Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Antigen release and presentation
T cells priming and activation
CTL trafficking and infiltration...
CTL recognition and killing of...
Combination nano-immunotherapy
Conclusions, challenges, and...
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(15):7471-7487. doi:10.7150/thno.59953 This issue Cite
Review
Nano-immunotherapy for each stage of cancer cellular immunity: which, why, and what?
Shiyi Zuo, Jiaxuan Song, Jingxuan Zhang, Zhonggui He, Bingjun Sun ![]() , Jin Sun
, Jin Sun ![]()
Department of Pharmaceutics, Wuya College of Innovation, Shenyang Pharmaceutical University, Shenyang 110016, China.
Received 2021-3-1; Accepted 2021-5-6; Published 2021-6-1
Abstract

Immunotherapy provides a new avenue for combating cancer. Current research in anticancer immunotherapy is primary based on T cell-mediated cellular immunity, which can be divided into seven steps and is named the cancer-immunity cycle. Unfortunately, clinical applications of cancer immunotherapies are restricted by inefficient drug delivery, low response rates, and unmanageable adverse reactions. In response to these challenges, the combination of nanotechnology and immunotherapy (nano-immunotherapy) has been extensively studied in recent years. Rational design of advanced nano-immunotherapies requires in-depth consideration of “which” immune step is targeted, “why” it needs to be further enhanced, and “what” nanotechnology can do for immunotherapy. However, the applications and effects of nanotechnology in the cancer-immunity cycle have not been well reviewed. Herein, we summarize the current developments in nano-immunotherapy for each stage of cancer cellular immunity, with special attention on the which, why and what. Furthermore, we summarize the advantages of nanotechnology for combination immunotherapy in two categories: enhanced efficacy and reduced toxicity. Finally, we discuss the challenges of nano-immunotherapy in detail and provide a perspective.
Keywords: Cancer immunotherapy, nanotechnology, cellular immunity, immunity cycle, combination therapy.
Introduction
Cancer is one of the most severe diseases threatening human health. Chemotherapy, surgery, and radiotherapy are the three standard clinical treatments for cancer. These conventional treatments can extend patient survival, but they are constantly challenged by intractable problems, including severe adverse reactions, inevitable tumor recurrence, and resistance [1]. In recent years, cancer immunotherapy has developed as the fourth treatment modality. Cancer immunotherapy evokes or boosts the inherent host immune system and then enhances antitumor immune responses, providing a new avenue to combat cancer [2].
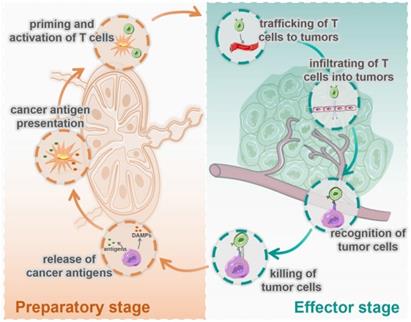
At present, most cancer immunotherapies are based on T cell-mediated cellular immunity [3, 4], which has been defined as the famous cancer-immunity cycle by Chen and Mellman [5]. As shown in Figure 1, this cycle includes seven steps: 1. release of cancer cell antigens; 2. cancer antigen presentation; 3. priming and activation of T cell; 4. trafficking of T cells to tumors; 5. infiltration of T cells into tumors; 6. recognition of cancer cells by T cells; and 7. killing of cancer cells. These seven steps can be divided into two stages: the preparatory stage (T cells responding) consisting of steps1 to 3, which mainly take place in the lymph nodes (LNs), and the effector stage (T cells killing) consisting of steps 4 to 7, which mainly take place in the tumor microenvironment (TME). In most tumors, the cancer-immunity cycle is blocked at one or more of these steps, resulting in restrained anticancer immune responses and tumor immune escape.
Seven immune actions in the cycle of cancer cellular immunity are divided into two stages, the preparatory stage and the effector stage. The preparatory stage (the left side) includes the release of cancer antigens (step 1), cancer antigen presentation (step 2), priming and activation of T cells (step 3), which mainly take place in the lymph nodes. The effector stage (the right side) includes trafficking of T cells into tumors (step 4), infiltration of T cells into tumors (step 5), recognition of cancer cells (step 6), and killing of cancer cells (step 7), which mainly happen in TME.
Despite huge breakthroughs, immunotherapy is still limited by unsatisfactory response rates, efficacy and safety [6, 7]. First of all, many immunotherapy agents suffer from low solubility, poor stability, and short half-lives [8]. Secondly, some immunotherapies can cause severe or even fatal allergy- and inflammation-related reactions [9]. These reactions happen when the immune systems not only fight cancer but also attack healthy cells and tissues in the body [10]. It is also challenging to deliver immune cells or agents into tumors through an immunosuppressive tumor microenvironment (iTME) [11]. Moreover, the low immunogenicity of tumor cells and the accumulation of immunosuppressive cells and cytokines in the iTME together limit the effects of immunotherapies [12]. Therefore, the rational design of advanced antitumor immunotherapies is still a great challenge.
Nano-drug delivery systems have been widely used in the field of anticancer therapy [13]. Nanoparticles (NPs) can increase the stability of drugs and protect them from being metabolized during blood circulation, thus enabling reduction of the administered dose and avoidence of high dose-related toxicities. Moreover, NPs can increase the accumulation of therapeutic agents in tumor tissue and LNs, leading to enhanced therapeutic effects and reduced side effects [14]. It is well known that tumors and LNs are the two main targets of immunotherapy [15, 16]. NPs can passively transport into tumor tissue through immature tumor vasculature and accumulate due to damaged lymphatic drainage, a phenomenon known as the enhanced permeability and retention (EPR) effect [17]. Also, NPs can actively target tumor cells after surface ligand modification [18]. Similarly, NPs can accumulate in LNs and deliver cancer vaccines to antigen-presenting cells (APCs) to activate an immune response [19-23]. What is more, NPs have distinct advantages for combination drug delivery, which can synergize multiple immunotherapy mechanisms to enhance the overall immune response [24-28]. Based on the above advantages, nano-immunotherapy has become a hot topic in recent years.
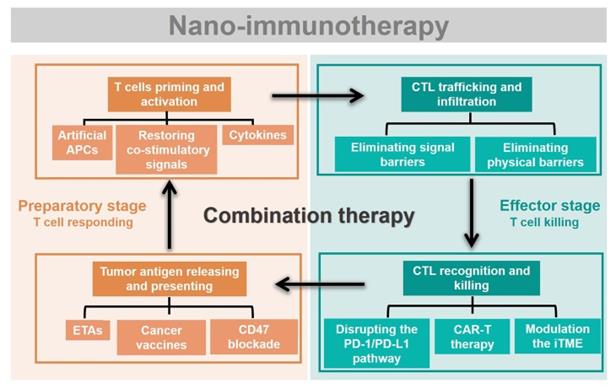
Rational design of advanced nano-immunotherapies requires in-depth consideration of “which” immune step is targeted, “why” it needs to be further enhanced, and “what” nanotechnology can do for immunotherapy. However, the applications and effects of nanotechnology in each stage of cancer immunity have not been systematically summarized. Herein, we streamline the seven steps of the cancer-immunity cycle into four parts according to their correlations (Figure 2): tumor antigen (TA) release and presentation (steps 1 and 2); T-cell priming and activation (step 3); cytotoxic T lymphocyte (CTL) trafficking and infiltration into the tumor site (steps 4 and 5); and CTL recognition and killing of tumor cells (steps 6 and 7). For each of these four parts, we summarize the current developments in nano-immunotherapy. In this way, which immune step is associated with a specific nano-immunotherapy is clear. In addition, the shortcomings of each immunotherapy are summarized based on preclinical and clinical research reports to explain why it needs to be further enhanced. The benefits and improvements that nanotechnology can provide to each immunotherapy are then presented. Moreover, recent preclinical and clinical studies have explored combinations of immunotherapies that target different pathways, which could be improved by nanotechnology [29]. Thus, we summarize the advantages of nanotechnology for combination immunotherapy. Finally, we discuss the challenges of nano-immunotherapy and provide our perspective on the future of the field.
Immunotherapies combing with nanotechnology. The seven steps of the cancer-immunity cycle are simplified into four parts. To present which immune step is associated, the current developments of nano-immunotherapy are divided into these four parts. Nano-immunotherapies acting on preparatory stage include tumor antigen releasing and presenting (by ETAs, cancer vaccines, CD47 blockade therapy), and T cells priming and activation (by artificial APCs, restoring co-stimulatory signals, cytokines therapy). The functions of nano-immunotherapies acting on the effector stage are CTL trafficking and infiltration (through eliminating physical barriers and signal barriers), and CTL recognition and killing (through disrupting the PD-1/PD-L1 pathway, CAR-T therapy and modulation the iTME). Besides, combination nano-immunotherapy consists of immunotherapies with different effects, making up for the deficiency of monotherapy in the immunity cycle.
Antigen release and presentation
Cellular immunity starts with the release and exposure of TAs, which are then captured by APCs such as dendritic cells (DCs) [30]. After migrating to draining LNs, DCs mature and subsequently present the antigens to naive T cells via major histocompatibility complex (MHC) I and II molecules [31]. Release and presentation of TAs are the preconditions of cellular immune response. However, many tumors have poor immunogenicity due to down-regulation of antigen expression, antigen loss, and antigen modulation [32, 33]. Also, antigen presentation to T cells by dysfunctional DCs induces antigen-specific immunotolerance. These traps impede initiation of T cell-mediated immunity. Plenty of immunotherapies have been developed to improve the release and presentation of TAs, including induction of endogenous tumor antigens (ETAs) [34, 35], cancer vaccines, [36] and blockade of the CD47 immune checkpoint [37].
Inducing endogenous tumor antigens
ETAs are able to arouse specific immune responses for personalized immunotherapy [38]. Treatments such as photodynamic therapy (PDT), photothermal therapy (PTT), certain chemotherapies, and radiotherapy have been reported to induce endoplasmic reticulum (ER) stress and immunogenic or necrotic tumor cell death, leading to release of ETAs from tumor cell residues [39]. However, the rapid immune clearance and inefficient delivery of these autologous antigens restrict stimulation of an antitumor immune response [40]. Rational design of nanocarriers can protect antigens from clearance and target their delivery to LNs. For example, Qian et al. developed mesoporous silica nanoparticles (MSNs) incorporating NP debris combined in situ with ETAs from tumor cells killed by PTT. Then, the nano-debris carried the antigens out of the necrotic tissue and selectively entered the immune organs for immunotherapy [41]. Similarly, Wang et al. found that ~10 nm iron oxide (Fe3O4) can transport ETAs from tumor to LNs because of their notable protein capture efficiency and LN-targeting ability. To ensure efficient combination of Fe3O4 with ETAs, a core-shell nanostructure (denoted as Ce6/Fe3O4) was developed to protect the Fe3O4 core from interaction with undesired proteins. Upon laser irradiation, reactive oxygen species (ROS) generated by the outer chlorin e6 (Ce6) photosensitizer triggered release of ETAs from cancer cells. Fe3O4 then captured the released ETAs and transported them to LNs via lymphatic drainage [34].
