Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Conclusion
Experimental Section
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2023; 13(2):472-482. doi:10.7150/thno.79452 This issue Cite
Research Paper
Simplified one-pot 18F-labeling of biomolecules with in situ generated fluorothiophosphate synthons in high molar activity
Hongzhang Yang1, Lei Zhang2, Huanhuan Liu1, Yunming Zhang1, Zhaobiao Mou1, Xueyuan Chen1, Jingchao Li1, Fengming He3, Zijing Li1 ![]()
1. Center for Molecular Imaging and Translational Medicine, State Key Laboratory of Molecular Vaccinology and Molecular Diagnostics, Department of Laboratory Medicine, School of Public Heath, Xiamen University, Xiamen, Fujian 361102, China.
2. Tianjin Engineering Technology Center of Chemical Wastewater Source Reduction and Recycling, School of Science, Tianjin Chengjian University, Tianjin 300384, China.
3. School of Pharmaceutical Sciences, Xiamen University, Xiamen, Fujian 361102, China.
Received 2022-10-1; Accepted 2022-11-2; Published 2023-1-1
Abstract

Rationale: Conventional 18F-labeling methods that demand substrate pre-modification or lengthy radiosynthesis procedures have impeded the visualization and translation of numerous biomolecules, as biomarkers or ligands, using modern positron emission tomography techniques in vivo. Moreover, 18F-labeled biomolecules in high molar activity (Am) that are indispensable for sensitive imaging could be only achieved under strict labeling conditions.
Methods: Herein, 18F-labeled fluorothiophosphate (FTP) synthons in high Am have been generated rapidly in situ in reaction solutions with < 5% water via nucleophilic substitution by wet [18F]F-, which required minimal processing from cyclotron target water.
Results: Various 18F-labeled FTP synthons have been prepared in 30 sec at room temperature with high radiochemical yields > 75% (isolated, non-decay-corrected). FTP synthons with unsaturated hydrocarbon or activated ester group can conjugate with typical small molecules, peptides, proteins, and metallic nanoparticles. 337-517 GBq μmol-1 Am has been achieved for 18F-labeled c(RGDyK) peptide using an automatic module with 37-74 GBq initial activity.
Conclusion: The combination of high 18F-fluorination efficiency of FTP synthons and following mild conjugation condition provides a universal simplified one-pot 18F-labeling method for broad unmodified biomolecular substrates.
Keywords: radiolabeling, radiosynthon, fluorine-18, fluorothiophosphate, positron emission tomography probe
Introduction
Positron emission tomography (PET) is a non-invasive, real-time functional imaging technique that provides abundant physiological and biochemical information. It relies on the spatiotemporal tracing of molecular probes composed of functional scaffolds and positron-emitting nuclides [1, 2]. Biomolecules, including small molecules, peptides and proteins, come from a treasury house of lead compounds with high specificity and low immunogenicity [3-5]. 18F (t1/2 = 109.7 min, 97% β+, maximum positron energy 0.64 MeV) is the most popular positron-emitting nuclide due to its favorable chemical, nuclide properties and production feasibility [6]. Numerous target-specific biomolecules merit 18F-labeling with optimum radiochemical parameters for pre-clinical and clinical PET imaging evaluation [7-10].
To achieve the mild 18F-labeling of biomolecules, two/multi-step/indirect approaches have been widely applied, which go through the incorporation of 18F into a prosthetic/linker synthon and then the gentle coupling of the synthon to the lead-compound biomolecules [4, 11-21] (Table 1). Since the hydration effect eliminates the reactivity/nucleophilicity of [18F]F- in aqueous media directly obtained from a cyclotron, radiosynthons with 18F at one end and an active group at the other end usually need to be prepared in a dried organic medium with heating, followed by deprotection. Alternative methods are one-step/direct, mild 18F-labeling via 18F/19F-exchange or Al18F chelation in aqueous solutions at a specific pH/temperature on non-carbon-centered prostheses that are pre-coupled to biomolecules [10, 22-24] (Table 1). Nevertheless, universally accessible, highly efficient 18F-labeling methods that do not require the pre-coupling of a prosthetic group, time-consuming alterations of reaction conditions, purification of intermediates, and use of inseparable precursors for high molar activity (Am) are still in open request [25].
Important parameters of selected 18F-labeling methods for biomolecules.
| Tracer | Labeling strategy | Mechanism | T (°C) | Solvent (v/v) | pH | Total synthesis time (min) | Precursor load (μmol) | RCC & RCY | Am (GBq μmol-1)/Starting activity (GBq) | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| [18F]FTP-c(RGDyK) | Instant FTP | SN[a] | RT | Borate buffer/DMSO (91/9) | 8.0 | 15[c] 60[d] | 0.2 | > 90%[f] & 34 ± 5%[g] | 49-107/3.7-5.6[c] 337-517/37-74[d] | - |
| [18F]FTP-HSA | Instant FTP | SN | RT | Borate buffer | 8.0 | 15[c] | 0.2 | 86 ± 5%[f] & 46 ± 9%[g] | 26.37-39.14/0.185-1.85[c] | - |
| [18F]FTP-5F7 | Instant FTP | SN | RT | Borate buffer | 8.0 | 15[c] | 0.2 | > 90%[f] & 29 ± 11%[g] | - | - |
| [18F]SFB-RGD | Multi-step | SN | RT | Borate buffer/DMSO | 7.4 | 130[d] | 13 | 13 ± 3%[g] | 14.06 ± 4.07[d]/-[h] | (14) |
| [18F]FBOM-GSH | Multi-step | SN | RT | PBS | 7.4 | > 60[d] | 30 | 22-30%[f] | -/- | (17) |
| (±)-[18F]AlFRESCA-HSA | One-step | Chelate reaction | RT | Ammonium acetate buffer | 4 | < 35[c] | 5.64 | 35-53%[f] | 79.92-85.10/1.258 [c] | (10) |
| [18F]SiFA-peptide | One-step | IEX[b] | RT | H2O/CH3CN (10/90) | 4 | 25[c] | 0.07 | 55-65%[f] | 2.96-5.18/0.178-0.248 [c] | (22) |
| [18F]AMBF3-TrisRGD | One-step | IEX | 80 | Pyridazine-HCl buffer | 2.5 | 12[e] | 0.05-0.08 | 23 ± 5%[g] | 111-148/29.6-37[d] | (23) |
| [18F]DBPOF-c(RGDyK) | One-step | IEX | RT | H2O/DMSO (95/5) | 7 | 25[c] | 1-3 | 15-25%[f] | 2.22-4.81/0.821-1.64[d] | (24) |
[a] Nucleophilic substitution. [b] Isotope exchange. [c] The probe was manually synthesized. [d] The probe was synthesized by automated radiosynthesis module. [e] The probe was synthesized manually, 12 min was reported for the 18F-labeling step alone. [f] RCCs were detected by radio-HPLC. [g] RCYs were isolated yields. [h] Not found.
