Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Organelle interactions
3. The role of organelle...
4. A strategy for delaying...
5. Conclusions
Abbreviation
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2022; 12(5):2445-2464. doi:10.7150/thno.70588 This issue Cite
Review
The relevance of organelle interactions in cellular senescence
Jiewu Huang1*, Ping Meng2*, Cheng Wang3, Yunfang Zhang4, Lili Zhou1,5 ![]()
1. State Key Laboratory of Organ Failure Research, National Clinical Research Center for Kidney Disease, Guangdong Provincial Clinical Research Center for Kidney Disease, Guangdong Provincial Key Laboratory of Nephrology, Division of Nephrology, Nanfang Hospital, Southern Medical University, Guangzhou, China.
2. Department of Central Laboratory, Huadu District People's Hospital, Southern Medical University, Guangzhou, China.
3. Division of nephrology, Department of medicine, the Fifth affiliated hospital of Sun Yat-Sen University, Zhuhai, Guangdong, China.
4. Department of Nephrology, Huadu District People's Hospital, Southern Medical University, Guangzhou, China.
5. Bioland Laboratory (Guangzhou Regenerative Medicine and Health Guangdong Laboratory), Guangzhou, China.
*These authors contribute equally to this work.
Received 2021-12-29; Accepted 2022-2-6; Published 2022-2-28
Abstract

Organelles are tiny structures with specific functions in eukaryotic cells. Since they are covered with membranes, different organelles can perform biological processes that are incompatible. Organelles can also actively communicate with each other to maintain cellular homeostasis via the vesicular trafficking pathways and membrane contact sites (MCSs), which allow the exchange of metabolites and other information required for normal cellular physiology. An imbalance in organelle interactions may result in multiple pathological processes. Growing evidence shows that abnormal organelle communication contributes to cellular senescence and is associated with organ aging. However, the key role of organelle interactions in aging has not yet been broadly reviewed and fully investigated. In this review, we summarize the role of organelle interactions in cellular senescence, and highlight their relevance for cellular calcium homeostasis, protein and lipid homeostasis, and mitochondrial quality control. Our review reveals important mechanisms of organelle interactions in cellular senescence and provides important clues for intervention strategies from a new perspective.
Keywords: organelle, interaction, communication, MCSs, cellular senescence
1. Introduction
Organelles, including the endoplasmic reticulum (ER), mitochondria, and endosomes, possess membranes that allow organelles to perform incompatible biological processes [1-3]. However, organelles also communicate and cooperate to accomplish information exchange and specific functions [1, 2]. Organelle interactions can be mediated by vesicle transmission and non-vesicular mechanisms [1, 4]. In this study, we focus on the latter. Membrane contact sites (MCSs) are formed by the close contact between two organelle membranes [5]. They are a major pathway for organelle interactions and act as hubs for signaling interactions [1, 6]. In addition to facilitating ion, lipid, and protein exchange, MCSs also play an important role in organelle dynamics [1, 6]. For example, lysosomes and ER both play a role in mitochondrial division through the MCSs [1, 3, 6]. MCSs are tethered by the parallel protein-protein or protein-lipid interactions, with a typical distance of 10-30 nm between organelles. MCS tethers are enriched with specific proteins or lipids that affect organelle function [5-8]. MCSs are often composed of several tethers, but they are dynamically expressed and regulated by modifications [6].
Cellular senescence can be defined as permanent cell cycle arrest. It plays a key role in embryogenesis, host immunity, tissue homeostasis [9, 10], inflammation, and malignancy [11]. Cellular senescence independently contributes to aging and age-related diseases, such as Alzheimer's disease and renal fibrosis [9, 10, 12, 13]. Organelle interactions are extensively involved in mitochondrial dysfunction and cellular calcium imbalance, which are the underlying mechanisms of cellular senescence. However, the relevance of organelle interactions in cellular senescence has not yet been elucidated. In this review, we discuss the key role of organelle interactions in cellular senescence and highlight their relevance in cellular energy metabolism. We also comment on intervention strategies to retard cellular senescence by modulating organelle interactions.
2. Organelle interactions
Organelle interactions occur across multiple organelles, including the ER, mitochondria, Golgi apparatus, peroxisomes, lipid droplets (LDs), endosomes, and lysosomes. As one of the largest organelles in eukaryotic cells, the ER plays a major role in synthesis, folding, modification, structural maturation, and targeted deployment of cellular proteomes [14, 15], and serves as a central hub of lipid metabolism [14]. Mitochondria are the master controllers of energy metabolism [16]. They possess two layers of plasma membranes [17], and their own circular genome, referred to as mitochondrial DNA (mtDNA) [17]. The Golgi apparatus, located adjacent to the nucleus, is a central membrane-bound organelle for trafficking, processing, and sorting of membrane proteins, secretory proteins, and lipids [18]. Peroxisomes have variable functions in different cells [19], ranging from fatty acid β-oxidation and hydrogen peroxide degradation to glycerol metabolism and maintenance of cellular integrity [19]. LDs store neutral lipids for energy or membrane synthesis and are primarily located in the cytoplasm [20]. There are two types of endosomes: early and late. Early endosomes are the cargo originating from the uptake of extracellular material by the plasma membrane (PM), which can be transported into the PM, ER, or late endosomes. Late endosomes go into lysosomes either via direct acidification or fusion with existing lysosomes [21]. Lysosomes digest extracellular or cell-surface cargos obtained via endocytosis or degrade intracellular components via autophagy [21]. Both endosomes and lysosomes play an important role in endocytosis and degradation, thereby forming an endolysosomal system [22, 23].
2.1 ER network
With extensive reticular structures [14], ER membranes can form abundant physical connections with the PM [24] and nearly all membrane-bound organelles [7, 25], such as mitochondria [26, 27], endosomes/lysosomes, phagosomes [28-32], Golgi apparatus [33], peroxisomes [34], and LDs [35]. The ER provides structural support for the transport of membrane-bound organelles [8], but is also connected with membraneless organelles [25, 36], thereby forming an intracellular network hub (Fig.1).
In eukaryotic cells, organelles widely interact with each other, forming networks such as the ER and mitochondrial networks. The ER interacts with the mitochondria via the IP3R-GRP75-VDAC-MCU complex. VAPs and OSBP play an important role in the interaction between the ER and the Golgi apparatus or endosomes. VAPs also play a significant role in ER-peroxisome interactions by coming in contact with ACBD5. FATP1 and DGAT2 bridge the ER to the LDs. The ER is also anchored to the PM by the STIM1-Orai1 interactions. Mitochondria interact with the Golgi apparatus and PM. However, the composition of mitochondria-Golgi apparatus and mitochondria-PM MCS tethers in mammalian cells remains unknown. ECI2/ACBD2 plays a significant role in the mitochondria-peroxisome MCSs. Rab7 plays an important role in the mitochondria-lysosome interaction. LDs can recruit mitochondria via DGAT2 and perilipin5. Syt7 in lysosomes interacts with PI(4,5)P2 in peroxisomes. Peroxisomes also come in contact with LDs via the spastin-ABCD1 interaction.
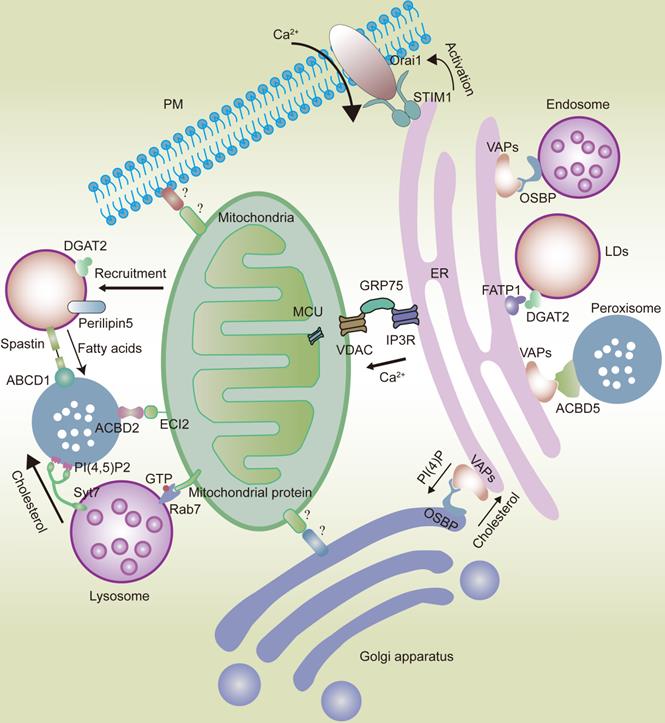
The stability of MCS requires tethering proteins, such as vesicle-associated membrane protein-associated proteins (VAPs), which play a key role in maintaining ER stability [6, 8, 37]. VAPs are highly conserved type II integral ER membrane proteins that can bind various intracellular proteins containing phenylalanine-phenylalanine acidic tract (FFAT) motifs [6, 38, 39].
Proteins containing FFAT motifs also have domains that can bind to lipids and other proteins [6]. For example, the oxysterol-binding protein-related protein family (ORPs) contains FFAT motifs and can be found in endosomes and the Golgi apparatus [39, 40]. VAPs can also bind to other proteins containing FFAT motifs, such as phosphatidylinositol transfer protein membrane associated 1 (PITPNM1, as known as Nir2) and ceramide-transfer protein (CERT) in the Golgi apparatus [41, 42], steroidogenic acute regulatory protein-related lipid transfer domain-3 (STARD3) and oxysterol-binding protein-related protein 1 (ORP1L) in endosomes [8, 43, 44], and acyl-CoA binding domain protein 5 (ACBD5) in peroxisomes [45]. The orthologues of VAPs in plants and yeast have similar effects [39, 46].
2.1.1 ER-PM interaction
2.1.1.1 ER-PM MCSs and lipid transport
Most lipids are synthesized in the ER and then are distributed to other membranes [47]. In mammalian cells, the ER and the PM form extensive MCSs, occupying approximately 2-5% of the cytoplasmic surface area [8]. It has been found that the extended synaptotagmin-like proteins (E-Syts), VAPs, transmembrane (TMEM) family, ORP5, and ORP8 contribute to ER-PM tethering [2, 8] and non-vesicular lipid transport between the ER and the PM [8, 48]. Notably, the absence of ER-PM tethers slows cell growth and reduces lipid content [47].
ER-PM MCSs play a key role in lipid transport. For example, ORP5 and ORP8 in the ER tether the ER to the PM through interactions with phosphatidylinositol 4-phosphate (PI(4)P) in the PM, thereby forming ER-PM MCSs to deliver PI(4)P to the ER for degradation and to deliver phosphatidylserine (PS) from the ER to the PM, to control PI(4)P levels and enrich PS in PM [8, 48, 49]. The E-Syts SMP domain can dimerize to form a long hydrophobic tunnel, allowing phospholipids to move between the ER and PM membranes [8, 37]. TMEM24 promotes ER-PM MCS formation to transfer phosphatidylinositol (PI) from the ER to the PM while decreasing Ca2+ concentration in the cytoplasm [8]. Nir2 in PM can bind to VAPs in the ER to form MCS, thus delivering PI from the ER to the PM and palmitic acid from the PM to the ER [8, 37, 50].
2.1.1.2 ER-PM MCSs and Ca2+ regulation
ER-PM MCSs also play a key role in Ca2+ regulation. The stromal interaction molecule 1 (STIM1)-Orai1 interaction in ER-PM MCSs plays an important role in calcium regulation [51, 52]. STIM1 in the ER is a sensor of Ca2+ concentration, and it translocates to ER-PM MCSs following the depletion of stored Ca2+, to bind to and activate PM-located Orai1, a hexameric Ca2+ release-activated Ca2+ (CRAC) channel [8, 53-55]. Orai1 mediates the influx of extracellular Ca2+, activates the sarco/ER Ca2+-ATPase (SERCA) pump, and guides Ca2+ into the ER lumen [8, 51]. The interaction between ryanodine receptor 1 (RyR1) in the ER and Cav1.1, a subunit of the voltage-dependent calcium channel in the PM, forms ER-PM MSCs and plays a significant role in controlling Ca2+ flux to drive cell contraction [8, 51]. When an action potential is generated in the PM, Cav1.1 undergoes a conformational change, leading to Ca2+ release via RyR1 into the cytoplasm [8, 56], which further induces cell contraction.
2.1.2 ER-mitochondria interaction
2.1.2.1 ER-mitochondria MCSs and lipid transport
The ER and the mitochondria can also form extensive MCSs, with a distance of approximately 10-25 nm, occupying approximately 2-5% of the mitochondrial surface area [8, 27]. Mitofusin 2 (Mfn2) is a major contributor in the formation of ER-mitochondria MCSs. ER-mitochondria MCSs are reduced by 40% when cells lack Mfn2 [27]. There are many protein interactions in ER-mitochondria MCSs, including ORP, tetrameric inositol 1,4,5-triphosphate receptor (IP3R), and B‐cell receptor‐associated protein 31 (BAP31) in the ER, protein tyrosine phosphatase interacting protein 51 (PTPIP51), voltage-dependent anion-selective channel protein (VDAC), and mitochondrial fission 1 (FIS1) in the mitochondria, and glucose‐regulated protein 75 (GRP75) in the cytoplasm [48, 57].
