Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results and Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2022; 12(5):1999-2014. doi:10.7150/thno.69119 This issue Cite
Research Paper
Anti-PD-L1 peptide-conjugated prodrug nanoparticles for targeted cancer immunotherapy combining PD-L1 blockade with immunogenic cell death
Yujeong Moon1,2,#, Man Kyu Shim1,#, Jiwoong Choi1,3, Suah Yang1,3, Jinseong Kim1,3, Wan Su Yun1,3, Hanhee Cho1,4, Jung Yeon Park3, Yongju Kim3, Joon-Kyung Seong2, Kwangmeyung Kim1,3 ![]()
1. Biomedical Research Institute, Korea Institute of Science and Technology (KIST), Seoul, 02792, Republic of Korea.
2. Department of Bioengineering, Korea University, Seoul, 02841, Republic of Korea.
3. KU-KIST Graduate School of Converging Science and Technology, Korea University, Seoul, 02841, Republic of Korea.
4. Department of Materials Science and Engineering, Seoul National University, Seoul, 08826, Republic of Korea.
#These authors contributed equally to this work.
Received 2021-11-16; Accepted 2022-1-4; Published 2022-1-31
Abstract

Rationale: Cancer immunotherapy combining immune checkpoint blockade (ICB) with chemotherapeutic drugs has provided significant clinical advances. However, such combination therapeutic regimen has suffered from severe toxicity of both drugs and low response rate of patients. In this study, we propose anti-PD-L1 peptide-conjugated prodrug nanoparticles (PD-NPs) to overcome these obstacles of current cancer immunotherapy.
Methods: The functional peptide, consisted of anti-PD-L1 peptide and cathepsin B-specific cleavable peptide, is conjugated to a doxorubicin (DOX), resulting in prodrug nanoparticles of PD-NPs via intermolecular interactions. The antitumor efficacy and immune responses with minimal side effects by PD-NPs combining PD-L1 blockade and ICD are evaluated in breast tumor models.
Results: The PD-NPs are taken up by PD-L1 receptor-mediated endocytosis and then induce ICD in cancer cells by DOX release. Concurrently, PD-L1 blockade by PD-NPs disrupt the immune-suppressing pathway of cancer cells, resulting in proliferation and reinvigoration of T lymphocytes. In tumor models, PD-NPs accumulate within tumor tissues via enhanced permeability and retention (EPR) effect and induce immune-responsive tumors by recruiting a large amount of immune cells.
Conclusions: Collectively, targeted tumor delivery of anti-PD-L1 peptide and DOX via PD-NPs efficiently inhibit tumor progression with minimal side effects.
Keywords: cancer immunotherapy, immune checkpoint blockade, prodrug nanoparticle, drug delivery, antitumor immune response
Introduction
Immune checkpoint blockade (ICB) immunotherapy has provided significant clinical advances to combat a variety of cancers, which provokes an antitumor immune response via monoclonal antibodies (mAbs) against programmed cell death protein 1 (PD-1), programmed death-ligand 1 (PD-L1) or cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) [1]. Furthermore, the therapeutic outcomes of ICB immunotherapy have dramatically improved when combined with immunogenic cell death (ICD)-inducing chemotherapeutic drugs in pre-clinical and clinical studies [2]. The underlying mechanism is that dying cells by ICD-inducing chemotherapeutic drugs release damage-associated molecular patterns (DAMPs) to promote the cross-presentation of tumor-associated antigens to the dendritic cells (DCs), and thus provokes DC maturation and T lymphocyte infiltration [3-6]. These cascade events of ICD turn the immunosuppressive tumor microenvironment (ITM) to the immune-responsive tumors, leading to increase the ICB immunotherapy efficiency [6, 7]. However, poor cancer-specificity of chemotherapeutic drugs can induce systemic toxicities as well as bone marrow suppression and damage to the host immune system, resulting in serious side effects and low response rate of patients [8]. In addition, immunogenicity of mAbs for ICB immunotherapy increase the risk of uncontrolled autoimmune toxicities, the so-called immune-related adverse events (irAEs), which are unpredictable, possibly permanent and occasionally fatal in almost every organ [9, 10].
Due to the potential ability to block the PD-1/PD-L1 interactions, small molecular PD-1/PD-L1 binding peptides have been received much attention in cancer immunotherapy [11]. For example, small peptide, NP-12, was reported as an anti-PD-1 peptide with 29 amino acid sequence to synergize with ICD for effective ICB immunotherapy [12]. In addition, antagonist peptide of PD-L1 (CLQKTPKQC) effectively blocked the PD-1/PD-L1 interactions via high binding affinity to PD-L1 and reinvigorated the T lymphocytes, thereby inducing potent ICB immunotherapy when combined with ICD-inducing chemotherapeutic drugs [13]. However, such PD-1/PD-L1 binding peptides have shown unfavorable therapeutic efficacy owing to the extensive proteolytic cleavage and short half-lives in vivo [14]. To overcome these drawbacks, anti-PD-1 or anti-PD-L1 peptides have broadly adopted to multifunctional nanomedicines, such as polymeric nanoparticles, liposomes and dendrimers [15-18]. Compared to mAbs for blockade of PD-1/PD-L1 interactions, these peptide antagonists have some advantages of amenability to chemical synthesis for design of various nanomedicine loading accurate contents due to their modifiable structure, and low production cost [16]. In particular, anti-PD-1/anti-PD-L1 peptide-modified nanoparticles containing chemotherapeutic drugs elicited potent antitumor immune responses by inducing ICD and disruption of PD-1/PD-L1 interactions [19]. The PD-1/PD-L1 blockade can remove the inhibitory signals for T lymphocyte activation and increase the T cell's recognition of cancer cells damaged by drugs, thereby eradicating heterogeneous and drug-resistant cancer cells by long-term durable and systemic immune responses [20]. However, their translation into the clinic is still difficult because of the innate features of nanoparticle or delivery carrier itself. Many preclinical studies have shown that tumor-targeting nanomedicines have unexpectedly poor delivery efficiency (> 1-3%) and non-specific drug release into the normal tissues and blood stream raise the risk of organ dysfunction and systemic immunosuppression [21]. Also, low drug loading efficiency, innate toxicity and difficulty in mass production of nano-sized carriers hinder their commercialization for ICB immunotherapy.
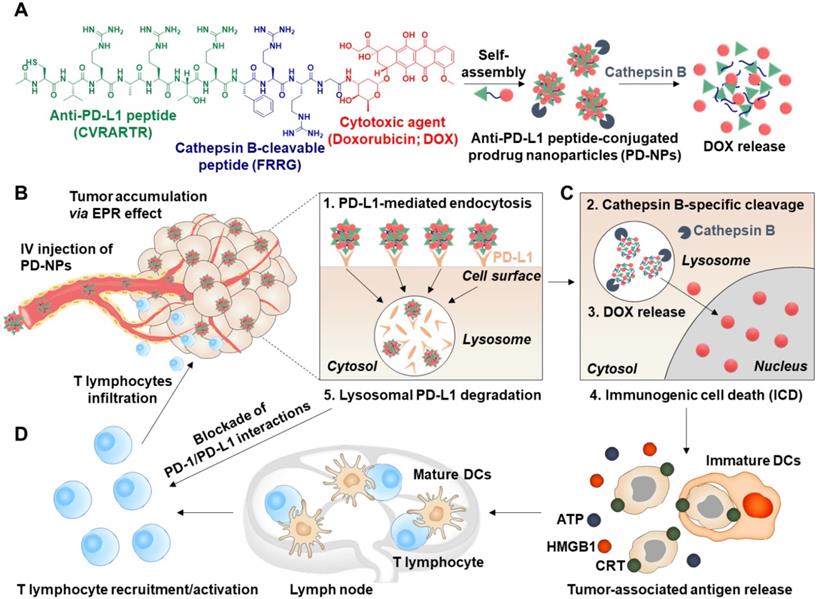
Herein, we propose anti-PD-L1 peptide-conjugated prodrug nanoparticles (PD-NPs) that consist of anti-PD-L1 peptide (CVRARTR; Cys-Val-Arg-Ala-Arg-Thr-Arg), cathepsin B-specific cleavable peptide (FRRG; Phe-Arg-Arg-Gly) and doxorubicin (DOX) to combine ICD with ICB immunotherapy (Scheme 1A). The functional peptide of CVRARTR-FRRG was directly conjugated to amine group of DOX via one-step amide bond reaction, and this direct conjugation of peptide and DOX can prevent non-specific drug release in off-target sites. This is because the target bioenzyme of cathepsin B overexpressed in cancer cells specifically cleave the -RRG- sequence in the PD-NPs to trigger DOX release in the targeted cancer cells, whereas the PD-NPs maintain non-toxic inactive state in normal cells with innately low cathepsin B expression [22-25]. Therefore, PD-NPs can reduce severe inflammatory responses in normal organs and damage to the host immune system compared to conventional nanoparticles encapsulating drugs non-covalently that cause negative immunologic effects by premature drug leakage [8]. The clinical outcomes of cancer immunotherapy rely importantly on the cross-talk between cancer cells and other cellular components, in particular immune cells in the tumor microenvironment [26]. Thus, the PD-NPs can lead safe and more effective cancer immunotherapy by inducing ICD preferentially in targeted cancer cells and minimizing the off-target toxicity via cathepsin B-specific drug release. Importantly, amphiphilic CVRARTRFRRG-DOX conjugates spontaneously self-assembled into anti-PD-L1 peptide-conjugated prodrug nanoparticles (PD-NPs) via intermolecular interactions [27, 28]. Therefore, PD-NPs achieve high and accurate anti-PD-L1 peptide and DOX loading contents (74.3 wt%) without any additional nano-sized carrier materials, which can solve the serious problems of conventional nanomedicines, such as low drug loading efficiency (<10 wt%), innate toxicity and difficulty in mass production. In tumor models, intravenously injected PD-NPs accumulate within targeted tumor tissues via enhanced permeability and retention (EPR) effect and then efficiently enter cancer cells through PD-L1 receptor-mediated endocytosis (Scheme 1B). Then, the DOX molecules released from PD-NPs in the cathepsin B-overexpressed cancer cells induce ICD, which promote high DAMP signals for DC maturation and cytotoxic T lymphocyte activation (Scheme 1C). In addition, PD-L1 binding of PD-NPs disrupt the immune-suppressing PD-1/PD-L1 interactions via lysosomal PD-L1 degradation, which enhance pre-existing antitumor immune responses of T lymphocytes to destruct cancer cells (Scheme 1D) [16]. Concurrently, toxicity against normal cells and immune cells is greatly reduced by their innately low cathepsin B expression, thereby increasing the safety of ICB immunotherapy. In this study, we investigate the cancer immunotherapy, which induce a potent in vivo antitumor efficacy and immune responses with minimal side effects via combination of PD-L1 blockade with ICD by PD-NPs in breast tumor models.
