Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results and Discussion
Conclusion
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2023; 13(2):673-684. doi:10.7150/thno.77417 This issue Cite
Research Paper
Enzymatically-mineralized double-network hydrogels with ultrahigh mechanical strength, toughness, and stiffness
Li Wang2, Wei Zhao1, Yining Zhao2, Wei Li1 ![]() , Guodong Wang1
, Guodong Wang1 ![]() , Qiang Zhang1,2
, Qiang Zhang1,2 ![]()
1. Department of Stomatology, Changzheng Hospital, Naval Medical University, Shanghai, 200003, P. R. China.
2. Shanghai Key Laboratory of Regulatory Biology, School of Life Sciences, East China Normal University, Shanghai, 200241, P.R. China.
Received 2022-7-24; Accepted 2022-12-7; Published 2023-1-1
Abstract

Background: Synthetic hydrogels are commonly mechanically weak which limits the scope of their applications.
Methods: In this study, we synthesized an organic-inorganic hybrid hydrogel with ultrahigh strength, stiffness, and toughness via enzyme-induced mineralization of calcium phosphate in a double network of bacterial cellulose nanofibers and alginate-Ca2+.
Results: Cellulose nanofibers formed the first rigid network via hydrogen binding and templated the deposition of calcium phosphate, while alginate-Ca2+ formed the second energy-dissipating network via ionic interaction. The two networks created a brick-mortar-like structure, in which the “tortuous fracture path” mechanism by breaking the interlaced calcium phosphate-coated bacterial cellulose nanofibers and the hysteresis by unzipping the ionic alginate-Ca2+ network made a great contribution to the mechanical properties of the hydrogels.
Conclusion: The optimized hydrogel exhibited ultrahigh fracture stress of 48 MPa, Young's modulus of 1329 MPa, and fracture energy of 3013 J/m2, which are barely possessed by the reported synthetic hydrogels. Finally, the hydrogel represented potential use in subchondral bone defect repair in an ex vivo model.
Keywords: hybrid hydrogel, double network, enzymatic mineralization, ultrahigh mechanical properties, subchondral bone defect repair
Introduction
Hydrogels have found wide applications in biomedical areas, such as drug delivery [1], tissue engineering [2], biosensors [3], and structural implants [4], owing to their nature of high-water content and three-dimensional network and their similarity to the extracellular matrix [5]. For practical uses, hydrogels with variable mechanical properties of strength, stiffness, toughness, and/or elongation are required for different application scenarios [6-8]. However, most synthetic hydrogels are mechanically weak [9]. They generally have low strength of ~100 kPa, stiffness of ~10 kPa, and toughness of ~10 J/m-2 [10]. In comparison, the natural load-bearing tissues like cartilage, tendons, skins, and muscles have high water contents of ~70% but are still mechanically strong and durable [11]. For instance, cartilages have a tensile strength of up to 25 MPa, Yong's modulus larger than 10 MPa, and fracture energy of around 1000 J/m-2 [12], and tendons possess a tensile strength of ~25 MPa and Yong's modulus of ~1.2 GPa [13]. Therefore, the weak mechanical properties of synthetic hydrogels severely limit their applications in biomedical areas.
In the past two decades, great efforts have been devoted to synthesizing hydrogels with improved mechanical properties. The tough hydrogels have been well made by introducing energy dissipation mechanisms that are majorly accessed via constructing a double network [14-17]. A typical case is that the hydrogels created by covalently and ionically crosslinked double networks achieve giant fracture energy of ~9000 J/cm2 [18]. However, the ultrahigh toughness is a compromised result of low strength and stiffness (156 kPa for fracture stress, and 29 kPa for Young's modulus) [18]. Historically, stiffness and toughness are considered mutually exclusive in a hydrogel, and thus the synthesis of hydrogels with both high stiffness and toughness is not easy. To stiffen hydrogels, diverse strategies, such as increasing solid contents and/or crosslink densities [19-21], forming crystallite regions [22-23], introducing hydrogen bonding [17], creating conjoined networks [24], adding hydrophobic interaction [25], and integrating macroscale fibers [26,27], are extensively exploited based on the double network structures. As a result, the stiffness of the hydrogels that possess high toughness has been well improved. In most cases, the elastic moduli of the tough hydrogels are enhanced up to 50 MPa [16,23,28]. Biomineralization is the major process in organisms to form ultra-stiff and tough tissues like bone and teeth [29], in which the organic components form a three-dimensional matrix and template the nucleation and growth of inorganics. Similarly, the organic-inorganic hybrid construction made via filling inorganic components and biomineralization has also been utilized to stiffen and strengthen hydrogels [30-32]. For instance, enzyme-induced calcium deposition in a polymer matrix is recently developed to prepare ultra-stiff and tough hydrogels [33]. The hybrid hydrogels represent ultrahigh Young's moduli up to 440 MPa and fracture energies larger than 1300 J/m2. Except for stiffness and toughness, strength is also a concern for synthetic hydrogels since certain natural tissues are ultra-strong like cartilages representing high strength of ~25 MPa [12]. However, few synthetic hydrogels can simultaneously possess ultrahigh strength, stiffness, and toughness. Even the mineralized hydrogels have ultrahigh stiffness and toughness, their tensile strength however is still relatively low (~1 MPa) [33].
In this study, we developed a new strategy to synthesize hybrid hydrogels with ultrahigh strength, stiffness, and toughness. In the method, a double network consisting of bacterial cellulose (BC) nanofibers and Ca2+-crosslinked alginate (alginate-Ca2+) was constructed, and then enzyme-induced calcium phosphate (CaP) mineralization occurred in the gel matrix. BC nanofibers acted as a stiff-reinforced component to create the first rigid network via hydrogen binding, and alginate-Ca2+ formed the second energy-dissipating network via ionic interaction. BC nanofibers also templated CaP deposition to form CaP-coated BC (BC@CaP). BC@CaP and alginate-Ca2+ intertwined in the hydrogels to form a brick-and-mortar-like structure. When hydrogel stretched, the “tortuous fracture path” mechanism by breaking interlaced BC@CaP nanostructures and the hysteresis by unzipping alginate-Ca2+networks made a great contribution to the mechanical properties, resulting in highly strong, stiff, and tough hydrogels. Finally, the hybrid hydrogel was well used for subchondral bone defect repair.
