Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Major signaling pathways
Other signaling pathways
Epigenetic alterations
Nuclear receptors
Other factors
Membrane targeting of NIS
Clinical trials
Future perspectives and...
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(13):6251-6277. doi:10.7150/thno.57689 This issue Cite
Review
Molecular mechanisms of radioactive iodine refractoriness in differentiated thyroid cancer: Impaired sodium iodide symporter (NIS) expression owing to altered signaling pathway activity and intracellular localization of NIS
Ji Min Oh1,2, Byeong-Cheol Ahn1,2,3 ![]()
1. Department of Nuclear Medicine, School of Medicine, Kyungpook National University, Daegu, Republic of Korea.
2. Department of Nuclear Medicine, Kyungpook National University Hospital, Daegu, Republic of Korea.
3. BK21 FOUR KNU Convergence Educational Program of Biomedical Sciences for Creative Future Talents, Department of Biomedical Science, School of Medicine, Kyungpook National University, Daegu, Republic of Korea.
Received 2020-12-30; Accepted 2021-3-22; Published 2021-4-15
Abstract

The advanced, metastatic differentiated thyroid cancers (DTCs) have a poor prognosis mainly owing to radioactive iodine (RAI) refractoriness caused by decreased expression of sodium iodide symporter (NIS), diminished targeting of NIS to the cell membrane, or both, thereby decreasing the efficacy of RAI therapy. Genetic aberrations (such as BRAF, RAS, and RET/PTC rearrangements) have been reported to be prominently responsible for the onset, progression, and dedifferentiation of DTCs, mainly through the activation of mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K)/AKT signaling pathways. Eventually, these alterations result in a lack of NIS and disabling of RAI uptake, leading to the development of resistance to RAI therapy. Over the past decade, promising approaches with various targets have been reported to restore NIS expression and RAI uptake in preclinical studies. In this review, we summarized comprehensive molecular mechanisms underlying the dedifferentiation in RAI-refractory DTCs and reviews strategies for restoring RAI avidity by tackling the mechanisms.
Keywords: radioactive iodine refractory thyroid cancer, redifferentiation, signaling pathways, sodium iodide symporter, membrane targeting
Introduction
Differentiated thyroid cancers (DTCs), including papillary thyroid cancer (PTC), follicular thyroid cancer (FTC), and Hűrthle cell cancer, arise from thyroid follicular cells and represent >90% of all types of thyroid cancer [1]. The development of PTC, accounting for >80% of all thyroid cancers, is majorly caused by BRAF (60%), RAS (15%), and RET/PTC rearrangements (12%), with these mutations being mutually exclusive, followed by refractoriness to radioactive iodine (RAI) owing to activated mitogen-activated protein kinase (MAPK) signaling pathway [2]. FTC, which represents 2%-5% of thyroid cancers, is associated with mostly mutually exclusive RAS mutation and PAX8-peroxisome proliferator-activated receptor ɣ (PPARɣ) fusion oncogene; however, Hűrthle cell cancer is genetically different from FTC [2, 3]. Although DTCs have a quite favorable prognosis via common therapeutic approaches such as thyroidectomy, RAI therapy, and thyroid-stimulating hormone (TSH) suppressive therapy, local recurrence and distant metastasis inevitably occur up to 20% and 10%, respectively [4]. Two-thirds of these patients exhibit loss of iodine-131 (131I) uptake initially or gradually, indicating dedifferentiation of the DTC known as RAI-refractory DTC [4, 5].
The 2015 ATA guideline suggested the following criteria as definition of RAI-refractory DTCs [6]: 1) patients with malignant/metastatic disease do not concentrate RAI at the time of the initial treatment; 2) patients whose tumors lose the ability to concentrate RAI after previous evidence of RAI-avid disease; 3) patients with RAI uptake concentrated in only some lesions; and 4) patients with metastatic disease that progresses despite substantial concentration of RAI. However, there were some differences regarding details of the definition among researchers owing to reasons such as the number of previous RAI administration, the cumulative dose of RAI, fluorodeoxyglucose (FDG) uptake of the lesion [7-10]. These trivial differences in the definition of RAI-refractory DTCs originate from the clinical perspective and can be further refined by clinical experience in the future [4, 11]. Cellular dedifferentiation in thyroid cancers eventually causes tumor progression through aggressiveness, metastasis, and reduced RAI uptake with no response to RAI therapy [12]. Consequentially, the 10-year survival rate for patients with DTC exhibiting RAI refractoriness is <10% [5].
An important element of RAI therapy is the ability of thyroid follicle cells to take up and concentrate iodide [13, 14]. This treatment exploits the unique function of the iodide-metabolizing machinery of thyroid follicular cells as well as the expression of thyroid-specific genes, such as sodium iodide symporter (NIS), thyroglobulin (Tg), TSH receptor (TSHR), and thyroperoxidase (TPO) in addition to thyroid-specific transcription factors such as paired box gene-8 (PAX-8) and thyroid transcription factor-1 (TTF-1) [15, 16]. However, the underlying molecular basis of the loss of RAI avidity in RAI-refractory thyroid cancer is the aberrant silencing of expression of both thyroid-specific genes and transcription factor [16]. Among these factors, decreased expression of NIS, diminished membrane targeting of NIS, or both, which are mainly caused by genetic and epigenetic alterations and dysregulated signaling pathways, are the major mechanisms underlying RAI refractoriness [1, 17, 18]. Genetic alterations in MAPK and phosphoinositide 3-kinase (PI3K)/AKT signaling pathways by point mutations or chromosomal rearrangements are fundamental drivers of the pathogenesis of thyroid cancers and RAI refractoriness [12]. In addition to the signaling pathways, epigenetic and genetic alterations of other pathways, including Wnt/ß-catenin and TGF-ß/Smad signaling pathways, are also related to the silencing of expression of thyroid-specific genes, resulting in the failure of RAI therapy [1, 16, 19].
This review comprehensively discusses the molecular mechanisms underlying RAI refractoriness in DTC and strategies for restoring RAI avidity.
Physiological NIS regulation
NIS expression can be regulated at both transcriptional and posttranslational levels [4]. Once TSH binds to TSHR, adenylyl cyclase is stimulated via the Gs-protein, with resulting increase in the levels of cyclic adenosine monophosphate (cAMP). Activated cAMP stimulates both protein kinase A (PKA)-dependent and PKA-independent signaling pathways [13, 20]. Once cAMP is activated, PKA increases and phosphorylates the cAMP-responsive element modulator, which is followed by an increase in NIS upstream enhancer (NUE) activity through the PKA-dependent signaling pathway [21]. Redox effector factor-1 (Ref-1) is activated through the PKA-independent signaling pathway, and then PAX-8 is stimulated to bind to NUE, resulting in NUE activation [12]. TSH-independent mechanisms also alter NIS expression by affecting the binding of PAX-8 to NUE. There are three primary signaling pathways that have been identified. First, activation of transforming growth factor-ß (TGF-ß) stimulates SMAD3, which inhibits the binding of PAX-8 to NUE [22, 23]. Second, toll-like receptor 4 (TLR4) stimulates nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) p65 which activates PAX-8 to bind NUE [24, 25]. Third, pituitary tumor-transforming gene 1 (PTTG1)-binding factor (PBF) interrupts the interaction between PAX-8 and NUE which suppresses NIS expression [26, 27].
Another crucial factor determining NIS functionality is the translocation of NIS from the cytoplasm to the plasma membrane by posttranslational modification [13]. Kogai et al. demonstrated that rat thyroid cells stimulated by TSH could induce NIS expression by more than 6-fold at 24 h compared with basal levels [28]. However, iodide uptake reached a maximum (>27-fold increase compared with basal levels) at 72 h, showing a time lag between NIS expression and iodide uptake owing to the posttranslational regulation of NIS by TSH stimulation. Additionally, TSH stimulation in rat thyroid cells revealed the localization of NIS on the cell membrane, whereas NIS is localized in the cytoplasm after the removal of TSH stimulation. Therefore, the presence of NIS in the cytoplasm is closely associated with posttranslational regulation, resulting in the reduction of iodide uptake.
Role of iodide metabolism in physiology
Uptake and concentration of iodide and synthesis of thyroid hormone are the main roles and functions of thyroid follicular cells [29]. The expression of thyroid-specific genes, such as NIS, TSHR, TPO, and Tg, work together to accumulate and retain iodide in the thyroid follicular cells [16]. NIS is localized in the basolateral membrane and mediates the active transport of iodide from the blood stream to the thyroid follicular cells [30]. TSHR plays a fundamental role in the regulation of thyroid cell proliferation, differentiation, and targeting for iodide uptake through TSH stimulation [31]. TSHR expression is impaired in thyroid cancer tissues, although to a lesser extent than other thyroid-specific genes [32]. TPO is a key thyroid enzyme for incorporation of iodide into Tg during thyroid hormone synthesis [16]. The decrease in TPO expression in thyroid cancer is related to the rapid loss of iodide caused by the cancer which results in the failure of RAI therapy [33, 34]. Tg is the iodoprotein precursor of thyroid hormone in thyroid follicular cells and is considered the best marker of thyroid cancer [35]. Thyroid-specific transcription factors such as PAX-8, TTF-1, and thyroid transcription factor-2 (TTF-2) play a crucial role in the regulation of gene transcription activity in thyroid follicular cells [16]. Several studies noted different expression levels of thyroid-specific genes and thyroid-specific transcription factors within human thyroid tissue [32, 36, 37]. Mostly, the expression of both thyroid-specific genes and thyroid-specific transcription factors decreased in dedifferentiated thyroid cancers. Therefore, the process of restoring RAI avidity to the thyroid cannot be achieved with NIS alone, and additional factors such as thyroid-specific genes and thyroid-specific transcription factors are also required.
Gene transfer with thyroid-specific genes or thyroid-specific transcription factors was used to convert dedifferentiation to redifferentiation in thyroid cancers cancers [38, 39]. Mu et al. demonstrated the possibility of gene transfer in thyroid cancers to provide effective RAI therapy [38]. The results indicated that cotransfer of TTF-1 and PAX-8 genes resulted in the induction of NIS, TPO, and Tg expression followed by RAI accumulation and retention. Another group showed that transfer of the TSHR gene into dedifferentiated thyroid cancer reinduced the expression of thyroid-specific genes followed by RAI uptake [39].
Major signaling pathways
MAPK signaling pathway
A MAPK can be classified into three main families: extracellular-signal-regulated kinases (ERKs), Jun amino-terminal kinases (JNKs), and p38/stress-activated protein kinases (SAPKs) [40]. The MAPK pathway plays a critical role in signaling cascades and transmits its extracellular signal to intracellular targets [40]. In thyroid cancer, the MAPK signaling pathway regulates cell proliferation, growth, survival, and dedifferentiation [41]. Approximately 70%-80% of human PTCs are connected via mutations of BRAF, RAS, RET/PTC rearrangement, or TRK [42-44]. Constitutive activation of the MAPK signaling pathway occurs through the oncoproteins encoded by these genes, which leads to the progression of thyroid cancer (Figure 1A).
Principal mechanisms underlying the pathogenesis of thyroid cancer and radioactive iodine (RAI)-refractory in differentiated thyroid cancer. (A) Mitogen-activated protein kinase (MAPK) signaling pathway. (B) Phosphoinositide 3-kinase (PI3K)/AKT signaling pathway. Abbreviations: 4E-BP: 4E-binding protein; DNMT1: DNA methyltransferase 1; mTORC1: mTOR complex 1; mTORC2: mTOR complex 2; P: phosphorylation; PTEN: phosphatase and tensin homolog; TERT: telomerase reverse transcriptase. Images were created with BioRender.com.
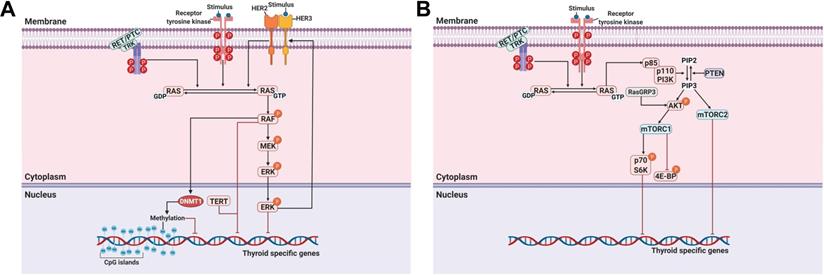
Among these mutations, mutation of BRAF, which is a highly prevalent oncogene, occurs in approximately 45% (range, 27%-87%) of PTCs and is critical for the aberrant activation of the MAPK signaling pathway [4, 45, 46]. In 2005, the first comprehensive multicenter study reported the association of the BRAF mutation with poor clinicopathological characteristics of PTC, such as extrathyroidal extension, lymph node metastasis, advanced tumor stage, tumor recurrence, RAI non-avidity, and treatment failure with RAI [47]. Subsequently, numerous studies also reported the association between the BRAF mutation and loss of RAI avidity with failure of RAI treatment in PTC [48-51]. The BRAF mutation is highly prevalent in recurrent or metastatic RAI-refractory PTC compared with primary PTC [47, 48, 52, 53]. Xing et al. noted that additional surgery or RAI therapy cured patients with recurrent PTC of wild-type BRAF [47]. However, recurrent PTC patients with the BRAF mutation had active disease despite repeated surgery or RAI therapy. Several studies have reported that BRAF expression in thyroid cells is closely associated with the impairment of thyroid-specific genes, including NIS; however, this impairment was not totally dependent on the activation of the MAPK signaling pathway [49, 54-56]. Liu et al. reported restoring thyroid-specific gene expression by inhibiting the MAPK signaling pathway using U0126 as a MEK inhibitor. This allowed effective RAI therapy against PTC harboring the BRAF mutation (Table 1) [56]. The results of treatment using BRAF inhibitors demonstrated restoration of thyroid-specific gene expression and RAI uptake in BRAF-mutated thyroid cancer cells in vitro [19, 57]. The in vitro study was expanded to an in vivo study, and it reported that suppression of the MAPK signaling pathway using BRAF and MEK inhibitors in transgenic mice with conditional BRAF activation also restored thyroid-specific gene expression and RAI uptake (Table 1) [58]. These results have potentially significant clinical ramifications and therapeutic possibilities for patients with RAI-refractory thyroid cancer.
Summary of preclinical studies with redifferentiation strategy for radioactive-iodine (RAI) refractoriness in differentiated thyroid cancer
| Target | Compound | Type of study | Result | Reference |
|---|---|---|---|---|
| Autophagy | Digitalis-like compounds (Digoxin, Digoxigenin, Proscillaridin A, Lanatoside C, Strophantin K) | In vitro | TTF-1, TTF-2, PAX-8, Tg ↑ NIS ↑ 125I uptake ↑ | [182, 183] |
| BRD4 | JQ1 | In vitro, In vivo | NIS ↑ 131I uptake ↑ | [186] |
| BRAF | PLX4720 | In vivo | NIS, TPO, Tg, TSHR, PAX-8, TTF-1 ↑ NIS ↑ 124I uptake ↑ 131I cytotoxicity ↑ | [58] |
| DNA methylation | 5-aza-2'-deoxycytidine | In vitro | NIS ↑ 125I uptake ↑ | [132] |
| 5-azacytidine | In vitro | NIS ↑ 125I uptake ↑ | [135] | |
| ERRɣ | Compound 35 | In vitro, In vivo | NIS, TPO, TSHR, Tg ↑ 124I, 125I uptake ↑ 131I cytotoxicity ↑ | [177] |
| DN200434 | In vitro, In vivo | NIS, TPO, TSHR, Tg ↑ 124I, 125I uptake ↑ 131I cytotoxicity ↑ | [176] | |
| GSK5182 | In vitro | NIS (membrane) ↑ 125I uptake ↑ 131I cytotoxicity ↑ | [175] | |
| HDAC | AB2, AB3 and AB10 | In vitro | NIS, PAX-8, TTF-1, TTF-2, TSHR ↑ | [152] |
| APHA compound 8 | In vitro | NIS ↑ 125I uptake ↑ | [148] | |
| Apicidine | In vitro | NIS ↑ 125I uptake ↑ | [148] | |
| Depsipeptide | In vitro, In vivo | NIS, Tg, TPO ↑ 125I uptake ↑ | [143, 144] | |
| Entinostat | In vitro | 125I uptake ↑ | [149] | |
| Panobinostat (LBH589) | In vitro | NIS ↑ 125I uptake ↑ 131I cytotoxicity ↑ | [145] | |
| Sodium butyrate | In vitro | NIS, PAX-8 ↑ | [150] | |
| Trichostatin A | In vitro | NIS ↑ NIS, Tg, TPO ↑ | [146, 150, 151] | |
| Valproic acid | In vitro | Tg ↑ NIS ↑ 125I uptake ↑ | [144, 147, 148] | |
| Vorinostat | In vitro | NIS, TSHR, TPO ↑ | [16] | |
| LXR | Dendrogenin A | In vitro | NIS, TPO, TSHR, Tg ↑ 123I, 131I uptake ↑ | [173] |
| MAPK | CTOM-DHP | In vitro, In vivo | NIS, TPO, Tg, TSHR, PAX-8, TTF-1 ↑ 123I, 125I uptake ↑ 131I cytotoxicity ↑ Tumor growth ↓ | [30] |
| K905-0266 | In vitro, In vivo | NIS, TPO, Tg, TSHR, PAX-8, TTF-1 ↑ 125I uptake ↑ 131I cytotoxicity ↑ Tumor burden ↓ | [84] | |
| MAPK, SAPK/JNK | Sunitinib | In vitro | NIS, TPO, Tg, TSHR, PAX-8↑ | [81] |
| MEK | CH5126766 | In vivo | NIS ↑ 124I uptake ↑ 131I cytotoxicity ↑ Tumor growth ↓ | [83] |
| PD0325901 | In vivo | NIS, TPO, Tg, TSHR, PAX-8, TTF-1 ↑ NIS ↑ 124I uptake ↑ 131I cytotoxicity ↑ | [58] | |
| Selumetinib | In vivo | NIS ↑ 124I uptake ↑ 131I cytotoxicity ↑ Tumor growth ↓ | [83] | |
| U0126 | In vitro | TSHR, NIS, Tg ↑ | [56] | |
| mTORC1 | Rapamycin | In vitro | NIS ↑ TTF-1, TTF-2, PAX-8, TSHR ↑ 125I uptake ↑ | [92] |
| mTORC1/mTORC2 | Torin2 | In vitro | NIS ↑ | [94] |
| Notch | Resveratrol | In vitro | NIS, TTF-1, TTF-2, PAX-8 ↑ | [96] |
| PI3K | LY294002 | In vitro | NIS ↑ 125I uptake ↑ | [89] |
| PPARɣ | Pioglitazone, Rosiglitazone | In vitro | 125I uptake ↑ | [168, 169] |
| Rosiglitazone | In vitro | 125I uptake ↑ | [148] | |
| Troglitazone | In vitro | NIS, Tg ↑ 125I uptake ↑ | [168, 169] | |
| Retinoic acid receptor | 13-cis retinoic acid | In vitro | 131I uptake ↑ | [163] |
| All-trans retinoic acid | In vitro | NIS, Tg ↑ NIS ↑ 125I uptake ↑ | [148, 164-166] | |
| All-trans retinol | In vitro | NIS ↑ 125I uptake ↑ | [148] | |
| Reverse transcriptase enzyme | Nevirapine | In vitro, In vivo | NIS, PAX-8, TSHR ↑ 125I uptake ↑ | [17, 201] |
| VEGFR, RET, MET, FLT3, AXL | Cabozantinib | In vitro | NIS, TPO, Tg, TSHR ↑ 125I uptake ↑ 18F-FDG uptake ↓ | [80] |
| VEGFR, PDGFR, RET, KIT, FLT | Sorafenib | In vitro | NIS, TPO, Tg, TSHR ↑ 125I uptake ↑ 18F-FDG uptake ↓ | [80] |
Abbreviations: BRD4: bromodomain-containing protein 4; ERRɣ: estrogen-related receptor ɣ; FLT: fms-related receptor tyrosine kinase; 18F-FDG: fluorodeoxyglucose (18F); FLT-3: fms-related receptor tyrosine kinase-3; HDAC: histone deacetylase; LXR: liver X receptor; MAPK: mitogen-activated protein kinase; MET: MNNG HOS transforming gene; NIS: sodium iodide symporter; PAX-8: paired box gene-8; PDGFR: platelet-derived growth factor receptor; PI3K: phosphoinositide 3-kinase; PPARɣ: peroxisome proliferator-activated receptor ɣ; Tg: thyroglobulin; TPO: thyroperoxidase; TSHR: thyroid-stimulating hormone (TSH) receptor; TTF-1: thyroid transcription factor-1; TTF-2: thyroid transcription factor-2; VEGFR: vascular endothelial growth factor receptor.
