Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(13):6491-6506. doi:10.7150/thno.55821 This issue Cite
Research Paper
Apilimod alters TGFβ signaling pathway and prevents cardiac fibrotic remodeling
Mathieu Cinato1, Laurie Guitou1, Amira Saidi1, Andrei Timotin1, Erwan Sperazza1, Thibaut Duparc1, Sergey N. Zolov2, Sai Srinivas Panapakkam Giridharan2, Lois S. Weisman2, Laurent O. Martinez1, Jerome Roncalli1,3, Oksana Kunduzova1, Helene Tronchere1*, Frederic Boal1 ![]() *
*
1. INSERM U1297 I2MC, Toulouse, France and Université Paul Sabatier, Toulouse, France.
2. Life Sciences Institute, University of Michigan, Ann Arbor, USA.
3. Department of Cardiology, Toulouse University Hospital, Toulouse, France.
* These authors equally contributed to this work.
Received 2020-11-12; Accepted 2021-4-2; Published 2021-4-19
Abstract

Rationale: TGFβ signaling pathway controls tissue fibrotic remodeling, a hallmark in many diseases leading to organ injury and failure. In this study, we address the role of Apilimod, a pharmacological inhibitor of the lipid kinase PIKfyve, in the regulation of cardiac pathological fibrotic remodeling and TGFβ signaling pathway.
Methods: The effects of Apilimod treatment on myocardial fibrosis, hypertrophy and cardiac function were assessed in vivo in a mouse model of pressure overload-induced heart failure. Primary cardiac fibroblasts and HeLa cells treated with Apilimod as well as genetic mutation of PIKfyve in mouse embryonic fibroblasts were used as cell models.
Results: When administered in vivo, Apilimod reduced myocardial interstitial fibrosis development and prevented left ventricular dysfunction. In vitro, Apilimod controlled TGFβ-dependent activation of primary murine cardiac fibroblasts. Mechanistically, both Apilimod and genetic mutation of PIKfyve induced TGFβ receptor blockade in intracellular vesicles, negatively modulating its downstream signaling pathway and ultimately dampening TGFβ response.
Conclusions: Altogether, our findings propose a novel function for PIKfyve in the control of myocardial fibrotic remodeling and the TGFβ signaling pathway, therefore opening the way to new therapeutic perspectives to prevent adverse fibrotic remodeling using Apilimod treatment.
Keywords: Heart failure, fibrotic remodeling, PIKfyve, Apilimod, TGFβ.
Introduction
Fibrosis is a conserved end-stage hallmark of a broad range of organ injuries and failure such as cardiac, kidney and pulmonary diseases [1]. To date, there are limited therapeutic treatments available to limit fibrotic remodeling progression, largely due to the heterogeneous evolution of fibrosis in different pathological stimuli and organs. For instance, myocardial fibrotic remodeling is a common pathological process in heart diseases and a determinant step of development and progression of heart failure, thereby participating to cardiac diastolic and systolic dysfunctions [2]. Fibrotic remodeling in the pathological heart is a highly orchestrated process in which cardiac fibroblasts are central cellular mediators [3]. Activated cardiac fibroblasts have increased proliferative, migrative and invasive capacities. They are characterized by α-SMA contractile fibres and excessive production and deposition of extracellular matrix (ECM) proteins, e.g. collagen type I and III [3]. Although TGFβ is recognized as one of the key mediators of fibroblast to myofibroblast transition, both in vitro and in vivo, the molecular control of its signaling and downstream effectors is still poorly understood [4]. To date, TGFβ-based anti-fibrotic therapies are limited and new strategies to target its downstream signaling are needed. The TGFβ signaling pathway is diverse and tightly regulated. At the molecular level, TGFβ receptor type 2 (TGFβ-R2) binds directly to the TGFβ, associates with the TGFβ receptor type 1 (TGFβ-R1) and induces its phosphorylation on serine and threonine residues [5]. This triggers the canonical signaling pathway through the Smad proteins, or the non-canonical pathways. At the plasma membrane, the TGFβ receptors have a rapid turnover, most likely linked to their internalization by endocytosis in a clathrin- or caveolin-dependent manner, determining the receptor fate and subsequent signaling [6, 7]. Once internalized to an early endosomal EEA1-positive compartment, the receptors can be either recycled back to the plasma membrane or directed to the late endosomal CD63-positive compartment, ultimately leading to their lysosomal degradation [8].
PIKfyve is an evolutionarily conserved phosphoinositide 5-kinase that generates PI(3,5)P2 and is responsible of most of the cellular pool of PI5P [9,10]. PIKfyve is a master regulator of membrane trafficking and signaling, mostly within the endosomal/endocytic system, where it controls the maturation of early endosomes to late endosomes/lysosomes through the conversion of PI3P into PI(3,5)P2 [9]. Indeed, inhibition of PIKfyve by overexpression of a dominant-negative mutant [11] or via its pharmacological inhibition [12] alters endomembrane homoeostasis, leading to extensive endosomal vacuolation. In vivo, the functions of PIKfyve have begun to be unraveled using genetically engineered mice or pharmacological manipulation. PIKfyve-null mice are embryonically lethal [13], but hypomorphic PIKfyveβ-geo/β-geo mice, which expressed residual PIKfyve activity are viable and develop defects within multiple organs, such as in the nervous, cardiopulmonary and renal systems [14]. Recently, a new potent and highly selective inhibitor of the kinase, Apilimod, has been identified through a high-throughput IL-12 inhibitor screening [15, 16]. Apilimod binds to the catalytic C-terminal domain of PIKfyve, most likely to the ATP binding pocket [15]. Recently, we took advantage of this inhibitor to unravel a novel role for PIKfyve in cardiomyocyte mitochondrial integrity, ROS production and apoptosis in response to stress, culminating in improved cardiac functions in a mouse model of diabetic cardiomyopathy [17]. Additionally, we observed a reduction in myocardial fibrosis following chronic PIKfyve inhibition, suggesting a potential role of PIKfyve in cardiac fibroblasts reprogramming. Here, we now demonstrate that PIKfyve is required for the fibrotic remodeling of cardiac tissue after chronic myocardial injury. We provide the first evidence that pharmacological inhibition of PIKfyve by Apilimod prevents excessive fibrotic development through the dampening of the TGFβ/Smad signaling pathway and ultimately improves cardiac function. Altogether, these results unveil PIKfyve as a novel regulator of fibroblast activation and propose Apilimod as a promising anti-fibrotic molecular therapy.
Methods
Reagents and antibodies
Antibodies used in this study are: anti-Hsp90 (sc-13119), anti-GAPDH (sc-32233) and anti-Smad2/3 (sc-133098) from Santa Cruz Biotechnology; anti-αSMA (A5228, for western-blot) and anti-flag M2 from Sigma; anti-CD68 (GeneTex GTX41864), anti-GM130 (#610822) from BD Biosciences; anti-EEA1 (GTX109638) and anti-caveolin-1 (GTX100205) from GeneTex; anti-CD63 (MCA2142) was from AbD Serotec; anti-EGFR (D38B1), anti-phospho-Smad2 (#3108) and 3 (#9520), anti-αSMA (#19245, for immunofluorescence), anti-COL1 (#91144) from Cell Signaling Technologies; anti-PI(3,5)P2 (Z-P035) from Echelon; anti-HA.11 (16B12, Covance). Fluorescent Alexa-coupled secondary antibodies and DAPI were from Life Technologies and HRP-coupled secondary antibodies from Cell Signaling Technology. Apilimod was purchased from Axon Medchem. The plasmids encoding GFP-ICAM1 and P2Y12-GFP were as described [18]. All other chemicals were from Sigma unless otherwise stated.
Generation of the 6X-HA-PIKfyve plasmid
6xHA PIKfyve was generated with a cDNA sequence that that is 100% identical to GenBank sequence AAR19397. Due to the large size of the cDNA (6.2 kb), PIKfyve was cloned into pCMV-HA vector (635690, Clontech Laboratories, Inc) in two stages. Citrine-PIKfyve [19] was digested with BsrGI, the internal fragment (nucleotides 2510-6213; 3703 bp) was isolated to reinsert later. The remaining plasmid corresponding to the N- and C-terminal regions of PIKfyve, was ligated and this deletion construct of PIKfyve was PCR amplified using primers: forward, acgcgtcgaccatggccacagatgataagacg and reverse: aaggaaaaaagcggccgctcagcaattcagacccaagc. The PCR fragment and pHA-CMV vector were cleaved with SalI and NotI and the PIKfyve deletion construct was inserted into the vector. A resultant clone with the correct sequence was further digested with BsrGI. The internal BsrGI digested fragment was re-ligated back to generate 1xHA-PIKfyve. This construct was further digested with SfII and SalI and an additional 5XHA -tag (gctcttatggccatggaggccTACCCATACGATGTTCCTGACTATGCGGGCTATCCCTATGACGTCCCGGACTATGCAGGATCCTATCCATATGACGTTCCAGATTACGCTtcaTACCCTTATGACGTGCCCGATTACGCCGGCagtTACCCTTACGATGTCCCAGATTACGCTCCGcggtcgaccatggccaca encoding 5xHA tag: YPYDVPDYA G YPYDVPDYA GS YPYDVPDYA S YPYDVPDYA GS YPYDVPDYA) was inserted at the N-terminus of PIKfyve using Gibson assembly to generate a 6xHA-PIKfyve wild type construct.