Bidirectional lipid transfer between the mitochondria and the ER plays an important role in maintaining the defined lipid composition of mitochondrial membranes [57]. Since the mitochondria cannot synthesize PS, it must be imported from the ER [57]. PS is transferred from the ER to the mitochondria by ORP5 and ORP8 interaction with PTPIP51 in ER-mitochondria MCSs [48]. After PS is converted into phosphatidylethanolamine (PE) in the mitochondria, PE is transferred from the mitochondria to the ER [57-59]. However, the role of ER-mitochondria MCSs in PE export from the mitochondria is unclear [58].
2.1.2.2 ER-mitochondria MCSs and Ca2+ regulation
ER-mitochondria MCSs can transfer Ca2+ from the ER lumen to the mitochondrial matrix [8, 60, 61]. The IP3R-GRP75-VDAC-mitochondrial calcium uniporter (MCU) complex is required in this process (a more detailed description is provided in Section 3.1.1) [62]. Other proteins are also important for ER-mitochondria MCSs. For instance, PDZ domain‐containing protein 8 (PDZD8), a protein found concentrated in ER-mitochondria MCSs, is necessary for ER-mitochondria MCS formation in mammalian cells and is required for Ca2+ uptake by the mitochondria after calcium release from the ER [63]. Mfn2 ablation leads to the absence of ER-mitochondria MCSs, which reduces the efficiency of mitochondrial Ca2+ uptake [27, 64]. PTPIP51 also plays an important role in Ca2+ uptake of the mitochondria from the ER, by binding to vesicle-associated membrane protein-associated protein B (VAPB) [65]. Moreover, it has been shown that Ca2+ transfer from the mitochondria to the ER occurs via an unidentified Na+/Ca2+ exchanger [66].
2.1.2.3 ER-mitochondria MCSs and mitochondrial dynamics
Mitochondria are dynamic organelles that maintain proper function, quality, and distribution through continuous fission and fusion [8, 67]. The regulatory mechanisms of the ER involved in mitochondrial dynamics via ER-mitochondria MCSs are described in detail in Section 3.3.2.
2.1.2.4 ER stress and unfolded protein response (UPR)
A more detailed description of ER stress and UPR is provided in Section 3.2.1.
2.1.2.5 Autophagosome formation
A more detailed description of autophagosome formation is provided in Section 3.2.2.
2.1.3 ER-Golgi apparatus interaction
The bidirectional vesicular trafficking between the ER and the Golgi apparatus is not the only mechanism of interaction between the two organelles. Previous studies have reported that non-vesicular transport, which mainly involves the transfer of lipids, also plays an important role in the interaction between the two organelles [33]. Vesicular trafficking mainly occurs on the nuclear side of the Golgi apparatus, while ER-Golgi apparatus MCSs are mainly located on the cytomembrane side of the Golgi apparatus, with a distance of approximately 10-20 nm between the ER and the Golgi apparatus [33].
In the ER-Golgi apparatus MCSs, ER protein VAPs bind to Nir2, CERT, and oxysterol-binding protein (OSBP) in the Golgi apparatus, all of which have FFAT motifs and play an important role in lipid transfer between the ER and the Golgi apparatus [8]. Nir2 transfers PI from the ER to the Golgi apparatus, decreasing PI concentration in the ER. Furthermore, PI is converted into PI(4)P, recruiting CERT and OSBP to ER-Golgi MCSs by binding to their PH domains [41]. CERT is responsible for the transport of ceramide between the ER (the site of its synthesis) and the Golgi apparatus (the site of its conversion to sphingomyelin) [33]. In addition, OSBP plays a significant role in the exchange of PI(4)P for cholesterol. In ER-Golgi apparatus MCSs, PI(4)P levels are low because of the ER phosphatase Sac1, which converts PI(4)P into PI. PI(4)P is transferred from the ER to the Golgi apparatus to form a concentration gradient which drives cholesterol transport [8, 33, 68, 69].
2.1.4 ER-peroxisomes interaction
Peroxisomes, originating from the division of existing peroxisomes or from the ER [1, 70, 71], play an essential role in lipid synthesis, breakdown and detoxification of fatty acids, and detoxification of reactive oxygen species (ROS) [8, 72]. To mediate these functions, peroxisomes interact with other organelles via vesicular trafficking or MCSs [72]. The interaction between peroxisomes and the ER is important for the biosynthesis of sterols, unsaturated fatty acids, and ether‐phospholipids [72]. For instance, the synthesis of ether-linked phospholipids is initiated in peroxisomes, but is completed in the ER [8].
Peroxisomes lack an appropriate enzyme inventory for autonomous lipid synthesis. Phospholipids essential for the growth and division of peroxisomes are provided by the ER [8, 73]. Lipid transfer between peroxisomes and the ER depends on ER-peroxisome MCSs rather than on vesicles [73]. Notably, the interaction between ACBD5 in peroxisomes and VAPs in the ER plays an important role in maintaining the ER-peroxisome MCSs and lipid transfer [74]. The overexpression of VAPs or ACBD5 increases the number and surface area of ER-peroxisome MCSs [8]. Moreover, the interaction between phosphatidylinositol‐4, 5‐bisphosphate (PI(4,5)P2) in peroxisomes and E-Syts in the ER plays an important role in cholesterol transfer from peroxisomes to the ER [75]. In addition, ER-peroxisome MCSs play a role in peroxisome mobility and distribution. The disruption of ER-peroxisome MCSs has been shown to increase the number and displacement of moving peroxisomes [8, 73].
2.1.5 ER-LD interaction
A more detailed description is provided in Section 3.4.
2.1.6 ER-endolysosomal organelle interaction
Both endosomes and lysosomes play an important role in the degradation of the cargo of cellular endocytosis, thereby forming an endolysosomal system [22, 23]. The ER-endolysosomal organelle MCSs regulate certain processes in these organelles [23]. The numbers of contacts between endosomes and the ER increase as the endosome matures. Approximately 50% of early endosomes and more than 99% of late endosomes are in contact with the ER, and their MCSs occupy approximately 5% of the endosome surface area.
Intracellular cholesterol mainly originates from endocytosis of low-density lipoprotein (LDL). LDL in early endosomes is delivered to the late endosomes/lysosomes and then hydrolyzed and transported by vesicular or non-vesicular mechanisms to downstream organelles, including the PM, ER, and mitochondria, to perform its functions [76]. Approximately 30% of LDLs from endosomes reach the ER via MCSs [23, 76]. VAPs and endosome-localized proteins, STARD3, ORP1L, and OSBP, play key roles in maintaining ER-endosome MCSs and regulating lipid transport. The interaction between VAPs and the late endosome cholesterol-binding protein ORP1L promotes cholesterol transport from the late endosomes to the ER. However, STARD3, which can bind cholesterol and transfer it between membranes, plays the opposite role [23]. VAP-OSBP interaction contributes to phospholipid homeostasis in ER-endosome MCSs by recruiting PI(4)P-phosphatase Sac1 to MCSs to further decrease PI(4)P level in endosomes [8, 23, 77]. A previous report has shown that high levels of PI(4)P block cargo trafficking from endosomes to the Golgi apparatus [8].
ER-endolysosomal organelle MCSs also play an important role in endolysosomal fission and positioning [23]. It has been reported that endosomal tubule fission occurs at ER-endosome MCSs in approximately 80% of cases [78]. The interaction between endosome-located IST1 factor associated with the endosomal sorting complex required for transport-III (IST1) and ER-located spastin links endosomes to the ER and regulates endosome fission [78]. Loss of spastin leads to impaired endosomal tubule fission and abnormal lysosomal morphology [23]. In addition, when cholesterol levels are high, ORP1L promotes endosome trafficking to the cell center, where they can fuse with lysosomes. However, when cholesterol levels are low, ORP1L leads to the accumulation of late endosomes at the cell periphery [23]. It has also been found that ER-endolysosomal organelle MCSs result in Ca2+ uptake from the ER. For example, IP3Rs have been found to be clustered at ER-lysosome MCSs and deliver Ca2+ to lysosomes [23].
2.2 Mitochondrion network
Mitochondria are the primary sites of ATP production. Therefore, mitochondrial homeostasis plays a crucial role in a variety of biological processes [79]. Mitochondria interact with other organelles to maintain cellular homeostasis. Mitochondria have extensive contact with PM [80] and other organelles [31, 57], such as the LDs [81], Golgi apparatus [82], lysosomes [83-85], and peroxisomes [86]. As a result, these organelles form a secondary intracellular reticulum network that acts as an important cellular signaling platform [31, 86].
2.2.1 Mitochondria-PM interaction
PM-mitochondria MCSs are better studied in the processes involved in mitochondrial fission and Ca2+ uptake [31]. Almost 10% of the PM is adjacent to mitochondria in animal cells [87]. Notably, PM-mitochondria MCSs also play a role in the distribution of the mitochondria during cell division [80, 87]. Subplasmalemmal mitochondria relay Ca2+ to the ER by modulating the activity of Ca2+-ATPases in the PM [88].
2.2.2 Mitochondria-lysosome interaction
Mitochondria and lysosomes are critical organelles involved in cellular homeostasis. Studies have shown that mitochondrial respiration dysfunction leads to lysosomal defects [89]. Lysosomes can regulate mitochondrial functions in a peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α)-independent manner through the biosynthesis of transcription factor EB (TFEB). Moreover, the dysfunction of lysosomal acidification leads to decreased mitochondrial respiration [85]. Mitochondria and lysosomes can also directly interact under stress conditions via mitochondrial-derived vesicles (MDVs) and mitophagy [85].
Mitochondria-lysosome MCSs are abundant and can be identified by electron microscopy in healthy cells [84]. Unlike MDVs fusing with lysosomes or mitophagy, mitochondria-lysosome MCSs do not result in bulk transfer of mitochondrial luminal content or mitochondrial degradation [84]. Mitochondria-lysosome MCSs have an average distance of 10 nm between the membranes and play an important role in modulating the dynamics of both organelles and ensuring Ca2+ transfer. Rab7, a GTPase, is an important protein that modulates the dynamics of mitochondria-lysosome MCS tethering and untethering [85]. Rab7 exhibits an active GTP-bound state under normal conditions, but shows an inactive GDP-bound state upon GTP hydrolysis [84]. The interaction between GTP-Rab7 in lysosomes and Rab7 effectors in the mitochondria plays a central role in maintaining the stabilization of mitochondria-lysosome MCSs. However, FIS1, the outer mitochondrial membrane (OMM) protein, recruits cytosolic TBC domain family member 15 (TBC1D15) to drive GTP-bound Rab7 hydrolysis to a GDP-bound state, thereby leading to untethering of MCSs [85].
Mitochondria can also modulate lysosomal retrograde and anterograde microtubule transport and coordinate lysosome dynamics via mitochondria-lysosome MCSs [84, 85]. These MCSs can also regulate mitochondrial dynamics (details are provided in Section 3.2.2). Mitochondria-lysosome MCSs also play a role in Ca2+ uptake by the mitochondria. The lysosomal channel transient receptor potential mucolipin 1 (TRPML1)-VDAC-MCU complex plays a significant role in the direct transfer of calcium from lysosomes to the mitochondria via mitochondria-lysosome MCSs [90]. The activation of TRPML1, a calcium-releasing channel on the lysosomal membrane, can increase the concentration of Ca2+ in mitochondria-lysosome MCSs [85, 90]. Furthermore, Ca2+ enters the mitochondrial matrix via the VDAC channel in the OMM and MCU channels in the inner mitochondrial membrane (IMM) [90]. TRPML1 activity is increased due to increased ROS [85].
2.2.3 Mitochondria-peroxisome interaction
Both mitochondria and peroxisomes play important roles in lipid and ROS metabolism [86], suggesting that there may be an interaction between these two organelles to balance this process. Peroxisomes and mitochondria exhibit a close functional interaction, including metabolic cooperation in regulating fatty acid β-oxidation and maintaining ROS homeostasis, sharing a redox-sensitive relationship, and cooperation in viral combat [91]. Mitochondria and peroxisomes interact with each other via MCSs, vesicular traffic, and biological messengers, such as ROS, lipids, and other metabolites [86, 91]. In yeast, Pex11p is required for mitochondria-peroxisome MCS formation [31]. Peroxisome-located acyl-CoA binding domain protein 2 (ACBD2) and mitochondria-located enoyl-CoA-δ isomerase 2 (ECI2) play an important role in the formation of mitochondria-peroxisome MCSs and steroids in mammals [92]. These two organelles also share key components of their division machinery in MCSs, including dynamin-related protein-1 (DRP1), fission factor 1, mitochondrial fission factor, and ganglioside-induced differentiation-associated protein 1 [91].
Mitochondrial function is impaired in senescent cells, and this can be compensated by peroxisomes via the mitochondrial retrograde (RTG) signaling pathway [93]. Following the increasing transcription of mitochondrial and peroxisomal genes related to carbohydrate and nitrogen metabolism, peroxisome proliferation is activated and fatty acids are converted into acetyl-CoA to replenish the tricarboxylic acid (TCA) cycle in mitochondria [93, 94].
2.2.4 Mitochondria-LD interaction
A more detailed description is provided in Section 3.4.