Although administration of BRAF inhibitors such as vemurafenib has shown promising results for RAI-refractory thyroid cancers, the occurrence of adverse effects and primary and/or acquired resistance often limit its applicability for targeted therapy [59]. The primary causes of resistance to BRAF inhibitors are mutational events or activation of alternative signaling pathways that reactivate ERK signaling [59]. Ofir et al. reported the case of a patient with PTC having RAI-refractory metastatic disease who was treated with vemurafenib for 43 months. Initially, there was a partial response but the disease progressed again. Genetic analysis following the occurrence of drug resistance revealed a new mutation in NRASQ61K [60]. Montero-Conde et al. reported that thyroid cancer with the BRAF mutation overexpressed human epidermal growth factor receptor 3 (HER3) via autocrine-secreted NRG1 secretion when treated with BRAF inhibitor. This phenomenon triggered the reactivation of both ERK and AKT in thyroid cancer [61]. Therefore, overcoming resistance to BRAF inhibitors is crucial for improving therapeutic responses. Cheng et al. applied this strategy for redifferentiation of PTC harboring the BRAF mutation and found that the HER inhibitor (lapatinib) prevented the reactivation of the MAPK signaling pathway and sensitized the BRAF/MEK inhibitor. They found that by treating with the HER inhibitor, PTC harboring the BRAF mutation was redifferentiated and the expression of thyroid-specific proteins and RAI uptake and therapy were restored (Table 2) [57]. Song et al. demonstrated that the effect of vemurafenib treatment was transient and followed by reactivation of the phosphorylation of ERK; however, a combination strategy with a MEK inhibitor overcame the limitations of a transient vemurafenib response in BRAF-mutated thyroid cancers [62]. Based on a previous study, the same group demonstrated synergistic effects following combination treatment with BRAF inhibitor and MEK inhibitor to increase NIS expression and restore RAI uptake by inhibiting reactivated phosphorylation of ERK in BRAF-mutated PTC cells [63].
Summary of preclinical studies with combination treatment for redifferentiation of radioactive-iodine (RAI) refractoriness in differentiated thyroid cancer
| Target | Compound | Type of study | Result | Reference |
|---|---|---|---|---|
| BRAF, HER, MEK | Dabrafenib, Lapatinib, Selumetinib, Lapatinib | In vitro | NIS, TPO, Tg, TSHR ↑ 125I uptake ↑ 131I cytotoxicity ↑ | [57] |
| BRAF, MEK | Vemurafenib, PD98059 | In vitro | NIS ↑ 125I uptake ↑ | [63] |
| DNA methylation, HDAC | 5-aza-2'-deoxycytidine, Sodium butyrate | In vitro | NIS ↑ 125I uptake ↑ | [156] |
| 5-aza-2'-deoxycytidine, Valproic acid | In vitro | NIS ↑ 125I uptake ↑ | [157] | |
| HDAC, MAPK | Vemurafenib, Vorinostat | In vitro | NIS, TPO, Tg, TSHR ↑ 125I uptake ↑ | [19] |
| Selumetinib, Dabrafenib, Panobinostat | In vitro | NIS, Tg, TSHR, TPO ↑ NIS ↑ 125I uptake ↑ 131I cytotoxicity ↑ | [154] | |
| HDAC, MAPK, PI3K/AKT | RDEA119, Temsirolimus, Perifosine, Vorinostat | In vitro | NIS, TSHR, TPO ↑ NIS ↑ 125I uptake ↑ | [16] |
| HDAC, Retinoic acid receptor | Tributyrin, Retinoic acid | In vitro | NIS ↑ 125I uptake ↑ | [155] |
| HMT, MAPK | Dabrafenib, Selumetinib, Tazemetostat | In vitro | NIS, TPO, Tg, TSHR, PAX-8, TTF-1 ↑ 125I uptake ↑ 131I therapy ↑ | [158] |
| MAPK, SAPK/JNK | Sunitinib, Forskolin | In vitro | NIS, TPO, Tg, TSHR, PAX-8 ↑ | [81] |
Abbreviations: HDAC: histone deacetylase; HER: human epidermal growth factor receptor; HMT: histone methyltransferase; MAPK: mitogen-activated protein kinase; NIS: sodium iodide symporter; PAX-8: paired box gene-8; PI3K: phosphoinositide 3-kinase; SAPK/JNK: stress-activated protein kinases/jun amino-terminal kinases; Tg: thyroglobulin; TPO: thyroperoxidase; TSHR: thyroid-stimulating hormone (TSH) receptor; TTF-1: thyroid transcription factor-1.
It is well-known that TSH stimulation has the ability to enhance NIS expression and RAI uptake. Increased levels of TSH are necessary before starting RAI therapy, which can be accomplished either by administering human recombinant TSH or withholding thyroid hormone supplementation. Kleiman et al. reported that BRAF silencing of BRAF-mutated thyroid cancer could restore thyroid-specific gene expression and RAI uptake with only TSH supplementation [55]. In addition, TSH stimulation, along with combination treatment with BRAF inhibitor and histone deacetylase (HDAC) inhibitor, maximized the synergistic effects of restoring thyroid-specific gene expression and RAI uptake in BRAF-mutated thyroid cancer [19].
TSH stimulates NIS expression through the accumulation of cAMP, which is a positive regulator of differentiation and proliferation of thyroid cells [64]. cAMP can activate the RAC1-MKK3/6-p38ɑ-CHOP signaling pathway for the induction of NIS expression in thyroid follicular cells [64-66]. Among these downstream factors, RAC1, known as a small GTP-binding protein, plays a role as a molecular switch in the proliferation, differentiation, and migration of cells [67]. Moreover, Faria et al. demonstrated the upregulation of NIS expression via overexpression of the constitutively active mutant G12V-RAC1 in PTC cells, suggesting that RAC1 activation upregulates NIS expression without TSH stimulation [68]. A RAC1b, an alternative splicing variant of RAC1, has been shown to be overexpressed in multiple cancers, suggesting its oncogenic role [69]. Several studies reported that the overexpression of RAC1b is closely associated with poor clinical outcomes in PTC and FTC [68, 70, 71]. In addition, a possible interaction between RAC1b and BRAF mutations may contribute to the downregulation of NIS expression as well as poor clinical outcomes in PTC [68, 71]. A possible interplay between RAC1b and BRAF mutations may contribute to the downregulation of NIS expression as well as poor clinical outcomes in PTC.
Telomerase reverse transcriptase (TERT), a catalytic protein subunit of telomerase, is a major oncogene in multiple cancers. The presence of TERT promoter mutations is found in thyroid cancers, except in medullary thyroid cancer (MTC), suggesting its role in thyroid tumorigenesis and poor clinical outcomes such as recurrence and mortality [72]. In addition, the frequency of TERT promoter mutations is associated with aggressive characteristics of thyroid cancers. The prevalence of TERT promoter mutations is higher in poorly differentiated thyroid cancer (PDTC) and anaplastic thyroid cancer (ATC) than in PTC and FTC [72, 73]. Several reports noted that concomitant BRAF and TERT mutations were closely associated with poor clinical outcomes in PTCs [74, 75]. Recently, Liu et al. investigated the genotype patterns of BRAF and TERT promoter mutations related to the loss of RAI avidity in recurrent PTC [76]. They found that the BRAF mutation and the concomitant BRAF and TERT promoter mutations were closely associated with the loss of RAI avidity and impaired the expression of thyroid-specific genes in recurrent PTC (Figure 1A).
Fuziwara et al. reported on the role of FAM83F, which is involved in the activation of the MAPK signaling pathway [77]. FAM83F is highly expressed in human PTC tissues, and normal thyroid cells overexpressing FAM83F showed loss of thyroid-specific gene expression following cross-regulation of TGF-ß and the MAPK signaling pathway with BRAF expression. A MEK1/2 inhibitor inhibited the expression of FAM83F with restoration of NIS expression. In the future, additional studies on the interplay between FAM83F and redifferentiation, including changes in thyroid-specific protein expression and the effect of RAI therapy, in thyroid cancers are necessary.
Conditional activation of RET/PTC1 or RET/PTC3 inhibited the expression of thyroid-specific genes promoting the differentiation process, and they also mediated the activation of the MAPK signaling pathway through Shc in thyroid cells [78]. Moreover, thyroid cells expressing RAS or MEK1 attenuated TSH-induced expression of Tg and NIS which was regained with MEK inhibitor treatment. These findings suggest that the MAPK signaling pathway promotes dedifferentiation in PDTC with constitutive activation of either RAS or RET/PTC [78]. In the context of the previous study, in thyroid cells expressing RET/PTC1, MEK inhibition modulated not only NIS expression but also RAI uptake [79]. Additionally, treatment with sorafenib or cabozantinib, as multikinase inhibitors (MKIs), restored RAI uptake and enhanced the expression of thyroid-specific genes in PTC cells harboring the RET/PTC1 rearrangement (Table 1) [80]. Sunitinib, which is an MKI, also inhibits MEK/ERK and SAPK/JNK signaling pathways and increases the expression of thyroid-specific genes, including NIS, in PTC cells harboring the RET/PTC1 rearrangement (Table 1) [81]. The combination of forskolin (adenylyl cyclase agonist) with sunitinib further increases the expression of thyroid-specific genes and transcription factors that bind to NUE in the cells (Table 2).
Initially, RAF and MEK inhibitors contributed to the suppression of the MAPK signaling pathway; however, this was followed by a marked reactivation of the signaling pathway to levels similar to that of controls [11]. Allosteric MEK inhibitor (CH5126766 and CKI) could be an alternative candidate to overcome the reactivation of the MAPK signaling pathway. CH5126766 selectively binds to the non-phosphorylated form of MEK and stabilizes the RAF/MEK complex which is inactivated. [82]. Thus, CH5126766 inhibits the feedback induction of MEK phosphorylation that occurs after ERK inhibition in cancer cells exposed to other MEK inhibitors. Nagarajah et al. reported that CH5126766, an allosteric MEK inhibitor, suppressed ERK in a sustained manner by blocking RAF reactivation, whereas selumetinib, an MEK inhibitor, suppressed ERK activation transiently in a tumor harboring the BRAF mutation [83]. Sustained ERK inhibition via CH5126766 treatment induced higher RAI uptake and 131I-mediated therapeutic effects (Table 1).
Our group employed the high-throughput NIS enhancer screening platform with a dual reporter gene system to excavate two tyrosine kinase inhibitors (TKIs; K905-0266 and CTOM-DHP) which enhance NIS promoter activity [30, 84]. Treatment with K905-0266 restored the expression of thyroid-specific proteins, including NIS, and promoted RAI uptake in ATC (Table 1) [84]. These results were followed by an increase in RAI-mediated cytotoxicity with 131I by pretreating with K905-0266. Mechanism analysis revealed that K905-0266 treatment inhibited ERK phosphorylation but not AKT phosphorylation. Additionally, in vivo results demonstrated the effect of K905-0266 with the enhancement of the NIS promoter followed by increased NIS protein expression and therapeutic effects. Another excavated compound, CTOM-DHP, also influenced ATC redifferentiation with restoration of thyroid-specific protein expression followed by RAI uptake and cytotoxicity effect (Table 1) [30]. Its effects were accomplished by reduction in the phosphorylation of both AKT and ERK. Moreover, nuclear imaging revealed an improvement in RAI avidity following CTOM-DHP treatment in in vivo study. CTOM-DHP also appeared to affect other thyroid cancers harboring KRAS and RET/PTC rearrangements, suggesting that they are promising candidates for restoring RAI avidity.
PI3K/AKT signaling pathway
In addition to the MAPK signaling pathway, the PI3K/AKT signaling pathway also plays a crucial role in cell growth and proliferation and thyroid tumorigenesis (Figure 1B) [85]. Several studies have reported that the stimulation of insulin-like growth factor-1 (IGF-1) inhibited iodide uptake in TSH-induced thyroid cells [86, 87]. Garcia et al. reported that IGF-1 could inhibit TSH/forskolin-induced NIS expression through activation of the PI3K/AKT signaling pathway in thyroid cells [88]. Moreover, it has been reported that inhibition of the PI3K/AKT signaling pathway could increase NIS expression and RAI uptake in thyroid cancer cells [16, 89]. Specifically, PTC cells which were engineered to constitutively express NIS restored NIS expression and RAI uptake by inhibiting the PI3K/AKT signaling pathway with LY294002 as a PI3K inhibitor regardless of blockade by insulin stimulation (Table 1) [89]. Interestingly, inhibition of AKT, which is downstream of PI3K, revealed that both NIS expression and RAI uptake were enhanced, exhibiting the mimicking effect of LY294002 in PTC cells. Song et al. reported that the prevalence of the RasGRP3 mutation was higher in metastatic, RAI-refractory thyroid cancer [90]. Moreover, RasGRP3 mutation led to the promotion of cell proliferation, migration, and invasion, whereas NIS and TSHR expression and RAI uptake declined through AKT activation in thyroid cancer cells. These results suggest that an activated AKT signaling pathway may be involved in RAI refractoriness by mediating RasGRP3 mutation in thyroid cancers (Figure 1B).
The serine-threonine protein kinase mechanistic target of rapamycin (mTOR), which is downstream of the PI3K/AKT signaling pathway, has been identified as a regulator of NIS expression, RAI uptake, and cell survival in both in vitro and in vivo studies (Figure 1B) [91]. Plantinga et al. reported that the increase in the expression of thyroid-specific genes and proteins and restoration of RAI uptake through mTOR inhibition in thyroid cancer cells with the BRAF mutation or PTEN deficiency depended on TTF-1 [92]. mTOR acts in two functionally distinct complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). mTORC1 comprises mTOR, raptor, mLST8, PRAS40, and DEPTOR and regulates cell growth through effectors such as p70 S6K (also called S6K1). mTORC2 contains mTOR, rictor, mLST8, mSIN1, PROTOC, and DEPTOR and phosphorylates AKT at Ser473 and protein kinase C-ɑ (PKC-ɑ) [91]. Tavares et al. reported that the role of mTOR phosphorylation as a marker of aggressiveness in PTC is closely associated with the absence of a tumor capsule, distant metastases, persistence of disease, and resistance to RAI therapy. Additionally, phosphorylation of mTOR significantly correlated with the frequency of the NRAS mutation but not the BRAF mutation [93]. However, tumors with S6 phosphorylation showed less aggressive characteristics, such as the presence of a tumor capsule, absence of extrathyroidal extension, and BRAF wild-type, although NRAS mutation was significantly higher [94]. Inhibition of mTORC1/mTORC2 by Torin2 treatment restored NIS expression by inhibiting the phosphorylation of AKT and S6; however, there was no effect with RAD001 treatment as an mTORC1 inhibitor in PTC (Table 1) [94]. Rapamycin, an mTOR inhibitor, is effective at inhibiting the phosphorylation of S6K (mTORC1). Interestingly, treatment with rapamycin led to increased expression of thyroid-specific genes and NIS protein in thyroid cancer cells with either the BRAF mutation or PTEN deficiency but not in PTC harboring a RET/PTC rearrangement (Table 1) [92]. Torin2, an mTORC1/mTORC2 inhibitor, increased NIS gene expression in PTC harboring the RET/PTC rearrangement instead of resistance to rapamycin treatment [94]. Considering the different responses of thyroid cancers depending on cell type and mutations, further studies are needed to elucidate accurate redifferentiation mechanisms for each specific dedifferentiated thyroid cancer.
Other signaling pathways
Notch signaling pathway
Notch is a multifunctional transmembrane receptor that regulates cell differentiation, proliferation, and survival and exerts oncogenic or tumor suppressor effects depending on cell types [95]. The Notch signaling pathway comprises 4 Notch receptors (Notch1-4) and 5 ligands (δ-like 1, 3, and 4, Jagged-1, and Jagged-2). Once Notch receptors are activated by binding to ligands, they promote two sequential proteolytic cleavages via the ɣ-secretase protease complex [96]. This results in the release of a cytoplasmic domain fragment (the second cleavage occurs within the transmembrane area) which moves from its transmembrane location into the nucleus and modulates gene expression (Figure 2A).
Molecular mechanisms involved in the regulation of thyroid-specific genes including sodium iodide symporter (NIS) in RAI-refractory differentiated thyroid cancer. (A) Notch signaling pathway. (B) TGF-ß/Smad signaling pathway. (C) Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathway. (D) Wnt/ß-catenin signaling pathway. Abbreviations: DNMT1: DNA methyltransferase 1; FoxP3: forkhead transcription factor 3; HIF1-ɑ: hypoxia-inducible factor-ɑ; IκB: Inhibitory κB; LRP5/6: low-density lipoprotein receptor-related protein 5/6; NECD: notch extracellular domain; NICD: notch intracellular domain; NOX4: NADPH oxidase 4; P: phosphorylation; ROS: reactive oxygen species; Ub: ubiquitination. Images were created with BioRender.com.
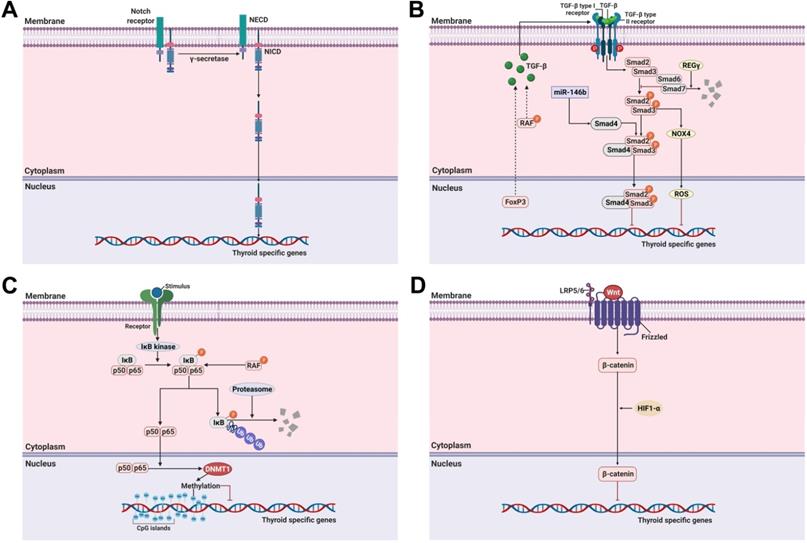
Ferretti et al. reported the correlation between the Notch signaling pathway and the differentiation of thyroid cancers from normal thyroid cells [95]. Overexpression of the Notch signaling pathway, including that of Notch1, Notch3, and Hes1, is directly associated with a differentiation phenotype (NIS, TPO) expression of both thyroid cancers and TSH-stimulated normal thyroid cells. Subsequently, Carre et al. reported the importance of Hes1 during mouse thyroid development and differentiation as with NIS [97]. Other studies also reported the importance of the Notch signaling pathway in the differentiation of thyroid cancers [96, 98]. Yu et al. identified that resveratrol, as a Notch1 activator, could restore NIS expression and thyroid-specific transcription factors (Table 1) [96]. Somnay et al. found that repressed Notch3 expression was inversely correlated with tumor size, extrathyroidal extension, and distant metastasis following dedifferentiation in clinicopathological features [98]. Overexpression of Notch intracellular domain 3 (NICD3), which is cleaved from the Notch receptor and then translocated from the membrane to the nucleus to regulate target genes, in FTC cells led to the functional activation of the genes involved in downstream signaling pathways such as Hes1, HEY1, and HEY2. Additionally, the gain of function of FTC cells with NICD3 overexpression induced the expression of thyroid-specific genes. In contrast, suppression of Notch3 by siRNA could silence the expression of thyroid-specific genes in thyroid cancers. These results suggest that the activation of Notch1 and Notch3 is a potential strategy for the redifferentiation of thyroid cancers.
TGF-ß/Smad signaling pathway
The TGF-ß signaling pathway regulates cellular functions such as proliferation, differentiation, adhesion, apoptosis, and survival in multiple cancers, including thyroid cancer [23, 99]. TGF-ß is a secreted protein with three isoforms, i.e., TGF-ß1, TGF-ß2, and TGF-ß3 [100]. Their effects are initiated by binding to a type II receptor on the cell membrane. The TGF-ß-type II receptor complex then recruits a type I receptor [101]. This new complex stimulates the phosphorylation of Smad2 and Smad3 (R-Smad) which is then combined with Smad4 (Co-Smad) This complex subsequently enters the nucleus where it participates in the regulation of gene expression. Inhibitory Smads comprise Smad6 and Smad7 which oppose the activation of the TGF-ß signaling pathway (Figure 2B) [101].