Induction of ER stress often leads to immunogenic cell death (ICD) [42, 43]. ICD can enhance adjuvanticity and antigenicity from dying cancer cells by releasing damage-associated molecular patterns (DAMPs), including calreticulin (CRT), high mobility group box 1 (HMGB1), and adenosine triphosphate (ATP) [44]. These DAMPs facilitate recruitment and maturation of DCs, thus improving antigen presentation. PDT is a common strategy to induce ICD of tumor cells. However, ICD induced by conventional PDT is reported to be less immunogenic than necrosis due to the slow release of DAMPs. Moreover, low PDT efficacy limits the release of ETAs. Zhang's group designed a tumor cell plasma membrane (PM)-targeted chimeric peptide, PpIX-C6-PEG8-KKKKKKSKTKC-OMe (PCPK), containing the photosensitizer protoporphyrin IX (PpIX) to achieve specific PM damage for enhanced photodynamic immunotherapy. In tumor cells, PCPK tightly anchored to the inner PM after protein farnesyltransferase (PFTase)-mediated enzymatic conversion. Under laser irradiation, PCPK generated cytotoxic ROS, which destroyed the structure of the PM. In addition, the selective rupture of the PM induced rapid release of ETAs and DAMPs, leading to significantly enhanced antitumor immune response compared to conventional PDT [45].
In summary, nanotechnology-based treatments inducing antigen release have been shown to enhance antigen production, retention and LN targeting.
Cancer vaccines
Cancer vaccines composed of TAs and adjuvants are widely used to induce an immune response [46, 47]. The major challenges of cancer vaccines include drainage of antigens to LNs, internalization of antigen by DCs, and cross-presentation of antigens by DCs for T-cell activation [36, 48, 49]. It has been found that NPs of a suitable size can accumulate in tumor-draining lymph nodes (TDLNs) following tumor accumulation by the EPR effect [19, 50]. Also, a negative surface charge can facilitate lymphatic uptake and retention of NPs through electrostatic interactions [51]. For example, MSNs have been extensively studied as antigen carriers because of their ultra-high surface area, easy surface modification, and adjustable particle size [52]. In addition, the surface of MSNs is negatively charged and contains hydrophilic silanol groups (Si-OH), making MSNs a potential LN-targeted carrier [53, 54]. Hong et al. constructed three types of MSNs with similar particle sizes (~80 nm) but different pore sizes and evaluated their ability to deliver a model antigen (ovalbumin, OVA) to TDLNs. The authors found that all three OVA@MSNs accumulated in TDLNs and were then internalized by LN-resident DCs to a similar degree, probably because their similar particle sizes. Interestingly, the MSNs with the largest pore size (MSNs-L) improved the cross-presentation of OVA. In addition, MSNs-L were degraded faster in TDLNs than the other MSNs, which may have facilitated OVA release and exposure. Rapid degradation of MSNs-L also helped to improve their safety in vivo. As a result, MSNs-L significantly enhanced the delivery efficiency of OVA and induced more robust immune responses than the MSNs with smaller pores [55].
Antigen presentation to T cells by dysfunctional DCs induces antigen-specific immunotolerance. Adjuvants, including particulate adjuvants (such as alum and emulsions) and pattern recognition receptors (PRRs) agonists, are applied to cancer vaccines to improve maturation of DCs [56-59]. Among them, PRRs agonists, the new-generation adjuvants, have been used to activate myeloid cells (macrophages and DCs) and induce antitumor immune responses [60-62]. However, application of PRRs agonists is restricted by serious adverse effects, which are mainly caused by their unfavorable pharmacokinetic profiles and biodistribution [63, 64]. Ni et al. reported a bi-adjuvant neoantigen nanovaccine (banNV) that co-delivered a peptide neoantigen (Adpgk) with the Toll-like receptor (TLR) 7/8 agonist R848 and the TLR9 agonist CpG. Through efficient co-delivery of the neoantigen and the dual synergistic adjuvants, the immunogenicity of the neoantigen was potentiated, and acute systemic toxicity was reduced [65].
To improve the targeting of cancer vaccines to APCs, NPs modified with specific receptors for APCs have been applied as carriers of cancer vaccine. For example, Affandi et al. described a nanovaccine targeting CD169/SIGLEC1+ DCs to drive antitumor T-cell responses. The authors used the natural ligands of CD169, gangliosides, as targeting ligands and constructed liposomal vaccine carriers to deliver TAs. Through targeted co-delivery of TA and TLR ligand to CD169+ monocyte-derived DCs and AXL+CD169+ DCs, the ganglioside-liposomes induce robust cross-presentation and activation of TA-specific CD8+ T cells [66]. Rajpu et al. engineered polymeric nanovaccines using inulin acetate (InAc) as the polymer material. DCs recognized InAc-NPs through TLR4-InAc interactions, which led to efficient vaccine uptake, antigen presentation, and TLR4-based signaling, improving DC activation and maturation [67]. Yang et al. constructed a nanovaccine (NP-R@M-M) by coating R837-loaded poly(lactic-co-glycolic acid) (PLGA) NPs with mannose-modified B16-OVA membranes. Owing to their strong interaction with DCs (mannose receptor positive), NP-R@M-M showed enhanced uptake by DCs and stimulation of DC maturation compared with untargeted NPs. Although NP-R@M-M exhibited strong antitumor efficacy in a B16-OVA melanoma tumor model, it did not show any antitumor efficacy in a 4T1 mouse breast tumor model, evidencing the specificity of the antitumor immunity induced by NP-R@M-M. Therefore, NP-R@M-M demonstrated a strong stimulation effect for triggering antitumor immune responses with good specificity and safety [21].
mRNA vaccines represent a promising alternative to conventional vaccines [68]. Instead of directly delivering pathogen proteins, mRNA vaccines trigger endogenous production of immunogenic proteins, thus inducing a strong MHC-I-mediated CD8+ T-cell response and reducing adverse reactions [69]. However, the delivery of mRNA vaccines to APCs is challenged by the following issues: (a) degradation by omnipresent endonucleases, (b) difficulty in accessing APCs, and (c) endosomal entrapment and degradation. Recently, ionizable lipid nanoparticles (LNPs) have emerged as promising platforms for efficient mRNA delivery [2, 70-72]. Oberli et al. developed LNPs encapsulating mRNA coding OVA. As lipids are the basic units of biomembranes, the LNPs had a good affinity with PM and facilitated cellular uptake of mRNA. Within acidic intracellular environments (such as lysosomes), the ionizable lipid core was positively charged, thereby integrating with the negatively charged mRNA and then protecting it from degradation. In addition, the positively charged LNPs facilitated escape of the mRNA from lysosomes via the proton sponge effect. Treatment of a B16-F10 melanoma tumor model with the LNPs resulted in tumor shrinkage and extended overall survival [73].
In brief, nanotechnology can improve the efficacy of cancer vaccines by protecting the antigen/adjuvant from clearance in the biological environment, promoting delivery of the antigen/adjuvant to LNs and APCs, and minimizing systemic toxicity.
CD47 blockade
CD47 transmits an inhibitory “don't eat me” signal upon ligation with its receptor signal regulatory protein alpha (SIRPα), which is mainly expressed on phagocytic cells, including macrophages and DCs [74]. Recent studies have demonstrated that CD47 blockade not only increases phagocytosis of cancer cells by macrophages but also promotes cross-presentation of TAs [75]. Blockade of the immune checkpoint with antibodies has been extensively evaluated in the clinical setting. Nanobodies have been developed to improve the therapeutic efficacy of conventional antibodies, with distinct advantages such as high stability, high affinity and specificity, and deep tissue penetration [10, 76]. Chowdhury et al. engineered a non-pathogenic Escherichia coli strain that specifically lysed within the TME and released an encoded nanobody antagonist of CD47 (CD47nb). Compared with the commonly used anti-mouse CD47 monoclonal antibody, the produced CD47nb had a higher binding affinity to CD47 on the surface of A20 lymphoma cells. Tumors treated with CD47nb showed increased proliferation of both FOXP3-CD4+ and CD8+ T cells. These results suggested that compared to conventional systemic monoclonal antibody therapy, localized release of the CD47nb increased the activation of tumor-infiltrating T cells and simultaneously prevented systemic toxicity [75].
T cells priming and activation
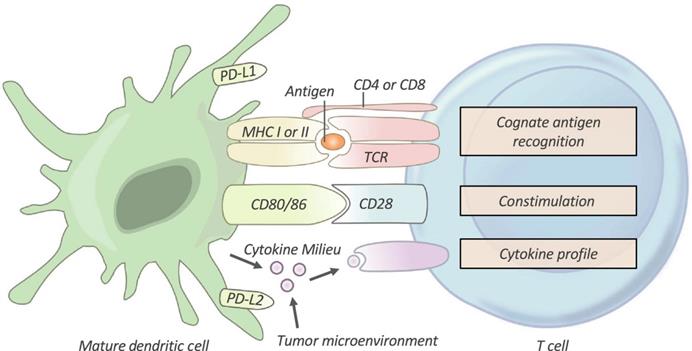
After being taken up by DCs, TAs are processed to peptide antigen-loaded major histocompatibility complex (pMHC). Immature T cells are then primed and activated to become effector T cells [5, 13]. As shown in Figure 3, priming and activation of T cells requires three signals from APCs: antigen recognition between T cells and APCs (signal 1), a co‐stimulatory signal (signal 2), and a cytokine signal (signal 3). However, in most cases, the T cells of cancer patients are activated without the combination of these signals. Therefore, the T cells usually become nonresponsive to further stimulation and stay in a state of anergy. Artificial APCs (aAPCs), restoration of co-stimulatory signals, and cytokine therapies, have been utilized to facilitate activation of T cells [4, 60, 77].
T-cell Activation. T cells require three signals from an antigen‐presenting cell (APC) in order to be effectively activated and primed. First, T cells must recognize their cognate antigen in the context of the correct MHC. Second, T cells require appropriate co-stimulation by an APC. Finally, the T cell receives instructive cytokines from immune cells and the tumor microenvironment which dictate its phenotypic differentiation. TCR, T-cell receptor; PD-L1/PD-L2, programmed death-ligands 1 and 2. Adapted with permission from [4], Copyright 2019 Clinical & Translational Immunology.
Artificial APCs
The inefficient presentation of TAs by immature APCs leads to deficiency of signal 1 and further restricts activation of CTLs [78]. aAPCs are microparticle- or NP-based biomimetic systems that contain pMHC complexes and positive co-stimulatory molecules on their surface. aAPCs can directly stimulate antigen-specific CTLs, bypassing the need for endogenous APCs [79, 80]. Hickey et al. demonstrated that aAPCs larger than 300 nm were more effective at activating CD8+ T cells than smaller aAPCs, presumably due to their ability to initiate clustering of pMHC-T cell receptor (TCR) complexes and costimulatory interactions [81]. Green's group showed that non-spherical aAPCs induced more robust antigen-specific T-cell responses than spherical aAPCs [80]. In addition, Majedi et al. combined mechanical forces with aAPCs and confirmed that exogenous mechanical forces increased the antigenic signal strength to T cells [82]. Magnetic aAPCs with an externally applied magnetic field can further improve T-cell activation. Zhang et al. developed biomimetic magnetosomes as versatile aAPCs. The magnetic nanoclusters were first coated with azide-engineered leucocyte membranes, then loaded with pMHC-I and the co-stimulatory ligand anti-CD28 for T-cell stimulation. These nano aAPCs exhibited a high performance for antigen-specific CTL expansion and stimulation and showed a good affinity for CTLs. Additionally, through magnetic resonance imaging and magnetic control, these biomimetic aAPCs efficiently guided reinfused CTLs to tumor tissues [83].