Results and Discussion
Fabrication of the organic-inorganic hybrid hydrogels
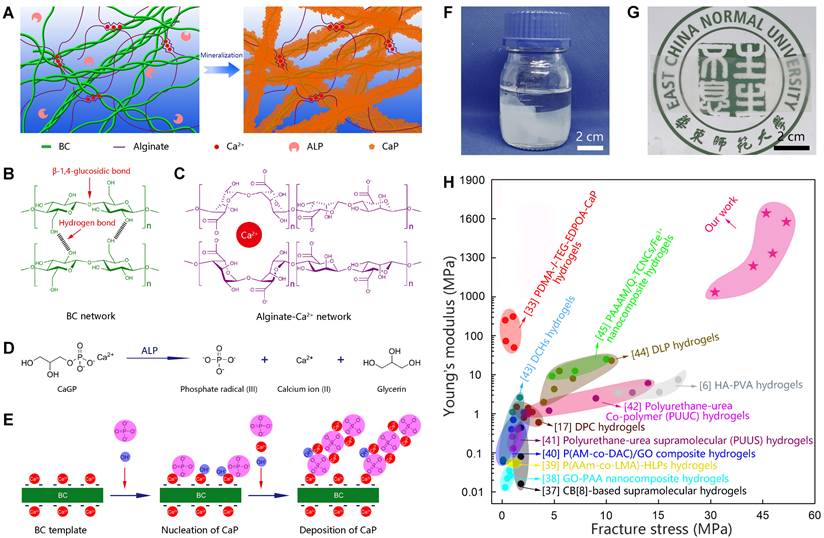
Biomineralization is the major process in organisms to form ultra-stiff and tough tissues like bone and teeth, in which the organic components form a three-dimensional matrix and serve as a template to control the nucleation and growth of inorganic components [29]. In this case, we prepared the hybrid hydrogels, that is BxA100-x-My hydrogels (B stands for BC, A does for alginate-Ca2+, and M does for mineralization; x means the weight percentage of BC in the dry film of BC and alginate, and y means the mineralization days), via enzyme-catalyzed mineralization of CaP in a double network of BC and alginate-Ca2+ (Figure 1A). The organic matrix was first prepared by mixing BC and alginate via a blade-casting method [34]. BC was used as the major component in the organic matrix due to its excellent mechanical properties of ultrahigh tensile stress and elastic modulus (Figure 1B) [35]. The alginate-Ca2+ network was introduced to enhance the toughness of hydrogels via an energy-dissipating mechanism (Figure 1C). Alkaline phosphatases (ALPs) were added to the matrix to catalyze the deposition of CaP [36]. The matrix was then immersed in a triethanolamine (TEA) buffer (0.2 M, pH=9.8) containing calcium glycerophosphate (CaGP, 5 g/L). ALPs catalyzed the decomposition of CaGP (Figure 1D). BC nanofibers had a negatively-charged surface (Figure S1), which absorbed Ca2+ and then templated the nucleation and deposition of CaP (Figure 1E). B80A20 hydrogel loaded with ALPs was immersed in TEA buffer for mineralization. However, the buffer solution became cloudy in 1-2 h (Figure S2A), because ALPs were too small (2.8 nm) to be trapped in the gel matrix [33]. To prevent enzyme leaking, ALPs were further crosslinked by different amounts of poly-glutaraldehyde (PGL) to form large-size PGL/ALP nanoparticles [33]. The enzyme activities of ALPs in PGL/ALP nanoparticles were further determined according to the calibration curve of p-nitrophenol, as ALPs catalyzed the decomposition of p-nitrophenol phosphonate to release p-nitrophenol. (Figure S3). The hydrodynamic sizes of PGL/ALP nanoparticles were increased along with the amounts of PGL, but the enzyme activities were gradually reduced (Figure S4 and S5). Finally, PGL/ALP nanoparticles at a weight ratio of ALP: PGL = 10: 1 were chosen for the following study due to that the nanoparticles had a relatively large size of 108 nm and also maintained 85% of the native enzyme activity (4.94 U/mg, Figure S4 and S5). As a result, B80A20 hydrogel loaded with PGL/ALP nanoparticles could well maintain the solution clear (Figure S2B). Even after mineralization for 6 days, the solution was still clear (Figure 1F), and a transparent mineralized B80A20-M6 hydrogel was obtained (Figure 1G). We compared the mechanical properties of our hydrogels with these of the strong hydrogels reported in the literature [6,17,33,37-45]. As shown in Figure 1H, our hydrogels' fracture stresses and Young's moduli were much larger than these of the reported hydrogels. The largest fracture stress of our hydrogels was 51.8±1.4 MPa and the largest Young's modulus was 1640.0±150.0 MPa (Figure 1H).
Enzyme catalyzed mineralization in BC/alginate-Ca2+ double-network hydrogel. (A) Schematic diagram of the mineralization process. (B and C) BC and alginate-Ca2+ formed the double-network hydrogel. (D) ALPs catalyze the transformation of CaGP into Ca2+, PO43-, and glycerin. (E) Ca2+ and PO43- deposit on the surface of BC nanofibers to form CaP nanostructures. (F) B80A20 hydrogel mineralized in 0.20 M TEA buffer containing 5 g/L of CaGP. (G) Photograph of B80A20-M6 hydrogel. (H) Ashby plot shows Young's modulus vs. fracture stress of B80A20-M6 hydrogel and other stiff and/or tough hydrogels reported in the literature.
Mechanical properties of the hybrid hydrogels
Initially, we employed BC as the exclusive component for the organic matrix. The as-obtained B100A0-M6 hydrogel represented Young's modulus of 360.8±125.7 MPa and work of fracture of 279.0±88.0 kJ/m3 (Figure 2A-B), which was highly stiff and tough but did not stand out compared with the mineralized polymer hydrogels [33]. Consequently, we incorporated BC with alginate-Ca2+ at different weight ratios to form double-network hydrogels (Figure S6), in which alginate-Ca2+ was used for dissipating energy via breaking the ionic crosslinking of guluronate blocks [18]. The proportions of alginate were tuned in a range of 0-60 wt% (dried content of BxA100-x hydrogels), and the mechanical properties of the hydrogels were measured. Among them, B80A20 hydrogels showed both relatively high Young's modulus (212.8±43.3 MPa) and work of fracture (386.1±119.3 KJ/m3, Figure S7 and S8). Furthermore, BxA100-x hydrogels containing different amounts of alginate were mineralized for 6 days. The mineralized hydrogels of B90A10-M6 and B80A20-M6 were quite transparent, while the other ones were cloudy (Figure S9), implying CaP nanocomposites formed in the two hydrogels of B90A10-M6 and B80A20-M6 were small [33]. The mechanical properties of B100A100-x-M6 hydrogels were further determined by testing the stress-strain curves. The fracture stresses were remarkably enhanced in the hydrogels containing alginate-Ca2+ (Figure 2A and S10). The tensile stresses were enhanced from 9.1±1.4 MPa in B100A0-M6 hydrogels to 40.1±5.3 MPa in B95A5-M6 ones and further increased to the largest value of 52.6±5.9 MPa in B60A40-M6 hydrogels (Figure S10). Meanwhile, the fracture elongations of the hydrogels containing alginate-Ca2+ were not compromised with the high stresses but also improved obviously (Figure 2A and S10). For instance, the strains were elongated from 4.1±0.6 in B100A0-M6 hydrogels to 6.1±0.2 % in B95A5-M6 ones (Figure S10). The Young's modulus was increased from 360.8±125.7 MPa in B100A0-M6 hydrogels to 848.3±85.8 MPa in B95A5-M6 ones, and the work of fracture did from 279.0±88.0 to 1339.2±175.3 KJ/m3 (Figure 2B). Along with the amount of alginate increased, Young's modulus of the hydrogels was increased and reached the highest value of 1671.0±141.5 MPa in B40A60-M6 hydrogels, and the work of fracture reached the highest value of 1690.9±161.9 KJ/m3 in B80A20-M6 hydrogels (Figure 2B). Additionally, the B0A100 hydrogel deformed during the mineralization process, and it became too brittle to perform a stress-strain test after mineralization for 6 days (Figure S11). The composite proportions in B100A100-x-M6 hydrogels were also determined. They all had similar component percentages of ~20 wt% organics, i.e. BC and alginate, ~30 wt% inorganics, and ~50 wt% water (Figure S12), which indicates alginate-Ca2+ played a critical role in the mechanical properties of the hydrogels.