2.3 Other organelle interactions
There are other organelle interactions, including peroxisome-lysosome [95, 96] and peroxisome-LD interactions [97]. Although the interactions of peroxisomes with lysosomes appears to be less frequent than their interaction with the ER or the mitochondria, peroxisome-lysosome interactions are still found in approximately 15% to 20% of the total peroxisomes in mammalian cells [73]. The lysosome-peroxisome MCSs are the principal pathway for cholesterol transfer from the lysosome to the peroxisome [76]. Synaptotagmin-7 (Syt7), on lysosomes, and PI(4,5)P2, on peroxisomes, form peroxisome-lysosome MCSs, which play an important role in cholesterol transport [34, 73, 76]. When cholesterol reaches peroxisomes, it participates in peroxisome biogenesis [76]. The disruption of lysosome-peroxisome MCSs decreases cholesterol levels in the PM and impairs cholesterol transport to the ER, suggesting that lysosome-peroxisome MCSs may be associated with cholesterol transport to other organelles [76]. MCSs may also regulate other functions of lysosomes and peroxisomes, such as autophagy and peroxisome biogenesis [76]. A more detailed description of LD-peroxisome interactions is provided in Section 3.4.
3. The role of organelle interaction in cellular senescence
Cellular senescence is a permanent cell cycle arrest induced by a series of stress factors, including telomere attrition, DNA damage, oxidative stress, oncogene activation, and organelle stress. Cellular senescence is characterized by chromatin remodeling, metabolic reprogramming, and senescence-associated secretory phenotype (SASP) [10, 98-101]. Senescent cells accumulate in organs during aging and secrete SASP molecules, including proinflammatory and fibrogenic molecules, to further promote a variety of age-related diseases [101, 102]. Senescence can be classified into the following types: replicative, DNA-damage-induced, stress-induced, and oncogene-induced [103].
3.1 Cellular calcium homeostasis and senescence
3.1.1 ER and mitochondrial calcium homeostasis
The ER is a dynamic calcium reservoir that maintains cellular calcium homeostasis [104]. In senescent cells, calcium is released from the ER to increase intracellular calcium levels [105]. Mitochondria and lysosomes also play crucial roles in cellular calcium homeostasis [85]. Hence, cellular calcium buffering is controlled by the interplay between ER, mitochondria, and lysosomes [106]. Studies have shown that an imbalance in ER calcium homeostasis not only induces the production of SASP molecules, but also causes cell cycle arrest by influencing mitochondrial calcium homeostasis [105]. In particular, appropriate calcium transfer from the ER to the mitochondria requires the correct distance between the two organelles [107] and accurate organelle interaction. ER-mitochondria MCS dysfunction can be found in senescent cells, which further leads to an imbalance in calcium control [108-110]. ER-mitochondria MCSs play an important role in Ca2+ homeostasis via the IP3R-GRP75-VDAC-MCU complex. Ca2+ is released through the IP3R channel in the ER, which then enters the mitochondrial matrix via the VDAC channel in the OMM and MCU channels in the IMM [8, 111]. The VDAC channel is crucial for mitochondrial Ca2+ uptake [90], and the MCU channel is considered to be the major transporter of Ca2+ into the mitochondrial matrix [90]. Notably, GRP75 plays a role in linking IP3R and VDAC to shorten the distance between the ER and the mitochondria [62]. In cellular senescence, IP3R-mediated calcium release from the ER is activated and the expression of MCU increases [105, 112], leading to the accumulation of mitochondrial calcium and contributing to a decrease in mitochondrial membrane potential and increased ROS production [105]. Excess ROS can induce DNA damage and lead to single-strand breaks in telomere regions, resulting in accelerated telomere shortening, both of which can activate the DNA damage response (DDR) pathway [100, 109, 113, 114]. Consequently, DDR performs cell cycle checkpoint functions and blocks cell cycle progression. After DNA damage, DDR factors accumulate at the sites of DNA damage and cause chromatin modifications, such as the phosphorylation of histone H2AX. Persistent DNA damage causes a prolonged DDR signaling pathway. It induces the activation of the tumor suppressor p53, contributing to the expression of the cyclin-dependent kinase inhibitor p21, to cooperatively induce cellular senescence [100]. It has been shown that IP3R knockdown can attenuate cellular senescence by reducing the number of ER-mitochondria MCSs and ER calcium release [115, 116]. Moreover, MCU knockdown can also prevent cellular senescence by attenuating mitochondrial calcium intake [117] (Fig.2).
The increased calcium released from the ER can be transferred to the mitochondria via the IP3R-GRP75-VDAC-MCU complex. This leads to the increased levels of ROS in the mitochondria, contributing to DNA damage and DDR, activating the p53/p21 pathway, and causing cellular senescence. Moreover, while the UPR is consistently activated, it also leads to calcium accumulation in the mitochondria, causing cellular senescence. On the one hand, dysfunctional mitochondria can repress the lysosomal calcium-releasing channel MCOLN1; on the other hand, there is increased calcium release from the ER via IP3R. Both these processes lead to calcium accumulation and an increase in pH in the lysosomes, resulting in dysfunction of the lysosome-autophagy proteolytic system and contributing to cellular senescence. Lysosomes can originate from the direct acidification of late endosomes. The Golgi apparatus deliver lysosomal enzymes to endosomes. Moreover, ER-mitochondria MCSs promote the formation of autophagosomes. Impaired autophagosomal formation and lysosomal function leads to the dysfunctional autophagy, thereby causing cellular senescence.
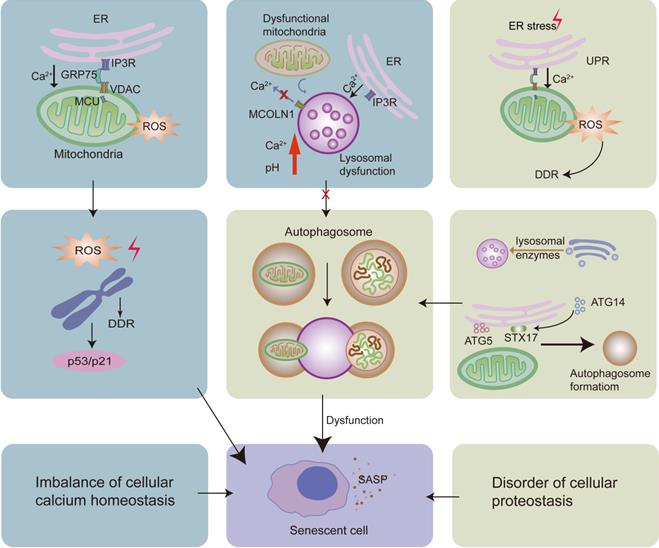
3.1.2 Lysosomal calcium homeostasis
Lysosomal calcium homeostasis plays an important role in maintaining lysosomal function. In senescent cells, the activated IP3R in the ER leads to increased ER calcium release [105] via ER-lysosome MCSs, resulting in lysosomal calcium accumulation. The imbalance of lysosomal calcium homeostasis induces disorders related to lysosomal acidification [118], which are associated with lysosomal proteolytic activity [119]. Furthermore, impaired proteolytic activity of lysosomes negatively affects the lysosome-mediated autophagy proteolytic system and causes cellular senescence [102, 120-123].
In senescent cells, damaged mitochondria accumulate [124]. Dysfunctional mitochondrial respiratory chain impairs lysosomal calcium homeostasis by repressing the lysosomal calcium-releasing channel mucolipin 1 (MCOLN1, also known as TRPML1) [125], plays an important role in regulating lysosomal biogenesis, exocytosis, and autophagy via the regulation of lysosomal calcium release [126]. The impairment of the function of TRPML1 leads to enlarged lysosomes, lysosomal calcium accumulation, and increased pH, which further contributes to lysosomal malfunction and cellular senescence [119, 126]. It has been reported that mitochondrial ROS can activate TRPML1, inducing lysosomal calcium release [85], further inducing autophagy and lysosomal biogenesis [127] and delaying cellular senescence. However, excess ROS levels in the mitochondria have the opposite effect, leading to lysosomal dysfunction and cellular senescence [126, 127].
Transmembrane BAX inhibitor motif containing 6 (TMBIM6), a calcium channel-like protein integral to the intracellular membranes of the ER, can also regulate lysosomal calcium [128]. Specifically, it increases lysosomal calcium release under stress conditions to restore cellular homeostasis by enhancing lysosomal calcium release-induced autophagy [128], thereby retarding cellular senescence [129].
3.2 Cellular protein homeostasis and senescence
Cellular protein homeostasis is crucial for maintaining cellular function. Dysregulation of cellular protein homeostasis contributes to cellular senescence and aging [130-134]. It has been shown that cellular proteostasis is decreased in senescent cells [135]. The imbalance of cellular protein homeostasis plays an important role in senescence-related pathologies [102], while improved proteostasis can delay aging [133]. Under persistent stress, cellular proteins are damaged, aggregated, misfolded, or no longer needed. The three primary pathways that are used by senescent cells to maintain cellular protein homeostasis are as follows: utilizing molecular chaperones, the proteasome proteolytic system, and the lysosome-autophagy proteolytic system [130, 133, 134, 136].
3.2.1 UPR and cellular senescence
Most secretory proteins in eukaryotic cells undergo chaperone-assisted folding in the ER to acquire an appropriate spatial structure [137]. When cells are under stress, unfolded or misfolded proteins accumulate in the ER lumen, contributing to ER stress [137]. To maintain ER function and cellular protein homeostasis, the UPR is activated by the ER-localized transmembrane stress sensors, namely inositol requiring enzyme 1 alpha (IRE1α), RNA-dependent protein kinase-like ER kinase (PERK), and activating transcription factor 6 alpha (ATF6α) [66, 137]. Both IRE1α and ATF6α play important roles in enhancing the degradation of misfolded proteins. Under ER stress, full-length ATF6 in the ER translocates to the Golgi apparatus, where it is cleaved into ATF6p50; then, it transitions into the nucleus to induce gene expression. PERK can induce a transient decrease in protein synthesis to relieve ER stress through the attenuated influx of newly synthesized proteins into the ER lumen [137].
During the early phases of ER stress, the UPR boosts pro-survival signaling [66], which increases and tightens the ER-mitochondria MCSs and increases mitochondrial Ca2+ uptake, thereby driving adaptive mitochondrial metabolic boost and ATP production to establish the metabolic basis for recovering cell homeostasis [57, 107, 138]. However, when stress is not resolved, the UPR leads to excess intake of mitochondrial Ca2+, causing cellular senescence or even cell death [99, 139, 140]. UPR activation occurs in almost all types of senescence [99]. The UPR is not only a consequence of cellular senescence, but also its driver [99]. However, the mechanism by which UPR induces senescence remains poorly understood.
It has been reported that a variety of ER chaperones involved in protein folding are enriched in ER-mitochondria MCSs [141], such as PERK and IRE1α [137]. PERK can facilitate the tethering of the ER to the mitochondria, leading to increased mitochondrial calcium intake and ROS production under ER stress [137]. Sustained ER stress leads to excess release of Ca2+ from the ER via IP3R. Calcium is enriched in ER-mitochondria MCSs and is taken up by the mitochondria via calcium uniporter. Large calcium influx leads to the loss of the mitochondrial membrane potential. Furthermore, cytochrome c translocates into the ER-mitochondria MCSs and binds to IP3R, which abolishes the calcium-mediated inhibition of IP3-associated calcium release and results in a feed-forward amplification of ER calcium release [66]. Finally, this causes an imbalance in mitochondrial Ca2+ homeostasis [139, 140] and increases ROS production [105, 142], thereby resulting in cellular senescence [109, 143].
3.2.2 Lysosome-autophagy proteolytic system and cellular senescence
Autophagy is a process of self-degradation of organelles and misfolded proteins by autophagosomes, which are then delivered to lysosomes [144]. The lysosome-autophagy proteolytic system plays a significant role in maintaining cellular protein homeostasis, and its imbalance contributes to cellular senescence [134, 145]. It has been reported that autophagy activation can prevent cellular senescence by downregulating p53 phosphorylation and p21 levels [146]. There are three main types of autophagy in mammalian cells that maintain the homeostasis of proteins and organelles: microautophagy, macroautophagy, and chaperone-mediated autophagy [145]. Lysosomal-mediated autophagosomal degradation plays an important role in maintaining cellular homeostasis under stress conditions [119, 128, 131]. Damaged proteins and organelles can induce cellular senescence [147]. Notably, autophagy plays a significant role in their degradation, providing adequate amino acids and maintaining energy levels to support cells [147-149]. Autophagy inhibition and insufficiency lead to cellular senescence [145], while increased autophagy delays aging [123, 150].
The formation of autophagosomes is the first step in the macroautophagy [148]. ER-mitochondria MCSs are the origins of autophagosomes [151]. When autophagy signals initiate, autophagosome formation-related proteins accumulate on the ER side of the ER-mitochondria MCSs [57, 152]. Autophagy-related gene 14 (ATG14), the pre-autophagosome marker, and autophagy-related gene 5 (ATG5), the autophagosome-formation marker, are enriched in ER-mitochondria MCSs after starvation. ATG14 is recruited to ER-mitochondria MCSs by syntaxin 17 (STX17), an ER-resident protein [151]. Mitochondria also supply membranes for autophagosome biogenesis [153].
Lysosomes play an important role in the final step of the lysosome-autophagy proteolytic system by fusing with autophagosomes. Lysosomal function affects the lysosome-autophagy proteolytic system. In senescent cells, lipofuscin accumulates in lysosomes, leading to diminished lysosomal activity [154]. In contrast, the activation of lysosomes restores the stemness of senescent stem cells [155]. Lysosomes can originate from the direct acidification of late endosomes [21]. In this process, lysosomal enzymes are transported from the Golgi apparatus to endosomes [156]. Previous studies have shown that IST1-spastin interaction in ER-endosome MCSs plays an important role in endosomal tubule fission and in the transfer of lysosomal enzymes from the Golgi apparatus to endosomes [78]. Lack of spastin or IST1 leads to lysosomal defects and abnormalities [78].