In summary, nanotechnology improved the LN targeting and safety of aAPCs. However, nanoscale aAPCs have a decreased surface area for contact with T cells compared with microscale aAPCs, which would affect their T-cell activation efficacy.
Restoring co-stimulatory signals
Interaction between the co-stimulatory molecules CD28 (on T cells) and B7 (on APCs) provides a co-stimulatory signal (signal 2) that is required for T-cell activation. Cytotoxic T lymphocyte antigen 4 (CTLA-4) small interfering RNA (siCTLA-4) utilizes endogenous RNA interference mechanisms to silence CTLA-4 expression, thereby promoting T-cell activation. However, the efficiency of siRNA is restricted by its large molecular weight, rapid degradation by plasmatic nucleases, and extracellular barriers. LNPs are capable of dealing with these problems. With their capacity to condense and protect siRNA and their good cellular internalization efficiency, LNPs have been developed as the gold standard gene vector for delivery of siRNA. Li et al. prepared cationic lipid-assisted poly(ethylene glycol)-polylactide (PEG-PLA) NPs to encapsulate siCTLA-4 (NPsiCTLA-4). The NPs effectively protected the siRNA from degradation and efficiently delivered it to T cells, consequently downregulating the expression of CTLA-4 on the T cell surface. Systemic delivery of NPsiCTLA-4 increased the number of CD4+ and CD8+ T cells and decreased the population of CD4+FOXP3+ regulatory T cells. Moreover, NPsiCTLA-4 effectively inhibited tumor growth and prolonged the survival of mice with melanoma tumors [84].
Programmed cell death protein -1 (PD-1) and its ligand (PD-L1) are also involved in T-cell activation through the CD28 pathway. Upon activation by PD-L1 overexpressed on DCs, PD-1 on T cells suppresses T-cell activation by inhibiting CD28 signaling [85]. Hobo et al. constructed siRNA-LNPs that mediated knockdown of PD-L1 on DCs. The engineered DCs boosted the expansion of both CD4+ T helper cell responses and CD8+ effector-memory T cells [86]. Hassannia et al. reported siRNA‐loaded chitosan-dextran sulfate NPs that silenced the expression of PD‐L1 on DCs and PD‐1 on T cells. The synthesized NPs demonstrated efficient cellular uptake and target gene silencing. Presentation of TAs by PD-L1-negative DCs to PD-1-silenced T cells induced potent T-cell responses [87].
Cytokines
The signal 3 cytokines, such as interleukin (IL) 12, IL‐1, and interferon (IFN) α/β, also play a crucial role in T-cell activation [88-91]. Due to their poor stability, cytokines must be administrated in large doses which often causes severe toxicities [8, 91]. To address this problem, Wang et al. conjugated IFNα to a class of biocompatible, biodegradable, and thermosensitive biopolymers called elastin-like polypeptides (ELPs). The obtained IFNα-ELPdiblock conjugates self-assembled into nanomicelles with IFNα tightly wrapped in the corona. Due to steric hindrance, IFNα was protected from degradation by proteases. In addition, the blood circulation of IFNα in the micelles was prolonged compared to free IFNα because of its improved proteolytic stability and increased size, effectively reduced renal clearance [92]. Tang et al. developed a TCR-signaling-responsive NP that controlled cytokine delivery in response to T-cell activation. The authors used human interleukin-15 super-agonist (IL-15SA) as a model drug cargo. The safe dose of IL-15SA was increased 8 times compared to free cytokine through the regulated drug release, increasing the therapeutic window for adjuvant cytokine therapy [93].
Activation of the stimulator of interferon genes (STING) pathway within tumor-resident DCs can induce the production of type I IFNs and adaptive immune responses against tumors [94, 95]. Current STING agonists, such as cyclic dimeric guanosine monophosphate (cdGMP), amidobenzimidazole, and 5,6-dimethylxanthenone-4-acetic acid (DMXAA), are limited by inefficient TDLN-targeting and uncontrollable systemic inflammation. Systemic administration of these immunotherapeutic agents can influence the function of immune cells at nontarget sites, breaking immune homeostasis and causing undesirable adverse events. As mentioned above, nanotechnology could reduce off-target distribution and so decrease systemic side effects. Hanson et al. encapsulated cdGMP within PEGylated LNPs (NP-cdGMP), which effectively improved the accumulation of cdGMP in TDLNs. Compared with free cdGMP, NP-cdGMP demonstrated increased CD8+ T-cell responses and enhanced therapeutic antitumor immunity [96].
In summary, nanotechnology greatly improves the efficacy and safety of cytokine therapies by protecting these vulnerable agents, reducing their non-specific biodistribution, and endowing them with specific LN-targeting ability.
CTL trafficking and infiltration into the tumor site
Once activated, effector T cells traffic through the bloodstream and infiltrate into the tumor site. This process is usually restricted by two types of obstacles. First, “signal barriers”, such as over-expression of immunosuppressive signals, or lack of recruiting cytokines, restrict the homing of CD8+ T cells to the tumor [60]. In addition, unlike hematological malignancies, solid tumors often build up “physical barriers”, such as deficient tumor vasculature, abundant cancer-associated fibroblasts (CAFs), and dense extracellular matrix (ECM) that restrain infiltrating CTLs [97, 98]. Given these obstacles, strategies aimed at eliminating signal barriers and physical barriers have been developed and are often used as adjuvant therapies to enhance checkpoint blockade immunotherapy.
Eliminating signal barriers
Chemokines are one of the major factors governing the homing of immune cells to tumors. For example, intratumoral infiltration of T cells can be inhibited by C-X-C motif chemokine ligand 12 (CXCL12) [99, 100]. Nanotechnology-based gene therapy has been widely used to modulate the expression of chemokines with the distinct advantages mentioned above. For example, Goodwin et al. developed a lipid calcium phosphate (LCP) NP to deliver plasmid DNA (pDNA) encoding an engineered CXCL12 protein trap (pCXCL12-trap). Liver-specific delivery and transient expression of pCXCL12-trap via the LCP nonviral vector directed the liver to resist infiltrating CXCR4+ metastatic cells. This strategy further inhibited the establishment of an iTME, allowing for enhanced cancer-specific CD8+ T-cell killing [101]. IL-10 is also considered an immunosuppressive cytokine due to its association with suppressive and regulatory cells, including tolerogenic DCs, regulatory CD4+T cells, M2 macrophages, and myeloid-derived suppressor cells (MDSCs). Huang's group utilized liposome-protamine-DNA (LPD) NPs to encapsulate plasmids encoding small trap proteins that target IL-10 and CXCL12. The IL-10 trap in combination with the CXCL12 trap, significantly reduced the establishment of an iTME [102, 103].
Eliminating physical barriers
The abnormal tumor vasculature is a critical barrier to T-cell infiltration because it provides inadequate blood perfusion and exhibits deregulated expression of the adhesion molecules required for T-cell extravasation. Therefore, treatment to normalize tumor vessels enhances CTL infiltration [104]. It has been reported that nitric oxide (NO) regulates angiogenesis and maintains vascular homeostasis [105]. However, most NO-delivery agents are limited by their short half-life, uncontrollable NO release, and poor tumor targeting. Sung et al. reported biodegradable lipid-PLGA NPs (denoted as NanoNO) encapsulating the NO donor dinitrosyl iron complex (DNIC). NanoNO avoided recognition by macrophages and interaction with serum proteins and demonstrated controlled release of NO. Moreover, NanoNO showed increased tumor accumulation compared with free DNIC due to the EPR effect. Thus, encapsulation of DNIC into NanoNO improved its stability, half-life, and tumor accumulation. In addition, after penetrating the TME and entering cancer cells, the pH-sensitive NanoNO specifically release NO in the acidic endosomes/lysosomes. The released NO reprogrammed the gene expression profile of endothelial cells, shifting them from a pro-angiogenic phenotype to a vascular-stabilizing signature. Therefore, treatment of mice bearing HCC tumors with NanoNO facilitated significant CD4+ and CD8+ T-cell infiltration [106].
CAFs are a subset of fibroblasts that are perpetually active in tumors and are one of the most crucial TME components [107]. The dense ECM produced by CAFs creates high interstitial fluid pressure, which is a physical barrier to T-cell infiltration [108]. CAFs also mediate T-cell exclusion, which prevents cancer cells from physically contacting CTLs. Therefore, killing CAFs could enhance the efficiency of immunotherapy [109]. Fibroblast-activation protein (FAP), which is overexpressed on the surface of CAFs, has been proposed as a universal tumor-targeting antigen. Zhen et al. reported a CAF-targeted nanoparticle-based photo-immunotherapy (nano-PIT). Ferritin, a compact nanoparticle protein cage, was exploited as a photosensitizer carrier and conjugated with a FAP-specific single-chain variable fragment (scFv) on its surface. These nanoconjugates selectively homed to CAFs in tumors. Upon laser irradiation, activated nano-PIT efficiently eliminated the CAFs. In response, the ECM surrounding the tumor was destroyed, leading to significantly enhanced CD8+ T-cell infiltration [110]. Many studies have demonstrated the benefit of CAF depletion for enhancing T-cell infiltration. Nonetheless, preclinical and clinical trials have also reported that depletion of CAFs could accelerate tumor progression and metastasis. These studies illustrate the need for a critical and comprehensive evaluation of CAF depletion [111, 112].
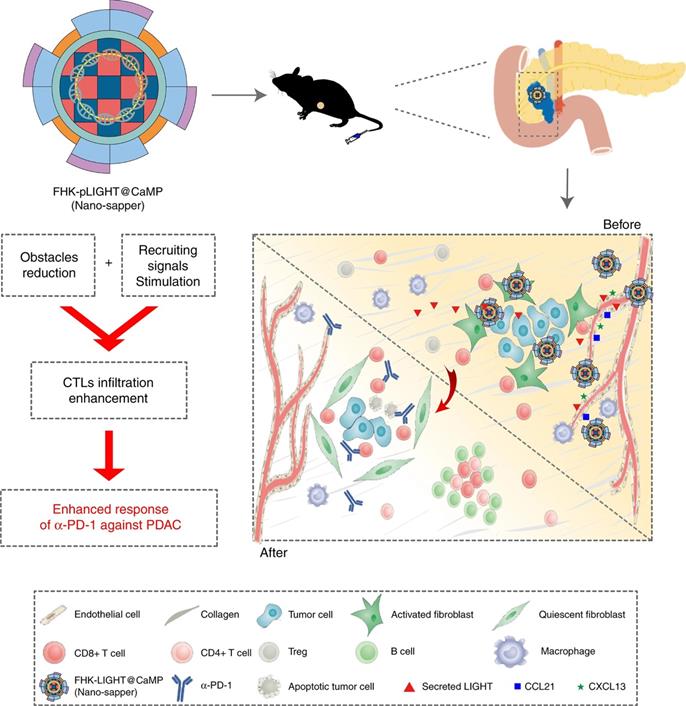
To optimally enhance the tumor infiltration of CTLs, strategies that address both signal barriers and physical barriers are needed. Huang et al. proposed an infiltration enhancement strategy that synergistically breaks the physical obstacles and increases recruiting signals in the iTME. This synergistic effect was achieved by combining phosphate-modified α-mangostin (MP) and a plasmid encoding a pleiotropic inflammatory cytokine (pLIGHT). MP is a natural small molecule that reduces fibrogenesis and decreases collagen deposition, while pLIGHT normalizes defective vessels and facilitates T-cell recruitment. MP and pLIGHT were co-loaded into an ECM glycoprotein (tenascin C) targeted peptide (FHK)-decorated calcium phosphate liposome (denoted as FHK-pLIGHT@CaMP), as shown in Figure 4. Decoration of the liposome with the FHK peptide enhanced the tumor retention of the loaded components. Then, MP reversed the CAFs activation, decreased collagen deposition, and relieved compressed vessels. The secreted LIGHT recovered vessel functions and stimulated the expression of lymphocyte-recruiting chemo-attractants. Accordingly, MP and LIGHT synergistically improved CTL infiltration and supported local generation of a tumor-specific immune response [113].
