Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(13):6393-6406. doi:10.7150/thno.53121 This issue Cite
Research Paper
Human endoglin-CD3 bispecific T cell engager antibody induces anti-tumor effect in vivo
Liping Zhong1,2*, Wei Shi3*, Lu Gan1*, Xiuli Liu1*, Yu Huo1, Pan Wu1, Zhikun Zhang1, Tao Wu2, Hongmei Peng2, Yong Huang1, Yongxiang Zhao1, Yulin Yuan1,4 ![]() , Zhiming Deng2
, Zhiming Deng2 ![]() , Hongliang Tang3,5
, Hongliang Tang3,5 ![]()
1. National Center for International Research of Bio-targeting Theranostics, Guangxi Key Laboratory of Bio-targeting Theranostics, Guangxi Medical University, Nanning, Guangxi 530021, China. Collaborative Innovation Center for Targeting Tumor Diagnosis and Therapy, Guangxi Medical University, Nanning, Guangxi 530021, China.
2. The First People's Hospital of Changde City, Changde, Hunan 41500, China
3. Department of Oncology, The First Affiliated Hospital, Guangxi University of Chinese Medicine, Nanning, Guangxi 530023, China.
4. Department of Laboratory Medicine, The People's Hospital of Guangxi Zhuang Autonomous Region, Nanning, Guangxi 530021, China.
5. Department of Scientific Research, The Affiliated Fangchenggang Hospital, Guangxi University of Chinese Medicine, Fangchenggang, Guangxi 538001, China.
* These authors contributed equally to this work.
Received 2020-9-11; Accepted 2021-3-12; Published 2021-4-19
Abstract

Rationale: Endoglin, also known as CD105, is a homo-dimeric membrane glycoprotein required for angiogenesis and serves as a marker for cancer vasculature. In this study, we constructed a bispecific T-cell engager (BiTE) antibody that targets human endoglin and CD3 (hEND-CD3/BiTE). We examined BiTE binding to endoglin-expressing cells and its effects on the cytolytic activity of T cells and cancer development.
Methods: The in vitro effects of hEND-CD3/BiTE, including binding to target cells, T-cell activation, proliferation, and cytotoxicity, were examined in endoglin-expressing 293T cells, human umbilical vascular endothelial cells, tumor-derived endothelial cells, and CD3+ T cells. An in vivo xenograft tumor model was established using A549 human lung cancer cells. The therapeutic efficacy of hEND-CD3/BiTE was assessed by monitoring tumor growth, angiogenesis, and mouse survival.
Results: hEND-CD3/BiTE specifically bound to endoglin-expressing cells and CD3+ T cells in vitro and stimulated T-cell activation, proliferation, and Th1 cytokine secretion, and promoted T-cell-mediated cytolysis of endoglin-expressing cells. The hEND-CD3/BiTE in vivo caused minimal toxicity to major organs, reduced tumor neoangiogenesis, inhibited tumor growth, and significantly improved mouse survival.
Conclusions: Our study demonstrated the therapeutic potential of hEND-CD3/BiTE and provided a novel approach to clinical cancer treatment.
Keywords: endoglin, bispecific T-cell engager antibody, neoangiogenesis, immune therapy.
Introduction
Surgery, chemotherapy, and radiation therapy used either alone or in combination remain the primary approaches in cancer treatment. Chemotherapy and radiotherapy can target recurrences and metastases that cannot be addressed using surgical excision; however, they lack the specificity to distinguish between normal and cancerous tissues, and are frequently associated with severe systemic toxicities and bystander effects on normal cells, and induce preferential growth of drug- or radiation-resistant tumor cells [1-3]. In contrast, immunotherapy has long been proposed as an ideal anti-cancer approach with promising efficacy and tolerability, by activating cancer-specific immunity for therapeutic efficacy [4, 5]. Immunotherapy includes passive approaches in which anti-cancer monoclonal antibodies or T cells are transferred to bypass activation of endogenous immunity, and directly fight cancer [6], and induce de novo cancer-specific immunity [7]. Antibodies targeting non-immunomodulatory cancer-related antigens (passive immunotherapy) have been well established for decades, including those involved in the growth or death of tumor cells and non-immune stromal cells, such as vascular endothelial cells and fibroblasts. However, recent clinical studies strongly supported the efficacy of active immunotherapy by antibodies targeting immune checkpoints. These included cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), programmed cell death protein 1 (PD-1), and chimeric antigen receptor therapy (CAR), resulting in significant cancer remission and survival benefits [8, 9]. The ultimate goal of cancer immunotherapy is to activate tumor-specific cytotoxic T lymphocytes (CTLs) and eradicate tumor cells.
Tumor cells develop multiple mechanisms to evade T cell surveillance during cancer development, resulting in deficient recognition of tumors by T cells, acquired resistance to T-cell-mediated killing, induction of T-cell anergy and apoptosis, and accumulation of immunosuppressive Tregs [10]. An ideal therapeutic strategy would, therefore, specifically enhance recognition of tumor cells by T cells and stimulate activation/expansion of CTLs. In this regard, bispecific T-cell engager (BiTE) antibody provides an attractive solution. BiTE is an artificial bispecific monoclonal antibody consisting of two single-chain variable fragments (scFv), one of which binds to T cells through the CD3 receptor and the other to a tumor-specific antigen. By linking T cells with tumor cells, BiTE recruits and activates T cell cytotoxicity to tumor sites in the absence of MHC-I or co-stimulatory molecules [11-13]. Blinatumomab, a CD19/CD3 BiTE, was the first BiTE antibody approved by the FDA in the clinic for refractory acute lymphoid leukemia treatment [14]. Several other BiTEs are currently in clinical trials for various human cancer types, all targeting tumor-specific antigens, including epithelial cell adhesion molecule (EpCAM), carcinoembryonic antigen, CD123, and CD20 [15].
Angiogenesis plays an essential role in supporting continuous tumor growth and metastasis, the latter accounting for more than 90% of cancer-related deaths. Targeting angiogenesis is thus a promising therapy and has been approved in cancer treatment. Most angiogenesis inhibitors in the clinic target vascular endothelial growth factors (VEGFs) or their receptors [16]. In contrast to tumor cells, which are highly heterogeneous and susceptible to mutations in response to microenvironmental alterations, chemotherapy or radiotherapy, vascular endothelial cells are genetically stable throughout the progression of most solid tumors, readily accessible to therapeutic agents, and less likely to develop resistance to anti-angiogenic therapy [17]. Furthermore, tumor vascular endothelial cells present differing phenotypes compared with normal vascular endothelial cells, enabling specific targeting of tumor vasculature [18, 19]. Intensive studies have been devoted to identifying and characterizing key biomarkers for tumor angiogenesis. Endoglin, also known as CD105, is a homo-dimeric cell membrane glycoprotein and co-receptor for transforming growth factor β (TGF-β) [20]. It is highly expressed on proliferating vascular endothelial cells, specifically tumor-associated vascular and lymphatic endothelium, and in response to hypoxia and inhibition of VEGF signaling [21-23]. These features make endoglin a critical marker for tumor angiogenesis and an ideal target for anti-angiogenic treatment, especially in combination with VEGF inhibitors [24]. In 2004, Korn et al. first constructed scDb EDGCD3 against endoglin, activating T cells to target killing of endoglin+ cells in vitro, but the effect was not studied in vivo [25].
In this study, we designed and generated a BiTE targeting human endoglin and CD3 (hEND-CD3/BiTE) and characterized the in vitro features, including binding to target cells and promoting T-cell activation, proliferation and cytolysis. We also examined in vivo biological activities of hEND-CD3/BiTE on cancer progression in a xenograft mouse model of lung cancer. Our goal was to extend the current BiTE strategy (linking T cells with tumor cells) to link T cells with other stromal cells and explore the combination immunotherapy potential with anti-angiogenic cancer treatments.