3.3 Mitochondrial quality control (MQC) and senescence
Mitochondrial dysfunction plays a crucial role in cellular senescence [109, 157]. Cellular senescence accelerates mitochondrial dysfunction and, conjointly, mitochondrial dysfunction is also a cause of cellular senescence [124, 158, 159]. Increased mitochondrial ROS production induces DNA damage, activates DDR, and contributes to cellular senescence. Furthermore, decreased NAD+/NADH ratio in dysfunctional mitochondria leads to the persistent activation of the energy sensor AMP-activated protein kinase (AMPK), which induces cellular senescence by phosphorylating p53 and stabilizing p16INK4a mRNA [159, 160].
MQC is an elaborate mechanism that maintains normal mitochondrial function. It includes two opposite processes: mitochondrial biogenesis and the removal of impaired mitochondria or their components [161]. Dysregulation of MQC induces the accumulation of damaged mitochondria, leading to cellular senescence [102]. Damaged proteins can be transported into lysosomes or peroxisomes in the form of mitochondrial-derived vesicles (MDVs) as the first defense against mitochondrial damage [102, 162]. The cargo delivered to lysosomes is ultimately degraded, however, the fate of the cargo delivered to peroxisomes is unclear [162]. When mitochondria are depolarized or dysfunctional, they are removed by mitophagy [102]. Cells can release damaged mitochondria into the extracellular space to maintain mitochondrial homeostasis [163, 164]. In addition, cells can acquire mitochondria via mitochondrial fission or by receiving extracellular healthy mitochondria [161, 165] (Fig.3).
Mitochondrial fission and fusion, mitophagy, MDVs, and mitochondrial transfer play important roles in MQC, and they are regulated by organelle interactions. Damaged mitochondrial proteins can be transported into lysosomes in the form of MDVs, and dysfunctional mitochondria can be degraded by lysosomes via mitophagy. Mitochondria can preserve shape and reduce damage via continuous fusion and fission cycles. ER tubules physically wrap around and constrict the mitochondria, promoting mitochondrial fission. Moreover, Golgi-derived vesicles containing PI(4)P are necessary for this process. Mitochondria-lysosome interactions also play an important role in mitochondrial fission. In addition, ER regulates mitochondrial fusion, and Mfn2 plays a significant role in this process. Dysfunctional mitochondria can also be released from cells and degraded by lysosomes of other cells. Furthermore, healthy cells can transfer healthy mitochondria into other cells via the ER tuber. Dysfunctional MQC leads to the accumulation of impaired mitochondria, leading to increased ROS levels and decreased NAD+/NADH ratios, both of which contribute to cellular senescence.
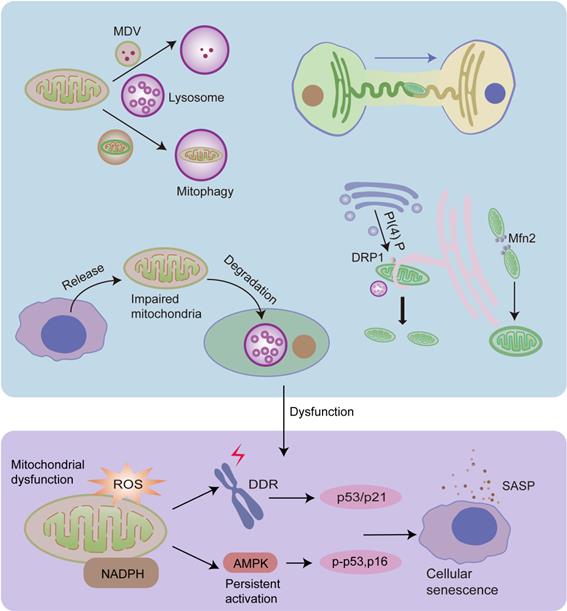
3.3.1 MDVs and mitophagy
Lysosomes play an important role in the final degradation of damaged proteins in MDVs and dysfunctional mitochondria during mitophagy [166]. Mitochondria-lysosome interactions play an important role in MQC [166]. Mitophagy plays a role in the removal of dysfunctional mitochondria. The removed mitochondria are eventually replenished via biogenesis [166]. Damaged mitochondrial components can also be degraded in the form of MDVs [166]. Dysregulation of mitophagy leads to the accumulation of damaged mitochondria, thus contributing to cellular senescence [166].
However, in senescent cells, dysfunctional mitochondria produce high levels of ROS, which contribute to the accumulation of lipofuscin in lysosomes and lysosomal dysfunction [154]. Moreover, damaged mitochondria also impair the calcium release of lysosomes, which further contributes to lysosomal dysfunction. Dysfunctional lysosomes cause impaired mitophagy and the function of degrading damaged mitochondrial proteins, leading to further mitochondrial malfunction. This feedback loop aggravates cellular senescence [154].
3.3.2 Mitochondrial dynamics
Mitochondria can preserve homeostasis via continuous fusion and fission cycles [166]. However, this process is impaired in senescent cells [109]. It has been reported that mitochondrial dynamics involve the interaction of multiple organelles, including the Golgi apparatus, ER, and lysosomes.
ER-mitochondria MCSs have been shown to play an important role in mitochondrial fission and fusion by providing a series of enzyme-like nodes [39, 57, 167]. Active mtDNA synthesis occurs at ER-mitochondria MCSs, and constriction of IMM is a priming event [67, 168]. This suggests that the initial mitochondrial fission signal comes from the mitochondrial matrix, which then recruits the ER [8]. During mitochondrial fission, ER tubules physically wrap around the mitochondria and lead to mitochondria constriction, facilitating the recruitment and assembly of DRP1, the main driver of mitochondrial fission [169]. DRP1 then moves from the cytoplasm to the OMM at MCSs, constricting mitochondria down to less than 50 nm, and finally, Dnm2, a dynamin protein, is recruited to MCSs to induce fission [8, 57, 169-171]. Furthermore, multiple studies have shown that Golgi-derived PI(4)P-containing vesicles are recruited to mitochondria-ER MCSs and can activate DRP1-mediated mitochondrial division. Disruption of PI(4)P production leads to the prevention of mitochondrial fission, and mitochondrial morphological defects [172]. Mitochondria-lysosome MCSs also play a significant role in mitochondrial fission. It has been shown that more than 80% of mitochondrial fission events are marked by the lysosomal markers lysosomal associated membrane protein 1 (LAMP1)-positive vesicles [84, 85, 173]. A study has also found that mitochondria-lysosome contacts play a role in ER-mediated mitochondrial fission [84]. ER-mitochondria MCSs play an important role in mitochondrial fusion [57, 174], but the underlying mechanism is still unclear. Mfn2 may play a significant role in this process, as Mfn2 not only plays a critical role in mitochondrial fusion, but also in the maintenance of ER-mitochondria MCSs [64, 174, 175].
3.3.3 Mitochondrial transfer
A novel MQC pathway involves the transfer of damaged mitochondria into the extracellular space for degradation [163]. For example, the mitochondria released from neurons can be degraded by lysosomes of neighboring glial cells [176]. This pathway is independent of mitophagy, and is activated by mitochondrial stress. When mitophagy is compromised, more mitochondria are released to compensate for the accumulation of damaged mitochondria [163].
Furthermore, intercellular mitochondrial transfer is a significant part of the MQC to rescue mitochondrial defects [163, 165]. It has been reported that donor cells, especially stem cells, can transfer healthy mitochondria into recipient cells, leading to the improvement of mitochondrial bioenergetics and reduction of mitochondrial ROS [165, 177]. There are four main methods for intercellular mitochondrial transfer, including utilizing tunneling nanotubes (TNTs), dendrites and microvesicles, and extrusion and internalization [165].
It has been shown that the mitochondrial transfer between osteocytes relies on the ER-mitochondria interaction [165]. The inhibition of ER-mitochondria tethering significantly inhibits the transfer of the mitochondria between osteocytes [178]. It has been hypothesized that the mitochondria are transferred along with the ER tuber into the terminus of osteocyte dendrites, where the ER contacts the PM. The arrival of mitochondria changes the lipid metabolism of the PM, which enables the ER of the donor to fuse with the ER extension of the recipient. Mitochondria are then transferred to the recipient along the fusing ER tubular network [178].
3.4 Lipid homeostasis and senescence
LDs play important roles in the maintenance of lipid homeostasis by coordinating lipid synthesis, storage, and secretion and lipolysis [179]. Under fed conditions, free fatty acids are converted into neutral lipids by ER, and then deposited in LDs [93, 180]. These neutral lipids include, but are not limited to, triacylglycerol, sterol esters, and esterified ceramides [179]. Under the stress conditions or energy insufficiency, lipases on the surface of LDs can convert triglycerides into fatty acids, which are then imported into the mitochondria for β-oxidation to satisfy energy demands [31, 81, 181, 182].
Lipotoxicity is caused by the accumulation of lipids, particularly fatty acids [179]. When cellular fatty acids exceed the requirements of synthesis and catabolism, they can be stored in the form of triacylglycerol in LDs to avoid lipotoxicity [179]. However, the capacity of triacylglycerol buffering is limited [179, 183]. When the limited triacylglycerol buffering capacity is saturated, accumulated lipids react with ROS, especially mitochondrial ROS, thereby producing large quantities of lipid peroxides. Interestingly, continuous production and accumulation of lipid peroxides cause serious lipid toxicity-related damage to mitochondrial structure and function, and induce a rapid increase in ROS levels, thereby contributing to cellular senescence [184]. Moreover, continuous production and accumulation of lipid peroxides can induce ER stress [179, 182], which also contributes to cellular senescence. It has been shown that triacylglycerol accumulates in replicative senescent cells. This is a cellular mechanism that prevents lipotoxicity in senescent cells [185]. As LDs play primary roles in lipid storage and homeostasis [179], the biogenesis and expansion of LDs, which are regulated by organelle interactions, play significant roles in cellular senescence. It has been shown that LD contact sites play an important role in fatty acid synthesis, storage, release, and breakdown [186]. Inhibition of LD biogenesis during rapid fatty acid release leads to fatty acid-mediated mitochondrial depolarization [81], which increases the release of mitochondrial ROS [187], and leads to cellular senescence (Fig.4).
LDs originate from the ER, and the ER can transport triacylglycerol synthesis enzymes required for LD growth and expansion via membrane bridges. Rab18-NRZ-SNARE interaction facilitates LD growth and maturation by regulating the synthesis of triglycerides in the ER. Moreover, FATP1 and DGAT2 complex maintain the ER-LD MCSs and facilitates LD expansion by coupling the synthesis and deposition of triglycerides into LDs. ORP5 plays an important role in transferring PC from the ER to LDs via ER-LD MCSs. Mitochondria-LD interactions also play an important role in maintaining lipid homeostasis. Perilipin5 and DGAT2 in LDs recruit the mitochondria to promote the expansion of LDs by providing ATP for triglyceride synthesis. In addition, the interaction of M1 spastin on LDs with ABCD1 on peroxisomes promotes fatty acid transfer from LDs to peroxisomes for β‐oxidation. Dysfunctional growth and expansion of LDs leads to the accumulation of fatty acids, which then react with ROS and produce lipid peroxides, thereby resulting in ER stress, increased mitochondrial ROS, and cellular senescence.
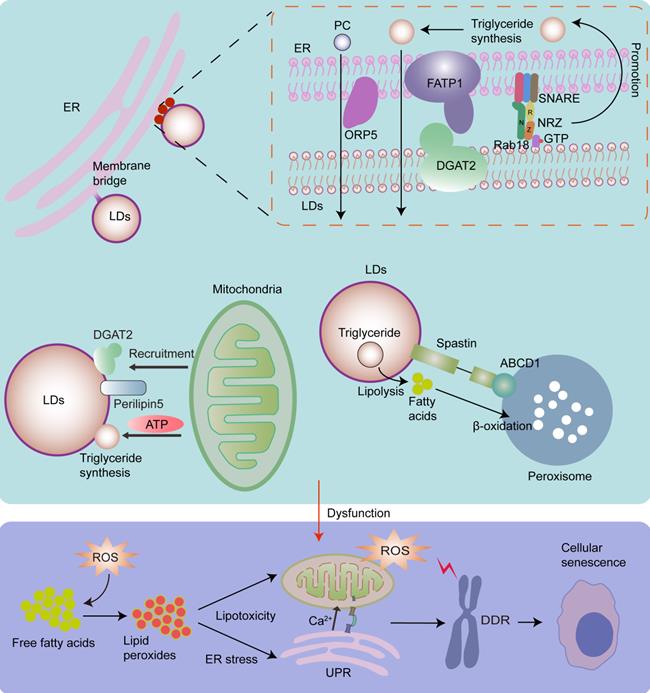
LDs originate from ER [182]. Most neutral lipid and phosphatidylcholine (PC) synthesizing enzymes are absent in LDs, while they exist in the ER, implying that the interaction between LDs and the ER is essential for lipid metabolism [35]. LDs widely interact with the ER via membrane bridges and MCSs. The ER-localized protein ORP5 plays an important role in the transfer of PC from the ER to LDs in ER-LD MCSs [188]. The tether proteins of ER-LD MCSs include ER-localized fatty acid transport protein 1 (FATP1) and LD-localized diacylglycerol acyltransferase 2 (DGAT2), the ER-associated NAG-RINT1-ZW10 (NRZ) tethering complex and their associated SNAREs (Syntaxin18, Use1, BNIP1), and LD-localized Rab18 [8, 35, 189]. Rab18 is associated with lipolysis and lipogenesis, and its overexpression induces a close apposition of the ER and LDs. It has been shown that the Rab18-NRZ-SNARE interaction facilitates LD growth and maturation by regulating the synthesis of triglycerides in the ER [35, 189]. The FATP1 and DGAT2 complexes maintain the ER-LD MCSs and facilitate LD expansion by coupling the synthesis and deposition of triglycerides into LDs [35, 190].