Targeted cancer immunotherapy by PD-NPs to combine PD-L1 blockade with ICD. A. PD-NPs are prepared by the self-assembly of anti-PD-L1 peptide (CVRARTR) and cathepsin B-specific cleavable peptide (FRRG), and doxorubicin (DOX) conjugates. B. The PD-NPs highly accumulate within targeted tumor tissues via EPR effect and then efficiently enter cancer cells through PD-L1 receptor-mediated endocytosis. C. Then, DOX molecules released from PD-NPs by cathepsin B overexpressed in the cancer cells induce effective ICD, which promote high DAMP signals for DC maturation and cytotoxic T lymphocyte activation. D. PD-NPs also disrupt the immune-suppressing PD-1/PD-L1 interactions via lysosomal PD-L1 degradation attributed from a potent binding affiinty to PD-L1, thereby enhancing pre-existing antitumor immune responses of T lymphocytes to destruct cancer cells.
Methods
Reagents
N-terminal acylated Cys-Val-Arg-Ala-Arg-Thr-Arg-Phe-Arg-Arg-Gly (Ac-CVRARTRFRRG-COOH) and Cys-Val-Arg-Ala-Arg-Thr-Arg (NH2-CVRARTR-COOH) peptides were synthesized from Peptron (Daejeon, Republic of Korea). Tem grid (Carbon Film 200 Mesh copper) was purchased from Electron Microscopy Sciences (Hatfield, PA, USA). Benzyloxycarbonyl-Phe-Ala-fluoromethylketone (Z-FA-FMK) and Monoclonal anti-mouse cathepsin B antibody were purchased from Santa Cruz Biotechnology (Dallas, Texas, USA). Cell counting kit-8 (CCK-8) was purchased from Vitascientific (Beltsville, MD, USA). CD8+ T cell enrichment column kit, recombinant cathepsin B, cathepsin D, cathepsin E, caspase-3, caspase-9 and TUNEL assays kit were purchased from R&D systems (Minneapolis, MN, USA). Doxorubicin hydrochloride (DOX) was purchased from Sigma Aldrich (Oakville, Ontario, USA). Dulbecco's Modified Eagle Medium (DMEM) high glucose medium, RPMI 1640 medium, fetal bovine serum (FBS), penicillin and streptomycin were purchased from WELGENE Inc. (Daegu, Republic of Korea). Anti-mouse calreticulin (CRT) antibody, Anti-mouse high mobility group box 1 (HMGB1) antibody, anti-β-actin antibody and IFN-γ ELISA Kit were purchased from Abcam (Hanam, Republic of Korea). Fluorescent dye-conjugated antibodies against mouse CD45.2, mouse CD8α, mouse CD3, mouse CD4, mouse CD25 and mouse CD86 (cat# 105005) were purchased from BioLegend (San Diego, CA, USA). Anti-mouse PD-L1 antibody (B7-H1) was purchased from BioXCell (Lebanon, NH, USA). 4T1 (human breast cancer cells) and H9C2 (rat BDIX heart myoblasts) cell lines were purchased from American Type Culture Collection (ATCC; Manassas, VA, USA). Tumor dissociation Kit and T Cell Activation/Expansion Kit were purchased from Miltenyi Biotechnology (Bergisch Gladbach, Germany). Six-week-old female Balb/c and Balb/c nu/nu mice were purchased from NaraBio, Inc (Seoul, Korea).
Molecular dynamics simulation
The preliminary conformers of the molecules were searched by the MacroModel module in Schrödinger Suites (Schrödinger) with the following parameters-force field: OPLS3; solvent: water; charged from force field; cutoff: none; minimization method: PRCG; maximum iterations: 2,500; converge on: gradient; convergence threshold: 0.05; conformational search method: mixed torsional/low-mode sampling; torsional sampling options: intermediate; maximum number of steps:1000; energy window for saving structures: 21 kJ/mol; eliminate redundant conformers using maximum atom deviation cutoff: 0.5.
The molecular dynamics simulations were performed to investigate the packing mode of the two or twenty molecules via the Desmond module in Schrödinger Suites with the following parameters- force field: OPLS3e; solvent model: TIP4PD; ion placement: chloride; boundary conditions: orthorhombic box shape, box size calculation method (buffer); simulation time: 20 ns or 100 ns; approximate number of frames: 1000; ensemble class: NPT; temperature: 300 K; pressure: 1.01325 bar; thermostat method: Nose-Hoover chain; coulombic interaction cutoff radius: 9.0 Å. Intermolecular interactions were analyzed at every 10 frames.
Cell preparation
4T1 (human breast cancer cells), HUVEC (human umbilical vein endothelial cells) and H9C2 (rat BDIX heart myoblasts) cells were maintained in RPMI-1640, Endothelial cell growth medium (EGM) or Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS), 1% streptomycin and 100 U/mL penicillin, respectively. Bone marrow-derived macrophages (BMDMs) and bone marrow-derived dendritic cells (BMDCs) were prepared by isolating bone marrow cells from BALB/c mice. Then, bone marrow cells were differentiated into BMDMs by incubating with M-CSF (Macrophage colony-stimulating factor, 20 ng/mL) for 7 days, or were differentiated into BMDCs by culturing for 7 days in the presence of GM-CSF (20 ng/mL), IL4 (20 ng/mL) and 0.1% β-mercaptoethanol. After isolation, BMDMs and BMDCs were maintained in RPMI-1640 medium supplemented with 10% (v/v) FBS, 1% streptomycin, and 100 U/mL penicillin.
Synthesis of anti-PD-L1 peptide-conjugated prodrug
The anti-PD-L1 peptide-conjugated prodrug was synthesized by one-step conjugation of N-terminal acylated Cys-Val-Arg-Ala-Arg-Thr-Arg-Phe-Arg-Arg-Gly peptide (Ac-CVRARTRFRRG-COOH) and doxorubicin (DOX) via EDC/NHS coupling chemistry. First, Ac-CVRARTRFRRG-COOH (1.8 g, 1.96 mmol), DOX (0.9 g, 1.66 mmol), NHS (400 mg, 3.48 mmol) and EDC (500 mg, 2.62 mmol) were dissolved in anhydrous DMF (100 mL) and then DIPEA (200 mg, 1.55 mmol) was subsequently added in mixture solution. After 6 h of incubation, prodrug was purified using high performance liquid chromatography (HPLC, Agilent Technologies, 1200 series, USA) and lyophilized at -90 oC and 5 mTorr for 24 h to obtain as a red powder (Freeze Dryer, ilShinBioBase, Republic of Korea).
Characterization of anti-PD-L1 peptide-conjugated prodrug nanoparticles
The purity and molecular weight were analyzed using HPLC and matrix-assisted laser desorption/ionization time of flight mass spectrometer (MALDI-TOF, AB Sciex TOF/TOF 5800 System, USA) with cyano-4-hydroycinnamic acid (CHCA) matrix, respectively. The average size of PD-NPs in saline (1 mg/mL) was determined via dynamic light scattering (DLS; Zetasizer Nano ZS Malvern Instruments, UK). The morphology of nanoparticles in distilled water (1 mg/mL) was observed by transmission electron microscope (TEM; CM-200, USA). As a control, the cathepsin B-cleaved PD-NPs were also observed using TEM, wherein PD-NPs (1mg/mL) were pre-incubated with cathepsin B (10 μg) at 37 oC for 24 h. Enzyme cleavage assays were performed by incubating PD-NPs (10 μM) with cathepsin B, cathepsin D, cathepsin E, caspase-3 and caspase-9 (10 μg) in MES buffer (0.1 M, pH 5.5). As a control experiment, PD-NPs (10 μM) were also pre-incubated with irreversible cathepsin B inhibitor (Z-FA-FMK; 50 μΜ) in MES buffer and then further incubated with cathepsin B. Enzyme reaction buffer was analyzed by HPLC using an acetonitrile: H2O gradient from 20:80 (0 min) to 80:20 (20 min). In vitro drug release from the PD-NPs was further investigated under both physiological and enzymatic conditions. For these analyses, PD-NPs dispersed in the mouse serum (1 mg/mL) were loaded in membrane (Molecular weight cut off: 2000 Da; Dialysis membrane Spectra/Por® 7 MWCO 2000, 18 mm; Carl Roth, Karlsruhe, Germany) in the presence or absence of cathepsin B enzyme (10 μg). After incubation, peptide-DOX or free DOX released to the outer membrane was analyzed using HPLC. Fluorescence profiles of PD-NPs (10 μM) in saline was analyzed by fluorescence spectrophotometer (F-7000, Hitachi).