Animal studies
Two-month old wild-type male C57BL6/J mice were purchased from Envigo. Transverse aortic constriction (TAC) was produced by constriction of the ascending aorta around a 26-gauge needle with the use of a 7-0 prolene suture, maintained for 4 weeks as described previously [20]. Sham-operated mice underwent a similar procedure without constriction of the ascending aorta. Animals were randomly divided into four groups: (i) Sham/vehicle (n = 7), (ii) Sham/Apilimod (n = 6), (iii) TAC/vehicle (n = 7), and (iv) TAC/Apilimod (n = 9). Mice were treated 4 days after surgery and then every day for 4 weeks with Apilimod (2 mg/kg/day, i.p.) or vehicle (DMSO), corresponding to a final DMSO concentration of 50% diluted in PBS as previously described [17]. For acute Apilimod treatment, mice were treated intraperitoneally with Apilimod (2 mg/kg/day) for 8 consecutive days. Cardiac fibroblasts were isolated as described below, let to adhere on gelatin-coated coverslips for 4 h, and process further for cell surface binding of fluorescent TGFβ. For the washout experiments, cells were extensively washed and further cultured for 24-48 h before being processed.
Echocardiography
Blinded echocardiography was performed as described [17] on isoflurane-anesthetized mice using a Vivid7 imaging system (General Electric Healthcare) equipped with a 14-MHz sectorial probe. Two-dimensional images were recorded in parasternal long- and short-axis projections, with guided M-mode recordings at the midventricular level in both views. Left ventricular (LV) dimensions and wall thickness were measured in at least five beats from each projection and averaged. Shortening fraction (SF) and ejection fraction (EF) were calculated from the two-dimensional images.
Morphology
Hematoxylin-eosin, Sirius red and fluorescent WGA stainings on 10 μm heart cryosections were performed according to standard methods. The extent of cardiac fibrosis was quantified using ImageJ software [17, 20]. Quantification of myocyte cross-sectional area was performed as described [17].
Cell culture, transfection and treatments
Mice primary cardiac fibroblasts were isolated and cultured according to the method described previously [20]. Briefly, the heart was isolated from adult male C57BL6/J mice, triturated and was then incubated with Digestion Buffer [HBSS (Sigma H8264); BSA (1 mg/mL); Pancreatin NB (Serva #31442; 0.5 mg/mL) and Collagenase NB4 (Serva #17454.01; 0.1 mg/mL)] at 37°C for four consecutive rounds of digestion. The suspension was centrifuged at 300 g for 5 min and the cell pellet was resuspended in DMEM F12 medium (Sigma D6434) supplemented with 10% FBS, penicillin (20 U/mL)-streptomycin (20 µg/mL) and 2 mM glutamine and cultured in a 37°C, 5% CO2 incubator. Primary cardiac fibroblasts were used up to passage 3. HeLa, HEK and MEF cells from hypomorphic PIKfyveβ-geo/β-geo mice were cultured in DMEM supplemented with 10% FBS, penicillin (20 U/mL)-streptomycin (20 µg/mL). For TGFβ treatment, the cells were pretreated for 30 min with Apilimod (100 nM) or DMSO (vehicle only) and then subjected to TGFβ stimulation (10 ng/mL) for 48 h unless otherwise stated. Cardiac fibroblasts, HeLa and HEK cells were transiently transfected using JetPRIME (Polyplus transfection), X-tremeGENE 9 (Roche) or TransIT-X2 (Mirus) according to manufacturers' instructions. After 24 h, the cells were serum-starved and treated with Apilimod 100 nM for 16 h. Cells were fixed with PFA, immunofluorescence was performed as described [21] and imaged on a Zeiss LSM-780 or a LSM900 confocal microscope. For PI5P staining, the previously described biotinylated PHD probe was used [22] in combination with a streptavidin-Alexa546. Colocalization studies were performed using ImageJ. The quantification of TGFβ-R2 internalization was essentially done as described [18]. Briefly, an outer ring just surrounding the cell was drawn, which yielded the total fluorescence intensity, i.e., the total amount of TGFβ-R2 expressed in the cell. An inner ring just below the plasma membrane was then drawn, which yielded the amount of TGFβ-R2 internalized into the cell. The ratio between the two values gave the amount of internalized TGFβ-R2, normalized against the total amount of receptor expressed in the cell.
Cell-surface binding of fluorescent TGFβ
TGFβ was fluorescently labeled using the AlexaFluor-555 protein Labeling Kit (A20174, Invitrogen) according to manufacturer's instructions. For cell-surface binding of fluorescent TGFβ, cells were washed with HBSS at 4°C and incubated with A555-TGFβ (500ng/mL) in HBSS at 4°C for 30min. Cells were extensively washed and processed further for immunofluorescence.
Cell migration, polarization and invasion assays
Cell migration was assessed using the wound healing assay. Briefly, a confluent fibroblast culture was scratched using a micropipet tip. Twenty-two hours later, cells were fixed and nuclei were stained with DAPI. The wound closure was measured using ImageJ. Cell polarization was assessed as described [23]. Briefly, fixed cells were stained using fluorescent phalloidin (to stain actin cytoskeleton) and an anti-GM130 antibody (to stain the Golgi apparatus). A cell was considered polarized if the Golgi apparatus was oriented within a 120° angle facing the wound. Cell invasion assay was performed in matrigel coated modified-Boyden chamber assay and assessed by spectrophotometric measurements as described [24].
Collagen gel contraction assay
Neonatal rat cardiac fibroblasts were isolated essentially as described [25], plated on gelatin-coated dishes and used at maximum passage 1. Collagen gel contraction assay was performed as described [26]. Briefly, cardiac fibroblasts were seeded in a collagen gel, treated, and the contraction of the gel was assessed after 72 h. After PFA-fixation, the gels were imaged on an Axio Observer Z1 microscope (Zeiss) using the mosaic function. The area of the gel was measured and normalized against the total well area.
Protein extraction and western blotting
Proteins from cardiac tissues or cells were extracted using RIPA buffer and quantified using the Bio‐Rad Protein Assay (Bio‐Rad). Proteins were loaded in Laemmli sample buffer, denaturated at 70°C for 15 min, and resolved by SDS-PAGE and Western blotting. Immunoreactive bands were detected by chemiluminescence with the Clarity Western ECL Substrate (Bio‐Rad) on a ChemiDoc MP Acquisition system (Bio‐Rad).
RNA Extraction and Quantitative Polymerase Chain Reaction (qRT-PCR)
Total RNAs were isolated from mice primary fibroblasts or tissues using the RNeasy mini kit (Qiagen) or the Direct-zol DNA/RNA miniprep kit (Zymo Research). Total RNAs were reverse transcribed using Superscript II reverse transcriptase (Invitrogen) or M-MLV Reverse Transcriptase (Promega) in the presence of a random hexamers. Real-time quantitative PCR was performed as previously described [27] using the MESA BLUE qPCR MasterMix Plus (Eurogentec) or the SsoFast™ EvaGreen® Supermix (Bio-Rad). The expression of target mRNA was normalized against house-keeping genes (Gapdh, Rps29, Rplpo or Hprt) mRNA expression. Primers used in this study are detailed in supplementary Table S1.
Data and Statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology [28]. Data are expressed as mean ± SEM. Comparison between two groups was performed by Student's two-tailed t-test while comparison of multiple groups was performed by one-way ANOVA followed by a Bonferroni's post hoc test using GraphPad Prism version 9.0.2 (GraphPad Software, Inc).
Study approval
The investigation conforms to the ARRIVE guidelines, the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1985) and was performed in accordance with the recommendations of the French Accreditation of the Laboratory Animal Care (approved by the local ethics committee).