Materials and Methods
Reagents
The cloning/expression plasmid pET-28a (+) was purchased from Invitrogen (Carlsbad, CA, USA). The following antibodies were used in this study: PerCP-conjugated anti-His-tag (ab117496), anti-endoglin (ab230925), and anti-CD34 (ab187282; Abcam, Cambridge, MA, USA); PE-conjugated anti-endoglin (12-1057), FITC-conjugated anti-CD4 (11-0048), PE-conjugated anti-CD8 (15-0088), PerCP-Cyanine5.5-conjugated anti-CD8a (45-0088-41), PE-conjugated anti-CD69 (12-0699), and PE-conjugated anti-CD25 (12-0259; eBioscience, San Diego, CA, USA); in addition to OKT3 (human CD3 monoclonal antibody; NDC: 59676-101) (Ortho Clinical Diagnostics, Raritan, NJ, USA); and anti-proliferating cell nuclear antigen (PCNA) (BM0104; Boster, Pleasanton, CA, USA). Recombinant human endoglin (1097-EN-25) and recombinant human interleukin-2 (IL-2; 202-IL-050) were purchased from R&D (Minneapolis, MN, USA). The fluorescent cell-staining dye carboxyfluorescein succinimidyl ester (CFSE; 00-6993) was from eBioscience, and the PKH26 Red Fluorescent Linker Kit was from Sigma (St. Louis, MO, USA).
Cells and experimental animals
The E. coli strain BL21 (DE3) was purchased from GenScript (Nanjing, China), and competent cells were prepared in our laboratory using the electro-transformation method.
The human lung adenocarcinoma cells A549 (v-054279), human umbilical vascular endothelial cells HUVEC (CRL-1730) and human embryonic kidney 293T cells were purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA) and cultured according to the ATCC instructions. Tumor-derived endothelial cells (Td-EC) were isolated from mouse A549 xenograft tumors using CD105 Microbeads (130-051-201, Miltenyi Biotech, San Diego, CA, USA) following the manufacturer's instructions. Human endoglin-expressing 293T cells (293T-hE) were purchased from GenScript and cultured according to the manufacturer's instructions. The peripheral blood mononuclear cells (PBMCs) were isolated from peripheral blood samples of healthy donors by density centrifugation on Ficoll gradient, as previously described [26]. Experiments involving human samples were approved by the Institutional Review Board of Guangxi Medical University (Guangxi, China).
Female specific antigen-free (SPF) immune-deficient, non-obese diabetic/severe combined immunodeficiency (NOD/SCID) mice (age 4-6 weeks) deficient in both B and T cells were purchased from Vital River (Beijing China) and maintained in the SPF experimental animal facility (Guangxi Medical University). All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of the University.
Construction, expression, purification, and characterization of hEND-CD3/BiTE and control hENDM-CD3/BiTE
We synthesized the coding sequences for VH and VL regions of human endoglin and human CD3 antibodies based on previously published sequences [27, 28]. We then used Primer 5.0 software (Primer Biosoft, Palo Alto, CA, USA) to design primers for the VH and VL regions of these two antibodies, and introduced the 15-amino-acid (GGGGS)3 linker sequence between the endoglin ScFv and CD3 ScFv fragments. Following the subsequent overlap replication-competent retrovirus (RCR), the linker-containing the hEND-CD3 PCR product was synthesized, an EcoRI restriction site was introduced into the upstream region of the endoglin VH sequence, and an XhoI restriction site was introduced into the downstream region of the CD3 VL sequence. The PCR product was cleaved with the restriction enzymes EcoRI and XhoI, cloned into the T7-promoter-containing PET-28a (+) plasmid (which added a His-tag to the N-terminal of the endoglin VH fragment), and was transformed into E. coli BL21(DE3) competent cells. E. coli cells containing the recombinant plasmids were inoculated into 5 mL of LB broth containing 100 µg/mL kanamycin and rocked at 200 rotations/min overnight at 30 ºC. The overnight cultures were then transferred at a 1:50 ratio to 50 mL of fresh LB medium and were grown at 30 ºC until an OD value at 600 nm of 0.6 to 0.8 was reached.
Isopropyl-β-D-thiogalactopyranoside (IPTG) was added to a final concentration of 0.2 mM, and cultures were grown for a further 4 h at 37 ºC. Cells were then harvested by centrifugation and re-suspended in 10 mM Tris-HCl pH 8.0. Subsequently, cells were sonicated on ice at 200 watts (4 s on and 6 s off cycles) for a total of 15 min and centrifuged at 13,000 rpm/min for 10 min at 4 ºC. After three washes with cold PBS, the inclusion body was dissolved using LE buffer at room temperature for 45 min. The supernatant was collected, filtered through a 0.45-μm strainer and purified using the AKTA pure chromatography system (GE Healthcare, Pittsburgh, PA, USA). The renaturation of the inclusion body was achieved through dialysis in a urea series with decreasing concentrations from 8 M to 6, 4, 2, 1, 0.5 and 0 M. As a control for hEND-CD3/BiTE, we introduced point mutations into the VH region of the endoglin antibody that disrupted the interaction with endoglin, referred to as hENDM-CD3/BiTE.
Flow cytometry-based in vitro assays
To examine the in vitro binding of hEND-CD3/BiTE to 293T, 293T-hE, HUVEC, Td-EC or PBMCs, target cells were re-suspended in RPMI1640 medium containing 10% fetal calf serum at 5×106-1×107/mL. For 293T, 293T-hE, HUVEC and Td-EC cells, 100 μL of the cell suspension was incubated with hEND-CD3/BiTE or hENDM/CD3-BiTE (final concentration 10 μg/mL) at 4 ºC for 30 min. The cells were then washed twice with PBS. For the detection of BiTE, a PerCP-conjugated anti-His tag antibody was used. Flow cytometry was performed with Epics XL (Beckman Coulter, Brea, CA, USA). For PBMCs, 100 μL of the cell suspension was incubated with hEND-CD3/BiTE or hENDM/CD3-BiTE at 4 ºC for 30 min. The cells were washed twice with PBS and further incubated with either FITC-conjugated anti-CD4+ PerCP-conjugated anti-His tag antibody or PE-conjugated anti-CD8+ PerCP-conjugated anti-His tag antibody at 4 ºC for 30 min. For co-targeting of both endothelial cells and T cells, endothelial cells (1×105) were stained with red fluorescent PKH26 dye according to the manufacturer's instructions and incubated with hEND-CD3/BiTE (final concentration 10 μg/mL) at 4 ºC for 2 h. After washing with PBS three times, 2×106 CFSE-labeled PBMCs (as previously described [29]) were added to endothelial cells and incubated at 37 ºC for 2-8 h. All cells were imaged under an inverted fluorescence microscope (Eclipse 80i, Nikon, Tokyo, Japan).
To examine T-cell activation, following the co-culture of PBMCs with 293T or 293T-hE cells in the presence of hEND-CD3/BiTE (final concentration 10 µg/mL) at 37 ºC for 48 h, the cells were incubated with FITC-conjugated anti-CD4, PerCP-Cyanine5.5-conjugated anti-CD8α, and PE-conjugated anti-CD69 or PE-conjugated anti-CD25 at 4 ºC for 30 min and analyzed by flow cytometry.
To analyze T-cell proliferation, BSA, hEND-CD3/BiTE, endoglin, or OKT3 (final concentration 10 μg/mL) were used to coat the 96-well plate before the CFSE-labeled PBMCs were seeded into the plate (1×106/well, triplicate for each condition). After incubation at 37 ºC for 72 h, the cells were examined using flow cytometry and analyzed with ModFit LT software (Verity Software, Topsham, ME, USA).
To measure T-cell-mediated cytolysis, PKH26-labeled cells (target cells) were incubated with freshly isolated PBMCs (effector cells) at E:T ratios of 0:1, 2.5:1, 5:1, 10:1, and 20:1. hEND/CD3-BiTE was added into the co-culture at 0, 10-3, 10-2, 10-1, 1, 10 and 100 μg/mL. Cells were co-cultured at 37 ºC for 18 h. The cells were incubated with propidium iodide, and PKH26+PI+ cells were detected by flow cytometry as apoptotic endothelial cells. The cytolysis percentage was calculated as follows: (PKH26+PI+% in the experimental group- PKH26+PI+% in the target cells alone group)/ (1-PKH26+PI+% in the target cells alone group) ×100%.