Mechanical properties of mineralized hydrogels. (A) The stress-strain curves of BxA100-x-M6 hydrogels containing different proportions of BC and alginate. (B) The Young's modulus and work of fracture for BxA100-x-M6 hydrogels (n=5). (C) The stress-strain curves of B80A20-My hydrogels mineralized for different days. (D) The fracture stress and fracture strain of B80A20-My hydrogels (n=5). (E) The Young's modulus and work of fracture of B80A20-My hydrogels (n=5). (F) The component proportions of B80A20-My hydrogels (n=5). (G) The fracture energies of B80A20-My hydrogels (n=5). (H) The loading-unloading curves of B100A0-M6, B80A20-M6, and B60A40-M6 hydrogels. (I) Schematic diagram shows the “tortuous fracture path” among BC@CaP nanostructures and the hysteresis of unzipping alginate/Ca2+networks when hydrogel stretched.
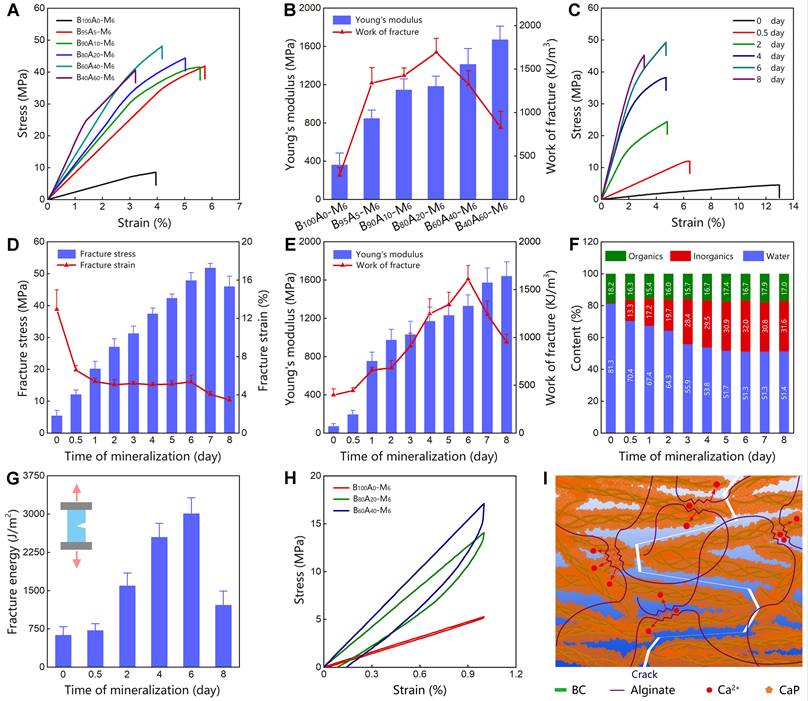
The B80A20-M6 hydrogels that possessed both high stiffness and toughness were further studied. The hydrogels mineralized for different days (B80A20-My) were measured for their mechanical properties. The initial hydrogels of B80A20-M0 showed a relatively large fracture strain (12.9±2.1 %) but very weak fracture stress (5.4±1.7 MPa, Figure 2C-D). When the mineralization time was prolonged, the fracture stresses of the hydrogels were gradually increased and reached a maximum of 51.8±1.4 MPa on the 7th day, and the fracture strains were gradually reduced in compromise (Figure 2C-D). The Young's modulus of the mineralized hydrogels was increased along with the mineralization days and reached the highest value of 1640.0±150.0 MPa on the 8th day (Figure 2E). The work of fracture of the mineralized hydrogels reached the highest value of 1608.4±144.5 KJ/m3 on the 6th day and then decreased due to the reduced fracture strains (Figure 2E). The compositions of hydrogels mineralized for different days were further determined. The weight percentages of CaP quickly increased at the first three days (from 0.5 wt% on 0th day to 28.4 wt% on the 3rd day), and then slowly reached a maximum ratio of 31.6 wt% on the 8th day (Figure 2F and S13). The fracture energies of the mineralized hydrogels were also increased along with the mineralization times and reached the highest value of 3012.7±306.4 J/m2 on the 6th day (Figure 2G). Overall, B80A20-M6 hydrogels possessed the optimized mechanical properties of fracture stress of 47.9±2.4 MPa, Young's modulus of 1329.0±116.1 MPa, work of fracture of 1608.4±144.5 KJ/m3, and fracture energy of 3012.7±306.4 J/m2. A small piece of B80A20-M6 hydrogel (0.2 x 8 x 60 mm) could even lift a 2 kg weight (Figure S14).
To explore the mechanism of high mechanical properties, the stress-strain curves of B100A0-M6, B80A20-M6, and B60A40-M6 hydrogels were tested for one loading-unloading cycle at 1% strain. The noticeable hysteresis was observed in B80A20-M6 and B60A40-M6 hydrogels, while B100A0-M6 hydrogel showed negligible hysteresis (Figure 2H). The dissipated energies of B80A20-M6 and B60A40-M6 hydrogels were 16.3±7.6 KJ/m3 and 23.24±9.2 KJ/m3, respectively, and that of B100A0-M6 hydrogels was only 1.7±0.8 KJ/m3. The data suggest alginate-Ca2+ network efficiently dissipated energies in the hybrid hydrogels. Moreover, BC@CaP and alginate-Ca2+ formed a brick-and-mortar-like structure. When stretched, BC@CaP nanostructures were broken in a “tortuous fracture path” fashion, and the alginate-Ca2+ network was unzipped (Figure 2I). Since BC@CaP nanostructures and alginate-Ca2+ were interwoven, BC@CaP nanostructures might additionally enlarge the dissipating energy by amplifying the unzipping zoom of the alginate-Ca2+ network (Figure 2I). Taken together, the “tortuous fracture path” mechanism and the hysteresis by unzipping ionic networks should make a great contribution to the mechanical properties of the hydrogels.