Due to the low heat of formation and high bond energy of P-F bonds, varied P(V) compounds exhibit a capacity for rapid mild 18F-labeling via 18/19F isotope exchange in both aprotic and protic solvents [24]. To challenge the low Am due to isotopic dilution, separable 18F-labeled fluorothiophosphates (FTPs) are synthesized via spontaneous [18F]F- nucleophilic substitution and thiirane elimination on oxydithiaphospholane 2-sulfide precursors in this study. Preliminary density functional theory (DFT) calculation predicts a thermodynamically favorable fluorination pathway overcoming low activation but high hydrolysis energy barriers and at room temperature (RT), thereby forming the basis for rapid site-specific substitution by hydrated F-. The inclusion of a -SH group is supposed to improve stability through coulombic repulsion and attribute to the moderate lipophilicity that can prevent the non-specific binding of the 18F-labeled biomolecules. Comprehensive screening for condition/substrate scopes provides further insights regarding the key parameters affecting the 18F-labeling efficiency.
With rapidly in situ generated FTP synthons in high Am, a simplified one-pot 18F-labeling procedure for unmodified biomolecules is ready to be adopted. The oxydithiaphospholane 2-sulfide precursors can be efficiently 18F-labeled during eluting with aqueous [18F]F- solution before mild conjugation with biomolecular substrates, skipping sophisticated automation. Typical peptide and protein biomolecules, such as c(RGDyK), human serum albumin (HSA) and nanobody 5F7, which are medically significant, are proof-of-concept lead compounds that exhibit solvent, temperature, or pH sensitivity. Selected 18F-labeled small molecular FTPs are also synthesized as phosphate tracers that exhibit a high level of similarity to the original phosphates, with regard to both stereo structures and biochemical interactions.
Results
Computational study
DFT calculations were performed at the B3LYP/6-311+G* level in water. A three-step addition-elimination pathway was identified as a plausible reaction pathway [26]. The rate-determining step of nucleophilic attacking by hydrated F- was predicted to overcome the free-energy barriers of 16.5 and 17.4 kcal mol-1 at 298.15 K for substrates 1b and 2b (Figure 1B, Figure S1).
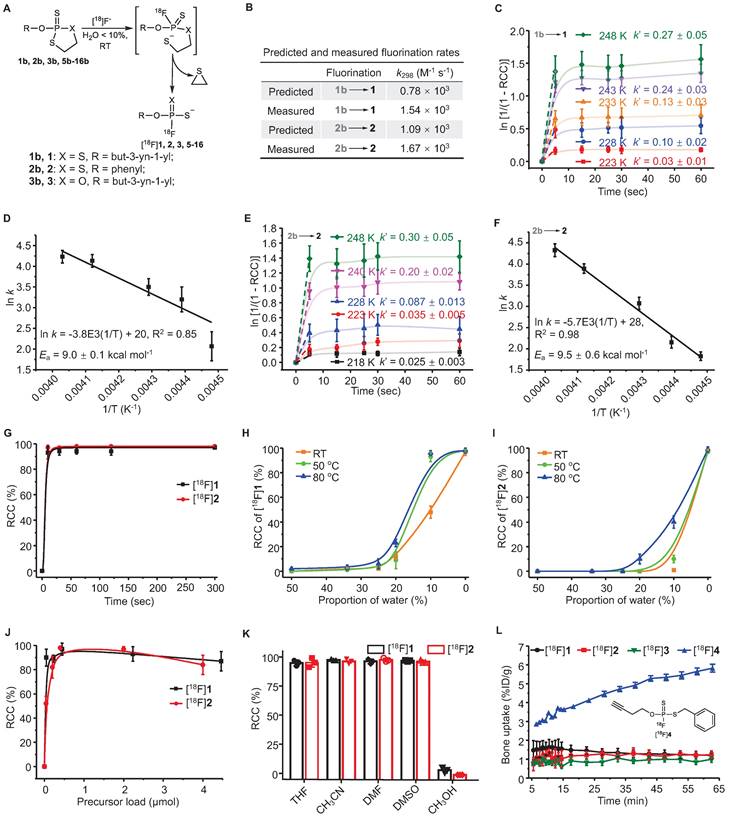
Analysis of rapid kinetics and condition scopes of the 18F-labeling of FTPs. (A) Reaction scheme showing the fluorination of FTPs. (B) Reaction rate constant (k) values that were predicted by density functional calculations at B3LYP/6-311+G* level of theory (at 298 K) and k values that were measured at 298 K. The pseudo-first order initial rate constant (k´) of [18F]1 (C) and [18F]2 (E) were determinated from the kinetic curves reflecting the change in ln [1/(1 - RCC)] in DMF at different temperatures. The Ea of [18F]1 (D) and [18F]2 (F) were determinated from Arrhenius plots of ln k against 1/T. (G) RCCs of [18F]1 and [18F]2 at different time periods from 10 to 300 sec in acetonitrile at RT. RCCs of [18F]1 (H) and [18F]2 (I) at different temperatures and water contents (The solvents are mixtures of acetonitrile and water, v/v. “0%” means no additional water is added.) post 30 sec. (J) RCCs of [18F]1 and [18F]2 with different precursor loads in acetonitrile post 30 sec at RT. (K) RCCs of [18F]1 and [18F]2 in different solvents post 30 sec at RT. (L) The time-bone uptake curves of [18F]1, [18F]2, [18F]3, and [18F]4 during 0-60 min post-injection. Bone uptake was measured with microPET in healthy mice (n = 3).
Chemistry
To synthesize the aliphatic (1, 5-9) and aromatic (2, 10-14) FTPs with either electron-donating (10-12) or electron-withdrawing (13, 14) substituents, 1,3,2-dithiaphospholanes (1a, 2a, 5a-14a) were first obtained through the phosphitylation of N,N-diethyl-1,3,2-dithiaphospholan-2-amine (19), in the presence of an alcohol/phenol and S-ethylthiotetrazole (Figure 1A, Scheme S1). Dithiaphospholanes were oxidized with elemental sulfur to obtain oxydithiaphospholane 2-sulfide substrates (1b, 2b, 5b-13b) at yields exceeding 50%. When these substrates were treated with an excess of tetrabutylammonium fluoride (TBAF) in tetrahydrofuran (THF), 1, 2, 5-13 were obtained at yields exceeding 95% in 2 min. In addition, 1b and 2b were entirely consumed upon treatment with an equivalent amount of TBAF in THF in less than 2 min, as shown by 31P NMR spectroscopy results (Figure S2). Same routes were used to synthesize 2-(but-3-yn-1-yloxy)-1,3,2-oxathiaphospholane 2-sulfide (3b) and O-(but-3-yn-1-yl) phosphorfluoridothioate (3, a monothio-derivative). The oxidation of 14a with elemental sulfur failed to obtain 14b. FTPs with typical active groups, N-hydroxysuccinimide ester (NHS) for 15, cyclooctene for 16, were fluorinated from 15b (Scheme S3) and 16b (Scheme S4) at yields exceeding 95%. 17b and 18b were treated with TBAF, and then deprotected to obtain 17-18, two bioactive phosphate analogs (Scheme S5, S6).