The expected effects of Nano-sapper synergized with immune-checkpoint inhibitor. Nano-sapper specifically modulate the TME of PDAC, which involves the reduced physical barrier (attenuated stroma and normalized vessels) and the release of chemoattractants recruiting lymphocytes (CCL21 and CXCL13). Once the TME has been reprogrammed by Nano-sapper, a variety of adaptive immune cells migrate to the PDAC and enhance the anti-tumor effects of α-PD-1. Adapted with permission from [113], Copyright 2020 Springer Nature.
In conclusion, NPs with good stability, long circulation, and efficient tumor targeting could greatly assist the tumor infiltration of T cells. These strategies could further improve the outcomes of immunotherapies such as adoptive T-cell therapy and anti-PD-1/anti-PD-L1 therapy, which constantly suffer from a low response rate due to the T-cell infiltration issue [98, 114].
CTL recognition and killing of tumor cells
Finally, depending on TCR and MHC interactions, effector T cells specifically recognize tumor cells and eliminate them by releasing granzymes and perforins [5]. However, once T cells finally infiltrate into the iTME, they are still prevented from effectively recognizing and killing tumor cells, due to binding of PD-L1 (expressed on the tumor cell surface) with PD-1 (expressed on the T cell surface). Additionally, the iTME may protect tumor cells against potent CTL recognition and killing, preventing CTLs from eliminating tumor. Strategies aimed at enhancing CTL recognition and killing of tumor cells have been assessed in clinical trials. These strategies include immune checkpoint blockade (ICB) of PD-1/PD-L1, chimeric antigen receptor (CAR) T-cell therapy (which endows T cells with antibody recognition specificity), and rebuilding the iTME.
Disrupting the PD-1/PD-L1 pathway
Except for attenuating T cell activation, the interaction between PD-1 on T cells with PD-L1 on tumor cells induces effector T cell apoptosis, anergy, and functional exhaustion [85]. PD-L1 on tumor cells is widely used as a biomarker for ICB [115]. Despite exciting clinical results, the clinically used PD-1/PD-L1-monoclonal antibody still faces considerable challenges, such as low response rate [116], low binding strength, and unmanageable side effects. Unfortunately, clinical translation of small molecular inhibitors is also restricted by their rapid clearance and poor tumor accumulation. For these reasons, nanotechnology has been applied to improve the therapeutic efficacy of PD-1/PD-L1 inhibitors. Bu et al. conjugated generation 7 (G7) poly(amidoamine) (PAMAM) dendrimers with PD-L1 targeting molecules to generate G7-aPD-L1 (Figure 5). The dendrimer NPs formed multiple binding pairs with PD-L1 proteins, creating significantly stronger interactions with the target receptors than free aPD-L1. This enhancement in binding kinetic increased the effects of the PD-L1 antagonist [117].
A schematic diagram illustrating the hypothesis that the dendrimer-mediated multivalent interaction would substantially increase the antagonist effect of ICIs as a result of increased binding kinetics. Adapted with permission from [117], Copyright 2020 American Chemical Society.
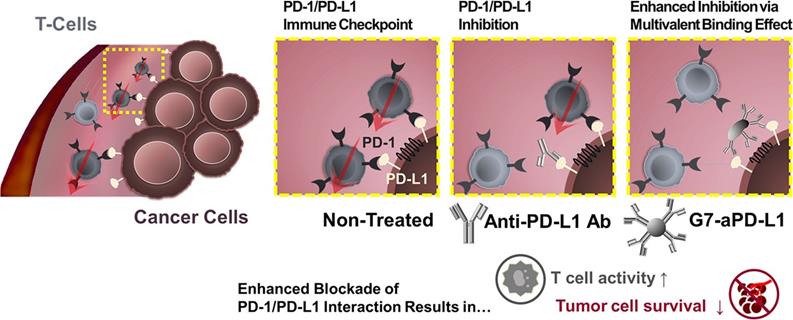
Systemic inhibition of PD-L1 risks breaking peripheral tolerance, causing autoimmune diseases and safety issues [18, 118, 119]. Thus, intelligent nanocarriers that can distinguish tumors from normal tissues have been developed to achieve tumor-specific drug delivery. Acidic microenviroment (pHe) and ROS overexpression, especially hydrogen peroxide (H2O2), are two typical characteristics of solid tumors. Zhang et al. constructed a dual-locking nanoparticle (DLNP) with clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated (Cas) enzyme Cas13a [120]. The DLNP had a core-shell structure with the CRISPR/Cas13a system pDNA targeting PD-L1 encapsulated inside the core, and a pHe/H2O2 dual-responsive polymer layer as the shell. In blood circulation and normal tissues, the polymer layer endowed the DLNP with a negatively charged and PEGylated surface, contributing to its stability. Upon reaching the TME, the polymer layer was degraded into a cationic polymer, facilitating cellular internalization and PD-L1 inhibition. This approach reduced the side effects of the immunotherapy by avoiding undesired PD-L1 inhibition in normal tissues [121].
Apart from PD-1/PD-L1, inhibitory molecules that restrain the activity of CTLs, include lymphocyte-activation gene-3 (Lag-3), T cell immunoglobulin-3 (TIM-3), T-cell immunoreceptor with Ig and ITIM domains (TIGIT), and V-domain Ig suppressor of T-cell activation (VISTA) [122-125]. Most of these are under development in clinical and preclinical studies and show synergistic effects in combination with blockade of the PD-1/PD-L1 pathway. However, such studies are rarely reported in combination with nanotechnology [33].
CAR-T therapy
Adoptive cell therapy (ACT) is a personalized therapy in which a patient's immune cells are expanded in vitro to large numbers and then reinfused to eradicate tumors [126]. Genetic modification of T cells with CARs is the most promising ACT strategy. In this strategy, a patient's T cells are transfected with a construct encoding an antibody against a tumor surface antigen (typically CD19), thereby endowing them with the specificity of antibody-like recognition [127]. However, the generation and storage of large numbers of CAR T cells is too complicated for industrial production and clinical application. Nanocarriers with simple preparation, good stability, and T cell-targeting ability could solve these problems. Hence, Smith et al. demonstrated that DNA-carrying NPs could efficiently introduce leukemia-targeted CAR genes into T-cell nuclei. The authors chose poly (β-amino ester) as the core material for the T cell-targeted nanocarriers. NPs carrying genes of CD19-specific CARs selectively and rapidly edited T-cell specificity in vivo. In addition, these polymer NPs were easy to manufacture in a stable form, which simplified storage and reduced costs, and demonstrated transfection efficacies comparable to that of the conventional adoptive transfer of laboratory-manufactured CAR T cells [128].
Modulation of iTME
The TME contains a network of immunosuppressive factors that pose a formidable barrier to CTLs [98]. Reprogramming of immunosuppressive factors in the iTME is essential. MDSCs and M2 macrophages are the main immunosuppressive immune cells promoting tumor growth. Inhibition of phosphoinositide-3-kinases (PI3Ks), which are pivotal for the function of myeloid cells, can effectively reshape the iTME. Zhang et al. designed aminoethylanisamide (AEAA)-modified polymeric NPs to encapsulate IPI-549, an oral PI3K-γ inhibitor in clinical development. The engineered IPI-549 NPs exhibited efficient cellular uptake, a long half-life, and strong tumor accumulation. The IPI-549 NPs induced the reduction of immunosuppressive cells, such as regulatory B cells and MDSCs, and changed the transcription factor and cytokines, such as IL-10 and IFN-γ, thereby remodeling the iTME [129]. Tumors often stimulate tumor-associated macrophages (TAMs) to display an immunosuppressive M2 phenotype, which supports tumor growth through the production of cytokines such as IL-10, instead of the anti-tumorigenic M1-like phenotype. A great amount of effort has been dedicated to either depleting M2-like TAMs or converting their phenotype into tumoricidal M1-like TAMs. Rodell et al. screened out a potent driver of the M1 phenotype, R848 (an agonist of TLR7/8). The authors constructed β-cyclodextrin nanoparticles (CDNPs) to deliver R848. CDNP-R848 displayed high TAM affinity and high drug-loading capacity due to the covalent crosslinking of the CD. Administration of CDNP-R848 in mice altered the functional orientation of TAMs towards an M1 phenotype, leading to controlled tumor growth and protection against tumor rechallenge [130].
In addition to their immunostimulatory functions, TLR7/8 agonists (TLR7/8a) can also modulate the iTME by transforming MDSCs into APCs such as DCs and macrophages, and polarizing TAMs from M2 to M1. Kim et al. encapsulated TLR7/8a in the core of a nanoemulsion (NE) to improve its pharmacokinetic properties and reduce systemic toxicity. Administration of NE (TLR7/8a) induced recruitment and activation of innate immune cells, infiltration of lymphocytes, and polarization of M2-like TAMs, thus reprogramming the iTME [131]. Liang et al. used triblock copolymer NPs to co-deliver the STING agonist DMXAA and SN38. The prepared NPs (named PS3D1@DMXAA) enabled efficient intracellular delivery of DMXAA. PS3D1@DMXAA enhanced antigen cross-presentation and induced conversion of the iTME to an immunogenic TME through the synergistic interaction between SN38 and STING activation [132].
Tumor acidity plays an immunosuppressive role in impeding effective antitumor T-cell immune responses. Specifically, CD8+ T cells tend to become anergic when exposed to a low pH environment. In addition, excessive lactate, the product of aerobic glycolysis, enhances the function of immunosuppressive cells and thereby blunts antitumor immune responses [133]. Hence, antagonizing tumor acidity may reverse the detrimental effects of lactate and recover the functions of antitumor T cells. As shown in Figure 6, Zhang et al. utilized vesicular cationic lipid-assisted nanoparticles (CLAN) to mediate the knockdown of lactate dehydrogenase A (LDHA) in tumor cells. CLAN prevented premature release of encapsulated siRNA during blood circulation and improved the tumor accumulation of siRNA. CLAN-mediated gene silencing efficiently downregulated LDHA expression, decreased lactate secretion, and raised the tumor pH. In immunocompetent syngeneic melanoma and breast tumor models, neutralization of tumor acidity increased infiltration of CD8+ T and NK cells, decreased the number of immunosuppressive T cells, and thus significantly inhibited tumor growth [134].
Nanotechnology-mediated reversion of tumor immunosuppressive microenvironment through tumor acidity modulation. The pH of the tumor microenvironment modulated the activation and proliferation of infiltrating immune cells and thereby regulated the balance between the immune surveillance and escape (also known as immune response and tolerance). Tumor cells overexpressing LDHA converted glucose into lactate, which blunted tumor surveillance by T cells. Nanotechnology-mediated knockdown of LDHA therapeutically reversed the tumor acidic immunosuppressive microenvironment, which decreased the number of immunosuppressive cells, increased infiltration of CD8+ T cells, and restored their anti-tumor functions. Tumor acidity neutralization prior to α-PD-1 checkpoint blockade therapy improved the anti-tumor response and produced synergistic effects. Adapted with permission from [134], Copyright 2019 American Chemical Society.