Surface plasmon resonance analysis
hEND-CD3/BiTE was dissolved in HBS-EP solution (0.01M HEPES, pH7.4, 0.15 M NaCl, 3 mM EDTA, and 0.005% P20) at different concentrations and injected continuously onto carboxymethylated dextran 5 (CM5) sensor chips (GE Healthcare, Pittsburgh, PA, USA) coated with antigen CD3ε or CD105. An antigen-free chip was used as a reference. The binding properties of BiTE were measured using Biacore T2000 (GE Healthcare).
Enzyme-linked immunosorbent assay (ELISA) for cytokine secretion
To examine the cytokine production of T cells in response to BiTE treatment, PBMCs (effector cells) were seeded into 96-well plates together with 293T or 293T-hE cells (target cells) at an E: T ratio of 20:1 (in triplicate for each condition). hEND-CD3/BiTE was then added into the above at a final concentration of 10 μg/mL. The supernatant was collected after 72 h incubation at 37 ºC, and the secretion of IL-10, interferon-γ (IFN-γ), and tumor necrosis factor α (TNF-α) were measured using ELISA kits for these cytokines (Neobioscience, Shenzhen, China).
In vivo xenograft tumor model
To establish a xenograft tumor model, A549 cells (3×106 in 100 μL PBS; target cells) were mixed with freshly isolated PBMCs (6×107 in 100 μL PBS; effector cells) at the E:T ratio of 20:1 and subcutaneously injected into the right axilla of the nude mice (Day 0). From Day 0 to Day 4, hEND-CD3/BiTE in 100 μL PBS was injected daily into the tail vein at 1, 10, or 100 μg. hENDM-CD3/BiTE; PBS was used as a control. To assess the in vivo specificity of hEND-CD3/BiTE, recombinant human endoglin was added into hEND-CD3/BiTE solution to a final concentration of 100 μg/mL prior to injection into the tail vein. The tumor length (L) and width (W) were measured every five days starting from Day 5 until Day 30, and the tumor volume (V) was calculated as V = L × W2/2.
To evaluate its in vivo safety, hEND-CD3/BiTE was intravenously injected into the mice receiving A549 and PBMCs at 20 mg/mL PBS daily for five consecutive days. Age-matched mice not receiving any injection or treatment were used as controls.
Immunohistochemistry (IHC) analysis
All mice were sacrificed on day 30 after A549 inoculation. To assess the in vivo safety of hEND-CD3/BiTE, the liver, heart, spleen, lung, and kidney were isolated from mice receiving 20 mg/mL of BiTE for five days, followed by their embedment in paraffin and staining with hematoxylin and eosin (HE).
For mice bearing xenograft tumors and receiving hEND-CD3/BiTE treatment, the tumor tissues were isolated, fixed in formalin, and embedded in paraffin. The anti-endoglin and anti-proliferating cell nuclear antigen (PCNA) antibodies were then used for IHC staining of endothelial cells and proliferating cells, respectively. For endoglin staining, slides were imaged at 200× (Eclipse 80i), and four fields containing the highest number of endoglin+ vessels from each slide were recorded. The average number of endoglin+ vessels for these four fields was defined as the microvascular density (MVD). For PCNA staining, slides were imaged at 400×, and the average number of PCNA+ cells from five random fields was recorded.
Statistical analysis
All statistical analyses were performed using SPSS 15.0 software (IBM, New York, NY, USA). Quantitative data are presented as the means ± SD. A P value of < 0.05 was considered statistically significant.
Results
Construction and characterization of hEND-CD3/BiTE
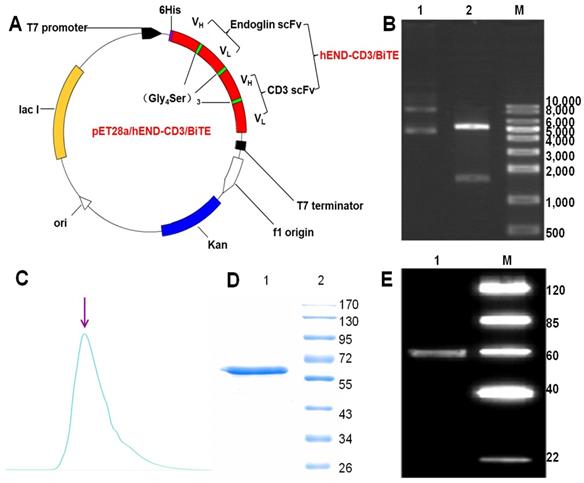
To generate a bispecific antibody targeting both human endoglin and CD3, we applied overlap PCR, constructed a sequential fusion of VHendoglin-VLendoglin-VHCD3-VLCD3 with a 15-amino-acid (GGGGS)3 linker sequence between each fragment and His tag at the 5'-end of the endoglin VH fragment. Subsequently, we cloned the fusion gene into the bacterial expression plasmid pET28a (+) (Figure 1A). The restriction digestion of the plasmid with EcoRI and XhoI generated two fragments of 5,369 and 1,641 base pairs (bps) (Figure 1B) and sequencing analysis of the 1,641-bp fragment confirmed the identity of the endoglin and CD3 scFv fragments (Table 1). Gene expression and protein purification parameters are shown in Supplemental Figure 1 and Western immunoblot. We acquired a single peptide of approximately 59.5 kDa (Figure 1C and D), which also reacted with anti-His-tag antibody (Figure 1E). The amino acid sequence of hEND-CD3/BiTE is shown in Table 2.
Design, expression, and purification of hEND-CD3/BiTE. (A) The schematic map of expression plasmid pET28a (+) containing human endoglin cDNA linked to human CD3 svFc fragment. (B) Electrophoretogram of parental pET28a (+) plasmid (lane 1) and pET26a (+)-hEND-CD3/BiTE plasmid (lane 2) digested with EcoRI and XhoI. M, size markers. (C) Detection of purified hEND-CD3/BiTE protein eluted from an immobilized metal-affinity chromatography column using a stepwise imidazole gradient. (D) Coomassie blue staining of SDS/PAGE gel of the eluted hEND-CD3/BiTE protein. 1. hEND-CD3/BiTE; 2. Protein molecular weight markers. (E) Western immunoblot analysis of hEND-CD3/BiTE using an anti-His-tag antibody. 1. hEND-CD3/BiTE; M. Protein molecular weight markers.
hEND-CD3/BiTE gene sequence.

hEND-CD3/BiTE amino acid sequence.
hEND-CD3/BiTE specifically binds to endoglin+ and CD3+ cells
We first optimized the concentration gradient of hEND-CD3/BiTE combined with endoglin and PBMCs (as seen in supplemental Figure 2). To examine the in vitro binding of hEND-CD3/BiTE to endothelial cells, we examined three different endoglin-expressing cells: HUVEC, Td-EC and 293T-hE. We found that hEND-CD3/BiTE bound to all three cell lines with high efficiency (99.8%, 98.7%, and 99.2% for HUVEC, Td-EC, and 293T-hE, respectively); however, there was no binding to non-endoglin-expressing 293T cells (Figure 2A).
Purified hEND-CD3/BiTE targets endoglin+ cells and CD3+ T lymphocytes. (A) hEND-CD3/BiTE (upper panels) or hENDM-CD3/BiTE (lower panels) were incubated with indicated cells, and binding was detected using PerCP-conjugated anti-His-tag antibody (green signal) or isotype-matched control IgG (grey signal) by flow cytometry. (B) hEND-CD3/BiTE was incubated with freshly isolated PBMCs, and its binding among CD4+ (left panel) or CD8+ lymphocytes was detected using flow cytometry. (C) Binding kinetics and affinity of hEND-CD3/BiTE to the corresponding antigens (endoglin and CD3+) were detected by surface plasmon resonance with the association rate constant Ka; dissociation rate constant Kd and equilibrium dissociation constant KD are shown. (D) hEND-CD3/BiTE-mediated linking of FITC-labeled T cells (green) with PKH26-labeled 293T, 293T-hE, HUVEC, or Td-EC was examined by fluorescence microscopy.