Measurement of fluorination kinetics and energetics
Two simple substrates, O-(But-3-yn-1-yl) [18F]phosphorofluoridodithioate ([18F]1) and O-phenyl [18F]phosphorofluoridodithioate ([18F]2), were used as model compounds to evaluate fluorination kinetics and activation energies (Ea). The pseudo-first order model is applied since the trace amount [18F]F- is negligible compared to the precursors. The pseudo-first order initial rate constant (k´) under different temperatures were calculated from the exponential fit equation (Figure 1C, 1E) and then divided by the concentration of 1b or 2b to determine the second-order rate constants (k). For the conversion of 1b to 1, k223 = 8.53 L mol-1 s-1, k228 = 25.66 L mol-1 s-1, k233 = 34.93 L mol-1 s-1, k243 = 61.20 L mol-1 s-1, k248 = 69.38 L mol-1 s-1. For the conversion of 2b to 2, k218 = 5.91 L mol-1 s-1, k223 = 8.60 L mol-1 s-1, k228 = 19.72 L mol-1 s-1, k240 = 47.85 L mol-1 s-1, and k248 = 66.20 L mol-1 s-1. The values of Ea were measured to be 9.0 ± 0.1 kcal mol-1 for the conversion of 1b to 1, and 9.5 ± 0.6 kcal mol-1 for the conversion of 2b to 2 from Arrhenius plots (ln k versus T-1) (Figure 1D-F).
Radiochemistry
18F-Labeling conditions were optimized in aprotic solvents containing graded proportions of water for different reaction durations (10-300 sec) under specific temperatures (RT-80 ℃). Non-decay-corrected radiochemical conversions (RCCs) were detected using both radio-TLC and radio-HPLC (F- adsorption might occur to radio-HPLC column) at continuous time points (n = 3). The activity adsorption by the vial/glass was measured to be ∼10% of the total initial activity. RCC values were 98 ± 5% for [18F]1 and 99 ± 4% for [18F]2 after incubation for 30 sec at RT in anhydrous acetonitrile, with > 90% of RCCs being achieved in just 10 sec (Figure 1G). Although the RCC values decreased with the increasing of solvent water contents, satisfactory RCC values of 48 ± 5% for [18F]1 and 10 ± 3% for [18F]2 could be achieved in a mixture of acetonitrile and water (v/v = 9/1) at RT. RCC values of 98 ± 2% for [18F]1 and 40 ± 5% for [18F]2 was achieved using a mixture of acetonitrile and water (v/v = 9/1) at 80 ℃ (Figure 1H-I). Substrate 2b showed higher sensitivity to water than 1b, in consistence with the calculated and experimental fluorination kinetics and energetics. After assessing precursor loads of 0.04-4.5 μmol/100 μL, the optimal precursor load was determined to be 0.20-2.00 μmol/100 μL (Figure 1J). This 18F-labeling method exhibited high efficiency in aprotic solvents (Figure 1K).
Stabilities of 18F-labeled FTPs and precursors
Each oxydithiaphospholane 2-sulfide substrate and representative FTP was incubated for 2 h in a mixture of acetonitrile and water (v/v = 1/9) with pH values of 1 to 13. The HPLC analysis results showed that the substrates could tolerate acids and weak bases but were unstable in strong alkaline solutions (Figure S33, S34). High stabilities of the FTP motif were observed in both acidic and alkaline solutions (pH values of 1 to 13, Figure S35, S36), which is critical for deprotection reaction in some occasion. The extent of defluorination (bone uptake, resistant to enzymatic hydrolysis in vivo) of [18F]1 and [18F]2 was insignificant in microPET imaging evaluation (n = 3) (Figure 1L, S48). While [18F]1 was detected only in a small percentage of the parent compound in the urine, 5 min after administering an i.v. injection, [18F]2 existed in the urine mainly in the form of the parent compound, as shown in Figure S45, S46. All procedures and animal use and care procedures have been approved by the Animal Care and Use Committee of Xiamen University.
A subsequent study showed that equally high stabilities were observed both in vitro and in vivo, if at least one O atom of phosphate was substituted by S atom, such as O-(but-3-yn-1-yl) [18F]phosphorofluoridothioate ([18F]3) and [18F]1. However, [18F]3 was cleared rapidly from the kidney probably due to its higher hydrophilicity, which might lead to insufficient internalization (Figure S47, S48). Defluorination occurred slowly in S-benzyl O-(but-3-yn-1-yl) [18F]phosphorofluoridothioate ([18F]4) in vitro and in vivo (bone uptake reached ~6 %ID g-1 at 60 min), where the -SH moiety was substituted intentionally with an alkylthio group (Figure 1L, S48). This suggests that the coulombic force contributes to the high stability of FTPs. The biodistribution study confirmed the high in vivo stabilities of 18F-labeled FTPs, where [18F]1, [18F]2, [18F]3, and [18F]15 ([18F]FTP-NHS, a typical N-hydroxysuccinimide ester synthon) all exhibited only background bone uptakes (0.9-1.1 %ID g-1) at 2 h post injection (Figure S49).