Results
Apilimod protects against myocardial interstitial fibrosis development and prevents pressure overload-induced cardiac dysfunction in mice
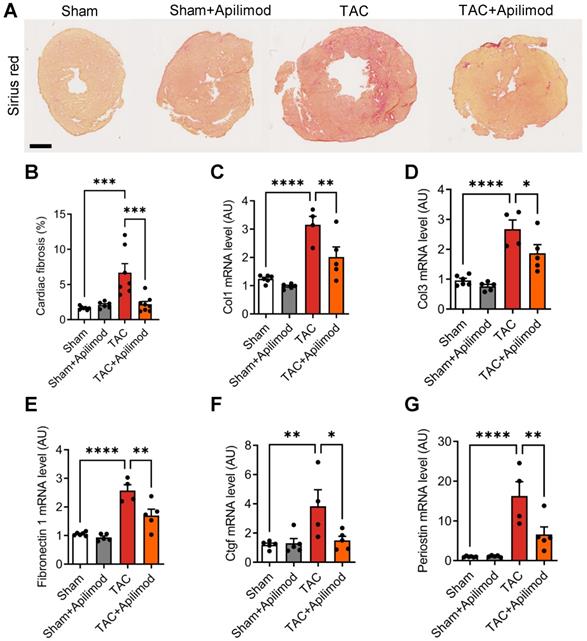
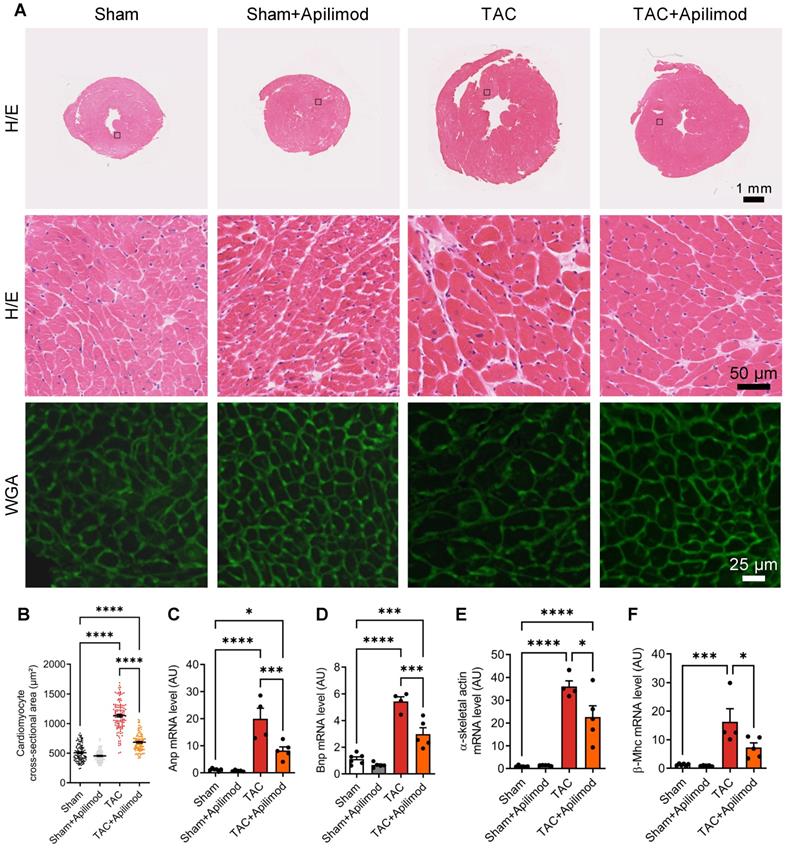
To address the effects of Apilimod in cardiac fibrotic remodeling, we used a mouse model of transverse aortic constriction (TAC). Cardiac pressure overload induced by four weeks TAC triggered a massive myocardial deposition of ECM proteins as shown by Sirius red staining, typical for end-stage fibrotic tissue remodeling (Figure 1A-B). Strikingly, Apilimod abrogated myocardial collagen fibre accumulation in TAC-mice (Figure 1A-B). Consistently, daily Apilimod treatment attenuated TAC-induced overexpression of cardiac pro-fibrotic genes including collagen 1 (Col1, Figure 1C), collagen 3 (Col3, Figure 1D), fibronectin 1 (Figure 1E) and connective tissue growth factor (Ctgf, Figure 1F). Moreover, upon Apilimod treatment, TAC-induced expression of periostin, a well-known marker of activated cardiac fibroblasts [29], was strongly inhibited (Figure 1G), suggesting that Apilimod prevents fibroblast activation in vivo. Consistently, TAC-stressed mice treated with Apilimod showed reduced level of α-SMA as demonstrated by immunofluorescent staining (Figure S1A). Moreover, we found that Apilimod decreased the recruitment of CD-68-positive macrophages (Figure S1B) and the production of the myocardial pro-inflammatory cytokines Il-6 (Figure S1C), Tnf-α (Figure S1D) and Ccl2 (Figure S1E). In addition, we found that Apilimod reduced cardiac hypertrophy induced by TAC, as shown morphologically (Figure 2A-B) and by the expression of the hypertrophic markers Anp (Figure 2C), Bnp (Figure 2D), α-skeletal actin (Figure 2E) and β-Mhc (Figure 2F). Consistently, echocardiography analysis showed that Apilimod reduced end-diastolic ventricular wall thickness, intraventricular septum thickness and left ventricular mass (Table 1) in TAC-stressed mice hearts. It has to be noted that Apilimod-treatment does not completely abrogate the hypertrophic response following TAC. Importantly, the myocardial anti-fibrotic effect of Apilimod culminated in the preservation of cardiac function in TAC-mice, as shown by improved EF and shortening fraction (Table 1 and Figure S1F, EF and SF respectively). These results suggest that Apilimod is a potent inhibitor of fibroblast activation and myocardial fibrosis development in vivo, leading to preserved cardiac performance following TAC surgery.
Echocardiograhic parameters of mice.
| Sham | Sham+Apilimod | TAC | TAC+Apilimod | |
|---|---|---|---|---|
| EF (%) | 64.33±1.05 | 62.17±4.03 | 40.00±8.07** | 59.13±3.29$ |
| SF (%) | 34.67±0.88 | 33.35±2.92 | 19.80±4.21** | 32.29±2.56$ |
| LVPWd (mm) | 0.83±0.61 | 0.89±0.07 | 1.23±0.03*** | 0.99±0.05$ |
| IVSd (mm) | 0.78±0.05 | 0.88±0.04 | 1.22±0.02**** | 1.09±0.02$,§§§§ |
| LV mass (mg) | 112.3±5.7 | 118.5±5.2 | 206.6±16.3**** | 167.3±10.4$,§§ |
EF: ejection fraction; SF: shortening fraction; LVPWd: end-diastolic ventricular wall thickness; IVSd: end-diastolic intraventricular septum thickness; LV mass: left ventricle mass. ANOVA followed by Bonferroni's post-hoc test: Sham vs. TAC **p < 0.01, ***p < 0.001, ****p < 0.0001; TAC vs. TAC+Apilimod $p < 0.05; Sham vs. TAC+Apilimod §§p < 0.01, §§§§p < 0.0001.
Apilimod prevents cardiac fibrosis development in mice. (A) Cardiac fibrosis was assessed on heart cryosections stained with Sirius red from mice sham-operated or subjected to TAC surgery treated intraperitoneally with Apilimod. Bar is 1 mm. (B) Quantification of cardiac fibrosis from (A). n = 6-8 mice per group. (C-G) Expression levels of myocardial collagen 1 (Col1, C), collagen 3 (Col3, D), fibronectin-1 (E), Ctgf (F) and periostin (G) by qRT-PCR. n = 6-8 mice per group. ANOVA followed by Bonferroni's post-hoc test, *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Apilimod blunts cardiac hypertrophic response in TAC-mice. (A) Heart cryosections of mice treated as indicated were stained with hematoxylin-eosin (H/E) or fluorescent WGA. (B) Quantification of cardiomyocytes cross-sectional area measured from (A). Quantification was performed from 3-4 images across 3-5 mice per group. (C-F) Expression levels of Anp, Bnp, α-skeletal actin and β-Mhc were measured by qRT-PCR in cardiac tissues. n = 4-6 mice per group. ANOVA followed by Bonferroni's post-hoc test, *p < 0.05; ***p < 0.001; ****p < 0.0001.
Apilimod reduces cardiac fibroblast activation and differentiation in vitro
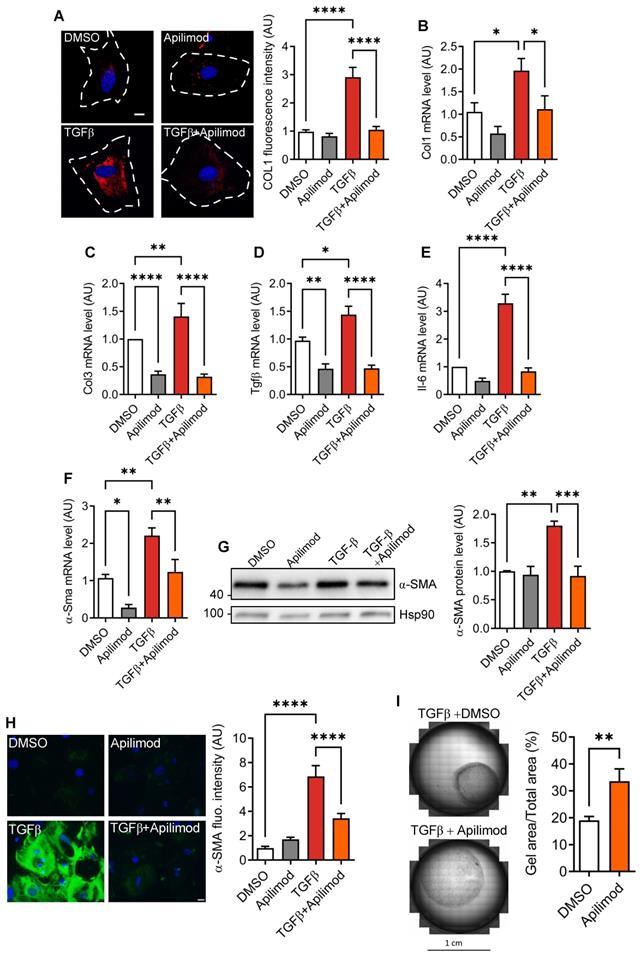
Fibroblast activation can be induced in vitro by TGFβ treatment [20]. After 48 h stimulation, we observed an increase in extracellular collagen fibre deposition as shown by collagen 1 immunostaining (Figure 3A). This was consistent with increased mRNA levels of pro-fibrotic factors collagen 1, collagen 3, Tgfβ (Figure 3B-D) and inflammatory cytokine Il-6 (Figure 3E). Strikingly, concomitant treatment of fibroblasts with Apilimod almost completely abrogated collagen accumulation (Figure 3A), pro-fibrotic (Figure 3B-D) and inflammatory Il-6 (Figure 3E) transcript levels. Next, we investigated the effect of Apilimod on the expression of the myofibroblast marker α-SMA. Apilimod treatment decreased both α-Sma mRNA (Figure 3F) and protein levels measured by western-blot and immunefluorescent staining (Figure 3G-H) in TGFβ-treated cardiac fibroblasts. Additionally, Apilimod markedly reduced the acquisition of the contractile phenotype of TGFβ stimulated cardiac myofibroblasts in a three-dimensional collagen gel contraction assay (Figure 3I).