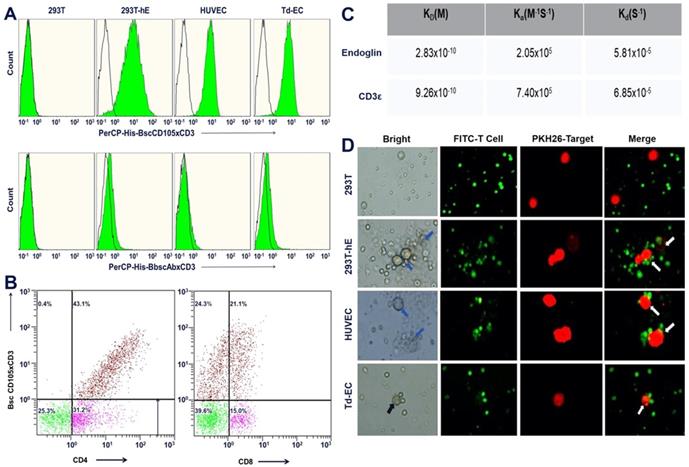
To evaluate the binding of hEND-CD3/BiTE to T cells, we isolated PBMCs from peripheral blood of healthy donors and detected hEND-CD3/BiTE+ cells using anti-His-tag antibody among CD4+ or CD8+ T cells by flow cytometry. As shown in Figure 2B, 43.1% and 21.1% of the CD4+ and CD8+ T cells, respectively, displayed positive binding to hEND-CD3/BiTE consistent with their relative proportions in total CD3+ T cells (Figure 2B), suggesting binding of hEND-CD3/BiTE to CD3 molecules on both CD4+ and CD8+ T cells.
We also used surface plasmon resonance to accurately measure the kinetics of the association rate constant Ka and dissociation rate constant Kd; the affinity was measured by the equilibrium dissociation constant KD of hEND-CD3/BiTE to the antigens, human endoglin and CD3ε. Following the 1:1 Langmuir kinetic model, the affinities of hEND/CD3-BiTE to both endoglin and CD3ε were of the same high magnitude, with binding to the former (KD=2.83×10-10) somewhat higher than the latter (KD=9.26×10-10) (Figure 2C).
By labeling the endoglin+ cells (HUVEC, Td-EC, and 293T-hE) with the red fluorescent probe PKH26 and T cells with green fluorescent CFSE, we showed that hEND-CD3/BiTE efficiently established direct contacts between T cells and endothelial cells but not between non-endoglin-expressing 293T and T cells (Figure 2D).
hEND-CD3/BiTE stimulates T-cell activation, proliferation, and Th1 cytokine production
We next examined hEND-CD3/BiTE effects on T-cell functions, including activation, proliferation, and cytokine production. As displayed in Figure 3A, hEND-CD3/BiTE in PBMC and 293T co-cultured cells did not activate either CD4+ or CD8+ T cells, as indicated by the absence of change in CD69 and CD25; however, hEND-CD3/BiTE stimulated the activation of both T-cell subtypes in the co-culture of PBMCs and 293T-hE cells, suggesting the specificity of this bispecific antibody.
hEND-CD3/BiTE activates T cells, stimulates T-cell proliferation, and increases secretion of Th1 cytokines. (A) PBMCs were co-cultured with 293T (grey signal) or 293T-hE (pink signal) cells in the presence of hEND-CD3/BiTE. The expression of T-cell activation markers CD69 (left panels) and CD25 (right panels) was detected in CD4+ (upper rows) and CD8+ (lower rows) by flow cytometry. (B) Proliferation of CFSE-labeled T cells in response to hEND-CD3/BiTE and other indicated reagents in the solid phase was measured using flow cytometry. The proliferation index was calculated using the ModFit LT software. (C) Supernatants were collected from the co-cultures of PBMCs with 293T (white bar) or 293T-hE cells (purple bar) in the presence of hEND-CD3/BiTE. The secretion of Th1 cytokines, including IFN-γ, and TNF-α, and Th2 cytokine IL10 was measured using ELISA assays. *P < 0.001.
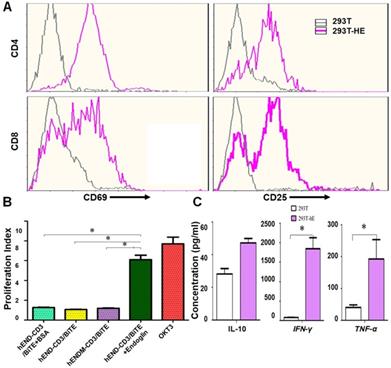
By measuring T-cell proliferation, we found that the presence of hEND-CD3/BiTE alone (1.07), hEND-CD3/BiTE+BSA (1.27), or control hENDM- CD3/BiTE+CD105 (1.19) in the solid could not induce T-cell proliferation. Only hEND-CD3/BiTE+CD105 (6.10) or anti-CD3 monoclonal antibody OKT3 (7.69) significantly stimulated T-cell proliferation (Figure 3B and Supplemental Figure 3).
Consistent with the activation and proliferation of T cells, we showed that pro-inflammatory Th1 cytokine (IFN-γ and TNF-α) secretion was robustly enhanced by hEND-CD3/BiTE acting on PBMC/293T-hE cocultured cells but not on the co-culture of PBMCs with 293T cells (Figure 3C).
hEND-CD3/BiTE promotes T-cell-mediated cytolysis of endoglin+ cells
Increased activation, proliferation, and cytokine production are prerequisite steps for T-cell-mediated cytolysis. By measuring the apoptosis in endoglin+ target cells (as represented by PKH26+PI+ cells by flow cytometry) in the co-culture with PBMCs, we found that the T cells' cytolytic activity increased with hEND-CD3/BiTE concentration (Supplemental Figure 4). The T-cell cytolysis presented a near-linear increase with an increased hEND-CD3/BiTE concentration from 10-4 to 10 μg/mL, and plateaued after 10 μg/mL (Figure 4A). The cytolysis increased with an increased E:T ratio between 0:1 to 20:1 (Figure 4B). We, therefore, chose to use 10 μg/mL END/CD3-BiTE and an E:T ratio of 20:1 in future experiments. We found that the cytolysis was specific for endoglin-expressing cells and not 293T cells; it was also effective in 293T-hE, HUVEC, and Td-EC cells (Figure 4C) in the presence of hEND-CD3/BiTE but not in the control hENDM-CD3/BiTE (Figure 4D).
hEND-CD3/BiTE stimulates T-cell-mediated cytolysis of endoglin+ cells in vitro. PBMCs (effector cells) were co-cultured with indicated target cells labeled with PKH26 in the presence of hEND-CD3/BiTE, and the lysis/apoptosis of target cells was analyzed using flow cytometry and compared with PKH26+PI+ cells. (A) PBMC-induced cytolysis of 293T (orange line) or 293T-hE cells (purple line) in response to various concentrations of hEND-CD3/BiTE. (B) PBMC-induced cytolysis of 293T-hE cells was assessed at different E:T ratios and hEND-CD3/BiTE concentrations. (C) PBMC-induced cytolysis of 293T (blue bar), 293T-hE (red bar), HUVEC (purple bar), and Td-EC (green bar) at an E:T ratio of 20:1 and/or in response to 10 μg/mL hEND-CD3/BiTE. (D) PBMC-induced cytolysis of 293T-hE cells was compared between hEND-CD3/BiTE and the control hENDM-CD3/BiTE. *P < 0.001.