Characterization of B80A20-My hydrogels
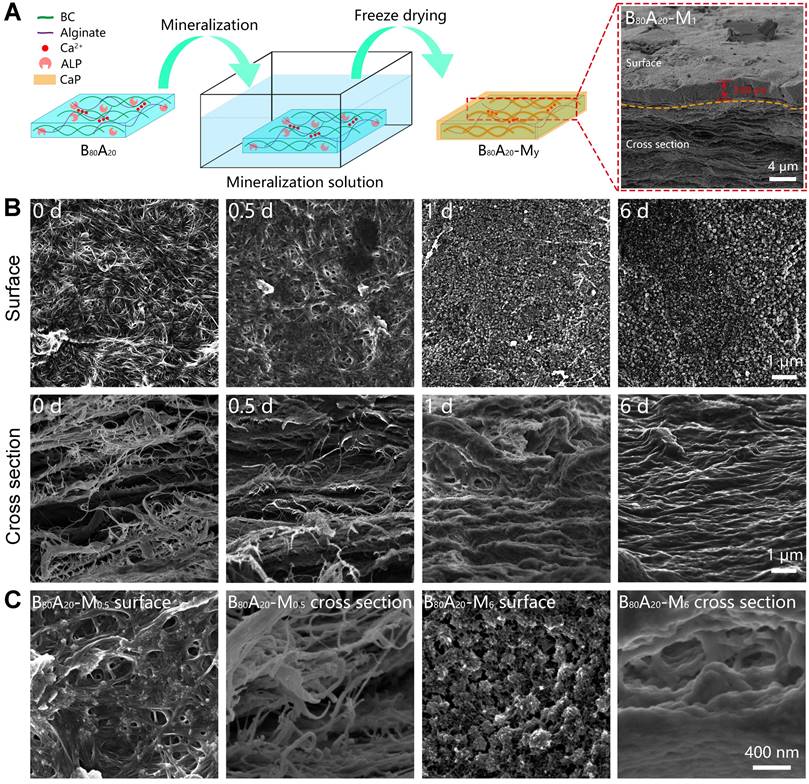
The microstructures of the mineralized hydrogels were observed. B80A20 hydrogels were immersed in the mineralization solution for different days and then were lyophilized for scanning electron microscopy (SEM) observation (Figure 3A). A whole SEM view of B80A20-M1 hydrogel reveals that there were two distinct morphologies exited in the mineralized hydrogel, a compact layer on the surface and a series of lamellar structures below the surface (Figure 3A). The compact surface layer was ~2.8 µm thick (Figure 3A). The energy-dispersive X‐ray spectroscopy (EDX) element mapping determines that the compact surface layer was composed of CaP (denoted by Ca and P elements) but not organic components (denoted by C element, Figure 4A). However, the lamellar structures were indicated to have both organic components and inorganic CaP (Figure 4A). The EDX element mapping from the top view of the hydrogel confirms that the surface of B80A20-M6 hydrogels was majorly composed of CaP (Figure S15). During the mineralization process, CaGP diffused from the buffer solution into hydrogels and was preferentially decomposed at their interfaces, leading to the formation of such a heterostructure. Furthermore, the surface topography of B80A20-M6 hydrogel was observed by using atomic force microscopy. Its representative topography image reveals that the material had a flat surface (Figure S16). The arithmetic mean and the root means square surface roughness of B80A20-M6 hydrogel were only 1.33±0.25 and 2.47±0.32 nm, respectively. The friction coefficient of B80A20-M6 hydrogel was measured to be 0.37 (Figure S17), in which CaP nanoparticles should contribute to the friction coefficient. The microstructures of B80A20 hydrogel after mineralization for different days were shown in Figure 3B. In B80A20 hydrogel, BC nanofibers were observed (Figure 3B). On the surface, BC intertwined to form a compact network, while on the cross-section profile, they formed lamellar sheets with large intervals (Figure 3B). The unique structure was probably due to the hydrogel prepared via a blade casting method, and BC nanofibers tended to be arranged in the plane when the film was dried. After mineralization for 0.5 days, the hydrogel was deposited with CaP components on the surface and inside of the gel matrix (Figure 3B). The zoom-in images reveal that clusters of CaP nanoparticles were formed on the surface of hydrogel, while in the hydrogels (cross-section view) CaP was deposited on the surface of BC nanofibers to form a continuous coating (Figure 3C). After mineralization for 1 day, the surface of the hydrogel was covered by the high density of CaP nanoparticles, while the cross-section of the hydrogel became denser, and BC nanofibers were conjoined by CaP (Figure 3B). In this case, the CaP structure was quite different from that observed in the polymer matrix [33]. The polymer hydrogels could not offer nucleating points for CaP. As a result, CaP nanoparticles were formed at the intervals of a polymer network [33]. In comparison, BC nanofibers provided the sites for CaP nucleation and deposition due to the negative charges on the surface (Figure 1E and S1). As a result, CaP nanoparticles were constantly deposited on the surface of BC nanofibers, and finally, a high density of BC@CaP nanostructures was formed on the 6th day (Figure 3B). The zoom-in images reveal that flower-like CaP nanoparticles were connected with internal bridges on the surface of B80A20-M6 hydrogel, and in the cross-section, BC@CaP nanofibers had fused to form a compact structure (Figure 3C).
Structural and compositional properties of mineralized hydrogels. (A) Schematic illustration of the mineralization process of B80A20 hydrogel. Inset is the SEM image of B80A20-M1 hydrogel. (B) SEM images show the surface and cross-section profiles of B80A20 hydrogels mineralized for different days. (C) The amplified SEM images of B80A20-M0.5 and B80A20-M6 hydrogels.
Characterization of B80A20-M6 hydrogel. (A) EDX element mapping of B80A20-M1 hydrogel (C stands for carbon element, P for phosphorus, and Ca for calcium). (B) FTIR spectra of B80A20-M6 hydrogel, BC, alginate, and CaP. (C) The X-ray diffraction spectra of B80A20 hydrogel, B80A20-M6 hydrogel, and CaP. CaP in b and c was produced via ALP-catalyzed deposition of CaGP in TEA buffer. (D and E) The thermogravimetric analysis and derivative thermogravimetric curves of B80A20, B80A20-M6 hydrogels, BC, and alginate.
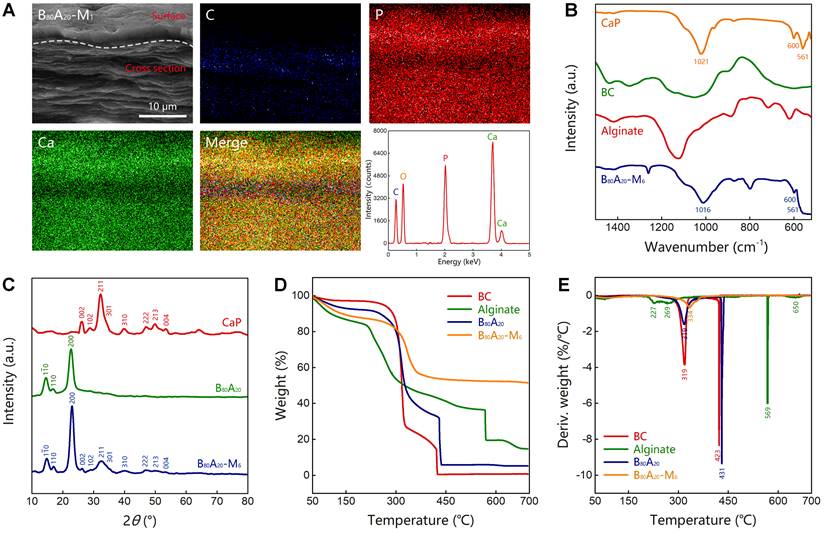
The Fourier transform infrared spectroscopy (FTIR) analysis of B80A20-M6 hydrogel and the individual components including CaP from ALP-catalyzed hydrolysis of CaGP, BC nanofibers, alginate was conducted (Figure 4B and S18). B80A20-M6 hydrogel possessed the characteristic peaks of all components. Especially, CaP power represented one sharp peak at 1021 cm-1 for asymmetric PO43- stretching vibrations, and two peaks at 600 and 561 cm-1 for asymmetric PO43- deformation vibrations, which indicates the mineralized CaP was made of hydroxyapatite [46]. B80A20-M6 hydrogel contained the typical peaks of hydroxyapatite, suggesting CaP deposited in the hydrogel should be hydroxyapatite. The X-ray diffraction spectra of CaP, B80A20 hydrogel, and B80A20-M6 one were also measured. The CaP powder showed multiple diffraction peaks at 26.1°, 28.5°, 32.3°, 33.3°, 34.1°, 39.7°, 46.9°, 48.2°, 50.0°, and 53.4° (Figure 4C), which were corresponding to the typical lattice plane of hydroxyapatite (002) m, (102) m, (211) m, (112) m, (301) m, (310) m, (222) m, (312) m, (213) m and (004) m [47]. B80A20 hydrogel showed the typical diffraction peaks of BC at 14.7°, 16.7°, and 22.7° (Figure 4C) [48]. B80A20-M6 hydrogel contained both the typical diffraction peaks of BC and hydroxyapatite (Figure 4C). Taken together, we confirmed that CaP in the hydrogel was hydroxyapatite. The dried B80A20 and B80A20-M6 hydrogels were further assessed by thermogravimetric analysis. The thermogravimetric curves show that the dried B80A20 hydrogel was majorly pyrolyzed at a temperature ranging from 250 to 435 °C, and the dried B80A20-M6 hydrogel did between 280 to 365 °C (Figure 4D). The dried B80A20 hydrogel was completely pyrolyzed due to its organic composition, while the dried B80A20-M6 hydrogel was pyrolyzed only 47.6 wt% (Figure 4D). The non-pyrolyzed component should be CaP (52.4 wt%, Figure 4D). The derivative thermogravimetric curves reveal that the maximum weight loss rate for dried B80A20 hydrogel was 430.6 °C and that for dried B80A20-M6 hydrogel was 334.1 °C (Figure 4E).