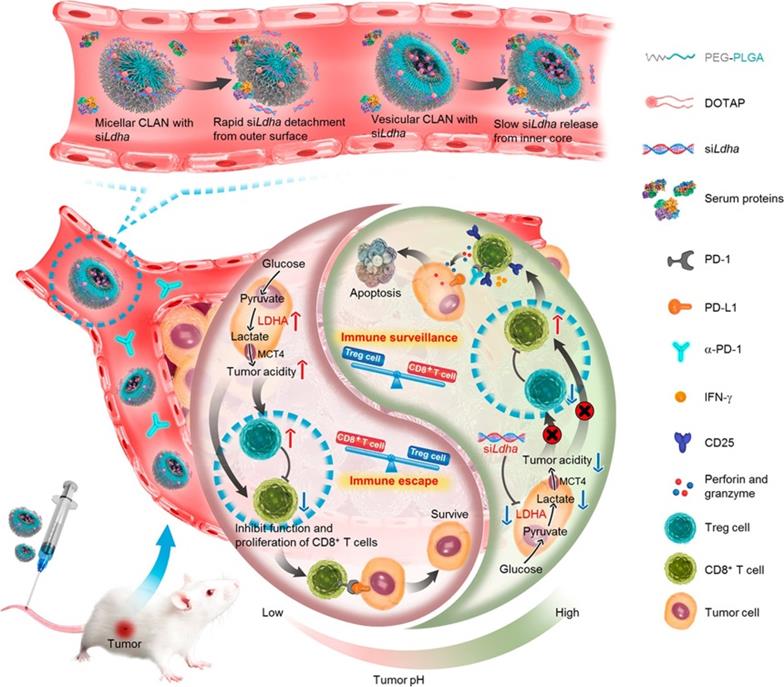
In summary, with advantages including long circulation, strong tumor accumulation, efficient cellular uptake, and intelligent drug release, advanced nanocarriers can be a great strategy to reduce immunosuppressive cytokines/cells, remodel the iTME, and ultimately restore T-cell function.
Combination nano-immunotherapy
Even though the above nano-immunotherapies have demonstrated efficiency in preclinical studies, only a minority of patients can benefit from them. This failure is mainly because most of these therapies only focus on a single step of the cancer-immunity cycle, which is insufficient when the cycle is affected by multiple factors. Combination immunotherapy is one way forward [135-137]. However, combination immunotherapy is challenged by inefficient co-delivery and dangerous autoimmune reactions [114]. In addition, since immunotherapy agents have different properties in terms of pharmacokinetics, biodistribution, and mechanism of action [6], it may not be easy to optimize the synergistic effect. NPs have emerged as a promising carrier for the co-delivery of multiple agents. Advanced nanocarriers can co-deliver multiple drugs with different physicochemical properties through physical adsorption, hydrogen bonding or chemical bonding [138-140]. Beyond this advantage, LN/tumor-targeted NPs can limit the exposure of drugs to normal tissues, thereby improving therapeutic efficacy while reducing systemic toxicity [141, 142].
Combination therapy with enhanced efficacy
NPs can deliver multiple drugs with different properties, contributing to effective synergetic therapy. Indoleamine 2,3-dioxygenase (IDO) inhibitors, like NLG-8189, have poor clinical efficacy due to their unsatisfactory pharmacokinetic profiles and tumor accumulation. Since, single use of IDO inhibitors has limited efficacy, combination with other therapies is necessary. Thus, NPs have been used to improve the bioactivity of IDO inhibitors and achieve effective co-delivery. Shen et al. prepared a bifunctional liposome to co-deliver intravenous oxaliplatin (Oxa [IV]) and NLG-919. To improve their encapsulation efficiencies, Oxa (IV) and NLG-919 were converted into prodrugs via conjugation with the phospholipid DSPE and dodecanoyl chloride, respectively. The amphiphilic Oxa (IV) prodrug and the hydrophobic NLG-919 prodrug self-assembled into aNLG/Oxa (IV)-Lip, achieving effective co-loading of NLG-919 and Oxa (IV). The obtained NPs released cytotoxic Oxa (IV) inside the reductive cytosol, triggering ICD of cancer cells. Additionally, the NPs efficiently retarded the degradation of tryptophan and reduced immunosuppressive kynurenine via NLG-919-mediated inhibition of IDO1. Furthermore, the NPs exhibited a long blood circulation time, thereby enabling efficient passive tumor targeting. As a result, the NPs presented synergistic antitumor efficacy, contributing to enhanced intratumoral infiltration of CD8+ T cells, secretion of cytotoxic cytokines, and downregulation of immunosuppressive regulatory T cells [143].
Combination therapy with reduced toxicity
NPs can reduce the distribution of immunotherapeutic drugs in normal tissues and allow them to specifically accumulate in target tissues, which is of great significance for reducing systemic toxicity. Chiang et al. reported a nanomedicine (IO@FuDex) composed of superparamagnetic iron oxide nanoparticles (IO), fucoidan (Fu), and aldehyde-functionalized dextran (Dex). IO@FuDex functionalized with anti-PD-L1 and the T-cell activators anti-CD3 and anti-CD28 (IO@FuDex3) achieved simultaneous PD-1/PD-L1 checkpoint inhibition and T-cell proliferation. Moreover, IO@FuDex3 could be localized to the desired site of action by application of an external magnetic field, which significantly reduced systemic accumulation. The combination of IO@FuDex3 and magnetic navigation decreased the occurrence of adverse events [144].
Conclusions, challenges, and perspectives
The ultimate goal of anticancer immunotherapy is to eliminate tumors and enable patients to maintain tumor immunity for a long time. At present, due to low efficacy and high risk of immune-mediated toxicities, anticancer immunotherapy is far from achieving this goal. The combination of nanotechnology and immunotherapy, named nano-immunotherapy, has brought opportunities to solve many of these problems. However, the complexity and variability of the host immune system pose great challenges to efficient nano-immunotherapy. Therefore, “which” immune step is targeted, “why” it needs to be further enhanced, and “what” nanotechnology can do for immunotherapy must be fully considered when developing nano-immunotherapies.
To answer these questions, we summarizd the current developments in nano-immunotherapy based on the concept of the cancer-immunity cycle. We divided cellular immunity into two stages and four parts. Immunotherapies in the preparatory stage cover the field of TA release and presentation (steps 1 and 2) and T-cell priming and activation (step 3), and include ICD induction, CD47 blockade, aAPCs, CTLA-4 blockade, and administration of inflammatory stimulating cytokines. These immunotherapies usually use sensitive antigens or proteins that are easily degraded or inactivated by enzymes in complex physiological environments. Nanotechnologies provide strong protection for these agents, increasing their half-lives, minimizing systemic toxicity, and promoting their delivery to APCs. Moreover, LNs are the “base camp” in the preparatory stage, where the anticancer immune response is initiated. Due to their suitable size and particular structure, NPs have the promising potential of achieving adequate drainage and retention in LNs, thus enhancing the immune response in the preparatory stage.
Nano-immunotherapies in the effector stage involve CTL trafficking and infiltration into the tumor site (steps 4 and 5) and CTL recognition and killing of tumor cells (steps 6 and 7), and include elimination of TME barriers, blockade of the PD-1/PD-L1 pathway, removal of iTME immunosuppression, and CAR-T therapy. NPs have prolonged systemic circulation and tumor targeting effect capabilities. In addition, specific NPs can control drug release in response to typical characteristics of the TME, such as hypoxia and low pH. All these advantages of nanotechnology could help to enhance the therapeutic effects of immunotherapies and decrease their side effects.
It has been reported that combination immunotherapy is more effective than monotherapy in cancer treatment. Combinations of tumor vaccines, immune checkpoint inhibitors, ACTs, and TME regulatory treatments have been well studied [57, 145, 146]. Despite the increased therapeutic benefits, the risks of autoimmune responses, toxicities, and adverse events are also greater for combination therapy than monotherapy. In addition, effective co-delivery of immunotherapy agents with different properties and mechanisms is also challenging. NPs that co-encapsulate multiple drug molecules are promising platforms to overcome the barriers of combination therapy. However, with synthetic carrier materials and complex preparation methods, combination nano-immunotherapy faces still greater challenges in clinical translation.
As new opportunities always come with new challenges, nanotechnology brings additional challenges to immunotherapy. The EPR effect has been regarded as a golden rule of anticancer nanomedicine and has been well-studied in animal models with xenografted tumors. However, xenograft tumors are significantly different from human cancers. Due to the complexity and heterogeneity of human tumors, as well as ethical issues with conducting experiments in patients, the EPR effect in human tumors has not yet been fully investigated and its clinical relevance remains controversial [17, 142]. Thus, whether the EPR effect in humans can effectively facilitate nano-immunotherapy needs to be reconsidered. In addition, nanotechnology increases the risk of overdriving the immune system, and nanomaterials also suffer from safety issues. The potential immunogenicity of nanomaterials is a double-edged sword for immunotherapy. On the one hand, some nanomaterials themselves can induce inflammatory responses and promote or enhance immune responses against cancer. On the other hand, if a nano-immunologic agent is recognized as foreign substance and opsonized by plasma proteins, the complement pathway is activated, resulting in rapid phagocytosis and clearance of the drug by the liver and spleen [147]. Complete activation of the immune response may lead to serious complications, including allergic reactions, hemolysis, thrombogenesis, and even disseminated intravascular coagulation [16, 98]. What is more, there are critical concerns about the quality of nano-immunologic agents, the fate of nanomedicines in the body, and interactions with immune organs and the TME, which need to be comprehensively studied and tested.
Abbreviations
LNs: lymph nodes; TME: tumor microenvironment; iTME: immunosuppressive tumor microenvironment; NPs: nanoparticles; EPR effect: the enhanced permeation and retention effect; APCs: antigen presenting cells; TAs: tumor antigens; CTL: cytotoxic T lymphocyte; DCs: dendritic cells; MHC: major histocompatibility complex; ETAs: endogenous tumor antigens; PDT: photodynamic therapy; PTT: photothermal therapy; ER: endoplasmic reticulum; MSNs: mesoporous silica nanoparticles; Fe3O4: iron oxide; ROS: reactive oxygen species; Ce6: chlorin e6; ICD: immunogenic cell death; DAMPs: damage associated molecular patterns; CRT: calreticulin; HMGB1: high mobility group box 1; ATP: adenosine triphosphate; Q: quercetin; A: alantolactone; PM: plasma membrane; PCPK: PpIX-C6-PEG8-KKKKKKSKTKC-OMe; PFTase: protein farnesyltransferase; TDLNs: tumor draining lymph nodes; Si-OH: hydrophilic silanol groups; OVA: ovalbumin; PRRs: pattern recognition receptors; TLR: Toll-like receptor; InAc: inulin acetate; PLGA: poly(lactic-co-glycolic acid); LNPs: lipid nanoparticles; SIRPα: signal regulatory proteinα; CD47nb: nanobody antagonist of CD47; pMHC: peptide-MHC; aAPCs: artificial APCs; TCR: T cell receptor; CTLA-4: cytotoxic T-lymphocyte associated antigen 4; siCTLA-4: CTLA-4 small interfering RNA; PEG-PLA: poly(ethylene glycol)-polylactide; PD-1: programmed cell death-1; PD-L1: programmed death-ligand 1; IL: interleukin; IFN: interferon; ELPs: elastin-like polypeptides; IL-15SA: IL-15 super-agonist; STING: Stimulator of Interferon Genes; cdGMP: cyclic di-GMP; DMXAA: 5,6-dimethylxanthenone-4-acetic acid; CAFs: carcinoma-associated fibroblasts; ECM: extracellular matrix; CXCL12: C-X-C motif chemokine ligand 12; LCP: lipid calcium phosphate; pDNA: plasmid DNA; MDSCs: myeloid-derived suppressor cells; LPD: liposome-protamine-DNA; NO: nitric oxide; DNIC: dinitrosyl iron complex; FAP: fibroblast-activation protein; nano-PIT: nanoparticle-based photo-immunotherapy; scFv: single-chain variable fragment; IL-10: Interleukin-10; MP: phosphate of α-mangostin; pLIGHT: plasmid encoding pleiotropic inflammatory cytokine; ICB: immune checkpoint blockade; CAR: chimeric antigen receptor; pHe: acidic microenvironment; H2O2: hydrogen peroxide; CRISPR: clustered regularly interspaced short palindromic repeat; Lag-3: lymphocyte activation gene-3; TIM-3: T cell immunoglobulin-3; TIGIT: T cell immunoreceptor with Ig and ITIM domains; VISTA: V-domain Ig suppressor of T cell activation; ACT: adoptive cell therapy; PI3Ks: phosphoinositide-3-kinases; AEAA: aminoethyl anisamide; TAMs: tumor-associated macrophages; CDNPs: cyclodextrin nanoparticles; CLAN: cationic lipid-assisted nanoparticles; LDHA: lactate dehydrogenase A; TLR7/8a: TLR7/8 agonists; IDO: indoleamine 2,3-dioxygenase.