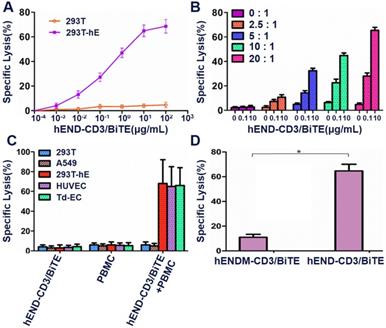
HEND-CD3/BiTE could induce T cells to kill 293T-HE cells expressing endoglin with a killing index of 66.9%. The killing effect of the negative control antibody hENDM-CD3/BiTE on 293T-HE cells was not obvious with the killing index of 13.59%; the difference was statistically significant. These results suggested that only double-specific single-chain antibodies with antigen-binding sites targeting endoglin could kill endoglin-expressing 293T-HE cells (Supplemental Figure 5).
hEND-CD3/BiTE reduces tumor neoangiogenesis and tumor cell proliferation
To assess the in vivo cytolytic effect of hEND-CD3/BiTE on tumor vascular endothelial cells, we measured the MVD by immunohistochemical staining of endoglin (Figure 5) and CD34 (Supplemental Figure 6). As shown in Figures 5A and 5B, co-administration of hEND-CD3/BiTE and PBMCs significantly reduced neoangiogenesis in the tumor (P<0.05 compared with all other groups). The addition of recombinant endoglin partially blocked neoangiogenesis inhibition.
hEND-CD3/BiTE inhibits A549 mouse xenograft tumor angiogenesis and tumor cell proliferation in vivo. (A) Vascular endothelial cells within tumors were detected by immunohistochemical staining of endoglin; representative IHC images (200 ×) from each group are presented. (B) Endoglin+ vessels in the tumors were quantified and compared between groups. (C) Tumor cell proliferation was examined by immunohistochemical staining of PCNA; representative IHC images (400 ×) from each group are presented. (D) PCNA+ cells in the tumors were quantified and compared between groups. a. PBS; b. hEND-CD3/BiTE; c. PBMC; d. hENDM-CD3/BiTE +PBMC; e. hEND-CD3/BiTE +PBMC + Endoglin; f. hEND-CD3/BiTE +PBMC. *P < 0.001.
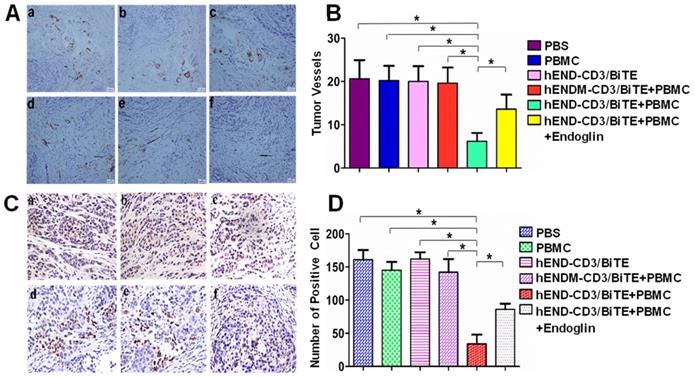
Angiogenesis is essential for continued tumor growth. Therefore, we analyzed tumor cell proliferation by PCNA immunohistochemical staining. Consistent with the changes in angiogenesis, co-administration of hEND-CD3/BiTE and PBMCs was associated with a significant reduction in PCNA+ proliferating cells (P<0.01 compared with all other groups; Figures 5C and 5D).
hEND-CD3/BiTE inhibits in vivo tumor growth and improves mouse survival
We established a xenograft tumor model in nude mice using human lung adenocarcinoma A549 cells with or without human PBMCs from healthy donors and characterized the in vivo activity of hEND-CD3/BiTE. We also administered hEND-CD3/BiTE or control hENDM-CD3/BiTE in mice via the tail vein.
By following tumor growth, we found that the co-injection of PBMCs with A549 without hEND-CD3/BiTE did not significantly affect tumor growth compared with mice receiving A549 cells alone (PBS group). The administration of 1 μg hEND-CD3/BiTE per day for five days significantly inhibited tumor growth. Moreover, treatment with hEND-CD3/BiTE at 10 μg/day for five days led to minimal tumor growth, whereas 100 μg of the antibody completely abolished tumor growth for the first 30 days after A549 injection (Figure 6A and B).
hEND-CD3/BiTE inhibits in vivo tumor growth and improves mouse survival. NOD/SCID mice (n = 6) were subcutaneously implanted with A549 cells without (PBS group) or with human PBMCs (PBMC group). Mice were also treated with hEND-CD3/BiTE or control hENDM-CD3/BiTE daily i.v. injections for the first five days. (A) and (B) Dose-response of tumor growth (A) and images of tumors (B) in mice receiving hEND-CD3/BiTE at indicated doses. 1, PBS; 2, PBMC; 3, PBMCs + 1 μg hEND-CD3/BiTE; 4, PBMCs + 10 μg hEND-CD3/BiTE; 5, PBMCs + 100 μg hEND-CD3/BiTE. (C) and (D) Tumor growth (C) and images (D) in the indicated groups of mice. 1, PBS group; 2, PBMC group; 3, hEND-CD3/BiTE group; 4, PBMC+ hENDM-CD3/BiTE; 5, PBMC+ hEND-CD3/BiTE; 6, PBMC+ hEND-CD3/BiTE + endoglin. (E) Kaplan-Meier survival analysis of mice from the indicated groups.
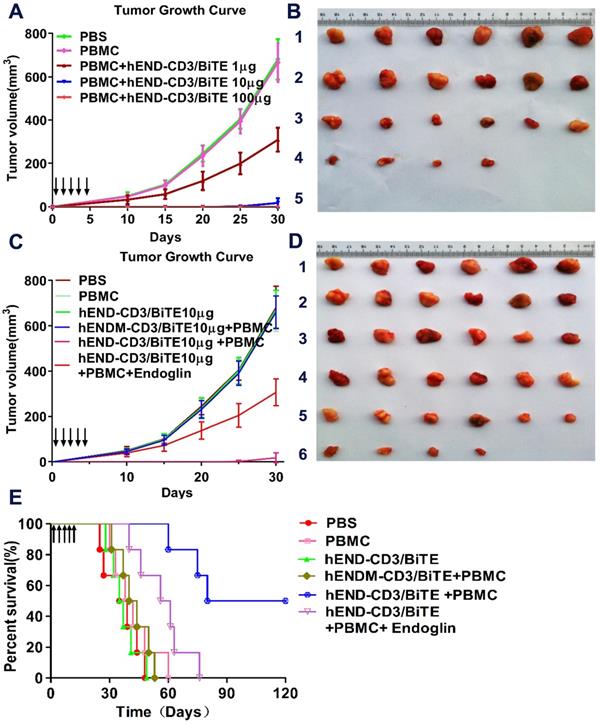
The tumor inhibitory activity of hEND-CD3/BiTE required the presence of both PBMCs and A549 cells because tumors in mice receiving hEND-CD3/BiTE alone continued to grow at a rate comparable with those in mice receiving only A549 cells (PBS group). The tumor inhibitory effect was also specific for hEND-CD3/BiTE because the control hENDM-CD3/BiTE did not slow tumor growth. Furthermore, co-administration of recombinant human endoglin partially yet significantly reduced tumor growth (Figure 6C and D).