Several reports have noted that TGF-ß plays a critical role in the growth and differentiation of normal thyroid cells [100-103]. Costamagna et al. reported that TGF-ß treatment decreases the expression and interaction of PAX-8 with Smad3 in response to TGF-ß stimulation, and PAX-8 decreases the DNA-binding activity of PAX-8 in normal thyroid cells [23]. Although inhibition of the MAPK signaling pathway, constitutively activated by BRAF, was considered a breakthrough, it is not always effective in resistant tumors, including RAI-refractory PTC. This suggests that other compensatory mechanisms affect adaptive resistance development in BRAF. Riesco-Eizaguirre et al. demonstrated that the BRAF oncogene activates the secretion of an autocrine TGF-ß loop leading to MEK-ERK-independent NIS diminution which operates in tandem with the MEK-ERK pathway in thyroid cancer (Figure 2B) [22]. Additionally, they noted that high TGF-ß expression is closely associated with extrathyroidal extension, nodal metastasis, and BRAF status in human PTC samples. Azouzi et al. reported that NADPH oxidase 4 (NOX4) expression is regulated by a BRAF mutation via the TGF-ß/Smad signaling pathway and that NOX4-dependent reactive oxygen species (ROS) production plays a critical role in the reduction of NIS expression in BRAF-mutated PTC (Figure 2B) [104].
Forkhead transcription factor 3 (FoxP3) is a key regulator of CD4+CD25+ regulatory T cells in maintaining immunologic tolerance [105, 106]. Its positive expression is associated with immune evasion of cancers, possibly by inducing the secretion of immunosuppressive cytokines such as TGF-ß1 and IL-10. Importantly, FoxP3 expression was correlated with extrathyroidal invasion, distant metastasis, and resistance to RAI therapy in PTC [107, 108]. Based on these findings, Ma et al. proposed that FoxP3 inhibits NIS expression by inducing TGF-ß1 secretion and subsequent activation of phosphorylation of Smad3 in thyroid cancer (Figure 2B) [108].
Recently, REGɣ, a proteasome activator, has been shown to enhance dedifferentiation by degrading Smad7, resulting in the activation of the TGF-ß/Smad signaling pathway in ATC with resistance to RAI therapy (Figure 2B) [109].
Platelet-derived growth factor receptor-ɑ
As noted previously, mTOR inhibition promotes the expression of thyroid-specific genes and proteins and RAI uptake with TTF1-dependent redifferentiation in thyroid cancers [92]. Lopez-Campistrous et al. reported the inverse relationship between platelet-derived growth factor receptor-ɑ (PDGFRɑ) and TTF-1 expression in PTC [110]. PDGFRɑ activation disrupts the transcriptional activity of TTF-1 and dephosphorylates TTF-1, resulting in its translocation from the nucleus to the cytoplasm. This phenomenon leads to a reduction in the expression of thyroid-specific genes and uptake of RAI in PTC. This finding suggests that PDGFRɑ blockade improves RAI therapy.
NF-κB pathway
The nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathway plays a pivotal role in the regulation of inflammatory responses involved in oncogenesis [111]. Most NF-κB dimers are sequestered in the cytoplasm in an inactive state by interaction with inhibitory κBs (IκBs) when not stimulated (Figure 2C). In response to stimulation, such as with lipopolysaccharide (LPS), tumor necrosis factor-ɑ (TNF-ɑ), and LTβR, degradation of IκBs is initiated by phosphorylation, ubiquitination, and degradation via the IκB kinase (IKK). NF-κB is then activated, resulting in its translocation to the nucleus [112].
Reale et al. highlighted the role of NF-κB in thyroid physiology by noting that genetic deletion of the NF-κB essential modulator locus could promote the downregulation of thyroid-specific genes (NIS, TPO, Tg, PAX-8, and TTF-1), resulting in the development of hypothyroidism [113]. Additionally, several articles reported that the NF-κB signaling pathway is related to the function and homeostasis of thyroid cells [24, 25, 114, 115]. In detail, NF-κB, especially the p65 subunit, mediates the effect of LPS to increase expression of TSH-induced Tg, NIS, and TPO and RAI uptake in normal thyroid cells. However, unlike LPS stimulation, TNF-ɑ stimulation downregulated NIS expression through an increase of IκB-ɑ in normal thyroid cells [28, 116]. One possible implication of the antagonistic effects between LPS and TNF-ɑ stimulation for NIS expression is that the intervention of additional signaling pathways may mediate the affinity of NF-κB transcription factors for other promoters. Elucidation of the exact molecular mechanisms of NIS expression by the NF-κB signaling pathway is still required for better understanding of thyroid physiology.
Increased NF-κB activation in thyroid cancer cell lines and human thyroid cancer tissues has been documented [117-119]. Moreover, this phenomenon leads to increased cell proliferation, growth, and resistance to drug-induced apoptosis through anti-apoptotic signaling pathways.
Starenki et al. initially reported that NF-κB inhibition could potentiate the therapeutic effect of ionizing radiation in thyroid cancer in in vitro and in vivo studies [120]. However, several reports on the effect of NF-κB inhibition to reinduce RAI uptake and therapy found no promising results [121, 122]. Meng et al. reported that 131I rather induced NF-κB activation followed by interruption of the therapeutic efficiency of RAI therapy [121]. Additionally, another study described that the cell survival rate declined following treatment with a combination of NF-κB gene silencing and 131I [122]. However, there was no improvement in RAI uptake via NF-κB inhibition. Finally, inhibition of NF-κB is not sufficient to enhance RAI uptake, and there are no results documenting a change in thyroid-specific gene expression. Therefore, in the future, it is necessary to consider other factors or signaling pathways that interact with the NF-κB signaling pathways with verification of changes in thyroid-specific gene expression in thyroid cancers.
Choi et al. reported that upregulated DNA methyltransferase 1 (DNMT1) via NF-κB activation caused by BRAF mutation suppresses the NIS expression caused by its promoter methylation (Figure 2C) [123]. In this manner, it is possible to infer that the interplay of other factors is involved in the activation of NF-κB, and overcoming this situation may be a better treatment strategy when weaker RAI uptake via NF-κB inhibition occurs.
Wnt/ß-catenin signaling pathway
The role of the Wnt/ß-catenin signaling pathway in the regulation of cell growth and proliferation and constitutive activation resulting in the development of cancer has been well-established [29]. After the canonical Wnt ligand binds to the Frizzled receptor and LRP5/6 as a co-receptor, ß-catenin is activated and translocated from the cytoplasm to the nucleus where it promotes the expression of various genes (Figure 2D).
Sastre-Perona et al. reported that Wnt-independent ß-catenin regulates cell proliferation and differentiation in normal thyroid cells [124]. Additionally, they elucidated that the direct interaction of ß-catenin with PAX-8 increases its transcriptional activity for NIS expression. These results suggested that ß-catenin is a positive regulator of NIS and RAI uptake under physiological conditions.
In contrast, it has been demonstrated that aberrant ß-catenin expression in the nucleus correlates with the dedifferentiation of thyroid cancers [125, 126]. Lan et al. demonstrated that HIF1-ɑ-induced ß-catenin modifies NIS subcellular localization and impairs RAI uptake in FTC cells (Figure 2D) [127]. Suppression of ß-catenin reversed these changes with RAI therapy and regulation of NIS localization in HIF1ɑ-overexpressing FTC cells.
Epigenetic alterations
DNA methylation
DNA methylation, one of the most commonly occurring epigenetic events, is the hallmark of human cancers, including thyroid cancer [29]. It involves covalent chemical modification, resulting in the addition of a methyl group (CH3) to the carbon residue at the 5'-position of the cytosine ring. DNA methylation typically occurs in CpG dinucleotides (Figure 3A) [128].
Molecular mechanisms involved in the regulation of thyroid-specific genes, including NIS in RAI-refractory differentiated thyroid cancer. (A) Epigenetic alterations, including DNA methylation and histone modification, and bromodomain-containing protein 4 (BRD4). (B) Nuclear receptors RAR, PPARɣ, LXRß and ERRɣ. (C) Autophagy. (D) MicroRNAs and PBF. Abbreviations: Ac: acetylation; BRD4: bromodomain-containing protein; DNMT1: DNA methyltransferase 1; ERRɣ: estrogen-related receptor ɣ; HDAC: histone deacetylase; HMGB1: high mobility group box 1; LXRß: liver X receptor ß; miR: microRNA; mTORC1: mTOR complex 1; PBF: pituitary tumor-transforming gene 1 (PTTG1)-binding factor; PPARɣ: peroxisome proliferator-activated receptor ɣ; RAR: retinoic acid receptor; ROS: reactive oxygen species; RXR: retinoid X receptor. Images were created with BioRender.com.
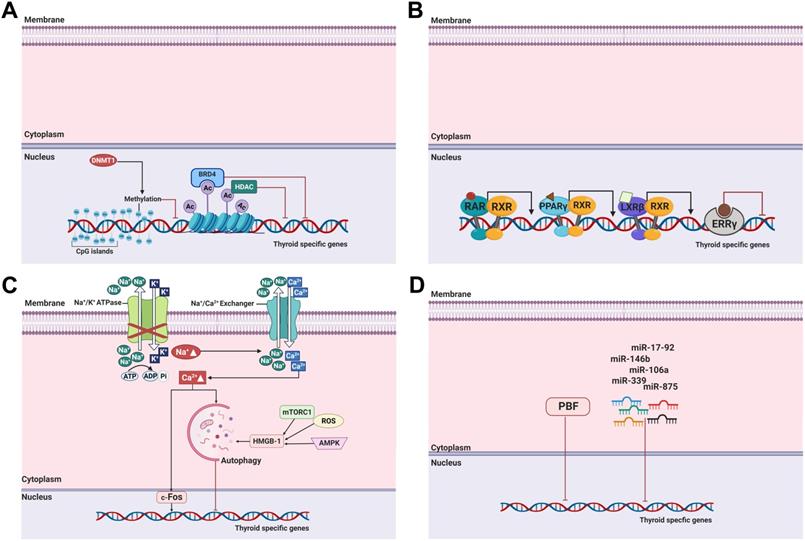
Previous studies demonstrated that aberrant NIS hypermethylation occurs in thyroid cancer regardless of cell type [129-131]. One study reported that the methylation level and frequency between benign and malignant thyroid tumors did not differ, and high NIS promoter methylation levels led to reduced NIS expression [131]. Later, Galrão et al. identified new CpG islands in the NIS proximal promoter by in silico screening and demonstrated that NIS hypermethylation had a significant inverse correlation with NIS messenger RNA (mRNA) expression [132]. Aberrant hypermethylation of thyroid-specific genes, namely, TSHR, Pendrin, and thyroid-specific transcription factors such as TTF-1, were also discovered in thyroid cancers [133, 134]. The application of DNA demethylating agents, such as 5-azacytidine and its deoxy derivative (5-aza-2'-deoxycytidine), to the aberrant NIS hypermethylated thyroid cancer restored RAI uptake following NIS expression (Table 1) [132, 135].
The BRAF mutation and its relation to aberrant gene methylation in thyroid cancer could be one of the possible targets for restoring the expression of thyroid-specific genes [136]. Khan et al. noted that the frequency of hypermethylation of the TSHR gene promoter was 73.3% in patients with BRAF mutation, whereas it was only 8.8% in patients with wild-type BRAF [137]. Liu et al. demonstrated the relationship between TSHR promoter methylation and the MAPK signaling pathway in thyroid cancer. In this study, the extent of TSHR promoter methylation was decreased by MEK inhibitor treatment, suggesting that the process for restoring the expression of thyroid-specific genes was linked with gene methylation [56]. Choi et al. reported that the BRAF mutation in thyroid cancer inhibits NIS expression by upregulating DNMT1, an inducer of promoter methylation in CpG islands (Figure 1A) [123].
Histone modification
Histone modifications, such as acetylation, methylation, phosphorylation, and ubiquitination, are critical determinants of the epigenetic state in addition to DNA methylation [138]. A nucleosome comprises 147bp of DNA wrapped around an octamer of histones H2A, H2B, H3, and H4 [85]. Histone modification can lead to either gene activation or inhibition depending on which residues are modified and the type of modification [139]. Overall, histone modification plays an important role in gene regulation and other chromatin-based processes [138].
Among histone modifications, histone acetylation typically occurs at lysine residues in the N-terminal tails of H3 and H4 by histone acetyltransferases that catalyze these processes. This leads to an open chromatin structure, resulting in the promotion of gene expression. Conversely, histone deacetylation is catalyzed by HDACs, resulting in the condensation of chromatin and then repression of gene expression [140]. Importantly, dysregulated histone acetyltransferase and HDAC activation are closely related to growth, proliferation, and differentiation of cells in cancers (Figure 3A) [138].
HDAC inhibitors may not only act specifically against limited types of HDACs (HDAC isoform-selective inhibitors) but also against all types of HDACs (pan-inhibitors). HDAC inhibitors are classified based on their specificity as members of five classes of compounds: (1) hydroxamic acids (hydroxamates), (2) short chain fatty (aliphatic) acids, (3) benzamides, (4) cyclic tetrapeptides, and (5) sirtuin inhibitors such as the pan-inhibitor nicotinamide and the specific SIRT1 and SIRT2 inhibitors sirtinol and cambinol, respectively [141].
Puppin et al. reported that modification of histone acetylation occurs in thyroid tumors [142]. They noted that the level of acetylated lysines 9 and 14 in H3 (H3K9-K14ac) was higher in follicular adenomas, PTC, FTC, and undifferentiated carcinomas than in normal tissue, and no difference in the H3 histone (H3K18ac) was found between normal tissue and undifferentiated carcinoma. It was suggested that modifications in histone acetylation is an early event in thyroid tumorigenesis, and acetylation of H3K18ac is switched off in the transition from differentiated to undifferentiated thyroid tumors.
Kitazono et al. were the first to demonstrate that treatment with depsipeptide (romidepsin, FR901228), a class I HDAC inhibitor, restored NIS expression and RAI uptake [143]. This suggests its utility as an HDAC inhibitor at in vitro levels (Table 1). Based on the results of this study, other preclinical studies have indicated that RAI avidity improved after reinduction of thyroid-specific gene expression and NIS localization to the cell membrane following treatment with the HDAC I and II inhibitors valproic acid, panobinostat (LBH589), and entinostat in in vitro and in vivo studies (Table 1) [144-150]. The use of pan-inhibitors such as vorinostat (suberoylanilide hydroxamic acid [SAHA]), trichostatin A, and sodium butyrate in thyroid cancer also resulted in the redifferentiation of cancer after enhanced thyroid-specific gene expression, leading to RAI accumulation (Table 1) [16, 146, 150, 151]. Jang et al. developed a new analog, HDAC inhibitor (AB1 to AB13), and excavated the effective compounds (AB2, AB3, and AB10) [152]. This allowed NIS, PAX-8, TTF-1, TTF-2, and TSHR gene expression to promote redifferentiation of thyroid cancers (Table 1).
Recently, oncogene-activated signaling pathways have been reported to also control histone posttranslational modification that affects the expression of thyroid-specific genes [153]. This finding supports attempts to convert dedifferentiated thyroid cancer into redifferentiated thyroid cancer by modulating histone acetylation and deacetylation.
Among the studies mentioned previously, Hou et al. reported the synergistic effects of vorinostat with MAPK, mTOR, and AKT inhibitors to upregulate thyroid-specific gene expression and RAI accumulation in thyroid cancers (Table 2) [16]. In addition, the same group demonstrated that BRAF-activated NIS silencing could be influenced by histone deacetylation at critical regulatory regions of the NIS promoter [140]. Cheng et al. also demonstrated that combination treatment of thyroid cancer cells with vorinostat and vemurafenib, a BRAF inhibitor, synergistically induced NIS expression and localization to the cell membrane and RAI uptake (Table 2) [19]. Remarkably, TSH stimulation during this treatment regimen revealed dramatic effects compared with treatment without TSH [16, 19]. Additionally, Fu et al. demonstrated that panobinostat, an HDAC inhibitor, induced a more robust redifferentiation when combined with selumetinib (a MEK inhibitor) or dabrafenib (a BRAF inhibitor) treatment regardless of BRAF mutation status (Table 2) [154]. Notably, tributyrin, a butyrate analog, is a trimer of butyric acid with retinoic acid (RA) that enhances RAI uptake by upregulating NIS expression in PDTC (Table 2) [155]. Moreover, combination treatment with DNA demethylating agents and HDAC inhibitors were used to increase thyroid-specific gene expression and restore RAI therapy effectiveness in thyroid cancer (Table 2) [156, 157]. Provenzano et al. reported that concurrent treatment of thyroid cancer cells with an HDAC inhibitor (sodium butyrate) and a DNA demethylating agent (5-aza-2'-deoxycytidine) promoted an increase in NIS expression and RAI uptake [156]. In the case of treatment with a single agent, although it may increase NIS expression, it did not affect RAI uptake owing to altered posttranslational modification. Another study also identified the effect of combination treatment with an HDAC inhibitor (valproic acid) and a DNA demethylating agent (5-aza-2'-deoxycytidine) to increase RAI uptake [157].
Recently, Fu et al. reported that dual inhibition of the MAPK signaling pathway and enhancement of zeste homolog 2 (EZH2) as a histone methyltransferase (HMT) had a synergistic effect in the redifferentiation of PTC harboring the BRAF mutation (Table 2) [158]. Specifically, the use of an MEK inhibitor (selumetinib) or a BRAF inhibitor (dabrafenib) with an EZH2 inhibitor (tazemetostat) improved thyroid-specific protein expression and enhanced the effect of RAI therapy.
Nuclear receptors
Retinoic acids
Retinoic acids (RAs), retinol, and their derivatives, are related to metabolites of vitamin A and act on the nuclear receptors RA receptor (RAR) and retinoid X receptor (RXR) (Figure 3B) [159]. There are three groups of RAs. The first group includes retinol, retinal, RA, and isotretinoin; the second group includes etretinate and acitretin; and the third group includes tazarotene and bexarotene [159].
Several studies have noted a correlation between treatment with RAs and differentiation of thyroid cancer cells [160-162]. Type I iodothyronine 5'-deiodinase activity, a differentiation marker, increased after treatment with RAs for thyroid cancer, whereas that of CD97, a dedifferentiation marker, decreased.
NIS expression in normal thyroid cells has been shown to decrease after RA treatment [163]. However, most thyroid cancers, such as FTC, PTC, and ATC, exhibit an elevation in NIS expression and NIS-mediated RAI uptake after RA treatment (Table 1) [148, 163-166]. Among these reports, Jeong et al. demonstrated that treatment of thyroid cancer with RA alters the expression of several genes involved in cell growth and differentiation and increased NIS and RAI uptake in a microarray experiment [166].
Peroxisome proliferator-activated receptor ɣ
Peroxisome proliferator-activated receptor (PPAR) is a nuclear receptor superfamily and comprises three forms: PPARɑ, PPARß, and PPARɣ [4]. It is involved in the regulation of cell proliferation and redifferentiation and is a ligand-dependent transcription factor that must form a heterodimer with RXR to bind to their response elements and activate gene expression (Figure 3B). The rearrangement of PAX-8/PPARɣ primarily occurs in FTC and follicular-variant PTC [167].
Studies have shown that the application of a PPARɣ agonist can cause redifferentiation in thyroid cancers [148, 168, 169]. Fröhlich et al. demonstrated the action of several thiazolidinediones, such as rosiglitazone, troglitazone, and pioglitazone, for redifferentiation, proliferation, and apoptosis in FTC (Table 1) [168]. In addition, Park et al. reported that treatment with the PPARɣ agonist troglitazone could upregulate NIS expression, whereas CD97, a marker of thyroid dedifferentiation, downregulated its expression. This suggests that PPARɣ agonists induce redifferentiation in different types of thyroid cancer (Table 1) [169].
Liver X receptors
Liver X receptors (LXRs) are members of the nuclear receptor family and are ligand-activated transcription factors (Figure 3B) [170]. LXRs exist as two isoforms: LXRɑ and LXRß, and they have a critical role in cholesterol transport, glucose and lipid metabolism, regulation of immune response, and cell differentiation and growth [171]. Additionally, the effects of LXRs have been studied as potential targets for cancer therapeutics [170].
Dendrogenin A (DDA), a class of LXR ligand, is a mammalian cholesterol-derived metabolite displaying the characteristics of a tumor suppressor [172]. Bauriaud-Mallet et al. reported the possibility of DDA inducing redifferentiation in thyroid cancer (Table 1) [173]. DDA and GW3965, a canonical LXR ligand, treatment could induce redifferentiation and enhanced thyroid-specific protein expression and RAI uptake in thyroid cancers.
Estrogen-related receptors
Estrogen-related receptors (ERRs) are an active nuclear receptor family and consist of three isoforms: ERRɑ, ERRß, and ERRɣ. They play an important role in disease development in cancer and metabolic disorders (Figure 3B) [174].
Singh et al. first demonstrated the effect of the ERRɣ inverse agonist GSK5182 to improve RAI therapy followed by an increase in membrane-localized NIS expression and RAI uptake (Table 1) [175]. These effects were accompanied by the downregulation of ERRɣ and activation of the ERK/MAPK signaling pathway. Recently, new ERRɣ inverse agonists have also shown efficacy at increasing RAI avidity in both in vitro and in vivo studies (Table 1) [176, 177].