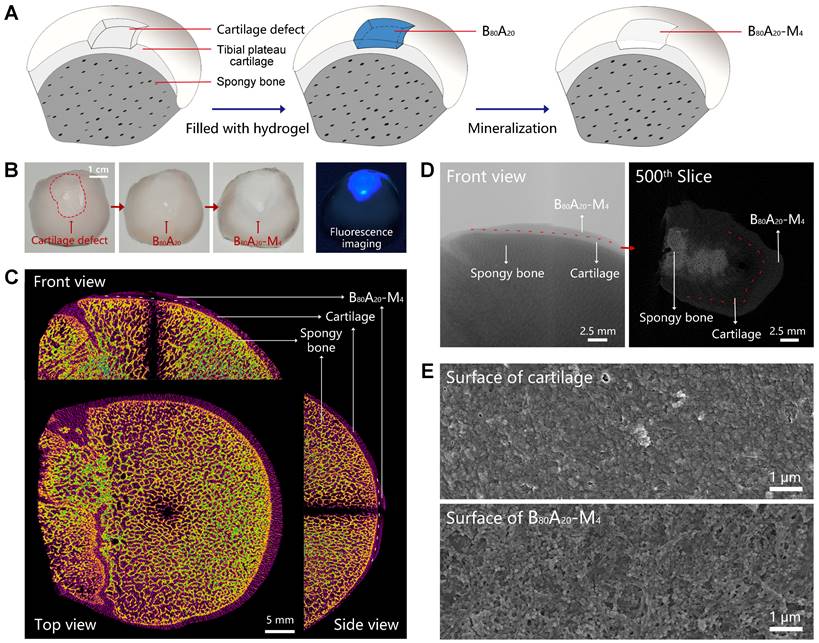
In situ mineralization in the cartilage defect
Cartilage defects can be divided into three types includes partial-thickness, full-thickness, and osteochondral defects. In the full-thickness and osteochondral defects, subchondral bone also needs to be repaired except for cartilage. Here, we employed B80A20-M4 hydrogel as a replacement material to fix the subchondral bone damage. The assay was carried out via in situ mineralization of CaP in the hydrogel in a man-made articular cartilage defect model ex vivo (Figure 5A). ALP is an enzyme that wildly exists in body tissue [49]. In bone tissue, ALPs regulate bone mineralization via hydrolyze pyrophosphate and also supply inorganic phosphate for the synthesis of hydroxyapatite via hydrolyze pyrophosphate and organic phosphomonoesters [50]. Therefore, ALP-catalyzed CaP mineralization in B80A20-M4 hydrogel might mimic the natural synthesis of CaP in bone tissues. Moreover, a partial-thickness cartilage defect was created instead of full-thickness or osteochondral defects, and thus the mineralized hydrogel directly adhered to the cartilage tissue in the defect, which facilitated the characterization of the mineralized hydrogel and its binding to the natural cartilage. The defect was filled with B80A20 hydrogel and then fed with the mineralization solution (Figure 5A). After repair, the defect region was deposited with CaP in the gel matrix, where the mineralized hydrogel perfectly integrated with the natural cartilage tissues at the boundary (Figure 5B). A fluorescent agent of calcofluor white stain was embedded in B80A20 hydrogel to indicate the location of repaired area (Figure 5B). Furtherly, the bone was imaged by micro-computerized tomography (micro-CT). The CT image reveals that the repaired region had a similar microstructure and density compared with the natural bone tissue (Figure 5C). The section CT further reveals that B80A20-M4 hydrogel was tightly incorporated with natural cartilage tissue (Figure 5D). The SEM images reveal that the in situ formed B80A20-M4 hydrogel was composed of BC nanofibers and CaP nanoparticles, while the natural cartilage showed a dense structure that was probably the extracellular matrix (Figure 5E and S19). The assay demonstrates that the hydrogel could be used for subchondral bone repair in the cartilage defect.
In-situ mineralization in the cartilage defect. (A) The scheme shows the mineralization process in a man-made articular cartilage defect ex vivo. (B) Photographs of the cartilage defect before and after being filled with B80A20-M4 hydrogel (labeled with calcofluor white stain). (C) The orthographic views of the cartilage defect filled with B80A20-M4 hydrogel (front, top, and side view). (D) The representative micro-CT images of the cartilage defect filled with B80A20-M4 hydrogel (front view and the 500th slice from the top down). The red line represents the boundary between B80A20-M4 hydrogel and natural cartilage. (E) SEM images of the surface of natural cartilage and B80A20-M4 hydrogel in the defect.
Conclusion
In summary, we have prepared a hydrogel with ultrahigh strength, stiffness, and toughness. In the hydrogel, BC and alginate-Ca2+ formed a crosslinked double network, and then ALPs induced CaP deposition in the organic matrix. BC nanofibers were used as the main component of the gel matrix instead of molecular polymers, which themselves were strong enough to reinforce the hydrogel, and also templated the deposition of CaP to form BC@CaP nanostructures. Then, BC@CaP and alginate-Ca2+ formed a brick-and-mortar-like structure. When stretched, the “tortuous fracture path” breaking among BC@CaP nanostructures and the hysteresis by unzipping the alginate-Ca2+ network worked synergistically, resulting in great improvement over the mechanical properties of the hydrogels. The best hydrogel possessed ultrahigh fracture stress of 48 MPa, Young's modulus of 1329 MPa, and fracture energy of 3013 J/m2. All three properties are superior to the parameters of most of the reported synthetic hydrogels with high mechanical behaviors. Moreover, the successful case for the ex vivo repair of subchondral bone defect suggests that this hydrogel has great promise in bone tissue engineering.