Apilimod inhibits cardiac fibroblast activation. (A) Cardiac fibroblasts were treated with TGFβ in the presence of Apilimod or DMSO, fixed and stained with an anti-collagen 1 antibody (shown in red). Nuclei were stained with DAPI (in blue). Bar is 10 µm. Quantification if shown on the right, n = 41-82 cells across 3 independent experiments. (B-E) Expression levels of collagen 1 (Col1, B), collagen 3 (Col3, C), Tgfβ (D) and Il-6 (E) were assessed by qRT-PCR on treated cardiac fibroblasts. (F-H) Expression level of α-Sma was assessed by qRT-PCR (F), western-blot (G) and immunofluorescence (H) on treated cardiac fibroblasts. (I) Collagen gel contraction assay on TGFβ activated cardiac fibroblasts treated with Apilimod or DMSO for 72 h. n = 4-6 independent experiments. ANOVA followed by Bonferroni's post-hoc test or Student's t-test (unpaired, two-tailed), *p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001.
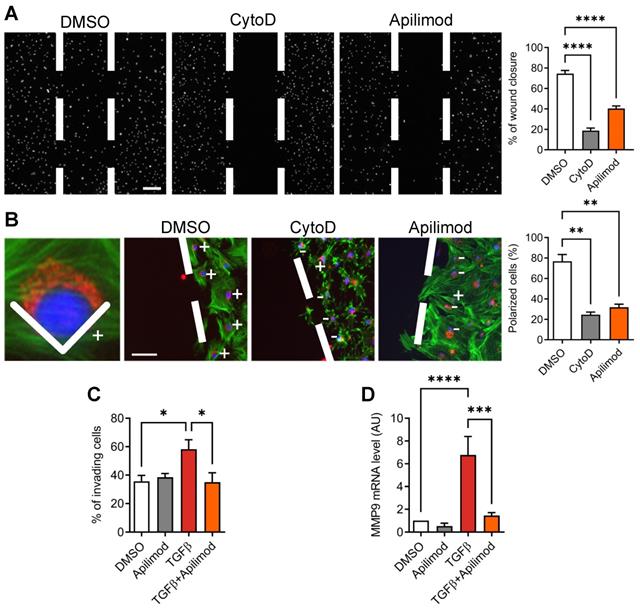
The activation of cardiac fibroblasts is characterized by a migratory and invasive phenotype. Therefore, we looked at the effect of Apilimod on the migrative properties of TGFβ stimulated cardiac fibroblasts. Apilimod treatment abolished TGFβ-induced cell migration in a 2D wound-healing assay, as potently as cytochalasin-D, an inhibitor of actin cytoskeleton and cell migration (Figure 4A). These data indicate that Apilimod inhibits directed cell migration of cardiac fibroblasts. Cell polarization is a prerequisite to cell migration and can be monitored by the positioning of the Golgi apparatus between the nucleus and the leading edge of the cell toward the wounded area. As shown in Figure 4B, while in DMSO-treated cells most of the fibroblasts became polarized towards the wounded area, Apilimod completely abrogated this phenomenon, similarly to cytochalasin D treatment. Next, we investigated the effect of Apilimod on activated fibroblasts invasive properties in a more physiologically relevant model, i.e. a matrigel-coated modified Boyden chamber assay. As shown in Figure 4C, while TGFβ enhanced the number of invading cells, Apilimod treatment dramatically reduced TGFβ-stimulated invasion of primary cardiac fibroblasts. Consistently, we observed a down-regulation of the matrix metalloproteinase Mmp9, a key ECM remodeling enzyme, in Apilimod-treated TGFβ-stimulated fibroblasts (Figure 4D). Collectively, these results identify Apilimod as a potent inhibitor of cardiac fibroblast activation in response to TGFβ.
Apilimod reduces cardiac fibroblasts migration and invasion. (A) Migrative properties of treated cardiac fibroblasts were assessed using the wound-healing assay. Nuclei were stained with DAPI (shown in white). Bar is 200 µm. Results are from 3 independent experiments. (B) Cell polarity was scored by the positioning of the Golgi apparatus (stained with GM130, shown in red) towards the migration front. Phalloidin staining of the actin cytoskeleton was used as a counterstain (shown in green). Cell nuclei were stained with DAPI (in blue). Bar is 100 µm. Results are from n = 19-43 cells across 3 independent experiments. (C) Cardiac fibroblasts invasive properties were assessed using a modified Boyden chamber invasion assay. Results are from 3 independent experiments. (D) Expression level of Mmp9 was measured by qRT-PCR in treated cardiac fibroblasts. Results are from 3 independent experiments. ANOVA followed by Bonferroni's post-hoc test, *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Apilimod inhibits cardiac fibroblast activation through vacuolar sequestration of TGFβ-R2
To gain further insight into the molecular mechanisms involved, we examined whether Apilimod was affecting TGFβ signaling pathway. After binding the TGFβ ligand, TGFβ receptor type 2 (TGFβ-R2) associates with TGFβ-R1, and the resulting complex is internalized to the endosomal compartment, as a requisite for its signaling [30]. As shown in Figure 5A, in resting control cardiac fibroblasts, the TGFβ-R2 was localized at the plasma membrane, as expected for the endogenous receptor. Strikingly, we found that Apilimod massively induced TGFβ-R2 receptor relocalization into intracellular vesicles independently from any TGFβ stimulation (Figure 5A). This intracellular retention of the TGFβ-R2 was also observed in HeLa cells (Figure 5B). Apilimod-driven TGFβ receptor internalization was further confirmed in a cell-surface labelling of TGFβ receptors. Indeed, Apilimod significantly reduced the cell-surface binding of fluorescent TGFβ, both in primary cardiac fibroblasts (Figure 5C) and HeLa cells (Figure 5D), indicating a reduction in the amount of surface available receptors in Apilimod-treated cells. Interestingly, Apilimod did not alter the localization of the tyrosine-kinase EGF receptor, the G-coupled receptor P2Y12 or of the adhesion molecule ICAM1 (Figure S2), suggesting that Apilimod targets specifically the TGFβ receptor localization.
PIKfyve inactivation triggers steady-state TGFβ-R2 internalization. (A-B) Isolated primary cardiac fibroblasts (A) or HeLa cells (B) expressing flag-TGFβ-R2 were serum starved and treated with Apilimod or DMSO as indicated. Cells were fixed and stained with an anti-flag antibody. Bar is 10 µm. Results are from n = 9-16 cells across 3 independent experiments. (C-D) Cell surface binding of fluorescent TGFβ (TGFβ-A555, shown in red) on treated- cultured primary cardiac fibroblasts (C) or HeLa cells (D). Nuclei were stained with DAPI (shown in blue). Bar is 10 µm. n = 43-110 cells across 3 independent experiments. Student's t-test (unpaired, two-tailed), ****p < 0.0001. (E) Mouse embryonic fibroblasts (MEF) isolated from PIKfyveβgeo/βgeo mice or wild-type littermates were transfected to express flag-TGFβ-R2, serum-starved and treated with Apilimod as indicated. Cells were fixed and stained with a flag antibody (green) and imaged by confocal microscopy. Bar is 10 µm. Right panel shows the quantification of flag-TGFβ-R2 internalization. n = 4-8 cells across 3 independent experiments. ANOVA followed by Bonferroni's post-hoc test or Student's t-test (unpaired, two-tailed), *p < 0.05; ****p < 0.0001. (F) HeLa cells were transfected with plasmids encoding flag-TGFβ-R2 and empty HA or HA-PIKfyveWT vector as indicated. Cells were treated either with DMSO, YM201636 or Apilimod (100 nM) for 48 h, fixed and stained with anti-flag (shown in green) and anti-HA (in red) antibodies. Nuclei were stained with DAPI (in blue). Bar is 10 µm.
