochemicals in cancer prevention.
| Phytochemicals | Cell line/tumor model | Anti-cancer effects |
|---|---|---|
| Curcumin | Pancreatic cancer cells (BxPC3 and PANC1) | Suppresses cancer cell migration and invasion [218] |
| Breast cancer cells (MDA-MB-231, BT-483, and MCF7) | Suppresses cancer cell proliferation and invasion [219, 220] | |
| NSCLC cell line (A549 and H460) | Decreases in vitro metastatic progression and increased apoptosis [221, 222] | |
| Prostate cancer cells (PC12) | Inhibits cancer cell invasion [223] | |
| UV-induced skin injury model | Reduces UV-induced cytotoxicity [224] | |
| Dalton's lymphoma bearing mice | Reduces tumor invasiveness [225] | |
| BaP-induced forestomach tumorigenesis | Reduces tumor growth [226] | |
| EGCG | Mammary epithelial cells (MCF10A) | Enhances antioxidant defense capacity [227] |
| Liver cancer cells (HepG2) | Reduces exogenous oxidative stress [228] | |
| Orthotropic mouse model of colon cancer | Reduces primary tumor growth and its metastasis to liver and lungs [229] | |
| Carcinogen-induced mouse model of colon carcinogenesis | Protective role against colon carcinogenesis [179] | |
| Immortalized human keratinocyte (HaCaT) | Protects skin against ionizing-radiation against DNA damage [230] | |
| Resveratrol | Mammary epithelial cells (MCF10A) | Protects against (4-OHE2)- induced migration and transformation [231] |
| Pancreatic cancer cells (BxPC3 and PANC1) | Inhibits ROS-induced proliferation and migration [232] | |
| Prostate cancer cells | Inhibits DHT-induced progression [233] | |
| Estrogen induced breast carcinogenesis | Protects from oxidative stress and its associated DNA damage [234, 235] | |
| Rat model of hepatocarcinogenesis | Suppresses oxidative stress [236] | |
| Hesperidin | Azoxymethane induced Liver carcinogenesis | Inhibits burden of Hepatic tumors [237] |
| NSCLC cell line (A549) | Suppresses migration and invasion in vitro [238] | |
| 2'-hydroxyflavanone | Renal cancer | Inhibits survival of cancer cells in vitro and tumors in vivo [239, 240] |
| Lung cancer cell lines | Inhibits growth in nude mouse xenograft models [241] | |
| Breast cancer | Inhibits cancer cell survival, cell cycle in vitro and tumor progression in vivo [242] | |
| Quercetin | Lung cancer | Suppresses metastasis [243] |
| Breast cancer | Incudes apoptosis and necroptosis [244] | |
| Liver cancer | Inhibits proliferation and induces apoptosis [245] |
The roles of pro-oxidants in cancer therapy
It is widely recognized that the antitumor effects of pro-oxidants are attributed to the induction of excessive oxidative stress and subsequent ROS-dependent cell death, which can be achieved by ROS-induction or antioxidant-inhibition therapies. As summarized in Table 2, multiple drugs with direct or indirect effects on ROS accumulation have been used clinically for cancer treatment. A more detailed investigation of the influences of these drugs on redox metabolism would be informative for designing individualized treatments with fewer side effects and a lower propensity of drug resistance.
Pro-oxidative drugs in antineoplastic therapies.
| Drug | Pharmacological mechanism | Cancer types | Phase (status) | Clinical trial ID |
|---|---|---|---|---|
| Directly affect redox metabolism | ||||
| NOV-002 | Glutathione disulphide mimetic; alters intracellular GSSG/GSH ratio [246] | Non-Small Cell Lung Carcinoma | Phase Ⅲ (Completed) | NCT00347412 |
| Ovarian Cancer | Phase Ⅱ (Completed) | NCT00345540 | ||
| Leukemias / Myelodysplastic Syndrome | Phase Ⅱ (Withdrawn) | NCT00960726 | ||
| L-asparaginase | Depletes glutamine; reduces GSH [247] | Acute Lymphoblastic Leukaemias (ALL) | Phase Ⅳ (Completed) | NCT00494897 |
| Peripheral T Cell Lymphoma (PTCL) | Phase Ⅳ (Recruiting) | NCT03071822 | ||
| Acute Myeloid Leukemia (AML) | Phase Ⅲ (Recruiting) | NCT04293562 | ||
| Adenocarcinoma of the Pancreas | Phase Ⅲ (Recruiting) | NCT03665441 | ||
| Sulphasalazine | Inhibitor of system XC-; reduces intracellular transport of cystine required for GSH synthesis [200] | Glioblastomas | Phase Ⅰ (Recruiting) | NCT04205357 |
| Buthionine sulphoximine (BSO) | Glutamate-cysteine ligase complex inhibitor; inhibits de novo GSH synthesis [195] | Neuroblastomas | Phase Ⅰ (Completed) | NCT00002730 |
| Melanoma | Phase Ⅰ (Withdrawn) | NCT00661336 | ||
| Arsenic trioxide (As2O3) | Reacts with cysteine residues on crucial proteins; inhibits mitochondrial respiratory function, thereby increasing ROS generation [248] | Acute Promyelocytic Leukemia (APL) | Phase Ⅳ (Active Not Recruiting) | NCT01987297 |
| Neuroblastoma | Phase Ⅱ (Completed) | NCT00024258 | ||
| Small Cell Lung Cancer (SCLC) | Phase Ⅱ (Completed) | NCT01470248 | ||
| Cervical Cancers | Phase Ⅱ (Completed) | NCT00005999 | ||
| Liver Cancer | Phase Ⅱ (Completed) | NCT00128596 | ||
| Indirectly affect redox metabolism | ||||
| Celecoxib | A selective cyclooxygenase-2 (COX-2) inhibitor; induction of ROS by inhibiting mitochondrial oxygen consumption [249] | Colorectal Cancers | Phase Ⅳ (Completed) | NCT00473980 |
| Hepatocellular Carcinoma (HCC) | Phase Ⅳ (Completed) | NCT02961998 | ||
| Biliary-pancreas Cancer | Phase Ⅳ (Active Not Recruiting) | NCT01111591 | ||
| Breast Cancer | Phase Ⅲ (Completed) | NCT02429427 | ||
| Lung Cancers | Phase Ⅲ (Terminated) | NCT01041781 | ||
| Nelfinavir | Originally developed as HIV protease inhibitor but it also induces mitochondrial ROS production [250] | Cervical cancer | Phase Ⅲ (Recruiting) | NCT03256916 |
| Adenoid Cystic Cancer of the Head and Neck | Phase Ⅱ (Completed) | NCT01065844 | ||
| AIDS-Related Kaposi's Sarcoma | Phase Ⅱ (Completed) | NCT00003008 | ||
| NSCLC | Phase Ⅱ (Terminated) | NCT01108666 NCT00791336 | ||
| Bortezomib | Proteasome inhibitor; induces ROS owing to mitochondrial dysregulation and ER stress [251] | Multiple Myeloma | Phase Ⅳ (Completed) | NCT02268890 |
| Acute Myeloid Leukemia (AML) | Phase Ⅲ (Active Not Recruiting) | NCT01371981 | ||
| Anthracyclines (doxorubicin) | Induce the generation of ROS through two main pathways: a non-enzymatic pathway that utilizes iron, and an enzymatic mechanism that involves the mitochondrial respiratory chain [252] | Follicular Lymphoma | Phase Ⅳ (Active Not Recruiting) | NCT03817853 |
| Breast Cancer | Phase Ⅳ (Active Not Recruiting) | NCT02419742 | ||
| Multiple Myeloma | Phase Ⅳ (Active Not Recruiting) | NCT02577783 | ||
| Non-Hodgkin's Lymphoma (NHL) | Phase Ⅳ (Completed) | NCT00969462 | ||
| HCC | Phase Ⅲ (Active Not Recruiting) | NCT01015833 | ||
| 2-methoxyestradiol | Metabolite of estradiol-17β; induces ROS through the loss of mitochondrial membrane potential [253] | Renal Cell Carcinoma | Phase Ⅱ (Completed) | NCT00444314 |
| Ovarian Cancer | Phase Ⅱ (Completed) | NCT00400348 | ||
| Prostate Cancer | Phase Ⅱ (Completed) | NCT00394810 | ||
| Fenretinide (4-hydroxyphenyl retinamide) | Synthetic retinoid derivative; induces apoptosis through the production of ROS and mitochondrial disruption [254] | Breast Cancer | Phase Ⅲ (Completed) | NCT00002646 |
| Bladder Cancer | Phase Ⅲ (Completed) | NCT00004154 | ||
| Prostate Cancer | Phase Ⅱ (Completed) | NCT00080899 | ||
| Head and Neck Carcinoma | Phase Ⅱ (Completed) | NCT00006471 | ||
| Lung Cancers | Phase Ⅱ (Completed) | NCT00009971 | ||
| Renal Cancers | Phase Ⅱ (Completed) | NCT00011973 | ||
ROS-inducing therapies
Traditional antineoplastic therapies, including chemotherapy and radiotherapy, are currently used to induce excessive levels of oxidative stress to selectively kill cancer cells. Patients who receive these treatments exhibit signs of ROS-induced cell death, DNA damage and lipid peroxidation.