Except for ER-LD MCSs, there are membrane bridges where the LD monolayer appears to be directly continuous with the ER bilayer, playing a significant role in maintaining the structure and biogenesis of LDs and facilitating the incorporation of neutral lipids and membrane proteins into LDs [8, 35]. Notably, triacylglycerol synthesis enzymes are required for LD growth and expansion, and they can be transferred from the ER into LDs via membrane bridges [180]. Moreover, the ER protein seipin plays an important role in protein transfer from the ER to LDs, LD biogenesis, and the regulation of the local lipid environment [1, 35, 191].
The interactions between the mitochondria and LDs also play an important role in maintaining lipid homeostasis. In the presence of excess fatty acids in the cell, LD coat proteins perilipin5, and DGAT2, which catalyze the final step in triglyceride synthesis [192], recruit mitochondria to LDs to provide ATP to promote the expansion of LDs, thereby protecting against lipotoxic insult [81, 193].
It has been shown that 10% of LDs form contacts with peroxisomes [73]. The peroxisome-LD MCSs may link LD-mediated lipolysis to fatty acid β‐oxidation in peroxisomes. LDs protein M1 spastin interacts with ATP binding cassette subfamily D member 1 (ABCD1), a peroxisome protein, to promote peroxisome-LD MCS formation and facilitate direct channeling of fatty acids across the boundaries of organelles to protect against lipotoxic insult [93, 97].
4. A strategy for delaying senescence based on organelle interaction
4.1 Maintaining calcium homeostasis
Mitochondrial calcium homeostasis plays an important role in maintaining mitochondrial function. Abnormal organelle interactions lead to excessive calcium intake into the mitochondria, thereby leading to cellular senescence. Thus, maintaining mitochondrial calcium homeostasis may be a reasonable way to retard cellular senescence. Notably, knockdown of MCU can prevent cellular senescence [115]. Furthermore, IP3-induced IP3R activation induces premature senescence [117], whereas IP3R absence decreases the progression of aging [115] (Fig.5).
There are some strategies for delaying senescence with respect to organelle interaction, including maintaining calcium homeostasis, maintaining protein homeostasis, enhancing MQC, and improving anti-lipotoxicity effects. We can decrease mitochondrial calcium intake by repressing the expression of IP3R, MCU or PACS-2, and increasing the expression of calreticulin. Moreover, we can inhibit the overactivated UPR by 4-PBA to delay cellular senescence. Promoting lysosome-mediated autophagy by rapamycin is also effective. Furthermore, we can increase mitochondrial fission or mitophagy via DRP1 or PRKN, respectively, to enhance MQC. Moreover, by overexpressing DGA1 (encodes DGAT homologue in yeast) and LRO1 (encodes phospholipid:diacylglycerol O-acyltransferase in yeast), we can increase the number of LDs to decrease mitochondrial ROS and mitochondrial fragmentation caused by high amounts of fatty acids.
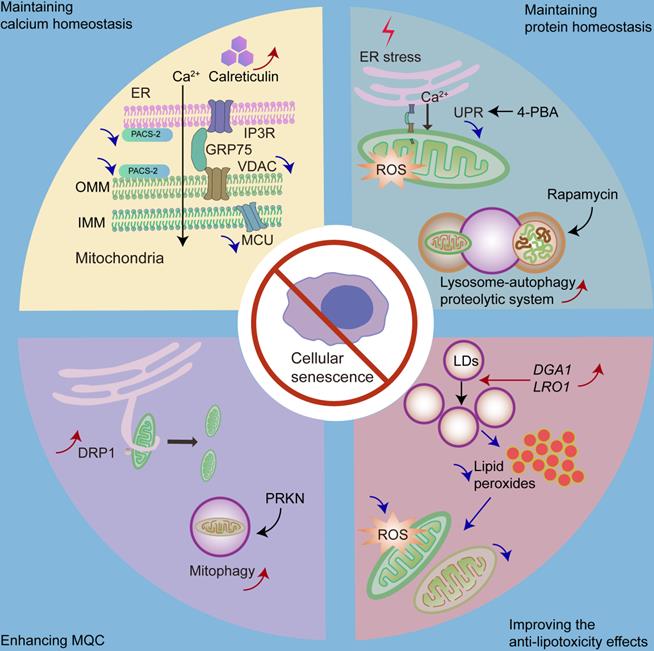
Mitochondrial calcium homeostasis can be regulated through ER-mitochondria MCSs. The increased number and area of ER-mitochondria MCSs leads to enhanced calcium transfer from the ER to the mitochondria. It has been shown that the downregulation of phosphofurin acidic cluster sorting protein 2 (PACS-2), a key regulator of ER-mitochondria MCSs, retards mitochondrial calcium accumulation [194]. While an increase in the ER-mitochondria contact leads to cellular senescence [116], the ablation of ER-mitochondria MCS tethers delays cellular senescence [116].
ER is the major storage compartment for intercellular calcium [107]. ER calcium homeostasis is closely related to mitochondrial calcium homeostasis [195]. There is an inverse relationship between calcium storage in the ER and the mitochondria [195]. Notably, a large amount of calcium is buffered by calcium-binding proteins, such as calsequestrin or calreticulin, in the ER [196]. It has been shown that decreased expression of calreticulin is related to aging [197], while overexpression of calreticulin enhances ER calcium storage and inhibits the activity of IP3R, thereby regulating mitochondrial calcium homeostasis [195].
4.2 Maintaining protein homeostasis
Overactivation of the UPR is positively associated with senescence [99]. One possible mechanism could be that UPR leads to an imbalance in mitochondrial calcium homeostasis. UPR activation is associated with cellular protection; however, overactivation of the UPR is associated with pathological processes. UPR inhibition also shows therapeutic value [195]. It has been shown that the inhibition of UPR retards cellular senescence [99]. For example, the chemical chaperone 4-phenylbutyric acid (4-PBA), a UPR inhibitor, reduces the number of senescent cells in a premature tubular epithelial cell senescence model [198].
In addition, lysosome-mediated autophagy plays an important role in maintaining cellular protein homeostasis and preventing cellular senescence; however, it is impaired in senescent cells [199]. Autophagy activation has been shown to inhibit stress-induced senescence [146, 200]. As a result, enhancing the function of lysosome-autophagy proteolytic system may be a strategy to delay senescence. It has been shown that restoring autophagy in stem cells can prevent cellular senescence and maintain stemness [144]. Furthermore, rapamycin, an autophagy inducer, can protect against oxidative stress-induced senescence [146].
4.3 Enhancing MQC
Mitochondrial dysfunction and MQC failure play significant roles in cellular senescence [166, 201]. Thus, enhancing MQC may be a therapeutic strategy against cellular senescence. Of note, DRP1 plays a significant role in mitochondrial fission by generating a constriction ring around the mitochondria [166]. Increased p53 levels in senescent cells inhibit DRP1 translocation into the mitochondria [202], while a previous report has shown that the increased expression of DRP1 promotes mitochondrial fission and delays cellular senescence [203, 204]. In addition, Parkin RBR E3 ubiquitin protein ligase (PRKN) plays a crucial role in regulating mitophagy [205]; hence, overexpression of PRKN could prevent cellular senescence by increasing mitophagy [205].
4.4 Improving anti-lipotoxicity effects
Since LDs play important roles against lipotoxicity, increasing the number of LDs may be an appropriate way to ameliorate cellular senescence. The overexpression of DGA1 (encodes DGAT homologue in yeast) and LRO1 (encodes phospholipid:diacylglycerol O-acyltransferase in yeast) [206], both of which are associated with triacylglycerol synthesis, leads to an increased number of LDs and reduced mitochondrial fragmentation as well as mitochondrial ROS production, ultimately contributing to the alleviation of replicative senescence [207].
5. Conclusions
The essential reactions in eukaryotic cells are compartmentalized in membrane-bound organelles, which allow the segregation of incompatible biological processes with tailored microenvironments [1, 25]. However, the cell is a complicated biological system, and organelles are not isolated entities [86]; they need to actively communicate and interact with each other to maintain cell homeostasis [1, 208]. For example, peroxisomes convert glyoxylate and alanine into glycine and pyruvate; then, glycine is transported into the mitochondria for oxidation and the detoxification of glyoxylate [91]. The exchange of metabolites and information between organelles depends on vesicular trafficking pathways and MCSs [1].
Cellular senescence is a process of permanent cell-cycle arrest. It involves cellular calcium homeostasis, protein homeostasis, and MQC and is regulated by organelle interactions. In this review, we provide important clues to the intervention strategies for cellular senescence from a new perspective, including maintaining calcium homeostasis, maintaining the protein homeostasis, enhancing MQC, and improving anti-lipotoxicity effects.
Dysfunctional organelle interactions not only lead to cellular senescence, but also affect other important processes, such as the imbalance of cellular energy metabolism. Mitochondria are the main energy producers [209]. Their activities, especially ATP production, are regulated by calcium signaling [209]. In particular, IP3R-mediated calcium transfer in ER-mitochondria MCSs plays an important role in maintaining basal mitochondrial metabolism [209], and the inhibition of IP3R activity leads to the impaired cellular energy metabolism under normal cellular conditions [209]. Peroxisomes and mitochondria act together in the degradation of fatty acids [91]. Dysfunctional mitochondria can replenish the TCA cycle via the RTG pathway, where peroxisomes convert fatty acids into acetyl-CoA [93, 94]. Defects in peroxisomes have an impact on mitochondrial pathology due to the impairment of fatty acid metabolism [210]. Thus, the organelles cooperatively form an interactive network to maintain cell homeostasis and accomplish a series of biological processes. Further understanding of the relevance of organelle interactions would certainly broaden our horizons for the understanding of mechanical and therapeutic exploration of complicated cell behavior. However, this is not limited to cellular senescence. Hence, our review provides important clues for a deeper understanding of cell function from a new perspective.
Abbreviation
ABCD1: ATP binding cassette subfamily D member 1; ACBD2: acyl-CoA binding domain protein 2; ACBD5: acyl-CoA binding domain protein 5; AMPK: AMP-activated protein kinase; ATF6α: activating transcription factor 6 alpha; ATG5: autophagy-related gene 5; ATG14: autophagy-related gene 14; BAP31: B‐cell receptor‐associated protein 31; CERT: ceramide-transfer protein; CRAC: Ca2+ release-activated Ca2+; DDR: DNA damage response; DGAT2: diacylglycerol acyltransferase 2; DRP1: dynamin-related protein-1; ECI2: enoyl-CoA-δ isomerase 2; ER: endoplasmic reticulum; E-Syts: extended synaptotagmin-like proteins; FATP1: fatty acid transport protein 1; FFAT: phenylalanine-phenylalanine acidic tract; FIS1: mitochondrial fission 1; GRP75: glucose‐regulated protein 75; IMM: inner mitochondrial membrane; IP3R: inositol 1,4,5-triphosphate receptor; IRE1α: inositol requiring enzyme 1 alpha; IST1: IST1 factor associated with the endosomal sorting complex required for transport-III; LAMP1: lysosomal associated membrane protein 1; LDL: low-density lipoprotein; LDs: lipid droplets; MCOLN1: mucolipin 1; MCS: membrane contact site; MCU: mitochondrial calcium uniporter; MDV: mitochondrial-derived vesicle; Mfn2: mitofusin 2; MQC: mitochondrial quality control; mtDNA: mitochondrial DNA; NRZ: NAG-RINT1-ZW10; OMM: outer mitochondrial membrane; ORPs: oxysterol-binding protein-related protein family; OSBP: oxysterol-binding protein; ORP1L: oxysterol-binding protein-related protein 1; PACS-2: phosphofurin acidic cluster sorting protein 2; PC: phosphatidylcholine; PDZD8: PDZ domain‐containing protein 8; PE: phosphatidylethanolamine; PERK: RNA-dependent protein kinase-like ER kinase; PGC-1α: proliferator-activated receptor gamma coactivator 1-alpha; PI: phosphatidylinositol; PITPNM1: phosphatidylinositol transfer protein membrane associated 1; PI(4)P: phosphatidylinositol 4-phosphate; PI(4,5)P2: phosphatidylinositol‐4,5‐bisphosphate; PM: plasma membrane; PRKN: Parkin RBR E3 ubiquitin protein ligase; PS: phosphatidylserine; PTPIP51: protein tyrosine phosphatase interacting protein 51; ROS: reactive oxygen species; RTG: retrograde; RyR1: ryanodine receptor 1; SASP: senescence-associated secretory phenotype; SERCA: sarco/ER Ca2+-ATPase; STARD3: steroidogenic acute regulatory protein-related lipid transfer domain-3; STIM1: stromal interaction molecule 1; STX17: syntaxin 17; Syt7: Synaptotagmin-7; TBC1D15: TBC domain family member 15; TCA: tricarboxylic acid; TFEB: transcription factor EB; TMBIM6: transmembrane BAX inhibitor motif containing 6; TMEM: transmembrane; TNTs: tunneling nanotubes; TRPML1: transient receptor potential mucolipin 1; UPR: unfolded protein response; VAPB: vesicle-associated membrane protein-associated protein B; VAPs: vesicle-associated membrane protein-associated proteins; VDAC: voltage-dependent anion-selective channel protein; 4-PBA: 4-phenylbutyric acid.