Structure scope of 18F-labeled FTPs
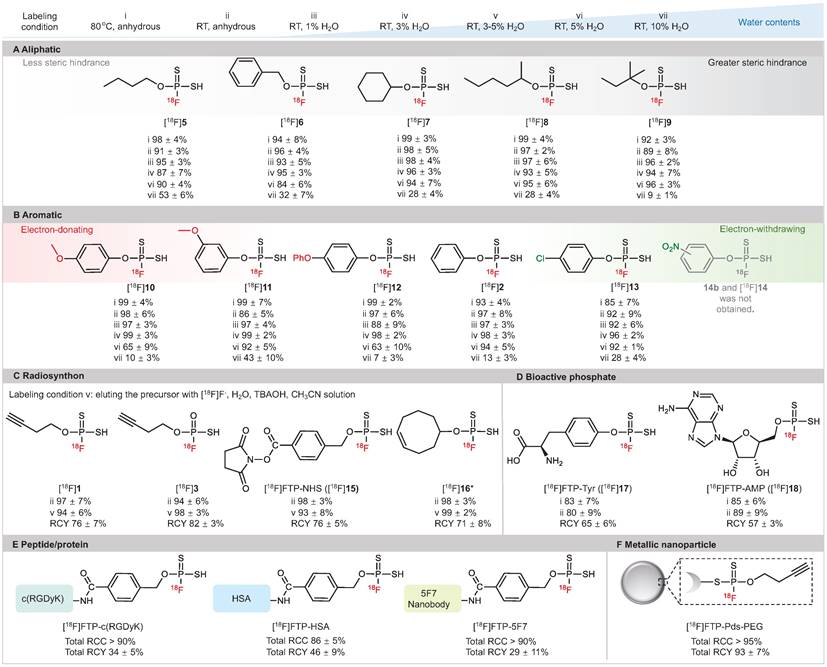
As illustrated in Figure 2, high RCC values of 88-98% were observed in anhydrous acetronitrile with a wide substrate scope of 18F-labeled FTPs, including alkanes, alkynes, heterocycles, halides, amino acid derivatives and nucleotide derivatives (condition i, ii). Notably, despite the absence of a phase transfer reagent, RCCs of 87-99% were observed with the use of wet [18F]F- in solvents with 1-3% water (condition iii, iv). The RCCs decreased gradually with an increase in the water content; when the water content was 5% (condition vi) and 10% (condition vii), RCCs were 63-96% and 7-53%, respectively. 18F-Labeled FTPs with functional groups, such as [18F]1, [18F]3, [18F]15 and [18F]16, could be coupled to biomolecules and acted as ready-to-use radiosynthons. High RCC values > 95% and high radiochemical yield (RCY, isolated yields, non-decay-corrected, n = 3) values > 75% were observed for these FTP synthons that were in situ generated with wet [18F]F- at RT (condition v, in solvents with 3-5% water). The Am of [18F]15 ([18F]FTP-NHS) reached 128.2 GBq μmol-1 by manual labeling with 3.7-5.6 GBq initail activity. Biologically active phosphate analogs, such as the 18F-labeled L-Tyr phosphate (p-Tyr) mimic ([18F]FTP-Tyr, [18F]17) and the 18F-labeled adenosine monophosphate mimic ([18F]FTP-AMP, [18F]18), were labeled directly with RCYs > 55%.
Substrate structure scope of 18F-labeled FTPs. Labeling condition i: 0.1 mg precursor, [18F]KF/K222 (1-5 mCi), 100 μL anhydrous acetonitrile, 80 °C, 30 sec. Labeling condition ii: 0.1 mg precursor, [18F]KF/K222 (1-5 mCi), 100 μL anhydrous acetonitrile, RT, 30 sec. Labeling condition iii: 0.1 mg precursor, [18F]F- in cyclotron target water (1 μL, 0.3-0.5 mCi), 100 μL acetonitrile, RT, 30 sec. Labeling condition iv: 0.1 mg precursor, [18F]F- in cyclotron target water (3 μL, 1-3 mCi), 100 μL acetonitrile, RT, 30 sec. Labeling condition v: 0.1 mg precursor, [18F]F- in cyclotron target water (3 μL, 1-3 mCi), tetrabutylammonium hydroxide (0.52 mg), 100 μL acetonitrile, RT, 30 sec. Labeling condition vi: 0.1 mg precursor, [18F]F- in cyclotron target water (5 μL, 1-5 mCi), 100 μL acetonitrile, RT, 30 sec. Labeling condition vii: 0.1 mg precursor, [18F]F- in cyclotron target water (10 μL, 1-5 mCi) and acetonitrile 90 μL, RT, 30 sec. Each 18F-labeling reaction was performed thrice with all reported RCCs determined by radio-HPLC or radio-TLC. RCCs mean ± standard deviation values. (A) Aliphatic substrates with different levels of steric hindrance. (B) Aromatic substrates with different electron density distributions. (C) 18F-Labeled FTPs as radiosynthons. (D) Small molecular FTPs as phosphate analogs. (E) 18F-Labeled peptide/protein biomolecules generated using FTP radiosynthons. (F) 18F-Labeled metallic nanoparticle generated using an FTP radiosynthon. * Cis-cyclooctene was used as a substitute.
[18F]FTP-c(RGDyK)
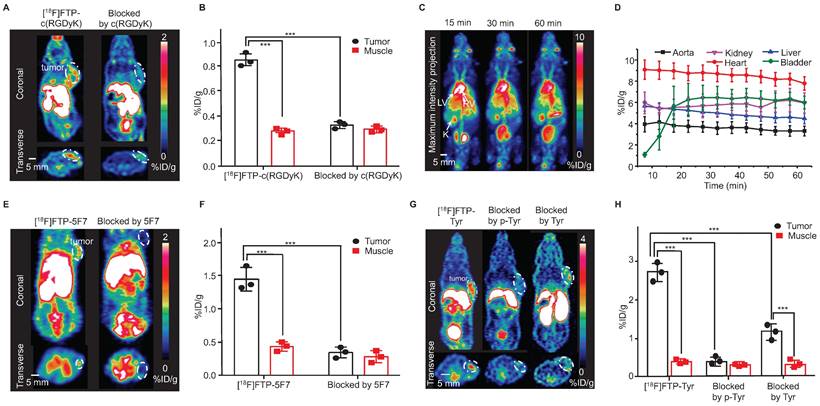
The Arg-Gly-Asp (RGD) sequence exhibits high affinity and selectivity for alpha(v)beta3 integrin, which is a significant peptide-based ligand [27]. Under mild coupling conditions, i.e., 10 min, 37 ℃, borate buffer (pH = 8.0), [18F]FTP-NHS was efficiently conjugated with c(RGDyK) to obtain [18F]FTP-c(RGDyK). The total RCC values were ~90% determined by radio-HPLC, and the average RCY value was 34 ± 5% and Am value was 107 GBq μmol-1 after manually labeling with 5.6 GBq initial activity (Figure S25, S31). The purification of [18F]FTP-c(RGDyK) using a C18 cartridge followed by HPLC resulted in > 98% radiochemical purity (RCP) (Figure S20). An automated procedure involving a commercial multifunctional radiosynthesis module was established (Figure S24). The total auto-synthesis time was ~60 min, and the RCY was 39 ± 8%, with an Am value of 337-517 GBq μmol-1 (initial activity 37-74 GBq). The microPET/CT imaging data indicated that [18F]FTP-c(RGDyK) was accumulated specifically within 4T1 xenografts. The average tumor uptake of this monomeric RGD tracer, based on the whole tumor region of interest (ROI), was 1.30 ± 0.28 %ID g-1, with a tumor-to-muscle ratio of 2.38 at 15 min post-injection (Figure 3A-B), in comparison to a value of roughly 1.8 %ID g-1 for [18F]AMBF3 labeled trimeric RGD [19].