Other factors
Autophagy
Autophagy is one of the important biological processes for cancer development and progression [178]. It may be triggered by hypoxia, nutritional deprivation, DNA damage, and other factors to facilitate the degradation of cytoplasmic materials such as damaged organelles and misfolded proteins [179]. These degraded materials are then engulfed into autophagosomes with a double membrane structure and degraded by fusion with lysosomes into autolysosomes, with the purpose of homeostasis maintenance and survival for cell recycling and various other stressors within the cell [178, 179]. Lin et al. suggested autophagy as a new target for thyroid cancer therapy [180]. Treatment with doxorubicin and irradiation exposure for thyroid cancer inhibited cell survival; however, combination treatment with 3-methyladenin, an autophagy-specific inhibitor, triggered chemoresistance and irradiation resistance. Those resistances were reversed by treatment with the autophagy activator RAD001 that sensitizes thyroid cells to doxorubicin and irradiation exposure. Moreover, they determined that the effects of autophagic activation are modulated mainly by Met inhibition [181]. Plantinga et al. reported that autophagic activity is associated with membranous NIS expression and response to RAI therapy in patients with non-MTC [179]. The same group then noted that upregulation of NIS expression and restoration of RAI uptake were mediated by autophagy-activating compounds, known as digitalis-like compounds (DLCs), by the accumulation of intracellular Ca2+ and FOS activation (Figure 3C, Table 1) [182]. Interestingly, DLCs induced redifferentiation with enhanced NIS expression in several thyroid cancer cell lines irrespective of mutations such as BRAF, PTEN loss, or RET/PTC rearrangement. Based on the preceding research showing that DLCs had a potent effect on redifferentiation in RAI-refractory PTC, the effects of DLCs in ATC were also demonstrated with increased NIS expression and RAI uptake activity although autophagy activity was lower than in PTC [183]. Recently, Chai et al. noted the function of the high mobility group box 1 (HMGB1), a nuclear protein, that plays a role in autophagy-mediated NIS degradation via ROS, AMPK, and mTOR signaling pathways (Figure 3C) [184]. Preclinical studies examined the association of HMGB1 with clinical pathological features. The results indicated that tumor size, lymph node metastasis, and clinical stage are closely connected with high HMGB1 expression. The results of these studies suggest that the activation of autophagy serves as a promising therapeutic option to restore RAI avidity and to increase therapeutic response to 131I.
Bromodomain-containing protein 4
Bromodomain-containing protein 4 (BRD4) is an epigenetic regulator and a member of the bromodomain and extra-terminal domain families. It plays a critical role in the genesis and development of various diseases, including cancer [185]. It affects gene transcription by binding to acetylated histones [186].
Gao et al. evaluated the degree of BRD4 expression levels in thyroid cancers and the possibility of BRD4 inhibition for cell viability, NIS expression, and RAI uptake (Figure 3A) [186]. Primarily, BRD4 was overexpressed in PTC specimens compared with that in normal tissues from patients and cell lines, suggesting the function of BRD4 in cancer progression. Treatment of thyroid cancer cells with JQ1, which inhibits the acetyl-lysine recognition site of BRD4, enhances NIS expression and RAI uptake and apoptosis (Table 1).
MicroRNAs
MicroRNAs (miRNAs) are small, non-coding RNAs containing approximately 19-25 nucleotides. They regulate gene expression by binding to the 3'-untranslated regions of mRNA at the posttranscriptional level and blocking the translation or cleaving targeted mRNA [187, 188]. A single miRNA can bind to numerous gene mRNAs; therefore, the regulation of miRNA in mRNA plays a critical role in many biological functions [189]. In thyroid cancer, miRNA is important for cancer initiation and progression, and it may serve as a diagnostic and prognostic biomarker.
To date, several miRNAs have been excavated to modulate NIS expression and RAI accumulation in both normal thyroids and cancers (Figure 3D). Among these miRNAs, miR-146b is one of the most studied miRNAs in thyroid cancer. Riesco-Eizaguirre et al. suggested that miR-146b, which is overexpressed in patients with PTC, is a key strategy for the conversion from dedifferentiation to differentiation by regulating the miR-146b/PAX-8/NIS circuit for the reinduction of RAI accumulation [190]. miR-146b expression was negatively regulated by HDAC3, and miR-146b antagonists have been shown to reinduce the expression of thyroid-specific genes (NIS, TSHR, and TPO) followed by an increase in RAI uptake in dedifferentiated FTC [191]. Apart from NIS, other thyroid-specific genes such as iodothyronine deiodinase 2 (DIO2), which converts T4 into T3, and iodotyrosine dehalogenase 1 (DEHAL1), which controls the recycling of iodide for thyroid hormone synthesis, were also inversely regulated by miR-146b [190, 192]. Additionally, Geraldo et al. reported that disruption of the TGF-ß/Smad4 signaling pathway by overexpression of miR-146b could deregulate the expression of the thyroid-specific genes NIS, Tg, TPO, and TSHR [193]. Recently, Hou et al. demonstrated the miR-146b could modulate NIS expression with translocation to the membrane by targeting MUC20 through the MET signaling pathway in dedifferentiated thyroid cancer [194]. Therefore, the importance of miR-146b regulation as a potential strategy for cell redifferentiation, restoration of RAI uptake, and modulation of several signaling pathways is clear.
Ricarte-Filho et al. reported the role of miR-let-7 as a positive regulator for the thyroid-specific genes TTF-1 and Tg by inhibiting MAPK activation in PTC harboring the RET/PTC rearrangement; however, NIS expression was not detected even with miR-let-7f overexpression [188]. In patients' samples, hsa-let-7f was found to be generally overexpressed in thyroid cancers regardless of type, whereas has-let-7b expression patterns differed among thyroid cancer types [195]. This suggests it is a valid diagnostic biomarker for the classification of PTC and ATC.
Shen et al. assessed serum miRNA profiles in patients with PTC having non-RAI-avid or RAI-avid lung metastasis and found that miRNA-106a was upregulated in patients with PTC with non-RAI-avid lung metastasis [196]. Furthermore, they found that miRNA-106a directly targeted RARß and could regulate NIS and TSHR expression and RAI uptake in thyroid cancers, suggesting miRNA-106a as a new diagnostic and therapeutic target.
In addition to the previously mentioned miRNAs, miR-339, miR-875, and miR-17-92 have been found to improve RAI uptake and NIS expression by modulating the upregulated miRNA in thyroid cancers [197, 198].
Recently, the effect of TKI on the expression of miRNAs has been assessed in thyroid cancer cells [199]. Treatment with selumetinib, a MEK inhibitor, increased NIS expression and downregulated both hsa-let-7f and miR-146b expression in PTCs, whereas ATCs only produced a partial response.
Others
As a non-coding RNA group, the role of SLC6A9 long non-coding RNA was also used to convert RAI resistance into RAI sensitivity in PTC via PARP-1 activity, suggesting that SLC6A9 is essential to maintain RAI sensitivity [200]. However, the change of thyroid-specific genes and protein expression was not assessed in this study.
Dong et al. suggested the role of non-nucleoside reverse transcriptase inhibitors for the redifferentiation of thyroid cancer [201]. In ATC, nevirapine, a non-nucleoside reverse transcriptase inhibitor, upregulated the expression of NIS and TSHR, and TSH co-stimulation further increased NIS mRNA expression. However, the improvement in RAI uptake was disappointing (Table 1). Recently, the same group reported the effect of nevirapine on dedifferentiated thyroid cancer for conversion to a redifferentiated status (Table 1) [17]. The thyroid-specific genes TSHR, PAX-8, and NIS were significantly upregulated, and improved RAI uptake was noted, whereas expression of CD97, an undifferentiation marker, declined in dedifferentiated thyroid cancers. Moreover, cytoplasmic and membrane fraction expression of NIS increased with the latter being more effective. Interestingly, it was increased by the TSHR/cAMP/cAMP response element-binding protein (CREB)/PAX-8 signaling pathway, suggesting the therapeutic potential of non-nucleoside reverse transcriptase inhibitors to restore RAI uptake.
AXL is a tyrosine kinase receptor that regulates cell proliferation, survival, invasiveness, and chemoresistance in several cancers. AXL stimulation via its ligand triggers multiple signaling pathways, such as MAPK, PI3K/AKT, Jak/Stat, and NF-κB [202]. Collina et al. demonstrated that elevated expression of AXL closely correlated with RAI refractoriness and disease persistence or recurrence in PTCs and a decrease in NIS expression and iodide uptake in an in vitro model [203]. Additionally, the concurrent presence of the BRAF mutation and high AXL expression significantly correlated with the frequency of RAI refractoriness and disease recurrence in patients with PTC.
Membrane targeting of NIS
Membrane protein is produced in the endoplasmic reticulum and then moves to the cellular membrane via the golgi complex [204]. Posttranslational modification of proteins, such as phosphorylation and glycosylation, is required for the process of membrane localization of proteins (Figure 4) [205].
Schematic diagram of factors related to NIS membrane targeting in RAI-refractory differentiated thyroid cancer. Abbreviations: ARF4: ADP-ribosylation factor 4; ERAD: endoplasmic-reticulum-associated protein degradation; LARG: leukemia-associated RhoA guanine exchange factor; PBF: pituitary tumor-transforming gene 1 (PTTG1)-binding factor; PIGU: phosphatidylinositol glycan anchor biosynthesis class U; VCP: valosin-containing protein. Image was created with BioRender.com.
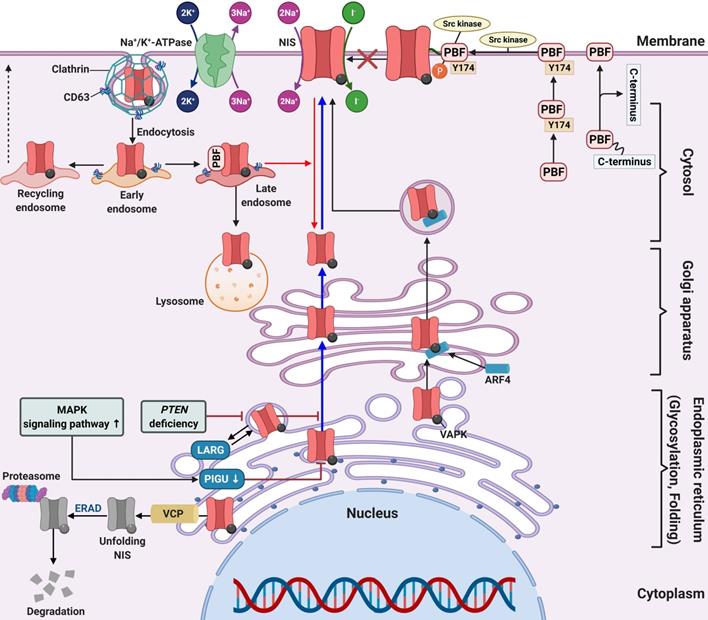
In previous studies, immunohistochemistry of patient samples revealed that approximately 70%-80% of thyroid cancers expressed or even overexpressed NIS [206, 207]. However, expression was primarily in the cytoplasm and not targeted to the plasma membrane. Regarding posttranslational regulation, impairment of NIS function is not limited to decreased or absent expression, but may be the result of impaired membrane targeting of NIS and insufficient retention of NIS in the membrane [4, 159]. Therefore, NIS must be expressed, targeted, and maintained in the plasma membrane of thyroid cells for proper iodide transport.
The tumor microenvironment (TME) includes vessels, immune cells, fibroblasts, signaling molecules, and extracellular matrix [208]. Tumors can affect the TME by releasing extracellular signals which promotes angiogenesis of the tumor and induces peripheral immune tolerance [209]. These signals can induce a change in the TME, and conversely, the TME can influence the growth or metastasis of tumors. Eventually, a tumor's heterogeneity will naturally increase owing to the various factors existing in the TME. Recently, a study highlighted that changes in the TME such as hypoxia or quiescence could impair NIS expression at the plasma membrane and induce distinct patterns of NIS subcellular localization followed by a reduction in NIS-mediated RAI uptake in exogenous NIS-expressing cancer cells [210]. These results suggest that there are diverse underlying molecular mechanisms that bring about change to NIS subcellular localization. Although this study used non-thyroid cancer cells to investigate the molecular mechanisms underlying impairment of NIS membrane targeting in a hypoxic and quiescent environment, the authors postulated that NIS localization on the plasma membrane could be impaired in DTCs by similar molecular mechanisms. Lan et al. reported that the overexpression of HIF1-ɑ, an hypoxia factor, could influence expression and localization of NIS followed by a decline of RAI uptake in FTC [127]. Therefore, appropriate understanding of the TME is also needed to affect the RAI refractoriness.
In a series of 60 human PTC samples, tumors harboring the BRAF mutation showed low expression and poor membrane targeting of NIS in comparison with those without the mutation. This suggests that the BRAF mutation is an obstacle for membrane targeting [49]. Additionally, treatment with a MEK inhibitor partially increased NIS expression, but did not allow NIS migration to the cell membrane in thyroid cells with the BRAF mutation.
A clinical study suggested that patients with significant expression of the proto-oncogene PTTG were closely associated with lymph node metastasis, distant metastasis, and advanced stages of thyroid cancer [211]. Additionally, Heaney et al. reported PTTG overexpression in thyroid cancer tissue, which caused cellular transformation and dedifferentiation such as NIS repression and inhibition of RAI uptake [212]. In thyroid cancer, PBF is upregulated and correlates with tumor growth, development, recurrence, and overall poorer disease outcome [28, 212, 213]. There are two mechanisms for the repression of NIS expression and RAI uptake in overexpressed PBF. First, PBF overexpression represses NIS expression at the transcriptional level and reduces RAI uptake in vitro and in vivo (Figure 3D) [26, 214]. Second, PBF overexpression causes the redistribution of NIS from the plasma membrane to the cytoplasm (Figure 4) [215]. Smith et al. reported that binding and induction of Src kinase led to increased phosphorylation of PBF at residue Y174, triggering the repression of NIS retention in the plasma membrane and RAI uptake (Figure 4) [216]. Abrogation of the Y174 residue by the Src kinase inhibitor (PP1) diminishes the interaction between PBF and NIS and increases RAI uptake.
Amit et al. reported the role of phosphatidylinositol glycan anchor biosynthesis class U (PIGU) expression for NIS trafficking to the cell membrane via the MAPK signaling pathway in PTC (Figure 4) [217]. Glycosylphosphatidylinositol transamidase (GPIT) catalyzes the addition of glycosylphosphatidylinositol anchor to substrate proteins in the endoplasmic reticulum [218]. A PIGU is a protein in the GPIT complex, and its expression was low in primary PTC and PTC cell lines compared with normal thyroid cells [217]. The downregulation of PIGU inhibited NIS glycosylation and cell membrane targeting followed by impaired RAI uptake in PTC. Overexpression of PIGU by blockade of the MAPK signaling pathway restored NIS function. Moreover, PIGU-positive tumors in patients with tumor recurrence who were treated with RAI therapy correlated with positive RAI uptake and moderate-to-strong NIS expression and high Tg levels compared with patients with PIGU-negative tumors, suggesting it to be a novel predictor for response to RAI.
Lacoste et al. described sequestered NIS in the cytoplasm or impaired NIS movement to the cell membrane through an interaction between NIS and leukemia-associated RhoA guanine exchange factor (LARG) with sequestered NIS increasing tumor cell motility and invasion (Figure 4) [219]. Recently, Feng et al. reported that PTEN alteration, a common somatic mutation, and upregulation of the PI3K/AKT signaling pathway likely inhibits NIS glycosylation which impairs its plasma membrane localization. This results in increased cytoplasmic NIS expression [220, 221]. Additionally, PTEN alteration increases LARG expression and RhoA activation, resulting in enhanced cytoplasmic NIS expression through its interaction with NIS-LARG.
Fletcher et al. identified two novel substances that interact with NIS, ADP-ribosylation factor 4 (ARF4) and valosin-containing protein (VCP). Both have important roles in NIS movement to the plasma membrane and enhancement of RAI uptake (Figure 4) [222]. ARF4 recognizes the VAPK motif in the NIS C-terminus and moves vesicular NIS to the plasma membrane. VCP, a component of ER-associated degradation, governs NIS proteolysis. The application of VCP inhibitors abrogated the inhibition of NIS function mediated by VCP and increased NIS expression at the plasma membrane followed by increased RAI uptake in thyroid cancer cells and primary thyrocytes isolated from a mouse model.
Huc-Brandt et al. suggested the dimerization of NIS based on electrophoresis patterns, size exclusion chromatography, and light scattering analyses [223]. Additionally, Thompson et al. demonstrated the role of NIS dimerization for its movement to the plasma membrane, suggesting new mechanisms to be considered in treating patients with RAI-refractory thyroid cancer in the future [224].
Clinical trials
Several clinical trials have been conducted that focus on dedifferentiation mechanisms to reinduce RAI avidity in progressive RAI-refractory thyroid cancers (Table 3).
Update of clinical trials of redifferentiation strategy for radioactive-iodine (RAI) refractory differentiated thyroid cancers
| Classification | Drug | Type of study | Patients (n) | Dose | Duration of treatment before RAI | Main Result | Reference |
|---|---|---|---|---|---|---|---|
| BRAF inhibitor | Dabrafenib | Interventional study | 10 | 150 mg | 25 days | Increase of RAI uptake (6/10); PR (2/6) and SD (4/6) after RAI therapy | [250] |
| Vemurafenib | Pilot study | Total 12 10 evaluable | 960 mg | 4 weeks | Increase of RAI uptake (4/10); Tumor regression after RAI therapy (3/4) | [251] | |
| HDAC inhibitor | Romidepsin (Depsipeptide) | Phase I | 11 | 1 to 9 mg/m2 | 6-112 weeks (21 days cycle) | Faintly increase of RAI uptake (2/6) | [247] |
| Phase II | 20 | 13 mg/m2 | 0.46-12 months (28 days cycle) | Increase of RAI uptake (2/16) | [248] | ||
| Valproic acid | Phase II | 13 | 500 mg once for 3 days followed by 500 mg | 10 weeks | Increase of RAI uptake (0/10) | [249] | |
| Vorinostat | Phase I | 6 | 200-400 mg | 12-37+ months (4 weeks cycle) | Increase of RAI uptake (1/3) | [246] | |
| - | Lithium | Pilot study | 12 | 1200 mg | Unknown duration (During and after 2nd RAI therapy) | Increase of RAI uptake (5/12); *Mixed pattern of RAI uptake (2/12); Response for RAI therapy (0/12). | [254] |
| MEK inhibitor | Selumetinib | Pilot study | Total 24 20 evaluable | 75 mg | 4 weeks | Increase of RAI uptake (12/20); Reached dosimetry threshold for RAI therapy (8/12); PR (5/8) and SD (3/8) after RAI therapy. | [7] |
| Non-nucleoside reverse transcriptase inhibitor | Nevirapine | Case report | 1 | 200 mg | Unknown duration | Increase of RAI uptake in metastatic sites | [253] |
| PPARɣ agonist | Pioglitazone | Case report | 5 | 30 mg followed by 45 mg | 6 months (30 mg dose for 2 weeks followed by 45 mg dose) | Increase of RAI uptake (0/5) | [245] |
| Rosiglitazone | Case report | 1 | 8 mg | 3 months | Increase of RAI uptake | [239] | |
| Case report | 1 | 8 mg | 2 months | Increase of RAI uptake; Tumor regression after RAI therapy. | [240] | ||
| Phase II | 10 | 4 mg or 8 mg | 1 week for 4 mg dose; 7 weeks for 8 mg dose | Increase of RAI uptake (4/10) | [241] | ||
| Phase II | 20 | 4 mg or 8 mg | 1 week for 4 mg dose; 7 weeks for 8 mg dose | PR for RAI uptake and Tg level (5/20) | [242] | ||
| PPARɣ agonist | Rosiglitazone | Pilot study | 5 | 4 mg or 8 mg | 1 month for 4 mg dose; 2 months for 8 mg dose | Faintly increase of RAI uptake (1/5) | [238] |
| Pilot study | 23 | 8 mg | 6 weeks | Increase of RAI uptake (5/23) | [243] | ||
| Pilot study | 9 | 4 mg followed by 8 mg | 6 months (4 mg dose for 2 weeks followed by 8 mg dose) | Increase of RAI uptake (5/9); Tumor regression after RAI therapy (3/9) | [244] | ||
| Retinoic acids | Bexarotene | Pilot study | 12 | 300 mg | 6 weeks | PR for RAI uptake (8/11) | [234] |
| Isotretinoin | Pilot study | 10 | 1.5 mg/kg | 6 weeks | Increase of RAI uptake (4/10) | [225] | |
| Pilot study | 50 | 1.5 mg/kg | 5 weeks | Increase of RAI uptake (21/50); Tumor regression after RAI therapy (6/37) | [226] | ||
| Pilot study | 25 | 1 mg/kg | 3 months | Increase of RAI uptake (5/25) | [227] | ||
| Pilot study | 5 | 1.0 to 1.5 mg/kg | 5 weeks | Increase of RAI uptake (3/5); Decrease of tumor size (1/5) | [228] | ||
| Pilot study | 11 | 1.5 mg/kg | 8 weeks | Increase of RAI uptake (2/11) | [229] | ||
| Pilot study | 27 | 0.66-1.5 mg/kg | 5-12 weeks | Optimal positive RAI uptake (9/27); Suboptimal RAI uptake (10/27); Tumor regression or stabilization after RAI therapy (7/17) | [230] | ||
| Pilot study | 47 | 1-1.5 mg/kg | 6 weeks | CR for RAI uptake (1/47); PR for RAI uptake (9/47) | [231] | ||
| Phase II | 53 | 1.0 mg/kg/day for 1 week followed by 1.5 mg/kg | 6 weeks | Increase of RAI uptake (9/53) | [232] | ||
| Phase II | 16 | 1.5 mg/kg | 8 weeks | Increase of RAI uptake (1/16) | [233] | ||
| Tretinoin | Pilot study | 13 | 1.5 mg/kg | 1.5-18 months | Faintly increase of RAI uptake (6/13); PR for RAI therapy (1/13) | [236] | |
| Retrospective study | 11 | 1.00±0.09 mg·kg-1·d-1 | 30 or 60 days | Re-induction of RAI uptake (4/11); PR for RAI therapy (5/11) | [235] | ||
| Combination | Dabrafenib ± Trametinib, Vemurafenib, Trametinib, Investigational MEK inhibitor | Retrospective study | 13 | Unknown | Median duration of treatment: 14.3 months (range, 0.9 to 76.4) | Increase of RAI uptake (9/13); PR (3/9) and SD (6/9) after RAI therapy | [252] |
| Trametinib ± Dabrafenib, Vemurafenib + Cobimetinib | Retrospective, cohort study | 6 | Unknown | 4 weeks | Increase of RAI uptake (4/6); PR (3/4) and SD (1/4) after RAI therapy | [10] |
Abbreviations: CR: complete response; HDAC: histone deacetylase; N/A: not available; PPARɣ: peroxisome proliferator-activated receptor; PR: partial response; RAI: radioactive iodine; SD: stable disease. *Mixed pattern means 2 regions of interest with different effects of lithium.