Acknowledgements
We are grateful to financial support by Liaoning Revitalization Talents Program (No. XLYC1808017), Shenyang Youth Science and Technology Innovation Talents Program (No. RC190454), China Postdoctoral Innovative Talents Support Program (No. BX20190219) and China Postdoctoral Science Foundation (No. 2019M661134).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Beil DR, Wein LM. Sequencing surgery, radiotherapy and chemotherapy: insights from a mathematical analysis. Breast Cancer Res Treat. 2002;74:279-86
2. Riley RS, June CH, Langer R, Mitchell MJ. Delivery technologies for cancer immunotherapy. Nat Rev Drug Discov. 2019;18:175-96
3. Yang W, Zhou Z, Lau J, Hu S, Chen X. Functional T cell activation by smart nanosystems for effective cancer immunotherapy. Nano Today. 2019;27:28-47
4. Neeve SC, Robinson BW, Fear VS. The role and therapeutic implications of T cells in cancer of the lung. Clin Transl Immunology. 2019;8:e1076
5. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1-10
6. Jiang W, Wang Y, Wargo JA, Lang FF, Kim BYS. Considerations for designing preclinical cancer immune nanomedicine studies. Nat Nanotechnol. 2021;16:6-15
7. Cao J, Huang D, Peppas NA. Advanced engineered nanoparticulate platforms to address key biological barriers for delivering chemotherapeutic agents to target sites. Adv Drug Deliv Rev. 2020;167:170-88
8. Waldmann TA. Cytokines in Cancer Immunotherapy. Cold Spring Harb Perspect Biol. 2018 10
9. Yong SB, Chung JY, Song Y, Kim J, Ra S, Kim YH. Non-viral nano-immunotherapeutics targeting tumor microenvironmental immune cells. Biomaterials. 2019;219:119401
10. Wang Y, Fan Z, Shao L, Kong X, Hou X, Tian D. et al. Nanobody-derived nanobiotechnology tool kits for diverse biomedical and biotechnology applications. Int J Nanomedicine. 2016;11:3287-303
11. Chen Z, Wang Z, Gu Z. Bioinspired and Biomimetic Nanomedicines. Acc Chem Res. 2019;52:1255-64
12. Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature. 2019;575:299-309
13. Yang Z, Ma Y, Zhao H, Yuan Y, Kim BYS. Nanotechnology platforms for cancer immunotherapy. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2020;12:e1590
14. Luo C, Sun J, Sun B, He Z. Prodrug-based nanoparticulate drug delivery strategies for cancer therapy. Trends Pharmacol Sci. 2014;35:556-66
15. Sautes-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer. 2019;19:307-25
16. Li Y, Ayala-Orozco C, Rauta PR, Krishnan S. The application of nanotechnology in enhancing immunotherapy for cancer treatment: current effects and perspective. Nanoscale. 2019;11:17157-78
17. Ding Y, Xu Y, Yang W, Niu P, Li X, Chen Y. et al. Investigating the EPR effect of nanomedicines in human renal tumors via ex vivo perfusion strategy. Nano Today. 2020;35:100970
18. Galstyan A, Markman JL, Shatalova ES, Chiechi A, Korman AJ, Patil R. et al. Blood-brain barrier permeable nano immunoconjugates induce local immune responses for glioma therapy. Nat Commun. 2019;10:3850
19. Rao DA, Forrest ML, Alani AWG, Kwon GS, Robinson JR. Biodegradable PLGA Based Nanoparticles for Sustained Regional Lymphatic Drug Delivery. J Pharm Sci. 2010 99(4); 2018-2031
20. Koker SD, Cui J, Vanparijs N, Albertazzi L, Grooten J, Caruso F. et al. Engineering Polymer Hydrogel Nanoparticles for Lymph Node Targeted Delivery. Angew Chem Int Ed. 2016;55:1334-13339
21. Yang R, Xu J, Xu L, Sun X, Chen Q, Zhao Y. et al. Cancer Cell Membrane-Coated Adjuvant Nanoparticles with Mannose Modification for Effective Anticancer Vaccination. ACS Nano. 2018;12:5121-9
22. Fu C, Zhou L, Mi QS, Jiang A. DC-Based Vaccines for Cancer Immunotherapy. Vaccines (Basel). 2020;8:706-22
23. Fang RH, Hu CM, Luk BT, Gao W, Copp JA, Tai Y. et al. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014;14:2181-8
24. Zhang Y, Chen H, Wang H, Wang T, Pan H, Ji W. et al. A synergistic cancer immunotherapy nano-system for preventing tumor growth. Chemical Engineering Journal. 2020;380:122472
25. Zhu R, Su L, Dai J, Li Z-W, Bai S, Li Q. et al. Biologically Responsive Plasmonic Assemblies for Second Near-Infrared Window Photoacoustic Imaging-Guided Concurrent Chemo-Immunotherapy. ACS Nano. 2020;14:3991-4006
26. Traini G, Ruiz-de-Angulo A, Blanco-Canosa JB, Zamacola Bascaran K, Molinaro A, Silipo A. et al. Cancer Immunotherapy of TLR4 Agonist-Antigen Constructs Enhanced with Pathogen-Mimicking Magnetite Nanoparticles and Checkpoint Blockade of PD-L1. Small. 2019;15:e1803993
27. Feng Z, Guo J, Liu X, Song H, Zhang C, Huang P. et al. Cascade of reactive oxygen species generation by polyprodrug for combinational photodynamic therapy. Biomaterials. 2020;255:120210
28. Goldberg MS. Improving cancer immunotherapy through nanotechnology. Nature Reviews Cancer. 2019;19:587-602
29. Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20:651-668
30. Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12:265-77
31. Le Gall CM, Weiden J, Eggermont LJ, Figdor CG. Dendritic cells in cancer immunotherapy. Nat Mater. 2018;17:474-5
32. Guevara ML, Persano F, Persano S. Nano-immunotherapy: Overcoming tumour immune evasion. Semin Cancer Biol. 2019;69:238-48
33. Tang S, Ning Q, Yang L, Mo Z, Tang S. Mechanisms of immune escape in the cancer immune cycle. Int Immunopharmacol. 2020;86:106700
34. Wang B, An J, Zhang H, Zhang S, Zhang H, Wang L. et al. Personalized Cancer Immunotherapy via Transporting Endogenous Tumor Antigens to Lymph Nodes Mediated by Nano Fe3 O4. Small. 2018;14:e1801372
35. Yang W, Zhang F, Deng H, Lin L, Wang S, Kang F. et al. Smart Nanovesicle-Mediated Immunogenic Cell Death through Tumor Microenvironment Modulation for Effective Photodynamic Immunotherapy. ACS Nano. 2020;14:620-31
36. Finn OJ. Cancer vaccines: between the idea and the reality. Nat Rev Immunol. 2003;3:630-41
37. Zhang Z, Liu S, Zhang B, Qiao L, Zhang Y, Zhang Y. T Cell Dysfunction and Exhaustion in Cancer. Front Cell Dev Biol. 2020;8:17
38. Guo Y, Lei K, Tang L. Neoantigen Vaccine Delivery for Personalized Anticancer Immunotherapy. Front Immunol. 2018;9:1499
39. Chao Y, Liang C, Tao H, Du Y, Wu D, Dong Z. et al. Localized cock-tail chemo-immunotherapy after in situ gelation to trigger robust systemic antitumor immune responses. Sci Adv. 2020;6:eaaz4204
40. Mao D, Hu F, Yi Z, Kenry K, Xu S, Yan S. et al. AIEgen-coupled upconversion nanoparticles eradicate solid tumors through dual-mode ROS activation. Sci Adv. 2020;6:eabb2712
41. Qian M, Chen L, Du Y, Jiang H, Huo T, Yang Y. et al. Biodegradable Mesoporous Silica Achieved via Carbon Nanodots-Incorporated Framework Swelling for Debris-Mediated Photothermal Synergistic Immunotherapy. Nano Lett. 2019;19:8409-17
42. Cruickshank B, Giacomantonio M, Marcato P, McFarland S, Pol J, Gujar S. Dying to Be noticed: epigenetic Regulation of immunogenic Cell Death for Cancer immunotherapy. Frontiers in Immunology. 2018 9
43. Li W, Yang J, Luo L, Jiang M, Qin B, Yin H. et al. Targeting photodynamic and photothermal therapy to the endoplasmic reticulum enhances immunogenic cancer cell death. Nat Commun. 2019;10:3349
44. Lu J, Liu X, Liao YP, Salazar F, Sun B, Jiang W. et al. Nano-enabled pancreas cancer immunotherapy using immunogenic cell death and reversing immunosuppression. Nat Commun. 2017;8:1811
45. Zhang C, Gao F, Wu W, Qiu WX, Zhang L, Li R. et al. Enzyme-Driven Membrane-Targeted Chimeric Peptide for Enhanced Tumor Photodynamic Immunotherapy. ACS Nano. 2019;13:11249-62
46. Tran T, Blanc C, Granier C, Saldmann A, Tanchot C, Tartour E. Therapeutic cancer vaccine: building the future from lessons of the past. Semin Immunopathol. 2019;41:69-85
47. Liu Q, Zhu H, Liu Y, Musetti S, Huang L. BRAF peptide vaccine facilitates therapy of murine BRAF-mutant melanoma. Cancer Immunol Immunother. 2018;67:299-310
48. Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S. et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577:561-5
49. Reuven EM, Leviatan Ben-Arye S, Yu H, Duchi R, Perota A, Conchon S. et al. Biomimetic Glyconanoparticle Vaccine for Cancer Immunotherapy. ACS Nano. 2019;13:2936-47
50. Irvine DJ, Hanson MC, Rakhra K, Tokatlian T. Synthetic Nanoparticles for Vaccines and Immunotherapy. Chem Rev. 2015;115:11109-46
51. Nakamura T, Harashima H. Dawn of lipid nanoparticles in lymph node targeting: Potential in cancer immunotherapy. Adv Drug Deliv Rev. 2020;167:78-88
52. AbouAitah K, Hassan HA, Swiderska-Sroda A, Gohar L, Shaker OG, Wojnarowicz J. et al. Targeted Nano-Drug Delivery of Colchicine against Colon Cancer Cells by Means of Mesoporous Silica Nanoparticles. Cancers (Basel). 2020;12:144
53. Fontana F, Fusciello M, Groeneveldt C, Capasso C, Chiaro J, Feola S. et al. Biohybrid Vaccines for Improved Treatment of Aggressive Melanoma with Checkpoint Inhibitor. ACS Nano. 2019;13:6477-90
54. Huang X, Teng X, Chen D, Tang F, He J. The effect of the shape of mesoporous silica nanoparticles on cellular uptake and cell function. Biomaterials. 2010;31:438-48