Selected 18F-labeled biomolecules via FTPs and their microPET imaging. (A) MicroPET images of [18F]FTP-c(RGDyK) showing specific accumulation in 4T1 xenografts at 15 min post-injection (The white dotted line indicates the tumor margin). (B) Quantitative PET imaging results. Data represent the mean ± standard deviation values (n = 3), ***P < 0.001. (C) Dynamic microPET images of healthy female mice reconstructed at 15, 30, and 60 min after administering an [18F]FTP-HSA injection. LV: left ventricle; RV: right ventricle; K: kidneys. (D) Time-activity curves of [18F]FTP-HSA in indicated organs. Uptake values are presented in terms of mean ± standard deviation values (n = 3). (E) MicroPET images of [18F]FTP-5F7 showing specific accumulation in MDA-MB-453 xenografts at 60 min post-injection (The white dotted line indicates the tumor margin). (F) Quantitative PET imaging results. Data represent the mean ± standard deviation values (n = 3), ***P < 0.001. (G) MicroPET images showing the specific accumulation of [18F]FTP-Tyr in B16 xenografts at 15 min post-injection (the white dotted line indicates the tumor margin). (H) Quantitative PET imaging results. Data represent the mean ± standard deviation values (n = 3), ***P < 0.001.
[18F]FTP-HSA
HSA, a heat-sensitive protein, has routinely been radiolabeled and used to evaluate plasma distribution volumes in specific organs, quantify cardiac function-related parameters, and assess vascular “leakage” in pathological tissues [28]. [18F]FTP-HSA was obtained via [18F]FTP-NHS after 10 min conjugation in borate buffer (pH = 8.0) at 40 ℃, with an overall RCC of 83 ± 5% (n = 3). The average RCY value was 46 ± 9% (RCP > 99%) after purification by a PD-10 column (GE Healthcare Bio-Science AB) (Figure S21). As shown by the dynamic microPET/CT images in Figure 3C, the central vasculature, including the cardiac ventricular chambers, were clearly visualized. There was no apparent radioactivity in the skeleton, which suggested the absence of significant amounts of free [18F]F- and other metabolites (Figure 3D).
[18F]FTP-5F7
Nanobodies (VHH, 12-15 kDa), the antigen-binding fragments of heavy-chain-only antibodies derived from Camelidae, have biological half-lives of 1-2 h that are comparative with 18F [29, 30]. Herein, we explored the feasibility of utilizing the 18F-labeled 5F7 anti-HER2 nanobody as a probe for evaluating the HER2 expression. [18F]FTP-5F7 was obtained via [18F]FTP-NHS after 10 min conjugation in borate buffer (pH = 8.0) at RT. Overall RCCs values were determined via radio-HPLC to be ~90%, and the average RCY value was 29 ± 11% (RCP > 95%) after purification by a size exclusion chromatography column (Figure S22). MicroPET/CT images of mice with HER2-overexpressing MDA-MB-453 xenografts demonstrated the rapid tumor accumulation (1.40 ± 0.22%) and clearance of [18F]FTP-5F7 from the background. Pre-treatment with excessive amounts of 5F7 substantially reduced the tumor uptake in the background (Figure 3E-F).
[18F]FTP-Pds-PEG
The use of radiolabeled nanoparticles for molecular imaging has attracted broad attention due to their large functional surface area and easy-to-control surface chemistry, which allows them to be tailored for the purpose of personalized cancer management [31]. The sulfhydryl groups on FTPs can form stable coordinate covalent bonds, which can be used to attach various metal nanoparticles efficiently. PEGylated Pd nanosheets (Pd-PEGs) with an average size of 40 nm were conjugated with [18F]1 upon stirring the reaction mixture for 15 min at RT. The overall RCYs of [18F]FTP-Pds-PEG were > 90% after ultrafiltration (same as measured by radio-TLC, RCP > 99%, Figure S23). 2 h after administering an i.v. injection, a significant tumor uptake of 4.87 ± 0.31 %ID g-1 based on the whole tumor ROI was illustrated by microPET/CT imaging, as well as a high tumor-to-muscle ratio of 3.4 in subcutaneous 4T1 tumors (Figure S56).
[18F]FTP-Tyr
Evidence has shown that greater uptake rate, solubility, and oxidative stability were observed during the uptake of L-DOPA phosphate and p-Tyr by melanomas than those observed with L-DOPA and L-Tyr [32, 33]. Thus, they were recruited as melanogenesis markers for the early diagnosis of melanomas. In this study, [18F]FTP-Tyr was synthesized as a non-hydrolytic (overcoming the in vivo enzymatic-instabilities of phosphates) p-Tyr analog with an RCC of 80 ± 9% and Am of > 3.91 GBq μmol-1 (manual labeling; initial activity 1.48-1.85 GBq; FTP-Tyr exhibited weak UV absorption to determine the Am; Figure S29). The results of the in vitro cell uptake study revealed that the significant specific uptake of [18F]FTP-Tyr by a B16 tumor within 120 min, with maximum uptake occurring at 30 min. This uptake could be either entirely inhibited by p-Tyr or partially inhibited by L-Tyr (Figure S53). More evidence for the similarity between FTP-Tyr and p-Tyr was provided in the supplementary material (Figure S50-S52, S54, S55). MicroPET/CT imaging of [18F]FTP-Tyr was performed in B16 xenograft mice, and specific accumulation in tumors and a tumor-to-muscle ratio of 6.4 were observed at 30 min after administering an i.v. injection. This specific accumulation was blocked by p-Tyr and only partially blocked by Tyr as well, indicating the specific uptake of [18F]FTP-Tyr by melanomas (Figure 3G-H).
Discussion
The easily oxidized trivalent phosphorus intermediates (19, 20, 1a-3a, 5a-18a, monitored by 31P NMR) were not isolated and directly treated with sulfur to obtain the separable pentavalent precursors liable to oxidation (1b-3b, 5b-13b, 15b-18b) with total yields ranged from 10-70%. Functional groups exhibiting higher steric repulsion and stronger electron withdrawal are unfavorable for precursor synthesis, e.g., the unformed 14b and 14. Although the predicted three-step pathway mechanism requires further experimental evidence, the extremely rapid fluorination kinetics were in accordance with the experimental rate constants and pseudo-first order analysis. Ultimately, the high 18F-fluorination efficiency at RT was attributed to the low free-energy barriers and trace amount [18F]F-. Favorable energy barriers also resulted in a higher selectivity for F- over other nucleophiles, such as, OH-, which is many-fold more in quantity in wet solvents. Thus, the routine azeotropic drying processing of [18F]F- can be simplified to evaporating processing to obtain reaction solutions that contain less than 5% water in automatic production.
Non-structure-biased 18F-labeled FTPs were generated at high RCCs within seconds in solvents with a water content of < 5%, using a very low molar quantity/concentration of storage-stable precursors (0.1 mg/0.22-0.45 μmol per 100 μL). Varied 18F-labeled FTP synthons with respective functional/linker groups, e.g., unsaturated hydrocarbon or activated ester group, are accessible. Insignificant levels of defluorination but multiple degradation products were observed in vivo for [18F]1 and [18F]2. The in vivo metabolic stabilities of 18F-labeled FTPs are attributed not only to the coulombic repulsion between the FTP group and anions, which protects the P-F bond from hydrolysis, but also the substitution of P=O with P=S, which enhances its resistance to enzymatic hydrolysis (Figure 1L, S48, S51).