Chemotherapy
Conventional chemotherapeutics such as anthracyclines and platinum coordination complexes generate extremely high levels of ROS [181]. For example, doxorubicin (DOX), an anthracycline antibiotic, induces oxidative DNA damage and caspase-dependent apoptosis initiated by direct H2O2 generation through NOX activation in human leukemia cells [182]. Moreover, in p53-null human osteosarcoma Saos-2 cells, ROS levels were increased, with subsequent mitochondrial membrane depolarization and Cyt-c release after 48 h of DOX treatment, and CAT abolished the proapoptotic effects of DOX [183]. In addition, it was found that DOX was indiscriminatingly localized at the mitochondria of normal cells and competes with coenzyme Q10 in ETC to induce ROS production, which is the basis of its cardiotoxicity [184]. Cisplatin, another classical conventional anticancer drug recognized as an agent that induces a mitochondrial-dependent ROS response, which significantly enhances the cytotoxic effect caused by mitochondrial DNA damage [185]. In HCT116 colon carcinoma cells, the apoptotic activity of cisplatin is dependent on p53-associated ROS accumulation and downstream p38 MAPK activation [186]. Treatment with antioxidants (ascorbic acid and dehydroascorbic acid) or a p53 inhibitor (pifithrin-α) can block cisplatin-induced apoptosis and reduce the generation of ROS. Similar to DOX, oxidative stress exacerbation by cisplatin occurs not only in cancer cells but also in normal cells. For instance, hearing loss, one of the common side effects caused by the ototoxicity of cisplatin, is induced by excessive generation of ROS in cochlear cells and can be effectively prevented or alleviated by the application of antioxidants [187]. In addition to DOX and cisplatin, 2-methoxyestradiol (2ME2), a metabolite of estradiol-17 beta, is also known to induce apoptosis in tumor cells via ROS generation. Mechanistically, 2ME2 inhibits mitochondrial respiration by inactivating complex I and shunts electrons leaked from ETC to form superoxide, subsequently resulting in Cyt-c release and caspase-9 activation [188].
Radiotherapy
Early in the 1950s, it was indeed well known that a high local concentration of molecular oxygen enhanced the efficacy of radiotherapy, as DNA lesions caused by ROS generated during H2O radiolysis can react with O2 to form superoxide anions [189]. Recent studies have shown that endogenous ROS, whether generated by the mitochondrial ETC or NADPH oxidases, can be activated by radiation exposure, leading to persistent oxidative stress in tumor cells [190, 191]. Because the ROS level is the critical mediator of irradiation-induced cell death, the mechanisms of radiation resistance are associated with impaired ROS production or a powerful antioxidant system. It has been shown that pharmacologic depletion of GSH by BSO in cancer stem cells (CSCs) significantly decreases their clonogenicity and results in radiosensitization [192]. Specifically, increasing ROS levels in tumor cells during radiotherapy can significantly enhance the efficiency and decrease the dosage of radiation, subsequently reducing nonselective killing of normal cells and severe systemic side effects on bystander organs. With this insight, Chen et al. (2019) developed a Gd-doped titania nanosensitizer that targets mitochondria to achieve efficient radiotherapy by triggering a "domino effect" of ROS accumulation, mitochondrial permeability transition, Cyt-c release and caspase-dependent cell apoptosis [193].
Antioxidant-inhibiting therapies
GSH and TRX are central players in antioxidant systems by transmitting reducing equivalents from NADPH to oxidized molecules through PRX- and GPX-dependent intracellular enzymatic redox reactions. Thus, antineoplastic therapeutic strategies selectively targeting GSH and TRX metabolism in cancer cells are effective measures to enhance the potency of ROS modulation.
6.2.1 Drugs that affect GSH metabolism
Compared to normal cells, cancer cells with high GSH content seem to be more sensitive to a selective GSH depletion strategy. As noted above, GCL is the rate-limiting enzyme in GSH synthesis and has been considered as an antineoplastic target for over 30 years [194]. Buthionine sulfoximine (BSO), an irreversible inhibitor of GCL, is most frequently used as an adjunct to synergize with other chemotherapies. Gana et al. (2019) proved that multidrug resistance protein 1 (MRP1) modulators (verapamil and apigenin) synergize with BSO to collaterally sensitize MRP1-expressing cancer cells to arsenic trioxide and selectively kill them [195]. In high-risk neuroblastoma, increased cellular GSH induces resistance to melphalan, a myeloablative drug. In 2016, a phase I trial of BSO and melphalan with autologous stem cells for recurrent/refractory neuroblastoma was conducted by Villablanca et al. The results showed that a combination of BSO plus melphalan was feasible and effective in the treatment of neuroblastoma [196]. Moreover, in a preclinical animal trial, the cytotoxic effect of azathioprine plus BSO was found to be effective for localized treatment of hepatocellular carcinoma, and further in vitro studies suggested that GSH depletion by BSO gave rise to Cyt-c release and mitochondrial-dependent cell death [197].
Cystine imported into the cytosol by system Xc- is an important material for GSH synthesis. Sulfasalazine, an anti-inflammatory drug used for various types of arthritis [198], has been found to specifically inhibit system Xc- activity [199]. Treatment with sulfasalazine led to reduced uptake of cystine and subsequent depletion of GSH, which inhibited the proliferation of human pancreatic cancer cells both in vitro and in vivo [200]. In human small cell lung cancer cells, it was proven that sulfasalazine was potentially useful as a target for therapies based on GSH depletion [201].
6.2.2 Drugs that affect TRX metabolism
As mentioned above, TRX, the molecular substrate of peroxiredoxin (PRX), participates in the scavenging of H2O2 to maintain redox balance. In addition, the reduced form of TRX-(SH)2 can directly reduce disulfide groups in oxidized proteins, and the oxidized form of TRX-(S)2 is then reduced by thioredoxin reductase (TRXR) in an NADPH-dependent manner. In recent years, TRXR/TRX has been recognized as an important modulator of tumor development; hence, targeting TRXR/TRX is a promising strategy for antitumor therapy. Auranofin, a gold compound initially utilized for the treatment of rheumatoid arthritis [202], is a strong inhibitor of TRXR in both the cytosol and mitochondria [203]. In 2014, Fiskus et al. reported the identification of auranofin in repurposing for the treatment of chronic lymphocytic leukemia (CLL) by inhibiting TRXR activity and increasing intracellular ROS levels [204]. Similarly, Pessetto et al. (2013) attempted to reposition auranofin to treat metastatic gastrointestinal stromal tumor (GIST). They found that auranofin dramatically inhibited GIST cell growth and suggested the clinical benefit of auranofin in GIST patients, particularly those with imatinib resistance [205]. Ethaselen (1,2-[bis(1,2-benzisoselenazolone-3(2H)-ketone)]ethane, BBSKE), is a novel TRXR-targeted organoselenium compound and is currently under investigation in clinical trial NCT02166242 for the treatment of advanced non-small cell lung cancer (NSCLC) [206]. In previous studies, ethaselen has shown antineoplastic effects against prostate, lung, colon and gastric cancer both in vivo and in vitro by suppressing TRXR activity, and elevating ROS levels, and subsequently inducing cell death [207-210].
PX-12 (1-methylpropyl 2-mercaptoimidazolyl disulfide), also called IV-2 in early references, was initially identified as a competitive inhibitor of TRXR-mediated reduction of TRX in 1998 [211]. Recently, PX-12 has been proven to inhibit the growth and induce the apoptosis of lung, cervical, liver, gastric and colorectal cancer cells by decreasing reduced TRX expression and increasing ROS levels [212-216]. Although PX-12 did not appear to be clinical efficacy in a randomized phase II study in pancreatic cancer, its potential as a clinical anticancer candidate is still promising [217].