Acknowledgements
This work was supported by National Key R&D Program of China (2020YFC2005000), National Natural Science Foundation of China Grant 82070707, 91949114; and the project of Innovation team of chronic kidney disease with integrated traditional Chinese and Western Medicine (2019KCXTD014); Frontier Research Program of Guangzhou Regenerative Medicine and Health Guangdong Laboratory (2018GZR110105004) and Outstanding Scholar Program of Guangzhou Regenerative Medicine and Health Guangdong Laboratory (2018GZR110102004); and Outstanding youth cultivation program in Nanfang Hospital (2021J001) and the Presidential Foundation of Nanfang Hospital (Grant No. 2019Z006).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Cohen S, Valm AM, Lippincott-Schwartz J. Interacting organelles. Curr Opin Cell Biol. 2018;53:84-91
2. Henne WM. Organelle remodeling at membrane contact sites. J Struct Biol. 2016;196:15-9
3. Helle SC, Kanfer G, Kolar K, Lang A, Michel AH, Kornmann B. Organization and function of membrane contact sites. Biochim Biophys Acta. 2013;1833:2526-41
4. Levine T. Short-range intracellular trafficking of small molecules across endoplasmic reticulum junctions. Trends Cell Biol. 2004;14:483-90
5. Toulmay A, Prinz WA. Lipid transfer and signaling at organelle contact sites: the tip of the iceberg. Curr Opin Cell Biol. 2011;23:458-63
6. Prinz WA. Bridging the gap: membrane contact sites in signaling, metabolism, and organelle dynamics. J Cell Biol. 2014;205:759-69
7. Phillips MJ, Voeltz GK. Structure and function of ER membrane contact sites with other organelles. Nat Rev Mol Cell Biol. 2016;17:69-82
8. Wu H, Carvalho P, Voeltz GK. Here, there, and everywhere: The importance of ER membrane contact sites. Science. 2018;361:eaan5835
9. He S, Sharpless NE. Senescence in health and disease. Cell. 2017;169:1000-11
10. McHugh D, Gil J. Senescence and aging: Causes, consequences, and therapeutic avenues. J Cell Biol. 2018;217:65-77
11. Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of cellular senescence. Trends Cell Biol. 2018;28:436-53
12. Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. 2015;21:1424-35
13. Xu J, Zhou L, Liu Y. Cellular senescence in kidney fibrosis: pathologic significance and therapeutic strategies. Front Pharmacol. 2020;11:601325
14. Stevenson J, Huang EY, Olzmann JA. Endoplasmic reticulum-associated degradation and lipid homeostasis. Annu Rev Nutr. 2016;36:511-42
15. Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol. 2015;10:173-94
16. Madreiter-Sokolowski CT, Ramadani-Muja J, Ziomek G, Burgstaller S, Bischof H, Koshenov Z. et al. Tracking intra- and inter-organelle signaling of mitochondria. FEBS J. 2019;286:4378-401
17. Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145-59
18. Li J, Ahat E, Wang Y. Golgi structure and function in health, stress, and diseases. Results Probl Cell Differ. 2019;67:441-85
19. Smith JJ, Aitchison JD. Peroxisomes take shape. Nat Rev Mol Cell Biol. 2013;14:803-17
20. Walther TC, Chung J, Farese RV Jr. Lipid droplet biogenesis. Annu Rev Cell Dev Biol. 2017;33:491-510
21. Du J, Ji Y, Qiao L, Liu Y, Lin J. Cellular endo-lysosomal dysfunction in the pathogenesis of non-alcoholic fatty liver disease. Liver Int. 2020;40:271-80
22. Toth AE, Nielsen SSE, Tomaka W, Abbott NJ, Nielsen MS. The endo-lysosomal system of bEnd.3 and hCMEC/D3 brain endothelial cells. Fluids Barriers CNS. 2019;16:14
23. Lee CA, Blackstone C. ER morphology and endo-lysosomal crosstalk: Functions and disease implications. Biochim Biophys Acta Mol Cell Biol Lipids. 2020;1865:158544
24. Deng X, Wang Y, Zhou Y, Soboloff J, Gill DL. STIM and Orai: dynamic intermembrane coupling to control cellular calcium signals. J Biol Chem. 2009;284:22501-5
25. Lee JE, Cathey PI, Wu H, Parker R, Voeltz GK. Endoplasmic reticulum contact sites regulate the dynamics of membraneless organelles. Science. 2020;367:eaay7108
26. Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS. et al. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science. 2009;325:477-81
27. Wiedemann N, Meisinger C, Pfanner N. Cell biology. Connecting organelles. Science. 2009;325:403-4
28. Wilhelm LP, Wendling C, Védie B, Kobayashi T, Chenard MP, Tomasetto C. et al. STARD3 mediates endoplasmic reticulum-to-endosome cholesterol transport at membrane contact sites. EMBO J. 2017;36:1412-33
29. Levin-Konigsberg R, Grinstein S. Phagosome-endoplasmic reticulum contacts: Kissing and not running. Traffic. 2020;21:172-80
30. Levin-Konigsberg R, Montaño-Rendón F, Keren-Kaplan T, Li R, Ego B, Mylvaganam S. et al. Phagolysosome resolution requires contacts with the endoplasmic reticulum and phosphatidylinositol-4-phosphate signalling. Nat Cell Biol. 2019;21:1234-47
31. Gatta AT, Levine TP. Piecing together the patchwork of contact sites. Trends Cell Biol. 2017;27:214-29
32. Nunes P, Cornut D, Bochet V, Hasler U, Oh-Hora M, Waldburger JM. et al. STIM1 juxtaposes ER to phagosomes, generating Ca²⁺ hotspots that boost phagocytosis. Curr Biol. 2012;22:1990-7
33. De Matteis MA, Rega LR. Endoplasmic reticulum-Golgi complex membrane contact sites. Curr Opin Cell Biol. 2015;35:43-50
34. Farré JC, Mahalingam SS, Proietto M, Subramani S. Peroxisome biogenesis, membrane contact sites, and quality control. EMBO Rep. 2019;20:e46864
35. Salo VT, Ikonen E. Moving out but keeping in touch: contacts between endoplasmic reticulum and lipid droplets. Curr Opin Cell Biol. 2019;57:64-70
36. Lee S, Bahmanyar S. The endoplasmic reticulum regulates membraneless organelles through contact sites. Biochemistry. 2020;59:1716-7
37. Hewlett B, Singh NP, Vannier C, Galli T. ER-PM contact sites - SNARING actors in emerging functions. Front Cell Dev Biol. 2021;9:635518
38. Loewen CJ, Roy A, Levine TP. A conserved ER targeting motif in three families of lipid binding proteins and in Opi1p binds VAP. EMBO J. 2003;22:2025-35
39. Murphy SE, Levine TP. VAP, a versatile access point for the endoplasmic reticulum: review and analysis of FFAT-like motifs in the VAPome. Biochim Biophys Acta. 2016;1861:952-61
40. Dong R, Saheki Y, Swarup S, Lucast L, Harper JW, De Camilli P. Endosome-ER contacts control actin nucleation and retromer function through VAP-dependent regulation of PI4P. Cell. 2016;166:408-23
41. Peretti D, Dahan N, Shimoni E, Hirschberg K, Lev S. Coordinated lipid transfer between the endoplasmic reticulum and the Golgi complex requires the VAP proteins and is essential for Golgi-mediated transport. Mol Biol Cell. 2008;19:3871-84
42. Keinan O, Kedan A, Gavert N, Selitrennik M, Kim S, Karn T. et al. The lipid-transfer protein Nir2 enhances epithelial-mesenchymal transition and facilitates breast cancer metastasis. J Cell Sci. 2014;127:4740-9
43. Alpy F, Rousseau A, Schwab Y, Legueux F, Stoll I, Wendling C. et al. STARD3 or STARD3NL and VAP form a novel molecular tether between late endosomes and the ER. J Cell Sci. 2013;126:5500-12
44. Zhao K, Foster J, Ridgway ND. Oxysterol-binding protein-related protein 1 variants have opposing cholesterol transport activities from the endolysosomes. Mol Biol Cell. 2020;31:793-802
45. Costello JL, Castro IG, Hacker C, Schrader TA, Metz J, Zeuschner D. et al. ACBD5 and VAPB mediate membrane associations between peroxisomes and the ER. J Cell Biol. 2017;216:331-42
46. Siao W, Wang P, Voigt B, Hussey PJ, Baluska F. Arabidopsis SYT1 maintains stability of cortical endoplasmic reticulum networks and VAP27-1-enriched endoplasmic reticulum-plasma membrane contact sites. J Exp Bot. 2016;67:6161-71
47. Quon E, Sere YY, Chauhan N, Johansen J, Sullivan DP, Dittman JS. et al. Endoplasmic reticulum-plasma membrane contact sites integrate sterol and phospholipid regulation. PLoS Biol. 2018;16:e2003864
48. Galmes R, Houcine A, van Vliet AR, Agostinis P, Jackson CL, Giordano F. ORP5/ORP8 localize to endoplasmic reticulum-mitochondria contacts and are involved in mitochondrial function. EMBO Rep. 2016;17:800-10
49. Chung J, Torta F, Masai K, Lucast L, Czapla H, Tanner LB. et al. Intercellular transport. PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER-plasma membrane contacts. Science. 2015;349:428-32
50. Kim YJ, Guzman-Hernandez ML, Wisniewski E, Balla T. Phosphatidylinositol-phosphatidic acid exchange by Nir2 at ER-PM contact sites maintains phosphoinositide signaling competence. Dev Cell. 2015;33:549-61
51. Zaman MF, Nenadic A, Radojičić A, Rosado A, Beh CT. Sticking with It: ER-PM membrane contact sites as a coordinating nexus for regulating lipids and proteins at the cell cortex. Front Cell Dev Biol. 2020;8:675
52. Navarro-Borelly L, Somasundaram A, Yamashita M, Ren D, Miller RJ, Prakriya M. STIM1-Orai1 interactions and Orai1 conformational changes revealed by live-cell FRET microscopy. J Physiol. 2008;586:5383-401
53. Luik RM, Wang B, Prakriya M, Wu MM, Lewis RS. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature. 2008;454:538-42
54. Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S. et al. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. 2009;136:876-90
55. Luik RM, Wu MM, Buchanan J, Lewis RS. The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J Cell Biol. 2006;174:815-25
56. Hernández-Ochoa EO, Pratt SJP, Lovering RM, Schneider MF. Critical role of intracellular RyR1 calcium release channels in skeletal muscle function and disease. Front Physiol. 2015;6:420
57. Gordaliza-Alaguero I, Cantó C, Zorzano A. Metabolic implications of organelle-mitochondria communication. EMBO Rep. 2019;20:e47928
58. Vance JE. MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim Biophys Acta. 2014;1841:595-609
59. Tatsuta T, Scharwey M, Langer T. Mitochondrial lipid trafficking. Trends Cell Biol. 2014;24:44-52
60. Cárdenas C, Miller RA, Smith I, Bui T, Molgó J, Müller M. et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell. 2010;142:270-83
61. Olson ML, Chalmers S, McCarron JG. Mitochondrial Ca2+ uptake increases Ca2+ release from inositol 1,4,5-trisphosphate receptor clusters in smooth muscle cells. J Biol Chem. 2010;285:2040-50
62. Lee S, Min KT. The interface between ER and mitochondria: molecular compositions and functions. Mol Cells. 2018;41:1000-7
63. Hirabayashi Y, Kwon SK, Paek H, Pernice WM, Paul MA, Lee J. et al. ER-mitochondria tethering by PDZD8 regulates Ca(2+) dynamics in mammalian neurons. Science. 2017;358:623-30
64. de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605-10
65. De Vos KJ, Mórotz GM, Stoica R, Tudor EL, Lau KF, Ackerley S. et al. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum Mol Genet. 2012;21:1299-311
66. Simmen T, Lynes EM, Gesson K, Thomas G. Oxidative protein folding in the endoplasmic reticulum: tight links to the mitochondria-associated membrane (MAM). Biochim Biophys Acta. 2010;1798:1465-73
67. Cho B, Cho HM, Jo Y, Kim HD, Song M, Moon C. et al. Constriction of the mitochondrial inner compartment is a priming event for mitochondrial division. Nat Commun. 2017;8:15754
68. Mesmin B, Bigay J, Moser von Filseck J, Lacas-Gervais S, Drin G, Antonny B. A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi tether OSBP. Cell. 2013;155:830-43
69. Mesmin B, Kovacs D, D'Angelo G. Lipid exchange and signaling at ER-Golgi contact sites. Curr Opin Cell Biol. 2019;57:8-15
70. Tabak HF, van der Zand A, Braakman I. Peroxisomes: minted by the ER. Curr Opin Cell Biol. 2008;20:393-400
71. Joshi AS, Nebenfuehr B, Choudhary V, Satpute-Krishnan P, Levine TP, Golden A. et al. Lipid droplet and peroxisome biogenesis occur at the same ER subdomains. Nat Commun. 2018;9:2940