A simplified 18F-labeling strategy has been consequently developed via the rapid in situ generation of instant FTP synthons that could couple with delicate biomolecules. This strategy does not require the pre-modification of biomolecules, preparation of dry [18F]F-, exposure of biomolecules to harsh conditions, and purification of radioactive intermediates. The negligible time-related costs associated with FTP synthon formation and application of the same mild conditions used for the conjugation step during multi-step labeling results in the high 18F-labeling efficiency. Typical medically significant biomolecules, such as c(RGDyK), HSA, and nanobody 5F7, which exhibit solvent/temperature/pH sensitivity, have been successfully labeled via FTP synthons either manually or automatically.
In contrast to the existing direct 18F-labeling methods that are mostly based on 18F/19F-isotope-exchange, due to the significant polarity difference between the more hydrophilic FTPs and the less hydrophilic precursors, they can be separated feasibly using chromatography, thus higher Ams values can be achieved for receptor imaging. Compared to the satisfactory Am obtained via 18F/19F-exchange with high initial activity, 18F-labeled FTPs have been generated via nucleophilic substitution to give > 300 GBq mol-1 Am with 37-74 GBq initial activity18-20. The ratio of Am to initial activity (Am achieved with certain amount of initial activity of 18F) under a certain molar amount of the precursor, instead of Am, was used an important parameter to reflect the 18F-fluorination capability of the labeling methods with certain chemical motif or precursor (Table 1). Enhanced hydrophilicity may also lead to an improved pharmacokinetic profile of the radiolabeled peptides by shifting hepatobiliary to renal clearance.
Conclusion
Mild and efficient nucleophilic fluorination with [18F]F- and separable precursors that allows site-specific 18F-labeling in high Am are long pursued for highly sensitive PET imaging. Water-tolerant [18F]F- nucleophilic fluorination, which was previously accessible mainly with isotope-exchange reactions, would reduce the failure risk of costly 18F-labeling in contrast to the 18F-labeling methods demanding strictly anhydrous condition. Taking this opportunity, this study describes a rapid, [18F]F- nucleophilic radiolabeling method via in situ generated FTPs in non-anhydrous solvent, and the introduction of high-Am FTPs as radiosynthons or tracers to facilitate the development of PET tracers from unmodified biomolecules and phosphate-based biomarkers. This sufficiently simple radiofluorination method in partially aqueous media using disposable and cheap labware may enable us to overcome the drawbacks associated with both indirect and direct 18F-labeling, and facilitate the development of a broadly applicable kit-like protocols for 18F-labeling of biomolecules.
Experimental Section
Materials
All the reagents we used in the synthesis and biology experiment were purchased from Energy Chemical Co., Ltd. (China) or J&K Co., Ltd. (China) and were used without further purification. Column chromatography purification was performed on silica gel (54-74 μm, Qingdao Haiyang Chemical Co., Ltd., China). Anhydrous dichloromethane, anhydrous tetrahydrofuran (THF), anhydrous dimethyl sulfoxide (DMSO), anhydrous acetonitrile and anhydrous dimethylformamide (DMF) were purchased from Energy Chemical Co., Ltd. (China) and used without further drying.
Fluorination kinetics
In order to determine the fluorination kinetics and Ea for 18F-fluorination process, we decided to carry out a series of experiments at different temperatures. We found experimentally that the optimal temperature range to monitor the labeling reaction rate of [18F]1 and [18F]2 is between -55 °C and -25 °C, since the reaction is too fast at higher temperatures and too slow at lower temperatures to measure. Concentrations of precursors and in DMF solutions were kept constant at 3.82 × 10-3 M and 4.01 × 10-3 M, respectively. The RCCs under each individual temperature was monitored by radio-TLC. Dynamic RCCs were able to be monitored by TLC because we took 10 μL of the reaction mixture at the indicated time points, quenched it in 1.0 mL of water before TLC developing. Labeling efficiency graphs could be converted to line graphs of reaction time versus ln [1/(1-RCC)], whose slopes represent specific rate constants at specific temperatures. Pseudo-first order initial rate constants at different temperatures were calculated from the exponential fit equation, and then k' was divided by the concentration of 1b or 2b to determine the actual second-order rate constants. These rate constants were used to create an Arrhenius plot (ln k versus T-1) to calculate the Ea, which was found to be 8.41 kcal mol-1 for 18F-labeling reaction of [18F]1 and 8.98 kcal mol-1 for 18F-labeling reaction of [18F]2.
General manual 18F-labeling procedures
Labeling procedure I: [18F]F- was azeotropically dried as previously described [14]. Briefly, [18F]F- was produced via the 18O(p, n)18F reaction and delivered as [18F]F- in [18O]H2O. [18F]F- (0.185-1.85 GBq) was separated from 18O-enriched-water using QMA and subsequently released with by a solution of 8.0 mg kryptofix 222 (K222) and 1.0 mg K2CO3 in 1.0 mL of acetonitrile/H2O (4/1, v/v). The solution was azeotropically dried for three times (300 μL acetonitrile × 3) at 100 ℃ in a clean glass vial. 0.2 μmol precursor was dissolved in 100 μL acetonitrile (anhydrous or non-anhydrous) and added into the glass vial containing [18F]F-. The mixture was incubated at RT for 30 sec before quenching by 500 μL water. RCCs were analyzed by radio-HPLC or radio-TLC (n = 3). Labeling procedure II (with wet [18F]F-): 0.2 μmol precursor was dissolved in 50 μL acetonitrile. Aqueous [18F]F- solution (diluted from target water, no K222 and K2CO3 added, 0.185-1.85 GBq) in appropriate volume was added into the solution. Then total reaction volume was adjusted to 100 μL with pure water and acetonitrile. The reaction vial was gently shaken at RT for 30 sec before quenching by adding 500 μL water. RCCs were analyzed by radio-HPLC or radio-TLC (n = 3).
Radiosynthesis of [18F]FTP-Tyr
[18F]17c was synthesized from 17b following labeling procedure I. [18F]17c was then dissolved in 100 μL MeOH followed by adding 100 μL NaOH (2.0 M) for 15 min. The reaction mixture was acidified by 1.0 M HCl and was dried under a stream of nitrogen. The resulting residue was dissolved in acetonitrile (100 μL). 5.0 M HCl or TFA (100 μL) was added and shaken at RT for 10 min. The solution was then neutralized to pH 7 by adding NaOH (1.0 M) and diluted with PBS and acetonitrile before being analyzed and purified on HPLC. The collected product was then put on the rotary evaporator to remove excess methanol from the solution. [18F]FTP-Tyr was dissolved in saline for PET imaging studies.