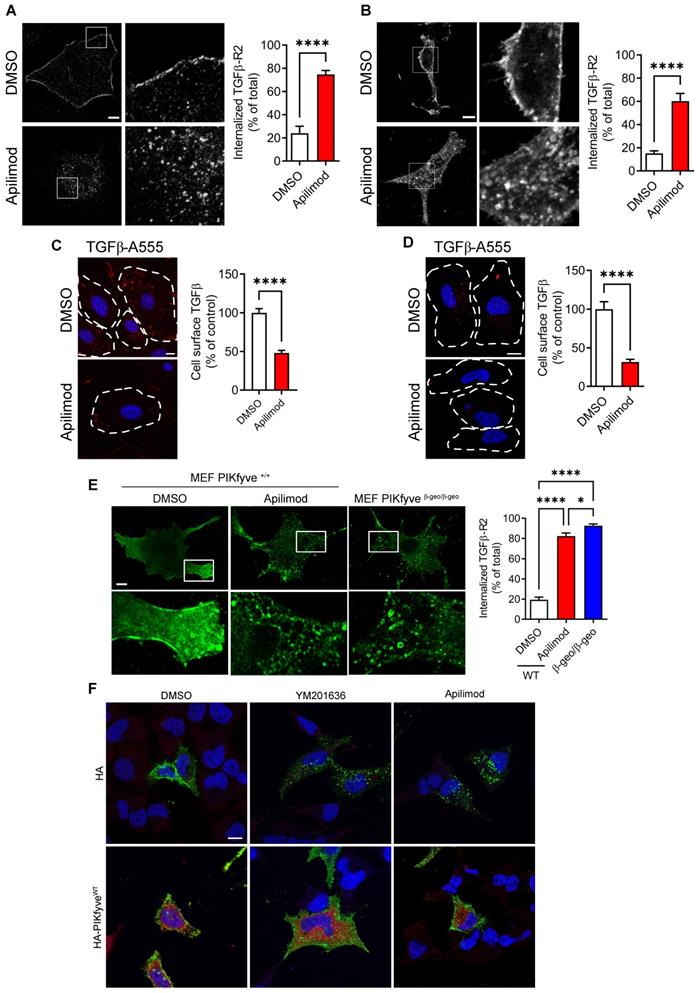
Apilimod has been shown to specifically inhibit the lipid kinase PIKfyve [15, 19]. This inhibitory effect was validated in our in vitro (Figure S3A) and in vivo models (Figure S3B) by monitoring the amount of PIKfyve products PI5P and PI(3,5)P2. To further confirm the implication of PIKfyve, we used mouse embryonic fibroblasts (MEF) isolated from WT or hypomorphic PIKfyveβ-geo/β-geo mice [14]. As shown in Figure 5E, TGFβ-R2 was restricted to the plasma membrane in resting DMSO-treated PIKfyve+/+ MEF, while Apilimod induced the intracellular sequestration of TGFβ-R2, as observed in cardiac fibroblasts and HeLa cells. Similarly, in MEF isolated from PIKfyveβ-geo/β-geo mice, TGFβ-R2 was absent from the cell surface and essentially localized in intracellular vesicles (Figure 5E). The implication of PIKfyve was also confirmed by the use of a structurally distinct inhibitor, the YM201636 compound [12], which induced TGFβ-R2 internalization in treated cells, similarly to Apilimod-treatment (Figure 5F). Moreover, overexpression of PIKfyveWT ablated the effect of both Apilimod and YM201636 on TGFβ-R2 localization (Figure 5F). This strongly suggests that Apilimod effect on TGFβ-R2 localization is due to its specific inhibition of PIKfyve.
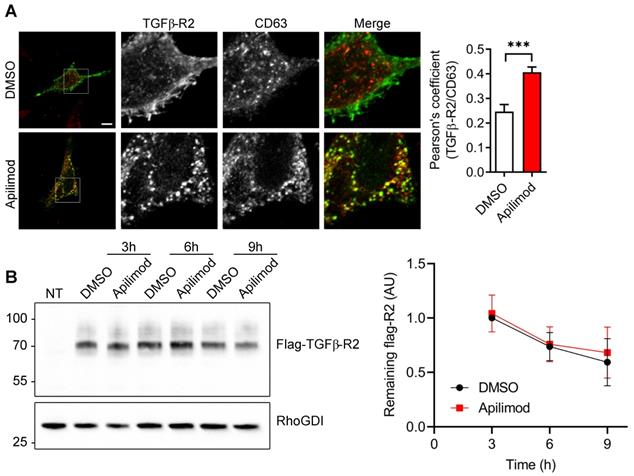
Next, we sought to identify the subcellular compartment in which TGFβ-R2 receptor is internalized upon Apilimod treatment. As shown in Figure S4A, Apilimod provoked an extensive vacuolation of EEA1-endosomes, but did not induce the internalization of TGFβ-R2 in this compartment. Similarly, Apilimod did not induce accumulation of TGFβ-R2 in caveolae (Figure S4B). In comparison, Apilimod increased significantly the colocalization of TGFβ-R2 with the late endosomal marker CD63 (Figure 6A), indicating that PIKfyve inhibition triggered the sequestration of TGFβ-R2 in a CD63-positive late endosomal compartment. Interestingly, we found that Apilimod did not alter the half-life of TGFβ-R2 (Figure 6B), suggesting that internalized TGFβ-R2 was not targeted for lysosomal degradation and that PIKfyve is more likely to be implicated in the receptor recycling back to the plasma membrane.
Apilimod induces TGF-βR2 mislocalization to late-endosomes. (A) Serum-starved HeLa cells expressing flag-TGFβ-R2 were treated as indicated with Apilimod or DMSO, fixed and stained with an anti-flag antibody (green) and an anti-CD63 antibody (red). Bar is 10 µm. Right panel shows the quantification of colocalization. n = 17-37 cells across 3 independent experiments. Student's t-test (unpaired, two-tailed) ***p < 0.001. (B) Non-transfected (NT) or HEK cells expressing flag-TGFβ-R2 were serum-starved and treated with DMSO or Apilimod for the indicated times. Cell lysates were blotted with an anti-flag antibody to assess the amount of remaining flag-TGFβ-R2. RhoGDI was used as a loading control. n = 3 independent experiments.
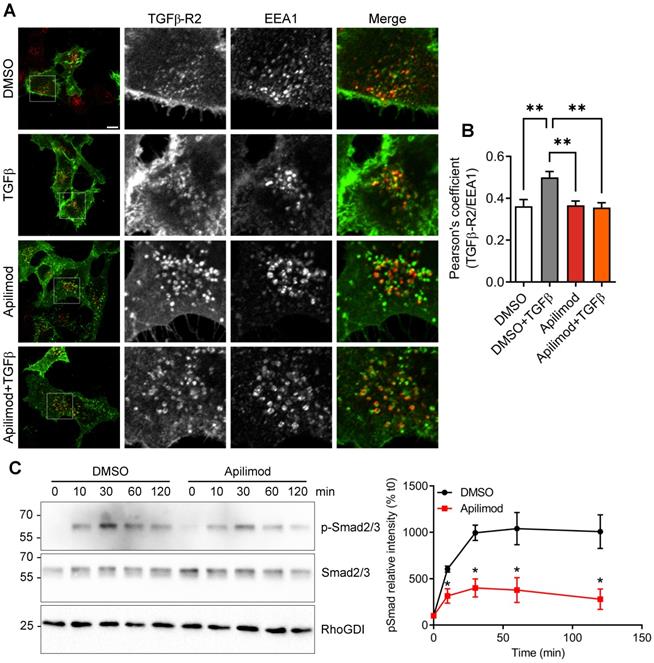
Apilimod dampens TGFβ signaling pathway
Upon binding to its ligand, TGFβ receptor is known to be first internalized to EEA1-positive endosomal compartment, mediating the downstream signaling pathway [8]. Indeed, in DMSO-treated cells, TGFβ stimulation induced the internalization of the receptor in EEA1-positive vesicles (Figure 7A-B). In contrast, this ligand-mediated internalization was lost in Apilimod-treated cells (Figure 7A-B). Furthermore, upon TGFβ stimulation, a fraction of the receptor is internalized in CD-63-positive late endosomes (Figure S5). Interestingly, in Apilimod-treated cells challenged with TGFβ, we did not observe a cumulative effect on the colocalization of TGFβ-R2 and CD63 (Figure S5), indicating that the internalized receptor is not responsive to TGFβ stimulation. Consequently, we found that Apilimod reduced TGFβ-dependent phosphorylation of the canonical downstream effectors Smad2/3 in cardiac fibroblasts (Figure 7C). Collectively, these results show that PIKfyve controls TGFβ receptor trafficking and regulates its downstream signaling.
Apilimod prevents TGFβ downstream signaling. (A) Serum-starved HeLa cells expressing flag-TGFβ-R2 were treated with Apilimod or DMSO as indicated, stimulated with TGFβ, fixed and stained with anti-flag (green) and anti-EEA1 (red) antibodies. Bar is 10 µm. (B) Quantification of colocalization between TGFβ-R2 and EEA1. n = 16-37 cells across 3 independent experiments (C) Primary cardiac fibroblasts were serum-starved and pre-treated with DMSO or Apilimod, before stimulation with TGFβ for the indicated time. Cell lysates were blotted for phosphorylated Smad2/3 (p-Smad2/3), total Smad2/3, or RhoGDI as a loading control. Quantification if shown on the right panel. n = 3 independent experiments. ANOVA followed by Bonferroni's post-hoc test or Student's t-test (unpaired, two-tailed), **p < 0.01.