Consistent with its effect on tumor growth, hEND-CD3/BiTE also significantly improved mouse survival (mean survival time, 100 days; P < 0.05) compared with the PBS (mean survival time, 37 d), hEND-CD3/BiTE alone (mean survival time, 36 d), PBMCs alone (mean survival time, 40 d), and hENDM-CD3/ BiTE+PBMCs (mean survival time, 42 d) groups. The addition of recombinant human endoglin to hEND-CD3/BiTE partially extended the mean mouse survival to 58.5 d, which was still significantly shorter than that achieved by hEND-CD3/BiTE, suggesting the blocking effect of recombinant endoglin (Figure 6E).
hEND-CD3/BiTE causes minimal toxicity to major organs
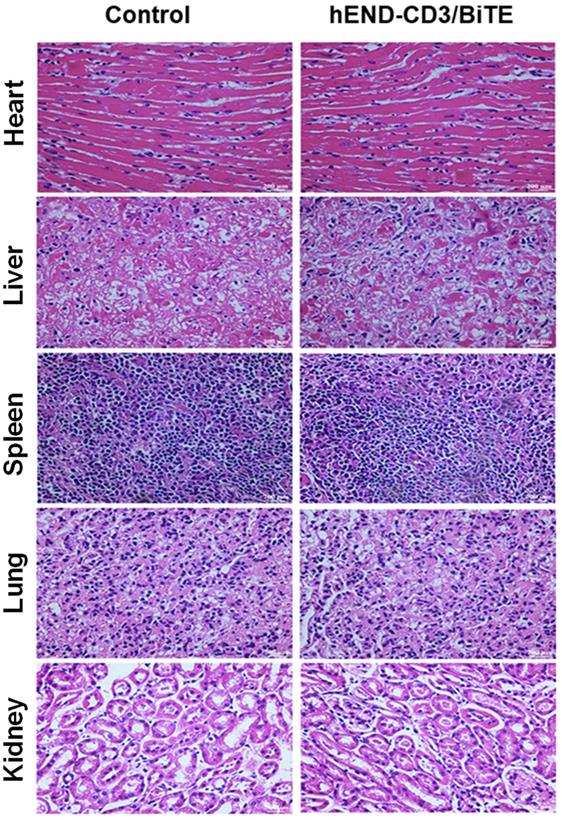
Lastly, we assessed the in vivo safety of hEND-CD3/BiTE after intravenous injection at 2 mg/day for five days, which was 20-fold higher than the biologically active dose demonstrated above. By analyzing several organs, including the heart, liver, spleen, lung, and kidney, we did not detect tumor metastasis, significant lymphocyte infiltration, or structural alterations in these organs compared with control mice not receiving any injections or treatments (Figure 7).
The in vivo toxicity of hEND-CD3/BiTE. Mice (n = 6) were subcutaneously implanted with A549 cells and human PBMCs and i.v. injected with 2 mg/day hEND-CD3/BiTE for the first five days. Untreated mice were used as controls. On day 30 after A549 injection, all mice were sacrificed, and the heart, liver, spleen, lung, and kidney isolated, stained with hematoxylin and eosin, and comparisons were made between the two groups; magnification, 400 ×.
Discussion
In this study, we generated a bispecific antibody targeting CD3+ T cells and endoglin+ endothelial cells. We showed that the hEND-CD3/BiTE was not only active in binding to the target cell types but was also biologically significant in activating the T-cell-mediated cytolysis of endothelial cells. These in vitro activities also translated to in vivo functions by reducing tumor angiogenesis, inhibiting tumor growth, and extending mouse survival.
During tumor progression, angiogenesis provides nutrients to support continuous tumor growth and also creates conduits for the distant dissemination of tumor cells [30]. Therefore, targeting angiogenesis becomes a significant strategy in cancer therapy and is under intensive investigation [31]. Continued research on the molecular mechanisms controlling tumor angiogenesis has and will continue to reveal important therapeutic targets leading to the development of several approaches, including targeting the key molecules (the most important being VEGFs and their receptors), gene therapy, and neutralizing monoclonal antibodies. Although exogenous genes delivered via a gene therapy strategy have several advantages and show efficacy in various proof-of-principle animal experiments, they must overcome impediments at multiple steps from transcription, translation, post-translational modifications to functional proteins targeting angiogenesis. This approach has not shown clinical efficacy in experimental animals, as demonstrated by in vitro studies [32, 33].
Neutralizing monoclonal antibodies, on the other hand, directly target endogenous proangiogenic factors and encounter reduced obstacles. The success of this strategy was first validated by FDA approval of Avastin, a humanized VEGF-neutralizing monoclonal antibody for metastatic colorectal cancer in combination with standard care [34] and subsequently for the treatment of non-small-cell lung cancer and metastatic breast cancer [35, 36]. Despite the initial promising performance of neutralizing monoclonal antibodies, especially those targeting VEGF, their clinical benefits are short-term, and patients eventually demonstrate a lack of response or resistance to the anti-angiogenesis therapy. Several mechanisms underlying the resistance phenotype have been proposed and extensively studied [37]. One significant mechanism is that tumor cells and other stromal cells may selectively stimulate alternative pro-angiogenic signaling, thereby contributing to anti-VEGF therapy resistance [38-41]. Therefore, it is important to identify the mechanisms of alternative pro-angiogenic factors that induce and mediate resistance to anti-VEGF therapy. In this context, the size of the macromolecular antibody may limit its bioavailability and effective concentration in the tumor mass reducing the efficacy of neutralizing antibody [42, 43], which may be addressed by antibody engineering or designing an alternative delivery vehicle. This report proposed a novel strategy to target both mechanisms that may lead to anti-VEGF therapy resistance.
Endoglin, a co-receptor of TGF-β, is highly expressed in tumor-associated vascular and lymphatic endothelium [21, 23, 44]. In vitro studies demonstrated that anti-tumor therapy targeting endoglin inhibited proliferation, migration, and adhesion and induced apoptosis of endothelial cells by disrupting TGF-β signaling [20, 45, 46]. Moreover, endoglin plays a significant role in the development of resistance to VEGF therapy. In mice bearing pancreatic xenografts and treated with anti-VEGF antibody, endoglin was among the few genes that showed significant up-regulation [47]. Tumors in mice with one copy of the endoglin gene exhibited a delayed onset of resistance to anti-VEGF treatment [48]. Also, high endoglin expression correlated with a worse prognosis in various human solid tumors, including lung, colorectal, prostate, and hepatocellular cancers. [49]. Therefore, anti-endoglin therapy combined with VEGF inhibitors would likely provide improved benefits.
Anti-endoglin therapy has consistently demonstrated positive therapeutic outcomes in experimental animal models. Seon et al. showed that anti-endoglin monoclonal antibodies (mAbs) inhibited tumor growth and suppressed metastasis by stimulating T-cell immunity resulting from antibody-dependent cell-mediated cytotoxicity and inhibited endothelial growth while promoting tumor cell apoptosis through Fas/FasL signaling [50]. Additionally, anti-endoglin mAbs are associated with minimal in vivo toxicity, supporting its potential in clinical therapy. Also, in immunocompetent mice, the anti-tumor activity of anti-endoglin mAbs was higher than in immunocompromised SCID mice; the depletion of CD4+ and/or CD8+ T cells abrogated the anti-tumor efficacy of anti-endoglin mAbs, indicating that T-cell immunity may synergize with the anti-tumor activity of anti-endoglin mAbs [51, 52].
These findings prompted us to develop an approach to boost T-cell immunity and target endoglin simultaneously. Herein, we showed that the bispecific hEND-CD3/BiTE antibody we developed specifically targeted endoglin+ cells, including those ectopically expressing endoglin (293T-hE) and normal, as well as tumor endothelial cells expressing endogenous endoglin (HUVEC and Td-EC). This process generated no toxicity and did not induce significant lymphocyte infiltration in the major organs of nude mice even at a dose 20-fold higher than a biologically active dose. However, the further work still needs to study cross-reactions in different species in near future.
BiTE is a bispecific antibody composed of scFv from two monoclonal antibodies, including one targeting CD3. The connection between the two scFv by a short peptide linker enables free rotation of the two arms and enhances the binding affinity to the corresponding antigens. The smaller size due to the minimal binding domains (Fv fragments) of two monoclonal antibodies reduces its antigenicity, thereby increasing its tissue penetration and ensuring the proximity between T cells and target cells. The simple design of BiTE also enables high-yield production for various biomedical applications [53]. Most BiTEs developed thus far target tumor-specific antigens in addition to T cells [11, 54]. Herein, we utilized BiTE properties to design a bispecific antibody targeting T cells and endoglin. We expected this approach to boost the anti-tumor activity of endoglin inhibitor by activating T-cell immunity and specifically induce the cytolysis of endothelial cells via T-cell immunity.