Future perspectives
A major question in the field of cancer redox biology is whether ROS can function as specific weapons to destroy tumor cells and not in the dualistic role, indiscriminately damaging normal cells. This question is the basis of many controversies in the field of redox biology and accounts for the conflicting results of clinical trials and experimental studies. Although measures to globally elevate ROS to cytotoxic levels have promising availability for killing cancer cells, such strategies inevitably induce systemic toxicity much like conventional chemo- and radiotherapeutic regimens. Leveraging ROS modulation for the development of safe and efficient anticancer therapies necessitates experimental delineation of the distinct redox signaling pathways that exclusively contribute to cancer cell growth and survival. In this regard, further elucidation of ROS-related cysteine modifications and their functional consequences will be fundamental to advancing our understanding of the selective effects of ROS on cancer and normal cells. New molecular probes that allow monitoring of ROS with temporal and spatial specificity will shed further light on the complicated regulatory relationship between different redox couples and their downstream influences on different subcellular organelles. In spite of scientific quandaries and technical challenges, the prospect of interdisciplinary collaborations between oncologists, pharmacists and biochemists towards a better understanding of cancer-specific redox signaling events holds promise for overcoming the controversial side effects of pro-oxidant antineoplastic therapies.
Acknowledgements
This research was supported by National Natural Science Foundation of China (No. 82073286; No. 81672440; No.81972625); The Construction of Liaoning Cancer Research Center (Lung Cancer) (2019JH6/10200011); Technological Special Project of Liaoning Province of China (2019020176-JH1/103); Central financial fund for promoting medical service and safeguarding capability (Capability construction of medical and health organizations) -subsidy to the Construction of Provincial Key Specialty; Research grant to introduced talents of Liaoning Cancer Hospital.
Author Contributions
Writing-review and editing: Yawei Wang, Hai-long Piao, Hong-Xu Liu; Supervision and supplement: Huan Qi, Yu Liu, Chao Duan, Xiaolong Liu, Tian Xia, Di Chen. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lushchak VI. Free radicals, reactive oxygen species, oxidative stress and its classification. Chemico-biological interactions. 2014;224:164-75
2. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014;94:909-50
3. Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu Y. et al. ROS and ROS-Mediated Cellular Signaling. Oxidative medicine and cellular longevity. 2016;2016:4350965
4. Scialò F, Fernández-Ayala DJ, Sanz A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Frontiers in physiology. 2017;8:428
5. El-Kenawi A, Ruffell B. Inflammation, ROS, and Mutagenesis. Cancer cell. 2017;32:727-9
6. Moloney JN, Cotter TG. ROS signalling in the biology of cancer. Seminars in cell & developmental biology. 2018;80:50-64
7. Reczek CR, Chandel NS. ROS Promotes Cancer Cell Survival through Calcium Signaling. Cancer cell. 2018;33:949-51
8. Aggarwal V, Tuli HS, Varol A, Thakral F, Yerer MB, Sak K. et al. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules. 2019 9
9. Anbar AD. Oceans. Elements and evolution. Science (New York, NY). 2008;322:1481-3
10. Mittler R. ROS Are Good. Trends Plant Sci. 2017;22:11-9
11. Niu BY, Li WK, Li JS, Hong QH, Khodahemmati S, Gao JF, et al. Effects of DNA Damage and Oxidative Stress in Human Bronchial Epithelial Cells Exposed to PM(2.5) from Beijing, China, in Winter. International journal of environmental research and public health. 2020; 17
12. Alberg A. The influence of cigarette smoking on circulating concentrations of antioxidant micronutrients. Toxicology. 2002;180:121-37
13. Wang G, Zhang T, Sun W, Wang H, Yin F, Wang Z. et al. Arsenic sulfide induces apoptosis and autophagy through the activation of ROS/JNK and suppression of Akt/mTOR signaling pathways in osteosarcoma. Free radical biology & medicine. 2017;106:24-37
14. He J, Wang M, Jiang Y, Chen Q, Xu S, Xu Q. et al. Chronic arsenic exposure and angiogenesis in human bronchial epithelial cells via the ROS/miR-199a-5p/HIF-1α/COX-2 pathway. Environmental health perspectives. 2014;122:255-61
15. Urso L, Cavallari I, Sharova E, Ciccarese F, Pasello G, Ciminale V. Metabolic rewiring and redox alterations in malignant pleural mesothelioma. British journal of cancer. 2020;122:52-61
16. de Jager TL, Cockrell AE, Du Plessis SS. Ultraviolet Light Induced Generation of Reactive Oxygen Species. Advances in experimental medicine and biology. 2017;996:15-23
17. Lee SY, Jeong EK, Ju MK, Jeon HM, Kim MY, Kim CH. et al. Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation. Molecular cancer. 2017;16:10
18. Xue C, Li X, Liu G, Liu W. Evaluation of Mitochondrial Respiratory Chain on the Generation of Reactive Oxygen Species and Cytotoxicity in HaCaT Cells Induced by Nanosized Titanium Dioxide Under UVA Irradiation. International journal of toxicology. 2016;35:644-53
19. Iatsenko I, Boquete JP, Lemaitre B. Microbiota-Derived Lactate Activates Production of Reactive Oxygen Species by the Intestinal NADPH Oxidase Nox and Shortens Drosophila Lifespan. Immunity. 2018;49:929-42.e5
20. Del Río LA, López-Huertas E. ROS Generation in Peroxisomes and its Role in Cell Signaling. Plant & cell physiology. 2016;57:1364-76
21. Yoboue ED, Sitia R, Simmen T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell death & disease. 2018;9:331
22. Paggio A, Checchetto V, Campo A, Menabò R, Di Marco G, Di Lisa F. et al. Identification of an ATP-sensitive potassium channel in mitochondria. Nature. 2019;572:609-13
23. Xiao W, Wang RS, Handy DE, Loscalzo J. NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxidants & redox signaling. 2018;28:251-72
24. Crompton M. The mitochondrial permeability transition pore and its role in cell death. The Biochemical journal. 1999;341( Pt 2):233-49
25. Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science (New York, NY). 2006;312:1882-3
26. Bienert GP, Møller AL, Kristiansen KA, Schulz A, Møller IM, Schjoerring JK. et al. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. The Journal of biological chemistry. 2007;282:1183-92
27. Miller EW, Dickinson BC, Chang CJ. Aquaporin-3 mediates hydrogen peroxide uptake to regulate downstream intracellular signaling. Proc Natl Acad Sci U S A. 2010;107:15681-6
28. Sousa JS, D'Imprima E, Vonck J. Mitochondrial Respiratory Chain Complexes. Sub-cellular biochemistry. 2018;87:167-227
29. Venditti P, Di Stefano L, Di Meo S. Mitochondrial metabolism of reactive oxygen species. Mitochondrion. 2013;13:71-82
30. Barja G. Mitochondrial oxygen radical generation and leak: sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. Journal of bioenergetics and biomembranes. 1999;31:347-66
31. Muller FL, Liu Y, Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. The Journal of biological chemistry. 2004;279:49064-73
32. Muller F, Crofts AR, Kramer DM. Multiple Q-cycle bypass reactions at the Qo site of the cytochrome bc1 complex. Biochemistry. 2002;41:7866-74
33. Raimondi V, Ciccarese F, Ciminale V. Oncogenic pathways and the electron transport chain: a dangeROS liaison. British journal of cancer. 2020;122:168-81
34. Missirlis F, Hu J, Kirby K, Hilliker AJ, Rouault TA, Phillips JP. Compartment-specific protection of iron-sulfur proteins by superoxide dismutase. The Journal of biological chemistry. 2003;278:47365-9
35. Brand MD. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free radical biology & medicine. 2016;100:14-31
36. Bertero E, Maack C. Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circulation research. 2018;122:1460-78
37. Zhu Z, Kawai T, Umehara T, Hoque SAM, Zeng W, Shimada M. Negative effects of ROS generated during linear sperm motility on gene expression and ATP generation in boar sperm mitochondria. Free radical biology & medicine. 2019;141:159-71