The TGFβ signaling pathway is a common feature among several fibrotic pathologies such as pulmonary, renal or hepatic diseases [31]. Therefore, we wondered whether long-term Apilimod treatment might affect other tissues beside the heart. We then performed qPCR analysis to address the hepatic fibrotic and inflammatory status Apilimod-treated mice subjected to TAC. As shown in Figure S6, 4 weeks TAC did not increase the hepatic fibrosis status (as demonstrated by the expression levels of collagen 1 and TGFβ), nor the inflammatory status of the liver (as shown by IL-6, TNF-α, F4/80 and CCL2 expression levels). Moreover, in sham animals, Apilimod treatment had no consequences on the hepatic fibrotic or inflammatory status (Figure S6), showing that systemic administration of Apilimod has no consequences on other tissues.
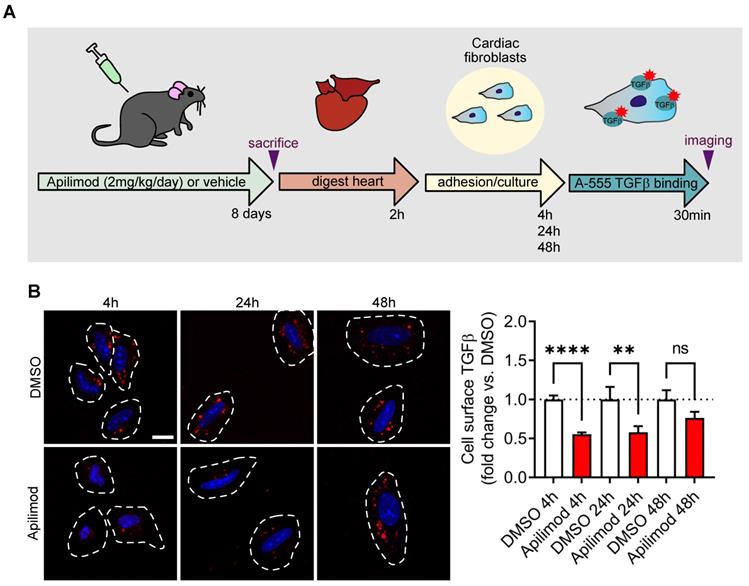
Apilimod controls TGFβ receptor localization in vivo
Lastly, in order to investigate whether Apilimod controls TGFβ receptor localization in vivo, mice were treated with the inhibitor for 8 consecutive days, and cardiac fibroblasts were isolated and let to adhere on a gelatine matrix. We then used the cell-surface labelling of TGFβ receptors in order to investigate the amount of available receptors. As shown in Figure 8, in cardiac fibroblasts isolated from DMSO-treated mice, we observed a punctate labelling, similar to what was observed in vitro (see Figure 5C-D). Strikingly, in cardiac fibroblast isolated from Apilimod-treated mice, a dramatic reduction in cell surface TGFβ labelling was observed (Figure 8B), suggesting that PIKfyve inhibition induced TGFβ receptor internalization in vivo. Interestingly, this inhibition was lost upon establishment of the culture (i.e. after 48 h), indicating that the effect of Apilimod on TGFβ receptor is reversible.
Apilimod induces TGFβ receptor internalization in vivo. (A) Schematic of the experimental setting. (B) Cardiac fibroblasts isolated from vehicle or Apilimod-treated mice were plated and processed for cell surface binding of fluorescent TGFβ (TGFβ-A555, shown in red) after the indicated time in culture. Nuclei were stained with DAPI (shown in blue). Bar is 10 µm. Quantification is shown on the right from 34-265 cells from 3 technical replicates from 2-4 mice per group. ANOVA followed by Bonferroni's post-hoc test, **p < 0.01; ****p < 0.0001; ns, non-significant.
Altogether, our results demonstrate for the first time that Apilimod controls TGFβ receptor trafficking and its subsequent signaling pathway and that Apilimod treatment opens the way to new anti-fibrotic therapeutic strategies.
Discussion
Fibrotic remodeling is a common pathological hallmark in numerous acute and chronic diseases leading to organ injury and failure. Activation of naive fibroblasts and their transition to myofibroblasts is an adaptative and necessary phenomenon in the short term but highly deleterious if prolonged, resulting in irreversible scar formation, organ stiffness and failure [2, 31]. In this context, we show that pharmacological inhibition of PIKfyve by Apilimod limits myocardial fibrosis development upon injury, culminating in the preservation of organ function. In vitro, our work demonstrates that Apilimod dampens TGFβ-mediated cell migration and invasion and fibroblast activation. Importantly, at the molecular level, we revealed that Apilimod induces the accumulation of TGFβ-R2 in intracellular vesicles of late endosomal nature. Given the fact that TGFβ-R2 continuously recycles between plasma membrane and endosomes [30], it is tempting to postulate that PIKfyve could be involved in the control of this recycling process. We propose that PIKfyve inhibition dramatically alters endosomal sorting of the TGFβ-R2, therefore re-routing it from the recycling pathway to the degradative pathway. However, Apilimod did not induce TGFβ-R2 degradation, which can be explained by the defect in lysosomal acidification typically observed upon PIKfyve-inhibition by the less-potent inhibitor YM201636 [32-34]. Interestingly, it has been shown in podocytes that PIKfyve knock-out induces an arrest of trafficking of the recycling pathway, causing extensive accumulation of early, late and recycling endosomes, namely EEA1, Rab5 and Rab11 vesicles [35]. Consistently, it has been shown that PIKfyve inhibition by YM201636 blocks the recycling of the tight junction adhesion molecules Claudin-1 and Claudin-2 in epithelial cells [36], and that PIKfyve regulates the Rab11-dependent recycling of two potassium channels [37]. Alternatively, one might hypothesize that PIKfyve inhibition triggers TGFβ-R2 internalization from the plasma membrane. Although this cannot be ruled out, such an early function for PIKfyve would be unprecedented, as the enzyme is typically involved in late endosomal maturation. Moreover, whether PIKfyve inhibition alters the dimerization of the TGFβ receptors R1 and R2, their phosphorylated or ubiquitinated status, remain to be addressed in future work. Interestingly, it has been shown that PIKfyve inhibition by YM201636 or siRNA-mediated knock-down does not alter EGF receptor internalization [38, 39], which is consistent with our results. However, this has been challenged by several studies [12, 40, 41]. In any case, all these studies refer to ligand-induced receptor internalization. Here, however, we show that PIKfyve inhibition alters the steady-state localization of TGFβ-R2, independently from any ligand. Altogether, these findings suggest that PIKfyve functions in the control of the endosomal system are diverse and receptor-specific.
The lipid kinase PIKfyve produces the two signaling lipids PI(3,5)P2 and PI5P. Although the role of PI(3,5)P2 in the endosomal maturation has been extensively documented [14, 42-44], the role of endogenous PI5P is scarcely described [45, 46]. We previously demonstrated that tampering with PI5P by artificially increasing its levels altered the trafficking and steady-state localization of different cell-surface proteins, like the EGF receptor [22, 47] and the adhesion molecule ICAM1 [18]. This is reminiscent of what we observe here on TGFβ-R2, with the internalization of the receptor in the absence of its ligand, although the situation is quite different. Indeed, the pharmacological inhibition of PIKfyve would certainly decrease the levels of PI5P. This suggests that a tightly controlled balance in PIKfyve activity is needed to ensure a proper trafficking of these receptors through the endosomal system.
TGFβ receptors internalization is known to play either a positive or a negative role in downstream signaling [8]. Our results show that the vacuolar retention of TGFβ-R2 induced by PIKfyve inhibition is in favour of a negative regulation of the downstream signaling pathway. Indeed, we found that Apilimod induced the TGFβ-R2 retention in late endosomes, a non-signaling compartment as opposed to early/signaling endosomes. Moreover, we show that Apilimod reduces the phosphorylation of Smad2/3, crucial downstream effectors of the TGFβ signaling pathway. Finally, Apilimod reduces TGFβ response, ultimately leading to a reduction in the expression of TGFβ pro-fibrotic and pro-inflammatory target genes.
Although TGFβ is the main route for fibroblast activation, the contribution of other routes, like inflammatory factors, cannot be ruled out. Apilimod has been shown to possess strong anti-inflammatory properties [15, 48, 49]. Consistently, we found in our study that Apilimod reduces the production of several key pro-inflammatory cytokines and the infiltration of inflammatory macrophages within the myocardium. This is reminiscent of what we observed in a diabetic cardiomyopathy mouse model, although we found that Apilimod treatment failed to reduce systemic inflammation [17]. The specific contribution of both pathways (pro-fibrotic vs pro-inflammatory) is difficult to address, particularly in vivo. Although we clearly demonstrate here that PIKfyve plays a direct role on TGFβ response in cardiac fibroblasts, we cannot rule out an effect of Apilimod on other cardiac cell types. Indeed, we found previously [17] and as demonstrated in this study that Apilimod administration reduces cardiomyocyte hypertrophic response. Interestingly, we show here that Apilimod administration does not completely abrogate the hypertrophic response in TAC mice. Lastly, although we previously showed that Apilimod acts directly on cardiomyocytes, we cannot rule out the existence of a paracrine signaling from fibroblasts on myocytes, particularly important in vivo.