Simon et al. administered 13-cis RA (isotretinoin) to patients with RAI-refractory thyroid cancer and found that RAI uptake was reinduced in 4 of 10 patients [225]. Based on results of that trial, several additional studies also reported approximately 20%-40% improvement in RAI uptake after isotretinoin treatment [226-231]. In addition, Handkiewicz-Junak et al. reported that increased RAI uptake was confirmed in 9 of 53 patients; however, no clinical benefit such as inducing tumor regression or preventing disease progression was found [232]. Short et al. conducted an open-label, non-randomized, phase II trial with isotretinoin and found that RAI uptake increased in only 1 of 16 patients, showing a different response from that noted in other studies of isotretinoin application [233]. Clinical trials with administration of other RAs such as RXR activator (bexarotene) and all-trans-RA (tretinoin) to patients with RAI-refractory thyroid cancer have also been reported by several groups [234-236]. An open, prospective, intervention study reported that 8 of 11 patients with RAI-refractory DTC having metastases responded to bexarotene administration with restoration of RAI uptake [234]. However, there was limited clinical relevance due to heterogenous responses of different metastases even in a patient with a low degree of RAI uptake. In 11 patients with advanced DTC, tretinoin treatment increased RAI uptake in 4 patients with a partial response of target lesions in 5 patients [235]. In another study with RAI-refractory DTC patients, tretinoin treatment slightly increased RAI uptake in 6 patients, whereas 7 patients experienced no RAI uptake [236]. A meta-analysis using 14 studies of RA treatment in RAI-refractory DTC revealed a pooled disease response rate assessed by RAI scintigraphy to be 27.6% [237]. Moreover, the pooled response rate was only 17.0% using the Response Evaluation Criteria in Solid Tumours (RECIST) criteria and 30.8% using the World Health Organization criteria. These results suggest that only a few patients with RAI-refractory thyroid cancer respond to RA treatment.
The pilot study to demonstrate increased RAI uptake with rosiglitazone, a PPARɣ agonist which is a member of the thiazolidinedione class, was conducted by Philips et al., and it was found that only 1 of 5 patients had a slightly increased RAI uptake [238]. Two case reports described the restoration of RAI uptake in RAI-refractory thyroid cancer following rosiglitazone treatment and decreased serum Tg and tumor size after RAI treatment [239, 240]. In a phase II trial, Kebebew et al. reported that 4 of 10 patients with RAI-refractory thyroid cancer had positive RAI uptake following 7 weeks of rosiglitazone administration [241]. In addition, another phase II trial by the same group reported that 5 of 20 patients had positive RAI uptake after rosiglitazone treatment [242]. Tepmongkol et al. reported that 7 of 23 patients exhibited strongly positive staining for PPARɣ in thyroid biopsies after rosiglitazone administration [243]. Among the 7 patients with positive PPARɣ staining, 5 had increased RAI uptake on RAI scan. Rosenbaum-Krumme et al. examined whether both rosiglitazone and pioglitazone have the ability to restore RAI uptake in patients with progressive RAI-refractory DTC [244, 245]. They found that 5 of 9 patients exhibited improved RAI uptake. Of the 4 patients treated with RAI, 3 had a partial response when the RECIST 1.1 criteria were used [244]. However, another study on pioglitazone treatment in 5 patients with progressive DTC found no increased RAI uptake [245].
Kelly et al. performed a phase I study with SAHA (vorinostat) as an HDAC inhibitor for advanced thyroid cancer patients and found that 1 of 3 patients with PTC exhibited increased RAI uptake after SAHA treatment [246]. The results of romidepsin administration to patients with RAI-refractory thyroid cancer have also been reported [247, 248]. In a phase I trial by Amiri-Kordestani et al., 2 of 6 patients with thyroid cancer exhibited slightly increased RAI uptake after romidepsin administration; however, the degree of RAI uptake was not sufficient to allow RAI therapy [247]. In another phase II trial of romidepsin, 2 of 16 patients with RAI-refractory thyroid cancer had a reversal of RAI uptake; however, RAI treatment had no clinical benefit [248]. Nilubol et al. tested the effect of restoring RAI avidity by valproic acid for patients with RAI-refractory thyroid cancer, and the results revealed that none of the 10 patients had increased RAI uptake [249].
Ho et al. reported promising outcomes with the administration of selumetinib as an MEK inhibitor to RAI-refractory DTC patients. They noted that 12 of 20 patients' recovered RAI uptake, and 8 of these 12 patients reached the dosimetry threshold for RAI therapy [7]. Among these patients, 5 harbored the NRAS mutation and only 1 had the BRAF mutation, suggesting that the recovery of RAI avidity was higher in patients with NRAS mutations than in those with the BRAF mutation. Rothenberg et al. reported clinical trials on dabrafenib as a BRAF inhibitor to overcome RAI refractoriness in patients with BRAF-mutated thyroid cancer [250]. Six weeks after the administration of dabrafenib to patients with BRAF-mutated thyroid cancer, 6 of 10 patients had restored RAI uptake, 2 exhibited a partial response, and 4 had stable disease. Dunn et al. published a pilot trial on utilizing vemurafenib for approximately 4 weeks in 12 patients with BRAF-mutated RAI-refractory PTC or PDTC [251]. Of the 10 patients evaluated, 4 exhibited restored RAI uptake and RAI therapy was initiated, resulting in tumor regression. Moreover, the effect of vemurafenib to inhibit the MAPK signaling pathway was closely associated with thyroid differentiation score and RAI uptake. Jaber et al. analyzed the restoration of RAI uptake after the administration of BRAF and MEK inhibitors in 13 patients with RAI-refractory DTC or PDTC [252]. Among these patients, 9 had the BRAF mutation, 3 had RAS mutations, and 1 had wild-type. Notably, 9 of 13 patients exhibited increased RAI uptake. In particular, the best responders were RAS-mutated patients (3 of 3 RAS-mutated patients, 5 of 9 BRAF-mutated patients, and 1 of 1 wild-type patients). Iravani et al. reviewed the restoration of RAI avidity by TKIs in patients with RAI-refractory DTC [10]. MEK inhibitor alone was given to 3 patients with the NRAS mutation, and combined MEK and BRAF inhibitors were given to 3 patients with the BRAF mutation. This study found that 4 of 6 patients exhibited restoration of RAI avidity, and patients with the BRAF mutation exhibited a better response by restoring RAI avidity than patients with the NRAS mutation (3 of 3 vs. 1 of 3). Following RAI therapy, 3 patients exhibited a partial response and 1 patient had stable disease. Several clinical studies with multiple drugs that target the MAPK signaling pathway are currently ongoing (NCT02145143, NCT04462471, NCT03244956, NCT02152995 and ISRCTN17468602) (Table 4).
Currently ongoing clinical trials of redifferentiation strategy for radioiodine refractoriness in differentiated thyroid cancer
| Identifier | Target | Drug | Type of Study | Enrolled Patients | Primary Outcome | Status |
|---|---|---|---|---|---|---|
| NCT02145143 | BRAF | Vemurafenib | Pilot study, Single arm, Open label | 12 (BRAF mutant, RAI-refractory thyroid cancers) | *Duration of overall response | Estimated study completion date: May, 2021 |
| NCT04462471 | BRAF, PI3K | Vemurafenib+ Copanlisib | Phase I, Single group assignment, Open label | 22 (BRAF mutant, RAI-refractory thyroid cancers) | MTD | Estimated study completion date: June, 2022 |
| NCT03244956 | MEK, BRAF | Trametinib or Dabrafenib | Phase II, Parallel assignment, Open label | 87 (one for patients with RAS mutation and one for patients with BRAFV600E mutation) | ORR | Estimated study completion date: December, 2022 |
| NCT02152995 | MEK | Trametinib | Phase II, Single arm, Open label | 35 (RAS mutant or RAS/RAF wild-type, RAI-refractory recurrent and/or metastatic thyroid cancers) | Proportion of patients alive following treatment with trametinib and 124I, PFS, ORR, Iodine incorporation | Estimated primary completion date: June 30, 2021 |
| ISRCTN17468602 | MEK | Selumetinib | Phase II, Single arm, | 80 (Differentiated thyroid cancer) | PFS | Closed (Overall trial end date: 18/01/2020) |
| NCT03469011 | PDGFRα | Imatinib | Phase I, Sequential assignment, Open label | 18 (Metastatic thyroid cancer) | Restore iodine uptake | Estimated study completion date: December 31, 2020 |
Abbreviations: MTD: maximum tolerated dose; ORR: objective response rate; PDGFRɑ: platelet-derived growth factor receptor-ɑ; PFS: progression-free survival; PI3K: phosphoinositide 3-kinase; RAI: radioactive iodine. * The duration of overall response is measured from the time measurement criteria are met for complete response or partial response (whichever is first recorded) until the first date that recurrent or progressive disease is objectively documented.
In addition to abovementioned clinical trials with regard to the MAPK signaling pathway, clinical trials targeting new therapeutic targets have also been conducted by several groups. Modoni et al. treated a patient with RAI-refractory PTC with nevirapine, a non-nucleoside reverse transcriptase inhibitor, and reported restoration of RAI uptake (Table 3) [253]. Liu et al. reported that 7 of 12 patients with DTC exhibited increased RAI uptake after lithium treatment, but adding lithium to RAI therapy had no clinical benefits (Table 3) [254]. Based on a preclinical study for regulating cell redifferentiation along with blockade of PDGFRɑ [110], a clinical trial is ongoing with the administration of imatinib as an anti-cancer drug by blocking PDGFRɑ function in patients with advanced thyroid cancer (NCT03469011) (Table 4).
Future perspectives and conclusion
For a long time, RAI therapy has been successfully used as a treatment for thyroid cancer. It is administered after thyroidectomy for ablation of any remaining thyroid tissue and residual or metastatic cancer, and it has contributed to the excellent prognosis of DTCs. However, dedifferentiated thyroid cancers that lose RAI avidity are not responsive to RAI therapy and have a poor prognosis. Restoring RAI avidity via thyroid-specific gene expression is a promising strategy to convert RAI-refractory thyroid cancers into RAI-sensitive thyroid cancers. Thus, various compounds have been examined to restore RAI uptake for RAI non-avid thyroid cancers in both preclinical studies and clinical trials by multiple researchers. Unfortunately, there are no definitive successful clinical trials that meet the expectation of pharmacologic redifferentiation effects yet. There are several shortcomings and limitations of current studies regarding the development of innovative compounds for understanding the mechanisms underlying RAI-refractory thyroid cancer. First, most studies examined and focused on a specific signaling pathway but not comprehensive signaling pathways. However, RAI refractoriness is complex and closely associated with multiple signaling pathways. Therefore, sometimes, blockade of a single signaling pathway was insufficient to completely block a target. In addition, resistance or reactivation of specific signaling pathways after pharmacologic treatment could also be an obstacle for redifferentiation efficacy. As noted previously, treatment with a single drug could fail to meet the redifferentiation efficacy owing to reactivation of the signaling pathway followed by drug resistance [57, 63]. Therefore, strategies with combination treatments to overcome resistance and decreased efficacy of drugs should be considered. Second, the TME can disturb the target drug response. Recently, an article reported that TME (hypoxia and quiescence) could influence NIS expression in exogenous NIS-expressing cancer cells. Specifically, immunohistochemistry with tumor tissue from an animal model showed heterogeneous characteristics within the peripheral region revealing stronger NIS expression with lower HIF-1ɑ levels than internal regions [210]. In addition, most in vitro studies employed 2-dimensional monolayer culture with cancer cells. This environment may not reflect the heterogenous characteristics of cancer with a change in TME. Therefore, the development of experimental protocols using an environment similar to that of actual cancer tissue can be critical to the discovery of valuable drugs for RAI-refractory thyroid cancers. To address this concern, we should consider the following approaches for enhancing RAI uptake via redifferentiating agents in RAI-refractory thyroid cancers: (1) tailored redifferentiation strategy depending on genetic and epigenetic alteration because RAI-refractory thyroid cancers are quite heterogeneous genetically and epigenetically and their responses to redifferentiation therapy also are variable by their inborn nature; (2) combination strategy with >2 compounds for redifferentiation because most RAI-refractory thyroid cancers develop dedifferentiation by multiple factors; and (3) optimization of redifferentiation strategy for dosing and duration. Understanding the detailed molecular mechanisms for dedifferentiation of individual RAI-refractory thyroid cancers, such as genetic mutations, epigenetic alteration, change of signaling pathways, and posttranslational protein modification, is needed to develop the optimal personalized redifferentiation strategy (Figures 4 and 5). In the future, pharmacologic redifferentiation followed by RAI treatment can be one of the main therapeutic strategies for dedifferentiated RAI-refractory thyroid cancers.
Comprehensive molecular mechanisms related to RAI refractoriness in differentiated thyroid cancer. Abbreviations: 4E-BP: 4E-binding protein; Ac: acetylation; BRD4: bromodomain-containing protein; DNMT1: DNA methyltransferase 1; ERRɣ: estrogen-related receptor ɣ; FoxP3: forkhead transcription factor 3; HDAC: histone deacetylase; HMGB1: high mobility group box 1; IκB: inhibitory κB; LXRß: liver X receptor ß; TERT: telomerase reverse transcriptase; TSHR: TSH receptor; miR: microRNA; mTORC1: mTOR complex 1; mTORC2: mTOR complex 2; P: phosphorylation. NECD: notch extracellular domain; NICD: notch intracellular domain; NOX4: NADPH oxidase 4; P: phosphorylation; PBF: pituitary tumor-transforming gene 1 (PTTG1)-binding factor; PTEN: phosphatase and tensin homolog; RAR: retinoic acid receptor; PPARɣ: peroxisome proliferator-activated receptor ɣ; ROS: reactive oxygen species; RAR: retinoic acid receptor; RXR: retinoid X receptor; TERT: telomerase reverse transcriptase; Ub: ubiquitination. Image was created with BioRender.com.
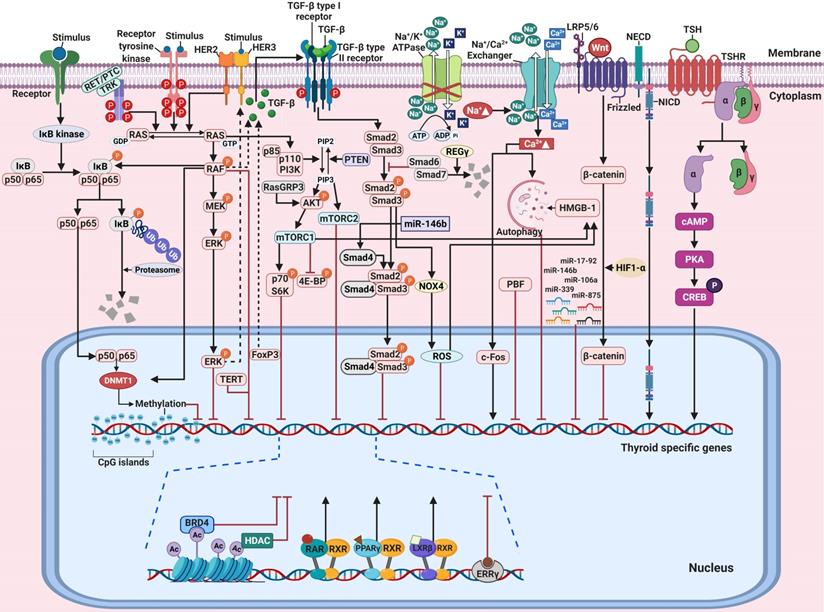
Abbreviations
4E-BP1: 4E-binding protein 1; ARF4: ADP-ribosylation factor 4; ATC: anaplastic thyroid cancer; BRD4: bromodomain-containing protein 4; cAMP: cyclic adenosine monophosphate; CREB: cAMP response element-binding protein; DEHAL1: iodotyrosine dehalogenase 1; DIO2: iodothyronine deiodinase 2; DLC: digitalis-like compound; DNMT1: DNA methyltransferase 1; DTC: differentiated thyroid cancers; ERRs: estrogen-related receptors; ERKs: extracellular-signal-regulated kinases; FDG: fluorodeoxyglucose; FoxP3: Forkhead transcription factor 3; FTC: follicular thyroid cancer; GPIT: glycosylphosphatidylinositol transamidase; H3K18ac: H3 histone; H3K9-K14ac: acetylated lysines 9 and 14 in H3; HDAC: histone deacetylase; HER3: human epidermal growth factor receptor 3; HMGB1: high mobility group box 1; IκB: inhibitory κB; IKK: IκB kinase; JNK: Jun amino-terminal kinase; LARG: leukemia-associated RhoA guanine exchange factor; LPS: lipopolysaccharide; LXRs: liver X receptors; MAPK: mitogen-activated protein kinase; miRNA: microRNA; mRNA: messenger RNA; MTC: medullary thyroid cancer; mTOR: mechanistic target of rapamycin; mTORC1: mTOR complex 1; mTORC2: mTOR complex 2; NECD: notch extracellular domain; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; NICD3: notch intracellular domain 3; NIS: sodium iodide symporter; NOX4: NADPH oxidase 4; NUE: NIS upstream enhancer; SAPK: stress-activated protein kinase; PAX-8: paired box gene-8; PBF: PTTG1-binding factor; PDGFRɑ: platelet-derived growth factor receptor-ɑ; PDTC: poorly differentiated thyroid cancer; PI3K: phosphoinositide 3-kinase; PIGU: phosphatidylinositol glycan anchor biosynthesis class U; PKA: protein kinase A; PPARɣ: PAX8-peroxisome proliferator-activated receptor ɣ; PTC: papillary thyroid cancer; PTTG1: pituitary tumor-transforming gene 1; RA: retinoic acid; RAI: radioactive iodine; RAR: RA receptor; RECIST: response evaluation criteria in solid tumors; Ref-1: redox effector factor-1; ROS: reactive oxygen species; RXR: retinoid X receptor; TERT: telomerase reverse transcriptase; Tg: thyroglobulin; TGF-ß: transforming growth factor-ß; TLR4: toll-like receptor 4; TME: tumor microenvironment; TNF-ɑ: tumor necrosis factor-ɑ; TPO: thyroperoxidase; TSH: thyroid-stimulating hormone; TSHR: TSH receptor; TTF-1: thyroid transcription factor-1; TTF-2: thyroid transcription factor-2; VCP: valosin-containing protein.