38. Yang Y, Karakhanova S, Hartwig W, D'Haese JG, Philippov PP, Werner J. et al. Mitochondria and Mitochondrial ROS in Cancer: Novel Targets for Anticancer Therapy. Journal of cellular physiology. 2016;231:2570-81
39. Babior BM, Kipnes RS, Curnutte JT. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. The Journal of clinical investigation. 1973;52:741-4
40. Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245-313
41. Block K, Gorin Y. Aiding and abetting roles of NOX oxidases in cellular transformation. Nature reviews Cancer. 2012;12:627-37
42. Buvelot H, Jaquet V, Krause KH. Mammalian NADPH Oxidases. Methods in molecular biology (Clifton, NJ). 2019;1982:17-36
43. Baehner RL, Nathan DG. Leukocyte oxidase: defective activity in chronic granulomatous disease. Science (New York, NY). 1967;155:835-6
44. de Boer M, de Klein A, Hossle JP, Seger R, Corbeel L, Weening RS. et al. Cytochrome b558-negative, autosomal recessive chronic granulomatous disease: two new mutations in the cytochrome b558 light chain of the NADPH oxidase (p22-phox). American journal of human genetics. 1992;51:1127-35
45. Singel KL, Segal BH. NOX2-dependent regulation of inflammation. Clin Sci (Lond). 2016;130:479-90
46. Belambri SA, Rolas L, Raad H, Hurtado-Nedelec M, Dang PM, El-Benna J. NADPH oxidase activation in neutrophils: Role of the phosphorylation of its subunits. European journal of clinical investigation. 2018;48(Suppl 2):e12951
47. Bánfi B, Maturana A, Jaconi S, Arnaudeau S, Laforge T, Sinha B. et al. A mammalian H+ channel generated through alternative splicing of the NADPH oxidase homolog NOH-1. Science (New York, NY). 2000;287:138-42
48. Ago T, Kitazono T, Kuroda J, Kumai Y, Kamouchi M, Ooboshi H. et al. NAD(P)H oxidases in rat basilar arterial endothelial cells. Stroke. 2005;36:1040-6
49. Cheng G, Diebold BA, Hughes Y, Lambeth JD. Nox1-dependent reactive oxygen generation is regulated by Rac1. The Journal of biological chemistry. 2006;281:17718-26
50. Paffenholz R, Bergstrom RA, Pasutto F, Wabnitz P, Munroe RJ, Jagla W. et al. Vestibular defects in head-tilt mice result from mutations in Nox3, encoding an NADPH oxidase. Genes & development. 2004;18:486-91
51. Bánfi B, Malgrange B, Knisz J, Steger K, Dubois-Dauphin M, Krause KH. NOX3, a superoxide-generating NADPH oxidase of the inner ear. The Journal of biological chemistry. 2004;279:46065-72
52. Ueno N, Takeya R, Miyano K, Kikuchi H, Sumimoto H. The NADPH oxidase Nox3 constitutively produces superoxide in a p22phox-dependent manner: its regulation by oxidase organizers and activators. The Journal of biological chemistry. 2005;280:23328-39
53. Cheng G, Ritsick D, Lambeth JD. Nox3 regulation by NOXO1, p47phox, and p67phox. The Journal of biological chemistry. 2004;279:34250-5
54. Geiszt M, Kopp JB, Várnai P, Leto TL. Identification of renox, an NAD(P)H oxidase in kidney. Proc Natl Acad Sci U S A. 2000;97:8010-4
55. Shiose A, Kuroda J, Tsuruya K, Hirai M, Hirakata H, Naito S. et al. A novel superoxide-producing NAD(P)H oxidase in kidney. The Journal of biological chemistry. 2001;276:1417-23
56. Martyn KD, Frederick LM, von Loehneysen K, Dinauer MC, Knaus UG. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cellular signalling. 2006;18:69-82
57. Prior KK, Leisegang MS, Josipovic I, Löwe O, Shah AM, Weissmann N. et al. CRISPR/Cas9-mediated knockout of p22phox leads to loss of Nox1 and Nox4, but not Nox5 activity. Redox biology. 2016;9:287-95
58. Hilenski LL, Clempus RE, Quinn MT, Lambeth JD, Griendling KK. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arteriosclerosis, thrombosis, and vascular biology. 2004;24:677-83
59. Kuroda J, Nakagawa K, Yamasaki T, Nakamura K, Takeya R, Kuribayashi F. et al. The superoxide-producing NAD(P)H oxidase Nox4 in the nucleus of human vascular endothelial cells. Genes to cells: devoted to molecular & cellular mechanisms. 2005;10:1139-51
60. Bánfi B, Molnár G, Maturana A, Steger K, Hegedûs B, Demaurex N. et al. A Ca(2+)-activated NADPH oxidase in testis, spleen, and lymph nodes. The Journal of biological chemistry. 2001;276:37594-601
61. Bánfi B, Tirone F, Durussel I, Knisz J, Moskwa P, Molnár GZ. et al. Mechanism of Ca2+ activation of the NADPH oxidase 5 (NOX5). The Journal of biological chemistry. 2004;279:18583-91
62. Magnani F, Nenci S, Millana Fananas E, Ceccon M, Romero E, Fraaije MW. et al. Crystal structures and atomic model of NADPH oxidase. Proc Natl Acad Sci U S A. 2017;114:6764-9
63. De Deken X, Wang D, Many MC, Costagliola S, Libert F, Vassart G. et al. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. The Journal of biological chemistry. 2000;275:23227-33
64. Dupuy C, Ohayon R, Valent A, Noël-Hudson MS, Dème D, Virion A. Purification of a novel flavoprotein involved in the thyroid NADPH oxidase. Cloning of the porcine and human cdnas. The Journal of biological chemistry. 1999;274:37265-9
65. Ameziane-El-Hassani R, Morand S, Boucher JL, Frapart YM, Apostolou D, Agnandji D. et al. Dual oxidase-2 has an intrinsic Ca2+-dependent H2O2-generating activity. The Journal of biological chemistry. 2005;280:30046-54
66. Fischer H. Mechanisms and function of DUOX in epithelia of the lung. Antioxidants & redox signaling. 2009;11:2453-65
67. Augsburger F, Filippova A, Jaquet V. Methods for Detection of NOX-Derived Superoxide Radical Anion and Hydrogen Peroxide in Cells. Methods in molecular biology (Clifton, NJ). 2019;1982:233-41
68. Tomita K, Takashi Y, Ouchi Y, Kuwahara Y, Igarashi K, Nagasawa T. et al. Lipid peroxidation increases hydrogen peroxide permeability leading to cell death in cancer cell lines that lack mtDNA. Cancer science. 2019;110:2856-66
69. Jeon SA, Kim DW, Cho JY. Neural precursor cell-expressed, developmentally down-regulated 4 (NEDD4) regulates hydrogen peroxide-induced cell proliferation and death through inhibition of Hippo signaling. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2019;33:14772-83
70. Marlein CR, Zaitseva L, Piddock RE, Robinson SD, Edwards DR, Shafat MS. et al. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood. 2017;130:1649-60
71. Wang C, Wang Z, Liu W, Ai Z. ROS-generating oxidase NOX1 promotes the self-renewal activity of CD133+ thyroid cancer cells through activation of the Akt signaling. Cancer letters. 2019;447:154-63
72. Tang CT, Lin XL, Wu S, Liang Q, Yang L, Gao YJ. et al. NOX4-driven ROS formation regulates proliferation and apoptosis of gastric cancer cells through the GLI1 pathway. Cellular signalling. 2018;46:52-63
73. Nguyen NH, Tran GB, Nguyen CT. Anti-oxidative effects of superoxide dismutase 3 on inflammatory diseases. Journal of molecular medicine (Berlin, Germany). 2020;98:59-69
74. Rhee SG. Overview on Peroxiredoxin. Molecules and cells. 2016;39:1-5
75. Zachara BA. Selenium in Complicated Pregnancy. A Review. Advances in clinical chemistry. 2018;86:157-78
76. Gebicka L, Krych-Madej J. The role of catalases in the prevention/promotion of oxidative stress. Journal of inorganic biochemistry. 2019;197:110699
77. Rhee SG, Woo HA, Kil IS, Bae SH. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. The Journal of biological chemistry. 2012;287:4403-10
78. Couto N, Wood J, Barber J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free radical biology & medicine. 2016;95:27-42
79. Sepasi Tehrani H, Moosavi-Movahedi AA. Catalase and its mysteries. Prog Biophys Mol Biol. 2018;140:5-12
80. Miller CG, Holmgren A, Arner ESJ, Schmidt EE. NADPH-dependent and -independent disulfide reductase systems. Free radical biology & medicine. 2018;127:248-61
81. Ju HQ, Lin JF, Tian T, Xie D, Xu RH. NADPH homeostasis in cancer: functions, mechanisms and therapeutic implications. Signal transduction and targeted therapy. 2020;5:231
82. Cho ES, Cha YH, Kim HS, Kim NH, Yook JI. The Pentose Phosphate Pathway as a Potential Target for Cancer Therapy. Biomolecules & therapeutics. 2018;26:29-38
83. Clark O, Yen K, Mellinghoff IK. Molecular Pathways: Isocitrate Dehydrogenase Mutations in Cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2016;22:1837-42