TGFβ triggers a canonical signaling pathway involved in many organs, regulating both physiological and pathological states [31]. In our study, although we observed a potent effect of Apilimod on the pathological heart, we found that long-term treatment does not affect the hepatic fibrotic or inflammatory status. This is consistent with the fact that Apilimod is well tolerated in human, as observed during the first phases of several clinical trials [50, 51]. Clinically, Apilimod possesses a real advantage as the oral formulation provides a clear improvement over injectable therapies. Although tested in two different clinical trials, against Crohn's disease and rheumatoid arthritis [50, 51], it failed to demonstrate an effect above placebo, suggesting that systemic administration is not potent to decrease the overall inflammation inherent to these pathologies. Moreover, TGFβ signaling dysregulation is often observed in several cancers, mostly linked to fibrosis development and cancer-associated fibroblasts [4]. Recently, Apilimod has been shown to possess anticancer activity against B-cell non-Hodgkin lymphoma [52] and is currently tested in a clinical trial, and different inhibitors of PIKfyve are promising anticancer drugs [53].
It has been shown that PIKfyve is required for the entry of several pathogen bacteria and viruses, including Ebola and Marburg viruses [54, 55] and Legionella pneumophila [56]. More recently, PIKfyve has been implicated in the entry of the SARS-CoV-2, responsible for the newly emerged pandemic zoonosis [57]. Of note, Apilimod was one of the first hit in a large-scale compound repurposing screen, demonstrating strong antiviral efficacy [58]. Although previous studies for PIKfyve function in these processes were centred on its role on phagocytosis and pathogen clearing [56], the recent work from Ou and colleagues suggests that PIKfyve could regulate the recycling of angiotensin convertase enzyme 2 (ACE2), the cell surface receptor of the virus [57]. While the authors did not provide direct evidence that PIKfyve controls ACE2 recycling, their data strongly suggest this is the case, which is reminiscent of what we observe in our study for the TGFβ receptor.
Conclusions
In conclusion, we demonstrate that Apilimod dampens cardiac fibrotic activation, and propose this compound as a regulator of TGFβ signaling. Therefore, PIKfyve pharmacological inhibition by Apilimod may provide new strategies to control fibrotic remodeling associated with chronic diseases.
Abbreviations
TGFβ, transforming growth factor beta; PIKfyve: phosphoinositide kinase, FYVE-type zinc finger containing; ECM: extracellular matrix; SMA: smooth muscular actin; IL: interleukin; ROS: reactive oxygen species; Smad: Mothers Against Decapentaplegic Homolog; TAC: transverse aortic constriction; CTGF: connective tissue growth factor; TNF-α: tumour necrosis factor-alpha; ANP: natriuretic peptide A; BNP: brain natriuretic peptide; β-MHC: β-myosin heavy chain; CCL2: C-C motif chemokine ligand 2; mRNA: messenger ribonucleic acid; DMSO: dimethyl sulfoxide; PBS: phosphate buffered saline; WGA: wheat germ agglutinin; HBSS: Hank's Balanced Salt Solution; BSA: bovine serum albumin; DMEM: Dulbecco's Modified Eagle Medium; FBS: fetal bovine serum; PFA: paraformaldehyde; RIPA buffer: Radioimmunoprecipitation assay; ECL: electrochemiluminescence; qRT-PCR: Quantitative Polymerase Chain Reaction; ANOVA: analysis of variance; MMP9: matrix metalloprotease 9; EGF: epidermal growth factor; ICAM1: intercellular adhesion molecule 1; MEF: mouse embryonic fibroblast; HEK: human embryonic kidney; EEA1: early endosome antigen 1; PI(3,5)P2: Phosphatidylinositol 3,5-bisphosphate; PI5P: Phosphatidylinositol 5-phosphate; ACE2: angiotensin I converting enzyme 2.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank the Rangueil imaging platform at the I2MC, Stéphane Bodin (CRBM-CNRS Montpellier, France) for kindly providing the flag-TGFβR2 plasmid, Denis Calise from the Anexplo UMS006 platform. We are grateful to Florence Tortosa (I2MC Toulouse, France) for the preparation of neonatal rat cardiac fibroblasts.
This work was funded by INSERM, Fondation Lefoulon-Delalande, Région Occitanie Midi-Pyrénées, Fédération Française de Cardiologie, and supported in part by NIH R01 NS099340 to LSW.
Authors' contributions
FB, JR, OK and HT conceived and designed the experiments and analyzed the data. FB, MC, AT, AS, ES, LG, HT and OK performed the experiments. SNZ, SSPG and LSW provided the MEF from WT and PIKfyveβ-geo/β-geo mice and the PIKfyve plasmid. TD and LOM designed and performed the experiments on mouse livers. FB and HT co-wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Weiskirchen R, Weiskirchen S, Tacke F. Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications. Mol Aspects Med. 2019;65:2-15
2. González A, Schelbert EB, Díez J, Butler J. Myocardial Interstitial Fibrosis in Heart Failure. J Am Coll Cardiol. 2018;71:1696-706
3. Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac Fibrosis. Circ Res. 2016;118:1021-40
4. Hu H-H, Chen D-Q, Wang Y-N, Feng Y-L, Cao G, Vaziri ND. et al. New insights into TGF-β/Smad signaling in tissue fibrosis. Chem Biol Interact. 2018;292:76-83
5. Vander Ark A, Cao J, Li X. TGF-β receptors: In and beyond TGF-β signaling. Cell Signal. 2018;52:112-20
6. Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL. Distinct endocytic pathways regulate TGF-β receptor signalling and turnover. Nat Cell Biol. 2003;5:410-21
7. He K, Yan X, Li N, Dang S, Xu L, Zhao B. et al. Internalization of the TGF-β type I receptor into caveolin-1 and EEA1 double-positive early endosomes. Cell Res. 2015;25:738-52
8. Chen Y-G. Endocytic regulation of TGF-β signaling. Cell Res. 2009;19:58-70
9. Shisheva A. PIKfyve: Partners, significance, debates and paradoxes. Cell Biol Int. 2008;32:591-604
10. McCartney AJ, Zhang Y, Weisman LS. Phosphatidylinositol 3,5-bisphosphate: Low abundance, high significance. BioEssays. 2014;36:52-64
11. Ikonomov OC, Sbrissa D, Shisheva A. Mammalian Cell Morphology and Endocytic Membrane Homeostasis Require Enzymatically Active Phosphoinositide 5-Kinase PIKfyve. J Biol Chem. 2001;276:26141-7
12. Jefferies HBJ, Cooke FT, Jat P, Boucheron C, Koizumi T, Hayakawa M. et al. A selective PIKfyve inhibitor blocks PtdIns(3,5)P 2 production and disrupts endomembrane transport and retroviral budding. EMBO Rep. 2008;9:164-70
13. Ikonomov OC, Sbrissa D, Delvecchio K, Xie Y, Jin J-P, Rappolee D. et al. The Phosphoinositide Kinase PIKfyve Is Vital in Early Embryonic Development. J Biol Chem. 2011;286:13404-13
14. Zolov SN, Bridges D, Zhang Y, Lee W-WW, Riehle E, Verma R. et al. In vivo, Pikfyve generates PI(3,5)P2, which serves as both a signaling lipid and the major precursor for PI5P. Proc Natl Acad Sci U S A. 2012;109:17472-7
15. Cai X, Xu Y, Cheung AK, Tomlinson RC, Alcázar-Román A, Murphy L. et al. PIKfyve, a class III PI kinase, is the target of the small molecular IL-12/IL-23 inhibitor apilimod and a player in Toll-like receptor signaling. Chem Biol. 2013;20:912-21
16. Wada Y, Lu R, Zhou D, Chu J, Przewloka T, Zhang S. et al. Selective abrogation of Th1 response by STA-5326, a potent IL-12/IL-23 inhibitor. Blood. 2007;109:1156-64
17. Tronchere H, Cinato M, Timotin A, Guitou L, Villedieu C, Thibault H. et al. Inhibition of PIKfyve prevents myocardial apoptosis and hypertrophy through activation of SIRT3 in obese mice. EMBO Mol Med. 2017;9:770-85
18. Boal F, Puhar A, Xuereb J-M, Kunduzova O, Sansonetti PJ, Payrastre B. et al. PI5P Triggers ICAM-1 Degradation in Shigella Infected Cells, Thus Dampening Immune Cell Recruitment. Cell Rep. 2016;14:750-9