Acknowledgements
This work was supported by Biomedical Research Institute grant, Kyungpook National University Hospital (2020). All figures were created with BioRender.com.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Spitzweg C, Bible KC, Hofbauer LC, Morris JC. Advanced radioiodine-refractory differentiated thyroid cancer: the sodium iodide symporter and other emerging therapeutic targets. Lancet Diabetes Endocrinol. 2014;2:830-42
2. Fagin JA, Wells SA Jr. Biologic and Clinical Perspectives on Thyroid Cancer. N Engl J Med. 2016;375:2307
3. Landa I, Ganly I, Chan TA, Mitsutake N, Matsuse M, Ibrahimpasic T. et al. Frequent somatic TERT promoter mutations in thyroid cancer: higher prevalence in advanced forms of the disease. J Clin Endocrinol Metab. 2013;98:E1562-6
4. Liu J, Liu Y, Lin Y, Liang J. Radioactive Iodine-Refractory Differentiated Thyroid Cancer and Redifferentiation Therapy. Endocrinol Metab (Seoul). 2019;34:215-25
5. Durante C, Haddy N, Baudin E, Leboulleux S, Hartl D, Travagli JP. et al. Long-term outcome of 444 patients with distant metastases from papillary and follicular thyroid carcinoma: benefits and limits of radioiodine therapy. J Clin Endocrinol Metab. 2006;91:2892-9
6. Haugen BR, Alexander EK, Bible KC, Doherty GM, Mandel SJ, Nikiforov YE. et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid. 2016;26:1-133
7. Ho AL, Grewal RK, Leboeuf R, Sherman EJ, Pfister DG, Deandreis D. et al. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N Engl J Med. 2013;368:623-32
8. Brose MS, Nutting CM, Jarzab B, Elisei R, Siena S, Bastholt L. et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet. 2014;384:319-28
9. Schlumberger M, Tahara M, Wirth LJ, Robinson B, Brose MS, Elisei R. et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N Engl J Med. 2015;372:621-30
10. Iravani A, Solomon B, Pattison DA, Jackson P, Ravi Kumar A, Kong G. et al. Mitogen-Activated Protein Kinase Pathway Inhibition for Redifferentiation of Radioiodine Refractory Differentiated Thyroid Cancer: An Evolving Protocol. Thyroid. 2019;29:1634-45
11. Hong CM, Ahn BC. Redifferentiation of Radioiodine Refractory Differentiated Thyroid Cancer for Reapplication of I-131 Therapy. Front Endocrinol (Lausanne). 2017;8:260
12. Aashiq M, Silverman DA, Na'ara S, Takahashi H, Amit M. Radioiodine-Refractory Thyroid Cancer: Molecular Basis of Redifferentiation Therapies, Management, and Novel Therapies. Cancers (Basel). 2019 11
13. Kogai T, Brent GA. The sodium iodide symporter (NIS): regulation and approaches to targeting for cancer therapeutics. Pharmacol Ther. 2012;135:355-70
14. Ahn BC. Sodium iodide symporter for nuclear molecular imaging and gene therapy: from bedside to bench and back. Theranostics. 2012;2:392-402
15. Chung JK. Sodium iodide symporter: its role in nuclear medicine. J Nucl Med. 2002;43:1188-200
16. Hou P, Bojdani E, Xing M. Induction of thyroid gene expression and radioiodine uptake in thyroid cancer cells by targeting major signaling pathways. J Clin Endocrinol Metab. 2010;95:820-8
17. Shang H, Zhao J, Yao J, Wang H, Dong J, Liao L. Nevirapine Increases Sodium/Iodide Symporter-Mediated Radioiodide Uptake by Activation of TSHR/cAMP/CREB/PAX8 Signaling Pathway in Dedifferentiated Thyroid Cancer. Front Oncol. 2020;10:404
18. Buffet C, Wassermann J, Hecht F, Leenhardt L, Dupuy C, Groussin L. et al. Redifferentiation of radioiodine-refractory thyroid cancers. Endocr Relat Cancer. 2020;27:R113-R32
19. Cheng W, Liu R, Zhu G, Wang H, Xing M. Robust Thyroid Gene Expression and Radioiodine Uptake Induced by Simultaneous Suppression of BRAF V600E and Histone Deacetylase in Thyroid Cancer Cells. J Clin Endocrinol Metab. 2016;101:962-71
20. Kogai T, Taki K, Brent GA. Enhancement of sodium/iodide symporter expression in thyroid and breast cancer. Endocr Relat Cancer. 2006;13:797-826
21. Fenton MS, Marion KM, Hershman JM. Identification of cyclic adenosine 3',5'-monophosphate response element modulator as an activator of the human sodium/iodide symporter upstream enhancer. Endocrinology. 2008;149:2592-606
22. Riesco-Eizaguirre G, Rodriguez I, De la Vieja A, Costamagna E, Carrasco N, Nistal M. et al. The BRAFV600E oncogene induces transforming growth factor beta secretion leading to sodium iodide symporter repression and increased malignancy in thyroid cancer. Cancer Res. 2009;69:8317-25
23. Costamagna E, Garcia B, Santisteban P. The functional interaction between the paired domain transcription factor Pax8 and Smad3 is involved in transforming growth factor-beta repression of the sodium/iodide symporter gene. J Biol Chem. 2004;279:3439-46
24. Nicola JP, Nazar M, Mascanfroni ID, Pellizas CG, Masini-Repiso AM. NF-kappaB p65 subunit mediates lipopolysaccharide-induced Na(+)/I(-) symporter gene expression by involving functional interaction with the paired domain transcription factor Pax8. Mol Endocrinol. 2010;24:1846-62
25. Nicola JP, Velez ML, Lucero AM, Fozzatti L, Pellizas CG, Masini-Repiso AM. Functional toll-like receptor 4 conferring lipopolysaccharide responsiveness is expressed in thyroid cells. Endocrinology. 2009;150:500-8
26. Boelaert K, Smith VE, Stratford AL, Kogai T, Tannahill LA, Watkinson JC. et al. PTTG and PBF repress the human sodium iodide symporter. Oncogene. 2007;26:4344-56
27. Stratford AL, Boelaert K, Tannahill LA, Kim DS, Warfield A, Eggo MC. et al. Pituitary tumor transforming gene binding factor: a novel transforming gene in thyroid tumorigenesis. J Clin Endocrinol Metab. 2005;90:4341-9
28. Kogai T, Endo T, Saito T, Miyazaki A, Kawaguchi A, Onaya T. Regulation by thyroid-stimulating hormone of sodium/iodide symporter gene expression and protein levels in FRTL-5 cells. Endocrinology. 1997;138:2227-32
29. Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat Rev Cancer. 2013;13:184-99
30. Oh JM, Baek SH, Gangadaran P, Hong CM, Rajendran RL, Lee HW. et al. A Novel Tyrosine Kinase Inhibitor Can Augment Radioactive Iodine Uptake Through Endogenous Sodium/Iodide Symporter Expression in Anaplastic Thyroid Cancer. Thyroid. 2020;30:501-18
31. Rowe CW, Paul JW, Gedye C, Tolosa JM, Bendinelli C, McGrath S. et al. Targeting the TSH receptor in thyroid cancer. Endocr Relat Cancer. 2017;24:R191-R202
32. Lazar V, Bidart JM, Caillou B, Mahe C, Lacroix L, Filetti S. et al. Expression of the Na+/I- symporter gene in human thyroid tumors: a comparison study with other thyroid-specific genes. J Clin Endocrinol Metab. 1999;84:3228-34
33. Takamatsu J, Hosoya T, Tsuji M, Yamada M, Murakami Y, Sakane S. et al. Peroxidase and coupling activities of thyroid peroxidase in benign and malignant thyroid tumor tissues. Thyroid. 1992;2:193-6
34. Tanaka T, Umeki K, Yamamoto I, Sugiyama S, Noguchi S, Ohtaki S. Immunohistochemical loss of thyroid peroxidase in papillary thyroid carcinoma: strong suppression of peroxidase gene expression. J Pathol. 1996;179:89-94
35. Prpic M, Franceschi M, Romic M, Jukic T, Kusic Z. Thyroglobulin as a Tumor Marker in Differentiated Thyroid Cancer - Clinical Considerations. Acta Clin Croat. 2018;57:518-27
36. Ros P, Rossi DL, Acebron A, Santisteban P. Thyroid-specific gene expression in the multi-step process of thyroid carcinogenesis. Biochimie. 1999;81:389-96
37. Fabbro D, Di Loreto C, Beltrami CA, Belfiore A, Di Lauro R, Damante G. Expression of thyroid-specific transcription factors TTF-1 and PAX-8 in human thyroid neoplasms. Cancer Res. 1994;54:4744-9
38. Mu D, Huang R, Li S, Ma X, Lou C, Kuang A. Combining transfer of TTF-1 and Pax-8 gene: a potential strategy to promote radioiodine therapy of thyroid carcinoma. Cancer Gene Ther. 2012;19:402-11
39. Feng F, Wang H, Hou S, Fu H. Re-induction of cell differentiation and (131)I uptake in dedifferentiated FTC-133 cell line by TSHR gene transfection. Nucl Med Biol. 2012;39:1261-5
40. Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12:9-18
41. Zaballos MA, Santisteban P. Key signaling pathways in thyroid cancer. J Endocrinol. 2017;235:R43-R61
42. Frattini M, Ferrario C, Bressan P, Balestra D, De Cecco L, Mondellini P. et al. Alternative mutations of BRAF, RET and NTRK1 are associated with similar but distinct gene expression patterns in papillary thyroid cancer. Oncogene. 2004;23:7436-40
43. Soares P, Trovisco V, Rocha AS, Lima J, Castro P, Preto A. et al. BRAF mutations and RET/PTC rearrangements are alternative events in the etiopathogenesis of PTC. Oncogene. 2003;22:4578-80
44. Kimura ET, Nikiforova MN, Zhu Z, Knauf JA, Nikiforov YE, Fagin JA. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003;63:1454-7
45. Xing M. BRAF mutation in papillary thyroid cancer: pathogenic role, molecular bases, and clinical implications. Endocr Rev. 2007;28:742-62
46. Lee JH, Lee ES, Kim YS. Clinicopathologic significance of BRAF V600E mutation in papillary carcinomas of the thyroid: a meta-analysis. Cancer. 2007;110:38-46
47. Xing M, Westra WH, Tufano RP, Cohen Y, Rosenbaum E, Rhoden KJ. et al. BRAF mutation predicts a poorer clinical prognosis for papillary thyroid cancer. J Clin Endocrinol Metab. 2005;90:6373-9
48. Ricarte-Filho JC, Ryder M, Chitale DA, Rivera M, Heguy A, Ladanyi M. et al. Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res. 2009;69:4885-93
49. Riesco-Eizaguirre G, Gutierrez-Martinez P, Garcia-Cabezas MA, Nistal M, Santisteban P. The oncogene BRAF V600E is associated with a high risk of recurrence and less differentiated papillary thyroid carcinoma due to the impairment of Na+/I- targeting to the membrane. Endocr Relat Cancer. 2006;13:257-69
50. Mian C, Barollo S, Pennelli G, Pavan N, Rugge M, Pelizzo MR. et al. Molecular characteristics in papillary thyroid cancers (PTCs) with no 131I uptake. Clin Endocrinol (Oxf). 2008;68:108-16
51. Durante C, Puxeddu E, Ferretti E, Morisi R, Moretti S, Bruno R. et al. BRAF mutations in papillary thyroid carcinomas inhibit genes involved in iodine metabolism. J Clin Endocrinol Metab. 2007;92:2840-3
52. Xing M. BRAF mutation in thyroid cancer. Endocr Relat Cancer. 2005;12:245-62
53. Henderson YC, Shellenberger TD, Williams MD, El-Naggar AK, Fredrick MJ, Cieply KM. et al. High rate of BRAF and RET/PTC dual mutations associated with recurrent papillary thyroid carcinoma. Clin Cancer Res. 2009;15:485-91
54. Mitsutake N, Knauf JA, Mitsutake S, Mesa C Jr, Zhang L, Fagin JA. Conditional BRAFV600E expression induces DNA synthesis, apoptosis, dedifferentiation, and chromosomal instability in thyroid PCCL3 cells. Cancer Res. 2005;65:2465-73
55. Kleiman DA, Buitrago D, Crowley MJ, Beninato T, Veach AJ, Zanzonico PB. et al. Thyroid stimulating hormone increases iodine uptake by thyroid cancer cells during BRAF silencing. J Surg Res. 2013;182:85-93
56. Liu D, Hu S, Hou P, Jiang D, Condouris S, Xing M. Suppression of BRAF/MEK/MAP kinase pathway restores expression of iodide-metabolizing genes in thyroid cells expressing the V600E BRAF mutant. Clin Cancer Res. 2007;13:1341-9
57. Cheng L, Jin Y, Liu M, Ruan M, Chen L. HER inhibitor promotes BRAF/MEK inhibitor-induced redifferentiation in papillary thyroid cancer harboring BRAFV600E. Oncotarget. 2017;8:19843-54
58. Chakravarty D, Santos E, Ryder M, Knauf JA, Liao XH, West BL. et al. Small-molecule MAPK inhibitors restore radioiodine incorporation in mouse thyroid cancers with conditional BRAF activation. J Clin Invest. 2011;121:4700-11
59. Crispo F, Notarangelo T, Pietrafesa M, Lettini G, Storto G, Sgambato A. et al. BRAF Inhibitors in Thyroid Cancer: Clinical Impact, Mechanisms of Resistance and Future Perspectives. Cancers (Basel). 2019 11
60. Ofir Dovrat T, Sokol E, Frampton G, Shachar E, Pelles S, Geva R. et al. Unusually long-term responses to vemurafenib in BRAF V600E mutated colon and thyroid cancers followed by the development of rare RAS activating mutations. Cancer Biol Ther. 2018;19:871-4
61. Montero-Conde C, Ruiz-Llorente S, Dominguez JM, Knauf JA, Viale A, Sherman EJ. et al. Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov. 2013;3:520-33
62. Song H, Zhang J, Ning L, Zhang H, Chen D, Jiao X. et al. The MEK1/2 Inhibitor AZD6244 Sensitizes BRAF-Mutant Thyroid Cancer to Vemurafenib. Med Sci Monit. 2018;24:3002-10
63. Zhang H, Chen D. Synergistic inhibition of MEK/ERK and BRAF V600E with PD98059 and PLX4032 induces sodium/iodide symporter (NIS) expression and radioiodine uptake in BRAF mutated papillary thyroid cancer cells. Thyroid Res. 2018;11:13
64. Pomerance M, Abdullah HB, Kamerji S, Correze C, Blondeau JP. Thyroid-stimulating hormone and cyclic AMP activate p38 mitogen-activated protein kinase cascade. Involvement of protein kinase A, rac1, and reactive oxygen species. J Biol Chem. 2000;275:40539-46
65. Pomerance M, Carapau D, Chantoux F, Mockey M, Correze C, Francon J. et al. CCAAT/enhancer-binding protein-homologous protein expression and transcriptional activity are regulated by 3',5'-cyclic adenosine monophosphate in thyroid cells. Mol Endocrinol. 2003;17:2283-94
66. Kogai T, Liu YY, Mody K, Shamsian DV, Brent GA. Regulation of sodium iodide symporter gene expression by Rac1/p38beta mitogen-activated protein kinase signaling pathway in MCF-7 breast cancer cells. J Biol Chem. 2012;287:3292-300
67. Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690-701
68. Faria M, Felix D, Domingues R, Bugalho MJ, Matos P, Silva AL. Antagonistic effects of RAC1 and tumor-related RAC1b on NIS expression in thyroid. J Mol Endocrinol. 2019;63:309-20
69. Zhou C, Licciulli S, Avila JL, Cho M, Troutman S, Jiang P. et al. The Rac1 splice form Rac1b promotes K-ras-induced lung tumorigenesis. Oncogene. 2013;32:903-9
70. Faria M, Capinha L, Simoes-Pereira J, Bugalho MJ, Silva AL. Extending the Impact of RAC1b Overexpression to Follicular Thyroid Carcinomas. Int J Endocrinol. 2016;2016:1972367
71. Silva AL, Carmo F, Bugalho MJ. RAC1b overexpression in papillary thyroid carcinoma: a role to unravel. Eur J Endocrinol. 2013;168:795-804
72. Liu R, Xing M. TERT promoter mutations in thyroid cancer. Endocr Relat Cancer. 2016;23:R143-55
73. Liu X, Bishop J, Shan Y, Pai S, Liu D, Murugan AK. et al. Highly prevalent TERT promoter mutations in aggressive thyroid cancers. Endocr Relat Cancer. 2013;20:603-10
74. Xing M, Liu R, Liu X, Murugan AK, Zhu G, Zeiger MA. et al. BRAF V600E and TERT promoter mutations cooperatively identify the most aggressive papillary thyroid cancer with highest recurrence. J Clin Oncol. 2014;32:2718-26
75. Liu R, Bishop J, Zhu G, Zhang T, Ladenson PW, Xing M. Mortality Risk Stratification by Combining BRAF V600E and TERT Promoter Mutations in Papillary Thyroid Cancer: Genetic Duet of BRAF and TERT Promoter Mutations in Thyroid Cancer Mortality. JAMA Oncol. 2017;3:202-8
76. Liu J, Liu R, Shen X, Zhu G, Li B, Xing M. The Genetic Duet of BRAF V600E and TERT Promoter Mutations Robustly Predicts Loss of Radioiodine Avidity in Recurrent Papillary Thyroid Cancer. J Nucl Med. 2020;61:177-82
77. Fuziwara CS, Saito KC, Leoni SG, Waitzberg AFL, Kimura ET. The Highly Expressed FAM83F Protein in Papillary Thyroid Cancer Exerts a Pro-Oncogenic Role in Thyroid Follicular Cells. Front Endocrinol (Lausanne). 2019;10:134
78. Knauf JA, Kuroda H, Basu S, Fagin JA. RET/PTC-induced dedifferentiation of thyroid cells is mediated through Y1062 signaling through SHC-RAS-MAP kinase. Oncogene. 2003;22:4406-12
79. Vadysirisack DD, Venkateswaran A, Zhang Z, Jhiang SM. MEK signaling modulates sodium iodide symporter at multiple levels and in a paradoxical manner. Endocr Relat Cancer. 2007;14:421-32
80. Ruan M, Liu M, Dong Q, Chen L. Iodide- and glucose-handling gene expression regulated by sorafenib or cabozantinib in papillary thyroid cancer. J Clin Endocrinol Metab. 2015;100:1771-9
81. Fenton MS, Marion KM, Salem AK, Hogen R, Naeim F, Hershman JM. Sunitinib inhibits MEK/ERK and SAPK/JNK pathways and increases sodium/iodide symporter expression in papillary thyroid cancer. Thyroid. 2010;20:965-74
82. Ishii N, Harada N, Joseph EW, Ohara K, Miura T, Sakamoto H. et al. Enhanced inhibition of ERK signaling by a novel allosteric MEK inhibitor, CH5126766, that suppresses feedback reactivation of RAF activity. Cancer Res. 2013;73:4050-60
83. Nagarajah J, Le M, Knauf JA, Ferrandino G, Montero-Conde C, Pillarsetty N. et al. Sustained ERK inhibition maximizes responses of BrafV600E thyroid cancers to radioiodine. J Clin Invest. 2016;126:4119-24
84. Oh JM, Kalimuthu S, Gangadaran P, Baek SH, Zhu L, Lee HW. et al. Reverting iodine avidity of radioactive-iodine refractory thyroid cancer with a new tyrosine kinase inhibitor (K905-0266) excavated by high-throughput NIS (sodium iodide symporter) enhancer screening platform using dual reporter gene system. Oncotarget. 2018;9:7075-87
85. Omur O, Baran Y. An update on molecular biology of thyroid cancers. Crit Rev Oncol Hematol. 2014;90:233-52
86. Zakarija M, McKenzie JM. Variations in the culture medium for FRTL5 cells: effects on growth and iodide uptake. Endocrinology. 1989;125:1253-9
87. Saji M, Kohn LD. Insulin and insulin-like growth factor-I inhibit thyrotropin-increased iodide transport in serum-depleted FRTL-5 rat thyroid cells: modulation of adenosine 3',5'-monophosphate signal action. Endocrinology. 1991;128:1136-43
88. Garcia B, Santisteban P. PI3K is involved in the IGF-I inhibition of TSH-induced sodium/iodide symporter gene expression. Mol Endocrinol. 2002;16:342-52
89. Kogai T, Sajid-Crockett S, Newmarch LS, Liu YY, Brent GA. Phosphoinositide-3-kinase inhibition induces sodium/iodide symporter expression in rat thyroid cells and human papillary thyroid cancer cells. J Endocrinol. 2008;199:243-52