Cellular uptake
To assess the intracellular behavior of PD-NPs, 2 × 105 4T1, HUVEC and H9C2 cells were seeded in glass-bottom confocal dishes. After 24 h of stabilization, each cell was incubated with free DOX or PD-NPs (2 μM) at 37 °C. As a control, 4T1, HUVEC and H9C2 cells were pre-incubated with anti-PD-L1 antibody for 24 h to block the PD-L1 on cell surface. Then, cells were washed with DPBS, fixed with 4% paraformaldehyde for 10 min, and stained with 4',6-diamidino-2-phenylindole (DAPI) for 5 min. Fluorescence imaging was performed using a Leica TCS SP8 confocal laser-scanning microscope (CLSM; Leica Microsystems GmbH; Wetzlar, Germany).
Cytotoxicity assay
The cytotoxicity of PD-NPs was assessed by cell counting kit-8 (CCK-8) assays. First, 1 x 104 4T1, HUVEC, H9C2, BMDMs, BMDCs or CD8+ T cells were seeded in 96-well cell culture plates. After 24 h of stabilization, the PD-NPs or free DOX were treated with each cell for 24 h. Then, the cells were further incubated with culture medium containing CCK-8 solution (10%) for 20 min, and cell viability was measured by a microplate reader with 450 nm of wavelength (VERSAmaxTM; Molecular Devices Corp., USA).
Western blot
PD-L1 expression in 4T1 cells after different treatment was analyzed via western blot. Briefly, 1 × 106 4T1 cells were seeded in 6-well cell culture plates with RPMI medium containing 10% FBS and 1% antibiotics. After 24 h of stabilization, the cells were pre-treated with IFN-γ (20 ng/mL) for 24 h and further incubated with equal molar dose (2 μM) of free DOX, PD-NPs, anti-PD-L1 antibody or anti-PD-L1 peptide (NH2-CVRARTR-COOH) for 24 h. Then, each cell was solubilized with lysis buffer containing 1% protease inhibitors, and the lysates were centrifuged at 10,000 rpm for 15 min to remove cell debris. Proteins in lysates were quantified via BCA protein quantification kit (Thermo Fisher scientific, USA), and then separated by sodium dodecyl sulfate-polyacrylamide (SDS-PAGE) gel electrophoresis and transferred onto polyvinylidene difluoride (PVDF) membranes. Membranes were incubated with TBS-T containing 5% bovine serum albumin (BSA) for 50 min to block non-specific IgG binding and incubated with anti-mouse PD-L1 primary antibody (1000:1) for 12 h at 4 °C. Then, membranes were further incubated with HRP-conjugated anti-rabbit IgG antibody for 90 min at room temperature. Finally, immunoreactive bands were observed via enhanced chemiluminescence (ECL) system.
DAMPs analysis
DAMPs from 4T1 cells were assessed by analyzing CRT, HMGB1 and ATP after treatment. First, 1 x 105 4T1 cells were incubated with free DOX or PD-NPs (2 μM) for 24 h. Then, cells were stained with a APC fluorescent dye-conjugated CRT antibody for 12 h at room temperature, followed by an analysis using a confocal laser-scanning microscope. Second, supernatants from each well were harvested to measure HMGB1 and ATP, which were analyzed via western blot and ATP assay kit, respectively (Beyotime Biotechnology). To evaluate the ICD of PD-NPs for DC maturation, 1 x 105 4T1 cells were cultured in 100-pi culture dishes. After 12 h of stabilization, cells were treated with free DOX or PD-NPs (2 μM) for 24 h. Then, cell culture medium containing DAMPs released from 4T1 cells was co-cultured with immature mouse bone marrow-derived dendritic cells (BMDCs) for 24 h, and the mature DCs were analyzed via flow cytometer (BD FACSVerse, BD Bioscience, USA) after staining with CD11c, CD40 and CD86.
Co-culture of CD8+ T lymphocytes with cancer cells for analysis of T lymphocyte activity
CD8+ T cells were isolated from BALB/c mice using a CD8 enrichment column and then activated by culturing on plates coated with anti-mouse CD3 antibody (1 μg/mL) in culture medium containing anti-mouse CD28 antibody at 37 oC for 3 h. Activated T lymphocytes were co-cultured with 4T1 cells pre-treated with anti-PD-L1 peptide, anti-PD-L1 antibody or PD-NPs (2 μM) at 37 oC for 24 h. To analyze T lymphocyte activity, the secretion of IFN-γ and T lymphocyte proliferation were measured. The levels of IFN-γ in the cell culture medium was measured using a IFN-γ ELISA Kit (Hanam, Republic of Korea) and T lymphocyte proliferation was measured by flow cytometry as below.
CD8+ T lymphocytes were labeled with CellTraceTM Far Red dye (Thermo Fisher Scientific Inc., Rockford, IL, USA) resuspended in dimethyl sulfoxide at a concentration of 5 mM, and the dilution assays of CellTraceTM Far Red dye was performed to analyze T lymphocyte proliferation. Briefly, activated T lymphocytes were incubated with Far Red dye in serum-free culture medium at 37 oC for 10 min, and then cells were resuspended in RPMI1640 medium containing 10% FBS. After 24 h of co-culture with 4T1 cells pre-treated with anti-PD-L1 peptide, anti-PD-L1 antibody or PD-NPs, the decreased fluorescence intensities of daughter T lymphocytes was compared to parental cells after cell division due to the dilution of Far Red dye.
Biodistribution study in breast tumor models
The 6-week female BALB/c mice were purchased from NaraBio (Gyeonggi-do, Republic of Korea). Mice were bred under pathogen-free conditions in the Korea Institute of Science and Technology (KIST). All experiments with animals were performed in compliance with the relevant laws and institutional guidelines of Institutional Animal Care and Use Committee (IACUC; approved number of 2020-123) in Korea Institute of Science and Technology (KIST). The in vivo biodistribution of PD-NPs was assessed in 4T1 tumor-bearing mice, which were prepared by subcutaneous inoculation of 1 x 107 4T1 cells. When the tumor volumes were approximately 150 - 200 mm3, the mice were intravenously injected with free DOX or PD-NPs (3 mg/kg based on DOX contents). Noninvasive near-infrared fluorescence (NIRF) imaging was performed using an IVIS Lumina Series III system (PerkinElmer; Waltham, MA, USA) after 1, 6 and 24 h of injection. Fluorescence intensity in tumor regions was quantified using a Living Image software (PerkinElmer; Waltham, MA, USA). For ex vivo fluorescence imaging, mice were sacrificed after 24 h of injection, followed by extraction of liver, lung, spleen, kidney, heart and tumor tissues. Fluorescence intensity in tissues was analyzed using an IVIS Lumina Series III system. Tumor tissues were cut into 8-μm thick sections for histological assays. Slide-mounted tumor sections were stained with APC fluorescent dye-conjugated CD31 or PD-L1 antibody at 4 oC for 12 h, after which tumor tissues were analyzed using a Leica TCS SP8 confocal laser-scanning microscope.
In vivo tumor inhibition and immune response
4T1 tumor-bearing mice were randomly divided into four groups: (i) saline; (ii) free DOX; (iii) free DOX with anti-PD-L1 antibody; and (iv) PD-NPs. Then, mice were treated once every three days, with free DOX or PD-NPs (3 mg/kg based on DOX contents), at which time tumor volumes had reached 60 - 80 mm3. The anti-PD-L1 antibody (10 mg/kg) was i.p. injected simultaneously with free DOX into mice. Antitumor efficacy was assessed by measuring tumor volumes, calculated as largest diameter x smallest diameter2 x 0.53, every 2 days. For the analysis of antitumor immune responses, tumor tissues were collected on day 7, and single cells were isolated from tumor tissues using a Tumor Dissociation Kit. Following cell counting, the single cells were incubated with FcBlock at 37 oC for 5 min to avoid non-specific antibody binding. Then, multi-parameter staining was performed for 40 min to assess the following populations in tumor tissues; (i) PD-L1+ cancer cells (CD45-PD-L1+); (ii) CRT+ cancer cells (CD45-CRT+); (iii) CD8+ T cells (CD45+CD3+CD8+); and (iv) regulatory T cells (Tregs; CD45+CD3+CD4+CD25+). In order to assay the concentration of cytokines in the tumor microenvironment, the IFN-γ and TNF-α in the tumor supernatants were assessed using commercialized ELISA assay kits (Catalog # SMIF00 and SMTA00B, respectively, R&D systems).
Toxicity study
The in vivo toxicity of PD-NPs was assessed by hematological analyses. Briefly, free DOX or PD-NPs (3 mg/kg based on DOX contents) were intravenously injected once every three days, and anti-PD-L1 antibody (10 mg/kg) was simultaneously i.p. injected into mice. Then, blood samples were collected from mice on day 16. For complete blood counts (CBCs), each blood sample was mixed with EDTA and a portion of each blood sample was centrifuged at 1,000 xg for 20 min, and then supernatants were stored at 4 °C. The following factors in blood samples were measured; aspartate transaminase (AST), alanine aminotransferase (ALT), blood urea nitrogen (BUN), creatinine kinase-MB (CK-MB), white blood cell (WBC), neutrophil (NEUT) and lymphocytes (LYMP).