Methods
Materials
BC membrane was obtained from Hainan Yeguo Foods Co., Ltd (Hainan, China). Sodium alginate, TEA, glutaraldehyde (aqueous solution, 50 wt %), sodium hydroxide, and calcium chloride were purchased from Macklin Biochemical Co., Ltd (Shanghai, China). ALPs extracted from the calf intestine were obtained from AppliChem GmbH (Darmstadt, Germany). CaGP, 4-nitrophenyl phosphate disodium salt hexahydrate, and p-nitrophenol were bought from Alfa Aesar Chemicals Co., Ltd (Shanghai, China). Hydrochloric acid and dimethyl sulfoxide were obtained from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China).
BC slurry preparation
BC membrane was smashed into small pieces using a blender (MQ5025, Braun, Germany), and then treated by a high-pressure homogenizer (1000 bar, AH-PILOT 2018, ATS Engineering Limited, China) three times.
Synthesis of PGL
PGL was synthesized based on a reported method [51]. 20 mL glutaraldehyde aqueous solution (50 wt %) was added in a mix of 15 mL deionized (DI) water and 5 mL dimethyl sulfoxide. The pH value of the solution was adjusted to 10.5 by adding sodium hydroxide (1 M). After stirring for 30 min, the solution was neutralized with hydrochloric acid (1 M), and then diluted ten times with DI water. The PGL solution was stored at 4 °C for use.
Preparation of ALP/PGL nanoparticles
The ALP/PGL nanoparticles were prepared via a reported method [52]. 0.1 mL of freshly-prepared ALPs (10 mg/mL) in DI water was mixed with different amounts of PGL solution (23.8 mg/mL, 0, 4.2, 12.6, 25.2, 50.4, and 105 μL) using a vortex mixer. After standing for 5 min, ALP/PGL nanoparticles were obtained and stored at 4 °C for use.
Enzyme activity of ALP/PGL nanoparticles
The enzyme activities of ALP/PGL nanoparticles were determined via a standard method. Before the assay, the standard curve of p-nitrophenol (in 0.2 M TEA buffer, pH = 9.8) was determined by measuring its absorption (405 nm) at different concentrations using an ultraviolet spectrophotometer (Cary 60, Agilent Technologies, USA). To determine the enzyme activity, 1 mL of ALP/PGL suspension (5 µg/mL for ALPs, in 0.2 M TEA buffer, pH = 9.8) was added in a quartz cuvette, which was held with 1 mL of 4-nitrophenyl phosphate disodium salt hexahydrate solution (0.1 M, in 0.2 M TEA buffer, pH = 9.8). The absorbance of the solution at 405 nm was recorded by the ultraviolet spectrophotometer over time. Finally, the specific activity (U/mg) of ALP/PGL was calculated according to the definition of active unit (the amount of enzyme required to convert 1 μM of the substrate within 1 min is defined as an international unit of enzyme activity).
Preparation of Bx/A100-x hydrogels
In a typical preparation of B80A20 hydrogel, 16 mL of BC aqueous suspension (0.4 wt%) and 4 mL of sodium alginate aqueous solution (0.4 wt%) were mixed, and then 312.6 μL of ALP/PGL suspension (10.6 mg/mL) was added. After homogenously mixing with a vortex mixer, the bubbles trapped in the solution were removed by ultrasonic treatment in an ice bath. After that, the solution was poured into a homemade rectangular mold (4 x 8 x 1 cm) made of polymethyl methacrylate plate. The sample was dried in a fume and then immersed in calcium chloride solution (5 mg/mL) for 30 min. BxA100-x hydrogels with different weight ratios of BC and sodium alginate were prepared in the same way.
Mineralization of Bx/A100-x hydrogels
In a typical case, B80A20 hydrogel was immersed in 80 mL of mineralization solution at room temperature. The mineralization solution was replaced every day. After incubation for 6 days, B80A20-M6 hydrogel was obtained. The hydrogel was washed with DI water three times and then stored in DI water at 4 °C. The mineralization solution was prepared by dissolving 5 g of CaGP into 1 L of 0.2 M TEA buffer, and the pH value was adjusted to 9.8 by adding 5 M hydrochloric acid. BxA100-x-My hydrogels were prepared in the same way.
Characterization
The SEM images and EDX mapping of hydrogels were obtained by using a scanning electron microscope (S-4800, Hitachi, Japan) coupled with an energy-dispersive X‐ray spectroscope. The FTIR spectra were collected by using a Fourier transform infrared spectroscope (Nicolet iS50, Thermo Fisher Scientific, America). The thermogravimetric analysis was conducted by using a thermogravimetric analyzer (TGA4000, PerkinElmer, America). The X-ray diffraction spectra were determined by using an X-ray diffractometer (Smartlab SE, Rigaku, Japan) within a 2θ range from 5 to 140°. The crystallinity Index of BC in hydrogels was calculated according to the reported method.35 The hydrodynamic size and zeta potential of materials were measured by using a dynamic light scattering (Zetasizer Nano ZS90, Malvern, UK).
Composition of BxA100-x-My hydrogel
The composition of BxA100-x-My hydrogel was determined by a gravimetric method. The dry weight of BxA100-x hydrogel (m1), and the dry weight (m2) and wet weights (m3) of the mineralized BxA100-x-My hydrogel were measured. The percentages of the contents were calculated as follows:
(1)
(2)
(3)
Mechanical properties of hydrogels
The mechanical properties of BxA100-x-My hydrogel were evaluated by using an electronic tensile testing machine (HY-0580, Shanghai Hengyi Testing Instruments Co., Ltd, China). The hydrogels in 0.05-0.3 mm thickness were cut into the testing samples with a strip shape of 5 mm width x 25 mm length by using a surgical blade. The samples were fixed by using clamps with an initial distance of 16 mm and then were stretched at a speed of 20 mm/min to obtain the stress-strain curves. The strain (%) was regarded as the length change related to the initial length of the sample. The stress (MPa) was calculated by dividing the force by the initial cross-sectional area of the samples. The Young's modulus (MPa) was determined from the slope of the initial linear region of the stress-strain curves. The work of fracture (KJ/m3) was determined by the area below the stress-strain curves. The dissipated energy (KJ/m3) was estimated by the area between the loading-unloading curves.
The fracture energy (J/m2) of BxA100-x-My hydrogel was estimated with a standard method introduced by Rivlin and Thomas [53]. Briefly, two rectangular samples (8 mm width x 25 mm length) separated from the same hydrogel were used for measurement. One was intact, and the other was cut with a 4 mm notch using a surgical blade. The two samples were stretched at a speed of 20 mm/min to obtain the stress-strain curves. The corresponding fracture energy (Γ) of the unnotched samples was calculated as follows:
(4)
Γ: The fracture energy (J/m2)
εc: The fracture strain of the notched samples (%)
σ: The stress of the unnotched samples (MPa)
lc: The initial length of the samples (mm)
Surface roughness analysis
The surface topography of B80A20-M6 hydrogel was observed by using atomic force microscopy (Dimension Icon, BRUKER, America) and the arithmetic mean and the root means square surface roughness were analyzed by a professional analysis software NanoScope Analysis 3.0.