55. Hong X, Zhong X, Du G, Hou Y, Zhang Y, Zhang Z. et al. The pore size of mesoporous silica nanoparticles regulates their antigen delivery efficiency. Sci Adv. 2020;6:eaaz4462
56. Li S, Feng X, Wang J, He L, Wang C, Ding J. et al. Polymer nanoparticles as adjuvants in cancer immunotherapy. Nano Research. 2018;11:5769-86
57. Hou Y, Wang Y, Tang Y, Zhou Z, Tan L, Gong T. et al. Co-delivery of antigen and dual adjuvants by aluminum hydroxide nanoparticles for enhanced immune responses. J Control Release. 2020;326:120-30
58. Gorbet MJ, Ranjan A. Cancer immunotherapy with immunoadjuvants, nanoparticles, and checkpoint inhibitors: Recent progress and challenges in treatment and tracking response to immunotherapy. Pharmacol Ther. 2020;207:107456
59. Zhao J, Li J, Jiang Z, Tong R, Duan X, Bai L. et al. Chitosan, N,N,N-trimethyl chitosan (TMC) and 2-hydroxypropyltrimethyl ammonium chloride chitosan (HTCC): The potential immune adjuvants and nano carriers. Int J Biol Macromol. 2020;154:339-48
60. Hollingsworth RE, Jansen K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines. 2019;4:7
61. Shi S, Zhu H, Xia X, Liang Z, Ma X, Sun B. Vaccine adjuvants: Understanding the structure and mechanism of adjuvanticity. Vaccine. 2019;37:3167-78
62. Mottas I, Bekdemir A, Cereghetti A, Spagnuolo L, Yang YS, Muller M. et al. Amphiphilic nanoparticle delivery enhances the anticancer efficacy of a TLR7 ligand via local immune activation. Biomaterials. 2019;190-191:111-20
63. Nuhn L, De Koker S, Van Lint S, Zhong Z, Catani JP, Combes F. et al. Nanoparticle-Conjugate TLR7/8 Agonist Localized Immunotherapy Provokes Safe Antitumoral Responses. Adv Mater. 2018;30:e1803397
64. Hossain MK, Wall KA. Use of Dendritic Cell Receptors as Targets for Enhancing Anti-Cancer Immune Responses. Cancers (Basel). 2019 11
65. Ni Q, Zhang F, Liu Y, Wang Z, Yu G, Liang B. et al. A bi-adjuvant nanovaccine that potentiates immunogenicity of neoantigen for combination immunotherapy of colorectal cancer. Sci Adv. 2020;6:eaaw6071
66. Affandi AJ, Grabowska J, Olesek K, Venegas ML, Barbaria A, Rodríguez E. et al. Selective tumor antigen vaccine delivery to human CD169+ antigen-presenting cells using ganglioside-liposomes. Proc Natl Acad Sci. 2020;117:27528-39
67. Mrigendra K. S. Rajput SSK, Sunny Kumar, Pratik Muley, Susmitha Narisetty, and Hemachand Tummala. Dendritic Cell-Targeted Nanovaccine Delivery System Prepared with an Immune-Active Polymer. ACS Appl Mater Interfaces. 2018;10:27589-602
68. Pardi N, Hogan MJ, Porter FW, Weissman D. mRNA vaccines — a new era in vaccinology. Nat Rev Drug Discov. 2018;18:261-79
69. Wang Y, Zhang L, Xu Z, Miao L, Huang L. mRNA Vaccine with Antigen-Specific Checkpoint Blockade Induces an Enhanced Immune Response against Established Melanoma. Mol Ther. 2018;26:420-34
70. Billingsley MM, Singh N, Ravikumar P, Zhang R, June CH, Mitchell MJ. Ionizable Lipid Nanoparticle-Mediated mRNA Delivery for Human CAR T Cell Engineering. Nano Lett. 2020;20:1578-89
71. Carmona-Ribeiro AM, Perez-Betancourt Y. Cationic Nanostructures for Vaccines Design. Biomimetics (Basel). 2020 5; 32-77
72. Liu L, Wang Y, Miao L, Liu Q, Musetti S, Li J. et al. Combination Immunotherapy of MUC1 mRNA Nano-vaccine and CTLA-4 Blockade Effectively Inhibits Growth of Triple Negative Breast Cancer. Mol Ther. 2018;26:45-55
73. Oberli MA, Reichmuth AM, Dorkin JR, Mitchell MJ, Fenton OS, Jaklenec A. et al. Lipid Nanoparticle Assisted mRNA Delivery for Potent Cancer Immunotherapy. Nano Lett. 2017;17:1326-35
74. Iribarren K, Buque A, Mondragon L, Xie W, Levesque S, Pol J. et al. Anticancer effects of anti-CD47 immunotherapy in vivo. Oncoimmunology. 2019;8:1550619
75. Chowdhury S, Castro S, Coker C, Hinchliffe TE, Arpaia N, Danino T. Programmable bacteria induce durable tumor regression and systemic antitumor immunity. Nat Med. 2019;25:1057-63
76. Tillib SV. “Camel nanoantibody” is an efficient tool for research, diagnostics and therapy. Molecular Biology. 2011;45:66-73
77. Yao S, Zhu Y, Zhu G, Augustine M, Zheng L, Goode DJ. et al. B7-h2 is a costimulatory ligand for CD28 in human. Immunity. 2011;34:729-40
78. Matsumoto A, Asuka M, Takahashi Y, Takakura Y. Antitumor immunity by small extracellular vesicles collected from activated dendritic cells through effective induction of cellular and humoral immune responses. Biomaterials. 2020;252:120112
79. Kim JV, Latouche JB, Riviere I, Sadelain M. The ABCs of artificial antigen presentation. Nat Biotechnol. 2004;22:403-10
80. Meyer RA, Sunshine JC, Perica K, Kosmides AK, Aje K, Schneck JP. et al. Biodegradable nanoellipsoidal artificial antigen presenting cells for antigen specific T-cell activation. Small. 2015;11:1519-25
81. Hickey JW, Vicente FP, Howard GP, Mao HQ, Schneck JP. Biologically Inspired Design of Nanoparticle Artificial Antigen-Presenting Cells for Immunomodulation. Nano Lett. 2017;17:7045-54
82. Majedi FS, Hasani-Sadrabadi MM, Thauland TJ, Li S, Bouchard LS, Butte MJ. Augmentation of T-Cell Activation by Oscillatory Forces and Engineered Antigen-Presenting Cells. Nano Lett. 2019;19:6945-54
83. Zhang Q, Wei W, Wang P, Zuo L, Li F, Xu J. et al. Biomimetic Magnetosomes as Versatile Artificial Antigen-Presenting Cells to Potentiate T-Cell-Based Anticancer Therapy. ACS Nano. 2017;11:10724-32
84. Li SY, Liu Y, Xu CF, Shen S, Sun R, Du XJ. et al. Restoring anti-tumor functions of T cells via nanoparticle-mediated immune checkpoint modulation. J Control Release. 2016;231:17-28
85. Peng Q, Qiu X, Zhang Z, Zhang S, Zhang Y, Liang Y. et al. PD-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat Commun. 2020;11:4835
86. Hobo W, Novobrantsev TL, Fredrix H, Wong J, Milstein S, Epstein-Barash H. et al. Improving dendritic cell vaccine immunogenicity by silencing PD-1 ligands using siRNA-lipid nanoparticles combined with antigen mRNA electroporation. Cancer Immunol Immunother. 2013;62:285-97
87. Hassannia H, Chaleshtari MG, Atyabi F, Nosouhian M, Masjedi A, Hojjat-Farsangi M. et al. Blockage of immune checkpoint molecules increases T-cell priming potential of dendritic cell vaccine. Immunology. 2019;159:75-87
88. Shetab Boushehri MA, Stein V, Lamprecht A. Cargo-free particles of ammonio methacrylate copolymers: From pharmaceutical inactive ingredients to effective anticancer immunotherapeutics. Biomaterials. 2018;166:1-12
89. Fan L, Zhang B, Xu A, Shen Z, Guo Y, Zhao R. et al. Carrier-Free, Pure Nanodrug Formed by the Self-Assembly of an Anticancer Drug for Cancer Immune Therapy. Mol Pharm. 2018;15:2466-78
90. Guo Z, Khattar M, Schroder PM, Miyahara Y, Wang G, He X. et al. A dynamic dual role of IL-2 signaling in the two-step differentiation process of adaptive regulatory T cells. J Immunol. 2013;190:3153-62
91. Curtsinger JM, Mescher MF. Inflammatory cytokines as a third signal for T cell activation. Curr Opin Immunol. 2010;22:333-40
92. Wang Z, Guo J, Liu X, Sun J, Gao W. Temperature-triggered micellization of interferon alpha-diblock copolypeptide conjugate with enhanced stability and pharmacology. J Control Release. 2020;328:444-53
93. Tang L, Zheng Y, Melo MB, Mabardi L, Castano AP, Xie YQ. et al. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat Biotechnol. 2018;36:707-16
94. Corrales L, Gajewski TF. Molecular Pathways: Targeting the Stimulator of Interferon Genes (STING) in the Immunotherapy of Cancer. Clin Cancer Res. 2015;21:4774-9
95. Flood BA, Higgs EF, Li S, Luke JJ, Gajewski TF. STING pathway agonism as a cancer therapeutic. Immunol Rev. 2019;290:24-38
96. Hanson MC, Crespo MP, Abraham W, Moynihan KD, Szeto GL, Chen SH. et al. Nanoparticulate STING agonists are potent lymph node-targeted vaccine adjuvants. J Clin Invest. 2015;125:2532-46
97. Stylianopoulos T, Poh MZ, Insin N, Bawendi MG, Fukumura D, Munn LL. et al. Diffusion of particles in the extracellular matrix: the effect of repulsive electrostatic interactions. Biophys J. 2010;99:1342-9
98. Martin JD, Cabral H, Stylianopoulos T, Jain RK. Improving cancer immunotherapy using nanomedicines: progress, opportunities and challenges. Nat Rev Clin Oncol. 2020;17:251-66
99. Jason W. Griffith CLS, Andrew D. Luster. Chemokines and Chemokine Receptors: Positioning Cells for Host Defense and Immunity. Annu Rev Immunol. 2014;32:659-702
100. Zinal S. Chheda RKS, Venkatakrishna R. Jala, Andrew D. Luster, Bodduluri Haribabu. Chemoattractant Receptors BLT1 and CXCR3 Regulate Antitumor Immunity by Facilitating CD8+ T Cell Migration into Tumors. The Journal of Immunology. 2016;197:2016 -
101. Goodwin TJ, Zhou Y, Musetti SN, Liu R, Huang L. Local and transient gene expression primes the liver to resist cancer metastasis. Sci Transl Med. 2016;8:364ra153
102. Shen L, Li J, Liu Q, Song W, Zhang X, Tiruthani K. et al. Local Blockade of Interleukin 10 and C-X-C Motif Chemokine Ligand 12 with Nano-Delivery Promotes Antitumor Response in Murine Cancers. ACS Nano. 2018;12:9830-41