84. Cordeiro AT. NADPH Producing Enzymes as Promising Drug Targets for Chagas Disease. Current medicinal chemistry. 2019;26:6564-71
85. Ronchi JA, Francisco A, Passos LA, Figueira TR, Castilho RF. The Contribution of Nicotinamide Nucleotide Transhydrogenase to Peroxide Detoxification Is Dependent on the Respiratory State and Counterbalanced by Other Sources of NADPH in Liver Mitochondria. The Journal of biological chemistry. 2016;291:20173-87
86. Yang M, Vousden KH. Serine and one-carbon metabolism in cancer. Nature reviews Cancer. 2016;16:650-62
87. Balsa E, Perry EA, Bennett CF, Jedrychowski M, Gygi SP, Doench JG. et al. Defective NADPH production in mitochondrial disease complex I causes inflammation and cell death. Nature communications. 2020;11:2714
88. Palmieri F. The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflugers Archiv: European journal of physiology. 2004;447:689-709
89. Ciccarese F, Ciminale V. Escaping Death: Mitochondrial Redox Homeostasis in Cancer Cells. Frontiers in oncology. 2017;7:117
90. Deshmukh P, Unni S, Krishnappa G, Padmanabhan B. The Keap1-Nrf2 pathway: promising therapeutic target to counteract ROS-mediated damage in cancers and neurodegenerative diseases. Biophysical reviews. 2017;9:41-56
91. Chan JY, Cheung MC, Moi P, Chan K, Kan YW. Chromosomal localization of the human NF-E2 family of bZIP transcription factors by fluorescence in situ hybridization. Human genetics. 1995;95:265-9
92. Motohashi H, O'Connor T, Katsuoka F, Engel JD, Yamamoto M. Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene. 2002;294:1-12
93. Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T. et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Molecular and cellular biology. 2004;24:7130-9
94. Furukawa M, Xiong Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Molecular and cellular biology. 2005;25:162-71
95. Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes to cells: devoted to molecular & cellular mechanisms. 2011;16:123-40
96. Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes & development. 2013;27:2179-91
97. Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H. et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer cell. 2012;22:66-79
98. DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D. et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nature genetics. 2015;47:1475-81
99. Chio IIC, Tuveson DA. ROS in Cancer: The Burning Question. Trends in molecular medicine. 2017;23:411-29
100. Barnes RP, Fouquerel E, Opresko PL. The impact of oxidative DNA damage and stress on telomere homeostasis. Mechanisms of ageing and development. 2019;177:37-45
101. Chang MC, Lin LD, Wu MT, Chan CP, Chang HH, Lee MS. et al. Effects of Camphorquinone on Cytotoxicity, Cell Cycle Regulation and Prostaglandin E2 Production of Dental Pulp Cells: Role of ROS, ATM/Chk2, MEK/ERK and Hemeoxygenase-1. PloS one. 2015;10:e0143663
102. Srinivas US, Tan BWQ, Vellayappan BA, Jeyasekharan AD. ROS and the DNA damage response in cancer. Redox biology. 2019;25:101084
103. Bravard A, Vacher M, Gouget B, Coutant A, de Boisferon FH, Marsin S. et al. Redox regulation of human OGG1 activity in response to cellular oxidative stress. Molecular and cellular biology. 2006;26:7430-6
104. Kotsantis P, Petermann E, Boulton SJ. Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place. Cancer discovery. 2018;8:537-55
105. An AR, Kim KM, Park HS, Jang KY, Moon WS, Kang MJ. et al. Association between Expression of 8-OHdG and Cigarette Smoking in Non-small Cell Lung Cancer. Journal of pathology and translational medicine. 2019;53:217-24
106. Nanashima A, Izumino H, Sumida Y, Tominaga T, Wakata K, Hidaka S. et al. Relationship Between Urinary 8-hydroxydeoxyguanine (8-OHdG) Levels and Clinicopathological Findings in Hepatobiliary Malignancies. Anticancer research. 2016;36:3899-903
107. Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, Fearon ER. et al. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science (New York, NY). 1997;275:1649-52
108. Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005;11:1306-13
109. Cao L, Xu X, Cao LL, Wang RH, Coumoul X, Kim SS. et al. Absence of full-length Brca1 sensitizes mice to oxidative stress and carcinogen-induced tumorigenesis in the esophagus and forestomach. Carcinogenesis. 2007;28:1401-7
110. Bae I, Fan S, Meng Q, Rih JK, Kim HJ, Kang HJ. et al. BRCA1 induces antioxidant gene expression and resistance to oxidative stress. Cancer research. 2004;64:7893-909
111. Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. The Journal of biological chemistry. 2002;277:20336-42
112. Salmeen A, Andersen JN, Myers MP, Meng TC, Hinks JA, Tonks NK. et al. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769-73
113. Satooka H, Hara-Chikuma M. Aquaporin-3 Controls Breast Cancer Cell Migration by Regulating Hydrogen Peroxide Transport and Its Downstream Cell Signaling. Molecular and cellular biology. 2016;36:1206-18
114. Pérez-Ramírez C, Cañadas-Garre M, Molina M, Faus-Dáder MJ, Calleja-Hernández M. PTEN and PI3K/AKT in non-small-cell lung cancer. Pharmacogenomics. 2015;16:1843-62
115. Hu M, Zhu S, Xiong S, Xue X, Zhou X. MicroRNAs and the PTEN/PI3K/Akt pathway in gastric cancer (Review). Oncology reports. 2019;41:1439-54
116. Calvo-Ochoa E, Sánchez-Alegría K, Gómez-Inclán C, Ferrera P, Arias C. Palmitic acid stimulates energy metabolism and inhibits insulin/PI3K/AKT signaling in differentiated human neuroblastoma cells: The role of mTOR activation and mitochondrial ROS production. Neurochemistry international. 2017;110:75-83
117. Peluso I, Yarla NS, Ambra R, Pastore G, Perry G. MAPK signalling pathway in cancers: Olive products as cancer preventive and therapeutic agents. Seminars in cancer biology. 2019;56:185-95
118. Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y. et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. The EMBO journal. 1998;17:2596-606
119. Liu H, Nishitoh H, Ichijo H, Kyriakis JM. Activation of apoptosis signal-regulating kinase 1 (ASK1) by tumor necrosis factor receptor-associated factor 2 requires prior dissociation of the ASK1 inhibitor thioredoxin. Molecular and cellular biology. 2000;20:2198-208
120. Burgoyne JR, Madhani M, Cuello F, Charles RL, Brennan JP, Schröder E. et al. Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science (New York, NY). 2007;317:1393-7
121. Frigolet ME, Thomas G, Beard K, Lu H, Liu L, Fantus IG. The bradykinin-cGMP-PKG pathway augments insulin sensitivity via upregulation of MAPK phosphatase-5 and inhibition of JNK. American journal of physiology Endocrinology and metabolism. 2017;313:E321-e34
122. Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649-61
123. Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006;12:895-904
124. Pani G, Galeotti T, Chiarugi P. Metastasis: cancer cell's escape from oxidative stress. Cancer Metastasis Rev. 2010;29:351-78
125. Chatterjee R, Chatterjee J. ROS and oncogenesis with special reference to EMT and stemness. European journal of cell biology. 2020;99:151073
126. Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629-35
127. Hall A. Rho GTPases and the control of cell behaviour. Biochemical Society transactions. 2005;33:891-5
128. Moldovan L, Irani K, Moldovan NI, Finkel T, Goldschmidt-Clermont PJ. The actin cytoskeleton reorganization induced by Rac1 requires the production of superoxide. Antioxidants & redox signaling. 1999;1:29-43
129. Chiarugi P, Pani G, Giannoni E, Taddei L, Colavitti R, Raugei G. et al. Reactive oxygen species as essential mediators of cell adhesion: the oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. The Journal of cell biology. 2003;161:933-44
130. Nimnual AS, Taylor LJ, Bar-Sagi D. Redox-dependent downregulation of Rho by Rac. Nature cell biology. 2003;5:236-41
131. Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovascular research. 2006;69:562-73
132. Del Carlo M, Schwartz D, Erickson EA, Loeser RF. Endogenous production of reactive oxygen species is required for stimulation of human articular chondrocyte matrix metalloproteinase production by fibronectin fragments. Free radical biology & medicine. 2007;42:1350-8