Friction testing
The friction coefficient of B80A20-M6 hydrogel was analyzed by using high-speed reciprocating friction and wear tester (MDW-02G, Jinan Yihua Tribology Testing Technology Co., Ltd, China) operated in reciprocating sliding mode. To simulate the frictional environment in vivo, the entire testing process was performed in PBS. In all of the tests, the sliding speed was 0.5 mm/s, the length of the wear track was 10 mm, and the normal load was 10 N, resulting in an average contact pressure of 0.1 MPa. In addition, the glass disk was ultrasonically cleaned after each test to ensure a fresh surface. The friction coefficient was calculated as follows:
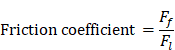
 : The friction force (N)
: The friction force (N)
 : The normal load (N).
: The normal load (N).
Ex-vivo repair of subchondral bone in the cartilage defect
The Ex-vivo model of cartilage defect was established on the tibial plateau cartilage of pork bone, which was bought from the supermarket. The defect was made by excising the cartilage section using a surgical blade. The size of the defect was approximately 1 cm width × 1.5 cm length × 0.5 mm depth. The mixture of BC, sodium alginate, and ALP/PGL for B80A20 hydrogel was prepared as above, and 50 μL calcofluor white stain was added. It was filled into the defect and then infiltrated by 2 mL of 5 mg/mL calcium chloride solution for 30 min. After that, the defect covered with hydrogel was flushed with DI water three times. Finally, 2 mL of mineralization solution was added at the defect location. The mineralization solution was changed twice a day, and the sample was mineralized for 4 days.
Micro-CT analysis
The micro-CT analysis of bone was performed by using an x-ray microtomography (SKYSCAN 1272, BRUKER, Germany). The samples were scanned at a source voltage of 60.0 kV and a source current of 100 μA for 70 min with a camera pixel size resolution of 22 μm. The region of interest was located from 1.232 mm (the 56th slice) to 16.874 mm (the 767th slice) where the bottom of the tibial plateau cartilage was defined as 0 mm. 3D reconstruction images were produced with CTVOL software (version 1.1.10, Bruker micro-CT).
Abbreviations
BC: bacterial cellulose; CaP: calcium phosphate; TEA: triethanolamine; ALPs: alkaline phosphatases; CaGP: calcium glycerophosphate; PGL: poly-glutaraldehyde; DI: deionized; SEM: scanning electron microscopy; FTIR: Fourier transform infrared; EDX: energy-dispersive X‐ray spectroscope; micro-CT: micro-computerized tomography.
Supplementary Material
Supplementary figures.
Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (31871010; 32071383). The authors acknowledge East China Normal University (ECNU) Electron Microscopy Center for the nanoparticle characterization and ECNU Multifunctional Platform for Innovation (011) for the animal experiments.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Vermonden T, Censi R, Hennink WE. Hydrogels for protein delivery. Chem Rev. 2012;112:2853-2888
2. Guo J, Kim YS, Xie V, Smith BT, Watson E, Lam J. et al. Modular, tissue-specific, and biodegradable hydrogel cross-linkers for tissue engineering. Sci Adv. 2019;5:eaaw7396
3. Yuk H, Lu B, Zhao X. Hydrogel bioelectronics. Chem Soc Rev. 2019;48:1642-1667
4. Lee JB, Kim DH, Yoon JK, Park DB, Kim HS, Shin YM. et al. Microchannel network hydrogel induced ischemic blood perfusion connection. Nat Commun. 2020;11:615
5. Burdick JA, Murphy WL. Moving from static to dynamic complexity in hydrogel design. Nat Commun. 2012;3:1269
6. Hua M, Wu S, Ma Y, Zhao Y, Chen Z, Frenkel I. et al. Strong tough hydrogels via the synergy of freeze-casting and salting out. Nature. 2021;590:594-599
7. Lei Y, Schaffer DV. A fully defined and scalable 3D culture system for human pluripotent stem cell expansion and differentiation. Proc Natl Acad Sci USA. 2013;110:5039-5048
8. Wang H, Heilshorn SC. Adaptable hydrogel networks with reversible linkages for tissue engineering. Adv Mater. 2015;27:3717-3736
9. Lin S, Liu J, Liu X, Zhao X. Muscle-like fatigue-resistant hydrogels by mechanical training. Proc Natl Acad Sci USA. 2019 116, 10244-10249
10. Li J, Suo Z, Vlassak JJ. Stiff, strong, and tough hydrogels with good chemical stability. J Mater Chem B. 2014;2:6708-6713
11. Mehrali M, Thakur A, Pennisi CP, Talebian S, Arpanaei A, Nikkhah M. et al. Nanoreinforced hydrogels for tissue engineering: biomaterials that are compatible with load-bearing and electroactive tissues. Adv Mater. 2017;29:1603612
12. Little CJ, Bawolin NK, Chen X. Mechanical properties of natural cartilage and tissue-engineered constructs. Tissue Eng Part B Rev. 2011;17:213-227
13. Constantinos NM, John PP. In vivo human tendon mechanical properties. J Physiol. 1999;521:307-313
14. Gong J, Katsuyama Y, Kurokawa T, Osada Y. Double-network hydrogels with extremely high mechanical strength. Adv Mater. 2003;15:1155-1158
15. Hu X, Vatankhah-Varnoosfaderani M, Zhou J, Li Q, Sheiko SS. Weak hydrogen bonding enables hard, strong, tough, and elastic hydrogels. Adv Mater. 2015;27:6899-6905
16. Means AK, Shrode CS, Whitney LV, Ehrhardt DA, Grunlan MA. Double network hydrogels that mimic the modulus, strength, and lubricity of cartilage. Biomacromolecules. 2019;20:2034-2042
17. Hu Y, Du Z, Deng X, Wang T, Yang Z, Zhou W. et al. Dual physically cross-linked hydrogels with high stretchability, toughness, and good self-recoverability. Macromolecules. 2016;49:5660-5668
18. Sun J, Zhao X, Illeperuma WR, Chaudhuri O, Oh KH, Mooney DJ. et al. Highly stretchable and tough hydrogels. Nature. 2012;489:133-136
19. Wang Y, Zhang X, Song Y, Zhao Y, Chen L, Su F. et al. Ultrastiff and tough supramolecular hydrogels with a dense and robust hydrogen bond network. Chem Mater. 2019;31:1430-1440
20. Li J, Illeperuma WRK, Suo Z, Vlassak JJ. Hybrid hydrogels with extremely high stiffness and toughness. ACS Macro Lett. 2014;3:520-523
21. Lin P, Ma S, Wang X, Zhou F. Molecularly engineered dual-crosslinked hydrogel with ultrahigh mechanical strength, toughness, and good self-recovery. Adv Mater. 2015;27:2054-2059