Statistics
The statistical significance between two groups was analyzed using Student's t-test. One-way analysis of variance (ANOVA) was performed for comparisons of more than two groups, and multiple comparisons were analyzed using Tukey-Kramer post-hoc test. Survival data was plotted as Kaplan-Meier curves and analyzed using log-rank test. Statistical significance was indicated with asterisk (*P < 0.05, **P < 0.01, ***P < 0.001) in the figures.
Data availability
All relevant data are available with the article and its Supplementary Information files, or available from the corresponding authors upon reasonable request.
Results and Discussion
Characterization of anti-PD-L1 peptide-conjugated prodrug nanoparticles (PD-NPs)
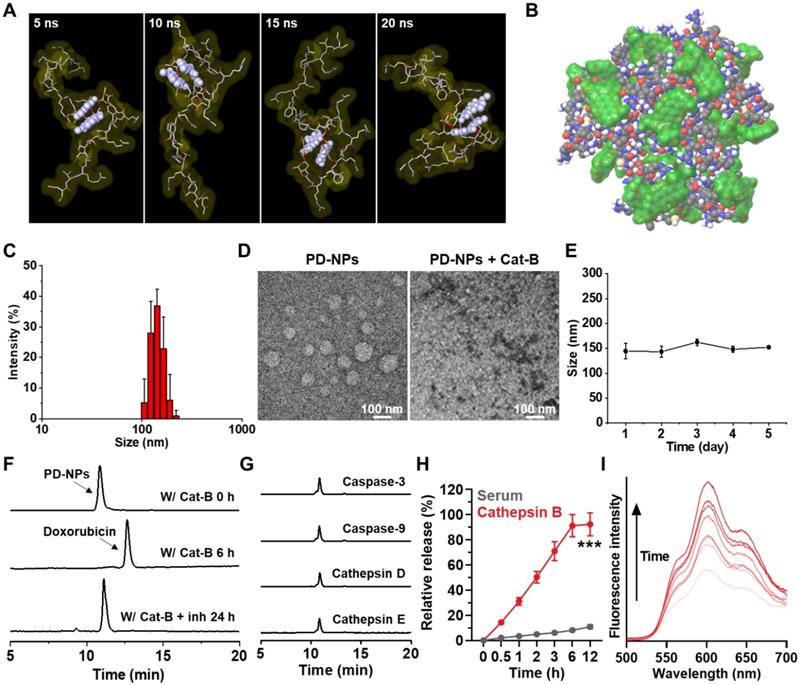
The anti-PD-L1 peptide-conjugated prodrug nanoparticles (PD-NPs) that consist of anti-PD-L1 peptide (CVRARTR; Cys-Val-Arg-Ala-Arg-Thr-Arg), cathepsin B-specific cleavable peptide (FRRG; Phe-Arg-Arg-Gly) and doxorubicin (DOX) were designed for targeted cancer immunotherapy combining PD-L1 blockade with ICD. The anti-PD-L1 peptide of CVRARTR had a potent binding affinity (KD) of 281 ± 92 nM, and it was further modified with cathepsin B-specfic cleavable peptide of FRRG [13]. It was also reported that -RRG- sequence in FRRG peptide was recognized and cleaved by cathepsin B enzyme [22]. To prepare anti-PD-L1 peptide-conjugated prodrug, N-terminal acylated functional peptide (Ac-CVRARTRFRRG-COOH) was directly conjugated to amine group of DOX via a one-step EDC/NHS reaction (Figure S1). After the reaction, 99% of peptide-DOX conjugates was purified with HPLC (Figure S2A). The molecular weight of peptide-DOX conjugate was calculated to be 1945.19 Da for C85H129N27O24S, and measured to be 1945.8717 m/z [M] via MALDI-TOF mass spectrometer (Figure S2B). Importantly, the amphiphilic peptide-DOX conjugates spontaneously self-assembled into nanoparticles in aqueous condition via intermolecular interactions [27, 28]. Therefore, the resulting PD-NPs exhibited a high drug loading contents of 74.3 wt% including 46.4% of anti-PD-L1 peptide (CVRARTR; 903.08 m/z) and 27.9% of DOX (543.53 m/z) in the peptide-DOX conjugates (1945.8717 m/z). The molecular dynamics (MD) simulation results of two molecules of peptide-DOX conjugates indicated that aromatic segments of DOX maintain the average distance of 5.5 Å for 20 ns, which demonstrate that driving force to promote the self-assembly of peptide-drug conjugates is aromatic interactions of DOX molecules (Figure 1A and S3). In addition, all atom MD simulation of twenty molecules of peptide-DOX conjugates further showed a micellar aggregate via π-π stacking, π-cation interactions, and hydrogen bonds, respectively (Figure 1B and S4). The PD-NPs showed an average size of 157.4 ± 12.1 nm in saline, as confirmed by dynamic light scattering (DLS; Figure 1C). TEM images also indicated the spherical structure of PD-NPs, and their nano-sized structure was dissociated after incubation with cathepsin B at 37 oC for 24 h (Figure 1D). In addition, PD-NPs were well-dispersed in mouse serum without significant change in particle size for 5 days (Figure 1E). These stable nanoparticle characteristics of PD-NPs in blood are expected that PD-NPs can increase tumor accumulation via EPR effect in vivo [29]. The target enzyme-specific cleavage of PD-NPs was assessed after incubation with various enzymes. When PD-NPs were incubated with cathepsin B (10 μg) in MES buffer (pH 5.5), nanoparticles began to release free DOX after 1 h of incubation, and approximately 99% was cleaved at 6 h post-incubation (Figure 1F). This was clearly supported by MALDI-TOF result that confirm molecular weights of free DOX (calculated mass: 543.52 Da, measured mass: 543.009 m/z [M], 549.947 m/z [M+Li], 567.9522 m/z [M+Na]) at a newly apperared characteristic peak (12 min) after incubation with cathepsin B enzyme in HPLC spectrum (Figure S5). In contrast, PD-NPs were not cleaved in the cathepsin B-suppressed condition that cathepsin B is pre-incubated with irreversible cathepsin B inhibitor (Figure 1F). Similar results were also observed when PD-NPs were incubated with other enzymes, such as caspase-3, caspase-9, cathepsin D and cathepsin E for 24 h of incubation (Figure 1G). The in vitro drug release of PD-NPs under physiological and enzymatic conditions was further investigated, wherein the PD-NPs dispersed in mouse serum (1 mg/mL) were loaded in the membranes (Molecular weight cut off: 2000 Da) in the presense or absence of cathepsin B enzyme. The results showed that only approximately 10% of peptide-DOX was released to the outer membrane, but over 90% of free DOX was observed in same condition in the presence of cathepsin B, indicating high target enzyme-specificity and stability in physiological condition (Figure 1H). As a control, the cathepsin B-scrambled functional peptide-conjugated DOX (CVRARTR-FGRG-DOX) was not cleaved by cathepsin B, indicating high target enzyme specificity of -RRG- peptide sequence (Figure S6). Finally, the fluorescence intensities of PD-NPs in saline were gradually increased in a time-dependent manner after incubation with cathepsin B (Figure 1I). These results demonstrate that PD-NPs maintain self-quenched inactive state as a stable nano-sized structure, but release free DOX by enzymatic degradation in the presence of cathepsin B.
Physicochemical characterization of PD-NPs. A. All atom molecular dynamics (MD) simulation results of two molecules of peptide-drug conjugates. Aromatic segment of DOX was highlighted by light purple, for clarity. B. MD simulation of twenty molecules of peptide-drug conjugates. Molecular surfaces of DOX were colored by translucent green. C. Size distribution of PD-NPs in saline was confirmed using dynamic light scattering (DLS). D. TEM image of PD-NPs in distilled water. As a control, PD-NPs incubated with cathepsin B (10 μg) at 37 oC for 24 h were observed via TEM. E. Size stability of PD-NPs in mouse serum for 5 days. F-G. HPLC spectrum when the PD-NPs were incubated with F. cathepsin B (pH 5.5) or cathepsin B with inhibitor (Z-FA-FMK), and G. other enzymes. H. In vitro drug release from PD-NPs in serum in the presence or absence of cathepsin B enzyme. I. Time-dependent fluorescence spectra of PD-NPs in the presence of cathepsin B for 48 h. Y-axis indicates the fluorescence intensity from PD-NPs. The DOX fluorescence (590 nm) from PD-NPs was gradually increased in a time-dependent manner after incubation with cathepsin B owing to de-quenching effect by enzymatic degradation. Significance was determined by Student's t test (H).
PD-L1 binding and cancer cell-specific cytotoxicity of PD-NPs
The in vitro binding between PD-NPs and PD-L1 on the cell surface was assessed in breast cancer cells. This is because breast cancer express significantly higher levels of PD-L1 compared to other cancers in clinic [30]. Moreover, breast cancer cells (4T1) expressed 5.01 ± 0.48-fold and 10.23 ± 1.66-fold high levels of PD-L1 in comparison to normal cells of human umbilical vein endothelial cells (HUVEC) and rat BDIX cardiomyocytes (H9C2; Figure 2A and S7). In cultured condition, PD-NPs (red color) were efficiently taken up by 4T1 cells in an incubation time-dependent manner (Figure 2B). However, pretreatment with anti-PD-L1 antibody to block the PD-L1 on cell surface significantly reduced the cellular uptake of PD-NPs, indicating that PD-NPs enter cancer cells through PD-L1 receptor-mediated endocytosis (Figure 2B). Quantitatively, cellular uptake of PD-NPs was 5.314 ± 0.481-fold higher in naive 4T1 cells than in anti-PD-L1 antibody-pretreated 4T1 cells after 24 h of incubation (Figure 2C). Moreover, these fluorescence imaging also showed that intracellular PD-NPs remained to the cytosol until 6 h of incubation, whereas DOX fluorescence was clearly observed in the nuclei from 9 h of incubation. Upon enhanced internalization driven by PD-L1-binding, DOX delivery into the nuclei was 3.04 ± 0.28-fold higher in naive 4T1 cells than in anti-PD-L1 antibody-pretreated 4T1 cells (Figure S8). The mode of action (MOA) of DOX is intercalating within DNA base pairs to cause breakage of DNA strand and inhibition of both RNA and DNA replication. Therefore, DOX inside the nuclei of the cells is an important indicator for its potent cytotoxicity. As control experiments, the non-specific binding of PD-NPs on HUVEC was evaluated because the PD-L1 expression levels of HUVEC cells are significantly higher than other normal cells of H9C2. However, the low cellular binding and uptake of PD-NPs were observed in HUVEC and H9C2, and those were similar irrespective of the pretreatment of anti PD-L1 antibody owing to much low PD-L1 expression than 4T1 cancer cells (Figure S9).