72. Schuldiner M, Zalckvar E. Incredibly close-A newly identified peroxisome-ER contact site in humans. J Cell Biol. 2017;216:287-9
73. Schrader M, Kamoshita M, Islinger M. Organelle interplay-peroxisome interactions in health and disease. J Inherit Metab Dis. 2020;43:71-89
74. Hua R, Cheng D, Coyaud É, Freeman S, Di Pietro E, Wang Y. et al. VAPs and ACBD5 tether peroxisomes to the ER for peroxisome maintenance and lipid homeostasis. J Cell Biol. 2017;216:367-77
75. Xiao J, Luo J, Hu A, Xiao T, Li M, Kong Z. et al. Cholesterol transport through the peroxisome-ER membrane contacts tethered by PI(4,5)P(2) and extended synaptotagmins. Sci China Life Sci. 2019;62:1117-35
76. Chu BB, Liao YC, Qi W, Xie C, Du X, Wang J. et al. Cholesterol transport through lysosome-peroxisome membrane contacts. Cell. 2015;161:291-306
77. Miao G, Zhang Y, Chen D, Zhang H. The ER-localized transmembrane protein TMEM39A/SUSR2 regulates autophagy by controlling the trafficking of the PtdIns(4)P phosphatase SAC1. Mol Cell. 2020;77:618-32.e5
78. Allison R, Edgar JR, Pearson G, Rizo T, Newton T, Günther S. et al. Defects in ER-endosome contacts impact lysosome function in hereditary spastic paraplegia. J Cell Biol. 2017;216:1337-55
79. Akbari M, Kirkwood TBL, Bohr VA. Mitochondria in the signaling pathways that control longevity and health span. Ageing Res Rev. 2019;54:100940
80. Westermann B. The mitochondria-plasma membrane contact site. Curr Opin Cell Biol. 2015;35:1-6
81. Benador IY, Veliova M, Liesa M, Shirihai OS. Mitochondria bound to lipid droplets: where mitochondrial dynamics regulate lipid storage and utilization. Cell Metab. 2019;29:827-35
82. Dolman NJ, Gerasimenko JV, Gerasimenko OV, Voronina SG, Petersen OH, Tepikin AV. Stable Golgi-mitochondria complexes and formation of Golgi Ca(2+) gradients in pancreatic acinar cells. J Biol Chem. 2005;280:15794-9
83. Das A, Nag S, Mason AB, Barroso MM. Endosome-mitochondria interactions are modulated by iron release from transferrin. J Cell Biol. 2016;214:831-45
84. Wong YC, Ysselstein D, Krainc D. Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature. 2018;554:382-6
85. Wong YC, Kim S, Peng W, Krainc D. Regulation and function of mitochondria-lysosome membrane contact sites in cellular homeostasis. Trends Cell Biol. 2019;29:500-13
86. Fransen M, Lismont C, Walton P. The peroxisome-mitochondria connection: how and why? Int J Mol Sci. 2017;18:1126
87. Szymański J, Janikiewicz J, Michalska B, Patalas-Krawczyk P, Perrone M, Ziółkowski W. et al. Interaction of mitochondria with the endoplasmic reticulum and plasma membrane in calcium homeostasis, lipid trafficking and mitochondrial structure. Int J Mol Sci. 2017;18:1576
88. Frieden M, Arnaudeau S, Castelbou C, Demaurex N. Subplasmalemmal mitochondria modulate the activity of plasma membrane Ca2+-ATPases. J Biol Chem. 2005;280:43198-208
89. Baixauli F, Acín-Pérez R, Villarroya-Beltrí C, Mazzeo C, Nuñez-Andrade N, Gabandé-Rodriguez E. et al. Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell Metab. 2015;22:485-98
90. Peng W, Wong YC, Krainc D. Mitochondria-lysosome contacts regulate mitochondrial Ca(2+) dynamics via lysosomal TRPML1. Proc Natl Acad Sci U S A. 2020;117:19266-75
91. Schrader M, Costello J, Godinho LF, Islinger M. Peroxisome-mitochondria interplay and disease. J Inherit Metab Dis. 2015;38:681-702
92. Fan J, Li X, Issop L, Culty M, Papadopoulos V. ACBD2/ECI2-mediated peroxisome-mitochondria interactions in Leydig cell steroid biosynthesis. Mol Endocrinol. 2016;30:763-82
93. Titorenko VI, Terlecky SR. Peroxisome metabolism and cellular aging. Traffic. 2011;12:252-9
94. Jazwinski SM. The retrograde response links metabolism with stress responses, chromatin-dependent gene activation, and genome stability in yeast aging. Gene. 2005;354:22-7
95. Hu A, Zhao XT, Tu H, Xiao T, Fu T, Wang Y. et al. PIP4K2A regulates intracellular cholesterol transport through modulating PI(4,5)P(2) homeostasis. J Lipid Res. 2018;59:507-14
96. Jin Y, Strunk BS, Weisman LS. Close encounters of the lysosome-peroxisome kind. Cell. 2015;161:197-8
97. Chang CL, Weigel AV, Ioannou MS, Pasolli HA, Xu CS, Peale DR. et al. Spastin tethers lipid droplets to peroxisomes and directs fatty acid trafficking through ESCRT-III. J Cell Biol. 2019;218:2583-99
98. Calcinotto A, Kohli J, Zagato E, Pellegrini L, Demaria M, Alimonti A. Cellular senescence: aging, cancer, and injury. Physiol Rev. 2019;99:1047-78
99. Pluquet O, Pourtier A, Abbadie C. The unfolded protein response and cellular senescence. A review in the theme: cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. Am J Physiol Cell Physiol. 2015;308:C415-25
100. Di Micco R, Krizhanovsky V, Baker D, d'Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. 2021;22:75-95
101. Ziegler DV, Wiley CD, Velarde MC. Mitochondrial effectors of cellular senescence: beyond the free radical theory of aging. Aging Cell. 2015;14:1-7
102. Cavinato M, Madreiter-Sokolowski CT, Büttner S, Schosserer M, Zwerschke W, Wedel S. et al. Targeting cellular senescence based on interorganelle communication, multilevel proteostasis, and metabolic control. FEBS J. 2021;288:3834-54
103. Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15:482-96
104. Chernorudskiy AL, Zito E. Regulation of calcium homeostasis by ER redox: a close-up of the ER/mitochondria connection. J Mol Biol. 2017;429:620-32
105. Martin N, Bernard D. Calcium signaling and cellular senescence. Cell Calcium. 2018;70:16-23
106. Santoni G, Maggi F, Amantini C, Marinelli O, Nabissi M, Morelli MB. Pathophysiological role of transient receptor potential mucolipin channel 1 in calcium-mediated stress-induced neurodegenerative diseases. Front Physiol. 2020;11:251
107. Marchi S, Patergnani S, Missiroli S, Morciano G, Rimessi A, Wieckowski MR. et al. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium. 2018;69:62-72
108. Yeh CH, Chou YJ, Kao CH, Tsai TF. Mitochondria and calcium homeostasis: Cisd2 as a big player in cardiac ageing. Int J Mol Sci. 2020;21:9238
109. Habiballa L, Salmonowicz H, Passos JF. Mitochondria and cellular senescence: Implications for musculoskeletal ageing. Free Radic Biol Med. 2019;132:3-10
110. Li J, Zhang D, Brundel B, Wiersma M. Imbalance of ER and mitochondria interactions: prelude to cardiac ageing and disease? Cells. 2019;8:1617
111. Hayashi T, Rizzuto R, Hajnoczky G, Su TP. MAM: more than just a housekeeper. Trends Cell Biol. 2009;19:81-8
112. Calvo-Rodríguez M, García-Durillo M, Villalobos C, Núñez L. In vitro aging promotes endoplasmic reticulum (ER)-mitochondria Ca(2+) cross talk and loss of store-operated Ca(2+) entry (SOCE) in rat hippocampal neurons. Biochim Biophys Acta. 2016;1863:2637-49
113. Birch J, Barnes PJ, Passos JF. Mitochondria, telomeres and cell senescence: Implications for lung ageing and disease. Pharmacol Ther. 2018;183:34-49
114. Correia-Melo C, Marques FD, Anderson R, Hewitt G, Hewitt R, Cole J. et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016;35:724-42
115. Ahumada-Castro U, Puebla-Huerta A, Cuevas-Espinoza V, Lovy A, Cardenas JC. Keeping zombies alive: The ER-mitochondria Ca(2+) transfer in cellular senescence. Biochim Biophys Acta Mol Cell Res. 2021;1868:119099
116. Ziegler DV, Vindrieux D, Goehrig D, Jaber S, Collin G, Griveau A. et al. Calcium channel ITPR2 and mitochondria-ER contacts promote cellular senescence and aging. Nat Commun. 2021;12:720
117. Wiel C, Lallet-Daher H, Gitenay D, Gras B, Le Calvé B, Augert A. et al. Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat Commun. 2014;5:3792
118. Coen K, Flannagan RS, Baron S, Carraro-Lacroix LR, Wang D, Vermeire W. et al. Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J Cell Biol. 2012;198:23-35
119. Fernandez-Mosquera L, Yambire KF, Couto R, Pereyra L, Pabis K, Ponsford AH. et al. Mitochondrial respiratory chain deficiency inhibits lysosomal hydrolysis. Autophagy. 2019;15:1572-91
120. Atakpa P, Thillaiappan NB, Mataragka S, Prole DL, Taylor CW. IP(3) receptors preferentially associate with ER-lysosome contact sites and selectively deliver Ca(2+) to lysosomes. Cell Rep. 2018;25:3180-93.e7
121. Ghislat G, Knecht E. Ca²⁺-sensor proteins in the autophagic and endocytic traffic. Curr Protein Pept Sci. 2013;14:97-110
122. Madureira M, Connor-Robson N, Wade-Martins R. LRRK2: autophagy and lysosomal activity. Front Neurosci. 2020;14:498
123. Fujii S, Hara H, Araya J, Takasaka N, Kojima J, Ito S. et al. Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. Oncoimmunology. 2012;1:630-41
124. Korolchuk VI, Miwa S, Carroll B, von Zglinicki T. Mitochondria in cell senescence: Is mitophagy the weakest link? EBioMedicine. 2017;21:7-13
125. Zhang X, Cheng X, Yu L, Yang J, Calvo R, Patnaik S. et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun. 2016;7:12109
126. Di Paola S, Scotto-Rosato A, Medina DL. TRPML1: The Ca((2+))retaker of the lysosome. Cell Calcium. 2018;69:112-21
127. Santoni G, Amantini C, Nabissi M, Maggi F, Arcella A, Marinelli O. et al. Knock-down of mucolipin 1 channel promotes tumor progression and invasion in human glioblastoma cell lines. Front Oncol. 2021;11:578928
128. Kim HK, Lee GH, Bhattarai KR, Lee MS, Back SH, Kim HR. et al. TMBIM6 (transmembrane BAX inhibitor motif containing 6) enhances autophagy through regulation of lysosomal calcium. Autophagy. 2021;17:761-78
129. Sachdeva K, Do DC, Zhang Y, Hu X, Chen J, Gao P. Environmental exposures and asthma development: autophagy, mitophagy, and cellular senescence. Front Immunol. 2019;10:2787
130. Leeman DS, Hebestreit K, Ruetz T, Webb AE, McKay A, Pollina EA. et al. Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science. 2018;359:1277-83
131. Rajawat YS, Hilioti Z, Bossis I. Aging: central role for autophagy and the lysosomal degradative system. Ageing Res Rev. 2009;8:199-213
132. Hamon MP, Ahmed EK, Baraibar MA, Friguet B. Proteome oxidative modifications and impairment of specific metabolic pathways during cellular senescence and aging. Proteomics. 2020;20:e1800421
133. Kaushik S, Cuervo AM. Proteostasis and aging. Nat Med. 2015;21:1406-15
134. Höhn A, Weber D, Jung T, Ott C, Hugo M, Kochlik B. et al. Happily (n)ever after: aging in the context of oxidative stress, proteostasis loss and cellular senescence. Redox Biol. 2017;11:482-501
135. Sabath N, Levy-Adam F, Younis A, Rozales K, Meller A, Hadar S. et al. Cellular proteostasis decline in human senescence. Proc Natl Acad Sci U S A. 2020;117:31902-13
136. Simic MS, Dillin A. The Lysosome, elixir of neural stem cell youth. Cell Stem Cell. 2018;22:619-20
137. Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. 2020;21:421-38
138. Bravo R, Vicencio JM, Parra V, Troncoso R, Munoz JP, Bui M. et al. Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J Cell Sci. 2011;124:2143-52
139. Martucciello S, Masullo M, Cerulli A, Piacente S. Natural products targeting ER stress, and the functional link to mitochondria. Int J Mol Sci. 2020;21:1905
140. Rainbolt TK, Saunders JM, Wiseman RL. Stress-responsive regulation of mitochondria through the ER unfolded protein response. Trends Endocrinol Metab. 2014;25:528-37
141. Paillusson S, Stoica R, Gomez-Suaga P, Lau DHW, Mueller S, Miller T. et al. There's something wrong with my MAM; the ER-mitochondria axis and neurodegenerative diseases. Trends Neurosci. 2016;39:146-57
142. Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287:C817-33