Radiosynthesis of [18F]FTP-AMP
[18F]18c was synthesized from 18b following labeling procedure I. Then 100 μL TFA was added to [18F]18c and stirred at RT for 5 min to deprotection. The solution was then neutralized to pH 7 by adding NaOH (1.0 M) and diluted with PBS and acetonitrile before being analyzed and purified on radio-HPLC. The collected product was then put on the rotary evaporator to remove excess acetonitrile from the solution. [18F]FTP-AMP was dissolved in saline for PET imaging studies.
Radiosynthesis of [18F]FTP-NHS
0.1 mg precursor 15b (0.2 μmol) was dissolved in dichloromethane (40 μL) and loaded onto a cotton ball (about 0.03 cm3). Then the dichloromethane was allowed to volatilize in a fume hood, and the small cotton carrying precursor 15b was fitted into a pipette tip (for manual labeling, any tube-like part for automatic modules. [18F]15 was synthesized from 18b following labeling procedure II. No-carrier-added [18F]F- was produced via the 18O(p, n)18F reaction and delivered as [18F]F- in [18O]H2O. [18F]F- (3 μL, 0.30-0.37 GBq) was added to 100 μL acetonitrile with tetrabutylammonium hydroxide (TBAOH, 0.52 mg/100 μL), which was used as the eluent later. The pipette tip carrying precursor 15b was eluted by this mixed solution into a polypropylene tube and subsequently dried under a stream of nitrogen to afford [18F]15 as a radiosynthon.
Radiosynthesis of [18F]FTP-c(RGDyK)
1.0 mg (1.6 μmol) c(RGDyK) was dissolved in a mixture of 10 μL DMSO and 100 μL of borate buffer (pH = 8.0). This c(RGDyK) solution was then added into the vial containing [18F]15. After 10 min reaction at 37 ℃, the reaction mixture was diluted with 10.0 mL of H2O and loaded onto a light C18 cartridge. The cartridge was flushed twice with 10.0 mL of pure water to remove the unreacted [18F]F- and 1.0 mL of ethanol to get crude product. The crude product was further purified by radio-HPLC. Purify condition: Waters XBridgeC-18 column (5 µm, 10 mm × 250 mm, USA). Phase A: PBS (0.02 mol L-1 pH = 7.4); phase B: HPLC grade acetonitrile; isocratic elution at 90% phase A and 10% phase B. Flow rate: 3.0 mL min-1. The HPLC fraction was dried under a stream of nitrogen at RT. [18F]FTP-c(RGDyK) was then dissolved in saline for injection.
Radiosynthesis of [18F]FTP-HSA
1.0 mg of HSA in 0.1 M borate buffer (pH = 8.0, 2 mg mL-1, 100 μL) was added to a glass vial containing dried [18F]15 and the mixture was incubated for 10 min at 40 ℃. [18F]FTP-HSA was purified by a PD-10 column (GE Healthcare Bio-Science AB) using PBS (pH = 7.4) as the eluent. A size exclusion chromatography (SEC) column was applied to analyze its RCP.
Radiosynthesis of [18F]FTP-5F7
0.1 mg of 5F7 was dissolved in 100 μL of borate buffer (pH = 8.0) and added to [18F]15. The reaction mixture was incubated for 10 min at RT. The product was purified and then analyzed by radio-HPLC equipped with an Xtimate SEC-300 column (Welch, China).
Radiosynthesis of [18F]FTP-Pds-PEG
18F-Labeled FTPs were prepared following the Labeling procedure I. Pd nanosheets with an average size of 40 nm (200 μg), as an example of metal nanoparticle, dissolved in thiol-polyethylene glycol (mPEG-SH) solution (2 mg in 100 μL water) to obtain PEGylated Pd nanosheets. Then, [18F]1 (taking [18F]1 as an example) was added to Pd-PEG and stirred for 15 min at RT to get [18F]FTP-Pds-PEG. RCCs was determined by radio-TLC analysis. [18F]1 labeled Pd nanosheets with a high efficiency (about 93%). Then the mixture was subject to ultrafiltration (13000 rpm for 10 min, repeated 3 times) to remove unlabeled [18F]1 and [18F]F-. RCPs was determined by radio-TLC analysis.
Abbreviations
Activation energy (Ea); Arg-Gly-Asp (RGD); density functional theory (DFT); dimethylformamide (DMF); dimethyl sulfoxide (DMSO); fluorothiophosphate (FTP); human serum albumin (HSA); N-hydroxysuccinimide ester (NHS); kryptofix 222 (K222); molar activity (Am); PEGylated Pd nanosheets (Pd-PEGs); positron emission tomography (PET); pseudo-first order initial rate constant (k´); radiochemical conversion (RCC); radiochemical purity (RCP); room temperature (RT); radiochemical yield (RCY); second-order rate constant (k); size exclusion chromatography (SEC); tetrabutylammonium fluoride (TBAF); tetrabutylammonium hydroxide (TBAOH); tetrahydrofuran (THF).
Supplementary Material
Supplementary materials and methods, figures, tables.
Acknowledgements
Data and materials availability
All data are available in the main text or the supplementary materials.
Funding
National Natural Science Foundation of China (81971674).