PD-L1 binding and cancer cell-specific cytotoxicity of PD-NPs. A. Relative PD-L1 expression in 4T1, HUVEC and H9C2 cells. B. Time-dependent cellular uptake of PD-NPs in 4T1 cells. As a control, anti-PD-L1 antibody was pre-incubated with 4T1 cells for 24 h before PD-NPs treatment. C. Quantification analysis on the amount of cellular uptake of PD-NPs in 4T1 or anti-PD-L1 antibody-treated 4T1 cells. D. Schematic illustration to explain the mechanism of PD-L1 blockade by PD-NPs. E. Fluorescence imaging of APC-conjugated anti-PD-L1 antibody-treated 4T1 cells after pretreatment of FRRG-DOX or PD-NPs for 2 h at 4 oC. F. Lysosome co-localization of PD-NPs in 4T1 cells. Intracellular lysosomes were labeled with Lamp1-RFP. G. Quantitative analysis of the co-localization of PD-NPs with lysosomes (Lamp1-RFP) or nuclei (DAPI) in the 4T1 cells. H. PD-L1 expression on the cell surface after different treatment was analyzed via western blot. I. Fluorescence imaging of 4T1, HUVEC and H9C2 cells treated with PD-NPs or free DOX for 24 h. J-K. Cytotoxicity of J. PD-NPs and K. free DOX in 4T1, HUVEC and H9C2 cells after 24 h treatment. Significance was determined by Student's t test (C) or Tukey-Kramer posthoc test (A, E, J).
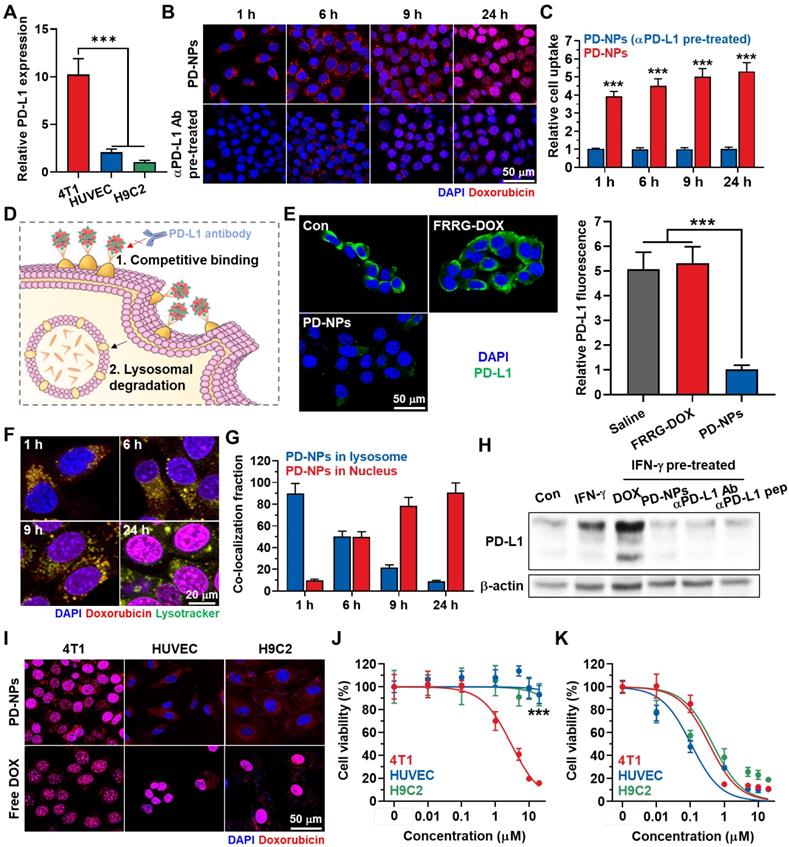
Next, we further assessed whether the binding to PD-L1 of PD-NPs can lead to blockade of PD-L1 via competitive binding and lysosomal PD-L1 degradation, and detail mechanisms were shown in Figure 2D. First, pretreatment with PD-NPs for 2 h at 4 oC significantly reduced APC-conjugated anti-PD-L1 antibody binding to PD-L1 on the cell surface via competitive binding, whereas pretreatment of FRRG-DOX that absence of anti-PD-L1 peptide (CVRARTR) did not affect to the PD-L1 antibody binding compared to naive cells (Figure 2E). We have also demonstrated substantial PD-NPs internalized into lysosomes, wherein the strong co-localization of PD-NPs (red color) and lysosome (green color) was clearly observed in 4T1 cells (Figure 2F). After 1 h of incubation, approximately 90% of PD-NPs was localized in the lysosomes, whereas DOX fluorescence in nuclei was gradually increased from 6 h post-incubation (Figure 2G). Recent study noted that controlled trafficking of PD-L1 to targeted degradation in the lysosome might be a key to prevent unwanted PD-L1 recycling, thus these intracellular fate of PD-NPs can also increase the lysosomal PD-L1 degradation [16]. As a result, the treatment of 4T1 cells with IFN-γ for 24 h resulted in significant PD-L1 upregulation, and subsequent treatment of free DOX further increased the PD-L1 expression (Figure 2H). However, PD-NPs significantly downregulated the PD-L1 expression levels of IFN-γ-treated 4T1 cells, and their treatment-induced downregulation of PD-L1 was nearly similar with anti-PD-L1 peptide (CVRARTR) or antibody. These results clearly indicate that PD-NPs successfully block the recognition of PD-L1 on cell surface by anti-PD-L1 antibody that can be considered a PD-1 of T lymphocytes via competitive binding and lysosomal degradation for PD-L1.
The cancer cell-specific cytotoxicity of PD-NPs premised on differential cathepsin B expression was assessed in 4T1, HUVEC and H9C2 cells. As expected, 4T1 cells expressed 26.13 ± 2.78-fold and 32.48 ± 3.14-fold of cathepsin B, and 4.39 ± 0.41-fold and 5.62 ± 0.581-fold of pro-cathepsin B compared to HUVEC and H9C2 cells, respectively (Figure S10). Three types of cells showed robust uptake of PD-NPs (red color) after 24 h of incubation as confirmed by confocal laser scanning microscope (CLSM; Figure 2I). Importantly, DOX fluorescence of PD-NPs remained limited to the cytoplasm of HUVEC and H9C2 cells, whereas that was observed in the nuclei of the 4T1 cells. In contrast, intracellular behavior of free DOX was similar regardless of cathepsin B expression in cells, wherein DOX fluorescence was clearly detected in the nuclei of three types of cells. Furthermore, such different intracellular behavior of PD-NPs resulted in cancer cell-specific cytotoxicity. The IC50 values of PD-NPs were measured to be 3.01 μM, 200 μM and > 200 μM in 4T1, HUVEC and H9C2 cells, respectively, which showed over a 70-fold difference that indicate greatly minimized cytotoxicity against normal cells (Figure 2J). In contrast, free DOX exhibited indiscriminate cytotoxicity with similar IC50 in three types of cells (Figure 2K). Taken together, these results indicate that PD-NPs specifically release free DOX into the nuclei of cathepsin B-overexpressed cancer cells, whereas maintain non-toxic inactive state in the cytosol of normal cells with innately low cathepsin B expression. In addition, these cancer cell-specific cytotoxicity of PD-NPs also suggest that PD-NPs can greatly mitigate the side effects of ICB immunotherapy by minimized toxicity towards normal tissues.