143. Boengler K, Kosiol M, Mayr M, Schulz R, Rohrbach S. Mitochondria and ageing: role in heart, skeletal muscle and adipose tissue. J Cachexia Sarcopenia Muscle. 2017;8:349-69
144. García-Prat L, Martínez-Vicente M, Perdiguero E, Ortet L, Rodríguez-Ubreva J, Rebollo E. et al. Autophagy maintains stemness by preventing senescence. Nature. 2016;529:37-42
145. Moreno-Blas D, Gorostieta-Salas E, Castro-Obregón S. Connecting chaperone-mediated autophagy dysfunction to cellular senescence. Ageing Res Rev. 2018;41:34-41
146. Li YF, Ouyang SH, Tu LF, Wang X, Yuan WL, Wang GE. et al. Caffeine protects skin from oxidative stress-induced senescence through the activation of autophagy. Theranostics. 2018;8:5713-30
147. Shirakabe A, Ikeda Y, Sciarretta S, Zablocki DK, Sadoshima J. Aging and autophagy in the heart. Circ Res. 2016;118:1563-76
148. Lapierre LR, Kumsta C, Sandri M, Ballabio A, Hansen M. Transcriptional and epigenetic regulation of autophagy in aging. Autophagy. 2015;11:867-80
149. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27-42
150. Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell. 2011;146:682-95
151. Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N. et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495:389-93
152. Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. 2013;14:759-74
153. Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK. et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656-67
154. Park JT, Lee YS, Cho KA, Park SC. Adjustment of the lysosomal-mitochondrial axis for control of cellular senescence. Ageing Res Rev. 2018;47:176-82
155. Audesse AJ, Webb AE. Enhancing lysosomal activation restores neural stem cell function during aging. J Exp Neurosci. 2018;12:1179069518795874
156. MacDonald E, Urbé S, Clague MJ. USP8 controls the trafficking and sorting of lysosomal enzymes. Traffic. 2014;15:879-88
157. Sun N, Youle RJ, Finkel T. The mitochondrial basis of aging. Mol Cell. 2016;61:654-66
158. Conduit SE, Hakim S, Feeney SJ, Ooms LM, Dyson JM, Abud HE. et al. β-catenin ablation exacerbates polycystic kidney disease progression. Hum Mol Genet. 2019;28:230-44
159. Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A. et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016;23:303-14
160. Lee SM, Dho SH, Ju SK, Maeng JS, Kim JY, Kwon KS. Cytosolic malate dehydrogenase regulates senescence in human fibroblasts. Biogerontology. 2012;13:525-36
161. Pickles S, Vigié P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. 2018;28:R170-r85
162. Sugiura A, McLelland GL, Fon EA, McBride HM. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J. 2014;33:2142-56
163. Choong CJ, Okuno T, Ikenaka K, Baba K, Hayakawa H, Koike M. et al. Alternative mitochondrial quality control mediated by extracellular release. Autophagy. 2021;17:2962-74
164. Lyamzaev KG, Nepryakhina OK, Saprunova VB, Bakeeva LE, Pletjushkina OY, Chernyak BV. et al. Novel mechanism of elimination of malfunctioning mitochondria (mitoptosis): formation of mitoptotic bodies and extrusion of mitochondrial material from the cell. Biochim Biophys Acta. 2008;1777:817-25
165. Liu D, Gao Y, Liu J, Huang Y, Yin J, Feng Y. et al. Intercellular mitochondrial transfer as a means of tissue revitalization. Signal Transduct Target Ther. 2021;6:65
166. Picca A, Calvani R, Coelho-Junior HJ, Landi F, Bernabei R, Marzetti E. Inter-organelle membrane contact sites and mitochondrial quality control during aging: a geroscience view. Cells. 2020;9:598
167. Abrisch RG, Gumbin SC, Wisniewski BT, Lackner LL, Voeltz GK. Fission and fusion machineries converge at ER contact sites to regulate mitochondrial morphology. J Cell Biol. 2020;219:e201911122
168. Lewis SC, Uchiyama LF, Nunnari J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science. 2016;353:aaf5549
169. Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. 2011;334:358-62
170. Westermann B. Organelle dynamics: ER embraces mitochondria for fission. Curr Biol. 2011;21:R922-4
171. Fonseca TB, Sánchez-Guerrero Á, Milosevic I, Raimundo N. Mitochondrial fission requires DRP1 but not dynamins. Nature. 2019;570:E34-e42
172. Nagashima S, Tábara LC, Tilokani L, Paupe V, Anand H, Pogson JH. et al. Golgi-derived PI(4)P-containing vesicles drive late steps of mitochondrial division. Science. 2020;367:1366-71
173. Cheng XT, Xie YX, Zhou B, Huang N, Farfel-Becker T, Sheng ZH. Revisiting LAMP1 as a marker for degradative autophagy-lysosomal organelles in the nervous system. Autophagy. 2018;14:1472-4
174. Marchi S, Patergnani S, Pinton P. The endoplasmic reticulum-mitochondria connection: one touch, multiple functions. Biochim Biophys Acta. 2014;1837:461-9
175. Naon D, Zaninello M, Giacomello M, Varanita T, Grespi F, Lakshminaranayan S. et al. Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc Natl Acad Sci U S A. 2016;113:11249-54
176. Davis CH, Kim KY, Bushong EA, Mills EA, Boassa D, Shih T. et al. Transcellular degradation of axonal mitochondria. Proc Natl Acad Sci U S A. 2014;111:9633-8
177. Chuang YC, Liou CW, Chen SD, Wang PW, Chuang JH, Tiao MM. et al. Mitochondrial transfer from Wharton's Jelly mesenchymal stem cell to MERRF cybrid reduces oxidative stress and improves mitochondrial bioenergetics. Oxid Med Cell Longev. 2017;2017:5691215
178. Gao J, Qin A, Liu D, Ruan R, Wang Q, Yuan J. et al. Endoplasmic reticulum mediates mitochondrial transfer within the osteocyte dendritic network. Sci Adv. 2019;5:eaaw7215
179. Chen FJ, Yin Y, Chua BT, Li P. CIDE family proteins control lipid homeostasis and the development of metabolic diseases. Traffic. 2020;21:94-105
180. Wilfling F, Wang H, Haas JT, Krahmer N, Gould TJ, Uchida A. et al. Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Dev Cell. 2013;24:384-99
181. Barbosa AD, Siniossoglou S. Function of lipid droplet-organelle interactions in lipid homeostasis. Biochim Biophys Acta Mol Cell Res. 2017;1864:1459-68
182. Geltinger F, Schartel L, Wiederstein M, Tevini J, Aigner E, Felder TK. et al. Friend or foe: lipid droplets as organelles for protein and lipid storage in cellular stress response, aging and disease. Molecules. 2020;25:5053
183. Slawik M, Vidal-Puig AJ. Lipotoxicity, overnutrition and energy metabolism in aging. Ageing Res Rev. 2006;5:144-64
184. Zhang H, Wang Y, Guan L, Chen Y, Chen P, Sun J. et al. Lipidomics reveals carnitine palmitoyltransferase 1C protects cancer cells from lipotoxicity and senescence. J Pharm Anal. 2021;11:340-50
185. Lizardo DY, Lin YL, Gokcumen O, Atilla-Gokcumen GE. Regulation of lipids is central to replicative senescence. Mol Biosyst. 2017;13:498-509
186. Renne MF, Hariri H. Lipid droplet-organelle contact sites as hubs for fatty acid metabolism, trafficking, and metabolic channeling. Front Cell Dev Biol. 2021;9:726261
187. Zhong H, Song R, Pang Q, Liu Y, Zhuang J, Chen Y. et al. Propofol inhibits parthanatos via ROS-ER-calcium-mitochondria signal pathway in vivo and vitro. Cell Death Dis. 2018;9:932
188. Du X, Zhou L, Aw YC, Mak HY, Xu Y, Rae J. et al. ORP5 localizes to ER-lipid droplet contacts and regulates the level of PI(4)P on lipid droplets. J Cell Biol. 2020;219:e201905162
189. Xu D, Li Y, Wu L, Li Y, Zhao D, Yu J. et al. Rab18 promotes lipid droplet (LD) growth by tethering the ER to LDs through SNARE and NRZ interactions. J Cell Biol. 2018;217:975-95
190. Xu N, Zhang SO, Cole RA, McKinney SA, Guo F, Haas JT. et al. The FATP1-DGAT2 complex facilitates lipid droplet expansion at the ER-lipid droplet interface. J Cell Biol. 2012;198:895-911
191. Salo VT, Belevich I, Li S, Karhinen L, Vihinen H, Vigouroux C. et al. Seipin regulates ER-lipid droplet contacts and cargo delivery. EMBO J. 2016;35:2699-716
192. McLaren DG, Han S, Murphy BA, Wilsie L, Stout SJ, Zhou H. et al. DGAT2 inhibition alters aspects of triglyceride metabolism in rodents but not in non-human primates. Cell Metab. 2018;27:1236-48.e6
193. Benador IY, Veliova M, Mahdaviani K, Petcherski A, Wikstrom JD, Assali EA. et al. Mitochondria bound to lipid droplets have unique bioenergetics, composition, and dynamics that support lipid droplet expansion. Cell Metab. 2018;27:869-85.e6
194. Arruda AP, Pers BM, Parlakgül G, Güney E, Inouye K, Hotamisligil GS. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat Med. 2014;20:1427-35
195. Burkewitz K, Feng G, Dutta S, Kelley CA, Steinbaugh M, Cram EJ. et al. Atf-6 regulates lifespan through ER-mitochondrial calcium homeostasis. Cell Rep. 2020;32:108125
196. Krebs J, Agellon LB, Michalak M. Ca(2+) homeostasis and endoplasmic reticulum (ER) stress: an integrated view of calcium signaling. Biochem Biophys Res Commun. 2015;460:114-21
197. Biwer LA, Askew-Page HR, Hong K, Milstein J, Johnstone SR, Macal E. et al. Endothelial calreticulin deletion impairs endothelial function in aged mice. Am J Physiol Heart Circ Physiol. 2020;318:H1041-h8
198. Liu J, Huang K, Cai GY, Chen XM, Yang JR, Lin LR. et al. Receptor for advanced glycation end-products promotes premature senescence of proximal tubular epithelial cells via activation of endoplasmic reticulum stress-dependent p21 signaling. Cell Signal. 2014;26:110-21
199. Tang C, Livingston MJ, Liu Z, Dong Z. Autophagy in kidney homeostasis and disease. Nat Rev Nephrol. 2020;16:489-508
200. Moreno-Blas D, Gorostieta-Salas E, Pommer-Alba A, Muciño-Hernández G, Gerónimo-Olvera C, Maciel-Barón LA. et al. Cortical neurons develop a senescence-like phenotype promoted by dysfunctional autophagy. Aging (Albany NY). 2019;11:6175-98
201. Picca A, Mankowski RT, Burman JL, Donisi L, Kim JS, Marzetti E. et al. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat Rev Cardiol. 2018;15:543-54
202. Kim YY, Um JH, Yoon JH, Lee DY, Lee YJ, Kim DH. et al. p53 regulates mitochondrial dynamics by inhibiting Drp1 translocation into mitochondria during cellular senescence. FASEB J. 2020;34:2451-64
203. Cho HM, Ryu JR, Jo Y, Seo TW, Choi YN, Kim JH. et al. Drp1-Zip1 interaction regulates mitochondrial quality surveillance system. Mol Cell. 2019;73:364-76.e8
204. Ren X, Chen L, Xie J, Zhang Z, Dong G, Liang J. et al. Resveratrol ameliorates mitochondrial elongation via Drp1/Parkin/PINK1 signaling in senescent-like cardiomyocytes. Oxid Med Cell Longev. 2017;2017:4175353
205. Araya J, Tsubouchi K, Sato N, Ito S, Minagawa S, Hara H. et al. PRKN-regulated mitophagy and cellular senescence during COPD pathogenesis. Autophagy. 2019;15:510-26
206. Iwasa S, Sato N, Wang CW, Cheng YH, Irokawa H, Hwang GW. et al. The phospholipid:diacylglycerol acyltransferase Lro1 is responsible for hepatitis c virus core-induced lipid droplet formation in a yeast model system. PLoS One. 2016;11:e0159324
207. Kovacs M, Geltinger F, Verwanger T, Weiss R, Richter K, Rinnerthaler M. Lipid droplets protect aging mitochondria and thus promote lifespan in yeast cells. Front Cell Dev Biol. 2021;9:774985
208. Michel AH, Kornmann B. The ERMES complex and ER-mitochondria connections. Biochem Soc Trans. 2012;40:445-50
209. Rossi A, Pizzo P, Filadi R. Calcium, mitochondria and cell metabolism: a functional triangle in bioenergetics. Biochim Biophys Acta Mol Cell Res. 2019;1866:1068-78
210. Thoms S, Grønborg S, Gärtner J. Organelle interplay in peroxisomal disorders. Trends Mol Med. 2009;15:293-302
Author contact
![]() Corresponding author: Dr. Lili Zhou, Division of Nephrology, Nanfang Hospital, 1838 North Guangzhou Ave, Guangzhou 510515, China, E-mail: jinli730edu.cn.
Corresponding author: Dr. Lili Zhou, Division of Nephrology, Nanfang Hospital, 1838 North Guangzhou Ave, Guangzhou 510515, China, E-mail: jinli730edu.cn.