Following gene expression and protein purification, most proteins were in the inclusion bodies, probably due to their high-level transcription driven by the T7 promoter in BL21(DE3), leading to numerous mis-folded proteins and thus aggregation into inclusion bodies. To renature the proteins, we applied urea with decreasing concentrations from 8 M to 6, 4, 2, 1, 0.5 and 0 M, with each dialysis step lasting for 2 h. We tested the biological activity of the purified and renatured BiTEs by examining their binding to the target cells. The specific binding to endoglin-expressing 293T cells but not to parental 293T cells confirmed its specificity for endoglin. The binding analysis also showed that, among the total PBMCs, 43.1% were positive for CD4 and hEND-CD3/BiTE and 21.1% for CD8 and hEND-CD3/BiTE. The total percentage of these two populations (64.2%) was consistent with the percentage of T cells in PBMCs. Furthermore, their relative ratio reflected the CD4+/CD8+ cell ratio within the T-cell population, indicating the specific binding of hEND-CD3/BiTE to the two major T cell subpopulations. In the co-culture system containing both PBMCs and endoglin-expressing cells, we directly visualized contacts between the two cell types mediated by hEND-CD3/BiTE but not between PBMCs and non-endoglin-expressing 293T cells.
Further in vitro functional studies showed that the hEND-CD3/BiTE could stimulate T-cell activation, proliferation, Th1 cytokine production, and cytolysis; these processes were dependent on the hEND-CD3/BiTE binding to endoglin in a specific cellular context. Consistent with our findings, Choi et al. reported that BiTE preferentially stimulated Th1 cytokine production [55]. The stimulation of multiple T-cell responses suggested that BiTE activity not only affected CD4+ but also CD8+ T cells. Future studies should explore the mechanisms underlying specific stimulation of Th1 response by BiTE.
The in vivo analysis focused on lung cancer because of its high morbidity and mortality and the significance of angiogenesis and endoglin in lung cancer progression and metastasis [56-58]. Consistent with the in vitro findings, hEND-CD3/BiTE administration significantly reduced tumor angiogenesis and inhibited tumor cell proliferation, leading to retarded tumor growth and improved mouse survival. In another xenograft model of HepG2-derived hepatocellular carcinoma in mice, we observed potent anti-tumor activity of hEND/CD3/BiTE (data not shown). Furthermore, the in vivo effects of hEND-CD3/BiTE were specifically and partially blocked by free recombinant human endoglin, supporting the specificity of this antibody.
It has been reported that in some hematological malignancies, BiTEs, such as blinatumomab, caused the cytokine release syndrome and adverse central nervous system events in a small number of patients [59, 60]. No obvious side effects of BiTEs in patients with solid tumors were observed [61], consistent with our results of hEND-CD3/BiTE in solid tumor vessels.
In summary, we proposed a novel design of the BiTE antibody for cancer therapy by targeting CD3+ T cells in conjunction with endothelial cells instead of tumor cells. We demonstrated the specificity, efficacy, and safety of this new approach using in vitro and in vivo analyses. We anticipate that this strategy will lead to promising therapies for various cancers that rely on angiogenesis for continuous growth and metastasis. Specifically, as a combinatorial treatment, this strategy may avoid the development of resistance to anti-VEGF therapy.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by the Programs for Changjiang Scholars and Innovative Research Team in University (No. IRT_15R13), the State Project for Essential Drug Research and Development (No. 2019ZX09301132), Guangxi Key Research and Development Project (No. AB20117001) and Guangxi Natural Science Foundation (2018JJA140524), and Guangxi Science and Technology Base and Talent Special Project (No. AD17129062).
Authors' Contributions
LZ, YY and WS designed and conceived the study. XL, GL, TW and YH performed the experiments and methodology. YZ, YH and PW organized and supervised the whole project. ZZ, HP and ZD, YY and HT analyzed data and wrote the manuscript, which was approved by all authors.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chan SL. Microbiome and cancer treatment: Are we ready to apply in clinics? Prog Mol Biol Transl Sci. 2020;171:301-8
2. Aggarwal U, Goyal AK, Rath G. Development of Drug Targeting and Delivery in Cervical Cancer. Curr Cancer Drug Targets. 2018;18:792-806
3. Tran P, Lee S-E, Kim D-H, Pyo Y-C, Park J-S. Recent advances of nanotechnology for the delivery of anticancer drugs for breast cancer treatment. Journal of Pharmaceutical Investigation. 2019;50:261-70
4. Kelly PN. The Cancer Immunotherapy Revolution. Science. 2018;359:1344-5
5. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350-5
6. Marin-Acevedo JA, Soyano AE, Dholaria B, Knutson KL, Lou Y. Cancer immunotherapy beyond immune checkpoint inhibitors. J Hematal Oncol. 2018;11:8
7. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480-9
8. Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science. 2013;342:1432-3
9. Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012;12:278-87
10. Töpfer K, Kempe S, Müller N, Schmitz M, Bachmann M, Cartellieri M. et al. Tumor evasion from T cell surveillance. J Biomed Biotechnol. 2011;2011:918471
11. Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93:290-6
12. Suryadevara CM, Gedeon PC, Sanchez-Perez L, Verla T, Alvarez-Breckenridge C, Choi BD. et al. Are BiTEs the "missing link" in cancer therapy? Oncoimmunology. 2015;4:e1008339
13. Walter RB. Biting back: BiTE antibodies as a promising therapy for acute myeloid leukemia. Expert Rev Hematol. 2014;7:317-9
14. Wu J, Fu J, Zhang M, Liu D. Blinatumomab: a bispecific T cell engager (BiTE) antibody against CD19/CD3 for refractory acute lymphoid leukemia. J Hematol Oncol. 2015;8:104
15. Sheridan C. Amgen's bispecific antibody puffs across finish line. Nat Biotechnol. 2015;33:219-21
16. Sennino B, McDonald DM. Controlling escape from angiogenesis inhibitors. Nat Rev Cancer. 2012;12:699-709
17. Jászai J, Schmidt MHH. Trends and Challenges in Tumor Anti-Angiogenic Therapies. Cells. 2019 8
18. Kasprzak A, Adamek A. Role of Endoglin (CD105) in the Progression of Hepatocellular Carcinoma and Anti-Angiogenic Therapy. Int J Mol Sci. 2018 19
19. Lugano R, Ramachandran M, Dimberg A. Tumor angiogenesis: causes, consequences, challenges and opportunities. Cell Mol Life Sci. 2020;77:1745-70
20. Nassiri F, Cusimano MD, Scheithauer BW, Rotondo F, Fazio A, Yousef GM. et al. Endoglin (CD105): a review of its role in angiogenesis and tumor diagnosis, progression and therapy. Anticancer Res. 2011;31:2283-90
21. Tual-Chalot S, Garcia-Collado M, Redgrave RE, Singh E, Davison B, Park C. et al. Loss of Endothelial Endoglin Promotes High-Output Heart Failure Through Peripheral Arteriovenous Shunting Driven by VEGF Signaling. Circ Res. 2020;126:243-57
22. Tian H, Huang JJ, Golzio C, Gao X, Hector-Greene M, Katsanis N. et al. Endoglin interacts with VEGFR2 to promote angiogenesis. Faseb j. 2018;32:2934-49