19. McCartney AJ, Zolov SN, Kauffman EJ, Zhang Y, Strunk BS, Weisman LS. et al. Activity-dependent PI(3,5)P 2 synthesis controls AMPA receptor trafficking during synaptic depression. Proc Natl Acad Sci. 2014;111:E4896-905
20. Pchejetski D, Foussal C, Alfarano C, Lairez O, Calise D, Guilbeau-Frugier C. et al. Apelin prevents cardiac fibroblast activation and collagen production through inhibition of sphingosine kinase 1. Eur Hear J. 2012;33:2360-9
21. Boal F, Stephens DJJ. Specific functions of BIG1 and BIG2 in endomembrane organization. PLoS One. 2010;5:e9898
22. Ramel D, Lagarrigue F, Pons V, Mounier J, Dupuis-Coronas S, Chicanne G. et al. Shigella flexneri infection generates the lipid PI5P to alter endocytosis and prevent termination of EGFR signaling. Sci Signal. 2011;4:ra61
23. Oppelt A, Lobert VH, Haglund K, MacKey AM, Rameh LE, Liestol K. et al. Production of phosphatidylinositol 5-phosphate via PIKfyve and MTMR3 regulates cell migration. EMBO Rep. 2013;14:57-64
24. Laurent V, Guérard A, Mazerolles C, Le Gonidec S, Toulet A, Nieto L. et al. Periprostatic adipocytes act as a driving force for prostate cancer progression in obesity. Nat Commun. 2016;7:10230
25. Villarreal FJ, Kim NN, Ungab GD, Printz MP, Dillmann WH. Identification of functional angiotensin II receptors on rat cardiac fibroblasts. Circulation. 1993;88:2849-61
26. Pincha N, Saha D, Bhatt T, Zirmire R, Jamora C. Activation of Fibroblast Contractility via Cell-Cell Interactions and Soluble Signals. BIO-PROTOCOL. 2018 8
27. Alfarano C, Foussal C, Lairez O, Calise D, Attané C, Anesia R. et al. Transition from metabolic adaptation to maladaptation of the heart in obesity: role of apelin. Int J Obes. 2015;39:312-20
28. Curtis MJ, Alexander S, Cirino G, Docherty JR, George CH, Giembycz MA. et al. Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. Vol. 175, British Journal of Pharmacology. John Wiley and Sons Inc. 2018
29. Ivey MJ, Tallquist MD. Defining the cardiac fibroblast. Vol. 80, Circulation Journal. 2016
30. Massagué J. How cells read TGF-β signals. Nat Rev Mol Cell Biol. 2000;1:169-78
31. Rockey DC, Bell PD, Hill JA. Fibrosis — A Common Pathway to Organ Injury and Failure. Longo DL, Ed. N Engl J Med. 2015;372:1138-49
32. Odorizzi G, Babst M, Emr SD. Fab1p PtdIns(3)P 5-Kinase Function Essential for Protein Sorting in the Multivesicular Body. Cell. 1998;95:847-58
33. Rusten TE, Rodahl LMW, Pattni K, Englund C, Samakovlis C, Dove S. et al. Fab1 phosphatidylinositol 3-phosphate 5-kinase controls trafficking but not silencing of endocytosed receptors. Gruenberg J, Ed. Mol Biol Cell. 2006;17:3989-4001
34. Bissig C, Hurbain I, Raposo G, van Niel G. PIKfyve activity regulates reformation of terminal storage lysosomes from endolysosomes. Traffic. 2017;18:747-57
35. Venkatareddy M, Verma R, Kalinowski A, Patel SR, Shisheva A, Garg P. Distinct Requirements for Vacuolar Protein Sorting 34 Downstream Effector Phosphatidylinositol 3-Phosphate 5-Kinase in Podocytes Versus Proximal Tubular Cells. J Am Soc Nephrol. 2016;27:2702-19
36. Dukes JD, Whitley P, Chalmers AD. The pikfyve inhibitor YM201636 blocks the continuous recycling of the tight junction proteins claudin-1 and claudin-2 in MDCK cells. PLoS One. 2012;7:1-10
37. Seebohm G, Strutz-Seebohm N, Birkin R, Dell G, Bucci C, Spinosa MR. et al. Regulation of Endocytic Recycling of KCNQ1/KCNE1 Potassium Channels. Circ Res. 2007;100:686-92
38. Ikonomov OC, Sbrissa D, Mlak K, Deeb R, Fligger J, Soans A. et al. Active PIKfyve Associates with and Promotes the Membrane Attachment of the Late Endosome-to-trans-Golgi Network Transport Factor Rab9 Effector p40. J Biol Chem. 2003;278:50863-71
39. Rutherford AC, Traer C, Wassmer T, Pattni K, Bujny M V, Carlton JG. et al. The mammalian phosphatidylinositol 3-phosphate 5-kinase (PIKfyve) regulates endosome-to-TGN retrograde transport. J Cell Sci. 2006;119:3944-57
40. de Lartigue J, Polson H, Feldman M, Shokat K, Tooze SA, Urbe S. et al. PIKfyve regulation of endosome-linked pathways. Traffic. 2009;10:883-93
41. Er EE, Mendoza MC, Mackey AM, Rameh LE, Blenis J. AKT facilitates EGFR trafficking and degradation by phosphorylating and activating PIKfyve. Sci Signal. 2013;6:ra45
42. Dove SK, Cooke FT, Douglas MR, Sayers LG, Parker PJ, Michell RH. Osmotic stress activates phosphatidylinositol-3,5-bisphosphate synthesis. Nature. 1997;390:187-92
43. Whiteford CC, Brearley CA, Ulug ET. Phosphatidylinositol 3,5-bisphosphate defines a novel PI 3-kinase pathway in resting mouse fibroblasts. Biochem J. 1997;323:597-601
44. Ikonomov OC, Sbrissa D, Shisheva A. Localized PtdIns 3,5-P 2 synthesis to regulate early endosome dynamics and fusion. Am J Physiol Physiol. 2006;291:C393-404
45. Viaud J, Boal F, Tronchère H, Gaits-Iacovoni F, Payrastre B. Phosphatidylinositol 5-phosphate: A nuclear stress lipid and a tuner of membranes and cytoskeleton dynamics. BioEssays. 2014;36:260-72
46. Hasegawa J, Strunk BS, Weisman LS. PI5P and PI(3,5)P2: Minor, but Essential Phosphoinositides. Cell Struct Funct. 2017;42:49-60
47. Boal F, Mansour R, Gayral M, Saland E, Chicanne G, Xuereb J-MM. et al. TOM1 is a PI5P effector involved in the regulation of endosomal maturation. J Cell Sci. 2015;128:815-27
48. Billich A. Drug evaluation: apilimod, an oral IL-12/IL-23 inhibitor for the treatment of autoimmune diseases and common variable immunodeficiency. IDrugs. 2007;10:53-9
49. Wada Y, Cardinale I, Khatcherian A, Chu J, Kantor AB, Gottlieb AB. et al. Apilimod inhibits the production of IL-12 and IL-23 and reduces dendritic cell infiltration in psoriasis. PLoS One. 2012;7:e35069
50. Sands BE, Jacobson EW, Sylwestrowicz T, Younes Z, Dryden G, Fedorak R. et al. Randomized, double-blind, placebo-controlled trial of the oral interleukin-12/23 inhibitor apilimod mesylate for treatment of active Crohn's disease. Inflamm Bowel Dis. 2010;16:1209-18
51. Burakoff R, Barish CF, Riff D, Pruitt R, Chey WY, Farraye FA. et al. A phase 1/2A trial of STA 5326, an oral interleukin-12/23 inhibitor, in patients with active moderate to severe Crohn's disease. Inflamm Bowel Dis. 2006;12:558-65
52. Gayle S, Landrette S, Beeharry N, Conrad C, Hernandez M, Beckett P. et al. Identification of apilimod as a first-in-class PIKfyve kinase inhibitor for treatment of B-cell non-Hodgkin lymphoma. Blood. 2017;129:1768-78
53. Ikonomov OC, Sbrissa D, Shisheva A. Small molecule PIKfyve inhibitors as cancer therapeutics: Translational promises and limitations. Toxicol Appl Pharmacol. 2019 114771
54. Nelson EA, Dyall J, Hoenen T, Barnes AB, Zhou H, Liang JY. et al. The phosphatidylinositol-3-phosphate 5-kinase inhibitor apilimod blocks filoviral entry and infection. Kashanchi F, Ed. PLoS Negl Trop Dis. 2017;11:e0005540
55. Qiu S, Leung A, Bo Y, Kozak RA, Anand SP, Warkentin C. et al. Ebola virus requires phosphatidylinositol (3,5) bisphosphate production for efficient viral entry. Virology. 2018;513:17-28
56. Buckley CM, Heath VL, Guého A, Bosmani C, Knobloch P, Sikakana P. et al. PIKfyve/Fab1 is required for efficient V-ATPase and hydrolase delivery to phagosomes, phagosomal killing, and restriction of Legionella infection. Luo Z-Q, Ed. PLOS Pathog. 2019;15:e1007551
57. Ou X, Liu Y, Lei X, Li P, Mi D, Ren L. et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun. 2020;11:1620
58. Riva L, Yuan S, Yin X, Martin-Sancho L, Matsunaga N, Pache L. et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature. 2020;586:113-9
Author contact
![]() Corresponding author: frederic.boalfr; INSERM U1297 I2MC, Toulouse, France and Université Paul Sabatier, Toulouse, France. +33(0)5 31 22 41 17.
Corresponding author: frederic.boalfr; INSERM U1297 I2MC, Toulouse, France and Université Paul Sabatier, Toulouse, France. +33(0)5 31 22 41 17.