133. Binker MG, Binker-Cosen AA, Richards D, Oliver B, Cosen-Binker LI. EGF promotes invasion by PANC-1 cells through Rac1/ROS-dependent secretion and activation of MMP-2. Biochemical and biophysical research communications. 2009;379:445-50
134. Choudhry H, Harris AL. Advances in Hypoxia-Inducible Factor Biology. Cell metabolism. 2018;27:281-98
135. Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Molecular cell. 2008;30:393-402
136. Huang C, Han Y, Wang Y, Sun X, Yan S, Yeh ET. et al. SENP3 is responsible for HIF-1 transactivation under mild oxidative stress via p300 de-SUMOylation. The EMBO journal. 2009;28:2748-62
137. Gerald D, Berra E, Frapart YM, Chan DA, Giaccia AJ, Mansuy D. et al. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118:781-94
138. Gao P, Zhang H, Dinavahi R, Li F, Xiang Y, Raman V. et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer cell. 2007;12:230-8
139. Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770-6
140. Meynier S, Rieux-Laucat F. FAS and RAS related Apoptosis defects: From autoimmunity to leukemia. Immunological reviews. 2019;287:50-61
141. Minchenko OH, Tsymbal DO, Minchenko DO, Ratushna OO. The role of the TNF receptors and apoptosis inducing ligands in tumor growth. Ukrainian biochemical journal. 2016;88:18-37
142. Kiraz Y, Adan A, Kartal Yandim M, Baran Y. Major apoptotic mechanisms and genes involved in apoptosis. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2016;37:8471-86
143. Safa AR. c-FLIP, a master anti-apoptotic regulator. Experimental oncology. 2012;34:176-84
144. Wilkie-Grantham RP, Matsuzawa S, Reed JC. Novel phosphorylation and ubiquitination sites regulate reactive oxygen species-dependent degradation of anti-apoptotic c-FLIP protein. The Journal of biological chemistry. 2013;288:12777-90
145. Burke PJ. Mitochondria, Bioenergetics and Apoptosis in Cancer. Trends in cancer. 2017;3:857-70
146. Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA. et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nature chemical biology. 2005;1:223-32
147. Dorstyn L, Akey CW, Kumar S. New insights into apoptosome structure and function. Cell death and differentiation. 2018;25:1194-208
148. Enoksson M, Fernandes AP, Prast S, Lillig CH, Holmgren A, Orrenius S. Overexpression of glutaredoxin 2 attenuates apoptosis by preventing cytochrome c release. Biochemical and biophysical research communications. 2005;327:774-9
149. Zuo Y, Xiang B, Yang J, Sun X, Wang Y, Cang H. et al. Oxidative modification of caspase-9 facilitates its activation via disulfide-mediated interaction with Apaf-1. Cell research. 2009;19:449-57
150. Kalkavan H, Green DR. MOMP, cell suicide as a BCL-2 family business. Cell death and differentiation. 2018;25:46-55
151. Pan JS, Hong MZ, Ren JL. Reactive oxygen species: a double-edged sword in oncogenesis. World journal of gastroenterology. 2009;15:1702-7
152. Luanpitpong S, Chanvorachote P, Stehlik C, Tse W, Callery PS, Wang L. et al. Regulation of apoptosis by Bcl-2 cysteine oxidation in human lung epithelial cells. Molecular biology of the cell. 2013;24:858-69
153. Li D, Ueta E, Kimura T, Yamamoto T, Osaki T. Reactive oxygen species (ROS) control the expression of Bcl-2 family proteins by regulating their phosphorylation and ubiquitination. Cancer science. 2004;95:644-50
154. Luanpitpong S, Chanvorachote P, Nimmannit U, Leonard SS, Stehlik C, Wang L. et al. Mitochondrial superoxide mediates doxorubicin-induced keratinocyte apoptosis through oxidative modification of ERK and Bcl-2 ubiquitination. Biochemical pharmacology. 2012;83:1643-54
155. Galluzzi L, Kroemer G. Necroptosis: a specialized pathway of programmed necrosis. Cell. 2008;135:1161-3
156. Hsu SK, Chang WT, Lin IL, Chen YF, Padalwar NB, Cheng KC. et al. The Role of Necroptosis in ROS-Mediated Cancer Therapies and Its Promising Applications. Cancers. 2020 12
157. Ogretmen B. Sphingolipid metabolism in cancer signalling and therapy. Nature reviews Cancer. 2018;18:33-50
158. Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ, Chen X. et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nature communications. 2017;8:14329
159. Shang L, Ding W, Li N, Liao L, Chen D, Huang J. et al. The effects and regulatory mechanism of RIP3 on RGC-5 necroptosis following elevated hydrostatic pressure. Acta biochimica et biophysica Sinica. 2017;49:128-37
160. Yang Z, Wang Y, Zhang Y, He X, Zhong CQ, Ni H. et al. RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nature cell biology. 2018;20:186-97
161. Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC. et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science (New York, NY). 2009;325:332-6
162. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X. et al. Ferroptosis: process and function. Cell death and differentiation. 2016;23:369-79
163. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ. et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell. 2017;171:273-85
164. Friedmann Angeli JP, Krysko DV, Conrad M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nature reviews Cancer. 2019;19:405-14
165. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060-72
166. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS. et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317-31
167. Yang WS, Stockwell BR. Ferroptosis: Death by Lipid Peroxidation. Trends in cell biology. 2016;26:165-76
168. Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW. et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxidants & redox signaling. 2013;18:522-55
169. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H. et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57-62
170. Yin L, Kosugi M, Kufe D. Inhibition of the MUC1-C oncoprotein induces multiple myeloma cell death by down-regulating TIGAR expression and depleting NADPH. Blood. 2012;119:810-6
171. Han X, Li J, Brasky TM, Xun P, Stevens J, White E. et al. Antioxidant intake and pancreatic cancer risk: the Vitamins and Lifestyle (VITAL) Study. Cancer. 2013;119:1314-20
172. Wright ME, Mayne ST, Stolzenberg-Solomon RZ, Li Z, Pietinen P, Taylor PR. et al. Development of a comprehensive dietary antioxidant index and application to lung cancer risk in a cohort of male smokers. American journal of epidemiology. 2004;160:68-76
173. Stice CP, Liu C, Aizawa K, Greenberg AS, Ausman LM, Wang XD. Dietary tomato powder inhibits alcohol-induced hepatic injury by suppressing cytochrome p450 2E1 induction in rodent models. Archives of biochemistry and biophysics. 2015;572:81-8
174. Pandurangan AK, Saadatdoust Z, Esa NM, Hamzah H, Ismail A. Dietary cocoa protects against colitis-associated cancer by activating the Nrf2/Keap1 pathway. BioFactors (Oxford, England). 2015;41:1-14
175. Gao YT, McLaughlin JK, Blot WJ, Ji BT, Dai Q, Fraumeni JF Jr. Reduced risk of esophageal cancer associated with green tea consumption. Journal of the National Cancer Institute. 1994;86:855-8
176. Chikara S, Nagaprashantha LD, Singhal J, Horne D, Awasthi S, Singhal SS. Oxidative stress and dietary phytochemicals: Role in cancer chemoprevention and treatment. Cancer letters. 2018;413:122-34
177. Patel SS, Acharya A, Ray RS, Agrawal R, Raghuwanshi R, Jain P. Cellular and molecular mechanisms of curcumin in prevention and treatment of disease. Critical reviews in food science and nutrition. 2020;60:887-939
178. Li W, Su ZY, Guo Y, Zhang C, Wu R, Gao L. et al. Curcumin Derivative Epigenetically Reactivates Nrf2 Antioxidative Stress Signaling in Mouse Prostate Cancer TRAMP C1 Cells. Chemical research in toxicology. 2018;31:88-96
179. Yuan JH, Li YQ, Yang XY. Protective effects of epigallocatechin gallate on colon preneoplastic lesions induced by 2-amino-3-methylimidazo[4,5-f ] quinoline in mice. Molecular medicine (Cambridge, Mass). 2008;14:590-8
180. Bettuzzi S, Brausi M, Rizzi F, Castagnetti G, Peracchia G, Corti A. Chemoprevention of human prostate cancer by oral administration of green tea catechins in volunteers with high-grade prostate intraepithelial neoplasia: a preliminary report from a one-year proof-of-principle study. Cancer research. 2006;66:1234-40
181. Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931-47
182. Mizutani H, Tada-Oikawa S, Hiraku Y, Kojima M, Kawanishi S. Mechanism of apoptosis induced by doxorubicin through the generation of hydrogen peroxide. Life sciences. 2005;76:1439-53
183. Tsang WP, Chau SP, Kong SK, Fung KP, Kwok TT. Reactive oxygen species mediate doxorubicin induced p53-independent apoptosis. Life sciences. 2003;73:2047-58
184. Chen PY, Hou CW, Shibu MA, Day CH, Pai P, Liu ZR. et al. Protective effect of Co-enzyme Q10 On doxorubicin-induced cardiomyopathy of rat hearts. Environmental toxicology. 2017;32:679-89
185. Marullo R, Werner E, Degtyareva N, Moore B, Altavilla G, Ramalingam SS. et al. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PloS one. 2013;8:e81162
186. Bragado P, Armesilla A, Silva A, Porras A. Apoptosis by cisplatin requires p53 mediated p38alpha MAPK activation through ROS generation. Apoptosis: an international journal on programmed cell death. 2007;12:1733-42
187. Sheth S, Mukherjea D, Rybak LP, Ramkumar V. Mechanisms of Cisplatin-Induced Ototoxicity and Otoprotection. Frontiers in cellular neuroscience. 2017;11:338
188. Hagen T, D'Amico G, Quintero M, Palacios-Callender M, Hollis V, Lam F. et al. Inhibition of mitochondrial respiration by the anticancer agent 2-methoxyestradiol. Biochemical and biophysical research communications. 2004;322:923-9
189. Gray LH, Conger AD, Ebert M, Hornsey S, Scott OC. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. The British journal of radiology. 1953;26:638-48
190. Yoshida T, Goto S, Kawakatsu M, Urata Y, Li TS. Mitochondrial dysfunction, a probable cause of persistent oxidative stress after exposure to ionizing radiation. Free radical research. 2012;46:147-53
191. Mortezaee K, Goradel NH, Amini P, Shabeeb D, Musa AE, Najafi M. et al. NADPH Oxidase as a Target for Modulation of Radiation Response; Implications to Carcinogenesis and Radiotherapy. Current molecular pharmacology. 2019;12:50-60
192. Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN. et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780-3
193. Chen Y, Li N, Wang J, Zhang X, Pan W, Yu L. et al. Enhancement of mitochondrial ROS accumulation and radiotherapeutic efficacy using a Gd-doped titania nanosensitizer. Theranostics. 2019;9:167-78
194. Griffith OW. Mechanism of action, metabolism, and toxicity of buthionine sulfoximine and its higher homologs, potent inhibitors of glutathione synthesis. The Journal of biological chemistry. 1982;257:13704-12
195. Gana CC, Hanssen KM, Yu DMT, Flemming CL, Wheatley MS, Conseil G. et al. MRP1 modulators synergize with buthionine sulfoximine to exploit collateral sensitivity and selectively kill MRP1-expressing cancer cells. Biochemical pharmacology. 2019;168:237-48
196. Villablanca JG, Volchenboum SL, Cho H, Kang MH, Cohn SL, Anderson CP. et al. A Phase I New Approaches to Neuroblastoma Therapy Study of Buthionine Sulfoximine and Melphalan With Autologous Stem Cells for Recurrent/Refractory High-Risk Neuroblastoma. Pediatric blood & cancer. 2016;63:1349-56
197. Hernández-Breijo B, Monserrat J, Ramírez-Rubio S, Cuevas EP, Vara D, Díaz-Laviada I. et al. Preclinical evaluation of azathioprine plus buthionine sulfoximine in the treatment of human hepatocarcinoma and colon carcinoma. World journal of gastroenterology. 2011;17:3899-911
198. Suarez-Almazor ME, Belseck E, Shea B, Wells G, Tugwell P. Sulfasalazine for rheumatoid arthritis. The Cochrane database of systematic reviews. 2000;1998:Cd000958
199. Gout PW, Buckley AR, Simms CR, Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia. 2001;15:1633-40
200. Lo M, Ling V, Low C, Wang YZ, Gout PW. Potential use of the anti-inflammatory drug, sulfasalazine, for targeted therapy of pancreatic cancer. Current oncology (Toronto, Ont). 2010;17:9-16
201. Guan J, Lo M, Dockery P, Mahon S, Karp CM, Buckley AR. et al. The xc- cystine/glutamate antiporter as a potential therapeutic target for small-cell lung cancer: use of sulfasalazine. Cancer chemotherapy and pharmacology. 2009;64:463-72
202. Suarez-Almazor ME, Spooner CH, Belseck E, Shea B. Auranofin versus placebo in rheumatoid arthritis. The Cochrane database of systematic reviews. 2000: Cd002048.
203. Onodera T, Momose I, Kawada M. Potential Anticancer Activity of Auranofin. Chemical & pharmaceutical bulletin. 2019;67:186-91
204. Fiskus W, Saba N, Shen M, Ghias M, Liu J, Gupta SD. et al. Auranofin induces lethal oxidative and endoplasmic reticulum stress and exerts potent preclinical activity against chronic lymphocytic leukemia. Cancer research. 2014;74:2520-32
205. Pessetto ZY, Weir SJ, Sethi G, Broward MA, Godwin AK. Drug repurposing for gastrointestinal stromal tumor. Molecular cancer therapeutics. 2013;12:1299-309
206. Zheng X, Chen Y, Bai M, Liu Y, Xu B, Sun R. et al. The antimetastatic effect and underlying mechanisms of thioredoxin reductase inhibitor ethaselen. Free radical biology & medicine. 2019;131:7-17
207. Shi C, Yu L, Yang F, Yan J, Zeng H. A novel organoselenium compound induces cell cycle arrest and apoptosis in prostate cancer cell lines. Biochemical and biophysical research communications. 2003;309:578-83
208. Zheng X, Zhang Y, Zhang L, Xu W, Ma W, Sun R. et al. Synergistic inhibition of sunitinib and ethaselen against human colorectal cancer cells proliferation. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2016;83:212-20
209. Zheng X, Xu W, Sun R, Yin H, Dong C, Zeng H. Synergism between thioredoxin reductase inhibitor ethaselen and sodium selenite in inhibiting proliferation and inducing death of human non-small cell lung cancer cells. Chemico-biological interactions. 2017;275:74-85
210. Wu W, Yang Z, Xiao X, An T, Li B, Ouyang J. et al. A thioredoxin reductase inhibitor ethaselen induces growth inhibition and apoptosis in gastric cancer. Journal of Cancer. 2020;11:3013-9
211. Kirkpatrick DL, Kuperus M, Dowdeswell M, Potier N, Donald LJ, Kunkel M. et al. Mechanisms of inhibition of the thioredoxin growth factor system by antitumor 2-imidazolyl disulfides. Biochemical pharmacology. 1998;55:987-94
212. You BR, Shin HR, Park WH. PX-12 inhibits the growth of A549 lung cancer cells via G2/M phase arrest and ROS-dependent apoptosis. International journal of oncology. 2014;44:301-8
213. Shin HR, You BR, Park WH. PX-12-induced HeLa cell death is associated with oxidative stress and GSH depletion. Oncology letters. 2013;6:1804-10
214. Li GZ, Liang HF, Liao B, Zhang L, Ni YA, Zhou HH. et al. PX-12 inhibits the growth of hepatocelluar carcinoma by inducing S-phase arrest, ROS-dependent apoptosis and enhances 5-FU cytotoxicity. American journal of translational research. 2015;7:1528-40
215. Shang W, Xie Z, Lu F, Fang D, Tang T, Bi R. et al. Increased Thioredoxin-1 Expression Promotes Cancer Progression and Predicts Poor Prognosis in Patients with Gastric Cancer. Oxidative medicine and cellular longevity. 2019;2019:9291683
216. Wang F, Lin F, Zhang P, Ni W, Bi L, Wu J. et al. Thioredoxin-1 inhibitor, 1-methylpropyl 2-imidazolyl disulfide, inhibits the growth, migration and invasion of colorectal cancer cell lines. Oncology reports. 2015;33:967-73
217. Ramanathan RK, Abbruzzese J, Dragovich T, Kirkpatrick L, Guillen JM, Baker AF. et al. A randomized phase II study of PX-12, an inhibitor of thioredoxin in patients with advanced cancer of the pancreas following progression after a gemcitabine-containing combination. Cancer chemotherapy and pharmacology. 2011;67:503-9
218. Cao L, Liu J, Zhang L, Xiao X, Li W. Curcumin inhibits H2O2-induced invasion and migration of human pancreatic cancer via suppression of the ERK/NF-κB pathway. Oncology reports. 2016;36:2245-51
219. Liu Q, Loo WT, Sze SC, Tong Y. Curcumin inhibits cell proliferation of MDA-MB-231 and BT-483 breast cancer cells mediated by down-regulation of NFkappaB, cyclinD and MMP-1 transcription. Phytomedicine: international journal of phytotherapy and phytopharmacology. 2009;16:916-22