23. Jin Y, Muhl L, Burmakin M, Wang Y, Duchez AC, Betsholtz C. et al. Endoglin prevents vascular malformation by regulating flow-induced cell migration and specification through VEGFR2 signalling. Nat Cell Biol. 2017;19:639-52
24. Rathouska J, Vecerova L, Strasky Z, Slanarova M, Brcakova E, Mullerova Z. et al. Endoglin as a possible marker of atorvastatin treatment benefit in atherosclerosis. Pharmacol Res. 2011;64:53-9
25. Xiong C, Mao Y, Wu T, Kang N, Zhao M, Di R. et al. Optimized Expression and Characterization of a Novel Fully Human Bispecific Single-Chain Diabody Targeting Vascular Endothelial Growth Factor165 and Programmed Death-1 in Pichia pastoris and Evaluation of Antitumor Activity In Vivo. Int J Mol Sci. 2018 19
26. Nazarpour R, Zabihi E, Alijanpour E, Abedian Z, Mehdizadeh H, Rahimi F. Optimization of Human Peripheral Blood Mononuclear Cells (PBMCs) Cryopreservation. Int J Mol Cell Med. 2012;1:88-93
27. Wu H, Yao L, Chou L, Yang JH, Zhang YX, Li XL. et al. Construction and functional analysis of an anti-human cervical carcinoma/anti-human CD3 single-chain bispecific antibody. Mol Med Rep. 2016;14:804-10
28. Jin X, Chen Y, Chen H, Fei S, Chen D, Cai X. et al. Evaluation of Tumor-Derived Exosomal miRNA as Potential Diagnostic Biomarkers for Early-Stage Non-Small Cell Lung Cancer Using Next-Generation Sequencing. Clin Cancer Res. 2017;23:5311-9
29. Quah BJ, Parish CR. The use of carboxyfluorescein diacetate succinimidyl ester (CFSE) to monitor lymphocyte proliferation. J Vis Exp. 2010
30. Bielenberg DR, Zetter BR. The Contribution of Angiogenesis to the Process of Metastasis. Cancer J. 2015;21:267-73
31. Gensicka M, Głowacka A, Dzierzbicka K, Cholewinski G. Inhibitors of Angiogenesis in Cancer Therapy - Synthesis and Biological Activity. Curr Med Chem. 2015;22:3830-47
32. Fallah A, Heidari HR, Bradaran B, Sisakht MM, Zeinali S, Molavi O. A gene-based anti-angiogenesis therapy as a novel strategy for cancer treatment. Life Sci. 2019;239:117018
33. Huang D, Lan H, Liu F, Wang S, Chen X, Jin K. et al. Anti-angiogenesis or pro-angiogenesis for cancer treatment: focus on drug distribution. Int J Clin Exp Med. 2015;8:8369-76
34. Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W. et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335-42
35. Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A. et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542-50
36. Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA. et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357:2666-76
37. Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8:592-603
38. Horikawa N, Abiko K, Matsumura N, Baba T, Hamanishi J. Anti-VEGF therapy resistance in ovarian cancer is caused by GM-CSF-induced myeloid-derived suppressor cell recruitment. Br J Cancer. 2020;122:778-88
39. Mitamura T, Pradeep S, McGuire M, Wu SY, Ma S, Hatakeyama H. et al. Induction of anti-VEGF therapy resistance by upregulated expression of microseminoprotein (MSMP). Oncogene. 2018;37:722-31
40. Schiffmann LM, Fritsch M, Gebauer F, Günther SD, Stair NR, Seeger JM. et al. Tumour-infiltrating neutrophils counteract anti-VEGF therapy in metastatic colorectal cancer. Br J Cancer. 2019;120:69-78
41. Jaiprasart P, Dogra S, Neelakantan D, Devapatla B, Woo S. Identification of signature genes associated with therapeutic resistance to anti-VEGF therapy. Oncotarget. 2020;11:99-114
42. Patel NR, Pattni BS, Abouzeid AH, Torchilin VP. Nanopreparations to overcome multidrug resistance in cancer. Adv Drug Deliv Rev. 2013;65:1748-62
43. Rakel N, Galm L, Bauer KC, Hubbuch J. Influence of macromolecular precipitants on phase behavior of monoclonal antibodies. Biotechnol Prog. 2015;31:145-53
44. Kim SK, Henen MA. Structural biology of betaglycan and endoglin, membrane-bound co-receptors of the TGF-beta family. Exp Biol Med. 2019;244:1547-58
45. Afshar Moghaddam N, Mahsuni P, Taheri D. Evaluation of Endoglin as an Angiogenesis Marker in Glioblastoma. Iran J Pathol. 2015;10:89-96
46. Jarosz M, Szala S. [Endoglin as a target of antitumor therapy]. Postepy Hig Med Dosw (Online). 2013;67:79-89
47. Dolezelova E, Sa ICI, Prasnicka A, Hroch M, Hyspler R, Ticha A. et al. High soluble endoglin levels regulate cholesterol homeostasis and bile acids turnover in the liver of transgenic mice. Life Sci. 2019;232:116643
48. Sennino B, Ishiguro-Oonuma T, Schriver BJ, Christensen JG, McDonald DM. Inhibition of c-Met reduces lymphatic metastasis in RIP-Tag2 transgenic mice. Cancer Res. 2013;73:3692-703
49. Rosen LS, Gordon MS, Robert F, Matei DE. Endoglin for targeted cancer treatment. Curr Oncol Rep. 2014;16:365
50. Seon BK, Haba A, Matsuno F, Takahashi N, Tsujie M, She X. et al. Endoglin-targeted cancer therapy. Curr Drug Deliv. 2011;8:135-43
51. Li T, Kang G, Wang T, Huang H. Tumor angiogenesis and anti-angiogenic gene therapy for cancer. Oncol Lett. 2018;16:687-702
52. Uneda S, Toi H, Tsujie T, Tsujie M, Harada N, Tsai H. et al. Anti-endoglin monoclonal antibodies are effective for suppressing metastasis and the primary tumors by targeting tumor vasculature. Int J Cancer. 2009;125:1446-53
53. Desplancq D, Rinaldi AS, Stoessel A, Sibler AP, Busso D, Oulad-Abdelghani M. et al. Single-chain Fv fragment antibodies selected from an intrabody library as effective mono- or bivalent reagents for in vitro protein detection. J Immunol Methods. 2011;369:42-50
54. Trabolsi A, Arumov A. T Cell-Activating Bispecific Antibodies in Cancer Therapy. J Immunol. 2019;203:585-92
55. Choi BD, Kuan CT, Cai M, Archer GE, Mitchell DA, Gedeon PC. et al. Systemic administration of a bispecific antibody targeting EGFRvIII successfully treats intracerebral glioma. Proc Natl Acad Sci U S A. 2013;110:270-5
56. Wang B, Zhang Z, Tang J, Tao H, Zhang Z. Correlation between SPARC, TGFβ1, Endoglin and angiogenesis mechanism in lung cancer. J Biol Regul Homeost Agents. 2018;32:1525-31
57. Qin SH, Lau ATY. Resveratrol Promotes Tumor Microvessel Growth via Endoglin and Extracellular Signal-Regulated Kinase Signaling Pathway and Enhances the Anticancer Efficacy of Gemcitabine against Lung Cancer. 2020; 12.
58. Sacco PC, Maione P, Rossi A, Sgambato A, Casaluce F, Palazzolo G. et al. New Antiangiogenetic Therapy Beyond Bevacizumab in the Treatment of Advanced Non Small Cell Lung Cancer. Curr Pharm Des. 2015;21:4763-72
59. Teachey DT, Rheingold SR, Maude SL, Zugmaier G, Barrett DM, Seif AE. et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood. 2013;121:5154-7
60. Portell CA, Wenzell CM, Advani AS. Clinical and pharmacologic aspects of blinatumomab in the treatment of B-cell acute lymphoblastic leukemia. Clin Pharmacol. 2013;5:5-11
61. Speck T, Heidbuechel JPW, Veinalde R, Jaeger D, von Kalle C, Ball CR. et al. Targeted BiTE Expression by an Oncolytic Vector Augments Therapeutic Efficacy Against Solid Tumors. Clin Cancer Res. 2018;24:2128-37
Author contact
![]() Corresponding authors: Hongliang Tang (tanghl204edu.cn), Zhiming Deng (dengzhmcdcom), Yulin Yuan (yuanyulincom).
Corresponding authors: Hongliang Tang (tanghl204edu.cn), Zhiming Deng (dengzhmcdcom), Yulin Yuan (yuanyulincom).