103. Cabel L, Proudhon C, Romano E, Girard N, Lantz O, Stern MH. et al. Clinical potential of circulating tumour DNA in patients receiving anticancer immunotherapy. Nat Rev Clin Oncol. 2018;15:639-50
104. Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D. et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91:1071-121
105. Feng Q, Li Y, Wang N, Hao Y, Chang J, Wang Z. et al. A Biomimetic Nanogenerator of Reactive Nitrogen Species Based on Battlefield Transfer Strategy for Enhanced Immunotherapy. Small. 2020;16:e2002138
106. Sung YC, Jin PR, Chu LA, Hsu FF, Wang MR, Chang CC. et al. Delivery of nitric oxide with a nanocarrier promotes tumour vessel normalization and potentiates anti-cancer therapies. Nat Nanotechnol. 2019;14:1160-9
107. Yeung TL, Leung CS, Yip KP, Sheng J, Vien L, Bover LC. et al. Anticancer Immunotherapy by MFAP5 Blockade Inhibits Fibrosis and Enhances Chemosensitivity in Ovarian and Pancreatic Cancer. Clin Cancer Res. 2019;25:6417-28
108. Seo N, Shirakura Y, Tahara Y, Momose F, Harada N, Ikeda H. et al. Activated CD8(+) T cell extracellular vesicles prevent tumour progression by targeting of lesional mesenchymal cells. Nat Commun. 2018;9:435
109. Kovacs D, Igaz N, Marton A, Ronavari A, Belteky P, Bodai L. et al. Core-shell nanoparticles suppress metastasis and modify the tumour-supportive activity of cancer-associated fibroblasts. J Nanobiotechnology. 2020;18:18
110. Zhen Z, Tang W, Wang M, Zhou S, Wang H, Wu Z. et al. Protein Nanocage Mediated Fibroblast-Activation Protein Targeted Photoimmunotherapy To Enhance Cytotoxic T Cell Infiltration and Tumor Control. Nano Lett. 2017;17:862-9
111. Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR. et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719-34
112. James Harper RCAS. Regulation of the anti-tumour immune response by cancer-associated fibroblasts. Seminars in Cancer Biology. 2014;25:69-77
113. Huang Y, Chen Y, Zhou S, Chen L, Wang J, Pei Y. et al. Dual-mechanism based CTLs infiltration enhancement initiated by Nano-sapper potentiates immunotherapy against immune-excluded tumors. Nat Commun. 2020;11:622
114. Marshall HT, Djamgoz MBA. Immuno-Oncology: Emerging Targets and Combination Therapies. Front Oncol. 2018;8:315
115. Rezazadeh H, Astaneh M, Tehrani M, Hossein-Nataj H, Zaboli E, Shekarriz R. et al. Blockade of PD-1 and TIM-3 immune checkpoints fails to restore the function of exhausted CD8(+) T cells in early clinical stages of chronic lymphocytic leukemia. Immunol Res. 2020
116. Tian Y, Wang X, Zhao S, Liao X, Younis MR, Wang S. et al. JQ1-Loaded Polydopamine Nanoplatform Inhibits c-MYC/Programmed Cell Death Ligand 1 to Enhance Photothermal Therapy for Triple-Negative Breast Cancer. ACS Appl Mater Interfaces. 2019;11:46626-36
117. Bu J, Nair A, Iida M, Jeong WJ, Poellmann MJ, Mudd K. et al. An Avidity-Based PD-L1 Antagonist Using Nanoparticle-Antibody Conjugates for Enhanced Immunotherapy. Nano Lett. 2020
118. Saleh R, Toor SM, Al-Ali D, Sasidharan Nair V, Elkord E. Blockade of PD-1, PD-L1, and TIM-3 Altered Distinct Immune- and Cancer-Related Signaling Pathways in the Transcriptome of Human Breast Cancer Explants. Genes (Basel). 2020 11
119. Riesenberg BP, Ansa-Addo EA, Gutierrez J, Timmers CD, Liu B, Li Z. Cutting Edge: Targeting Thrombocytes to Rewire Anticancer Immunity in the Tumor Microenvironment and Potentiate Efficacy of PD-1 Blockade. J Immunol. 2019;203:1105-10
120. Urena-Bailen G, Lamsfus-Calle A, Daniel-Moreno A, Raju J, Schlegel P, Seitz C. et al. CRISPR/Cas9 technology: towards a new generation of improved CAR-T cells for anticancer therapies. Brief Funct Genomics. 2020;19:191-200
121. Zhang Z, Wang Q, Liu Q, Zheng Y, Zheng C, Yi K. et al. Dual-Locking Nanoparticles Disrupt the PD-1/PD-L1 Pathway for Efficient Cancer Immunotherapy. Adv Mater. 2019;31:e1905751
122. Wuerdemann N, Pütz K, Eckel H, Jain R, Wittekindt C, Huebbers CU. et al. LAG-3, TIM-3 and VISTA Expression on Tumor-Infiltrating Lymphocytes in Oropharyngeal Squamous Cell Carcinoma—Potential Biomarkers for Targeted Therapy Concepts. International Journal of Molecular Sciences. 2020;22:379
123. Mulati K, Hamanishi J, Matsumura N, Chamoto K, Mise N, Abiko K. et al. VISTA expressed in tumour cells regulates T cell function. Br J Cancer. 2019;120:115-27
124. Solomon BL, Garrido-Laguna I. TIGIT: a novel immunotherapy target moving from bench to bedside. Cancer Immunol Immunother. 2018;67:1659-67
125. Wolf Y, Anderson AC, Kuchroo VK. TIM3 comes of age as an inhibitory receptor. Nat Rev Immunol. 2020;20:173-85
126. Guedan S, Ruella M, June CH. Emerging Cellular Therapies for Cancer. Annu Rev Immunol. 2019;37:145-71
127. Met O, Jensen KM, Chamberlain CA, Donia M, Svane IM. Principles of adoptive T cell therapy in cancer. Semin Immunopathol. 2019;41:49-58
128. Smith TT, Stephan SB, Moffett HF, McKnight LE, Ji W, Reiman D. et al. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat Nanotechnol. 2017;12:813-20
129. Zhang X, Shen L, Liu Q, Hou L, Huang L. Inhibiting PI3 kinase-gamma in both myeloid and plasma cells remodels the suppressive tumor microenvironment in desmoplastic tumors. J Control Release. 2019;309:173-80
130. Rodell CB, Arlauckas SP, Cuccarese MF, Garris CS, Li R, Ahmed MS. et al. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat Biomed Eng. 2018;2:578-88
131. Kim SY, Kim S, Kim JE, Lee SN, Shin IW, Shin HS. et al. Lyophilizable and Multifaceted Toll-like Receptor 7/8 Agonist-Loaded Nanoemulsion for the Reprogramming of Tumor Microenvironments and Enhanced Cancer Immunotherapy. ACS Nano. 2019;13:12671-86
132. Liang J, Wang H, Ding W, Huang J, Zhou X, Wang H, wt al. Nanoparticle-enhanced chemo-immunotherapy to trigger robust antitumor immunity. Sci Adv. 2020;6:eabc3646
133. Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L. et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat Immunol. 2016;17:95-103
134. Zhang Y, Zhao Y, Shen J, Sun X, Liu Y, Liu H. et al. Nanoenabled Modulation of Acidic Tumor Microenvironment Reverses Anergy of Infiltrating T Cells and Potentiates Anti-PD-1 Therapy. Nano Lett. 2019;19:2774-83
135. Ott PA, Hodi FS, Kaufman HL, Wigginton JM, Wolchok JD. Combination immunotherapy: a road map. J Immunother Cancer. 2017;5:16
136. Li X, Hou Y, Zhao J, Li J, Wang S, Fang J. Combination of chemotherapy and oxidative stress to enhance cancer cell apoptosis. Chemical Science. 2020;11:3215-22
137. Wu X, Li J, Connolly EM, Liao X, Ouyang J, Giobbie-Hurder A. et al. Combined Anti-VEGF and Anti-CTLA-4 Therapy Elicits Humoral Immunity to Galectin-1 Which Is Associated with Favorable Clinical Outcomes. Cancer Immunol Res. 2017;5:446-54
138. Chen W, Qin M, Chen X, Wang Q, Zhang Z, Sun X. Combining photothermal therapy and immunotherapy against melanoma by polydopamine-coated Al2O3 nanoparticles. Theranostics. 2018;8:2229-41
139. He C, Duan X, Guo N, Chan C, Poon C, Weichselbaum RR. et al. Core-shell nanoscale coordination polymers combine chemotherapy and photodynamic therapy to potentiate checkpoint blockade cancer immunotherapy. Nat Commun. 2016;7:12499
140. Deng H, Kan A, Lyu N, Mu L, Han Y, Liu L. et al. Dual Vascular Endothelial Growth Factor Receptor and Fibroblast Growth Factor Receptor Inhibition Elicits Antitumor Immunity and Enhances Programmed Cell Death-1 Checkpoint Blockade in Hepatocellular Carcinoma. Liver Cancer. 2020;9:338-57
141. Pan J, Hu P, Guo Y, Hao J, Ni D, Xu Y. et al. Combined Magnetic Hyperthermia and Immune Therapy for Primary and Metastatic Tumor Treatments. ACS Nano. 2020;14:1033-44
142. Savitsky K, Yu X. Combined strategies for tumor immunotherapy with nanoparticles. Clin Transl Oncol. 2019;21:1441-9
143. Shen F, Feng L, Zhu Y, Tao D, Xu J, Peng R. et al. Oxaliplatin-/NLG919 prodrugs-constructed liposomes for effective chemo-immunotherapy of colorectal cancer. Biomaterials. 2020;255:120190
144. Chiang CS, Lin YJ, Lee R, Lai YH, Cheng HW, Hsieh CH. et al. Combination of fucoidan-based magnetic nanoparticles and immunomodulators enhances tumour-localized immunotherapy. Nat Nanotechnol. 2018;13:746-54
145. Lu J, Liu X, Liao Y-P, Wang X, Ahmed A, Jiang W. et al. Breast Cancer Chemo-immunotherapy through Liposomal Delivery of an Immunogenic Cell Death Stimulus Plus Interference in the IDO-1 Pathway. ACS Nano. 2018;12:11041-61
146. Xie W, Deng WW, Zan M, Rao L, Yu GT, Zhu DM. et al. Cancer Cell Membrane Camouflaged Nanoparticles to Realize Starvation Therapy Together with Checkpoint Blockades for Enhancing Cancer Therapy. ACS Nano. 2019;13:2849-57
147. Chen VE, Greenberger BA, Taylor JM, Edelman MJ, Lu B. The Underappreciated Role of the Humoral Immune System and B Cells in Tumorigenesis and Cancer Therapeutics: A Review. Int J Radiat Oncol Biol Phys. 2020;108:38-45
Author contact
![]() Corresponding authors: sunbingjun_spycom; sunjinedu.cn
Corresponding authors: sunbingjun_spycom; sunjinedu.cn