In vitro immunogenic cell death and reinvigoration of T lymphocytes by PD-NPs
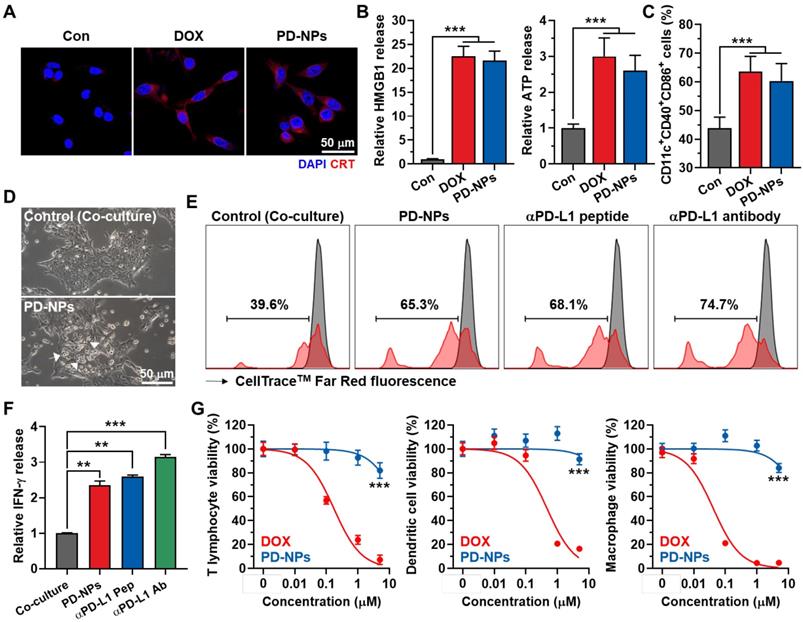
The immunogenic cell death (ICD) by PD-NPs in cancer cells was examined by measuring the amount of DAMPs, such as caleticulin (CRT) expression on the cell surface, extracellular release of high mobility group box 1 (HMGB1) and adenosine triphosphate (ATP). After 24 h of treatment, the CRT expression on the surface of 4T1 cells treated with PD-NPs was similar with free DOX (Figure 3A). In addition, there was no significant differences in extracellular release of HMGB1 and ATP from 4T1 cells treated with PD-NPs or free DOX, and those were significantly higher than naive cells (Figure 3B and S11). The effective ICD by PD-NPs was further evaluated in co-culture study. For these analyses, 4T1 cells were treated with free DOX or PD-NPs (2 μM) for 24 h, and cell culture medium containing DAMPs released from the cells was incubated with immature mouse bone marrow-derived dendritic cells (BMDCs) for 24 h. The results showed the equivalent levels of mature DCs (CD11c+CD40+CD86+) in BMDCs after each drug treatment, which is attributable to the DAMPs induced by strong ICD of the PD-NPs (Figure 3C and S12). To examine the ability of PD-NPs to reinvigorate T lymphocyte activity by blockade of interactions between PD-1 and PD-L1, we exploited the co-culture of T lymphocytes with 4T1 cells. For this study, CD8+ T lymphocytes were isolated from mice spleen and activated by culturing with medium containing anti-CD28 antibody on anti-CD3 antibody-coated plates, and subsequently co-cultured with 4T1 cells for 24 h. Interestingly, T lymphocytes located near 4T1 cells treated with PD-NPs was considerably increased than naive 4T1 cells after washout, suggesting the increase of cancer cell recognition by T lymphocytes (Figure 3D). As a result, co-culture with PD-NPs-treated 4T1 cells significantly increased the T lymphocyte proliferation and the secretion of IFN-γ from T lymphocytes (2.35 ± 0.13-fold compared to control), and those were similar with anti-PD-L1 peptide (CVRARTR) and antibody groups (Figure 3E-F). These results indicate that PD-NPs successfully block the interaction between PD-1 and PD-L1 to enhance T lymphocyte target recognition, which lead reinvigoration effect of T lymphocytes. We further assessed the cytotoxicity of PD-NPs against immune cells, such as T lymphocytes, dendritic cells (DCs) and macrophages. Because it has reported that immune cells express relatively lower cathepsin B compared to cancer cells, we expected that PD-NPs can reduce cytotoxicity towards immune cells to allerviate their dysfunction [5]. The results showed that cytotoxicity of PD-NPs was significantly reduced in T lymphocytes (P < 0.001), DCs (P < 0.001) and macrophages (P < 0.001) compared to free DOX (Figure 3G). Taken together, PD-NPs can elicit effective ICD in cancer cells by DOX release as well as promote proliferation and reinvigoration of T lymphocytes by PD-L1 blockade. In addition, PD-NPs based ICB immunotherapy may greatly reduce damage to host immune system owing to minimized toxicity against immune cells.
ICD and reinvigoration of T lymphocytes by PD-NPs. A. CRT expression in the 4T1 cells after saline, free DOX or PD-NPs treatment. B. Quantification analysis on the amount of HMGB1 and ATP released from 4T1 cells after saline, free DOX or PD-NPs treatment. C. The levels of mature DCs (CD11c+CD40+CD86+) in BMDCs after co-culture with culture medium containing DAMPs released from 4T1 cells treated with free DOX or PD-NPs (2 μM). D. Photographs of T lymphocytes co-cultured with naive or PD-NPs-treated 4T1 cells for 24 h. E. T lymphocyte proliferation assays after 24 h of co-culture with 4T1 cells treated with PD-NPs, anti-PD-L1 peptide (CVRARTR) or anti-PD-L1 antibody. F. Relative amount of IFN-γ in culture medium after co-culture of T lymphocyte with 4T1 cells treated with PD-NPs, anti-PD-L1 peptide (CVRARTR) or anti-PD-L1 antibody for 24 h. G. Cytotoxicity of PD-NPs and free DOX in T lymphocytes, DCs and macrophages after 24 h treatment. Significance was determined by Student's t test (G) or one-way ANOVA with the Tukey-Kramer posthoc test (B, C, F).
Antitumor efficacy and immune reponses by PD-NPs in breast tumor models
The tumor-targeting ability of PD-NPs was monitored in breast tumor models via noninvasive near-infrared fluorescence (NIRF) imaging. The 4T1 tumor-bearing mice were prepared by subcutaneous inoculation of 1 x 106 4T1 cells into the BALB/c mice. When the tumor volumes were approximately 150 - 200 mm3, PD-NPs or free DOX (3 mg/kg based on DOX contents) were intravenously injected into the mice. The NIRF intensity of free DOX is not large enough in vivo with limited tissue penetration to screen the fluorescence of PD-NPs in whole body, thus fluorescence imaging was performed by appointing the tumor areas. In case of free DOX, only weak DOX fluorescence was observed at targeted tumor tissues and then rapidly decreased after 6 h of injection (Figure 4A). In contrast, PD-NPs at tumor tissues were gradually increased until 6 h post-injection and sustainably retained for 24 h, which showed 4.01-4.48 times more accumulation compared to free DOX after 24 h injection (Figure S13). These NIRF imaging confirm efficient tumor accumulation of PD-NPs via nanoparticle-derived EPR effect. The histological assay of whole tumor tissues stained with CD31 also showed high tumor accumulation of PD-NPs than free DOX, wherein the strong DOX fluorescence of PD-NPs (red color) along to CD31-positive blood vessels (green color) was clearly observed (Figure 4B). In addition, tumor tissues stained with APC-conjugated anti-PD-L1 antibody showed low PD-L1 expression in PD-NPs group compared to free DOX group with PD-L1 upregulation, indicating the successful PD-L1 blockade by PD-NPs (Figure 4C).
Antitumor efficacy and immune responses by PD-NPs. A. Noninvasive NIRF images of 4T1 tumor-bearing mice treated with free DOX or PD-NPs (3 mg/kg based on DOX contents). B. Tumor tissues stained with APC-conjugated CD31 antibody after 24 h of free DOX or PD-NPs treatment. C. Tumor tissues stained with APC-conjugated PD-L1 antibody after 24 h of free DOX or PD-NPs treatment. D. Tumor growth curves of 4T1 tumor-bearing mice during saline, free DOX, free DOX with PD-L1 antibody, or PD-NPs treatment. Mice were treated once every three days. E. Tumor tissues stained with TUNEL to assess apoptosis on day 16 after treatment. F. Relative amount of HMGB1 and ATP in the tumor supernatants were analyzed on day 7 after treatment. G-I. Percentage of G. CRT-positive cancer cells (CD45-CRT+), PD-L1-positive cancer cells (CD45-PD-L1+), H. cytotoxic T lymphocytes (CD45+CD3+CD8+), I. Tregs (CD45+CD3+CD4+CD25+) and the ratio of CD8+ T lymphocytes to Tregs in the tumor tissues on day 7 after treatment. J. Relative concentration of the IFN-γ and TNF-α in the tumor supernatants from 4T1 tumor-bearing mice after 7 days of treatment. The asterisks in Figure indicate the significance in comparison to PD-NPs group. Significance was determined by one-way ANOVA with the Tukey-Kramer posthoc test.