90. Song J, Qiu W, Deng X, Qiu Z, Fan Y, Yang Z. A somatic mutation of RasGRP3 decreases Na(+)/I(-) symporter expression in metastases of radioactive iodine-refractory thyroid cancer by stimulating the Akt signaling pathway. Am J Cancer Res. 2018;8:1847-55
91. de Souza EC, Padron AS, Braga WM, de Andrade BM, Vaisman M, Nasciutti LE. et al. MTOR downregulates iodide uptake in thyrocytes. J Endocrinol. 2010;206:113-20
92. Plantinga TS, Heinhuis B, Gerrits D, Netea MG, Joosten LA, Hermus AR. et al. mTOR Inhibition promotes TTF1-dependent redifferentiation and restores iodine uptake in thyroid carcinoma cell lines. J Clin Endocrinol Metab. 2014;99:E1368-75
93. Tavares C, Coelho MJ, Melo M, da Rocha AG, Pestana A, Batista R. et al. pmTOR is a marker of aggressiveness in papillary thyroid carcinomas. Surgery. 2016;160:1582-90
94. Tavares C, Eloy C, Melo M, Gaspar da Rocha A, Pestana A, Batista R. et al. mTOR Pathway in Papillary Thyroid Carcinoma: Different Contributions of mTORC1 and mTORC2 Complexes for Tumor Behavior and SLC5A5 mRNA Expression. Int J Mol Sci. 2018 19
95. Ferretti E, Tosi E, Po A, Scipioni A, Morisi R, Espinola MS. et al. Notch signaling is involved in expression of thyrocyte differentiation markers and is down-regulated in thyroid tumors. J Clin Endocrinol Metab. 2008;93:4080-7
96. Yu XM, Jaskula-Sztul R, Ahmed K, Harrison AD, Kunnimalaiyaan M, Chen H. Resveratrol induces differentiation markers expression in anaplastic thyroid carcinoma via activation of Notch1 signaling and suppresses cell growth. Mol Cancer Ther. 2013;12:1276-87
97. Carre A, Rachdi L, Tron E, Richard B, Castanet M, Schlumberger M. et al. Hes1 is required for appropriate morphogenesis and differentiation during mouse thyroid gland development. PLoS One. 2011;6:e16752
98. Somnay YR, Yu XM, Lloyd RV, Leverson G, Aburjania Z, Jang S. et al. Notch3 expression correlates with thyroid cancer differentiation, induces apoptosis, and predicts disease prognosis. Cancer. 2017;123:769-82
99. Jakowlew SB. Transforming growth factor-beta in cancer and metastasis. Cancer Metastasis Rev. 2006;25:435-57
100. Pisarev MA, Thomasz L, Juvenal GJ. Role of transforming growth factor beta in the regulation of thyroid function and growth. Thyroid. 2009;19:881-92
101. Nicolussi A, D'Inzeo S, Santulli M, Colletta G, Coppa A. TGF-beta control of rat thyroid follicular cells differentiation. Mol Cell Endocrinol. 2003;207:1-11
102. Grubeck-Loebenstein B, Buchan G, Sadeghi R, Kissonerghis M, Londei M, Turner M. et al. Transforming growth factor beta regulates thyroid growth. Role in the pathogenesis of nontoxic goiter. J Clin Invest. 1989;83:764-70
103. Kawaguchi A, Ikeda M, Endo T, Kogai T, Miyazaki A, Onaya T. Transforming growth factor-beta1 suppresses thyrotropin-induced Na+/I- symporter messenger RNA and protein levels in FRTL-5 rat thyroid cells. Thyroid. 1997;7:789-94
104. Azouzi N, Cailloux J, Cazarin JM, Knauf JA, Cracchiolo J, Al Ghuzlan A. et al. NADPH Oxidase NOX4 Is a Critical Mediator of BRAF(V600E)-Induced Downregulation of the Sodium/Iodide Symporter in Papillary Thyroid Carcinomas. Antioxid Redox Signal. 2017;26:864-77
105. Niu J, Jiang C, Li C, Liu L, Li K, Jian Z. et al. Foxp3 expression in melanoma cells as a possible mechanism of resistance to immune destruction. Cancer Immunol Immunother. 2011;60:1109-18
106. Hinz S, Pagerols-Raluy L, Oberg HH, Ammerpohl O, Grussel S, Sipos B. et al. Foxp3 expression in pancreatic carcinoma cells as a novel mechanism of immune evasion in cancer. Cancer Res. 2007;67:8344-50
107. Ugolini C, Elisei R, Proietti A, Pelliccioni S, Lupi C, Borrelli N. et al. FoxP3 expression in papillary thyroid carcinoma: a possible resistance biomarker to iodine 131 treatment. Thyroid. 2014;24:339-46
108. Ma S, Wang Q, Ma X, Wu L, Guo F, Ji H. et al. FoxP3 in papillary thyroid carcinoma induces NIS repression through activation of the TGF-beta1/Smad signaling pathway. Tumour Biol. 2016;37:989-98
109. Jiao C, Li L, Zhang P, Zhang L, Li K, Fang R. et al. REGgamma ablation impedes dedifferentiation of anaplastic thyroid carcinoma and accentuates radio-therapeutic response by regulating the Smad7-TGF-beta pathway. Cell Death Differ. 2020;27:497-508
110. Lopez-Campistrous A, Adewuyi EE, Benesch MGK, Ko YM, Lai R, Thiesen A. et al. PDGFRalpha Regulates Follicular Cell Differentiation Driving Treatment Resistance and Disease Recurrence in Papillary Thyroid Cancer. EBioMedicine. 2016;12:86-97
111. Karin M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol. 2009;1:a000141
112. Park MH, Hong JT. Roles of NF-kappaB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches. Cells. 2016 5
113. Reale C, Iervolino A, Scudiero I, Ferravante A, D'Andrea LE, Mazzone P. et al. NF-kappaB Essential Modulator (NEMO) Is Critical for Thyroid Function. J Biol Chem. 2016;291:5765-73
114. Velez ML, Costamagna E, Kimura ET, Fozzatti L, Pellizas CG, Montesinos MM. et al. Bacterial lipopolysaccharide stimulates the thyrotropin-dependent thyroglobulin gene expression at the transcriptional level by involving the transcription factors thyroid transcription factor-1 and paired box domain transcription factor 8. Endocrinology. 2006;147:3260-75
115. Nazar M, Nicola JP, Velez ML, Pellizas CG, Masini-Repiso AM. Thyroid peroxidase gene expression is induced by lipopolysaccharide involving nuclear factor (NF)-kappaB p65 subunit phosphorylation. Endocrinology. 2012;153:6114-25
116. Faria M, Domingues R, Paixao F, Bugalho MJ, Matos P, Silva AL. TNFalpha-mediated activation of NF-kappaB downregulates sodium-iodide symporter expression in thyroid cells. PLoS One. 2020;15:e0228794
117. Visconti R, Cerutti J, Battista S, Fedele M, Trapasso F, Zeki K. et al. Expression of the neoplastic phenotype by human thyroid carcinoma cell lines requires NFkappaB p65 protein expression. Oncogene. 1997;15:1987-94
118. Pacifico F, Mauro C, Barone C, Crescenzi E, Mellone S, Monaco M. et al. Oncogenic and anti-apoptotic activity of NF-kappa B in human thyroid carcinomas. J Biol Chem. 2004;279:54610-9
119. Mitsiades CS, McMillin D, Kotoula V, Poulaki V, McMullan C, Negri J. et al. Antitumor effects of the proteasome inhibitor bortezomib in medullary and anaplastic thyroid carcinoma cells in vitro. J Clin Endocrinol Metab. 2006;91:4013-21
120. Starenki D, Namba H, Saenko V, Ohtsuru A, Yamashita S. Inhibition of nuclear factor-kappaB cascade potentiates the effect of a combination treatment of anaplastic thyroid cancer cells. J Clin Endocrinol Metab. 2004;89:410-8
121. Meng Z, Lou S, Tan J, Xu K, Jia Q, Zheng W. Nuclear factor-kappa B inhibition can enhance apoptosis of differentiated thyroid cancer cells induced by 131I. PLoS One. 2012;7:e33597
122. Chen F, Yin S, Zhu J, Jia L, Zhang H, Yang C. et al. Effects of nuclear factorkappaB on the uptake of 131iodine and apoptosis of thyroid carcinoma cells. Mol Med Rep. 2018;17:4959-64
123. Choi YW, Kim HJ, Kim YH, Park SH, Chwae YJ, Lee J. et al. B-RafV600E inhibits sodium iodide symporter expression via regulation of DNA methyltransferase 1. Exp Mol Med. 2014;46:e120
124. Sastre-Perona A, Santisteban P. Wnt-independent role of beta-catenin in thyroid cell proliferation and differentiation. Mol Endocrinol. 2014;28:681-95
125. Garcia-Rostan G, Tallini G, Herrero A, D'Aquila TG, Carcangiu ML, Rimm DL. Frequent mutation and nuclear localization of beta-catenin in anaplastic thyroid carcinoma. Cancer Res. 1999;59:1811-5
126. Garcia-Rostan G, Camp RL, Herrero A, Carcangiu ML, Rimm DL, Tallini G. Beta-catenin dysregulation in thyroid neoplasms: down-regulation, aberrant nuclear expression, and CTNNB1 exon 3 mutations are markers for aggressive tumor phenotypes and poor prognosis. Am J Pathol. 2001;158:987-96
127. Lan L, Basourakos S, Cui D, Zuo X, Deng W, Huo L. et al. Inhibiting beta-catenin expression promotes efficiency of radioiodine treatment in aggressive follicular thyroid cancer cells probably through mediating NIS localization. Oncol Rep. 2017;37:426-34
128. Das PM, Singal R. DNA methylation and cancer. J Clin Oncol. 2004;22:4632-42
129. Smith JA, Fan CY, Zou C, Bodenner D, Kokoska MS. Methylation status of genes in papillary thyroid carcinoma. Arch Otolaryngol Head Neck Surg. 2007;133:1006-11
130. Stephen JK, Chitale D, Narra V, Chen KM, Sawhney R, Worsham MJ. DNA methylation in thyroid tumorigenesis. Cancers (Basel). 2011;3:1732-43
131. Galrao AL, Sodre AK, Camargo RY, Friguglietti CU, Kulcsar MA, Lima EU. et al. Methylation levels of sodium-iodide symporter (NIS) promoter in benign and malignant thyroid tumors with reduced NIS expression. Endocrine. 2013;43:225-9
132. Galrao AL, Camargo RY, Friguglietti CU, Moraes L, Cerutti JM, Serrano-Nascimento C. et al. Hypermethylation of a New Distal Sodium/Iodide Symporter (NIS) enhancer (NDE) is associated with reduced NIS expression in thyroid tumors. J Clin Endocrinol Metab. 2014;99:E944-52
133. Xing M, Usadel H, Cohen Y, Tokumaru Y, Guo Z, Westra WB. et al. Methylation of the thyroid-stimulating hormone receptor gene in epithelial thyroid tumors: a marker of malignancy and a cause of gene silencing. Cancer Res. 2003;63:2316-21
134. Kondo T, Nakazawa T, Ma D, Niu D, Mochizuki K, Kawasaki T. et al. Epigenetic silencing of TTF-1/NKX2-1 through DNA hypermethylation and histone H3 modulation in thyroid carcinomas. Lab Invest. 2009;89:791-9
135. Venkataraman GM, Yatin M, Marcinek R, Ain KB. Restoration of iodide uptake in dedifferentiated thyroid carcinoma: relationship to human Na+/I-symporter gene methylation status. J Clin Endocrinol Metab. 1999;84:2449-57
136. Zhang K, Li C, Liu J, Tang X, Li Z. DNA methylation alterations as therapeutic prospects in thyroid cancer. J Endocrinol Invest. 2019;42:363-70
137. Khan MS, Pandith AA, Masoodi SR, Wani KA, Ul Hussain M, Mudassar S. Epigenetic silencing of TSHR gene in thyroid cancer patients in relation to their BRAF V600E mutation status. Endocrine. 2014;47:449-55
138. Kurdistani SK. Histone modifications as markers of cancer prognosis: a cellular view. Br J Cancer. 2007;97:1-5
139. Catalano MG, Fortunati N, Boccuzzi G. Epigenetics modifications and therapeutic prospects in human thyroid cancer. Front Endocrinol (Lausanne). 2012;3:40
140. Zhang Z, Liu D, Murugan AK, Liu Z, Xing M. Histone deacetylation of NIS promoter underlies BRAF V600E-promoted NIS silencing in thyroid cancer. Endocr Relat Cancer. 2014;21:161-73
141. Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int J Mol Sci. 2017 18
142. Puppin C, Passon N, Lavarone E, Di Loreto C, Frasca F, Vella V. et al. Levels of histone acetylation in thyroid tumors. Biochem Biophys Res Commun. 2011;411:679-83
143. Kitazono M, Robey R, Zhan Z, Sarlis NJ, Skarulis MC, Aikou T. et al. Low concentrations of the histone deacetylase inhibitor, depsipeptide (FR901228), increase expression of the Na(+)/I(-) symporter and iodine accumulation in poorly differentiated thyroid carcinoma cells. J Clin Endocrinol Metab. 2001;86:3430-5
144. Fortunati N, Catalano MG, Arena K, Brignardello E, Piovesan A, Boccuzzi G. Valproic acid induces the expression of the Na+/I- symporter and iodine uptake in poorly differentiated thyroid cancer cells. J Clin Endocrinol Metab. 2004;89:1006-9
145. Pugliese M, Fortunati N, Germano A, Asioli S, Marano F, Palestini N. et al. Histone deacetylase inhibition affects sodium iodide symporter expression and induces 131I cytotoxicity in anaplastic thyroid cancer cells. Thyroid. 2013;23:838-46
146. Furuya F, Shimura H, Suzuki H, Taki K, Ohta K, Haraguchi K. et al. Histone deacetylase inhibitors restore radioiodide uptake and retention in poorly differentiated and anaplastic thyroid cancer cells by expression of the sodium/iodide symporter thyroperoxidase and thyroglobulin. Endocrinology. 2004;145:2865-75
147. Shen WT, Wong TS, Chung WY, Wong MG, Kebebew E, Duh QY. et al. Valproic acid inhibits growth, induces apoptosis, and modulates apoptosis-regulatory and differentiation gene expression in human thyroid cancer cells. Surgery. 2005;138:979-84 discussion 84-5
148. Frohlich E, Brossart P, Wahl R. Induction of iodide uptake in transformed thyrocytes: a compound screening in cell lines. Eur J Nucl Med Mol Imaging. 2009;36:780-90
149. Altmann A, Eisenhut M, Bauder-Wust U, Markert A, Askoxylakis V, Hess-Stumpp H. et al. Therapy of thyroid carcinoma with the histone deacetylase inhibitor MS-275. Eur J Nucl Med Mol Imaging. 2010;37:2286-97
150. Puppin C, D'Aurizio F, D'Elia AV, Cesaratto L, Tell G, Russo D. et al. Effects of histone acetylation on sodium iodide symporter promoter and expression of thyroid-specific transcription factors. Endocrinology. 2005;146:3967-74
151. Zarnegar R, Brunaud L, Kanauchi H, Wong M, Fung M, Ginzinger D. et al. Increasing the effectiveness of radioactive iodine therapy in the treatment of thyroid cancer using Trichostatin A, a histone deacetylase inhibitor. Surgery. 2002;132:984-90 discussion 90
152. Jang S, Yu XM, Odorico S, Clark M, Jaskula-Sztul R, Schienebeck CM. et al. Novel analogs targeting histone deacetylase suppress aggressive thyroid cancer cell growth and induce re-differentiation. Cancer Gene Ther. 2015;22:410-6
153. D'Agostino M, Sponziello M, Puppin C, Celano M, Maggisano V, Baldan F. et al. Different expression of TSH receptor and NIS genes in thyroid cancer: role of epigenetics. J Mol Endocrinol. 2014;52:121-31
154. Fu H, Cheng L, Jin Y, Cheng L, Liu M, Chen L. MAPK Inhibitors Enhance HDAC Inhibitor-Induced Redifferentiation in Papillary Thyroid Cancer Cells Harboring BRAF (V600E): An In vitro Study. Mol Ther Oncolytics. 2019;12:235-45
155. Zhang M, Guo R, Xu H, Zhang M, Li B. Retinoic acid and tributyrin induce in-vitro radioiodine uptake and inhibition of cell proliferation in a poorly differentiated follicular thyroid carcinoma. Nucl Med Commun. 2011;32:605-10
156. Provenzano MJ, Fitzgerald MP, Krager K, Domann FE. Increased iodine uptake in thyroid carcinoma after treatment with sodium butyrate and decitabine (5-Aza-dC). Otolaryngol Head Neck Surg. 2007;137:722-8
157. Massimino M, Tirro E, Stella S, Frasca F, Vella V, Sciacca L. et al. Effect of Combined Epigenetic Treatments and Ectopic NIS Expression on Undifferentiated Thyroid Cancer Cells. Anticancer Res. 2018;38:6653-62
158. Fu H, Cheng L, Sa R, Jin Y, Chen L. Combined tazemetostat and MAPKi enhances differentiation of papillary thyroid cancer cells harbouring BRAF(V600E) by synergistically decreasing global trimethylation of H3K27. J Cell Mol Med. 2020;24:3336-45
159. Frohlich E, Wahl R. The current role of targeted therapies to induce radioiodine uptake in thyroid cancer. Cancer Treat Rev. 2014;40:665-74
160. Schreck R, Schnieders F, Schmutzler C, Kohrle J. Retinoids stimulate type I iodothyronine 5'-deiodinase activity in human follicular thyroid carcinoma cell lines. J Clin Endocrinol Metab. 1994;79:791-8
161. Hoang-Vu C, Bull K, Schwarz I, Krause G, Schmutzler C, Aust G. et al. Regulation of CD97 protein in thyroid carcinoma. J Clin Endocrinol Metab. 1999;84:1104-9
162. Aust G, Eichler W, Laue S, Lehmann I, Heldin NE, Lotz O. et al. CD97: a dedifferentiation marker in human thyroid carcinomas. Cancer Res. 1997;57:1798-806
163. Schmutzler C, Winzer R, Meissner-Weigl J, Kohrle J. Retinoic acid increases sodium/iodide symporter mRNA levels in human thyroid cancer cell lines and suppresses expression of functional symporter in nontransformed FRTL-5 rat thyroid cells. Biochem Biophys Res Commun. 1997;240:832-8
164. Van Herle AJ, Agatep ML, Padua DN 3rd, Totanes TL, Canlapan DV, Van Herle HM. et al. Effects of 13 cis-retinoic acid on growth and differentiation of human follicular carcinoma cells (UCLA R0 82 W-1) in vitro. J Clin Endocrinol Metab. 1990;71:755-63
165. Kurebayashi J, Tanaka K, Otsuki T, Moriya T, Kunisue H, Uno M. et al. All-trans-retinoic acid modulates expression levels of thyroglobulin and cytokines in a new human poorly differentiated papillary thyroid carcinoma cell line, KTC-1. J Clin Endocrinol Metab. 2000;85:2889-96
166. Jeong H, Kim YR, Kim KN, Choe JG, Chung JK, Kim MK. Effect of all-trans retinoic acid on sodium/iodide symporter expression, radioiodine uptake and gene expression profiles in a human anaplastic thyroid carcinoma cell line. Nucl Med Biol. 2006;33:875-82
167. Raman P, Koenig RJ. Pax-8-PPAR-gamma fusion protein in thyroid carcinoma. Nat Rev Endocrinol. 2014;10:616-23
168. Frohlich E, Machicao F, Wahl R. Action of thiazolidinediones on differentiation, proliferation and apoptosis of normal and transformed thyrocytes in culture. Endocr Relat Cancer. 2005;12:291-303
169. Park JW, Zarnegar R, Kanauchi H, Wong MG, Hyun WC, Ginzinger DG. et al. Troglitazone, the peroxisome proliferator-activated receptor-gamma agonist, induces antiproliferation and redifferentiation in human thyroid cancer cell lines. Thyroid. 2005;15:222-31
170. Bovenga F, Sabba C, Moschetta A. Uncoupling nuclear receptor LXR and cholesterol metabolism in cancer. Cell Metab. 2015;21:517-26
171. Lin CY, Vedin LL, Steffensen KR. The emerging roles of liver X receptors and their ligands in cancer. Expert Opin Ther Targets. 2016;20:61-71
172. de Medina P, Paillasse MR, Segala G, Voisin M, Mhamdi L, Dalenc F. et al. Dendrogenin A arises from cholesterol and histamine metabolism and shows cell differentiation and anti-tumour properties. Nat Commun. 2013;4:1840
173. Bauriaud-Mallet M, Vija-Racaru L, Brillouet S, Mallinger A, de Medina P, Rives A. et al. The cholesterol-derived metabolite dendrogenin A functionally reprograms breast adenocarcinoma and undifferentiated thyroid cancer cells. J Steroid Biochem Mol Biol. 2019;192:105390
174. Ariazi EA, Jordan VC. Estrogen-related receptors as emerging targets in cancer and metabolic disorders. Curr Top Med Chem. 2006;6:203-15