220. Zong H, Wang F, Fan QX, Wang LX. Curcumin inhibits metastatic progression of breast cancer cell through suppression of urokinase-type plasminogen activator by NF-kappa B signaling pathways. Molecular biology reports. 2012;39:4803-8
221. Fan Z, Duan X, Cai H, Wang L, Li M, Qu J. et al. Curcumin inhibits the invasion of lung cancer cells by modulating the PKCα/Nox-2/ROS/ATF-2/MMP-9 signaling pathway. Oncology reports. 2015;34:691-8
222. Pongrakhananon V, Nimmannit U, Luanpitpong S, Rojanasakul Y, Chanvorachote P. Curcumin sensitizes non-small cell lung cancer cell anoikis through reactive oxygen species-mediated Bcl-2 downregulation. Apoptosis: an international journal on programmed cell death. 2010;15:574-85
223. Du Y, Long Q, Zhang L, Shi Y, Liu X, Li X. et al. Curcumin inhibits cancer-associated fibroblast-driven prostate cancer invasion through MAOA/mTOR/HIF-1α signaling. International journal of oncology. 2015;47:2064-72
224. Ben Yehuda Greenwald M, Frušić-Zlotkin M, Soroka Y, Ben Sasson S, Bitton R, Bianco-Peled H. et al. Curcumin Protects Skin against UVB-Induced Cytotoxicity via the Keap1-Nrf2 Pathway: The Use of a Microemulsion Delivery System. Oxidative medicine and cellular longevity. 2017;2017:5205471
225. Das L, Vinayak M. Long term effect of curcumin in regulation of glycolytic pathway and angiogenesis via modulation of stress activated genes in prevention of cancer. PloS one. 2014;9:e99583
226. Singh SV, Hu X, Srivastava SK, Singh M, Xia H, Orchard JL. et al. Mechanism of inhibition of benzo[a]pyrene-induced forestomach cancer in mice by dietary curcumin. Carcinogenesis. 1998;19:1357-60
227. Na HK, Kim EH, Jung JH, Lee HH, Hyun JW, Surh YJ. (-)-Epigallocatechin gallate induces Nrf2-mediated antioxidant enzyme expression via activation of PI3K and ERK in human mammary epithelial cells. Archives of biochemistry and biophysics. 2008;476:171-7
228. Murakami C, Hirakawa Y, Inui H, Nakano Y, Yoshida H. Effect of tea catechins on cellular lipid peroxidation and cytotoxicity in HepG2 cells. Bioscience, biotechnology, and biochemistry. 2002;66:1559-62
229. Yuan JH, Li YQ, Yang XY. Inhibition of epigallocatechin gallate on orthotopic colon cancer by upregulating the Nrf2-UGT1A signal pathway in nude mice. Pharmacology. 2007;80:269-78
230. Zhu W, Xu J, Ge Y, Cao H, Ge X, Luo J. et al. Epigallocatechin-3-gallate (EGCG) protects skin cells from ionizing radiation via heme oxygenase-1 (HO-1) overexpression. Journal of radiation research. 2014;55:1056-65
231. Fernandez SV, Russo IH, Russo J. Estradiol and its metabolites 4-hydroxyestradiol and 2-hydroxyestradiol induce mutations in human breast epithelial cells. International journal of cancer. 2006;118:1862-8
232. Cao L, Chen X, Xiao X, Ma Q, Li W. Resveratrol inhibits hyperglycemia-driven ROS-induced invasion and migration of pancreatic cancer cells via suppression of the ERK and p38 MAPK signaling pathways. International journal of oncology. 2016;49:735-43
233. Jang YG, Go RE, Hwang KA, Choi KC. Resveratrol inhibits DHT-induced progression of prostate cancer cell line through interfering with the AR and CXCR4 pathway. The Journal of steroid biochemistry and molecular biology. 2019;192:105406
234. Banerjee S, Bueso-Ramos C, Aggarwal BB. Suppression of 7,12-dimethylbenz(a)anthracene-induced mammary carcinogenesis in rats by resveratrol: role of nuclear factor-kappaB, cyclooxygenase 2, and matrix metalloprotease 9. Cancer research. 2002;62:4945-54
235. Singh B, Shoulson R, Chatterjee A, Ronghe A, Bhat NK, Dim DC. et al. Resveratrol inhibits estrogen-induced breast carcinogenesis through induction of NRF2-mediated protective pathways. Carcinogenesis. 2014;35:1872-80
236. Bishayee A, Barnes KF, Bhatia D, Darvesh AS, Carroll RT. Resveratrol suppresses oxidative stress and inflammatory response in diethylnitrosamine-initiated rat hepatocarcinogenesis. Cancer prevention research (Philadelphia, Pa). 2010;3:753-63
237. Mahmoud AM, Mohammed HM, Khadrawy SM, Galaly SR. Hesperidin protects against chemically induced hepatocarcinogenesis via modulation of Nrf2/ARE/HO-1, PPARγ and TGF-β1/Smad3 signaling, and amelioration of oxidative stress and inflammation. Chemico-biological interactions. 2017;277:146-58
238. Xia R, Xu G, Huang Y, Sheng X, Xu X, Lu H. Hesperidin suppresses the migration and invasion of non-small cell lung cancer cells by inhibiting the SDF-1/CXCR-4 pathway. Life sciences. 2018;201:111-20
239. Nagaprashantha LD, Vatsyayan R, Singhal J, Lelsani P, Prokai L, Awasthi S. et al. 2'-hydroxyflavanone inhibits proliferation, tumor vascularization and promotes normal differentiation in VHL-mutant renal cell carcinoma. Carcinogenesis. 2011;32:568-75
240. Singhal SS, Singhal J, Figarola JL, Riggs A, Horne D, Awasthi S. 2'-Hydroxyflavanone: A promising molecule for kidney cancer prevention. Biochemical pharmacology. 2015;96:151-8
241. Awasthi S, Singhal SS, Singhal J, Nagaprashantha L, Li H, Yuan YC. et al. Anticancer activity of 2'-hydroxyflavanone towards lung cancer. Oncotarget. 2018;9:36202-19
242. Singhal J, Nagaprashantha L, Chikara S, Awasthi S, Horne D, Singhal SS. 2'-Hydroxyflavanone: A novel strategy for targeting breast cancer. Oncotarget. 2017;8:75025-37
243. Chang JH, Lai SL, Chen WS, Hung WY, Chow JM, Hsiao M. et al. Quercetin suppresses the metastatic ability of lung cancer through inhibiting Snail-dependent Akt activation and Snail-independent ADAM9 expression pathways. Biochimica et biophysica acta Molecular cell research. 2017;1864:1746-58
244. Khorsandi L, Orazizadeh M, Niazvand F, Abbaspour MR, Mansouri E, Khodadadi A. Quercetin induces apoptosis and necroptosis in MCF-7 breast cancer cells. Bratislavske lekarske listy. 2017;118:123-8
245. Ren KW, Li YH, Wu G, Ren JZ, Lu HB, Li ZM. et al. Quercetin nanoparticles display antitumor activity via proliferation inhibition and apoptosis induction in liver cancer cells. International journal of oncology. 2017;50:1299-311
246. Uys JD, Manevich Y, Devane LC, He L, Garret TE, Pazoles CJ. et al. Preclinical pharmacokinetic analysis of NOV-002, a glutathione disulfide mimetic. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2010;64:493-8
247. Batool T, Makky EA, Jalal M, Yusoff MM. A Comprehensive Review on L-Asparaginase and Its Applications. Applied biochemistry and biotechnology. 2016;178:900-23
248. Chayapong J, Madhyastha H, Madhyastha R, Nurrahmah QI, Nakajima Y, Choijookhuu N. et al. Arsenic trioxide induces ROS activity and DNA damage, leading to G0/G1 extension in skin fibroblasts through the ATM-ATR-associated Chk pathway. Environmental science and pollution research international. 2017;24:5316-25
249. Pritchard R, Rodríguez-Enríquez S, Pacheco-Velázquez SC, Bortnik V, Moreno-Sánchez R, Ralph S. Celecoxib inhibits mitochondrial O(2) consumption, promoting ROS dependent death of murine and human metastatic cancer cells via the apoptotic signalling pathway. Biochemical pharmacology. 2018;154:318-34
250. Xiang T, Du L, Pham P, Zhu B, Jiang S. Nelfinavir, an HIV protease inhibitor, induces apoptosis and cell cycle arrest in human cervical cancer cells via the ROS-dependent mitochondrial pathway. Cancer letters. 2015;364:79-88
251. Selimovic D, Porzig BB, El-Khattouti A, Badura HE, Ahmad M, Ghanjati F. et al. Bortezomib/proteasome inhibitor triggers both apoptosis and autophagy-dependent pathways in melanoma cells. Cellular signalling. 2013;25:308-18
252. Simůnek T, Stérba M, Popelová O, Adamcová M, Hrdina R, Gersl V. Anthracycline-induced cardiotoxicity: overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacological reports: PR. 2009;61:154-71
253. Zhang Q, Ma Y, Cheng YF, Li WJ, Zhang Z, Chen SY. Involvement of reactive oxygen species in 2-methoxyestradiol-induced apoptosis in human neuroblastoma cells. Cancer letters. 2011;313:201-10
254. Lai WL, Wong NS. ROS mediates 4HPR-induced posttranscriptional expression of the Gadd153 gene. Free radical biology & medicine. 2005;38:1585-93
Author contact
![]() Corresponding authors: Hong-Xu Liu, hxliuedu.cn; Hai-long Piao, hpiaoac.cn
Corresponding authors: Hong-Xu Liu, hxliuedu.cn; Hai-long Piao, hpiaoac.cn