Author contributions
Dr. Zijing Li conceived the study and coordinated all the research. Hongzhang Yang preformed the experiments. Zijing Li and Hongzhang Yang analyzed the results and prepared the manuscript. Computational chemistry was carried out by Dr. Lei Zhang. Molecular docking was carried out by Fengming He. Huanhuan Liu, Yunming Zhang, Zhaobiao Mou, Xueyuan Chen and Jingchao Li all offered help in biology and microPET imaging experiments.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Phelps ME. Positron emission tomography provides molecular imaging of biological processes. Proc Natl Acad Sci USA. 2000;97:9226-9233
2. Weissleder R. Scaling down imaging: molecular mapping of cancer in mice. Nat Rev Cancer. 2002;2:11-18
3. Vaneycken I, D'huyvetter M, Hernot S, De Vos J, Xavier C, Devoogdt N. et al. Immuno-imaging using nanobodies. Curr Opin Biotechnol. 2011;22:877-881
4. Richter S, Wuest F. 18F-Labeled peptides: the future is bright. Molecules. 2014;19:20536-20556
5. Chen R, Huang S, Lin T, Ma H, Shan W, Duan F. et al. Photoacoustic molecular imaging-escorted adipose photodynamic-browning synergy for fighting obesity with virus-like complexes. Nat Nanotechnol. 2021;16:455-465
6. Preshlock S, Tredwell M, Gouverneur V. 18F-Labeling of arenes and heteroarenes for applications in positron emission tomography. Chem Rev. 2016;116:719-766
7. Kumar K. 18F-AlF-Labeled biomolecule conjugates as imaging pharmaceuticals. J Nucl Med. 2018;59:1208-1209
8. Ilhan H, Todica A, Lindner S, Boening G, Gosewisch A, Wängler C. et al. First-in-human 18F-SiFAlin-TATE PET/CT for NET imaging and theranostics. Eur J Nucl Med Mol Imaging. 2019;46:2400-2401
9. Ilhan H, Lindner S, Todica A, Cyran CC, Tiling R, Auernhammer CJ. et al. Biodistribution and first clinical results of 18F-SiFAlin-TATE PET: a novel 18F-labeled somatostatin analog for imaging of neuroendocrine tumors. Eur J Nucl Med Mol Imaging. 2020;47:870-880
10. Cleeren F, Lecina J, Ahamed M, Raes G, Devoogdt N, Caveliers V. et al. Al18F-Labeling of heat-sensitive biomolecules for positron emission tomography imaging. Theranostics. 2017;7:2924-2939
11. Jacobson O, Kiesewetter DO, Chen X. Fluorine-18 radiochemistry, labeling strategies and synthetic routes. Bioconjug Chem. 2015;26:1-18
12. Olberg DE, Hjelstuen OK. Labeling strategies of peptides with ¹⁸F for positron emission tomography. Curr Top Med Chem. 2010;10:1669-1679
13. Richter S, Wuest M, Bergman CN, Way JD, Krieger S, Rogers BE. et al. Rerouting the metabolic pathway of 18F-labeled peptides: the influence of prosthetic groups. Bioconjug Chem. 2015;26:201-212
14. Thonon D, Goblet D, Goukens E, Kaisin G, Paris J, Aerts J. et al. Fully automated preparation and conjugation of N-succinimidyl 4-[18F]fluorobenzoate ([18F]SFB) with RGD peptide using a GEFASTlabTM synthesizer. Mol Imaging Biol. 2011;13:1088-1095
15. Olberg DE, Arukwe JM, Grace D, Hjelstuen OK, Solbakken M, Kindberg GM. et al. One step radiosynthesis of 6-[18F]fluoronicotinic acid 2,3,5,6-tetrafluorophenyl ester ([18F]F-Py-TFP): a new prosthetic group for efficient labeling of biomolecules with fluorine-18. J Med Chem. 2010;53:1732-1740
16. Kiesewetter DO, Jacobson O, Lang L, Chen X. Automated radiochemical synthesis of [18F]FBEM: a thiol reactive synthon for radiofluorination of peptides and proteins. Appl Radiat Isot. 2011;69:410-414
17. Wuest F, Köhler L, Berndt M, Pietzsch J. Systematic comparison of two novel, thiol-reactive prosthetic groups for 18F-labeling of peptides and proteins with the acylation agent succinimidyl-4-[18F]fluorobenzoate ([18F]SFB). Amino Acids. 2009;36:283-295
18. Schirrmacher R, Wängler B, Bailey J, Bernard-Gauthier V, Schirrmacher E, Wängler C. Small prosthetic groups in 18F-radiochemistry: useful auxiliaries for the design of 18F-PET tracers. Semin Nucl Med. 2017;47:474-492
19. Krishnan HS, Ma L, Vasdev N, Liang SH. 18F-Labeling of sensitive biomolecules for positron emission tomography. Chemistry. 2017;23:15553-15577
20. Van der Born D, Pees A, Poot AJ, Orru RVA, Windhorst AD, Vugts DJ. Fluorine-18 labelled building blocks for PET tracer synthesis. Chem Soc Rev. 2017;46:4709-4773
21. Cole EL, Stewart MN, Littich R, Hoareau R, Scott PJ. Radiosyntheses using fluorine-18: the art and science of late stage fluorination. Curr Top Med Chem. 2014;14:875-900
22. Schirrmacher R, Bradtmöller G, Schirrmacher E, Thews O, Tillmanns J, Siessmeier T. et al. 18F-Labeling of peptides by means of an organosilicon-based fluoride acceptor. Angew Chem Int Ed. 2006;45:6047-6050
23. Liu Z, Pourghiasian M, Radtke MA, Lau J, Pan J, Dias GM. et al. An organotrifluoroborate for broadly applicable one-step 18F-labeling. Angew Chem Int Ed. 2014;53:11876-11880
24. Hong H, Zhang L, Xie F, Zhuang R, Jiang D, Liu H. et al. Rapid one-step 18F-radiolabeling of biomolecules in aqueous media by organophosphine fluoride acceptors. Nat Conmmun. 2019;10:989
25. Sap JBI, Meyer CF, Ford J, Straathof NJW, Dürr AB, Lelos MJ. et al. [18F]Difluorocarbene for Positron Emission Tomography. Nature. 2022;606:102-108
26. Stec W, Grajkowski A, Kobylariska A, Karwowski B, KozioHciewicz M, Misiura K. et al. Diastereomers of Nucleoside 3'-O-(2-Thio-1,3,2-oxathia(selena)phospholanes): Building Blocks for Stereocontrolled Synthesis of Oligo(nucleoside phosphorothioate)s. J Am Chem Soc. 1995;117:12019-12029
27. Alipour M, Baneshi M, Hosseinkhani S, Mahmoudi R, Jabari Arabzadeh A, Akrami M. et al. Recent progress in biomedical applications of RGD-based ligand: From precise cancer theranostics to biomaterial engineering: A systematic review. J Biomed Mater Res A. 2020;108:839-850
28. Basuli F, Zhang X, Williams MR, Seidel J, Green MV, Choyke PL. et al. One-pot synthesis and bio-distribution of fluorine-18 labelled serum albumin for vascular imaging. Nucl Med Biol. 2018;62-63:63-70
29. Vaidyanathan G, McDougald D, Choi J, Koumarianou E, Weitzel D, Osada T. et al. Preclinical evaluation of 18F-labeled anti-HER2 nanobody conjugates for imaging HER2 receptor expression by immunoPET. J Nucl Med. 2016;57:967-973
30. Pruszynski M, Koumarianou E, Vaidyanathan G, Revets H, Devoogdt N, Lahoutte T. et al. Targeting breast carcinoma with radioiodinated anti-HER2 Nanobody. Nucl Med Biol. 2013;40:52-59
31. Singh AK, Pandey A, Tewari M, Kumar R, Sharma A, Pandey HP. et al. Prospects of nano-material in breast cancer management. Pathol Oncol Res. 2013;19:155-165
32. McLane J, Osber M, Pawelek JM. Phosphorylated isomers of L-DOPAL stimulate MSH binding capacity and responsiveness to MSH in cultured melanoma cells. Biochem Bioph Res Co. 1987;145:719-725
33. Pawelek JM, Murray M. Increase in melanin formation and promotion of cytotoxicity in cultured melanoma cells caused by phosphorylated isomers of L-dopa. Cancer Res. 1986;46:493-497
Author contact
![]() Corresponding author: Zijing Li Ph.D., Email: zijing.liedu.cn
Corresponding author: Zijing Li Ph.D., Email: zijing.liedu.cn