175. Singh TD, Jeong SY, Lee SW, Ha JH, Lee IK, Kim SH. et al. Inverse Agonist of Estrogen-Related Receptor gamma Enhances Sodium Iodide Symporter Function Through Mitogen-Activated Protein Kinase Signaling in Anaplastic Thyroid Cancer Cells. J Nucl Med. 2015;56:1690-6
176. Singh TD, Song J, Kim J, Chin J, Ji HD, Lee JE. et al. A Novel Orally Active Inverse Agonist of Estrogen-related Receptor Gamma (ERRgamma), DN200434, A Booster of NIS in Anaplastic Thyroid Cancer. Clin Cancer Res. 2019;25:5069-81
177. Kim J, Song J, Ji HD, Yoo EK, Lee JE, Lee SB. et al. Discovery of Potent, Selective, and Orally Bioavailable Estrogen-Related Receptor-gamma Inverse Agonists To Restore the Sodium Iodide Symporter Function in Anaplastic Thyroid Cancer. J Med Chem. 2019;62:1837-58
178. Yi H, Long B, Ye X, Zhang L, Liu X, Zhang C. Autophagy: A potential target for thyroid cancer therapy (Review). Mol Clin Oncol. 2014;2:661-5
179. Plantinga TS, Tesselaar MH, Morreau H, Corssmit EP, Willemsen BK, Kusters B. et al. Autophagy activity is associated with membranous sodium iodide symporter expression and clinical response to radioiodine therapy in non-medullary thyroid cancer. Autophagy. 2016;12:1195-205
180. Lin CI, Whang EE, Abramson MA, Jiang X, Price BD, Donner DB. et al. Autophagy: a new target for advanced papillary thyroid cancer therapy. Surgery. 2009;146:1208-14
181. Lin CI, Whang EE, Donner DB, Du J, Lorch J, He F. et al. Autophagy induction with RAD001 enhances chemosensitivity and radiosensitivity through Met inhibition in papillary thyroid cancer. Mol Cancer Res. 2010;8:1217-26
182. Tesselaar MH, Crezee T, Swarts HG, Gerrits D, Boerman OC, Koenderink JB. et al. Digitalis-like Compounds Facilitate Non-Medullary Thyroid Cancer Redifferentiation through Intracellular Ca2+, FOS, and Autophagy-Dependent Pathways. Mol Cancer Ther. 2017;16:169-81
183. Tesselaar MH, Crezee T, Schuurmans I, Gerrits D, Nagarajah J, Boerman OC. et al. Digitalislike Compounds Restore hNIS Expression and Iodide Uptake Capacity in Anaplastic Thyroid Cancer. J Nucl Med. 2018;59:780-6
184. Chai W, Ye F, Zeng L, Li Y, Yang L. HMGB1-mediated autophagy regulates sodium/iodide symporter protein degradation in thyroid cancer cells. J Exp Clin Cancer Res. 2019;38:325
185. Liu Z, Wang P, Chen H, Wold EA, Tian B, Brasier AR. et al. Drug Discovery Targeting Bromodomain-Containing Protein 4. J Med Chem. 2017;60:4533-58
186. Gao X, Wu X, Zhang X, Hua W, Zhang Y, Maimaiti Y. et al. Inhibition of BRD4 suppresses tumor growth and enhances iodine uptake in thyroid cancer. Biochem Biophys Res Commun. 2016;469:679-85
187. Chou CK, Liu RT, Kang HY. MicroRNA-146b: A Novel Biomarker and Therapeutic Target for Human Papillary Thyroid Cancer. Int J Mol Sci. 2017 18
188. Ricarte-Filho JC, Fuziwara CS, Yamashita AS, Rezende E, da-Silva MJ, Kimura ET. Effects of let-7 microRNA on Cell Growth and Differentiation of Papillary Thyroid Cancer. Transl Oncol. 2009;2:236-41
189. Boufraqech M, Klubo-Gwiezdzinska J, Kebebew E. MicroRNAs in the thyroid. Best Pract Res Clin Endocrinol Metab. 2016;30:603-19
190. Riesco-Eizaguirre G, Wert-Lamas L, Perales-Paton J, Sastre-Perona A, Fernandez LP, Santisteban P. The miR-146b-3p/PAX8/NIS Regulatory Circuit Modulates the Differentiation Phenotype and Function of Thyroid Cells during Carcinogenesis. Cancer Res. 2015;75:4119-30
191. Li L, Lv B, Chen B, Guan M, Sun Y, Li H. et al. Inhibition of miR-146b expression increases radioiodine-sensitivity in poorly differential thyroid carcinoma via positively regulating NIS expression. Biochem Biophys Res Commun. 2015;462:314-21
192. Riesco-Eizaguirre G, Santisteban P. ENDOCRINE TUMOURS: Advances in the molecular pathogenesis of thyroid cancer: lessons from the cancer genome. Eur J Endocrinol. 2016;175:R203-17
193. Geraldo MV, Yamashita AS, Kimura ET. MicroRNA miR-146b-5p regulates signal transduction of TGF-beta by repressing SMAD4 in thyroid cancer. Oncogene. 2012;31:1910-22
194. Hou S, Xie X, Zhao J, Wu C, Li N, Meng Z. et al. Downregulation of miR-146b-3p Inhibits Proliferation and Migration and Modulates the Expression and Location of Sodium/Iodide Symporter in Dedifferentiated Thyroid Cancer by Potentially Targeting MUC20. Front Oncol. 2020;10:566365
195. Damanakis AI, Eckhardt S, Wunderlich A, Roth S, Wissniowski TT, Bartsch DK. et al. MicroRNAs let7 expression in thyroid cancer: correlation with their deputed targets HMGA2 and SLC5A5. J Cancer Res Clin Oncol. 2016;142:1213-20
196. Shen CT, Qiu ZL, Song HJ, Wei WJ, Luo QY. miRNA-106a directly targeting RARB associates with the expression of Na(+)/I(-) symporter in thyroid cancer by regulating MAPK signaling pathway. J Exp Clin Cancer Res. 2016;35:101
197. Lakshmanan A, Wojcicka A, Kotlarek M, Zhang X, Jazdzewski K, Jhiang SM. microRNA-339-5p modulates Na+/I- symporter-mediated radioiodide uptake. Endocr Relat Cancer. 2015;22:11-21
198. Tang Y, Meng X, Yu X, Shang H, Chen S, Liao L. et al. Inhibition of microRNA-875-5p promotes radioiodine uptake in poorly differentiated thyroid carcinoma cells by upregulating sodium-iodide symporter. J Endocrinol Invest. 2020;43:439-50
199. Wachter S, Wunderlich A, Greene BH, Roth S, Elxnat M, Fellinger SA. et al. Selumetinib Activity in Thyroid Cancer Cells: Modulation of Sodium Iodide Symporter and Associated miRNAs. Int J Mol Sci. 2018 19
200. Xiang C, Zhang ML, Zhao QZ, Xie QP, Yan HC, Yu X. et al. LncRNA-SLC6A9-5:2: A potent sensitizer in 131I-resistant papillary thyroid carcinoma with PARP-1 induction. Oncotarget. 2017;8:22954-67
201. Dong JJ, Zhou Y, Liu YT, Zhang ZW, Zhou XJ, Wang HJ. et al. In vitro evaluation of the therapeutic potential of nevirapine in treatment of human thyroid anaplastic carcinoma. Mol Cell Endocrinol. 2013;370:113-8
202. Vouri M, Hafizi S. TAM Receptor Tyrosine Kinases in Cancer Drug Resistance. Cancer Res. 2017;77:2775-8
203. Collina F, La Sala L, Liotti F, Prevete N, La Mantia E, Chiofalo MG. et al. AXL Is a Novel Predictive Factor and Therapeutic Target for Radioactive Iodine Refractory Thyroid Cancer. Cancers (Basel). 2019 11
204. Chung T, Youn H, Yeom CJ, Kang KW, Chung JK. Glycosylation of Sodium/Iodide Symporter (NIS) Regulates Its Membrane Translocation and Radioiodine Uptake. PLoS One. 2015;10:e0142984
205. Beyer S, Lakshmanan A, Liu YY, Zhang X, Wapnir I, Smolenski A. et al. KT5823 differentially modulates sodium iodide symporter expression, activity, and glycosylation between thyroid and breast cancer cells. Endocrinology. 2011;152:782-92
206. Dohan O, Baloch Z, Banrevi Z, Livolsi V, Carrasco N. Rapid communication: predominant intracellular overexpression of the Na(+)/I(-) symporter (NIS) in a large sampling of thyroid cancer cases. J Clin Endocrinol Metab. 2001;86:2697-700
207. Wapnir IL, van de Rijn M, Nowels K, Amenta PS, Walton K, Montgomery K. et al. Immunohistochemical profile of the sodium/iodide symporter in thyroid, breast, and other carcinomas using high density tissue microarrays and conventional sections. J Clin Endocrinol Metab. 2003;88:1880-8
208. Baghban R, Roshangar L, Jahanban-Esfahlan R, Seidi K, Ebrahimi-Kalan A, Jaymand M. et al. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun Signal. 2020;18:59
209. Arneth B. Tumor Microenvironment. Medicina (Kaunas). 2019 56
210. Castillo-Rivera F, Ondo-Mendez A, Guglielmi J, Guigonis JM, Jing L, Lindenthal S. et al. Tumor microenvironment affects exogenous sodium/iodide symporter expression. Transl Oncol. 2021;14:100937
211. Saez C, Martinez-Brocca MA, Castilla C, Soto A, Navarro E, Tortolero M. et al. Prognostic significance of human pituitary tumor-transforming gene immunohistochemical expression in differentiated thyroid cancer. J Clin Endocrinol Metab. 2006;91:1404-9
212. Heaney AP, Nelson V, Fernando M, Horwitz G. Transforming events in thyroid tumorigenesis and their association with follicular lesions. J Clin Endocrinol Metab. 2001;86:5025-32
213. Boelaert K, McCabe CJ, Tannahill LA, Gittoes NJ, Holder RL, Watkinson JC. et al. Pituitary tumor transforming gene and fibroblast growth factor-2 expression: potential prognostic indicators in differentiated thyroid cancer. J Clin Endocrinol Metab. 2003;88:2341-7
214. Read ML, Lewy GD, Fong JC, Sharma N, Seed RI, Smith VE. et al. Proto-oncogene PBF/PTTG1IP regulates thyroid cell growth and represses radioiodide treatment. Cancer Res. 2011;71:6153-64
215. Smith VE, Read ML, Turnell AS, Watkins RJ, Watkinson JC, Lewy GD. et al. A novel mechanism of sodium iodide symporter repression in differentiated thyroid cancer. J Cell Sci. 2009;122:3393-402
216. Smith VE, Sharma N, Watkins RJ, Read ML, Ryan GA, Kwan PP. et al. Manipulation of PBF/PTTG1IP phosphorylation status; a potential new therapeutic strategy for improving radioiodine uptake in thyroid and other tumors. J Clin Endocrinol Metab. 2013;98:2876-86
217. Amit M, Na'ara S, Francis D, Matanis W, Zolotov S, Eisenhaber B. et al. Post-translational Regulation of Radioactive Iodine Therapy Response in Papillary Thyroid Carcinoma. J Natl Cancer Inst. 2017 109
218. Paladino S, Melillo RM. Editorial: Novel Mechanism of Radioactive Iodine Refractivity in Thyroid Cancer. J Natl Cancer Inst. 2017 109
219. Lacoste C, Herve J, Bou Nader M, Dos Santos A, Moniaux N, Valogne Y. et al. Iodide transporter NIS regulates cancer cell motility and invasiveness by interacting with the Rho guanine nucleotide exchange factor LARG. Cancer Res. 2012;72:5505-15
220. Feng F, Yehia L, Ni Y, Chang YS, Jhiang SM, Eng C. A Nonpump Function of Sodium Iodide Symporter in Thyroid Cancer via Cross-talk with PTEN Signaling. Cancer Res. 2018;78:6121-33
221. Feng F, Yehia L, Eng C. Pro-tumorigenic non-pump function of sodium iodide symporter: A reimagined Trojan horse? Oncotarget. 2019;10:688-9
222. Fletcher A, Read ML, Thornton CEM, Larner DP, Poole VL, Brookes K. et al. Targeting Novel Sodium Iodide Symporter Interactors ADP-Ribosylation Factor 4 and Valosin-Containing Protein Enhances Radioiodine Uptake. Cancer Res. 2020;80:102-15
223. Huc-Brandt S, Marcellin D, Graslin F, Averseng O, Bellanger L, Hivin P. et al. Characterisation of the purified human sodium/iodide symporter reveals that the protein is mainly present in a dimeric form and permits the detailed study of a native C-terminal fragment. Biochim Biophys Acta. 2011;1808:65-77
224. Thompson RJ, Fletcher A, Brookes K, Nieto H, Alshahrani MM, Mueller JW. et al. Dimerization of the Sodium/Iodide Symporter. Thyroid. 2019;29:1485-98
225. Simon D, Kohrle J, Schmutzler C, Mainz K, Reiners C, Roher HD. Redifferentiation therapy of differentiated thyroid carcinoma with retinoic acid: basics and first clinical results. Exp Clin Endocrinol Diabetes. 1996;104(Suppl 4):13-5
226. Simon D, Korber C, Krausch M, Segering J, Groth P, Gorges R. et al. Clinical impact of retinoids in redifferentiation therapy of advanced thyroid cancer: final results of a pilot study. Eur J Nucl Med Mol Imaging. 2002;29:775-82
227. Gruning T, Tiepolt C, Zophel K, Bredow J, Kropp J, Franke WG. Retinoic acid for redifferentiation of thyroid cancer-does it hold its promise? Eur J Endocrinol. 2003;148:395-402
228. Coelho SM, Corbo R, Buescu A, Carvalho DP, Vaisman M. Retinoic acid in patients with radioiodine non-responsive thyroid carcinoma. J Endocrinol Invest. 2004;27:334-9
229. Courbon F, Zerdoud S, Bastie D, Archambaud F, Hoff M, Eche N. et al. Defective efficacy of retinoic acid treatment in patients with metastatic thyroid carcinoma. Thyroid. 2006;16:1025-31
230. Fernandez CA, Puig-Domingo M, Lomena F, Estorch M, Camacho Marti V, Bittini AL. et al. Effectiveness of retinoic acid treatment for redifferentiation of thyroid cancer in relation to recovery of radioiodine uptake. J Endocrinol Invest. 2009;32:228-33
231. Oh SW, Moon SH, Park DJ, Cho BY, Jung KC, Lee DS. et al. Combined therapy with 131I and retinoic acid in Korean patients with radioiodine-refractory papillary thyroid cancer. Eur J Nucl Med Mol Imaging. 2011;38:1798-805
232. Handkiewicz-Junak D, Roskosz J, Hasse-Lazar K, Szpak-Ulczok S, Puch Z, Kukulska A. et al. 13-cis-retinoic acid re-differentiation therapy and recombinant human thyrotropin-aided radioiodine treatment of non-Functional metastatic thyroid cancer: a single-center, 53-patient phase 2 study. Thyroid Res. 2009;2:8
233. Short SC, Suovuori A, Cook G, Vivian G, Harmer C. A phase II study using retinoids as redifferentiation agents to increase iodine uptake in metastatic thyroid cancer. Clin Oncol (R Coll Radiol). 2004;16:569-74
234. Liu YY, Stokkel MP, Pereira AM, Corssmit EP, Morreau HA, Romijn JA. et al. Bexarotene increases uptake of radioiodide in metastases of differentiated thyroid carcinoma. Eur J Endocrinol. 2006;154:525-31
235. Zhang Y, Jia S, Liu Y, Li B, Wang Z, Lu H. et al. A clinical study of all-trans-retinoid-induced differentiation therapy of advanced thyroid cancer. Nucl Med Commun. 2007;28:251-5
236. Damle N, Patnecha M, Kumar P, Maharjan S, Bal C. Retinoic acid therapy in patients with radioiodine negative differentiated thyroid cancer and clinical or biochemical evidence of disease: An initial experience. Indian J Nucl Med. 2011;26:144-8
237. Pak K, Shin S, Kim SJ, Kim IJ, Chang S, Koo P. et al. Response of Retinoic Acid in Patients with Radioactive Iodine-Refractory Thyroid Cancer: A Meta-Analysis. Oncol Res Treat. 2018;41:100-4
238. Philips JC, Petite C, Willi JP, Buchegger F, Meier CA. Effect of peroxisome proliferator-activated receptor gamma agonist, rosiglitazone, on dedifferentiated thyroid cancers. Nucl Med Commun. 2004;25:1183-6
239. Elias AN, Lizotte P. Enhanced radioiodine uptake in a patient with poorly differentiated papillary thyroid cancer after treatment with rosiglitazone. Clin Nucl Med. 2006;31:517-9
240. Elola M, Yoldi A, Emparanza JI, Matteucci T, Bilbao I, Goena M. [Redifferentiation therapy with rosiglitazone in a case of differentiated thyroid cancer with pulmonary metastases and absence of radioiodine uptake]. Rev Esp Med Nucl. 2011;30:241-3
241. Kebebew E, Peng M, Reiff E, Treseler P, Woeber KA, Clark OH. et al. A phase II trial of rosiglitazone in patients with thyroglobulin-positive and radioiodine-negative differentiated thyroid cancer. Surgery. 2006;140:960-6 discussion 6-7
242. Kebebew E, Lindsay S, Clark OH, Woeber KA, Hawkins R, Greenspan FS. Results of rosiglitazone therapy in patients with thyroglobulin-positive and radioiodine-negative advanced differentiated thyroid cancer. Thyroid. 2009;19:953-6
243. Tepmongkol S, Keelawat S, Honsawek S, Ruangvejvorachai P. Rosiglitazone effect on radioiodine uptake in thyroid carcinoma patients with high thyroglobulin but negative total body scan: a correlation with the expression of peroxisome proliferator-activated receptor-gamma. Thyroid. 2008;18:697-704
244. Rosenbaum-Krumme SJ, Freudenberg LS, Jentzen W, Bockisch A, Nagarajah J. Effects of rosiglitazone on radioiodine negative and progressive differentiated thyroid carcinoma as assessed by (1)(2)(4)I PET/CT imaging. Clin Nucl Med. 2012;37:e47-52
245. Rosenbaum-Krumme SJ, Bockisch A, Nagarajah J. Pioglitazone therapy in progressive differentiated thyroid carcinoma. Nuklearmedizin. 2012;51:111-5
246. Kelly WK, O'Connor OA, Krug LM, Chiao JH, Heaney M, Curley T. et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol. 2005;23:3923-31
247. Amiri-Kordestani L, Luchenko V, Peer CJ, Ghafourian K, Reynolds J, Draper D. et al. Phase I trial of a new schedule of romidepsin in patients with advanced cancers. Clin Cancer Res. 2013;19:4499-507
248. Sherman EJ, Su YB, Lyall A, Schoder H, Fury MG, Ghossein RA. et al. Evaluation of romidepsin for clinical activity and radioactive iodine reuptake in radioactive iodine-refractory thyroid carcinoma. Thyroid. 2013;23:593-9
249. Nilubol N, Merkel R, Yang L, Patel D, Reynolds JC, Sadowski SM. et al. A phase II trial of valproic acid in patients with advanced, radioiodine-resistant thyroid cancers of follicular cell origin. Clin Endocrinol (Oxf). 2017;86:128-33
250. Rothenberg SM, McFadden DG, Palmer EL, Daniels GH, Wirth LJ. Redifferentiation of iodine-refractory BRAF V600E-mutant metastatic papillary thyroid cancer with dabrafenib. Clin Cancer Res. 2015;21:1028-35
251. Dunn LA, Sherman EJ, Baxi SS, Tchekmedyian V, Grewal RK, Larson SM. et al. Vemurafenib Redifferentiation of BRAF Mutant, RAI-Refractory Thyroid Cancers. J Clin Endocrinol Metab. 2019;104:1417-28
252. Jaber T, Waguespack SG, Cabanillas ME, Elbanan M, Vu T, Dadu R. et al. Targeted Therapy in Advanced Thyroid Cancer to Resensitize Tumors to Radioactive Iodine. J Clin Endocrinol Metab. 2018;103:3698-705
253. Modoni S, Landriscina M, Fabiano A, Fersini A, Urbano N, Ambrosi A. et al. Reinduction of cell differentiation and 131I uptake in a poorly differentiated thyroid tumor in response to the reverse transcriptase (RT) inhibitor nevirapine. Cancer Biother Radiopharm. 2007;22:289-95
254. Liu YY, van der Pluijm G, Karperien M, Stokkel MP, Pereira AM, Morreau J. et al. Lithium as adjuvant to radioiodine therapy in differentiated thyroid carcinoma: clinical and in vitro studies. Clin Endocrinol (Oxf). 2006;64:617-24
Author contact
![]() Corresponding authors: Prof. Byeong-Cheol Ahn., M.D., Ph.D. Department of Nuclear Medicine, School of Medicine, Kyungpook National University, Gukchaebosang-ro 680, Jung-Gu, Daegu-41944, Republic of Korea. Tel: 82-53-420-5583; Fax: 82-53-200-6447; E-mail: abc2000ac.kr.
Corresponding authors: Prof. Byeong-Cheol Ahn., M.D., Ph.D. Department of Nuclear Medicine, School of Medicine, Kyungpook National University, Gukchaebosang-ro 680, Jung-Gu, Daegu-41944, Republic of Korea. Tel: 82-53-420-5583; Fax: 82-53-200-6447; E-mail: abc2000ac.kr.