22. Zhao D, Huang J, Zhong Y, Li K, Zhang L, Cai J. High-strength and high-toughness double-cross-linked cellulose hydrogels: a new strategy using sequential chemical and physical cross-linking. Adv Funct Mater. 2016;26:6279-6287
23. Yang X, Abe K, Biswas SK, Yano H. Extremely stiff and strong nanocomposite hydrogels with stretchable cellulose nanofiber/poly(vinyl alcohol) networks. Cellulose. 2018;25:6571-6580
24. Xu L, Wang C, Cui Y, Li A, Qiao Y, Qiu D. Conjoined-network rendered stiff and tough hydrogels from biogenic molecules. Sci Adv. 2019;5:eaau3442
25. Zhang X, Wang Y, Sun S, Hou L, Wu P, Wu Z. et al. A tough and stiff hydrogel with tunable water content and mechanical properties based on the synergistic effect of hydrogen bonding and hydrophobic interaction. Macromolecules. 2018;51:8136-8146
26. Lin S, Cao C, Wang Q, Gonzalez M, Dolbow JE, Zhao X. Design of stiff, tough and stretchy hydrogel composites via nanoscale hybrid crosslinking and macroscale fiber reinforcement. Soft Matter. 2014;10:7519-7527
27. King DR, Sun T, Huang Y, Kurokawa T, Nonoyama T, Crosby AJ. et al. Extremely tough composites from fabric reinforced polyampholyte hydrogels. Mater Horizons. 2015;2:584-591
28. Cui K, Ye Y, Sun T, Chen L, Li X, Kurokawa T. et al. Effect of structure heterogeneity on mechanical performance of physical polyampholytes hydrogels. Macromolecules. 2019;52:7369-7378
29. Yao S, Jin B, Liu Z, Shao C, Zhao R, Wang X. et al. Biomineralization: from material tactics to biological strategy. Adv Mater. 2017;29:1605903
30. Xu B, Zheng P, Gao F, Wang W, Zhang H, Zhang X. et al. A mineralized high strength and tough hydrogel for skull bone regeneration. Adv Funct Mater. 2017;27:1604327
31. Gu Z, Chen L, Xu Y, Liu Y, Zhao Z, Zhao C. et al. General strategy to fabricate highly filled microcomposite hydrogels with high mechanical strength and stiffness. ACS Appl Mater Interfaces. 2018;10:4161-4167
32. Rauner N, Meuris M, Dech S, Godde J, Tiller JC. Urease-induced calcification of segmented polymer hydrogels-a step towards artificial biomineralization. Acta Biomater. 2014;10:3942-3951
33. Rauner N, Meuris M, Zoric M, Tiller JC. Enzymatic mineralization generates ultrastiff and tough hydrogels with tunable mechanics. Nature. 2017;543:407-410
34. Wang Q, Yao Q, Liu J, Sun J, Zhu Q, Chen H. Processing nanocellulose to bulk materials: a review. Cellulose. 2019;26:7585-7617
35. Hsieh YC, Yano H, Nogi M, Eichhorn SJ. An estimation of the Young's modulus of bacterial cellulose filaments. Cellulose. 2008;15:507-513
36. Yao J, Fang W, Guo J, Jiao D, Chen S, Ifuku S. et al. Highly mineralized biomimetic polysaccharide nanofiber materials using enzymatic mineralization. Biomacromolecules. 2020;21:2176-2186
37. Liu J, Tan C, Yu Z, Li N, Abell C, Scherman OA. Tough supramolecular polymer networks with extreme stretchability and fast room-temperature self-healing. Adv Mater. 2017;29:1605325
38. Zhong M, Liu Y, Xie X. Self-healable, super tough graphene oxide-poly(acrylic acid) nanocomposite hydrogels facilitated by dual cross-linking effects through dynamic ionic interactions. J Mater Chem B. 2015;3:4001-4008
39. Xia S, Song S, Ren X, Gao G. Highly tough, anti-fatigue and rapidly self-recoverable hydrogels reinforced with core-shell inorganic-organic hybrid latex particles. Soft Matter. 2017;13:6059-6067
40. Pan C, Liu L, Chen Q, Zhang Q, Guo G. Tough, stretchable, compressive novel polymer/graphene oxide nanocomposite hydrogels with excellent self-healing performance. ACS Appl Mater Interfaces. 2017;9:38052-38061
41. Cui Y, Tan M, Zhu A, Guo M. Non-covalent interaction cooperatively induced stretchy, tough and stimuli-responsive polyurethane-urea supramolecular (PUUS) hydrogels. J Mater Chem B. 2015;3:2834-2841
42. Yang N, Yang H, Shao Z, Guo M. Ultrastrong and tough supramolecular hydrogels from multiurea linkage segmented copolymers with tractable processablity and recyclability. Macromol Rapid Commun. 2017;38:1700275
43. Ye D, Chang C, Zhang L. High-strength and tough cellulose hydrogels chemically dual cross-linked by using low- and high-molecular-weight cross-linkers. Biomacromolecules. 2019;20:1989-1995
44. Qin Z, Niu R, Tang C, Xia J, Ji F, Dong D. et al. A dual-crosslinked strategy to construct physical hydrogels with high strength, toughness, good mechanical recoverability, and shape-memory ability. Macromol Mater Eng. 2018 303
45. Zhang T, Zuo T, Hu D, Chang C. Dual physically cross-linked nanocomposite hydrogels reinforced by tunicate cellulose nanocrystals with high toughness and good self-recoverability. ACS Appl Mater Interfaces. 2017;9:24230-24237
46. Mustafov SD, Sen F, Seydibeyoglu MO. Preparation and characterization of diatomite and hydroxyapatite reinforced porous polyurethane foam biocomposites. Sci Rep. 2020;10:13308
47. Feng P, Niu M, Gao C, Peng S, Shuai C. A novel two-step sintering for nano-hydroxyapatite scaffolds for bone tissue engineering. Sci Rep. 2014;4:5599
48. Rollini M, Musatti A, Cavicchioli D, Bussini D, Farris S, Rovera C. et al. From cheese whey permeate to Sakacin-A/bacterial cellulose nanocrystal conjugates for antimicrobial food packaging applications: a circular economy case study. Sci Rep. 2020;10:21358
49. Spoerke ED, Anthony SG, Stupp SI. Enzyme directed templating of artificial bone mineral. Adv Mater. 2009;21:425-430
50. Vimalraj S. Alkaline phosphatase: structure, expression and its function in bone mineralization. Gene. 2020;754:144855
51. Tanriseven A, Olcer Z. A novel method for the immobilization of glucoamylase onto polyglutaraldehyde-activated gelatin. Biochem Eng J. 2008;39:430-434
52. Sabrina Q, Ratri CR, Hardiansyah A, Lestariningsih T, Subhan A, Rifai A. et al. Preparation and characterization of nanofibrous cellulose as solid polymer electrolyte for lithium-ion battery applications. RSC Adv. 2021;11:22929-22936
53. Rivlin RS, Thomas AG. Rupture of rubber. I. Characteristic energy for tearing. J Polym Sci Polym Phys Ed. 1953;10:291-318
Author contact
![]() Corresponding authors: Q. Z. (E-mail: qzhangecnu.edu.cn); G. W. (E-mail: wangguodongedu.cn); W. L. (E-mail: li_wei_sh com).
Corresponding authors: Q. Z. (E-mail: qzhangecnu.edu.cn); G. W. (E-mail: wangguodongedu.cn); W. L. (E-mail: li_wei_sh com).