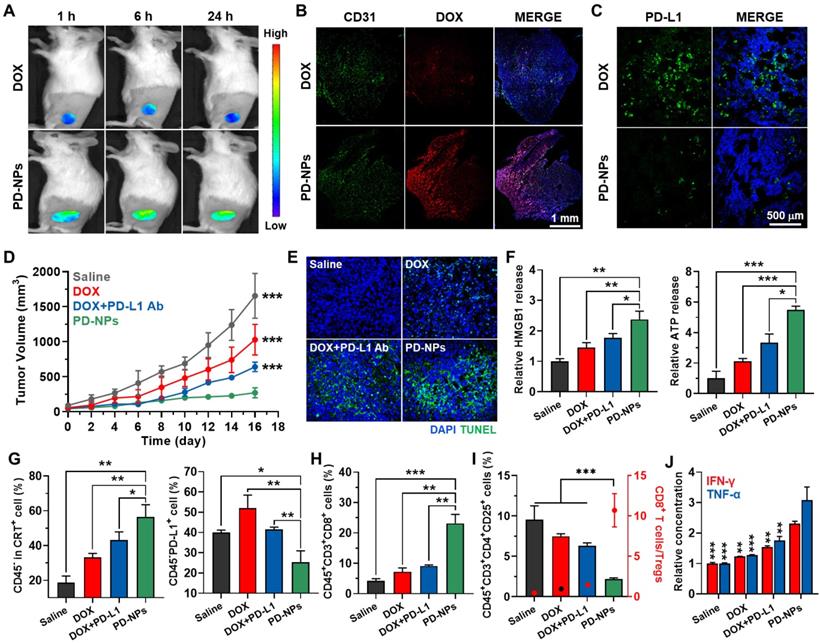
Next, the enhanced antitumor efficacy and immune responses by PD-NPs were assessed in 4T1 tumor-bearing mice. The mice were randomly divided into four groups of saline, free DOX (3 mg/kg), free DOX with anti-PD-L1 antibody (10 mg/kg), and PD-NPs (3 mg/kg based on DOX contents). Each drug was intravenously injected into mice once every three days, and anti-PD-L1 antibody was i.p. injected simultaneously with free DOX. Importantly, the PD-NPs group (271.15 ± 71.49 mm3) showed minimal tumor progression on day 16 compared to saline (1655.9 ± 319.94 mm3), free DOX (1028.73 ± 218.15 mm3), and free DOX with anti-PD-L1 antibody (641.45 ± 67.22 mm3) groups (Figure 4D). Tumor tissues stained with TUNEL clearly exhibited that PD-NPs treatment resulted in elevated apoptosis than other treatments, thereby confirming the enhanced antitumor efficacy by targeted tumor delivery of DOX and anti-PD-L1 peptide (Figure 4E). Next, we investigated the antitumor immune responses by measuring DAMPs from tumor tissues on day 7 after treatment. Firstly, extracellular release of HMGB1 and ATP into the tumor supernatants was significantly increased in the PD-NPs group than other groups (Figure 4F and S14). In addition, the population of CRT positive tumor cells, PD-L1 positive tumor cells, cytotoxic T lymphocytes and regulatory T lymphocytes in tumor tissues was analyzed by flow cytometry on day 7 after treatment (Figure S15). In case of free DOX, the population of CRT positive tumor cells (CD45-CRT+) was increased by inducing ICD, but PD-L1 positive tumor cells (CD45-PD-L1+) were also increased compared to saline group (Figure 4G). In contrast, free DOX with anti-PD-L1 antibody group showed significantly increased CRT positive tumor cells, whereas PD-L1 positive tumor cells were decreased compared to free DOX group. Most importantly, PD-NPs group showed a most high population of CRT positive tumor cells (CD45-CRT+), while PD-L1 positive tumor cells (CD45-PD-L1+) were dramatically decreased compared to other groups. These results indicate the effective ICD and PD-L1 blockade by PD-NPs, which induced immune-responsive tumors by recruiting a large amount of immune cells at the tumor tissues. Consequentially, the population of cytotoxic T lymphocytes (CD45+CD3+CD8+) in the tumor tissues was considerably increased in the PD-NPs group (Figure 4H). In contrast, the percentage of regulatory T cells (Tregs; CD45+CD3+CD4+CD25+) in the tumor tissues was significantly decreased in the PD-NPs group, wherein the ratio of CD8+ T lymphocytes to Tregs was greatly upregulated compared to other groups (Figure 4I). Finally, the IFN-γ and TNF-α in the tumor microenvironment were significantly increased in the PD-NPs group, indicating high activity of tumor-infiltrating lymphocytes (TILs) secreting cytokines (Figure 4J). Taken together, targeted delivery of anti-PD-L1 peptide and DOX by PD-NPs efficiently inhibit tumor progression by inducing strong antitumor immune responses, which are attributable to the effective ICD and PD-L1 blockade.
Safety of ICB immunotherapy by PD-NPs
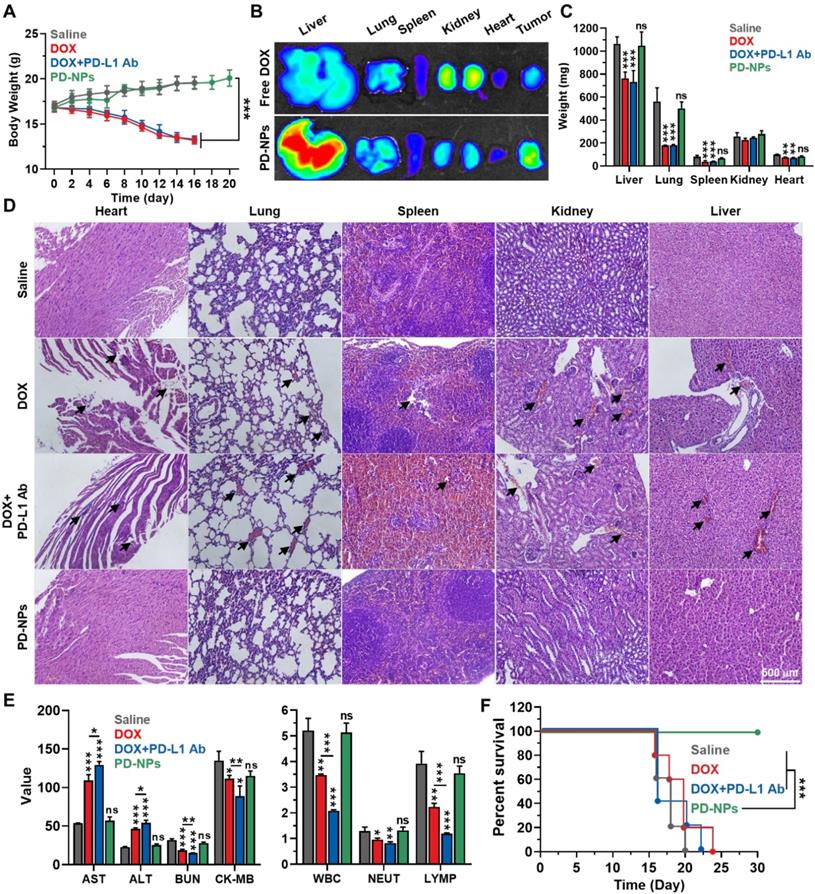
The in vivo safety of PD-NPs based ICB immunotherapy was compared to free DOX or free DOX with anti-PD-L1 antibody in 4T1 tumor-bearing mice. The mice were treated once every three days as in Figure 4. First, the body weights of mice in the PD-NPs group were gradually increased during treatment and similar with saline group, whereas free DOX and free DOX with anti-PD-L1 antibody groups showed severe body weight loss owing to the in vivo systemic toxicity (Figure 5A). After 24 h of injection, PD-NPs showed significantly higher accumulation in the liver tissues, and distribution in other organs was similar with free DOX as shown in ex vivo NIRF imaging (Figure 5B and S16). However, their toxicity towards organs was greatly minimized by high cathepsin B-specificity, as confirmed by histological and hematological analyses, on the 16 day that occurred serious body weight loss in free DOX and free DOX with anti-PD-L1 antibody groups. The free DOX and free DOX with anti-PD-L1 antibody caused serious reduction of organ weights, while those in the PD-NPs group were similar with saline group (Figure 5C). In addition, the major organs stained with H&E exhibited substantial damaged areas in free DOX and free DOX with anti-PD-L1 antibody groups, but significant structural abnormalities were not observed in the PD-NPs group (Figure 5D). The blood analyses exhibited severe cardiac, renal and hepatic toxicity in the free DOX group, as confirmed by significant changes in the hematological parameters, such as aspartate aminotransferase (AST), alanine transaminase (ALT), blood urea nitrogen (BUN) and creatinine kinase (CK; Figure 5E and S17). Importantly, organ toxicity more worsens in the free DOX with anti-PD-L1 antibody group, and severe leukopenia, neutropenia and lymphocytopenia were also observed. The extensive organ dysfunction accompanying hematological toxicity is representative immune-related adverse events (irAEs), thus these results show limitations of current ICB immunotherapy that is suffer from severe side effects [9, 31]. In contrast, those hematological parameters in the PD-NPs group were in normal range and similar with saline group. In agreement with the above toxicity analyses, the median survival of free DOX and free DOX with anti-PD-L1 antibody groups was measured to be 20 and 16 days, respectively, whereas PD-NPs-treated mice were survived over 30 days (Figure 5F). As a control, saline group showed the median survival of 18 days, wherein the mice were dead due to tumor progression. These findings demonstrate that PD-NPs efficiently mitigate the both of off-target toxicity and irAEs by chemotherapeutic drugs and mAbs for ICB immunotherapy.
Safety of ICB immunotherapy by PD-NPs. A. Body weights during treatment with free DOX (3 mg/kg), free DOX with PD-L1 antibody (10 mg/kg), or PD-NPs (3 mg/kg based on DOX contents) once every three days. B. NIRF imaging of major organs from mice treated with free DOX or PD-NPs after 24 h injection. C. Organ weights after 16 days of treatment. D. Major organ tissues stained with H&E after 16 days of treatment. Black arrows indicate structural abnormalities. E. Blood analyses of mice treated with free DOX, free DOX with PD-L1 antibody, or PD-NPs once every three days. Blood samples were collected after 16 days of treatment. F. Survival analysis of mice. Significance was determined by one-way ANOVA with the Tukey-Kramer posthoc test (A, C, E) or log-rank test (F). The asterisk (*P < 0.05, **P < 0.01, ***P < 0.001) in Figures indicate comparison with Saline group (C, E).
Conclusions
In summary, we proposed how overcoming the unfavorable limitations of current ICB immunotherapy with anti-PD-L1 peptide-conjugated prodrug nanoparticles (PD-NPs) via targeted tumor delivery of anti-PD-L1 peptide and DOX for combining PD-L1 blockade and ICD. The PD-NPs self-assembled of functional peptide-drug conjugates exhibited high and accurate drug loading contents and stable nanoparicle strcutre in physiological condition without additional carrier materials. Importantly, the PD-NPs selectively released free DOX in cathepsin B-overexpressed cancer cells after PD-L1-mediated endocytosis, which provoked strong antitumor immune responses through combining of PD-L1 blockade and ICD. The in vivo study further demonstrated that highly accumulated PD-NPs in the tumor tissues via EPR effect not only turn the immunosuppressive tumor microenvironment to the immune-responsive tumors by recruiting a large amount of immune cells through DOX-mediated ICD, but also enhance the pre-existing antitumor immune responses of T lymphocytes by anti-PD-L1 peptide-mediated blockade of PD-1/PD-L1 interactions. As a result, localized tumor delivery of anti-PD-L1 peptide and DOX by PD-NPs greatly inhibited the tumor progression with minimal side effects. This work provides an approach for effective and safe ICB immunotherapy, which may open avenues for advanced prodrug design and provide important insight into translational nanomedicine.
Abbreviations
DOX: doxorubicin; ICB: immune checkpoint blockade; mAbs: monoclonal antibodies; PD-1: programmed death-receptor 1; PD-L1: programmed death-ligand 1; CTLA-4: cytotoxic T lymphocyte associated protein 4; DAMPs: damage-associated molecular patterns; DCs: dendritic cells; ITM: immunosuppressive tumor microenvironment; irAEs: immune-related adverse events; EPR: enhanced permeability and retention; CRT: calreticulin; HMGB1: high mobility group box 1; ATP: adenosine triphosphate; AST: aspartate transaminase; ALT: alanine aminotransferase; BUN: blood urea nitrogen; CK-MB: creatinine kinase-MB; WBC: white blood cell; NEUT: neutrophil; LYMP: lymphocytes; CBCs: complete blood counts; HPLC: highperformance liquid chromatography; DLS: dynamic light scattering; TEM: transmission electron microscopy; H&E: hematoxylin-eosin; TUNEL: terminal deoxynucleotidyl transferase dUTP nick end labeling.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported from the National Research Foundation (NRF) of South Korea, funded by the Ministry of Science (NRF-2019R1A2C3006283) of Republic of Korea, the KU-KIST Graduate School of Converging Science and Technology (Korea University & KIST), and the Intramural Research Program of KIST.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wei SC, Duffy CR, Allison JP. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018;8:1069
2. Galluzzi L, Humeau J, Buqué A, Zitvogel L, Kroemer G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat Rev Clin Oncol. 2020;17:725-41
3. Zhao X, Yang K, Zhao R, Ji T, Wang X, Yang X. et al. Inducing enhanced immunogenic cell death with nanocarrier-based drug delivery systems for pancreatic cancer therapy. Biomaterials. 2016;102:187-97
4. Xiao X, Liang S, Zhao Y, Pang M, Ma Pa, Cheng Z. et al. Multifunctional carbon monoxide nanogenerator as immunogenic cell death drugs with enhanced antitumor immunity and antimetastatic effect. Biomaterials. 2021;277:121120
5. Yang S, Shim MK, Kim WJ, Choi J, Nam G-H, Kim J. et al. Cancer-activated doxorubicin prodrug nanoparticles induce preferential immune response with minimal doxorubicin-related toxicity. Biomaterials. 2021;272:120791
6. Yang K, Liu Y, Wang Y, Ren Q, Guo H, Matson JB. et al. Enzyme-induced in vivo assembly of gold nanoparticles for imaging-guided synergistic chemo-photothermal therapy of tumor. Biomaterials. 2019;223:119460
7. Xie L, Wang G, Sang W, Li J, Zhang Z, Li W. et al. Phenolic immunogenic cell death nanoinducer for sensitizing tumor to PD-1 checkpoint blockade immunotherapy. Biomaterials. 2021;269:120638
8. Mathios D, Kim JE, Mangraviti A, Phallen J, Park C-K, Jackson CM. et al. Anti-PD-1 Antitumor Immunity Is Enhanced by Local and Abrogated by Systemic Chemotherapy in GBM. Sci Transl Med. 2016;8:370ra180-370ra180
9. Postow MA, Sidlow R, Hellmann MD. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N Engl J Med. 2018;378:158-68
10. Huang J, Xiao Z, An Y, Han S, Wu W, Wang Y. et al. Nanodrug with dual-sensitivity to tumor microenvironment for immuno-sonodynamic anti-cancer therapy. Biomaterials. 2021;269:120636
11. Chang H-N, Liu B-Y, Qi Y-K, Zhou Y, Chen Y-P, Pan K-M. et al. Blocking of the PD-1/PD-L1 Interaction by a D-Peptide Antagonist for Cancer Immunotherapy. Angew Chem Int Ed. 2015;54:11760-4
12. Sasikumar PG, Ramachandra RK, Adurthi S, Dhudashiya AA, Vadlamani S, Vemula K. et al. A Rationally Designed Peptide Antagonist of the PD-1 Signaling Pathway as an Immunomodulatory Agent for Cancer Therapy. Mol Cancer Ther. 2019;18:1081
13. Gurung S, Khan F, Gunassekaran GR, Yoo JD, Poongkavithai Vadevoo SM, Permpoon U. et al. Phage display-identified PD-L1-binding peptides reinvigorate T-cell activity and inhibit tumor progression. Biomaterials. 2020;247:119984
14. Zaman R, Islam RA, Ibnat N, Othman I, Zaini A, Lee CY. et al. Current strategies in extending half-lives of therapeutic proteins. J Control Release. 2019;301:176-89
15. Cheng K, Ding Y, Zhao Y, Ye S, Zhao X, Zhang Y. et al. Sequentially Responsive Therapeutic Peptide Assembling Nanoparticles for Dual-Targeted Cancer Immunotherapy. Nano Lett. 2018;18:3250-8
16. Li L, Li Y, Yang C-H, Radford DC, Wang J, Janát-Amsbury M. et al. Inhibition of Immunosuppressive Tumors by Polymer-Assisted Inductions of Immunogenic Cell Death and Multivalent PD-L1 Crosslinking. Adv Funct Mater. 2020;30:1908961
17. Yang S, Sun I-C, Hwang HS, Shim MK, Yoon HY, Kim K. Rediscovery of nanoparticle-based therapeutics: boosting immunogenic cell death for potential application in cancer immunotherapy. J Mater Chem B. 2021;9:3983-4001
18. Lim S, Park J, Shim MK, Um W, Yoon HY, Ryu JH. et al. Recent advances and challenges of repurposing nanoparticle-based drug delivery systems to enhance cancer immunotherapy. Theranostics. 2019;9:7906-23
19. Jeon IS, Yoo JD, Gurung S, Kim M, Lee C, Park EJ. et al. Anticancer nanocage platforms for combined immunotherapy designed to harness immune checkpoints and deliver anticancer drugs. Biomaterials. 2021;270:120685
20. Armand P. Immune checkpoint blockade in hematologic malignancies. Blood. 2015;125:3393-400
21. Hare JI, Lammers T, Ashford MB, Puri S, Storm G, Barry ST. Challenges and Strategies in Anti-Cancer Nanomedicine Development: An Industry Perspective. Adv Drug Deliv Rev. 2017;108:25-38
22. Shim MK, Park J, Yoon HY, Lee S, Um W, Kim J-H. et al. Carrier-free nanoparticles of cathepsin B-cleavable peptide-conjugated doxorubicin prodrug for cancer targeting therapy. J Control Release. 2019;294:376-89
23. Shim MK, Moon Y, Yang S, Kim J, Cho H, Lim S. et al. Cancer-specific drug-drug nanoparticles of pro-apoptotic and cathepsin B-cleavable peptide-conjugated doxorubicin for drug-resistant cancer therapy. Biomaterials. 2020;261:120347
24. Choi J, Shim MK, Yang S, Hwang HS, Cho H, Kim J. et al. Visible-Light-Triggered Prodrug Nanoparticles Combine Chemotherapy and Photodynamic Therapy to Potentiate Checkpoint Blockade Cancer Immunotherapy. ACS Nano. 2021;15:12086-98
25. Kim J, Shim MK, Cho Y-J, Jeon S, Moon Y, Choi J. et al. The safe and effective intraperitoneal chemotherapy with cathepsin B-specific doxorubicin prodrug nanoparticles in ovarian cancer with peritoneal carcinomatosis. Biomaterials. 2021;279:121189
26. Baghdadi M, Wada H, Nakanishi S, Abe H, Han N, Putra WE. et al. Chemotherapy-Induced IL34 Enhances Immunosuppression by Tumor-Associated Macrophages and Mediates Survival of Chemoresistant Lung Cancer Cells. Cancer Res. 2016;76:6030
27. Huang L, Zhao S, Fang F, Xu T, Lan M, Zhang J. Advances and perspectives in carrier-free nanodrugs for cancer chemo-monotherapy and combination therapy. Biomaterials. 2021;268:120557
28. Li G, Sun B, Li Y, Luo C, He Z, Sun J. Small-Molecule Prodrug Nanoassemblies: An Emerging Nanoplatform for Anticancer Drug Delivery. Small. 2021: 2101460.
29. Torchilin V. Tumor Delivery of Macromolecular Drugs Based on the EPR Effect. Adv Drug Deliv Rev. 2011;63:131-5
30. Mittendorf EA, Philips AV, Meric-Bernstam F, Qiao N, Wu Y, Harrington S. et al. PD-L1 Expression in Triple-Negative Breast Cancer. Cancer Immunol Res. 2014;2:361
31. Ramos-Casals M, Brahmer JR, Callahan MK, Flores-Chávez A, Keegan N, Khamashta MA. et al. Immune-related adverse events of checkpoint inhibitors. Nat Rev Dis Primers. 2020;6:38
Author contact
![]() Corresponding author: K. Kim (E-mail: kimre.kr; address: Biomedical Research Institute, Korea Institute of Science and Technology (KIST), Seoul, 02792, Republic of Korea).
Corresponding author: K. Kim (E-mail: kimre.kr; address: Biomedical Research Institute, Korea Institute of Science and Technology (KIST), Seoul, 02792, Republic of Korea).